

Abstract
Atherosclerosis is an inflammatory disease characterized by plaques that can cause sudden myocardial infarction upon rupture. Such rupture-prone plaques have thin fibrous caps due to collagenase degradation, and a noninvasive diagnostic tool and targeted therapy that can identify and treat vulnerable plaques and may inhibit the onset of acute cardiac events. Toward this goal, monocyte-binding, collagenase-inhibiting, and gadolinium-modified peptide amphiphile micelles (MCG PAMs) are developed. Monocyte chemoattractant protein-1 (MCP-1) binds to C-C chemokine receptor-2 expressed on pathological cell types present within plaques. Through the peptide binding motif of MCP-1, MCG PAMs bind to monocytes and vascular smooth muscle cells in vitro. Moreover, using magnetic resonance imaging, MCG PAMs show enhanced targeting and successful detection of plaques in diseased mice in vivo and act as contrast agents for molecular imaging. Through the collagenase-cleaving peptide sequence of collagen [VPMS-MRGG], MCG PAMs can compete for collagenases that degrade the fibrous cap of plaques, providing therapy. MCG PAM-treated mice show increased fibrous cap thickness by 61% and 113% histologically compared to nontargeting micelle- or PBS-treated mice (p = 0.0075 and 0.001, respectively). Overall, this novel multimodal nanoparticle offers new theranostic opportunities for noninvasive diagnosis and treatment of atherosclerotic plaques.
Keywords: atherosclerosis, collagen, magnetic resonance imaging, theranostic nanoparticle, vulnerable plaque
1. Introduction
Ischemic heart disease and stroke have been the leading causes of death worldwide for the past 70 years.[1] Atherosclerosis, an asymptomatic inflammatory disease that results in plaque formation in the vasculature, has been marked as a major precursor to myocardial infarction and stroke.[2] Advanced atherosclerotic plaques can cause luminal narrowing that exacerbates normal blood flow. However, increasing evidence suggests that luminal stenosis alone is not a strong risk factor for acute cardiac events.[3,4] Instead, “vulnerable” plaques with a thin and unstable outer fibrous cap are predisposed to rupture and highly correlated with cardiac morbidity and mortality.[5,6]
Atherosclerotic plaques develop with endothelial cell activation and endothelial dysfunction which leads to lipoprotein accumulation in the subendothelium of lesion-prone areas in the vasculature.[6,7] Upon activation, endothelial cells release inflammatory cytokines, such as monocyte chemoattractant protein-1 (MCP-1).[8,9] The release of MCP-1 stimulates the recruitment of C-C chemokine receptor-2 (CCR2)-expressing monocytes that bind to MCP-1. Monocytes, in atherosclerotic lesions, differentiate into macrophages and phagocytose lipoproteins, eventually becoming foam cells.[10] Monocyte recruitment and excess numbers of foam cells further trigger the migration and proliferation of pathogenic vascular smooth muscle cells (VSMCs).[11–14] Proliferating VSMCs, which also express CCR2, synthesize extracellular matrix (ECM) components such as fibrin and collagen to form the protective fibrous cap of atherosclerotic plaques.[2,15,16]
Eventually however, prolonged inflammation induces the release of matrix metalloproteinases (MMPs) from inflammatory cells and apoptotic VSMCs.[17] Interstitial collagenases, such as MMP-1, MMP-8, and MMP-13, break down collagen which lead to fibrous cap thinning and plaque destabilization.[18] Specifically, MMP-1, or collagenase-1, expressed by macrophages and VSMCs, is shown to cleave type I and type III collagens, which are two major structural components of fibrous caps characteristic of stable plaques.[19] The cleavage site of collagens, identified by the peptide sequence [VPMS-MRGG], or Col-1 peptide, is recognized by MMP-1 and undergoes rapid degradation.[20] Without adequate tissue repair or inhibition of degradation, atherosclerotic plaques with thin fibrous caps that are less than 65 μm in thickness pose risks of rupture and thrombosis.[21] Thus, a potential therapeutic strategy to preserve structural integrity of plaques may be through competitive inhibition of MMP collagenases using the collagen cleavage recognition site sequence, Col-1 peptide.
To that end, we have incorporated Col-1 peptides into peptide amphiphile micelle (PAM) nanoparticles functionalized with the CCR2-binding motif of MCP-1 ([YNFTNRKISVQRLASYRRITSSK]) to target and treat unstable atherosclerotic plaques. In order to provide clinical relevance and adaptability, we also incorporated gadolinium (Gd) to allow for simultaneous molecular magnetic resonance (MR) imaging of plaques. We chose MR imaging specifically as it is a noninvasive imaging modality capable of producing high-resolution images, without the use of ionizing radiation that are characteristic to angiography, computed tomography (CT), and positron emission tomography (PET).[22–27] Moreover, although near-infrared fluorescence (NIRF) imaging is popular for small animal imaging, NIRF imaging alone is hindered by poor tissue penetration and high tissue autofluorescence, making it difficult for transition into the clinic.[28] Gd is an FDA-approved contrast agent that produces contrast-enhanced MR images with high signal-to-noise ratio (SNR), which makes it favorable for use in nanoparticle platforms.[29] Although as free ions, Gd3+ is a toxic heavy metal that has been shown to cause detrimental inhibitory effects on important voltage-gated calcium channels, we and others have chelated Gd3+ into diethylenetriaminepentaacetic acid (DTPA) and reported its utility and safety as molecular contrast agents upon incorporation into nanoparticles.[30–32]
Together, herein, we demonstrate that monocyte-binding, collagenase-inhibiting, and gadolinium-modified PAMs (MCG PAMs) are biocompatible nanoparticles that bind to VSMCs and monocytes and are molecular imaging tools that are able to detect plaques in vivo. Furthermore, MCG PAMs elicit therapeutic potency by increasing collagen and fibrous cap thickness which stabilizes atherosclerotic plaques from rupture. The potential of PAMs as a theranostic platform to image plaque stability and location in real-time, and competitively inhibit collagenase as a therapeutic strategy for atherosclerosis are discussed.
2. Results and Discussion
2.1. Particle Synthesis and Characterization
Nanoparticles have become favorable platforms for molecular imaging and targeted therapy. Previously, we have developed PAMs as tunable nanoparticles for targeted drug carriers and diagnostic agents.[30,33–35] PAMs are comprised of peptide amphiphile (PA) molecules, consisting of a hydrophobic lipid tail attached to a hydrophilic headgroup, that self-assemble into micelles. Multimodal theranostic MCG PAMs for targeted therapy and diagnostics of atherosclerotic plaques were developed using MCP-1 peptides for CCR2 targeting, therapeutic Col-1 peptides for competitive inhibition of collagenases, and Gd contrast agents for MR imaging.
Specifically, MCP-1, scrambled MCP-1, and Col-1 peptides were synthesized using standard Fmoc solid-phase synthesis methods and purified using high performance liquid chromatography (HPLC). Peptides were confirmed using matrix-assisted laser desorption/ionization time-of-flight mass spectrometer (MALDI-TOF, Figure S1, Supporting Information). An additional cysteine residue was added to the N-terminus of each peptide and conjugated to the DSPE-PEG(2000)-maleimide lipid tail via thioether linkage. To facilitate MR imaging contrast abilities, DSPE-PEG(2000)-DTPA-tetra was used to chelate Gd and incorporated into PAMs via thin film hydration. Furthermore, DSPE-PEG(2000)-Cy7 amphiphiles were added to MCG PAMs for its NIRF properties for in vitro and histological studies.
To construct MCG PAMs, MCP-1 PA, Col-1 PA, Gd amphiphiles, and DSPE-PEG(2000)-methoxy amphiphiles were mixed at 45:30:15:10 molar ratios, respectively, and used for nanoparticle characterization. DSPE-PEG(2000)-methoxy was replaced with Cy7 amphiphiles in later studies for cell binding and in vivo studies. A nontargeting control micelle was formulated with scrambled MCP-1 peptides to construct SCG PAMs.
MCG and SCG PAM size, charge, and polydispersity were measured using dynamic light scattering (DLS, Table 1; Figure S2, Supporting Information). The hydrodynamic diameter of MCG PAMs (100 μm) in Milli-Q (MQ) water was found to be 14.8 ± 0.6 nm and the particles were monodispersed as seen via a polydispersity index of 0.11 ± 0.03. Transmission electron micro-graphs (TEM) further verified the monodispersity of the particles and the size to be ≈15 nm (Figure 1A). Previous studies on permeable arterial endothelium in atherosclerosis reported leaky endothelial tight junctions to be ≈20–1330 nm range.[34,36–38] Given that MCG PAMs are smaller than 20 nm, our micelles have the capacity to extravasate past dysfunctional endothelium, allowing for passive targeting of plaques in addition to active targeting via the CCR2 receptor.[39] The zeta potential of 100 μm MCG PAMs in MQ water was found to be slightly positive (3.6 ± 2.1 mV), which is attributed to the net five positive charges of the MCP-1 peptide derived from the arginine and lysine residues. Nonetheless, consistent with previous reports published from our group using MCP-1 targeting peptides, zeta potential of MCG PAMs were within the threshold to be considered physiologically neutral, or within −10 and +10 mV, for prolonged circulation in vivo.[33,40–43] Similar to MCG PAMs, SCG PAMs were 13.2 ± 2.2 nm in diameter with a polydispersity index of 0.23 ± 0.01 and zeta potential of 5.3 ± 2.6 mV (Figure S2, Supporting Information).[44]
Table 1.
Characterization of MCG and SCG PAMs.
| Particle | Diameter [nm] | Polydispersity index | Zeta potential [mV] |
|---|---|---|---|
| MCC PAMs | 14.8 ± 0.6 | 0.11 ± 0.03 | 3.6 ± 2.1 |
| SCC PAMs | 13.2 ± 2.2 | 0.23 ± 0.01 | 5.3 ± 2.6 |
Figure 1.
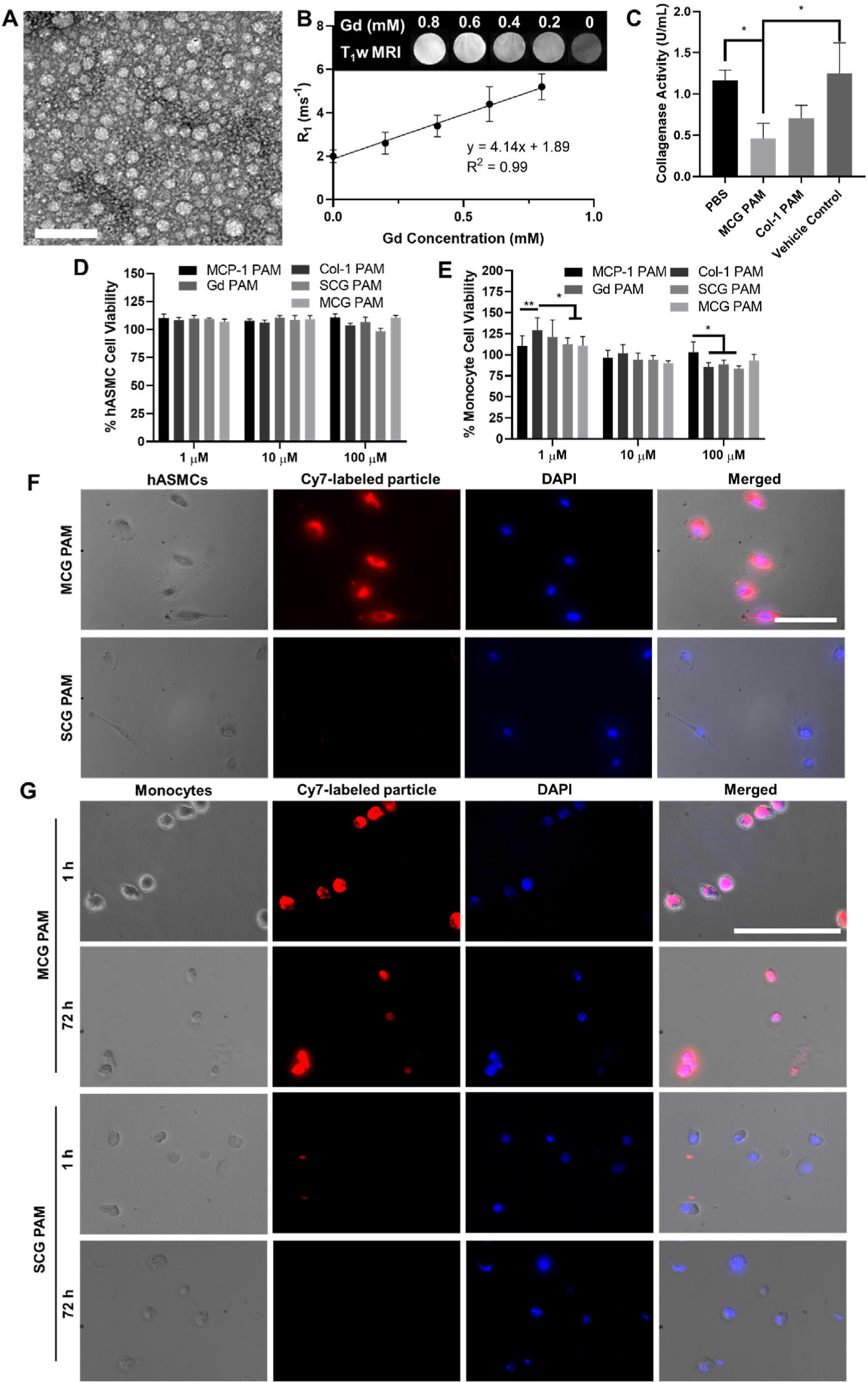
Physicochemical and in vitro analyses of MCG PAMs. A) MCG PAMs are monodisperse, spherical particles ≈15 nm as seen in FEG-TEM images. Scale bar 100 nm. B) Phantom scans (inset) of MCG PAMs demonstrate the nanoparticles have relaxivity of 4.14 ± 0.46 mm−1s−1 (N = 3). C) Collagenase inhibition measurements show MCG PAMs and Col-1 PAMs significantly reduce collagenase activity by 2.5-fold and 1.6-fold compared to PBS and vehicle controls (one-way ANOVA *p ≤ 0.05). PAMs have minimal cytotoxicity in D) hASMCs and E) monocytes (N = 9, one-way ANOVA *p ≤ 0.05, **p < 0.01). Binding analysis using fluorescence microscopy verify MCG PAMs selectively bind to F) hASMCs when compared to SCG PAMs (N = 3). G) MCG PAM incubation for 1 and 72 h with monocytes demonstrate that MCG PAMs bind to monocytes. SCG PAMs show no binding to monocytes (N = 3). Scale bars 100 μm.
We assessed the efficacy of MCG PAMs as MR contrast agents by measuring T1 relaxation times of MCG PAMs at varying concentrations with a 7T PET-MR system in order to determine particle relaxivity, r1 (Figure 1B). The relaxivity was calculated as 4.14 ± 0.46 mm−1 s−1, which is comparable to the commercially available Gd-DTPA contrast agent, Magnevist, that has been widely used for cardiovascular imaging.[45] To test the feasibility of competing with collagen to inhibit MMP activity, the therapeutic effect of MCG PAMs was initially tested in vitro using a collagenase activity assay. Specifically, the collagenase inhibitory effect of MCG PAMs (100 μm total particle concentration or 17 μm Col-1 PA) or Col-1 PAMs (PAMs consisting of 100% Col-1, 17 μm) was assessed by incubating PAMs with collagenase and fluorescently-labeled collagen and determining collagenase activity by the fluorescence emitted by cleaved collagens. After 1 h, MCG PAMs or Col-1 PAMs actively reduced collagenase activity by 2.5- or 1.6-fold, respectively, when compared to a PBS control or a blank micelle consisting of DSPE-PEG(2000)-methoxy micelles (p ≤ 0.05, Figure 1C). Hence, in vitro, MCG PAMs impaired MMP activity, showing potential of protecting collagen fibers from cleavage and degradation on fibrous caps in vivo.
We next evaluated the biocompatibility of MCG PAMs, SCG PAMs, MCP-1 PAMs, Col-1 PAMs, or Gd PAMs from 1 to 100 μm by incubating with WEHI 274.1 murine monocytes or human aortic smooth muscle cells (hASMCs) for 48 h and assessing cell viability via an MTS assay (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium). Viability for hASMCs remained close to 100% for all particles at all concentrations. For monocytes, cell viability from 1 to 100 μm concentrations of particles on monocytes remained above 80%, although some differences were seen between MCP-1 PAMs and Col-1 PAMs, Gd PAMs, and SCG PAMs at 100 μm, with a slightly lower viability observed for the latter three particle formulations. Nonetheless, overall, MCG PAMs and SCG PAMs were found to have minimal cytotoxic effects on hASMCs or monocytes (Figure 1D,E), similar to previous PAM reports from our group.[33,34,46]
To confirm CCR2 targeting, Cy7-labeled MCG PAM or SCG PAM (100 μm) binding on hASMCs was evaluated after 1 h of incubation using fluorescence microscopy. When compared with the nontargeting SCG PAMs that lack the CCR2 binding motif, MCG PAMs showed significant binding to hASMCs (Figure 1F). Furthermore, given that CCR2 is also expressed on circulating and recruited monocytes, we incubated monocytes with MCG and SCG PAMs for 1 and 72 h which may represent another method of targeting and drug delivery to plaques (Figure 1G). Unlike previous reports that showed nanoparticle endocytosis and phagocytosis via clustering in vesicles represented by punc-tate fluorescence, MCG PAMs decorated the surface of monocytes, confirming their ability to stay bound to CCR2 and the surface of monocytes.[47,48] These in vitro data suggest MCG PAMs show promise as atherosclerotic plaque-targeting nanoparticles via monocyte and smooth muscle binding without compromising biocompatibility.
2.2. MCG PAMs Detect Plaques In Vivo via MR Imaging
Next, we evaluated the ability of MCG PAMs to target and detect atherosclerotic plaque in vivo via MR imaging using murine atherosclerotic disease models. Angiotensin II infusion in ApoE−/− mice induces hypertension and the marked increase of rapid late-stage atherosclerotic plaque development.[49,50] Previously it was shown that when fed an atherogenic diet, angiotensin II-treated ApoE−/− mice have a higher incidence of acute plaque rupture compared to mice without angiotensin II infusion.[51] Furthermore, angiotensin II-infused mice showed overall decreases in fibrous cap thickness and increases in MMP activity in the plaque, making it an ideal model for our studies investigating the therapeutic effects of MCG PAMs in enhancing plaque stability through collagenase inhibition.[51–53]
In our study, 24-week-old ApoE−/− mice were fed an atherogenic diet for 3 weeks and treated with angiotensin II for an additional 3 weeks while on the atherogenic diet (Figure S3, Supporting Information). Concurrently with angiotensin II infusion and atherogenic diet, mice were treated with either MC PAMs, SC PAMs (without Cy7 or Gd, 1 mm, 100 μL), or PBS (100 μL) via tail vein injection once a week for 3 weeks. A weekly dosing regimen was selected based on previous studies from our group that demonstrated clearance of PAMs by 7 days.[35] At the end of 3 weeks, mice were injected with Cy7-labeled MCG PAMs, Cy7-labeled SCG PAMs, or PBS to assess plaque targeting capabilities via MR imaging and T1-weighted coronal and transverse MR scans were taken of the mice immediately before and after nanoparticle administration. Notable MR signal enhancements were observed in the aortic root and aorta in the coronal and transverse scans of mice treated with MCG PAMs (white arrows, Figure 2A,B). When SNR was measured and calculated from the aortas of MCG PAM-, SCG PAM-, and PBS-treated mice, MCG PAM treatment was found to exhibit greater SNR (MCG PAM: 16.7 ± 8.5, SCG PAM: 1.5 ± 0.3, and PBS: 1.5 ± 0.3, p ≤ 0.05 between MCG PAM and SCG PAM or PBS, Figure 2C), verifying the targeted molecular imaging capabilities of MCG PAMs to athero-prone regions.[53]
Figure 2.
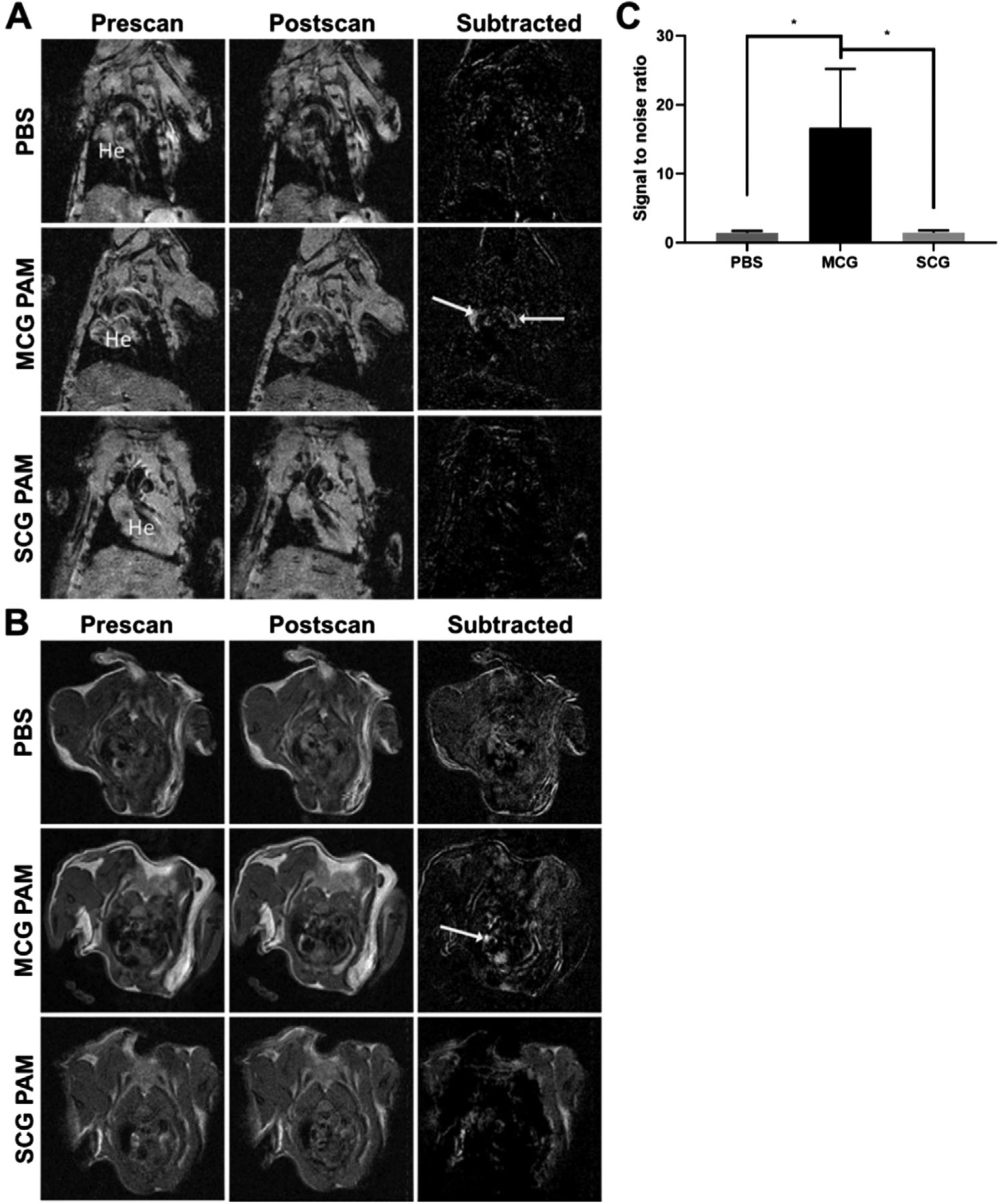
MR images of MCG PAM-, SCG PAM-, and PBS-treated mice. MCG PAMs show MR signal enhancement in atherosclerotic plaques in the aorta (arrows) as shown via A) coronal (He = heart) and B) transverse scans. No tissue enhancement is observed with SCG PAM and PBS treatment (N = 3 for PBS or 4 for MCG PAM and SCG PAM). C) MCG PAMs demonstrate significant increases in SNR compared to PBS and SCG PAMs (MCG PAM: 16.68, SCG PAM:1.52, PBS:1.46, N = 4 for MCG PAM and SCG PAM and N = 3 for PBS, one-way ANOVA *p ≤ 0.05).
To further assess and cross-verify the plaque-targeting abilities of MCG PAMs, 24 h after the MR scans, mice were euthanized and the aortic root, aortic arch, descending aorta, and brachiocephalic artery were analyzed histologically for plaque targeting. Aortic arch sections from mice depicted in the MR images from Figure 2 were stained for nuclei with DAPI and imaged for particle fluorescence (Figure 3A). Corroborating our MR images, Cy7 fluorescence signal from MCG PAMs was observed in the lesions in the aortic arch, while mice treated with SCG PAMs demonstrated trace amounts of fluorescence in the atherosclerotic plaque. The Cy7 particle fluorescence signal in atherosclerotic lesions across all mice were measured to quantify MCG PAM and SCG PAM targeting capabilities (Figure 3B). While no significant difference was observed between SCG PAM and PBS, MCG PAM-treated mice showed a 4.2-fold increase in fluorescence in atherosclerotic lesions found in the aorta and the brachiocephalic artery compared to SCG PAM-treated mice (MCG PAM: 1152 RFU/mm2 vs SCG PAM: 271.5 RFU/mm2, *p = 0.018).
Figure 3.
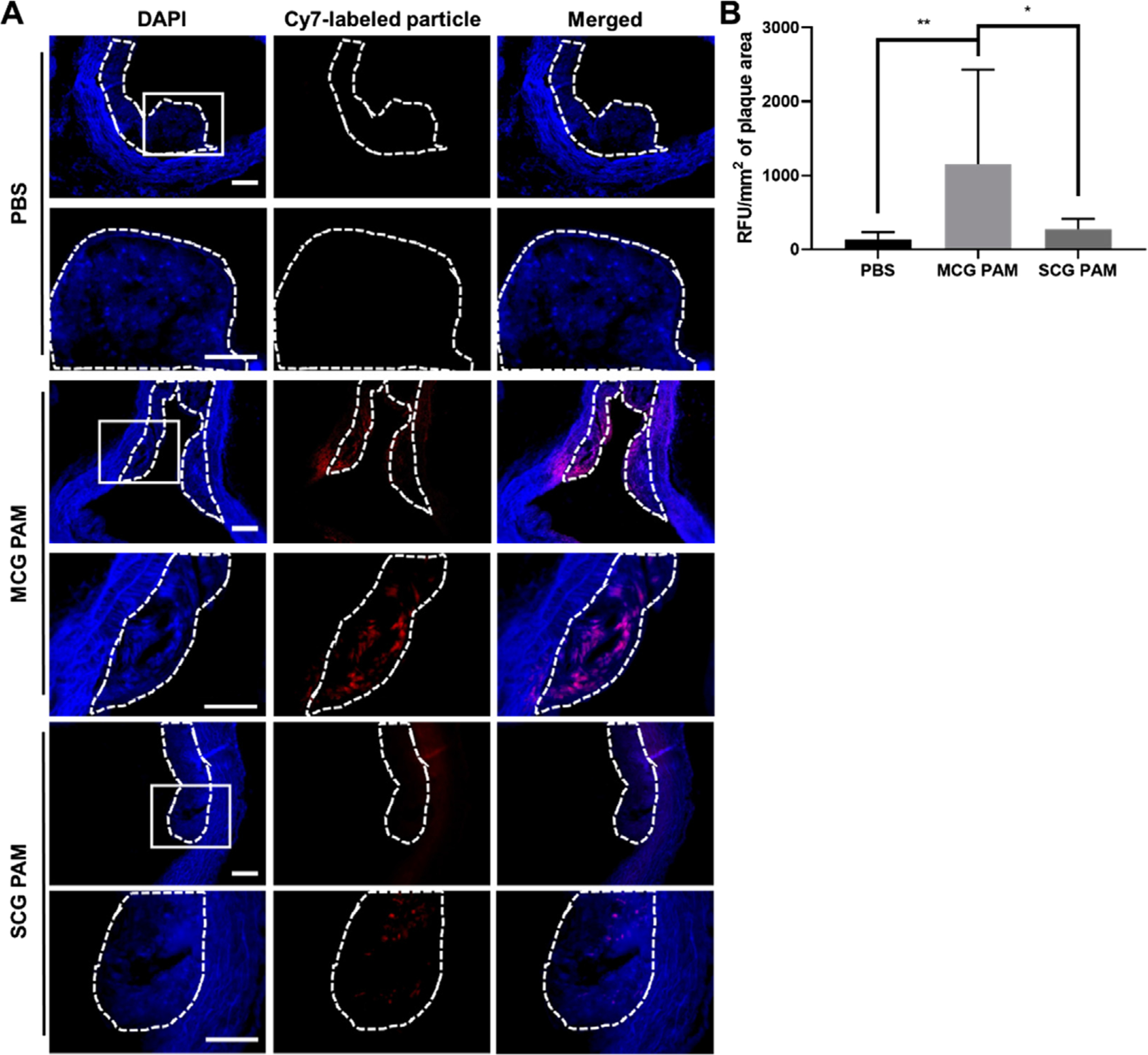
Plaque targeting in the aorta. A) Fluorescence images of DAPI-stained aortic arch sections indicate MCG PAMs (red) target plaques (dotted line) in the aortic arch. The second row of images are magnifications of the white box. SCG PAM- and PBS-treated mice show a minimal, background signal. Scale bars 100 μm. B) Cy7 fluorescence in plaques found in the aortic root, arch, descending aorta, and brachiocephalic arteries of each mouse quantified with ImageJ show increased fluorescence with MCG PAM treatment (N = 4 mice for MCG PAM and SCG PAM and N = 3 mice for PBS, one-way ANOVA *p ≤ 0.05, **p < 0.01).
Current diagnostic techniques using MR imaging and ultra-sound in the clinic can benefit from the use of targeted contrast agents such as MCG PAMs that allow for enhanced molecular imaging and a concentrated signal amplification that is highly specific to plaques. Previous studies using iron oxide or Gd paramagnetic micelles have been reported to image atherosclerotic plaque via macrophage targeting.[54,55] While these studies showed a modest twofold increase in SNR compared to controls, these differences were found to be most prominent after 24 h, leading to long post-contrast imaging delay times. Additionally, while ultrasmall superparamagnetic iron oxide (USPIO) particles have been popular as MR imaging contrast agents as they demonstrate nonspecific macrophage uptake in atherosclerotic plaques, currently, these formulations suffer from the lack of specificity, limited penetration into atherosclerotic plaques, iron oxide induced signal loss, and the need for high doses.[56,57] In contrast, MCG PAMs allow for rapid imaging, with significant SNR at atherosclerotic plaques within 1 h of particle injection, with high specificity for plaques as seen from the MR images and the histological sections. Taken together, our MR imaging and histological data indicate MCG PAMs have the capability to detect and target atherosclerotic plaques.
2.3. MCG PAMs Increase Fibrous Cap Thickness for Atherosclerosis Therapy
After verification of plaque targeting, we evaluated the therapeutic efficacy of MCG PAMs by examining changes in plaque composition. Plaques were analyzed by staining aortic roots, aortic arches, descending aorta, and brachiocephalic arteries with hematoxylin and eosin (H&E) and the fibrous cap thicknesses of plaques were measured after a picrosirius red collagen stain (Figure 4). Following previously reported methods, as the morphology of a fibrous cap is inconsistent across the entire atherosclerotic plaque, fibrous cap thicknesses were assessed by measuring the cap at three distinct locations for each lesion: at two shoulder regions and the center of the plaque to obtain an average cap thickness per lesion (Figure 4B,C; Figure S4, Supporting Information).[58] MCG PAM-treated mice had thick fibrous caps at ≈68 ± 29 μm for each lesion. This is compared to SCG PAM-treated mice that had cap thicknesses averaging 42 ± 12 μm and PBS-treated mice at 32 ± 16 μm (MCG PAM-treated mice vs SCG PAM-treated mice (p ≤ 0.01) or PBS-treated mice (p ≤ 0.001)). Hence, the targeting moiety of MCG PAMs significantly increased the potency of the therapy compared to nontargeting SCG PAMs. As seen in fluorescence images of aortic sections (Figure 3), MCG PAMs showed localization in the lesion, potentially increasing the concentration of therapeutic Col-1 at the disease site and allowing for competition with endogenous collagens found on the fibrous cap against enzymatic degradation. Currently, the weekly dosing strategy demonstrates efficacy in increasing fibrous cap thickness. However, future experiments will determine the optimal dosing regimen for maximum efficiency.
Figure 4.
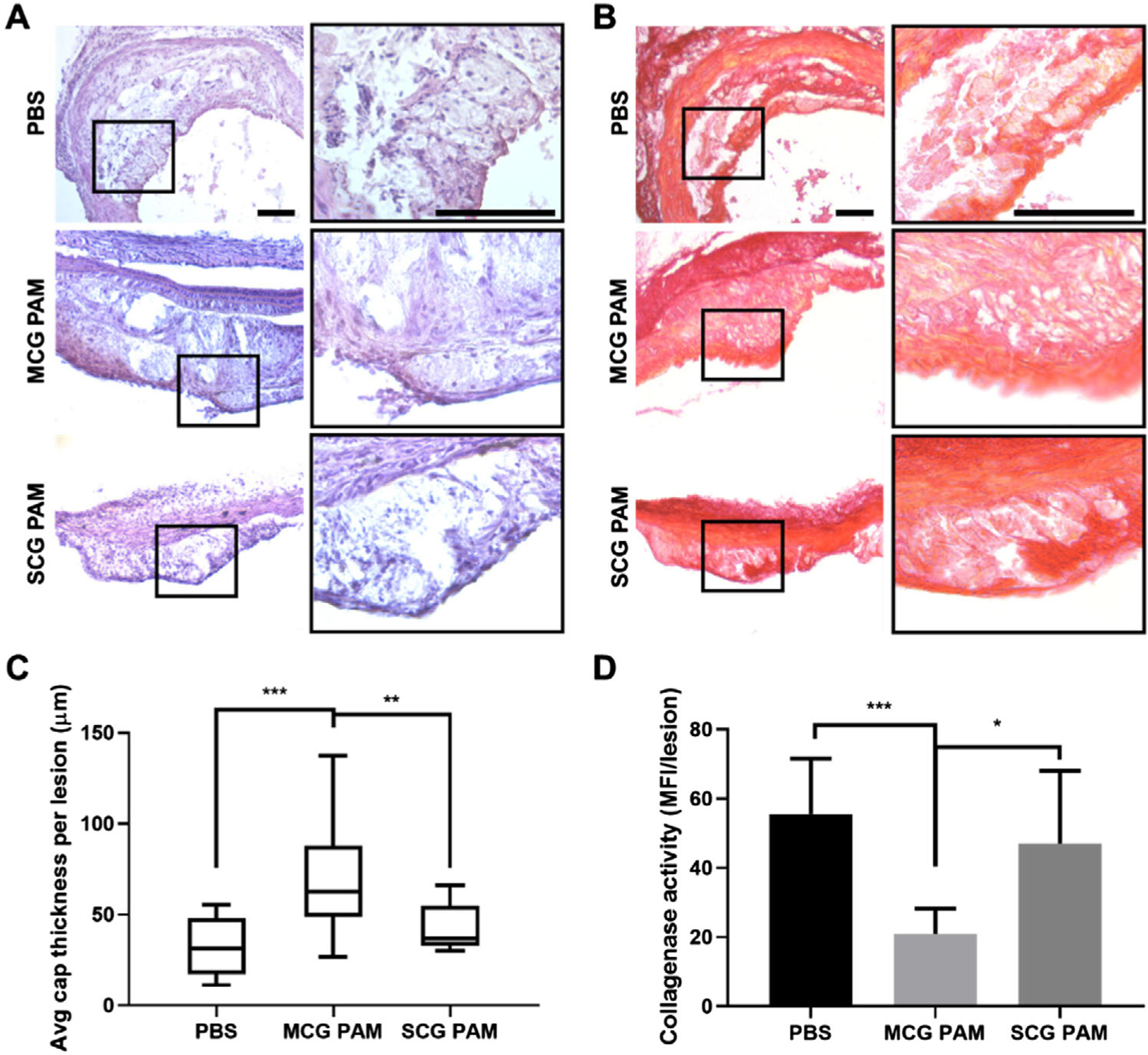
Histological analysis of fibrous caps and plaques in the aorta. MCG PAM treatment increases the thickness of the fibrous cap as found via histological analysis using A) H&E and B) picrosirius red staining. Images on the right column are magnifications of the black box. Scale bars 100 μm. C) Quantification of the fibrous cap thickness from lesions of the aortic root, arch, descending aorta, and brachiocephalic arteries indicate MCG PAM treatment (68 ± 29 μm) increases fibrous cap thickness by 61% compared to SCG PAM treatment (42 ± 12 μm), and by 113% when compared to PBS (32 ± 16 μm, one-way ANOVA **p < 0.01, ***p < 0.001). Fibrous cap thicknesses were measured at three points of each lesion, at the center and two shoulders, to obtain an average cap thickness per lesion. D) In situ zymography of aortic arches show MCG PAMs (20.9 ± 7.3 MFI/lesion) significantly reduce collagenase activity in atherosclerotic lesions by 55% versus SCG PAMs (47.0 ± 21.0 MFI/lesion) and by 62% versus PBS (55.5 ± 16.1 MFI/lesion, N = 4 mice for MCG PAM and SCG PAM and N = 3 mice for PBS, one-way ANOVA *p ≤ 0.05, ***p ≤ 0.001).
In order to verify the fibrous cap thickening by MCG PAMs was due to collagenase inhibition, MMP collagenase activity within the aortic atherosclerotic lesions was measured via in situ zymography.[58–61] Unfixed cryostat sections of aortic arches with atherosclerotic lesions were incubated with DQ-gelatin, which fluoresces when cleaved by collagenase. In correlation with cap thickness measurements, lesions in mice treated with MCG PAMs exhibited the least amount of fluorescence and collagenase activity per lesion (20.9 ± 7.3 mean fluorescence intensity (MFI)/lesion), followed by SCG PAM- and PBS-treated mice (47.0 ± 21.0 MFI/lesion and 55.5 ± 16.1 MFI/lesion, respectively). Differences between MCG PAMs and PBS were statistically significant with ***p ≤ 0.001 and *p ≤ 0.05 when compared with SCG PAMs. These results align with our in vitro studies demonstrating collagenase inhibiting effects of MCG PAMs and Col-1 PAMs (Figure 4D). Hence, MCG PAMs successfully shuttled into atherosclerotic lesions and impeded MMP collagenase activity, thereby, providing a fibrous cap thickening therapeutic outcome.
To our knowledge, this is the first use of a multimodal micelle nanoparticle system using dual MR and NIRF imaging of atherosclerotic plaques with targeted delivery of atherosclerotic plaque stabilizing therapy. While previous studies have investigated siRNA technology as well as annexin A1 mimetic peptides to alleviate inflammation as a means of stabilizing plaques, there is a dearth of studies that have developed combined capabilities of diagnostics and therapy.[58,62,63] By developing a multimodal nanoparticle platform with targeted MRI contrast agents, NIRF capabilities, and therapeutic cargo, MCG PAMs show efficacy as theranostic nanoparticles to detect and stabilize atherosclerotic plaques.
2.4. In Vivo Pharmacokinetics, Off-Target Effects, and Safety of MCG PAMs
MCG PAMs were further characterized for circulation and half-life in vivo as well as safety and off-target effects. To investigate particle circulation and half-life, MCG PAMs were injected into C57BL/6J mice and blood was collected at 5 min, 1, 3, 8, and 24 h post-injection before analysis via inductively coupled plasma-optical emission spectrometry (ICP-OES, Figures S5 and S6, Supporting Information). The t1/2α and t1/2β of MCG PAMs were determined to be 1.3 ± 0.1 and 23.9 ± 1.8 h, respectively, which is consistent with MCP-1, scrambled MCP-1, and Gd-based micelles previously reported and is a marked improvement over commercially available Gd contrast agents that undergo rapid elimination by the kidneys with a terminal plasma half-life of 15–20 min in murine models.[30,42,64–68]
As many nanoparticles are eliminated via renal and hepatic clearance, we assessed the possible off-target effects of MCG PAMs in inducing unwanted collagen expression in the liver and kidneys. Kidney and liver organs were resected from the PBS-, MCG PAM-, and SCG PAM-treated mice given angiotensin II and sectioned to look at collagen content in the organs via picrosirius red staining (Figure 5).[69] Although our biodistribution studies indicated MCG PAMs also accumulated in the liver, no significant differences in collagen content was observed for MCG PAM- and SCG PAM-treated mice when compared to PBS-treated controls, confirming MCG PAMs do not elicit off-target collagen expression.[70]
Figure 5.
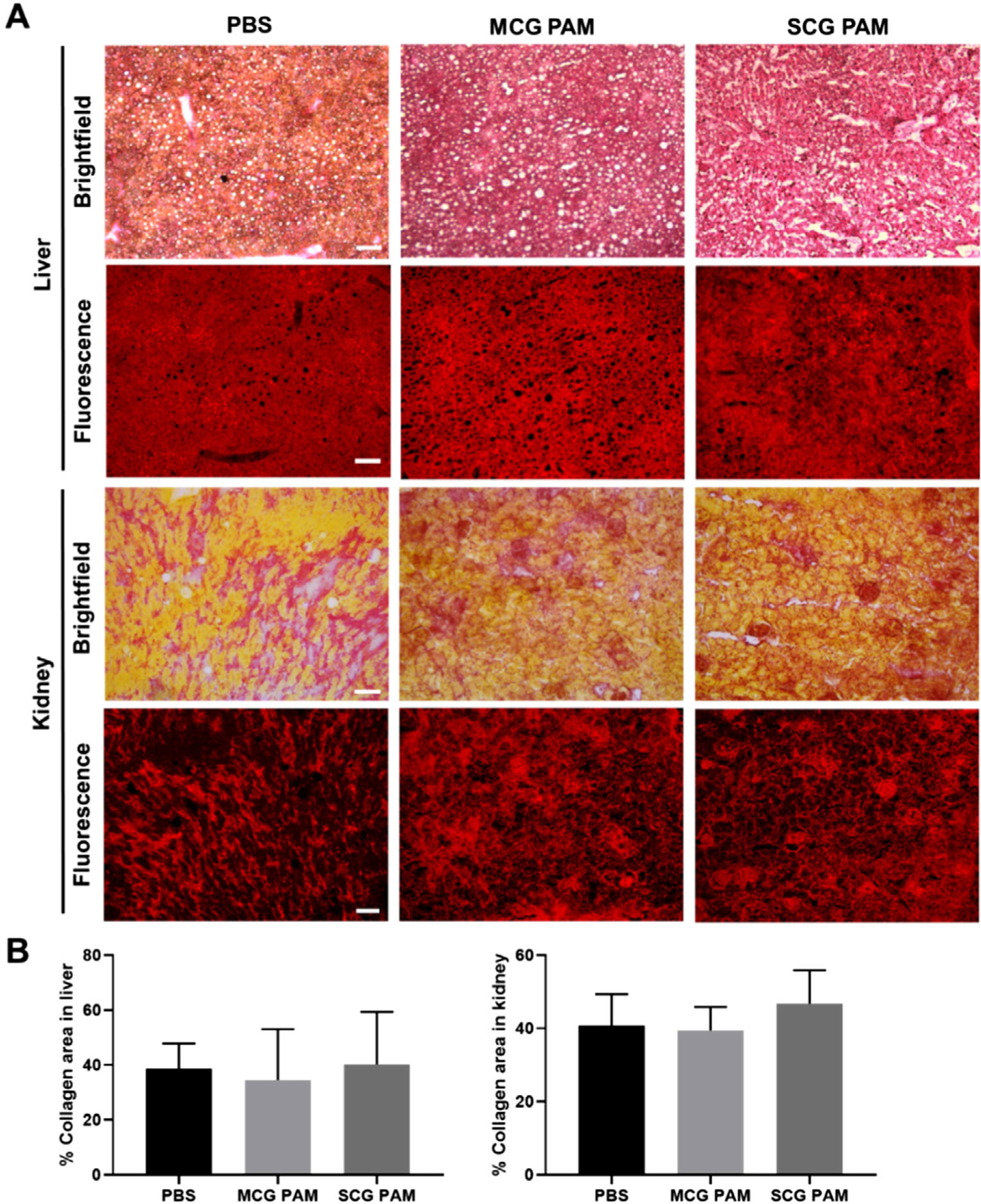
Off-target, collagen content assessment. A) Kidney and liver sections from PBS-, MCG PAM-, and SCG PAM-treated mice stained with picrosirius red show collagen (red) content. Scale bars 100 μm. B) Quantification of the collagen content demonstrate there are no significant differences or enhancements of collagen among MCG PAM- or SCG PAM-treated mice, indicating a lack of off-target effects from the nanoparticles (N = 4 mice for MCG PAM and SCG PAM and N = 3 mice for PBS).
To assess safety of MCG PAMs, organ toxicity was measured 24 h after systemic injection of MCG PAMs in C57BL/6J mice. The brain, heart, lungs, liver, spleen, and kidney of mice were resected, sectioned, and stained with H&E. H&E stains of the major organs demonstrated no significant differences in cell morphology or signs of cell death between PBS- and MCG PAM-treated mice (Figure 6A). Furthermore, since higher doses of Gd are known to cause renal toxicity, kidney function was assessed in mice treated with MCG PAMs.[71] Blood urea nitro gen (BUN), plasma creatinine, and urine creatinine were measured and found to fall within normal levels for mice (BUN: 28.4 ± 2.6 mg dL−1, plasma creatinine: 0.48 ± 0.06 mg dL−1, urine creatinine: 5.2 ± 2.9 mg dL−1, Table 2). In addition, BUN and creatinine levels between MCG PAM- and PBS-treated mice were not found to be statistically significant.
Figure 6.
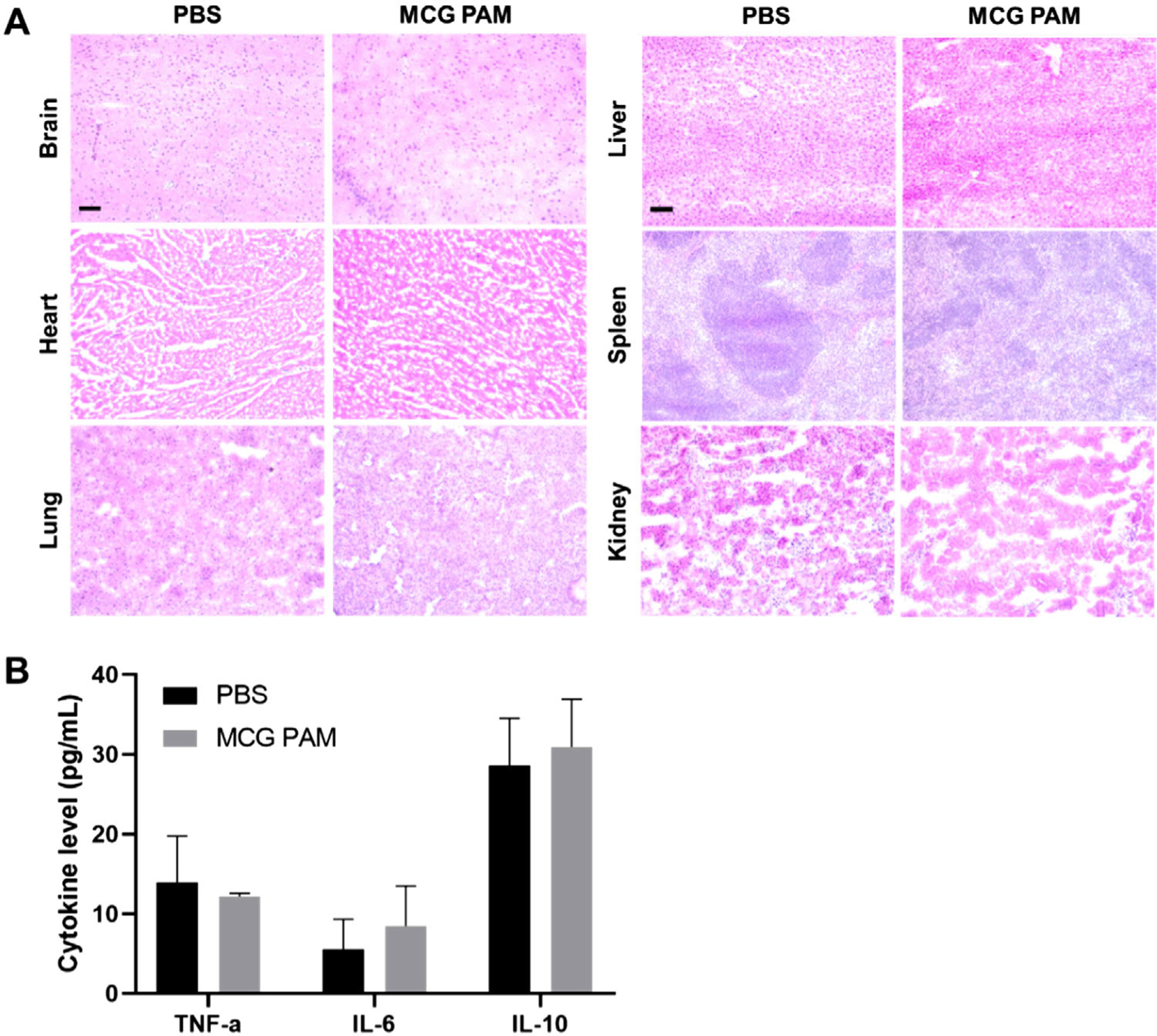
In vivo toxicity in MCG PAM-treated mice. A) Organs display no significant changes in morphology or toxicity with MCG PAMs. Scale bars 100 μm. B) Cytokine levels remained within normal ranges upon MCG PAM treatment (N = 3).
Table 2.
Renal function in MCG PAM- and PBS-treated mice.
| BUN [mg dL−1] | Plasma creatinine [mg dL−1] | Urine creatinine [mg dL−1] | |
|---|---|---|---|
| MCG PAMs | 28.4 ± 2.6 | 0.48 ± 0.06 | 5.2 ±2.9 |
| PBS | 27.2 ± 1.3 | 0.58 ±0.07 | 4.8 ± 1.0 |
Moreover, we measured the cytokine levels for tumor necrosis factor-alpha (TNF-α), interleukin-6 (IL-6), and interleukin-10 (IL-10) to ensure MCG PAMs did not induce an inflammatory response (Figure 6B). After 24 h, when comparing serum level cytokines with PBS-treated mice, we found no significant changes in inflammatory cytokine levels from MCG PAM treatment. Overall, when injected with MCG PAMs, cytokines remained within normal thresholds for mice (TNF-α: 12.2 ± 0.4 pg mL−1, IL-6: 8.5 ± 5.1 pg mL−1, IL-10: 30.9 ± 6.0 pg mL−1). Thus, MCG PAMs display biocompatibility in vivo and are safe theranostic nanoparticles for potential prolonged use in chronic diseases such as atherosclerosis.
3. Conclusion
In this study, we developed a novel theranostic micelle nanoparticle with the ability to target and image atherosclerotic plaques and provide therapeutic collagenase inhibition that allows for fibrous cap thickening and enhanced plaque stability. In vitro, MCG PAMs demonstrated biocompatibility with monocytes and VSMCs and selective binding. Moreover, by incorporating Gd for utilization as MR contrast agents, MCG PAMs successfully identified plaques in the aorta of diseased mice in real-time while histological analysis further confirmed MCG PAM successfully accumulated in lesions. Notably, through competitive inhibition of destructive collagenases, Col-1 peptides in MCG PAMs were able to preserve the fibrous cap to stabilize atherosclerotic plaques without causing any off-target collagen growth in the kidneys or liver. Overall, our studies show the potential of MCG PAMs in the targeting, diagnosis, and treatment of vulnerable plaques in atherosclerotic mice.
4. Experimental Section
Synthesis of MCP-1, Col-1, and Scrambled MCP-1 PAs:
MCP-1 ([CYNFTNRKISVQRLASYRRITSSK]), scrambled MCP-1 ([CYNSLVFRIRNSTQRKYRASIST]), and Col-1 ([CVPMSMRGG]) peptides were synthesized using standard Fmoc-mediated solid phase peptide synthesis on an automatic PS3 peptide synthesizer (Protein Technologies, Tucson, AZ, USA).[42,43,68] Col-1 peptide was N-capped with an acetyl group and cleaved from a rink amide resin using trifluoroacetic acid:1,2 ethanedithiol:H2O:triisopropylsilane at 94:2.5:2.5:1 volume ratios. MCP-1 and scrambled MCP-1 peptides were cleaved from Wang resin using the same chemical cocktail. Cleaved peptides were precipitated with ice cold diethyl ether, lyophilized, and stored at −20 °C until purification using reverse-phase HPLC (Phenomenex, Torrance, CA, USA; Shimadzu, Kyoto, Japan). Peptides were purified at 55 °C in 0.1% formic acid in acetonitrile/water mixture using a C8 column (Phenomenex, Torrance, CA, USA) and characterized using MALDITOF mass spectral analysis (Bruker, Billerica, MA, USA). Peptides were conjugated to 1,2 distearoyl-sn-glycero-3-phosphoethanolamine-N-[maleimide(polyethylene glycol)-2000], or DSPE-PEG(2000)-maleimide (Nanocs, New York, NY, USA) via a thioether linkage by adding a 10% molar excess of the peptide to lipid in water at a pH of 7. Conjugated amphiphiles were purified using a C4 column (Phenomenex, Torrance, CA, USA) and verified using MALDI-TOF.
Synthesis of DSPE-PEG(2000)-Gd:
To synthesize DSPE-PEG(2000)-DTPA(Gd), diethylenetriamine-N,N,N″,N″-tetra-tert-butyl acetate-N′-acetic acid (DTPA-tetra, Macrocyclics, Plano, TX, USA) and DSPEPEG(2000)-amine (Avanti Lipids, Alabaster, AL, USA) were reacted to form a peptide bond. Gd ions from Gd(Cl3) (Sigma-Aldrich, St Louis, MO, USA) were chelated onto a deprotected DSPE-PEG(2000)-DTPA-tetra and onto the amphiphile. Free Gd ions were separated from the amphiphile using a desalting column (GE Healthcare Life Science, Marlborough, MA, USA) and successful conjugation was confirmed by MALDI-TOF.
Micelle Formulation:
Six types of micelles were synthesized using self-assembly technique: 1) MCG micelles, 2) Cy7-labeled MCG micelles, 3) MC micelles, 4) SCG micelles, 5) Cy7-labeled SCG micelles, and 6) SC micelles (M: MCP-1 PA, C: Col-1 PA, S: scrambled MCP-1 PA, G: Gd amphiphile, Cy7: Cy7 amphiphile). Briefly, amphiphiles were mixed at a molar ratio of 45:30:15:10 for MCP-1/scrambled MCP-1:Col-1:Gd:Cy7 amphiphiles and dissolved in methanol. Particles without all the individual components were synthesized with the substitution of DSPE-PEG(2000)-methoxy.
Dynamic Light Scattering:
The diameter, zeta potential, and polydispersity index of the micelles were measured using DLS at a concentration of 100 μm in MQ water (N = 6). DLS measurements were taken at 90° and 637 nm using a Wyatt DynaPro Mobius (Wyatt, Santa Barbara, CA, USA).
Transmission Electron Microscopy:
MCG micelles (100 μm) in MQ water were placed onto a 400-mesh carbon TEM grid (Ted Pella, Redding, CA, USA) for 2 min before briefly washing with MQ water and negatively staining the particles with 2 wt% uranyl acetate solution (Polysciences, Warrington, PA, USA). The stain was removed and samples were dried overnight and imaged on a JEOL 2100F (JEOL, Tokyo, Japan, N = 3).
r1 Relaxivity of MCG PAMs:
T1 relaxation times of MCG PAMs (1.2, 2.4, 3.6, and 4.8 mm MCG PAM concentrations with 45:30:15:10 molar ratios for MCP-1:Col-1:Gd:Cy7 amphiphiles) were determined using an MR Solutions 7T PET-MR system (MR Solutions Ltd., Guildford, UK) at the Zilkha Neurogenetic Institute (University of Southern California, Los Angeles, CA, USA). A 9-cm diameter radiofrequency body coil was used (bore size ≈24-mm, up to 600 mT m−1 maximum gradient). A T1-weighted variable flip angle fast low angle shot (FLASH) gradient echo sequence was used with the following imaging parameters on a 7T scanner to determine particle T1 relaxation times: echo time (TE) = 5 ms, repetition time (TR) = 60 ms, flip angle (FA) array = (15°, 30°, 45°, 70°, and 80°), field of view (FOV) = 36 × 36 mm, slice thickness = 1 mm, image matrix = 192 × 192, number of averages (NA) = 4. A multi-echo multi-slice spine echo sequence (MEMS) was used to determine T2 relaxation times of the particle with the following parameters: ten echoes with 15 ms interval beginning with TE = 15, TR = 2500, FOV = 36 × 36 mm, slice thickness = 1 mm, matrix size = 256 × 256, NA = 1. Using ROCKETSHIP v.1.1 code in MATLAB (R2014b), T1 and T2 maps were generated through a pixel-by-pixel exponential fitting of signal intensities across the different FA and TE values, respectively.[72] A region of interest (ROI) was manually drawn around each concentration of MCG PAMs that were measured using ImageJ. R1 maps were generated by taking the inverse of the T1. Using R1 values, r1 relaxivity was determined (R1/concentration of Gd) over Gd concentrations of 0.2, 0.4, 0.6, and 0.8 mm.
Collagenase Assay:
The effect of MCG PAMs on collagenase activity was measured using a collagenase assay kit (Chondrex, Redmond, WA, USA, N = 3). Following the manufacturer’s protocol, particles were incubated with latent collagenase with an activator. Activated samples were then reacted with FITC-labeled collagen before denaturing and digesting cleaved collagen fragments with an enhancer. Fluorescence intensity of the extracted fragments were then measured at 520 nm to assess collagenase activity.
Cell Culture:
WEHI 274.1 murine monocytes were cultured in Dulbecco’s modified Eagle’s medium (ThermoFisher, Waltham, MA, USA) with 4.5 g L−1 glucose, 10% fetal bovine serum, 0.05 mm 2-mercaptoethanol, and 1% penicillin and streptomycin. Media was changed every 2–3 days. Only cells from passages 2–6 were used for experiments. Human aortic smooth muscle cells (hASMCs) were cultured in Media 231 supplemented with Smooth Muscle Growth Supplement (ThermoFisher, Waltham, MA, USA) and 1% penicillin and streptomycin. Media was changed every 2–3 days.
In Vitro Biocompatibility:
Murine monocytes were seeded and incubated with MCP-1 PAMs, Col-1 PAMs, Gd PAMs, SCG PAMs, MCG PAMs (1, 10, 100 μm), or PBS control for 48 h before biocompatibility was assessed using an MTS cell proliferation assay (Biovision, Milpitas, CA, USA). After 48 h of treatment, MTS reagent was added to the cells for 1 h at 37 °C to allow for reaction. Absorbance of the MTS reagent was measured using a plate reader at 490 nm (N = 9). Cell viability was calculated by comparing with PBS-treated cells.
Monocyte and SMC Binding:
WEHI 274.1 murine monocytes or hASMCs were seeded onto coverslips and incubated with Cy7-labeled MCG or SCG PAMs for 1 or 72 h. After 1 or 72 h, the PAM solution was removed, and cells were washed with PBS. Cells were then fixed in 4% paraformaldehyde for 15 min at room temperature and subsequently stained with DAPI for nuclei. Next, the cells were imaged using a fluorescence microscope (Leica DMi8, Leica, Wetzlar, Germany, N = 3).
Atherosclerotic Murine Model and Treatment:
Atherosclerotic diseased murine models (The Jackson Laboratories, Bar Harbor, Maine, USA) were developed using angiotensin II infusion in both male and female ApoE−/− mice. ApoE−/− mice were switched to an atherogenic, Western diet (21% (wt/wt) fat, 0.2% (wt/wt) cholesterol, 19.5% (wt/wt) casein, Teklad, Indianapolis, IN, USA) at 24 weeks old for 6 weeks. At age 27 weeks, Alzet osmotic minipumps (Model 2004; ALZA Scientific Products, Mountain View, CA, USA) were implanted subcutaneously into ApoE−/− mice. Pumps were filled with angiotensin II (Bachem, Torrance, CA, USA) following the manufacturer’s protocol to deliver subcutaneous angiotensin II at a rate of 1.9 mg kg−1 d−1 for 28 days.[52] Mice were anesthetized using ketamine/xylazine and isoflurane at flow rate of 2%. Pumps were placed into the subcutaneous space behind the right shoulder of the mice through a small incision that was closed with surgical glue. Buprenorphine SR was also administered subcutaneously at a dose of 1.0 mg kg−1 as an analgesic.
One day after implantation of the osmotic pump, mice were injected with MC PAMs, SC PAMs (1000 μm, 100 μL, N = 4), or PBS (100 μL, N = 3) via tail vein IV injection. Subsequent injections were administered once a week over 3 weeks for a total of four injections. The last injection consisted of Cy7-labeled MCG or SCG PAMs for use as imaging contrast agents.
MR Imaging of Mice:
Mice were anesthetized with 2% isoflurane at a flow rate of 250 μL min−1. Animals were transferred onto a heated scanner bed with anesthesia levels maintained between 1.5% and 2%. Temperature was monitored and maintained at 37 °C. Electrocardiogram (ECG) was monitored and used for gating by inserting two leads subcutaneously on the anterior side close to each axilla, and one lead was inserted subcutaneously on the lower right abdomen. A pneumatic pillow was placed underneath the animal for respiration monitoring and gating. The mice were positioned at the magnet bore isocenter using a motorized system. Temperature, respiration, and ECG were monitored using SAII equipment and accompanying PC-SAM software.
A bird cage whole mouse body coil, with an axial field of view of 60 cm and 35 mm inner diameter, was used during ECG and respiration gated cardiac imaging. A gradient echo scan was used to obtain three orthogonal slices for positioning. A series of multi slice T1-weighted FLASH (TE = 5 ms, TR = 170 ms, FA = 20°, FOV = 40 × 40 mm, slice thickness = 1 mm, matrix size = 192 × 192, NA = 1) sequences were used to achieve cross sections of the heart along the heart’s short and long axes. ImageJ was used to subtract the MR images and measure SNR.
Histological Analysis and Immunohistochemistry:
Mice were euthanized 24 h after MR imaging and the aorta, heart, kidneys, and livers were harvested. Aortas were cleaned of excess fat and perfused with PBS. Harvested tissues were embedded in Optimal Cutting Temperature (OCT) compound, flash frozen in 2-methylbutane and liquid nitrogen, and sectioned using a cryostat (10 μm thickness, Leica CM3050S, Leica, Wetzlar, Germany). Tissue sections of the aortic root, aortic arch, descending aorta, and brachiocephalic artery were then stained with hematoxylin and eosin (H&E) or picrosirius red (PR), mounted, and imaged (Leica DMi8). Fibrous cap thicknesses were measured at three distinct areas of the plaque: the middle and the two shoulders, and averaged and analyzed using ImageJ following previously reported methods.[58] Fibrous cap thicknesses were blindly measured by three individuals. To analyze Cy7-labeled particle localization in the tissue, sectioned aortas were stained with DAPI, mounted, and imaged using a fluorescence microscope. ROIs were drawn around plaques and quantified for relative fluorescence to determine plaque targeting using ImageJ. Collagenase activity in the atherosclerotic lesions was determined using the DQ Collagenase Assay Kit (ThermoFisher, Waltham, MA, USA) following the manufacturer’s protocol and previous methods.[58–61] MFI for individual lesions was measured with ImageJ to analyze collagenase.
Sections of the kidneys and livers were stained with picrosirius red to assess any off-target collagen deposition. Brightfield and fluorescence images were taken and used to quantify total collagen area with ImageJ.
In Vivo Biodistribution, Pharmacokinetics, and Safety:
Biodistribution and pharmacokinetics of the particle were assessed 24 h after C57BL/6J mice were injected with PBS or MCG PAMs (1000 μm, 100 μL, N = 3). Blood was collected via tail vein at times: 5 min, 1, 3, 8, and 24 h after injection. To determine the circulation half-life of the particle and assess biodistribution, plasma samples and resected organs including the brain, heart, lungs, liver, spleen, and kidneys were digested in nitric acid and diluted in 2% nitric acid for analysis of Gd content via ICP-OES.
To assess safety, C57BL/6J mice were injected with PBS and MCG PAMs (1000 μm, 100 μL, N = 3) as above and 24 h later, the major organs were resected and flash-frozen in OCT as mentioned earlier. Histological sections (10 μm) were taken from each organ and stained with H&E to check for changes in cell morphology or death. In addition, renal toxicity was assessed from blood urea nitrogen (BUN, Bioo Scientific, Austin, TX, USA), plasma creatinine, and urine creatinine (Crystal Chem, Elk Grove Village, IL, USA) levels in mouse serum, plasma, and urine following the manufacturer’s protocols. An ELISA kit (R&D Systems, Minneapolis, MN, USA) was used to measure cytokine levels of TNF-α, IL-10, and IL-6. All mice experiments were approved by and performed in compliance with the University of Southern California (USC) Institutional Animal Care and Use Committee (IACUC).
Statistical Analysis:
Quantification data are given as mean ± standard deviation. All statistical analyses were performed using GraphPad Prism 8 (San Diego, CA, USA). A Student’s t-test or analysis of variance (ANOVA) was used with a Tukey’s test for post-hoc analysis to determine statistical significance and p ≤ 0.05 was considered to be significant.
Supplementary Material
Acknowledgements
The authors would like to acknowledge the financial support from the American Heart Association Predoctoral Fellowship (19PRE34380998) awarded to D.D.C., and Women in Science and Engineering (WiSE), Gabilan Assistant Professorship, L. K. Whittier Foundation the National Heart, Lung, and Blood Institute (NHLBI, R00HL124279), and NIH New Innovator Award (DP2-DK121328) awarded to E.J.C. TEM images were taken with the aid of USC Center of Excellence in Nano Imaging.
Footnotes
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
Conflict of Interest
The authors declare no conflict of interest.
Contributor Information
Deborah D. Chin, Department of Biomedical Engineering, University of Southern California, Los Angeles USC 90089 CA, USA
Christopher Poon, Department of Biomedical Engineering, University of Southern California, Los Angeles USC 90089 CA, USA.
Noah Trac, Department of Biomedical Engineering, University of Southern California, Los Angeles USC 90089 CA, USA.
Jonathan Wang, Department of Biomedical Engineering, University of Southern California, Los Angeles USC 90089 CA, USA.
Jackson Cook, Department of Biomedical Engineering, University of Southern California, Los Angeles USC 90089 CA, USA.
Johan Joo, Department of Biomedical Engineering, University of Southern California, Los Angeles USC 90089 CA, USA.
Zhangjingyi Jiang, Department of Biomedical Engineering, University of Southern California, Los Angeles USC 90089 CA, USA.
Naomi Sulit Sta Maria, Department of Physiology and Neuroscience, Zilkha Neurogenetic, Institute and Keck School of Medicine, University of Southern California, Los Angeles 90033 CA, USA.
Russell E. Jacobs, Department of Physiology and Neuroscience, Zilkha Neurogenetic, Institute and Keck School of Medicine, University of Southern California, Los Angeles 90033 CA, USA
Eun Ji Chung, Department of Biomedical Engineering, University of Southern California, Los Angeles USC 90089 CA, USA; Eli and Edythe Broad Center for Regenerative Medicine and Stem Cell, Research, Keck School of Medicine, University of Southern California, Los Angeles 90033 CA, USA; Norris Comprehensive Cancer Center, Keck School of Medicine, University of Southern California, Los Angeles 90033 CA, USA; Department of Chemical Engineering and Materials Science, University of Southern California, Los Angeles USC 90089 CA, USA; Department of Medicine, Division of Nephrology and Hypertension, Keck School of Medicine, University of Southern California, Los Angeles 90033 CA, USA; Department of Surgery, Division of Vascular Surgery and Endovascular, Therapy, Keck School of Medicine, University of Southern California, Los Angeles 90033 CA, USA.
References
- [1].Heron M, Anderson RN, NCHS Data Brief 2016, 254, 1. [PubMed] [Google Scholar]
- [2].Libby P, Arterioscler., Thromb., Vasc. Biol 2012, 32, 2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Naghavi M, Libby P, Falk E, Casscells SW, Litovsky S, Rumberger J, Badimon JJ, Stefanadis C, Moreno P, Pasterkamp G, Fayad Z, Stone PH, Waxman S, Raggi P, Madjid M, Zarrabi A, Burke A, Yuan C, Fitzgerald PJ, Siscovick DS, de Korte CL, Aikawa M, Airaksinen KE, Assmann G, Becker CR, Chesebro JH, Farb A, Galis ZS, Jackson C, Jang IK, et al. , Circulation 2003, 108, 1772.14557340 [Google Scholar]
- [4].Fuster V, Badimon L, Badimon JJ, Chesebro JH, Engl N. J. Med 1992, 326, 242. [DOI] [PubMed] [Google Scholar]
- [5].Falk E, Nakano M, Bentzon JF, Finn AV, Virmani R, Eur. Heart J 2013, 34, 719. [DOI] [PubMed] [Google Scholar]
- [6].Finn AV, Nakano M, Narula J, Kolodgie FD, Virmani R, Arterioscler., Thromb., Vasc. Biol 2010, 30, 1282. [DOI] [PubMed] [Google Scholar]
- [7].Simionescu N, Vasile E, Lupu F, Popescu G, Simionescu M, Am. J. Pathol 1986, 123, 109. [PMC free article] [PubMed] [Google Scholar]
- [8].Szmitko PE, Wang CH, Weisel RD, de Almeida JR, Anderson TJ, Verma S, Circulation 2003, 108, 1917. [DOI] [PubMed] [Google Scholar]
- [9].Salcedo R, Ponce ML, Young HA, Wasserman K, Ward JM, Kleinman HK, Oppenheim JJ, Murphy WJ, Blood 2000, 96, 34. [PubMed] [Google Scholar]
- [10].Tabas I, Garcia-Cardena G, Owens GK, J. Cell Biol 2015, 209, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Swirski FK, Pittet MJ, Kircher MF, Aikawa E, Jaffer FA, Libby P, Weissleder R, Proc. Natl. Acad. Sci. USA 2006, 103, 10340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Kashiwagi M, Imanishi T, Tsujioka H, Ikejima H, Kuroi A, Ozaki Y, Ishibashi K, Komukai K, Tanimoto T, Ino Y, Kitabata H, Hirata K, Akasaka T, Atherosclerosis 2010, 212, 171. [DOI] [PubMed] [Google Scholar]
- [13].Fromen CA, Fish MB, Zimmerman A, Adili R, Holinstat M, Eniola-Adefeso O, Bioeng. Transl. Med 2016, 1, 103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Lewis DR, Petersen LK, York AW, Ahuja S, Chae H, Joseph LB, Rahimi S, Uhrich KE, Haser PB, Moghe PV, Cardiovasc. Res 2016, 109, 283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Spinetti G, Wang M, Monticone R, Zhang J, Zhao D, Lakatta EG, Arterioscler., Thromb., Vasc. Biol 2004, 24, 1397. [DOI] [PubMed] [Google Scholar]
- [16].Schecter AD, Berman AB, Yi L, Ma H, Daly CM, Soejima K, Rollins BJ, Charo IF, Taubman MB, J. Leukocyte Biol 2004, 75, 1079. [DOI] [PubMed] [Google Scholar]
- [17].Goetzl EJ, Banda MJ, Leppert D, J. Immunol 1996, 156, 1. [PubMed] [Google Scholar]
- [18].Libby P, J. Clin. Invest 2013, 123, 3201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Nikkari ST, O’Brien KD, Ferguson M, Hatsukami T, Welgus HG, Alpers CE, Clowes AW, Circulation 1995, 92, 1393. [DOI] [PubMed] [Google Scholar]
- [20].Turk BE, Huang LL, Piro ET, Cantley LC, Nat. Biotechnol 2001, 19, 661. [DOI] [PubMed] [Google Scholar]
- [21].Vengrenyuk Y, Carlier S, Xanthos S, Cardoso L, Ganatos P, Virmani R, Einav S, Gilchrist L, Weinbaum S, Proc. Natl. Acad. Sci. USA 2006, 103, 14678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Chin DD, Chowdhuri S, Chung EJ, Regener. Eng. Transl. Med 2019, 5, 74. [PMC free article] [PubMed] [Google Scholar]
- [23].Tarkin JM, Dweck MR, Evans NR, Takx RA, Brown AJ, Tawakol A, Fayad ZA, Rudd JH, Circ. Res 2016, 118, 750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Rudd JH, Hyafil F, Fayad ZA, Arterioscler., Thromb., Vasc. Biol 2009, 29, 1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Quillard T, Libby P, Circ. Res 2012, 111, 231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Choudhury RP, Fuster V, Badimon JJ, Fisher EA, Fayad ZA, Arterioscler., Thromb., Vasc. Biol 2002, 22, 1065. [DOI] [PubMed] [Google Scholar]
- [27].DiStasio N, Lehoux S, Khademhosseini A, Tabrizian M, Materials 2018, 11, 754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Kosaka N, Ogawa M, Choyke PL, Kobayashi H, Future Oncol 2009, 5, 1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Semelka RC, Ramalho M, AlObaidy M, Ramalho J, Am. J. Roentgenol 2016, 207, 229. [DOI] [PubMed] [Google Scholar]
- [30].Yoo SP, Pineda F, Barrett JC, Poon C, Tirrell M, Chung EJ, ACS Omega 2016, 1, 996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Sherry AD, Caravan P, Lenkinski RE, J. Magn. Reson. Imaging 2009, 30, 1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Reynolds CH, Annan N, Beshah K, Huber JH, Shaber SH, Lenkinski RE, Wortman JA, J. Am. Chem. Soc 2000, 122, 8940. [Google Scholar]
- [33].Poon C, Chowdhuri S, Kuo C-H, Fang Y, Alenghat FJ, Hyatt D, Kani K, Gross ME, Chung EJ, ACS Biomater. Sci. Eng 2017, 3, 3273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Chin DD, Wang J, Mel de Fontenay M, Plotkin A, Magee GA, Chung EJ, J. Mater. Chem. B 2019, 7, 6449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Chung EJ, Cheng Y, Morshed R, Nord K, Han Y, Wegscheid ML, Auffinger B, Wainwright DA, Lesniak MS, Tirrell MV, Biomaterials 2014, 35, 1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Lobatto ME, Calcagno C, Millon A, Senders ML, Fay F, Robson PM, Ramachandran S, Binderup T, Paridaans MP, Sensarn S, Rogalla S, Gordon RE, Cardoso L, Storm G, Metselaar JM, Contag CH, Stroes ES, Fayad ZA, Mulder WJ, ACS Nano 2015, 9, 1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Tarbell JM, Cardiovasc. Res 2010, 87, 320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Chen YL, Jan KM, Lin HS, Chien S, Atherosclerosis 1995, 118, 89. [DOI] [PubMed] [Google Scholar]
- [39].Kelley WJ, Safari H, Lopez-Cazares G, Eniola-Adefeso O, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol 2016, 8, 909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Yamamoto Y, Nagasaki Y, Kato Y, Sugiyama Y, Kataoka K, J. Controlled Release 2001, 77, 27. [DOI] [PubMed] [Google Scholar]
- [41].Blanco E, Shen H, Ferrari M, Nat. Biotechnol 2015, 33, 941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Chung EJ, Mlinar LB, Nord K, Sugimoto MJ, Wonder E, Alenghat FJ, Fang Y, Tirrell M, Adv. Healthcare Mater 2015, 4, 367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Poon C, Sarkar M, Chung EJ, J. Vis. Exp 2017, 129, e56625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Marques Neto LM, Kipnis A, Junqueira-Kipnis AP, Front. Immunol 2017, 8, 239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Rohrer M, Bauer H, Mintorovitch J, Requardt M, Weinmann HJ, Invest. Radiol 2005, 40, 715. [DOI] [PubMed] [Google Scholar]
- [46].Wang J, Poon C, Chin D, Milkowski S, Lu V, Hallows KR, Chung EJ, Nano Res 2018, 11, 5584. [Google Scholar]
- [47].Orr GA, Chrisler WB, Cassens KJ, Tan R, Tarasevich BJ, Markillie LM, Zangar RC, Thrall BD, Nanotoxicology 2011, 5, 296. [DOI] [PubMed] [Google Scholar]
- [48].Liu X, Jin Q, Ji Y, Ji J, J. Mater. Chem 2012, 22, 1916. [Google Scholar]
- [49].Weiss D, Kools JJ, Taylor WR, Circulation 2001, 103, 448. [DOI] [PubMed] [Google Scholar]
- [50].Weiss D, Sorescu D, Taylor WR, Am. J. Cardiol 2001, 87, 25C. [DOI] [PubMed] [Google Scholar]
- [51].Sato K, Nakano K, Katsuki S, Matoba T, Osada K, Sawamura T, Sunagawa K, Egashira K, J. Atheroscler. Thromb 2012, 19, 986. [DOI] [PubMed] [Google Scholar]
- [52].Lu H, Howatt DA, Balakrishnan A, Moorleghen JJ, Rateri DL, Cassis LA, Daugherty A, JoVE 2015, 103, e53191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Matoba T, Sato K, Egashira K, Curr. Opin. Lipidol 2013, 24, 419. [DOI] [PubMed] [Google Scholar]
- [54].Mulder WJ, Strijkers GJ, Briley-Saboe KC, Frias JC, Aguinaldo JG, Vucic E, Amirbekian V, Tang C, Chin PT, Nicolay K, Fayad ZA, Magn. Reson. Med 2007, 58, 1164. [DOI] [PubMed] [Google Scholar]
- [55].Lipinski MJ, Amirbekian V, Frias JC, Aguinaldo JG, Mani V, Briley-Saebo KC, Fuster V, Fallon JT, Fisher EA, Fayad ZA, Magn. Reson. Med 2006, 56, 601. [DOI] [PubMed] [Google Scholar]
- [56].Rogers WJ, Basu P, Atherosclerosis 2005, 178, 67. [DOI] [PubMed] [Google Scholar]
- [57].Kooi ME, Cappendijk VC, Cleutjens KB, Kessels AG, Kitslaar PJ, Borgers M, Frederik PM, Daemen MJ, van Engelshoven JM, Circulation 2003, 107, 2453. [DOI] [PubMed] [Google Scholar]
- [58].Fredman G, Kamaly N, Spolitu S, Milton J, Ghorpade D, Chiasson R, Kuriakose G, Perretti M, Farokzhad O, Tabas I, Sci. Transl. Med 2015, 7, 275ra20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Mook OR, Van Overbeek C, Ackema EG, Van Maldegem F, Frederiks WM, J. Histochem. Cytochem 2003, 51, 821. [DOI] [PubMed] [Google Scholar]
- [60].Gkantidis N, Blumer S, Katsaros C, Graf D, Chiquet M, PLoS One 2012, 7, e47762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Porto IM, Rocha LB, Rossi MA, Gerlach RF, J. Histochem. Cytochem 2009, 57, 615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Sager HB, Dutta P, Dahlman JE, Hulsmans M, Courties G, Sun Y, Heidt T, Vinegoni C, Borodovsky A, Fitzgerald K, Wojtkiewicz GR, Iwamoto Y, Tricot B, Khan OF, Kauffman KJ, Xing Y, Shaw TE, Libby P, Langer R, Weissleder R, Swirski FK, Anderson DG, Nahrendorf M, Sci. Transl. Med 2016, 8, 342ra80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Bejarano J, Navarro-Marquez M, Morales-Zavala F, Morales JO, Garcia-Carvajal I, Araya-Fuentes E, Flores Y, Verdejo HE, Castro PF, Lavandero S, Kogan MJ, Theranostics 2018, 8, 4710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Aime S, Caravan P, J. Magn. Reson. Imaging 2009, 30, 1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Ghaghada K, Hawley C, Kawaji K, Annapragada A, Mukundan S Jr., Acad. Radiol 2008, 15, 1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Briley-Saebo KC, Shaw PX, Mulder WJ, Choi SH, Vucic E, Aguinaldo JG, Witztum JL, Fuster V, Tsimikas S, Fayad ZA, Circulation 2008, 117, 3206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Poon C, Chin D, Joo J, Ong V, Jiang Z, Cheng K, Chang T, Chung EJ, Multifuctional Peptide Micelles for Gene Therapy in Atherosclerosis, SAGE Publishing, Washington, DC: 2019. [Google Scholar]
- [68].Chung EJ, Mlinar LB, Sugimoto MJ, Nord K, Roman BB, Tirrell M, Nanomedicine 2015, 11, 479. [DOI] [PubMed] [Google Scholar]
- [69].Vogel B, Siebert H, Hofmann U, Frantz S, MethodsX 2015, 2, 124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Chen LJ, Xu YL, Song B, Yu HM, Oudit GY, Xu R, Zhang ZZ, Jin HY, Chang Q, Zhu DL, Zhong JC, Peptides 2016, 79, 49. [DOI] [PubMed] [Google Scholar]
- [71].Rogosnitzky M, Branch S, BioMetals 2016, 29, 365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Barnes SR, Ng TSC, Santa-Maria N, Montagne A, Zlokovic BV, Jacobs RE, BMC Med. Imaging 2015, 15, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.