

Summary
The HCN1–4 channel family is responsible for the hyperpolarization-activated cation current If/Ih that controls automaticity in cardiac and neuronal pacemaker cells. We present cryoelectron microscopy (cryo-EM) structures of HCN4 in the presence or absence of bound cAMP, displaying the pore domain in closed and open conformations. Analysis of cAMP-bound and -unbound structures sheds light on how ligand-induced transitions in the channel cytosolic portion mediate the effect of cAMP on channel gating and highlights the regulatory role of a Mg2+ coordination site formed between the C-linker and the S4-S5 linker. Comparison of open/closed pore states shows that the cytosolic gate opens through concerted movements of the S5 and S6 transmembrane helices. Furthermore, in combination with molecular dynamics analyses, the open pore structures provide insights into the mechanisms of K+/Na+ permeation. Our results contribute mechanistic understanding on HCN channel gating, cyclic nucleotide-dependent modulation, and ion permeation.
Keywords: HCN4, pore, gating, selectivity, permeation, cAMP, metal ion, cryo-EM, molecular dynamics, pacemaker
Graphical abstract
Highlights
-
•
HCN4 structure is shown in ligand-free and ligand-bound state
-
•
Pore domain is shown in closed and in open configuration
-
•
Permeability and selectivity mechanisms of HCN channels are uncovered
-
•
A metal ion coordination site functionally couples cytoplasmic and transmembrane domains
HCN4 channels underlie the pacemaker current that controls heart rate. Saponaro et al. report the structure of HCN4 with the pore in closed and in open configuration and provide information on ion permeability and selectivity. In HCN4, a metal ion coordination site functionally connects the C-linker to the S4-S5 linker.
Introduction
Although HCN channels are members of the voltage-gated potassium channel (Kv) superfamily, they differ from Kv channels in three important ways. First, they are opened by hyperpolarization, not by depolarization. Second, they discriminate relatively poorly K+ from Na+. Third, their opening is facilitated by binding of cyclic nucleotides to a C-terminal cytosolic cyclic nucleotide binding domain (CNBD). A single-particle cryoelectron microscopy (cryo-EM) structure of HCN1 was recently reported at 3.5-Å resolution (Lee and MacKinnon, 2017) and confirmed that, similar to Kv channels, HCNs are tetrameric proteins. Each protomer contains six α-helixes (S1–S6) in the transmembrane (TM) domain, including a voltage sensor domain (VSD) composed of S1–S4 and a pore domain (PD) composed of S5, a re-entrant P region, and S6. HCN1 further presents folded domains at both cytosolic termini. At the N terminus, the HCN domain directly precedes the S1 TM segment of the VSD, while at the C terminus, a ∼80-amino acid C-linker connects CNBD and S6 TM segment. Akin to cyclic nucleotide-gated (CNG) and Ether-a-go-go (Eag) channels, the four subunits adopt a “non-domain-swapped” arrangement of the voltage sensor relative to the pore (James and Zagotta, 2018; Li et al., 2017; Whicher and MacKinnon, 2016). In contrast, the cytosolic domains form a “swapped” tetrameric arrangement in which the C-linker of one subunit rests on the C-linker of the adjacent one (Zagotta et al., 2003), while the HCN domain interacts with the adjacent and the opposite subunits (Porro et al., 2019). This distinctive tetrameric arrangement of the cytosolic domains is thought to contribute to mediating the transfer of the signal initiated by cAMP binding (in the CNBD) to the TM domain (Porro et al., 2019).
Of note, in all HCN1 structures solved thus far, the pore is closed, even when the alleged hyperpolarized conformation of S4 was imposed on the VSD by chemical crosslinking (Lee and MacKinnon, 2019).
Despite the significance of the HCN1 structural studies, several fundamental questions about HCN properties remain unsolved. The lack of an open-pore structure leaves unexplained the nature of the conformational change that gates the channel open. The HCN1 structures provide no insight into the mechanisms of ion permeation, particularly the 4:1 selectivity for K+ versus Na+ (Lyashchenko and Tibbs, 2008; Moroni et al., 2000), which is unusually low for Kv channels, and the remarkably low single-channel conductance (DiFrancesco, 1986; Johnson and Zagotta, 2005; Thon et al., 2013). Additionally, the HCN1 structures provide no insight into the mechanism of cAMP modulation of gating, because negligible conformational changes are seen upon cAMP binding, which may reflect the low cAMP efficacy on HCN1 gating relative to HCN2 or HCN4 (Chen et al., 2001b; Viscomi et al., 2001; Wainger et al., 2001). A systematic comparison of the structure of HCN1 with those of other HCN isoforms may help resolve such fundamental questions.
Here, we present a study of the HCN4 isoform, the predominant HCN channel isoform expressed in sinoatrial node myocytes and the major determinant of the cardiac pacemaker “funny” current (If) (Altomare et al., 2003; Brown et al., 1979; Shi et al., 1999). By comparing HCN4 structures obtained by cryo-EM in the absence and presence of cAMP, we were able to reconstruct the movements induced by ligand binding in the cytosolic C-linker/CNBD region and uncover the formation of a unique contact between the C-linker and the TM domain. Biochemical and electrophysiology experiments show that this contact is HCN4 specific and contributes to the marked cAMP response of this isoform. Two of the presented structures show the PD in an ostensibly open conformation, since in silico docking and molecular dynamics simulations demonstrate it is able to bind the open channel blocker ivabradine (Bucchi et al., 2006) and conduct Na+ and K+ ions at the expected rate and selectivity. Overall, our data advance the understanding of gating and permeation in HCN4 channels, in particular, and in HCN channels in general.
Results
Structure of the HCN4 channel
For single-particle cryo-EM structural studies, we employed a construct (hereafter termed HCN4 for simplicity; Figure S1) carrying an internal deletion that removes a poorly conserved region in the C terminus (Lee and MacKinnon, 2017). Functional characterization of this construct shows that it maintains the properties that distinguish HCN4 from HCN1 (slower kinetics, hyperpolarized V1/2, large response to cAMP; Figures S2A–S2C; Tables S1 and S2). This construct yielded high protein levels in HEK293 cells, following solubilization and detergent exchange, using either lauryl maltose neopentyl glycol with cholesteryl hemisuccinate (LMNG/CHS), a non-ionic detergent, or amphipols, membrane mimetic polymers (Figures S2D and S2E). HCN4 cryo-EM analyses were carried out to an overall resolution of 3.2 Å for LMNG/CHS-solubilized particles purified in cAMP-bound (holo) and cAMP-free (apo/LC) states (Figures S3–S6; Table 1) and to an overall resolution of 3.6 Å for amphipol reconstructed particles in the cAMP-free (apo/AM) state (Figures S7 and S8; Table 1).
Table 1.
Cryo-EM data collection, image processing, and model refinement statistics
| HCN4holo | HCN4apo/LC | HCN4apo/AM | |
| Data collection and image processing | |||
| Microscope | Titan Krios | Titan Krios | TALOS Arctica |
| Voltage (kV) | 300 | 300 | 200 |
| Camera | K2 Summit | K2 Summit | Falcon 3EC |
| Magnification | 29,000 | 29,000 | 120,000 |
| Total electron dose (e-/Å2) | 67.88 | 71.85 | 40.0 |
| Defocus range (μm) | −1 to −1.8 | −1.1 to −2 | −0.5 to −2.0 |
| Pixel size (Å) | 0.84 | 0.834 | 0.889 |
| Micrographs (no.) | 4,478 | 4,465 | 1,571 |
| Symmetry imposed | C4 | C4 | C4 |
| Initial particle images (no.) | 2,006,780 | 914,005 and 1,039,436 | 100,130 |
| Final particle images (no.) | 54,828 | 51,758 | 11,406 |
| Resolution (Å) (fourier shell correlation threshold) | 3.2 (0.143) | 3.2 (0.143) | 3.6 (0.143) |
| Sharpening B-factor (Å2) | −77.8 | −80.3 | −52 |
| EMDB code | EMD-12513 | EMD-12512 | EMD-12466 |
| Model refinement | |||
| Nonhydrogen atoms | 16,588 | 16,128 | 15,436 |
| Protein residues | 2,004 | 1,960 | 1,876 |
| cAMP molecules | 4 | – | – |
| N-acetyl-d-glucosamine molecules | – | 4 | – |
| Root-mean-square deviations (RMSDs) | |||
| Bond lengths (Å) | 0.008 | 0.008 | 0.010 |
| Bond angles (°) | 0.960 | 0.917 | 1.158 |
| Ramachandran plot | |||
| Favored (%) | 97.19 | 92.32 | 93.98 |
| Allowed (%) | 2.81 | 7.68 | 6.02 |
| Disallowed (%) | 0.00 | 0.00 | 0.00 |
| Validation | |||
| Molprobity score | 1.52 | 2.29 | 2.09 |
| Clashscore | 6.75 | 16.35 | 15.25 |
| Poor rotamers (%) | 0.22 | 1.35 | 0.88 |
| EM-Ringer Score | 3.75 | 2.64 | 1.55 |
| Map-model correlation | 0.84 | 0.82 | 0.80 |
| PDB code | 7NP4 | 7NP3 | 7NMN |
Figure 1 shows the holo structure of HCN4, which is a tetrameric protein overall similar in its 3D structure to cAMP-bound HCN1 (Lee and MacKinnon, 2017). The HCN4 tetramer (Figure 1A) shows the general architecture of HCN1, with the VSD of one subunit packed against the pore of the same subunit (non-swapped), while the cytosolic domains are swapped such that the C-linker of one protomer (green) is found under the TM domain of the adjacent one (blue). Figure 1B shows the HCN4 protomer, with the conserved structural features labeled. A more detailed comparison of HCN4 holo with HCN1 holo (PDB: 5U6P) highlights significant local differences between the Cα chains of the protomers (color-coded gray to red in Figure 1C). Structural deviations are observed in particular in the S4-S5 linker and in the C-linker helices A′ and B′ underneath.
Figure 1.

HCN4 structure and its comparison with that of HCN1
(A) Structure of the HCN4 channel tetramer in cAMP-bound state (holo), in a cross-membrane view. For clarity, only two subunits are shown in full (tan and blue), while for the other two subunits (black and green), only the cytosolic C-linker/CNBD domains are presented.
(B) Protomer of HCN4 holo with structural elements indicated.
(C) HCN4 comparison with HCN1. Only two subunits in the tetramer are shown in full, while for the other two subunits the cytosolic C-linker/CNBD domains are shown. HCN4 structure color-coded (gray-to-red) according to the local deviations observed for the Cα atoms of HCN4 versus HCN1 holo (PDB: 5U6P). Gray color represents 0.5-Å displacements, and red color represents displacements up to 5 Å, as shown by the bar.
An intersubunit metal ion coordination site connects the TM to the cytosolic domain
Despite the high sequence identity with HCN1, the S4-S5 linker adopts a distinct conformation in HCN4: the HCN4 S4 segment bends after residue I405 (highlighted in red in Figure 2A), and the S4-S5 linker develops a full helical turn. As a result, HCN4 H407 is exposed to the cytosolic side, while in HCN1 the equivalent histidine H286 is not. This conformation promotes in HCN4 the contact of the S4-S5 linker of one subunit with the underlying B′ helix in the C-linker of its neighboring subunit. Four residues, H407 and D411 from the S4-S5 linker and H553 and E557 from the B′ helix, build a sort of tetrahedral arrangement (hereafter “tetrad”) that coordinates a central ion clearly resolved as extra density in the HCN4 map (Figure 2B; density map file EMDB: EMD-12513; see also EMDB: EMD-0094, PDB: 6GYO). This arrangement is not present in HCN1, where H286 and D290 on the S4-S4 linker fall farther away from H432 and E436 of the C-linker B′ helix.
Figure 2.

Structural, biochemical, and functional evidence for the tetrad, connecting the transmembrane and cytosolic domains in HCN4
(A) Comparison of the S4-S5 linker of HCN1 (yellow) and HCN4 (blue). Backbones of I405 in HCN4 and the corresponding I284 in HCN1 are shown in red.
(B) Ribbon representation of S4-S5 linker of one subunit (blue) and underlying C-linker “elbow” (A′–B′ helices) of adjacent subunit (green) of HCN4. Four residues forming the ion coordination site (tetrad) in HCN4 are shown as sticks and labeled (H407, D411, H553, E557). Ion coordinated by the tetrad is represented as a purple sphere.
(C and D) Thermostability assay based on fluorescence-detection size-exclusion chromatography (fSEC-TS) was employed to quantify the change in melting temperature of purified protein in absence or presence of Mg2+ (see STAR Methods). (C) Representative melting curves for holo HCN4 protein purified in LMNG/CHS. Normalized fluorescence intensity (FI) plotted over pre-conditioning temperature (°C). The protein was incubated without (black) or with 10 mM EDTA (red), or, after EDTA removal, with 10 mM MgCl2 (blue). Wild-type protein (HCN4 WT) is shown on the left; double-mutant H407A/H553A (HCN4 HA/HA) is shown on the right. Data points are fitted with sigmoidal dose-response equation (see STAR Methods). (D) Mean denaturation midpoint temperature (Tm) for HCN4 WT in control (53.6°C ± 1.5°C), EDTA (47.8°C ± 0.8°C), + Mg2+ (53.7°C ± 0.4°C); for HCN4 HA/HA: control (44°C ± 0.5°C), EDTA (44.8°C ± 0.1°C), + Mg2+ (45.3°C ± 0.3°C). Values are mean of n = 3 experiments ± SEM. Statistical analysis performed with one-way ANOVA, followed by Fisher’s test (∗∗p < 0.01).
(E) Representative current traces recorded in HEK293 cells expressing WT and mutant E557A HCN4 channels. Patch-clamp recordings were performed in whole-cell configuration, with 30 μM cAMP in pipette solution where indicated. Voltage step protocol was from −30 to −150 mV with −15-mV increments; only currents recorded at −45, −90, and −135 mV are shown. Scale bars: 200 pA × 0.5 s.
(F) cAMP-induced shift (mV) on half activation voltages (V1/2) calculated by fitting a Boltzmann equation to the data (see STAR Methods). Current recordings and activation curves for all WT and mutant channels displayed are shown in Figures S9–S11. V1/2 values are reported in Table S1. Mutations H407A, H553A, and E557A introduced in HCN4, either alone or in combination, reduce the cAMP effect (Figure S9), whereas an equivalent mutation in HCN1 (E436A) does not (see also Figure S10). Addition of 60 mM EDTA to pipette solution of WT channels reduces cAMP response in HCN4 (Figure S9), but not in HCN1 and HCN2 (Figure S11). Values are means of n > 3 experiments ± SEM. Statistical analysis performed with one-way ANOVA, followed by Fisher’s test (∗p < 0.05).
The tetrad is found at the interface between the TM and the cytosolic domains, a key position in HCN channel gating (Chen et al., 2001a; Kwan et al., 2012; Porro et al., 2019; Weiβgraeber et al., 2017). We thus investigated whether the tetrad contributes to structural and functional properties of HCN4 and whether these effects are isoform specific.
The tetrad mediates the effect of cAMP on HCN4 channel gating
The combination of histidines and negatively charged residues in the tetrad is reminiscent of a Zn2+ ion coordination sphere, but it could also support Mg2+ coordination (Dokmaníc et al., 2008). Because our protein purification procedure takes place in 10 mM Mg2+ and millimolar concentrations of this ion are physiologically present in cells, we tested whether Mg2+ is the coordinating ion.
Metals are often present in many macromolecules to maintain their structural stability (Zheng et al., 2017a). We thus reasoned that Mg2+ coordination, by promoting formation of the tetrad, should affect HCN4 thermal stability. To test this hypothesis, we performed a thermal denaturation assay on the purified protein to evaluate whether the removal of Mg2+ could affect HCN4 melting temperature (Tm). Figure 2C (left) shows the melting curve for wild-type (WT) HCN4, the effect of 10 mM EDTA, a chelating agent for Mg2+, and the effect of 10 mM Mg2+ that was added after EDTA removal. Addition of EDTA destabilized the protein, decreasing the Tm by about 6°C. The effect of EDTA was fully rescued by addition of 10 mM Mg2+ following EDTA removal. Average values of Tm, reported in Figure 2D, were as follows: control = 53.6°C ± 1.5°C, EDTA = 47.8°C ± 0.8°C, + Mg2+ = 53.7°C ± 0.4°C. To confirm that the effect of Mg2+ removal is specifically related to tetrad stability, we disrupted the latter by point mutations. Given the major role of histidines in metal ion coordination sites (Dokmaníc et al., 2008), H407 and H553 were mutated into alanine, and the resulting double mutant (HA/HA) was tested for thermal stability as above. The double mutation clearly destabilized the protein, decreasing the Tm by about 10°C (44°C ± 0.5°C) (Figure 2C, right). Furthermore, the Tm was unaffected either by the addition of EDTA or by addition of Mg2+ after EDTA removal (EDTA = 44.8°C ± 0.1°C, + Mg2+ = 45.3°C ± 0.3°C), indicating that the double mutation prevents Mg2+ binding (Figure 2D).
We then examined the following hypothesis: if the tetrad favors coupling between cytosolic and TM domains, and given that this contact crucially controls cAMP response in HCN (Porro et al., 2019), then mutations perturbing its stability should affect the channel response to cAMP. HCN4 channels carrying H407A, D411, H553A, or E557A mutations, either individually or in combination, were expressed in HEK293 cells and tested for cAMP response by patch-clamp recording (Figure S9; Table S1). Measurements were performed in whole-cell configuration, with cAMP in the pipette solution where indicated (see STAR Methods). All mutants apart from D411A, which was not functional, generated HCN4-type currents, and in line with our prediction, their cAMP response was greatly reduced. Figure 2E shows, as an example for the behavior of all mutants, currents recorded from E557A. In the mutant channel, cAMP did neither accelerate activation kinetics nor increase current amplitude as much as in the WT. The latter effect is best visible at the intermediate voltage of −90 mV. The cAMP-induced shift in the activation curve of this mutant is roughly halved (WT = 18.4 ± 1.6 mV; E557A = 10 ± 2.6 mV), and a similar strong reduction was found in all mutants (Figure 2F; Figure S9; Table S1).
If the effect of the mutations is due to a weakening of the tetrad, then removal of Mg2+ should result in a quantitatively similar effect on the channel response to cAMP. In line with the prediction, addition of EDTA to the pipette, for removal of Mg2+ from the intracellular solution, reduced the effect of cAMP to a similar extent as the mutations (Figure 2F; Figure S9; Table S1).
The fact that the tetrad was not found in HCN1 structures (PDB: 5U6P and 5U6O) suggests that it might be an isoform-specific feature of HCN4. To address this question, we introduced the mutation equivalent to E557A in HCN1 (E436A) and tested the response to cAMP of this isoform (Figure 2F; Figure S10; Table S1). This mutation did not change the cAMP-induced effect in HCN1, supporting the structural evidence that the tetrad is not formed in this isoform. In HCN2 as well, the equivalent mutation E478A did not affect the channel response to cAMP (Porro et al., 2019). Finally, Mg2+ removal by EDTA in HCN1 and HCN2 did not alter cAMP sensitivity of the channels (Figure 2F; Figure S11; Table S1). All together, these experiments support the view that formation of the tetrad and its functional role are unique to HCN4.
Conformational transitions in the HCN4 C-linker and CNBD upon cAMP binding
In line with the reasoning and results presented above, the tetrad is not found in cAMP-free HCN4 structures (Figure 3B; Figure S12). In the apo/LC structure, the density for three amino acid side chains in the tetrad, H407 and D411 on the S4-S5 linker and E557 on the C-linker B′ helix, is missing. This can be ascribed to a different conformation adopted by the C-linker A′-B′ helices compared with holo HCN4. Superimposition of helices A′ and B′ from apo/LC and holo structures (Figure 3C) highlights a tilt of the two helices, leading to tetrad formation in the holo. By comparing the whole cytosolic domain (C-linker and CNBD) in the apo and holo structures, we were able to reconstruct the cAMP-induced movements leading to C-linker tilting. As described in previous studies (Akimoto et al., 2014; Lee and MacKinnon, 2017; Saponaro et al., 2014), upon cAMP binding, the β-roll of the CNBD rotates toward the C-helix (downward in Figure 3D), while the N-terminal helical bundle, comprising helices E′ and F′, moves in the opposite direction (upward in Figure 3D). In HCN4, such opposite shifts are forcefully transmitted to the A′ and B′ helices of the C-linker via contacts within and between subunits. In particular, the downward rotation of the CNBD pulls the B′ helix of the same subunit via the R552-D622 salt bridge (Craven and Zagotta, 2004; Craven et al., 2008) (circled in Figure 3E). The upward movement of the E′ and F′ helices is transmitted to the A′ helix of the C-linker of the adjacent subunit via an extended network of contacts between helices C′ and D′ of one subunit and helices A′ and B′ of the neighboring subunit, which build the well-characterized “elbow on shoulder” arrangement of the C-linker (Gross et al., 2018; Weiβgraeber et al., 2017; Zagotta et al., 2003). This readjusts the position of helices A′ and B′ (“the elbow”) (Figure 3C) relative to the S4-S5 linker allowing tetrad formation. More specifically, we observed a combined movement of helices A′ and B′, measured as an upward 2.9-Å displacement of the Cα of S540 at the tip of the elbow and a 3.8-Å lateral-downward displacement of the Cα of H558 at the end of the B′ helix. Similar cAMP-induced rearrangements are found in the HCN4 structures solved by the Structural Genomics Consortium (apo, PDB: 6GYN; holo, PDB: 6GYO). Such structural rearrangement differs largely from what is observed in HCN1, where C-linker displacement in response to cAMP binding is negligible, and the A′ and B′ helices do not come in contact with the TM domain portion of the channel (Video S1). The marked difference in the degree of movement induced by cAMP binding in HCN4 versus HCN1 correlates well with these isoforms’ functional response, wherein ligand binding shifts the voltage activation midpoint by ∼15–20 mV toward more depolarized potentials in HCN4, compared with only ∼5–7 mV for HCN1 (Chen et al., 2001b; Viscomi et al., 2001; Wainger et al., 2001). The tight connection between the C-linker and the TM domain induced by cAMP in HCN4 could conceivably improve the coupling between voltage sensor movement and pore opening. This is similar to the electromechanical coupling recently proposed for the hyperpolarization-activated KAT1 channels (Clark et al., 2020).
Figure 3.

cAMP-dependent rearrangement of the cytosolic region of HCN4 channels
(A) Ribbon representation of HCN4 apo/LC dimer in gray, along with C-linker/CNBD of a third subunit in the tetramer shown in orange.
(B) Ribbon representation of S4-S5 linker of one subunit and C-linker “elbow” (A′-B′ helices) of adjacent subunit of apo/LC HCN4. Density map of tetrad region is shown as gray mesh. Only one of the four residues forming the ion coordination site in HCN4 holo (tetrad) was modeled and is shown as sticks and labeled H553.
(C) Superimposition of C-linker “elbow” (A′, B′ helices) of HCN4 apo/LC (orange) and HCN4 holo (blue) shown in two orientations, rotated by 45 degrees. Dotted black lines indicate displacement of 2.9 Å in Cα of S540 (at the tip of the elbow) and of 3.8 Å in Cα of H558 at end of B′ helix; black arrows show direction of movement induced by cAMP binding to underlying CNBD.
(D) Superimposition of CNBDs of HCN4 in the apo (orange, apo/LC) and holo (blue) configurations. The structural elements are labeled (helices E′, F′, beta roll, helices A–C). Arrows indicate direction of movements induced by cAMP binding, downward (in this figure orientation) in beta roll and upward in helices E′ and F′, the so-called N-terminal helical bundle (Saponaro et al., 2014).
(E) Ribbon representation of C-linker/CNBD in two adjacent subunits (gray and orange) of HCN4 apo/LC. Helices forming C-linker “elbow” (A′, B′) and “shoulder” (C′, D′) are labeled. Residues involved in intrasubunit interaction between C-linker “elbow” (R552) and CNBD (D622) are shown as green sticks and circled with a dotted black line. Black arrows indicate concerted movements of C-linkers and CNBDs induced by cAMP binding.
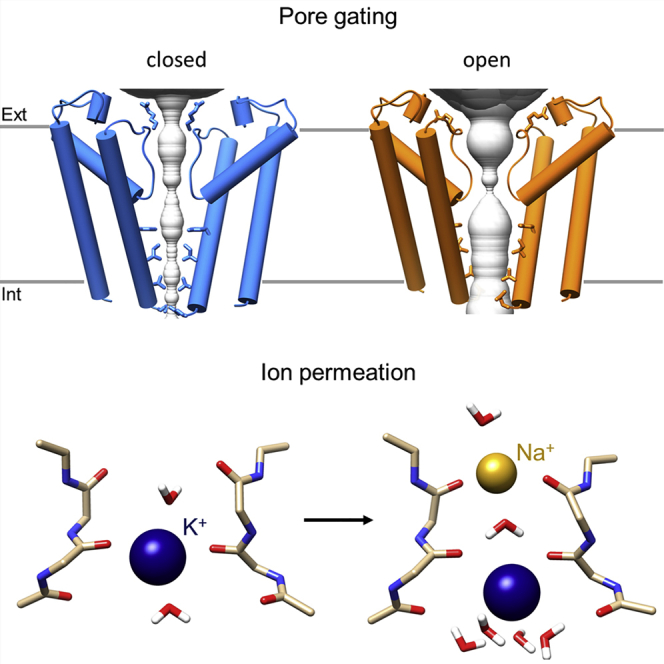
The TM region (S1–S6) displays closed and open pore conformations
The TM region (S1–S6) of HCN4 holo is overall similar to that of HCN1 (Figure 1B). Helices S4, S5, and S6 are found in a quasi-planar array (Figure 4A), tightly connected through a zipper of hydrophilic and hydrophobic interactions (Figure S13A). A crucial observation is, however, that, differently from HCN1, in HCN4 this parallel arrangement of TM helices seems to be further stabilized by lipids. Non-protein density features are visible in the holo structure map, which we assigned as the putative alkyl chains of phospholipids (highlighted in green, red, and yellow in Figure 4A). The green presumed lipid contacts the lower part of the S4 helix, where it connects to the S4-S5 linker, a key position for gating, as discussed before. Helix S5 is sandwiched in between two putative lipids: the red tubular density encircles the helix from one side, while the yellow feature contacts the helix lengthwise from the opposite site. Interestingly, a lipid encircling S5, very similar to the red density shown here, was described in another non-domain-swapped inward rectifier channel, KAT1, as an integral component of the gating machinery (Clark et al., 2020). Nearly identical non-protein densities are also observed in the HCN4 holo structure solved by the Structural Genomics Consortium (EMDB: EMD-0094, PDB: 6GYO).
Figure 4.

Transmembrane domain rearrangements in the three HCN4 structures
(A) Ribbon representation of S4, S5, and S6 TM helices in HCN4 holo (blue) in a cross-membrane view. For clarity, only two subunits are shown. Putative lipid densities contacting S4 (green mesh) and S5 (red and yellow mesh) are shown. Residues involved in polar interaction (dotted line) between S4 (E403) and S5 (N421) are shown as sticks and labeled on one subunit. TM helices S4, S5, and S6 are also labeled on one subunit.
(B and C) Ribbon representation of pore of HCN1 holo (yellow, PDB: 5U6P) (B) and HCN4 holo (blue) (C). For ease of view, only two opposite subunits in the assembled tetramer are shown. The pore diameter is shown as a gray surface. Residues facing pore inner cavity and residue of SF facing extracellular side (A363 in HCN1 and R484 in HCN4) are shown as sticks and labeled. The atoms of side chain of R484 whose density is not resolved are colored in gray.
(D) Ribbon representation of S4, S5, and S6 TM helices in HCN4 apo/LC (orange) in a cross-membrane view. For clarity, only two subunits of tetramer are shown. Lipids are absent, and polar interaction between E403 and N421 is lost. Inset: in comparison with HCN4 holo (blue), the apo/LC structure shows a tilting movement of S5 and rotation of S6, as shown by shift in the side chains of Y507, I511, and Q519.
(E) Superimposition of helices S4, S5, and S6 of HCN4 apo/AM (green) and HCN1 in hyperpolarized conformation (gray, PDB: 6UQF). In HCN4 apo/AM, the lipids are absent, and both the polar interaction E403-N421 and hydrophobic interactions between S4 and S5 are lost (see also Figure S10).
(F and G) Ribbon representation of pore of HCN4 apo/LC (orange) (F) and HCN4 apo/AM (green) (G). Labeling as in (B) and (C). Rotation in lower part of S6 relocates side chains of indicated residues away from cavity, widening solvent-accessible pathway (gray) from cytosolic side.
(H), Plot of pore radii, color-coded as in (B), (C), (F), and (G). The dotted line marks the radius of hydrated K+. Corresponding portions of selectivity filter, cavity, and gate are indicated by black vertical bars. Dotted black lines encompassing (B), (C), and (F)–(H) indicate, respectively, R484 in HCN4, the corresponding A363 residue in HCN1, and glutamine residues at the cytosolic gate (Q519 in HCN4, Q398 in HCN1).
Analysis of the PD (helices S5–S6) shows that the pore of the HCN4 holo structure is closed. Figures 4B and 4C show comparative profiles of the pore diameter of HCN4 and HCN1. The side chains of four residues (Y507, I511, T515, and Q519 in HCN4) are seen to impinge on the central cavity, with the point of minimal bore found at the level of Q519, the same as in HCN1. At the extracellular end, however, the pore of HCN4 appears overall narrower because R484, which substitutes A363 of HCN1, introduces a constriction at the pore entrance (shown by the pore radius plot; Figure 4H). Closer inspection highlights that the position of the R484 side chain further correlates with the conformation adopted by the side chain of residue Y482 in the GYG sequence (Figure S14). Although in HCN1 it points toward the filter inner region (“Y in”), in HCN4 holo, this tyrosine points away from the central axis (“Y out”) (Figures S14A and S14B). The “Y in” conformation is found, however, in other HCN4 pores solved by us (Figure S14C) and others (EMDB: EMD-0093, PDB: 6GYN). In the HCN4 “Y in” conformation, the R484 side chain is stabilized by contact with Y468 on the pore helix, thus rotating away from the central axis and removing the constriction on the pore entrance, which correlates with Y482 in the GYG sequence pointing inward (Figure S14C).
It is interesting to note that the arginine at position 484 is responsible for the sharp dependency of the HCN4 current on extracellular chloride, which is not seen in HCN1 (Wahl-Schott et al., 2005). Our results furthermore highlight a marked flexibility of the GYG sequence of HCN channels. This is in line with prior evidence showing that, differently from Kv channels, this sequence does not contribute to the ion binding sites of HCN channels (Lee and MacKinnon, 2017).
Two key differences are seen between the TM domains of the holo and the apo HCN4 structures presented here. First, there is no evidence for bound lipids in either apo/LC or apo/AM (Figures 4D and 4E). The reason for this is unclear, given that in one case (apo/LC) the purification procedure was the same of HCN4 holo, apart from the absence of cAMP. Intriguingly, in the lipid-free structures, the quasi-planar array of S4, S5, and S6 is lost, suggesting that the presence of lipids may contribute to stabilize the relative positioning of the helices. Thus, in the absence of lipids, the tight network of hydrophilic and hydrophobic interactions between S4 and S5 helices is lost, either partially (loss of hydrophilic interaction; Figure 4D; Figure S13B) or totally (loss of hydrophilic and hydrophobic interactions; Figure 4E; Figure S13C). In either case, these alterations include the disruption of the hydrogen bond between E403 (S4) and N421 (S5) (Figures 4D and 4E), which was previously shown by functional mutagenesis experiments to lead to channels with a large constitutive current component in HCN2 (Chen et al., 2001a) and the invertebrate Strongylocentrotus purpuratus spHCN (Flynn and Zagotta, 2018; Ramentol et al., 2020). Second, the lower end of S5 tilts upward, pointing out of the planar array. The movement of S5 in turn drives the rotation of the lower half of S6, which is connected to it through hydrophobic interactions (Video S2). The rotation of S6 is best seen in the outward movement of the side chains of Y507 and F510. The rotation of S6 displaces Q519, widening the pore cytosolic entrance (Figure 4D, inset; the two black arrows indicate the upward tilt of S5 and the rotation of S6). Analysis of the PD shows that the two lipid-free HCN4 structures indeed display a significant widening of the pore (Figures 4F and 4G) with the pore radius at the lower end of the S6 helices (i.e., at the bundle crossing) increasing from ∼1 to ∼5 Å (Figure 4H). The overall ∼8-Å increase in pore diameter is well above the 6-Å diameter of a solvated K+ ion, suggesting that the pore in the lipid-free structures may be in the open state despite the absence of cAMP and a voltage of 0 mV. In apo/LC, the pore radius at the level of T515 is below 4 Å, still above the cut-off of the radius of hydrated K+ (dotted vertical line, Figure 4H), but somewhat narrower than at the bundle crossing, because of a rotameric conformation adopted by the T515 side chain that is not seen in apo/AM (Figure 4G).
Support for the idea that these pores are open comes from comparison of the apo/LC and apo/AM structures with two recently obtained HCN1 structures, where the opening of the pore was attempted either by forcing S4 into the hyperpolarization-activated position through chemical linkage or by introducing a point mutation generating a constitutively open channel (Lee and MacKinnon, 2019). Although neither modification yielded an HCN1 open pore structure, an overlay of the respective TM regions shows that such approaches led to similar displacements of the S5 helix compared with the lipid-free HCN4 structures (Figures S15A–S15D). Of note, even the smaller displacement observed in the case of apo/LC (orange, Figure S15A) allows S6 rotation and widening at the bundle crossing, yielding the same pore conformation as apo/AM (Figures 4F and 4G) where the S5 displacements are larger.
Altogether, the above results strongly suggest that S5 displacement from the S4-S5-S6 planar array is conducive to pore opening in HCN channels, and that environmental factors, more specifically the interaction with distinct lipids, influence this process, at least in purified particles.
Molecular dynamics analysis of HCN channel ion conduction and selectivity
To test if the widening of the pore described above is sufficient to allow free passage of water and ions, we performed a series of molecular dynamics (MD) simulations.
In the absence of applied voltage, the cavity of the HCN4 structure with the narrow cytosolic entrance shows no internal solvation (Figure 5A). Pore entrance is occluded by the Q519 side chains, which prevents water entry. In contrast, the two HCN4 structures with the wider cytosolic entrance are fully solvated, because displacement of the Q519 side chain allows free passage of ions through the bundle crossing (Figure 5B). To further confirm that the widening corresponds to a physiologically meaningful open state of the channel, we performed docking experiments with ivabradine. This pore blocker reaches its site of action in the pore from the cytosolic side, and HCN4 channel opening is required for the drug to access its binding site (Bucchi et al., 2006). The docking poses in Figures 5C and 5D show that the structure with the narrow cytosolic entrance prevents docking of ivabradine inside the pore. In contrast, the structures with the wider opening offer free access of ivabradine into the cavity where the drug explores several poses, including one that positions it in close proximity to two residues, Y507 and I511, which were identified as the binding site for the blocker (Bucchi et al., 2013; Tanguay et al., 2019).
Figure 5.

Pore hydration and ivabradine docking in HCN4 structures
(A and B) Representative snapshot from unrestrained MD simulation of PD of HCN4 structure with narrow cytosolic entrance (blue, holo) (A) and with wider cytosolic entrance (B) (green, apo/AM; identical results were obtained with apo/LC). Two subunits of tetramer are shown for clarity. In (A), Y507, I511, T515, and Q519 side chains occupy cavity and do not allow passage of water molecules (red and white sticks and balls) and K+ ions (blue spheres) through bundle crossing. In (B), a different conformation of S6 allows water and K+ ions to pass through bundle crossing and enter inner pore cavity.
(C and D) Docking of ivabradine in two PDs (colored as above, with orange mesh representing ivabradine density). In (C), the narrow constriction created by Q519 prevents ivabradine entry into cavity. In (D), ivabradine enters in cavity and explores several positions, including one (ivabradine shown in orange sticks) in close proximity with Y507 and I511 (highlighted in red).
Both independent approaches confirm that the cytosolic entry is open. Moreover, successful docking with ivabradine suggests that these pore structures correspond to a physiologically relevant open state, at least in terms of the ability of this drug to reach its normal binding site.
The finding that the bundle crossing is open prompted us to investigate whether the entire pore structure is conductive in MD simulations with an applied voltage.
Figure 6A shows that the selectivity filter (SF) of the open HCN4 pore has a similar architecture to that of HCN1, displaying the same two putative ion binding sites (3 and 4) described for HCN1 (Lee and MacKinnon, 2017). Simulations performed on the open structures of HCN4 with an applied electric field (see STAR Methods and Table S3 for details) show that this conformation of the selectivity filters is indeed conductive. Under the conditions of our simulations, we were able to observe several passages of ions, both K+ and Na+, reproducing the experimentally determined properties of HCN channels, namely, low conductance and moderate K+/Na+ selectivity.
Figure 6.

Mechanism of ion permeation in HCN channels
(A) Comparison of selectivity filters of an open HCN4 pore (apo/LC) and HCN1. The sequence 479CIGYG481 (HCN4 numbering) is marked.
(B) Conduction trajectory for individual K+ ions traversing selectivity filter in a MD simulation of HCN4 in KCl solution. The positions occupied by the ion are identified by dotted lines, labeled a–c, and cross-referenced in (C). K+ ions entering filter at subsequent times are shown in different shades of blue. Positions of the carbonyl plane of C479 and I480 are indicated by black arrows.
(C) Snapshots of MD simulation cross-referenced by arrowheads in (B). The ion entering from extracellular side of SF (dark blue) occupies position a, half coordinated by water molecules from top and by carbonyl oxygen atoms of I480 at the bottom. Occupancy of position a is compatible with a second ion (light blue) in filter at position c, coordinated by carbonyl oxygen atoms of I480 from the top and by water molecules from the bottom (two-ion state). Next, the entering ion moves to position b, at plane of carbonyl oxygen atoms of C479, and the second ion moves out of the filter, into the cavity (one-ion state). Entrance of a new K+ ion in position a (purple) moves the second ion from position b to c and resets initial two-ion state.
(D) Conduction trajectory of Na+ (yellow) and K+ ions (blue) in a MD simulation with equimolar NaCl and KCl solution. Labeling as in (B) with ion positions cross-referenced in (E). K+ and Na+ ions entering the filter at subsequent times are shown in different shades of blue and orange, respectively.
(E) Snapshots of MD simulation cross-referenced by arrowheads in (D). A Na+ ion stably occupies position b, coordinated by carbonyl oxygen atoms of C479 (one-ion state) until entry of a K+ ion displaces it, with no appreciable resting time of ions in (A) or (C). Several K+ conduction events follow as described in (B) and (C) until the next Na+ ion enters (see Figure S13C for full trajectory and density plot).
(F) Density profiles of distance between opposite carbonyl oxygens of Cys 479 (tetramer subunits A and C, solid line; B and D, dotted line) in SF of HCN4 in MD simulation with pure KCl solution. Solid and dashed lines correspond to measured width between subunits A/C and B/D, respectively. Histograms were calculated over time frame indicated in Figure S16A. Inset: cartoon representation of C479 carbonyl distortion (and increase in width) when K+ ion enters in (B).
(G) Density profiles (as per F) in MD simulation with equimolar KCl:NaCl, calculated over time frame indicated in Figure S16C. Inset: entry of a Na+ in (b) does not induce changes in carbonyl plane.
cav, cavity; ext, extracellular side.
In KCl solution, we observed a total of nine K+ ion passages over four independent simulations with a total simulation time of 2,100 ns (Video S3; Figure S16A). This is compatible with the low picoSiemens (1–10 ions/μs) conductance of If and HCN channels (DiFrancesco, 1986; Johnson and Zagotta, 2005). In NaCl solution, no passage of ions was noted over multiple simulations lasting a total of 4,500 ns (Video S4; Figure S16B), in keeping with the finding that HCN channels do not carry an inward Na+ current in the absence of external K+ (Frace et al., 1992; Lyashchenko and Tibbs, 2008; Wollmuth and Hille, 1992). In simulations performed in mixed K+ and Na+ solution (Video S5; Figure S16C), we detected 11 K+ ions and only 4 Na+ ions passing through the pore during a total simulation time of 7,600 ns, closely matching the weak K+ selectivity (PK:PNa, ∼4:1) reported for HCN channels (Lyashchenko and Tibbs, 2008; Moroni et al., 2000).
Figure 6B shows an example trajectory of a typical inward passage of a K+ ion, in a simulation with K+-only solution. The simulation identifies three main binding sites for the ion, labeled a, b, and c, and cross-referenced in the snapshots in Figure 6C. Two main ion configurations alternate in the SF: “two ions” (in positions a and c) and “one ion” (in position b). A permeation event occurs when the ion in position a moves into b, and the ion in position c leaves the filter (“one-ion” configuration). Entry of a second ion in position a displaces the ion from position b to c, resetting the initial “two-ion” configuration. Conduction follows a soft “knock-on” mechanism whereby ions become only partially desolvated and water can co-permeate through the SF (Kopec et al., 2018). In binding site a, the K+ ion is coordinated by water molecules from above and by the carbonyl oxygens of I480 from below. In binding site b, the K+ is found, surprisingly, at the plane of carbonyl oxygens of C479, and water molecules are found both above and below the ion. In binding site c, the K+ ion is coordinated by carbonyls oxygens from C479 above and by water molecules below. This site corresponds to binding site 4, previously identified in HCN1 (Figure 6A). Remarkably, in our ion conduction simulations in HCN4, binding site 3 is only transiently occupied (<1 ns) by K+ during the transition from position a to b (see trajectory in Figure 6B). The fact that we observed the same trajectory and ion binding sites, independently of the voltage applied, ion concentration used, or force field employed (see STAR Methods) strongly suggests that in HCN channels, K+ ions are coordinated at the carbonyl plane of C479 rather than by the carbonyl cage formed at the more conventional K+ binding site 3.
Simulations in a mixed K+/Na+ solution show that Na+ ions bind to the same position b as K+ ions (Figures 6D and 6E). Of note, however, the substitution of the Na+ ion in position b by a new K+ ion entering the filter occurs directly, without adoption of the “two-ion” configuration (a, c) of the SF. If a second K+ ion enters the filter, then the scheme of Figure 6C is restored and K+ permeates according to it. If K+ is at position b, a new Na+ can enter the filter and bind to position a (Figures 6D and 6E) with the first K+ ion moving to position c.
During all mixed ion simulations, we observed a Na+ ion displacing a second Na+ ion only once. This observation suggests that the “knock-on” effect required for Na+ permeation is preferentially provided by K+. Moreover, despite our total simulation time in mixed solution being ∼3.5-fold greater than simulations in K+ only, we never observed a Na+ ion occupying position c. Thus, there can never be more than a single Na+ in the SF, as opposed to K+ ions that alternate between a two-ion and single-ion configuration. This phenomenon presumably contributes to the higher permeability for K+ over Na+ in HCN channels and provides an atomistic explanation for the experimental finding that “HCN channels require K+ to occupy one or more sites within the pore to maintain significant Na+ conductance” (Lyashchenko and Tibbs, 2008). The above hypothesis is further supported by the simulations in pure Na+ solution, where a single Na+ ion stably occupies the SF in position b leading to a non-conductive channel (see trajectory and density plot in Figure S16B and Table S3).
Finally, a close inspection of protein behavior during the simulations reveals that the SF of HCN channels is highly dynamic, adapting its dimensions to either K+ or Na+ ions. The probability density function of the width at carbonyls of C479 reveals two populations during ion transitions (Figure 6F). When a K+ ion occupies position b, the carbonyl oxygens widen by ∼0.5 Å, from 4.8 to 5.3 Å at C479 (Figure 6F). These distances are reduced by the same amount when the K+ ion leaves and/or when Na+ ion enters in this position (Figure 6G). The insets in Figures 6F and 6G further show that changes in dimensions are the result of carbonyl distortion more than backbone widening: carbonyl oxygens rotate laterally out of the SF, measurable by a higher distance between the oxygens.
Altogether, the findings reinforce the notion that the pore of the lipid-free structures represents a functional open state, supporting the view that the movements described for TM domains S5 and S6 resemble those occurring in response to hyperpolarization. The data furthermore uncover that the SF of an HCN channel is more flexible than that of a canonical K+ channel, which is in line with other studies that have proposed a causal relationship between plasticity of the filter and weak cation selectivity (Derebe et al., 2011; Napolitano et al., 2015; Shi et al., 2018).
Discussion
Here we present three cryo-EM structures of the HCN4 pacemaker channel at near-atomic resolution with novel information on HCN channel function in general and isoform-specific features in particular.
Recent work has highlighted the role of the N-terminal HCN domain in mediating the response to cAMP in HCN channels. Through its reciprocal interactions, the HCN domain connects the C-linker to the VSD, and mutations that prevent these interactions abolish cAMP response in HCN1, HCN2, and HCN4 (Porro et al., 2019). Our cryo-EM structures now highlight an additional pathway, likely unique to HCN4, whereby the S4-S5 linker acts as a facilitator for the cAMP effect between the C-linker and the VSD. Thus, in HCN4, but not in HCN1 and HCN2, a metal ion coordination site, which we refer to as the tetrad, connects the TM domain of one subunit to the cytosolic domain of its neighboring subunit.
Biochemical and functional experiments show that this metal coordination site supports Mg2+ binding, and that disruption of this site through depletion of Mg2+ and/or point mutations halves the effect of cAMP on voltage-dependent gating. Altogether, the structural and functional data advocate for a model according to which the coordination site provides an additional mechanical coupling between the cytoplasmic and the TM domains, which contributes to the transmission of the cAMP-induced conformational changes to the voltage sensor.
A second major finding of the present study is that structures with open pores were obtained using purification protocols that removed bound lipids associated with TM helices S4 and S5. Current data suggest that, at least in the case of HCN4, the kind of detergent used during the purification may affect the stability of the pore. Although in our study amphipols and LMNG/CHS promoted pore opening in the HCN4 apo protein, another detergent (n-Dodecyl β-D-maltoside [DDM]/CHS; https://www.thesgc.org/tep/hcn4) preserved the lipids associated with S4 and S5 and did not yield an open pore (EMDB: EMD-0093, PDB: 6GYN). We therefore speculate that such lipids stabilize the HCN4 structure in the closed pore state, similar to what has been suggested for the related inward-rectifying KAT1 channel (Clark et al., 2020).
In the absence of lipids, crucial hydrophobic and hydrophilic interactions between S4, S5, and S6 are partially or totally lost, S5 tilts upward and out of the plane, rotating S6 along and widening the constriction at the bundle crossing. Strikingly, a similar loss of contacts between S4 and S5, and tilting of S5 out of plane, was observed in HCN1, in which S4 was locked in the putative hyperpolarized position (Lee and MacKinnon, 2019). The fact that two independent strategies, which opened or attempted to open the HCN channel, resulted in the same structural rearrangement is a strong indication that the observed conformational changes are causally related to channel opening. A similar gating model in which the upward tilt of the S5 N terminus allows S6 the space needed for pore opening was also suggested by MD simulations of the HCN1 channel under hyperpolarization (Kasimova et al., 2019) and by transition metal ion fluorescence resonance energy transfer (tmFRET) in spHCN (Dai et al., 2019).
Our MD and docking data fully support the notion of an open pore in the HCN4 structures presented here. The bundle crossing widens and allows free passage of K+ and water in and out of the cavity, as well as the docking of ivabradine inside the pore. The pore is kept closed by a number of side chains protruding into the cavity (at residues Y507, I511, T515, and Q519), identifying the entry at the bundle crossing as the cytosolic gate in HCN channels (specifically Q519 in HCN4). Of note, a glutamine in this position is conserved in HCN-related channels, Ether-a-go-go1 (Eag1) and human Ether-a-go-go-Related Gene (hERG), and its displacement is similarly associated with opening of the permeation pathway (James and Zagotta, 2018; Wang and MacKinnon, 2017). Both in the case of HCN4 and Eag1/hERG, the movement of the lower half of the S6 helix hinges on the presence of a glycine residue positioned ∼15 residues (four helical turns) upstream of the bundle crossing glutamine. The conservation of this glycine residue extends to KAT1 channels (Clark et al., 2020), as well as CNG channels (Zheng et al., 2020), where it provides the main hinge point for the S6 twisting and splaying that leads to CNG channel opening upon ligand binding (G399 in TAX-4). All together, these observations suggest evolutionary preservation in the basic principles of S6 movement associated with gating in “non-domain-swapped” CNBD channels.
We further found that the channels displaying an open cytosolic gate are fully conductive, with MD simulations recapitulating experimental properties of HCN channels: very low unitary conductance, weak K+:Na+ selectivity, and no conductance in pure Na+. Therefore, this structure is a good model for understanding the peculiarities of HCN channels compared with other K+ selective channels.
Even so, we cannot conclusively know to what extent this configuration reflects the one that the channel acquires at negative voltages. The propensity to open in the absence of voltage reported here for HCN4 correlates with the known property of HCN channels to generate an instantaneous current (Iinst). This time- and voltage-independent component makes up ∼5%–10% of the total conductance (Proenza and Yellen, 2006; Proenza et al., 2002). The open structures may thus reflect an open state in the absence of voltage, further supported by the findings that (1) displacement of S5 in the lipid-free structures leads to disruption of the hydrogen bond between E403 and N421, which was shown to yield channels with a large constitutive current component in functional mutagenesis studies (Chen et al., 2001a; Ramentol et al., 2020); and (2) the displacement of S5 in the apo/LC structure is virtually identical to that seen in the structure of the constitutively open HCN1 mutant Y286D (Chen et al., 2001a; Lee and MacKinnon, 2019).
In summary, our data advance the understanding on HCN channel gating, cyclic nucleotide-dependent modulation, and ion permeation. Because HCNs represent the only class of “non-domain-swapped” CNBD channels known to respond both to voltage and ligand binding, our findings also provide further insight into mechanistic integration of voltage sensing and cyclic-nucleotide binding. The identification of HCN isoform-specific differences highlighted here will ultimately pave the way for the design of drugs with isoform selectivity, advancing the pharmacological treatment of selected cardiac and neurological disorders.
Limitations of the study
The open structures of HCN4 shown here were not obtained under conditions expected to promote an open channel, i.e., membrane hyperpolarization. Nonetheless, they display the expected functional properties in terms of conductance, selectivity, and ivabradine binding of an open HCN pore. In addition, the open structures of HCN4 (apo/LC and apo/AM) resemble, respectively, the HCN1 structure bearing a mutation that leads to channels with constitutive current and the HCN1 structure capturing S4 of the VSD in its active state through chemical crosslinking (Lee and MacKinnon, 2019).
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Experimental models: Organisms/strains | ||
| FreeStyle HEK293-F Cells | Thermo Fisher | Cat# R79007 |
| HEK293T | ATCC | RRID: CRL-3216 |
| Chemicals, peptides, and recombinant proteins | ||
| Turbofect transfection reagent | Thermo Fisher | Cat# R0533 |
| polyethyleneimine | Polysciences | Cat# 24765 |
| protease inhibitor cocktail | Roche | Cat# 04693124001 |
| DNase | Sigma | Cat# DN25 |
| RNase | Sigma | Cat# R4875 |
| PMSF | Sigma | Cat# P7626 |
| LMNG | Anatrace | Cat# NG310 |
| CHS | Anatrace | Cat# CH210 |
| Amphipol A8-35 | Anatrace | Cat# A835 |
| Adenosine 3′,5′-cyclophosphate | Sigma | Cat# A9501 |
| Critical commercial assays | ||
| QuikChange Lightning Site-Directed Mutagenesis | Thermo Fisher | Cat# 210518 |
| Recombinant DNA plasmids | ||
| pEGA: HCN4ΔC | This paper | N/A |
| pEGA: HCN4ΔC (H407A, H553A) | This paper | N/A |
| pCI:TRIP8b(1a-4) | Porro et al., 2020 | N/A |
| pCI:rbHCN4 | Porro et al., 2019 | N/A |
| pCI:rbHCN4 (H407A) | This paper | N/A |
| pCI:rbHCN4 (H553A) | This paper | N/A |
| pCI:rbHCN4 (E557A) | This paper | N/A |
| pCI:rbHCN4 (H407A, H553A) | This paper | N/A |
| pCI:rbHCN4 (H407A, H553A, E557A) | This paper | N/A |
| pCI:hHCN1 | Porro et al., 2019 | N/A |
| pCI:hHCN1 (E436A) | This paper | N/A |
| pCI:mHCN2 | Porro et al., 2019 | N/A |
| Deposited data | ||
| CryoEM map of HCN4holo | This paper | EMDB: EMD-12513 |
| CryoEM map of HCN4apo/LC | This paper | EMDB: EMD-12512 |
| CryoEM map of HCN4apo/AM | This paper | EMDB: EMD-12466 |
| Atomic model ofHCN4holo | This paper | PDB: 7NP4 |
| Atomic model ofHCN4apo/LC | This paper | PDB: 7NP3 |
| Atomic model ofHCN4apo/AM | This paper | PDB: 7NMN |
| Software and algorithms | ||
| RELION-3.0 | Zivanov et al., 2018 | https://www2.mrc-lmb.cam.ac.uk/relion/index.php/Main_Page |
| CryoSPARC v.2 | Punjani et al., 2017 | https://cryosparc.com |
| PHENIX | Adams et al., 2010 | https://phenix-online.org |
| Coot | Emsley et al., 2010 | https://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot/ |
| UCSF CHIMERA | Pettersen et al., 2004 | http://www.cgl.ucsf.edu/chimera |
| MOLPROBITY | Chen et al., 2010 | http://molprobity.biochem.duke.edu/ |
| HOLE | Smart et al., 1996 | http://www.holeprogram.org/ |
| AutoDock 4.2.6 in conjunction with AutoDockTools 1.5.6 | Morris et al., 2009 | http://autodock.scripps.edu/ |
| GROMACS 2019 | Abraham et al., 2015; Van Der Spoel et al., 2005 | https://manual.gromacs.org/documentation/2019/index.html# |
| LINCS | Hess et al., 1997 | https://dblp.uni-trier.de/rec/journals/jcc/HessBBF97.html |
| Biotite | Kunzmann and Hamacher, 2018 | https://github.com/biotite-dev/biotite |
| Other | ||
| Grids | Quantifoil | Three-hundred mesh holey gold R0.6/1 and R1.2/1.3 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Anna Moroni (anna.moroni@unimi.it).
Materials availability
All unique/stable reagents generated in this study are available from the Lead Contact without restriction.
Data and code availability
The cryo-EM density maps have been deposited in the Electron Microscopy Data Bank under the accession numbers: EMD-12466, EMD-12512, and EMD-12513. Coordinates have been deposited in the Protein Data Bank under the PDB accession numbers: 7NMN, 7NP3, and 7NP4.
Experimental model and subject details
HEK293-T cells (ATCC) were used for electrophysiology. Freestyle HEK293-F cell cultures (Thermo Fisher) were used for heterologous protein expression.
Method details
Constructs
The cDNA encoding rabbit HCN4 (GenBank: NM_001082707) carrying an internal deletion (from residues 783 to 1064), hereafter HCN4ΔC, was cloned into a modified pEG BacMam vector (Goehring et al., 2014) (hereafter pEGA) for large-scale protein purification from mammalian cells. The internal deletion eliminates a poorly conserved region in the C-terminal portion of the HCN channel protein, but preserves the extreme C-terminal SNL tripeptide sequence responsible for binding the auxiliary subunit TRIP8b (Santoro et al., 2004, 2011). The pEGA vector allows expression of the protein of interest with both eGFP and decahistidine tags at its N terminus. The tags can be cleaved using TEV enzyme, due to the presence of a TEV cleavage site between the tags and the protein of interest. For co-expression of HCN4 protein in the presence of TRIP8b, the cDNA encoding full-length mouse TRIP8b (splice variant 1a4) (GenBank: NM_001163516) was cloned into the pEGA vector and the HCN4ΔC cDNA into pCI vector (Promega Corporation). For whole-cell patch clamp analysis, the human HCN1 cDNA, the rabbit HCN4 cDNA and the mouse HCN2 cDNA were cloned into the pCDNA3.1 and in the pCI vector, respectively, as reported in Saponaro et al., 2018. All mutations were generated by site-directed mutagenesis (QuikChange site-directed mutagenesis kit; Agilent Technologies) and confirmed by sequencing.
Electrophysiology and data analysis
HEK293-T cells (Invitrogen) were cultured as described (Porro et al., 2019, 2020) and transiently transfected either with HCN4, HCN1 or HCN2 wild-type or mutant channels (1μg per transfection) using Turbofect transfection reagent (Thermo Fisher) according to the manufacturer recommended protocol. When needed, GFP (cloned in pMAX vector) was cotransfected to identify successfully transfected cells. All the experiments were performed at room temperature (about 25°C). Currents were recorded in whole-cell configuration with an Axopatch 200B amplifier (Molecular Devices, CA, USA) or with an ePatch amplifier (Elements, Cesena, Italy); data acquired with the Axopatch 200B amplifier were digitized with an Axon Digidata 1550B (Molecular Devices, CA, USA) converter. All data were analyzed offline with Axon pClamp 10.7. Patch pipettes (3–6 MΩ) were filled with a solution containing: 10 mM NaCl, 130 mM KCl, 1 mM egtazic acid (EGTA), 0.5 mM MgCl2, 2 mM ATP (Magnesium salt) and 5 mM HEPES–KOH buffer (pH 7.2). The extracellular bath solution contained 110 mM NaCl, 30 mM KCl, 1.8 mM CaCl2, 0.5 mM MgCl2 and 5 mM HEPES–KOH buffer (pH 7.4). The EDTA-containing pipette solution was prepared as follows: 60 mM EDTA-KOH, 20 mM KCl, 10 mM NaCl, 5 mM HEPES-NaOH buffer (pH 7.4).
Where indicated, Adenosine 3′,5′-cyclic monophosphate (cAMP, Sigma-Aldrich) was added to the pipette solution from a previously prepared stock solution.
For channel activation, hyperpolarizing steps were applied from a holding potential and current tails were measured upon return to a fixed voltage. The duration and the number of the steps used to activate the channels were adjusted for the different HCN isoform. For HCN1, holding potential was −20 mV (1 s), with steps from −20 mV to −120 mV (10 mV interval, 3.5 s) and tail currents recorded at −40 mV (3 s); for HCN4, holding potential was −20 mV (1 s), with steps from −30 mV to −165 mV (15 mV interval, 4.5 s) and tail currents were recorded at −40 mV (4.5 s); for HCN2, holding potential was −20 mV (1 s), with steps from −30 mV to −130 mV (10 mV interval, 4.5 s) and tail currents recorded at −40 mV (4.5 s).
Mean activation curves were obtained by fitting maximal tail current amplitude, plotted against the voltage step applied, with the Boltzmann equation:
where V is voltage, y the fractional activation, V1/2 the half-activation voltage, and k the inverse-slope factor. Mean V1/2 values were obtained by fitting individual curves from each cell to the Boltzmann equation and then averaging all the obtained values.
Activation and deactivation time constants (τ) were obtained by fitting a single exponential function:
to current traces obtained with the activation protocol described above. Deactivation time constants were obtained by fitting tail currents collected at −40 mV after a fully activation pulse at −135 mV.
Protein expression and membrane isolation
Freestyle HEK293-F cell cultures (Thermo Fisher) were transiently transfected with pEGA: HCN4ΔC (1μg per ml) at a cell density of 2 × 106 cells per ml using polyethyleneimine (PEI) (Polysciences). The transfected cells were harvested by centrifugation after 48 hours of growth in shaker flasks at 37°C, 5% CO2. Cell pellets were resuspended in low salt buffer (10 mM KCl, 10 mM MgCl2, 10 mM HEPES pH 7.5, 0.5 mM PMSF, EDTA-free complete protease inhibitor cocktail (Roche) (1:1000), 20μg/mL DNase, and 10μg/mL RNase) and lysed by gentle homogenization in a glass homogenizer. Membranes were isolated by ultracentrifugation (40 min at 17000 xg), resuspended by homogenization and washed two times with high salt buffer: 1 M NaCl, 10 mM KCl, 10 mM MgCl2, 10 mM HEPES pH 7.5, 0.5 mM PMSF, EDTA-free complete protease inhibitor tablet, 20μg/mL DNase, 10μg/mL RNase. Isolated membranes were resuspended by homogenization in the storage buffer: 200 mM NaCl, 20 mM HEPES, pH 7.5, 0.5 mM PMSF, EDTA-free complete protease inhibitor cocktail (1:1000) and stored at −80°C until use.
Protein purification in LMNG/CHS
The isolated membranes were thawed on ice and solubilized by the addition of a mixture of detergents (lauryl maltose neopentyl glycol (LMNG) with cholesteryl hemisuccinate (CHS) in a 5 to 1 ratio) to a final concentration of 1% (w/v), and gently agitated for 2 hours at 4°C. The solution was cleared by ultracentrifugation (40 min at 1700 xg). Pre-equilibrated Ni2+-NTA resin (QIAGEN) was added to the sample, together with 10 mM imidazole and the mixture allowed to gently rotate overnight at 4°C. After transferring the mixture to a column, the resin was washed in two steps: 1) 5 column volumes of buffer containing 50 mM imidazole; 2) 5 column volumes of buffer containing 75 mM imidazole. The proteins were eluted with 10 column volumes of the following buffer: 200 mM NaCl, 20 mM HEPES, pH 7.5, 300 mM imidazole. The eluted protein was loaded on a Superose 6 increase 10/300 GL SEC column (GE Healthcare Life Sciences) pre-equilibrated with buffer containing 200 mM NaCl, 20 mM HEPES pH 7.0 and detergent (LMNG-CHS) at the concentration of 0.002% (w/v). For the protein sample used to solve the structure of HCN4 bound to cAMP, the ligand (Sigma-Aldrich) was kept at a concentration of 0.2 mM in all steps of membrane isolation and protein purification procedure described above. The decahistidine-eGFP tag at the N terminus of HCN4 protein was not removed. Final yield of purified protein was about 1mg per 1 l of cells.
Protein purification in amphipols
For purification in amphipols, HCN4 protein was obtained following co-expression and co-purification with the auxiliary subunit TRIP8b. This approach was chosen to prevent any cellular cAMP from occupying the CNBD (Lolicato et al., 2011) due to the antagonistic nature of TRIP8b/cAMP binding (Bankston et al., 2017; Gross et al., 2018; Hu et al., 2013; Saponaro et al., 2018). Although we were unable to detect signal for the TRIP8b protein upon cryo-EM image analysis, potentially due to loss of TRIP8b during vitrification and/or variability in the number of TRIP8b subunits associated with the channel, this approach did yield a near atomic-resolution structure of the HCN4 channel protein stabilized by amphipols in the cAMP-unbound conformation.
Following co-transfection of pCI: HCN4ΔC (0.75μg per ml) and pEGA: TRIP8b (0.75μg per ml), membranes were isolated and the protein complex affinity purified with a Ni2+-NTA resin as described above. Proteins were then desalted by using PD-10 desalting columns (GE Healthcare) and reconstituted into amphipol A8-35 (Anatrace) at a protein: amphipol ratio of 1: 10 by weight. The protein – amphipol mixture was incubated for 4 hours with gentle rotation at 4°C. After 4 hours of incubation with amphipol, the detergent was removed by the addition of Bio-Beads (Bio-Rad) overnight at 4°C. The reconstituted protein was then loaded on a Superose 6 increase 10/300 GL SEC column pre-equilibrated in a buffer composed by 200 mM NaCl and 20 mM HEPES pH 7.0 without detergent in order to remove the excess of free amphipol and residual detergents. Final yield of purified HCN4ΔC in complex with GFP-TRIP8b was about 1mg per 1 l of cells.
Cryo-EM sample preparation and image acquisition (LMNG/CHS)
LMNG/CHS solubilized HCN4 alone (apo/LC) and in the presence 0.2 mM cAMP (holo) was concentrated to 0.5 and 0.6 mg ml-1, respectively, using a 100 kDa concentrator (Amicon) and 3 μL of protein solution was applied to freshly plasma-cleaned (Gatan Solarus) 0.6/1 μm 300 mesh holey gold grids (Quantifoil UltrAuFoil) and blotted for 2.5 s at blot force 3 using a Vitrobot (FEI) operating at 22°C and > 90% humidity prior to being immediately plunged into liquid ethane to vitrify the sample. Images were acquired on a Titan Krios electron microscope (FEI) equipped with a K2 summit detector (Gatan) operating in counting mode with a calculated pixel size of 0.83 Å per pixel. Automated data collection was achieved with the Leginon software package (Suloway et al., 2005). For HCN4 apo, 4465 exposure movies were collected with an estimated dose of 71.85 e- Å-2 for the 6 s exposure composed of 40 frames (150 ms per frame) using a defocus range of −1.1 to −2 μm. For HCN4 holo, 4478 exposure movies were obtained using the same exposure as HCN4 apo but with an estimated total dose of 67.88 e- Å-2 and a defocus range from −1 to −1.8 μm.
Single-particle analysis (LMNG/CHS)
For the HCN4 holo and apo/LC datasets, frame alignment was performed using the Relion 2.1 (Kimanius et al., 2016; Scheres, 2012) implementation of MotionCor2 (Zheng et al., 2017b) with 3 by 3 patches and a B-factor of 150 and estimation of the contrast transfer function (CTF) was done for each micrograph using CTFFind4 (Rohou and Grigorieff, 2015). Approximately 1500 particles were manually picked, extracted (binned by 4) and subjected to 2D classification in Relion to obtain templates for automated picking. Template picking runs were done in Relion using templates, low-pass filtered to 20 Å, which corresponded to top, bottom and side views resulting in 2,006,780 and 914,005 picked particles for holo and apo/LC, respectively. An additional template picking run using only side-view templates and a lower picking threshold was performed on the apo/LC dataset to obtain separate stack of 1,039,436 particles. All particles were extracted with a 68 pixel box at 3.32 Å per pixel and imported into CryoSPARC v.2 (Punjani et al., 2017) for initial classification. The pyem script (https://doi.org/10.5281/zenodo.3576630) was used for all conversions between Relion and CryoSPARC. Throughout the processing of these data we sought to determine if any deviations from C4 symmetry were present but ultimately did not observe any strong evidence. Therefore, unless specified differently, C4 symmetry was imposed for all reconstructions described below.
For HCN4 holo, several rounds of 2D classification in CryoSPARC resulted in a cleaned stack of 252,458 particles which were moved back into Relion and extracted with recentered coordinates and an unbinned box size of 272 pixels and moved back into CryoSPARC where an ab-initio reconstruction without symmetry constraints produced a volume with apparent C4 symmetry. Several rounds of 3D classification (ab-initio into multiple classes and heterogeneous refinement) resulted in 170,263 particles and 4 Å map with streaking and smearing of density observed in the transmembrane domain (TMD). To improve the quality of the map in this region, particles were moved back to Relion where maps were reconstructed, and signal subtraction performed using a mask that removed the detergent micelle and the lower half of the cytosolic domains. Subsequent classification of the signal-subtracted particles was done in CryoSPARC to obtain a 3.4 Å reconstruction with much improved density in the TMD from 67,583 particles. The signal-subtracted particles were converted back to the original particles in Relion followed by Bayesian polishing prior to a final round of 3D classification and non-uniform refinement in CryoSPARC where 54,828 particles produced the final reconstruction at 3.2 Å resolution.
For apo/LC, particles from the two template picking runs were initially processed independently using 2D and 3D classification in CryoSPARC resulting in cleaned particle stacks containing 132,630 and 120,030 particles from the template picking runs using templates with various orientations and only side view templates, respectively. The two particle stacks were merged in Relion and duplicate particles were removed by setting the minimum inter-particle distance to 50 pixels prior to unbinned extraction of recentered particles. This merged stack of 213,381 particles was imported back into CryoSPARC where an initial 3D refinement produced a 4 Å reconstruction that was used as an initial model for 4 rounds heterogeneous refinement with 3 to 5 classes. 85,512 particles from to the best classes from heterogeneous refinement were merged and a 3.5 Å reconstruction was obtained using non-uniform refinement. These particles were imported back into Relion where the volumes were reconstructed and CTF parameters refined. Following CTF refinement, 3D classification into 8 classes resulted in one class with 51,758 particles that contained the most structural features and cleanest density. These particles were imported into CryoSPARC where non-uniform refinement produced a 3.4 Å reconstruction. Finally, the particles were moved back into Relion for Bayesian polishing prior to non-uniform refinement in CryoSPARC to obtain the final map at 3.2 Å resolution.
Cryo-EM sample preparation and image acquisition (amphipols)
For cryo-EM, a 3.5 μL droplet of freshly purified sample, at final concentration of 0.3-0.7 mg/ml, was applied onto a UltrAu R1.2/1.3 300-mesh gold holey grid (Quantifoil), previously glow discharged for 30 s at 30mA using a GloQube system (Quorum Technologies). Following an incubation of 90 s at 4°C and 100% relative humidity, the grid was blotted for 2-5 s and plunge-frozen in liquid ethane using a Vitrobot Mk IV (Thermo Fischer Scientific).
All data were acquired on a 200 kV Talos Arctica (Thermo Fischer Scientific) transmission electron microscope aligned to operate in parallel illumination. Cryo-EM data were acquired using EPU automated data collection software (Thermo Fischer Scientific). Images were collected at nominal magnification of 120,000 ×, corresponding to a pixel size of 0.889 Å/pixel at the specimen level, with an applied defocus values between −0.5 and −2.0 μm. Movies were acquired using Falcon 3EC direct electron detector (Thermo Fischer Scientific) operating in electron counting mode (0.5 e-/pix/sec) with a total exposure time of ∼61 s and a total accumulated dose of 40 e-/A2, equally distributed over 40 movie fractions (1 e-/A2 per fraction).
Single-particle analysis (amphipols)
A total of 1,571 movies were collected, with frames aligned before summing in individual fractions within EPU software. Micrograph movies were imported in RELION-3.0 for all subsequent image processing tasks (Zivanov et al., 2018). Prior to particle picking, all images were subjected to motion-correction and dose-weighting using MotionCor2 (version 1.2.1) (Zheng et al., 2017b). Estimation of the contrast transfer function (CTF) was performed on aligned, not weighted sum images using CTFFIND4 (version 4.1.10) (Rohou and Grigorieff, 2015). Particles were automatically picked using Gautomatch (version 0.56) (https://www.mrc-lmb.cam.ac.uk/kzhang/). Coordinates were then imported in RELION for visual inspection and manual removal of erroneously picked contaminants and aggregates. A total of 100,130 particles were initially normalized and extracted in RELION-3.0. Particles set was subjected to several rounds of reference-free 2D classification to remove junk particles. An initial 3D auto-refinement was performed using as reference a low-resolution 3D model obtained by negative staining EM, low-pass filtered at 40 Å. Subsequent rounds of 3D classification were performed imposing C4 symmetry to isolate particles best contributing to isotropic, near atomic-resolution reconstructions. A final dataset of 11,146 particles were selected and subjected to two rounds of CTF refinement, beam tilt correction and 3D auto-refinement followed by Bayesian polishing as implemented in RELION-3.0. A final 3D auto-refinement was performed on polished particles by applying a soft-edged and extended mask to a 15Å low-pass filtered reference map and enabling solvent-flattened FSC calculation. Refined 3D reconstructions were sharpened using RELION-3.0 standard post-processing procedure applying a soft-edge and extended solvent mask. Overall resolutions estimates were calculated from Fourier shell correlations at 0.143 (applied B-factor and estimated resolution listed in Table 1). Estimates of local resolutions were performed using RELION-3.0 local resolution tool.
Model building, refinement and validation
The initial model of HCN4 apo/AM was based on the HCN1 apo cryo-EM structure (PDB: 5U6O) (Lee and MacKinnon, 2017). The initial model was rigid body fitted into the cryo-EM reconstructions using UCSF CHIMERA (Pettersen et al., 2004) and the resulting aligned model were subjected to real-space refinement using PHENIX (Adams et al., 2010) before sequence adjustment and manual model building with COOT (Emsley et al., 2010). Subsequent HCN4 apo/LC and HCN4 holo models were rigid body fitted with UCSF CHIMERA and real-space refinement with PHENIX using the previous HCN4 apo/AM coordinates as template. A polyalanine truncation and a subsequent rigid body refinement with non-crystallographic symmetry (NCS) restrains was carried out with each model to properly assess NCS using PHENIX. The full atomic models of HCN4 apo/AM, HCN4 apo/LC and HCN4 holo were subjected to multiple rounds of real-space refinement in PHENIX including global minimization and refinement of atomic displacement parameters, and applying secondary structures, Ramachandran and NCS restrains (Afonine et al., 2018).
Thermal denaturation assay
GFP-HCN4ΔC protein was purified following the procedure described in the section “Protein purification in LMNG/CHS.” The buffer in which the protein was eluted from the SEC column contained 0.2 mM cAMP and 0.002% (w/v) LMNG-CHS which were kept present in all incubation buffers. The purified protein (3 μM) was maintained for 48 h at 4°C before being used for thermal denaturation assay. For Mg2+ removal assay, GFP-HCN4ΔC was incubated 1h with 10 mM Ethylenediaminetetraacetic acid (EDTA, Sigma-Aldrich) pH 7. The buffer with EDTA was replaced by using PD-G25 desalting columns (GE Healthcare) with a buffer without EDTA +10 mM MgCl2. Aliquots of purified protein (5μg) were heated for 10 min over the following range of temperatures: 20, 30, 40, 50, 55, 60, 65, 70, 80°C. the samples were centrifuged (30 min, at 18000 xg) to remove precipitated protein and the supernatant was loaded on a Superose 6 increase 10/300 GL SEC column (GE Healthcare Life Sciences) pre-equilibrated with buffer containing 200 mM NaCl, 20 mM HEPES pH 7.0 and detergent (LMNG-CHS) at the concentration of 0.01% (w/v). Since we were following the emission signal (509 nm) of the excited GFP fused at the N terminus of HCN4ΔC, the SEC column was connected to a Prominence UFLC system (Shimadzu) fitted with an RF-10AXL fluorescence detector (Shimadzu). Thermal denaturation curves were obtained by measuring the height of the fluorescent SEC (fSEC) peak measured from samples at each of the different temperatures above reported. The height of the fSEC peak for sample incubated at 20°C was used as control for normalization. The thermal denaturation curves were fitted with a sigmoidal dose-response equation:
where Y is the fluorescence emission in arbitrary units, A1 the minimal fluorescence (from the sample heated at 80°C), A2 the maximal fluorescence (from the sample heated at 20°C), LOGx0 the melting temperature (Tm), x a given temperature, and p is the Hill slope. Mean Tm values were obtained by fitting individual curves from each experiment to the Boltzmann equation and then averaging all the obtained values.
Pore Analysis
The pore radius was calculated with HOLE (Smart et al., 1996) using Amber van-der-Waals radii.
Ivabradine docking
In-silico docking experiments were performed on HCN4 holo, apo/LC and apo/AM structures in the same manner (apo/LC and apo/AM yielding identical results). AutoDock 4.2.6 in conjunction with AutoDockTools 1.5.6 (Morris et al., 2009) was used for preparation of proteins and ligands. Ligands were drawn and energy-minimized with Avogadro (Hanwell et al., 2012) using the UFF force field (Rappe et al., 1992). All bound ligands, detergents and water molecules were removed from protein structures prior to model preparation. Gasteiger charges were used. A cubic box with x = 4.725nm and a grid spacing of 0.0375nm was centered below the selectivity filter to include the central pore module and C-linker. 250 docking attempts with 2,500,000 evaluations were performed per protein-ligand system and clustered based on RMSD. The density of ivabradine was calculated from docked poses as 3d histogram over the atomic coordinates with a grid spacing of 0.1nm. Only bins with a count of at least 3 were considered.
Molecular dynamics simulations
Molecular dynamics simulations were performed using the PD (residues 412-523) of HCN4 holo, apo/LC and apo/AM structures embedded into a pre-equilibrated 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) bilayer. Starting configurations were selected based on testing different variations in the initial positioning of the ions (either one or two K+/Na+ inside the SF, with or without a separating water molecule) prior to production runs. Based on results from these preliminary trial runs, the following configurations were selected: For simulations in pure KCl solution, we placed two K+ inside the SF at the height of binding sites a and c, separated by a single water molecule. For pure NaCl and mixed KCl:NaCl solutions, a single Na+ ion was placed inside the SF at the height of the C479 carbonyl oxygen plane with two water molecules above and below, due to the fact that the two ion configuration was not stable. Short trial simulations were also performed to test a range of applied voltages and ion solution concentrations. To increase the rate of sampling of conduction events during production runs, we applied artificial electric fields of −500mV or −700mV (Andersson et al., 2018; Köpfer et al., 2014; Stock et al., 2013). However, the same conduction mechanism was observed at hyperpolarizing voltages as low as −300mV and ion concentrations as low as 150 mV. Individual simulation conditions and the final composition of each system in the production runs are summarized in Table S3 (note that all production runs reported in Table S3 and described in Figure 6 were performed using HCN4 apo/LC). All simulations were carried out with GROMACS 2019 (Abraham et al., 2015; Van Der Spoel et al., 2005) in conjunction with the Amber99sb∗-ILDN force field (Best and Hummer, 2009; Lindorff-Larsen et al., 2010), the TIP3P water model (Jorgensen et al., 1983), Berger-derived POPC lipids (Cordomí et al., 2012) and ion parameters by Joung and Cheatham (Joung and Cheatham, 2008). Van-der-Waals interactions were cut-off at 1nm and electrostatics were treated by PME (Essmann et al., 1995) beyond 1nm. Temperature and Pressure were kept at 310K and 1bar using the V-Rescale Thermostat (Bussi et al., 2007) and Parrinello-Rahman Barostat (Parrinello and Rahman, 1981), respectively. All bonds were restraint using LINCS (Hess et al., 1997) and hydrogen atoms were represented as virtual sites to allow for an integration time step of 4fs (Feenstra et al., 1999). After system preparation, we performed 2000 steps energy minimization (steepest descent) and 20ns position-restrainted equilibration (Fc = 1,000 kJ/mol/nm2). Finally, restraints were gradually lifted over 3ns followed by 1,000-1,500 ns unrestrained simulation. A set of distance restraints were applied on the bottom half of the S6 helix to prevent the cytosolic gate from closing (Kopec et al., 2018). Molecular dynamics trajectories were analyzed using GROMACS tools and Biotite (Kunzmann and Hamacher, 2018).
Quantification and statistical analysis
As indicated in the figure legend of Figure 2D, the Thermal denaturation assays were repeated three times, and the data are represented as mean ± standard error of the mean (SEM) of the three independent experiments. Mean Tm values were compared using one-way ANOVA followed by Fisher’s test. Significance level was set to p = 0.05.
As indicated in the figure legend of Figure 2F, cAMP-induced shifts on half activation voltages (V1/2) are represented as mean ± SEM of independent experiments (n > 3; for details see Table S1). Mean activation and deactivation time constants shown in Figure S1 and listed in Table S3, were compared using Student’s t test. Significance level was set to p = 0.05. Mean V1/2 values, reported in Table S1 and derived from tail current activation curves shown in Figures S1 and S9–S11, were compared using one-way ANOVA followed by Fisher’s test or using Student’s t test. Significance level was set to p = 0.05.
All the above-described analyses were performed using Originpro software (Originlab, Northampton, MA, USA).
Cryo-EM data collection and refinement statistics as well as refinement of protein structure 3D models are listed in Table 1. Validation of the models’ geometry and all-atoms contacts were carried out with MOLPROBITY (Chen et al., 2010) and the RCSB PDB validation server.
Acknowledgments
Calculations were conducted on the Lichtenberg high-performance computer of the TU-Darmstadt. Part of the work was performed at the Centro di Ricerca Pediatrica Romeo e Enrica Invernizzi, Cryo-electron microscopy lab, University of Milano, and at the Columbia University Cryo-Electron Microscopy Center. This work was partially supported by Fondation Leducq TNE19CVD03 (to D.D.); Cariplo Young Investigator grant no. 2018–0231 (to A.S.); ERC Advanced grant noMAGIC 695078 (to A.M. and G.T.); and NIH grants R01-NS109366 (to S.A.S. and W.A.H.), P41-GM116799 (W.A.H.), and R01-GM107462 (W.A.H.). Some of the work was performed at the Simons Electron Microscopy Center and National Resource for Automated Molecular Microscopy located at the New York Structural Biology Center, supported by grants from the Simons Foundation (SF349247), NYSTAR, and the NIH (GM103310 and OD019994) with additional support from Agouron Institute (F00316).
Author contributions
A.S. performed the experiments, interpreted the data, prepared the figures, and wrote the first draft of the manuscript (ms); D.B. performed MD simulations and contributed to writing the first draft of the ms; M.H.G. solved the apo and holo structures in LMNG/CHS and contributed to fitting atomic models to density maps; P.S. solved the apo structure in AM; A.P. and F.G. prepared mutants and performed patch-clamp experiments; A.S.S. performed TD experiments; A.-C.S. fitted the models to the density maps; L.A., G.P., and G.C. established the protocols for protein purification; O.B.C. supervised cryo-EM experiments; K.H. supervised MD simulations; H.M.C. and F.M. supervised experiments and revised the ms; W.A.H., S.A.S., D.D., and M.B. contributed to interpretation of data and revised the ms; G.T. coordinated MD simulations, contributed to interpretation of data, and revised the ms; B.S. and A.M. conceived the study, designed the experiments, interpreted the data, and wrote the ms; A.M. coordinated the study.
Declaration of interests
The authors declare no competing interests.
Published: June 23, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.molcel.2021.05.033.
Contributor Information
Bina Santoro, Email: bs73@columbia.edu.
Anna Moroni, Email: anna.moroni@unimi.it.
Supplemental information
References
- Abraham M.J., Murtola T., Schulz R., Páll S., Smith J.C., Hess B., Lindah E. Gromacs: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX. 2015;1–2:19–25. [Google Scholar]
- Adams P.D., Afonine P.V., Bunkóczi G., Chen V.B., Davis I.W., Echols N., Headd J.J., Hung L.-W., Kapral G.J., Grosse-Kunstleve R.W. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Afonine P.V., Poon B.K., Read R.J., Sobolev O.V., Terwilliger T.C., Urzhumtsev A., Adams P.D. Real-space refinement in PHENIX for cryo-EM and crystallography. Acta Crystallogr. D Struct. Biol. 2018;74:531–544. doi: 10.1107/S2059798318006551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akimoto M., Zhang Z., Boulton S., Selvaratnam R., VanSchouwen B., Gloyd M., Accili E.A., Lange O.F., Melacini G. A mechanism for the auto-inhibition of hyperpolarization-activated cyclic nucleotide-gated (HCN) channel opening and its relief by cAMP. J. Biol. Chem. 2014;289:22205–22220. doi: 10.1074/jbc.M114.572164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altomare C., Terragni B., Brioschi C., Milanesi R., Pagliuca C., Viscomi C., Moroni A., Baruscotti M., DiFrancesco D. Heteromeric HCN1-HCN4 channels: a comparison with native pacemaker channels from the rabbit sinoatrial node. J. Physiol. 2003;549:347–359. doi: 10.1113/jphysiol.2002.027698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson A.E.V., Kasimova M.A., Delemotte L. Exploring the Viral Channel KcvPBCV-1 Function via Computation. J. Membr. Biol. 2018;251:419–430. doi: 10.1007/s00232-018-0022-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bankston J.R., DeBerg H.A., Stoll S., Zagotta W.N. Mechanism for the inhibition of the cAMP dependence of HCN ion channels by the auxiliary subunit TRIP8b. J. Biol. Chem. 2017;292:17794–17803. doi: 10.1074/jbc.M117.800722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Best R.B., Hummer G. Optimized molecular dynamics force fields applied to the helix-coil transition of polypeptides. J. Phys. Chem. B. 2009;113:9004–9015. doi: 10.1021/jp901540t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown H.F., DiFrancesco D., Noble S.J. How does adrenaline accelerate the heart? Nature. 1979;280:235–236. doi: 10.1038/280235a0. [DOI] [PubMed] [Google Scholar]
- Bucchi A., Tognati A., Milanesi R., Baruscotti M., DiFrancesco D. Properties of ivabradine-induced block of HCN1 and HCN4 pacemaker channels. J. Physiol. 2006;572:335–346. doi: 10.1113/jphysiol.2005.100776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bucchi A., Baruscotti M., Nardini M., Barbuti A., Micheloni S., Bolognesi M., DiFrancesco D. Identification of the molecular site of ivabradine binding to HCN4 channels. PLoS ONE. 2013;8:e53132. doi: 10.1371/journal.pone.0053132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bussi G., Donadio D., Parrinello M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007;126:014101. doi: 10.1063/1.2408420. [DOI] [PubMed] [Google Scholar]
- Chen J., Mitcheson J.S., Tristani-Firouzi M., Lin M., Sanguinetti M.C. The S4-S5 linker couples voltage sensing and activation of pacemaker channels. Proc. Natl. Acad. Sci. USA. 2001;98:11277–11282. doi: 10.1073/pnas.201250598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S., Wang J., Siegelbaum S.A. Properties of hyperpolarization-activated pacemaker current defined by coassembly of HCN1 and HCN2 subunits and basal modulation by cyclic nucleotide. J. Gen. Physiol. 2001;117:491–504. doi: 10.1085/jgp.117.5.491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen V.B., Arendall W.B., 3rd, Headd J.J., Keedy D.A., Immormino R.M., Kapral G.J., Murray L.W., Richardson J.S., Richardson D.C. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark M.D., Contreras G.F., Shen R., Perozo E. Electromechanical coupling in the hyperpolarization-activated K+ channel KAT1. Nature. 2020;583:145–149. doi: 10.1038/s41586-020-2335-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordomí A., Caltabiano G., Pardo L. Membrane Protein Simulations Using AMBER Force Field and Berger Lipid Parameters. J. Chem. Theory Comput. 2012;8:948–958. doi: 10.1021/ct200491c. [DOI] [PubMed] [Google Scholar]
- Craven K.B., Zagotta W.N. Salt bridges and gating in the COOH-terminal region of HCN2 and CNGA1 channels. J. Gen. Physiol. 2004;124:663–677. doi: 10.1085/jgp.200409178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craven K.B., Olivier N.B., Zagotta W.N. C-terminal movement during gating in cyclic nucleotide-modulated channels. J. Biol. Chem. 2008;283:14728–14738. doi: 10.1074/jbc.M710463200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai G., Aman T.K., DiMaio F., Zagotta W.N. The HCN channel voltage sensor undergoes a large downward motion during hyperpolarization. Nat. Struct. Mol. Biol. 2019;26:686–694. doi: 10.1038/s41594-019-0259-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derebe M.G., Sauer D.B., Zeng W., Alam A., Shi N., Jiang Y. Tuning the ion selectivity of tetrameric cation channels by changing the number of ion binding sites. Proc. Natl. Acad. Sci. USA. 2011;108:598–602. doi: 10.1073/pnas.1013636108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiFrancesco D. Characterization of single pacemaker channels in cardiac sino-atrial node cells. Nature. 1986;324:470–473. doi: 10.1038/324470a0. [DOI] [PubMed] [Google Scholar]
- Dokmanić I., Sikić M., Tomić S. Metals in proteins: correlation between the metal-ion type, coordination number and the amino-acid residues involved in the coordination. Acta Crystallogr. D Biol. Crystallogr. 2008;64:257–263. doi: 10.1107/S090744490706595X. [DOI] [PubMed] [Google Scholar]
- Emsley P., Lohkamp B., Scott W.G., Cowtan K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Essmann U., Perera L., Berkowitz M.L., Darden T., Lee H., Pedersen L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995;103:8577–8593. [Google Scholar]
- Feenstra K.A., Hess B., Berendsen H.J.C. Improving efficiency of large time-scale molecular dynamics simulations of hydrogen-rich systems. Journal of Computational Chemistry. 1999;20(8):786–798. doi: 10.1002/(SICI)1096-987X(199906)20:8<786::AID-JCC5>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- Flynn G.E., Zagotta W.N. Insights into the molecular mechanism for hyperpolarization-dependent activation of HCN channels. Proc. Natl. Acad. Sci. USA. 2018;115:E8086–E8095. doi: 10.1073/pnas.1805596115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frace A.M., Maruoka F., Noma A. External K+ increases Na+ conductance of the hyperpolarization-activated current in rabbit cardiac pacemaker cells. Pflugers Arch. 1992;421:97–99. [PubMed] [Google Scholar]
- Goehring A., Lee C.-H., Wang K.H., Michel J.C., Claxton D.P., Baconguis I., Althoff T., Fischer S., Garcia K.C., Gouaux E. Screening and large-scale expression of membrane proteins in mammalian cells for structural studies. Nat. Protoc. 2014;9:2574–2585. doi: 10.1038/nprot.2014.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross C., Saponaro A., Santoro B., Moroni A., Thiel G., Hamacher K. Mechanical transduction of cytoplasmic-to-transmembrane-domain movements in a hyperpolarization-activated cyclic nucleotide-gated cation channel. J. Biol. Chem. 2018;293:12908–12918. doi: 10.1074/jbc.RA118.002139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanwell M.D., Curtis D.E., Lonie D.C., Vandermeersch T., Zurek E., Hutchison G.R. Avogadro: an advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 2012;4:17. doi: 10.1186/1758-2946-4-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess B., Bekker H., Berendsen H.J.C., Fraaije J.G.E.M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997;18:1463–1472. [Google Scholar]
- Hu L., Santoro B., Saponaro A., Liu H., Moroni A., Siegelbaum S. Binding of the auxiliary subunit TRIP8b to HCN channels shifts the mode of action of cAMP. J. Gen. Physiol. 2013;142:599–612. doi: 10.1085/jgp.201311013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James Z.M., Zagotta W.N. Structural insights into the mechanisms of CNBD channel function. J. Gen. Physiol. 2018;150:225–244. doi: 10.1085/jgp.201711898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson J.P.J., Jr., Zagotta W.N. The carboxyl-terminal region of cyclic nucleotide-modulated channels is a gating ring, not a permeation path. Proc. Natl. Acad. Sci. USA. 2005;102:2742–2747. doi: 10.1073/pnas.0408323102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen W.L., Chandrasekhar J., Madura J.D., Impey R.W., Klein M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983;79:926–935. [Google Scholar]
- Joung I.S., Cheatham T.E., 3rd Determination of alkali and halide monovalent ion parameters for use in explicitly solvated biomolecular simulations. J. Phys. Chem. B. 2008;112:9020–9041. doi: 10.1021/jp8001614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasimova M.A., Tewari D., Cowgill J.B., Ursuleaz W.C., Lin J.L., Delemotte L., Chanda B. Helix breaking transition in the S4 of HCN channel is critical for hyperpolarization-dependent gating. eLife. 2019;8:e53400. doi: 10.7554/eLife.53400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimanius D., Forsberg B.O., Scheres S.H., Lindahl E. Accelerated cryo-EM structure determination with parallelisation using GPUs in RELION-2. eLife. 2016;5:e18722. doi: 10.7554/eLife.18722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopec W., Köpfer D.A., Vickery O.N., Bondarenko A.S., Jansen T.L.C., de Groot B.L., Zachariae U. Direct knock-on of desolvated ions governs strict ion selectivity in K+ channels. Nat. Chem. 2018;10:813–820. doi: 10.1038/s41557-018-0105-9. [DOI] [PubMed] [Google Scholar]
- Köpfer D.A., Song C., Gruene T., Sheldrick G.M., Zachariae U., de Groot B.L. Ion permeation in K+ channels occurs by direct Coulomb knock-on. Science. 2014;346:352–355. doi: 10.1126/science.1254840. [DOI] [PubMed] [Google Scholar]
- Kunzmann P., Hamacher K. Biotite: a unifying open source computational biology framework in Python. BMC Bioinformatics. 2018;19:346. doi: 10.1186/s12859-018-2367-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwan D.C.H., Prole D.L., Yellen G. Structural changes during HCN channel gating defined by high affinity metal bridges. J. Gen. Physiol. 2012;140:279–291. doi: 10.1085/jgp.201210838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C.-H., MacKinnon R. Structures of the Human HCN1 Hyperpolarization-Activated Channel. Cell. 2017;168:111–120.e11. doi: 10.1016/j.cell.2016.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C.-H., MacKinnon R. Voltage Sensor Movements during Hyperpolarization in the HCN Channel. Cell. 2019;179:1582–1589.e7. doi: 10.1016/j.cell.2019.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M., Zhou X., Wang S., Michailidis I., Gong Y., Su D., Li H., Li X., Yang J. Structure of a eukaryotic cyclic-nucleotide-gated channel. Nature. 2017;542:60–65. doi: 10.1038/nature20819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindorff-Larsen K., Piana S., Palmo K., Maragakis P., Klepeis J.L., Dror R.O., Shaw D.E. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins. 2010;78:1950–1958. doi: 10.1002/prot.22711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lolicato M., Nardini M., Gazzarrini S., Möller S., Bertinetti D., Herberg F.W., Bolognesi M., Martin H., Fasolini M., Bertrand J.A. Tetramerization dynamics of C-terminal domain underlies isoform-specific cAMP gating in hyperpolarization-activated cyclic nucleotide-gated channels. J. Biol. Chem. 2011;286:44811–44820. doi: 10.1074/jbc.M111.297606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyashchenko A.K., Tibbs G.R. Ion binding in the open HCN pacemaker channel pore: fast mechanisms to shape “slow” channels. J. Gen. Physiol. 2008;131:227–243. doi: 10.1085/jgp.200709868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moroni A., Barbuti A., Altomare C., Viscomi C., Morgan J., Baruscotti M., DiFrancesco D. Kinetic and ionic properties of the human HCN2 pacemaker channel. Pflugers Arch. 2000;439:618–626. doi: 10.1007/s004249900225. [DOI] [PubMed] [Google Scholar]
- Morris G.M., Huey R., Lindstrom W., Sanner M.F., Belew R.K., Goodsell D.S., Olson A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009;30:2785–2791. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Napolitano L.M.R., Bisha I., De March M., Marchesi A., Arcangeletti M., Demitri N., Mazzolini M., Rodriguez A., Magistrato A., Onesti S. A structural, functional, and computational analysis suggests pore flexibility as the base for the poor selectivity of CNG channels. Proc. Natl. Acad. Sci. USA. 2015;112:E3619–E3628. doi: 10.1073/pnas.1503334112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parrinello M., Rahman A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Physiol. 1981;52:7182–7190. [Google Scholar]
- Pettersen E.F., Goddard T.D., Huang C.C., Couch G.S., Greenblatt D.M., Meng E.C., Ferrin T.E. UCSF Chimera--a visualization system for exploratory research and analysis. J. Comput. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Porro A., Saponaro A., Gasparri F., Bauer D., Gross C., Pisoni M., Abbandonato G., Hamacher K., Santoro B., Thiel G., Moroni A. The HCN domain couples voltage gating and cAMP response in hyperpolarization-activated cyclic nucleotide-gated channels. eLife. 2019;8:e49672. doi: 10.7554/eLife.49672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porro A., Binda A., Pisoni M., Donadoni C., Rivolta I., Saponaro A. Rational design of a mutation to investigate the role of the brain protein TRIP8b in limiting the cAMP response of HCN channels in neurons. J. Gen. Physiol. 2020;152 doi: 10.1085/jgp.202012596. e202012596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proenza C., Yellen G. Distinct populations of HCN pacemaker channels produce voltage-dependent and voltage-independent currents. J. Gen. Physiol. 2006;127:183–190. doi: 10.1085/jgp.200509389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proenza C., Angoli D., Agranovich E., Macri V., Accili E.A. Pacemaker channels produce an instantaneous current. J. Biol. Chem. 2002;277:5101–5109. doi: 10.1074/jbc.M106974200. [DOI] [PubMed] [Google Scholar]
- Punjani A., Rubinstein J.L., Fleet D.J., Brubaker M.A. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods. 2017;14:290–296. doi: 10.1038/nmeth.4169. [DOI] [PubMed] [Google Scholar]
- Ramentol R., Perez M.E., Larsson H.P. Gating mechanism of hyperpolarization-activated HCN pacemaker channels. Nat. Commun. 2020;11:1419. doi: 10.1038/s41467-020-15233-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rappe A.K., Casewit C.J., Colwell K.S., Goddard W.A., Skiff W.M. UFF, a full periodic table force field for molecular mechanics and molecular dynamics simulations. J. Am. Chem. Soc. 1992;114:10024–10035. [Google Scholar]
- Rohou A., Grigorieff N. CTFFIND4: Fast and accurate defocus estimation from electron micrographs. J. Struct. Biol. 2015;192:216–221. doi: 10.1016/j.jsb.2015.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santoro B., Wainger B.J., Siegelbaum S.A. Regulation of HCN channel surface expression by a novel C-terminal protein-protein interaction. J. Neurosci. 2004;24:10750–10762. doi: 10.1523/JNEUROSCI.3300-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santoro B., Hu L., Liu H., Saponaro A., Pian P., Piskorowski R.A., Moroni A., Siegelbaum S.A. TRIP8b regulates HCN1 channel trafficking and gating through two distinct C-terminal interaction sites. J. Neurosci. 2011;31:4074–4086. doi: 10.1523/JNEUROSCI.5707-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saponaro A., Pauleta S.R., Cantini F., Matzapetakis M., Hammann C., Donadoni C., Hu L., Thiel G., Banci L., Santoro B., Moroni A. Structural basis for the mutual antagonism of cAMP and TRIP8b in regulating HCN channel function. Proc. Natl. Acad. Sci. USA. 2014;111:14577–14582. doi: 10.1073/pnas.1410389111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saponaro A., Cantini F., Porro A., Bucchi A., DiFrancesco D., Maione V., Donadoni C., Introini B., Mesirca P., Mangoni M.E. A synthetic peptide that prevents cAMP regulation in mammalian hyperpolarization-activated cyclic nucleotide-gated (HCN) channels. eLife. 2018;7:e35753. doi: 10.7554/eLife.35753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheres S.H.W. A Bayesian view on cryo-EM structure determination. J. Mol. Biol. 2012;415:406–418. doi: 10.1016/j.jmb.2011.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi W., Wymore R., Yu H., Wu J., Wymore R.T., Pan Z., Robinson R.B., Dixon J.E., McKinnon D., Cohen I.S. Distribution and prevalence of hyperpolarization-activated cation channel (HCN) mRNA expression in cardiac tissues. Circ. Res. 1999;85:e1–e6. doi: 10.1161/01.res.85.1.e1. [DOI] [PubMed] [Google Scholar]
- Shi C., He Y., Hendriks K., de Groot B.L., Cai X., Tian C., Lange A., Sun H. A single NaK channel conformation is not enough for non-selective ion conduction. Nat. Commun. 2018;9:717. doi: 10.1038/s41467-018-03179-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smart O.S., Neduvelil J.G., Wang X., Wallace B.A., Sansom M.S. HOLE: a program for the analysis of the pore dimensions of ion channel structural models. J. Mol. Graph. 1996;14:354–360, 376. doi: 10.1016/s0263-7855(97)00009-x. [DOI] [PubMed] [Google Scholar]
- Stock L., Delemotte L., Carnevale V., Treptow W., Klein M.L. Conduction in a biological sodium selective channel. J. Phys. Chem. B. 2013;117:3782–3789. doi: 10.1021/jp401403b. [DOI] [PubMed] [Google Scholar]
- Suloway C., Pulokas J., Fellmann D., Cheng A., Guerra F., Quispe J., Stagg S., Potter C.S., Carragher B. Automated molecular microscopy: the new Leginon system. J. Struct. Biol. 2005;151:41–60. doi: 10.1016/j.jsb.2005.03.010. [DOI] [PubMed] [Google Scholar]
- Tanguay J., Callahan K.M., D’Avanzo N. Characterization of drug binding within the HCN1 channel pore. Sci. Rep. 2019;9:465. doi: 10.1038/s41598-018-37116-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thon S., Schmauder R., Benndorf K. Elementary functional properties of single HCN2 channels. Biophys. J. 2013;105:1581–1589. doi: 10.1016/j.bpj.2013.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Der Spoel D., Lindahl E., Hess B., Groenhof G., Mark A.E., Berendsen H.J.C. GROMACS: fast, flexible, and free. J. Comput. Chem. 2005;26:1701–1718. doi: 10.1002/jcc.20291. [DOI] [PubMed] [Google Scholar]
- Viscomi C., Altomare C., Bucchi A., Camatini E., Baruscotti M., Moroni A., DiFrancesco D. C terminus-mediated control of voltage and cAMP gating of hyperpolarization-activated cyclic nucleotide-gated channels. J. Biol. Chem. 2001;276:29930–29934. doi: 10.1074/jbc.M103971200. [DOI] [PubMed] [Google Scholar]
- Wahl-Schott C., Baumann L., Zong X., Biel M. An arginine residue in the pore region is a key determinant of chloride dependence in cardiac pacemaker channels. J. Biol. Chem. 2005;280:13694–13700. doi: 10.1074/jbc.M413197200. [DOI] [PubMed] [Google Scholar]
- Wainger B.J., DeGennaro M., Santoro B., Siegelbaum S.A., Tibbs G.R. Molecular mechanism of cAMP modulation of HCN pacemaker channels. Nature. 2001;411:805–810. doi: 10.1038/35081088. [DOI] [PubMed] [Google Scholar]
- Wang W., MacKinnon R. Cryo-EM Structure of the Open Human Ether-à-go-go-Related K+ Channel hERG. Cell. 2017;169:422–430.e10. doi: 10.1016/j.cell.2017.03.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weißgraeber S., Saponaro A., Thiel G., Hamacher K. A reduced mechanical model for cAMP-modulated gating in HCN channels. Sci. Rep. 2017;7:40168. doi: 10.1038/srep40168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whicher J.R., MacKinnon R. Structure of the voltage-gated K+ channel Eag1 reveals an alternative voltage sensing mechanism. Science. 2016;353:664–669. doi: 10.1126/science.aaf8070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wollmuth L.P., Hille B. Ionic selectivity of Ih channels of rod photoreceptors in tiger salamanders. J. Gen. Physiol. 1992;100:749–765. doi: 10.1085/jgp.100.5.749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zagotta W.N., Olivier N.B., Black K.D., Young E.C., Olson R., Gouaux E. Structural basis for modulation and agonist specificity of HCN pacemaker channels. Nature. 2003;425:200–205. doi: 10.1038/nature01922. [DOI] [PubMed] [Google Scholar]
- Zheng H., Cooper D.R., Porebski P.J., Shabalin I.G., Handing K.B., Minor W. CheckMyMetal: a macromolecular metal-binding validation tool. Acta Crystallogr. D Struct. Biol. 2017;73:223–233. doi: 10.1107/S2059798317001061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng S.Q., Palovcak E., Armache J.-P., Verba K.A., Cheng Y., Agard D.A. MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods. 2017;14:331–332. doi: 10.1038/nmeth.4193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng X., Fu Z., Su D., Zhang Y., Li M., Pan Y., Li H., Li S., Grassucci R.A., Ren Z. Mechanism of ligand activation of a eukaryotic cyclic nucleotide-gated channel. Nat. Struct. Mol. Biol. 2020;27:625–634. doi: 10.1038/s41594-020-0433-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zivanov J., Nakane T., Forsberg B.O., Kimanius D., Hagen W.J., Lindahl E., Scheres S.H. New tools for automated high-resolution cryo-EM structure determination in RELION-3. eLife. 2018;7:e42166. doi: 10.7554/eLife.42166. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The cryo-EM density maps have been deposited in the Electron Microscopy Data Bank under the accession numbers: EMD-12466, EMD-12512, and EMD-12513. Coordinates have been deposited in the Protein Data Bank under the PDB accession numbers: 7NMN, 7NP3, and 7NP4.