

Abstract
Electrochemistry has recently gained increased attention as a versatile strategy for achieving challenging transformations at the forefront of synthetic organic chemistry. Electrochemistry’s unique ability to generate highly reactive radical and radical ion intermediates in a controlled fashion under mild conditions has inspired the development of a number of new electrochemical methodologies for the preparation of valuable chemical motifs. Particularly, recent developments in electrosynthesis have featured an increased use of redox-active electrocatalysts to further enhance control over the selective formation and downstream reactivity of these reactive intermediates. Furthermore, electrocatalytic mediators enable synthetic transformations to proceed in a manner that is mechanistically distinct from purely chemical methods, allowing for the subversion of kinetic and thermodynamic obstacles encountered in conventional organic synthesis. This review highlights key innovations within the past decade in the area of synthetic electrocatalysis, with emphasis on the mechanisms and catalyst design principles underpinning these advancements. A host of oxidative and reductive electrocatalytic methodologies are discussed and are grouped according to the classification of the synthetic transformation and the nature of the electrocatalyst.
1. Introduction
Since its inception in the 19th century, electrochemistry has matured to serve a variety of applications ranging from energy storage to metallurgy to chemical synthesis.1 The success of electrochemistry in these diverse contexts can be credited to the unique capabilities afforded by electrochemical systems. Principally, the use of an external applied potential provides a means to drive reactions far from thermodynamic equilibria, enabling access to reaction pathways and chemical intermediates that would otherwise be difficult to employ. Additionally, electrochemical systems often afford mild conditions and the potential for scalability, supporting their use in the industrial synthesis of bulk and fine chemicals. The use of electricity in place of environmentally deleterious chemical reagents such as stoichiometric oxidants and reductants also renders electrochemistry an attractive approach for improving the efficiency and sustainability of synthetic chemistry.
In recent years, electrochemistry has experienced an exciting and ongoing renaissance in the context of organic synthesis. Driven in part by the characteristic advantages of electrochemistry described above, this surge of interest has led to the development of an increasing number of electrochemical synthetic methodologies, including reactivities not directly attainable through traditional chemical methods alone.2,3 Specifically, single-electron oxidation and reduction events facilitated by electrochemistry are well-suited4 towards the creative use of radical intermediates in achieving synthetic transformations. When the formation and downstream reactivity of these intermediates are placed under the control of catalytic redox mediators, electrochemistry emerges as a powerful strategy for addressing prevailing trends in organic synthesis.
A conceptual analysis of an electrochemical reaction quickly reveals the enhanced capabilities offered by redox mediators in electrosynthesis. While electrochemistry can enable activation of substrates at the electrode surface (i.e., direct electrolysis), this mode of activation often requires substrate engineering to regulate reactivity and can forgo control over the selectivity of chemical steps following electron transfer.5 When electrochemistry is applied towards the construction and modification of complex organic molecules, the limitations of direct electrolysis can become more pronounced. However, such constraints can be addressed through the implementation of redox-active catalysts which facilitate efficient electron transfer events and offer increased selectivity relative to non-mediated processes.6
Electrocatalysis can either utilize molecular mediators as catalytic species (such as transition metal complexes or redox-active organic compounds; commonly referred to as homogenous or molecular electrocatalysis)7 or an electrode surface itself (referred to as heterogenous electrocatalysis8 herein). Traditionally, the term “electrocatalysis” is used to define the latter case involving an catalytic electrode, whereas systems featuring molecular mediators have been referred to as “molecular catalysis for electrochemical reactions.”9 In this review, electrocatalysis is described to encompass both homogeneous and heterogeneous cases, given that the defining feature of an electrocatalytic system is the use of a redox-active species which can facilitate a catalytic cycle and accelerate a specific reaction pathway.10,11 In many of the examples presented herein, electrocatalysis gives rise to new reactivities that are not accessible through non-electrocatalytic means.
The scope of electrocatalysis in chemical literature is extensive, with major areas of growth including the development of electrocatalytic systems for energy-related applications (such as water splitting12,13 and CO2 reduction14) and, more recently, synthetic chemistry.15,16 In the area of organic synthesis, continuing pressure to develop efficient and sustainable synthetic strategies has driven a tremendous surge of interest in electrocatalytic methodologies from within the broader organic chemistry community (Fig. 1).
Fig. 1.

Number of publications per year (indexed in SciFinder since 2000) in the research areas of organic electrochemistry and organic electrocatalysis.
This review highlights key innovations in the past decade (since ca. 2010) within the growing discipline of synthetic electrocatalysis. We aim to showcase the versatility of these electrocatalytic systems as well as illustrate the design principles used to develop the electrocatalytic mediators needed to power these systems. The electrocatalysts discussed herein can be subdivided into two general classes base on their role in mediating electrochemical transformations.13 The first category of electrocatalysts encompasses redox mediators which serve the sole purpose of facilitating electron transfer events between electrodes and substrates, accelerating redox reactions relative to a non-mediated system. The second category of electrocatalysts we discuss are electrocatalysts that shuttle both electrons and chemical information to substrates, placing reactivity and selectivity directly under the control of the catalyst itself. Consistent with the general definition of a catalyst, we primarily focus on reactions that utilize electrocatalysts in sub-stoichiometric amounts. However, to fully describe the reactivities enabled by electrochemical systems, we include some examples where an electrochemical mediator is used in higher loadings.
Emphasis is placed on the mechanistic details of a host of oxidative and reductive electrocatalytic transformations at the cutting edge of the discipline. We first discuss advancements occurring within the well-established realm of oxidative electrocatalysis, and then proceed to discuss emerging discoveries in the less explored areas of electroreductive transformations, paired electrolysis, and heterogenous electrocatalysis. Finally, we explore bioelectrocatalysis electrophotocatalysis to demonstrate the extension of electrocatalytic strategies into interdisciplinary applications. These recent developments illustrate many of the design principles used to realize a range of electrocatalytic methodologies and showcase the capabilities of electrochemistry in facilitating increasingly complex transformations.
Electrochemical glossary
Readers who are unfamiliar with electrochemical reactions can find valuable information on the basics of electrochemistry, including how to set up electrochemical reactions, in recent reviews by Hilt,17 Schotten and Willans,18 and in a recent account by Baran.19
In the present review, the following notation is used: a (+) sign denotes the anode; a (−) sign denotes the cathode; “|” denotes an undivided cell setup; “∥” denotes a divided cell setup. For example, “C(+)|Pt(−)” indicates that the electrolysis uses an C anode and a Pt cathode in an undivided cell. Current (i) or current density (j), electrode potential (Ec or Ea), cell voltage (Ucell) values are given when the electrolysis is conducted under constant current, constant potential, or constant cell voltage conditions, respectively. Potential values are listed with respect to a reference electrode such as ferrocene/ferrocenium (Fc/Fc+) or saturated calomel electrode (SCE). The conversion between Fc/Fc+ and SCE is E1/2(Fc/Fc+) = 0.40 V vs. SCE.
Additionally, throughout the text, mechanistic schemes are color-coded by the nature of the electrochemical transformation discussed: oxidative reactions are identified by a beige background, reductive reactions are shown with a blue background, and paired electrolysis reactions are presented with a grey background.
2. Anodic oxidation
Oxidative reactions are important tools for the synthesis of complex molecules, enabling the efficient construction of C–C bonds and the incorporation of heteroatoms into molecular architectures. Common chemical methods for oxidative reactions typically rely on the use of stoichiometric amounts of oxidizing agents, such as high-valent chromium and manganese complexes as well as hypervalent iodine species, which can often be hazardous and generate substantial amounts of waste. Recent methodological advances have led to milder reaction systems using more desirable oxidants such as O2 and H2O2.20,21 Nevertheless, finding “alternatives for oxidations” remains one of the key green chemistry research areas identified by the ACS Green Chemistry Institute Pharmaceutical Roundtable in their 2018 report.22 In this regard, the adoption of a catalyst that can be regenerated upon anodic oxidation is highly desirable from a green chemistry perspective, since waste generation is considerably reduced. Indeed, electrochemical oxidations are usually coupled with hydrogen evolution reaction to generate H2 gas as the major by-product.
Beyond its utility in recycling traditional chemical oxidants, anodic oxidation has been exploited towards the design of electrocatalytic systems to accomplish a wide range of chemical transformations. The scope of chemical mediators for these oxidative systems is extensive. For instance, the radical cations of triarylamines and ferrocenium ion (Fc+) have been employed for electron transfer. The phthalimide N-oxyl (PINO) radical, the radical cation from quinuclidine, chlorine radicals, and nitrate radicals have been used as hydrogen atom transfer (HAT) agents. Oxoammonium cations derived from nitroxyl radicals have been employed for formal hydride transfer. Further, anodic oxidation of metallic species to high valent states is often used to promote reductive elimination, which is commonly seen in C–H activation reactions. Recently, Earth-abundant 3d metals have been explored in a myriad of radical transformations. This diversity of electrooxidative reactions is discussed in the following subsections and is grouped based on the nature of the redox mediator.
Main group compounds as mediators
2.1. Halogen-based mediators (halides, hypervalent iodine)
Halide salts are among the simplest and most commonly used halogen-based mediators, which are non-toxic and inexpensive. Other less explored halogen-based mediators include iodate/periodate and hypervalent iodine compounds. Upon anodic oxidation, halides generate reactive halogen compounds (e.g., Cl2, Br2, I2), and when electrolysis is carried out in water, hypohalites can also form.
Well-established reactivities have been explored with oxidized halides, such as nucleophilic substitution and base-promoted elimination.23,24 Generation of radicals from the homolytic cleavage of weak heteroatom–halogen bonds is a less explored yet powerful approach for forging carbon–heteroatom bonds. Although halogen-based mediators can in theory be regenerated in electrochemical systems, in some cases they are used in stoichiometric amounts to ensure high reaction yields.
2.1.1. Activation of alkenes with halogens.
In 2016, Zeng, Sun, and co-workers generated 3-methoxyindolines via an electrochemical aminooxygenation reaction, mediated by iodide (Fig. 2).25 This reaction uses anodically generated iodine to produce an iodonium ion 2–3, which is subjected to an intramolecular SN2 reaction, followed by a second substitution with cathodically generated methoxide. LiClO4 can be removed from the system without affecting the yield, since the iodide source also functions as an electrolyte.
Fig. 2.

(A and B) Iodide-mediated aminooxygenation of alkenes.
In 2020, the formation of iodonium intermediates was explored by Lennox and co-workers for vicinal difluorination of alkenes (Fig. 3).26 The fluorine source arises from a mixture of commercially available Et3N·3HF and pyridine·9HF, to generate a ratio of 5.6 equivalents of HF per amine. The mechanism is proposed to start with formation of iodonium ion 3–3, followed by ring opening with fluoride and displacement of aryl iodide with a second fluoride ion. For alkenes that are more easily oxidized than the iodoarene, the authors first generated a pool of hypervalent iodine reagent and later added the alkene, demonstrating a broad functional group compatibility for this methodology.
Fig. 3.

(A and B) Hypervalent Iodine-mediated difluorination of alkenes.
2.1.2. α-Functionalization of nitriles and carbonyls.
Recently, the use of halides as electrochemical mediators for the α-functionalization of nitriles and carbonyls has been extensively explored. This reactivity relies on the α-deprotonation of these functional groups, generating stabilized anions, which can react with Br2 or I2 (see intermediate 4–3, Fig. 4). This newly installed halogen group can be easily displaced by a wide range of nucleophiles in an SN2 fashion. Appending a nucleophile in the same molecule has been employed to generate cyclic motifs intramolecularly (Fig. 4), such as cyclopropanes,27–30 cyclobutanes,31 cyclopentanes,32 β-lactams,33 and dihydrofurans.34 In the intermolecular realm, carbon, nitrogen, and sulfur nucleophiles have been explored, including nitrile anions,35 enolates,36 amines,37 sulfinates38 and carbamodithioates.39 Although several of these methods use high loadings or even stoichiometric amounts (see example 4–6, Fig. 4) of halide mediators, halide salts are fairly inexpensive. In 2021, the scalability of these methods was explored by Baran and Sabatini,25 where compound 4–5 was synthesized on a hundred-gram scale.
Fig. 4.

(A and B) Iodide-mediated cyclization.
During the 2010s, a different approach towards the electrocatalytic α-functionalization of ketones was reported by Zha, Wang, and co-workers, where the electrolysis of arylmethylketones and iodide is carried out under aerobic conditions (Fig. 5). The authors propose the intermediacy of a peroxyl radical (5–4), which could be later converted to different functionalities, such as α-keto-esters,40 α-keto-amides,41 enaminones,42 and isatins.43 Although the role of the iodide mediator is not fully understood for these reactions, we hypothesize that in situ generated iodine could react with an enolate, and the resultant C–I bond could undergo homolytic cleavage followed by O2 trapping to give the peroxyl radical intermediate.
Fig. 5.

(A and B) Iodide-mediated oxidation of methylketones.
Elinson and co-workers reported the electrochemical conversion of ketones to α-carbonyl-acetals.44–46 In this reaction, an enolate reacts with anodically generated iodine to form a C−I bond. Addition of cathodically generated methoxide to the carbonyl of 6–1 gives intermediate 6–3, which undergoes epoxide formation followed by methanolysis, releasing a phenol leaving group to generate 6–2. In 2017, a similar reactivity was explored by Zeng, Sun, and co-workers during the selective oxidation of lignin-type model compounds (Fig. 6).47
Fig. 6.

(A and B) Iodide-mediated oxidation of lignin model compounds.
2.1.3. Periodate as mediator.
The periodate anion is a versatile oxidant and is typically employed in the oxidative cleavage of diols or the in situ regeneration of catalysts such as osmium tetroxide or ruthenium tetroxide. Oxidative cleavage of vicinal diols, vicinal aminoalcohols, and α-hydroxyketones was studied by Pillai and co-workers using anodically generated periodate.48 The production of this hypervalent iodine reagent requires the use of a divided cell to prevent its unproductive cathodic reduction. This mediator was also employed by Schäfer and co-workers to promote regeneration of ruthenium tetroxide, which was used in the electrochemical oxidative cleavage of alkenes49 (Fig. 7). In 2020, Waldvogel and co-workers reported an efficient method for generating periodate electrochemically from iodide for ex situ applications.50
Fig. 7.

(A and B) Oxidative cleavage of alkenes using electrochemical generated periodate.
2.1.4. Oxidation of sulfur functional groups.
The use of halides and hypervalent iodine compounds as electrochemical mediators has been useful for the selective oxidation of different sulfur functional groups, including sulfinate salts, thiols, dithianes, and dithiolanes.51,52 For example, upon anodic oxidation of halides, sulfinate salts can form adducts with a labile heteroatom-halogen bond (O–I or S–I), which can be homolytically cleaved by light or heat. The newly generated sulfur-centered radicals (8–4) can then add to the π systems of alkynes53 or alkenes54 (Fig. 8), and this protocol can also be used for the construction of sulfonamides.55
Fig. 8.

(A and B) Radical cyclization triggered by electrochemical generated sulfur centered radicals.
In 2010, Fuchigami and co-workers reported an electrochemical approach to convert dithiolanes to geminal difluoride groups (Fig. 9). This transformation is mediated by 9–2, which can undergo anodic oxidation in the presence of Et3N-5HF electrolyte to generate hypervalent iodine species 9–4 with a tethered imidazolium ion. 9–4 then reacts with substrate 9–1 to give the gem-difluorinated product 9–3. At the end of the reaction, the aryl iodide mediator can be recovered with a simple extraction and reused.56 Fuchigami has also developed a fluorodesulfurization employing chloride as an electrochemical mediator, which can aid in the generation of hypervalent iodine species from polystyrene-supported iodobenzene.57
Fig. 9.
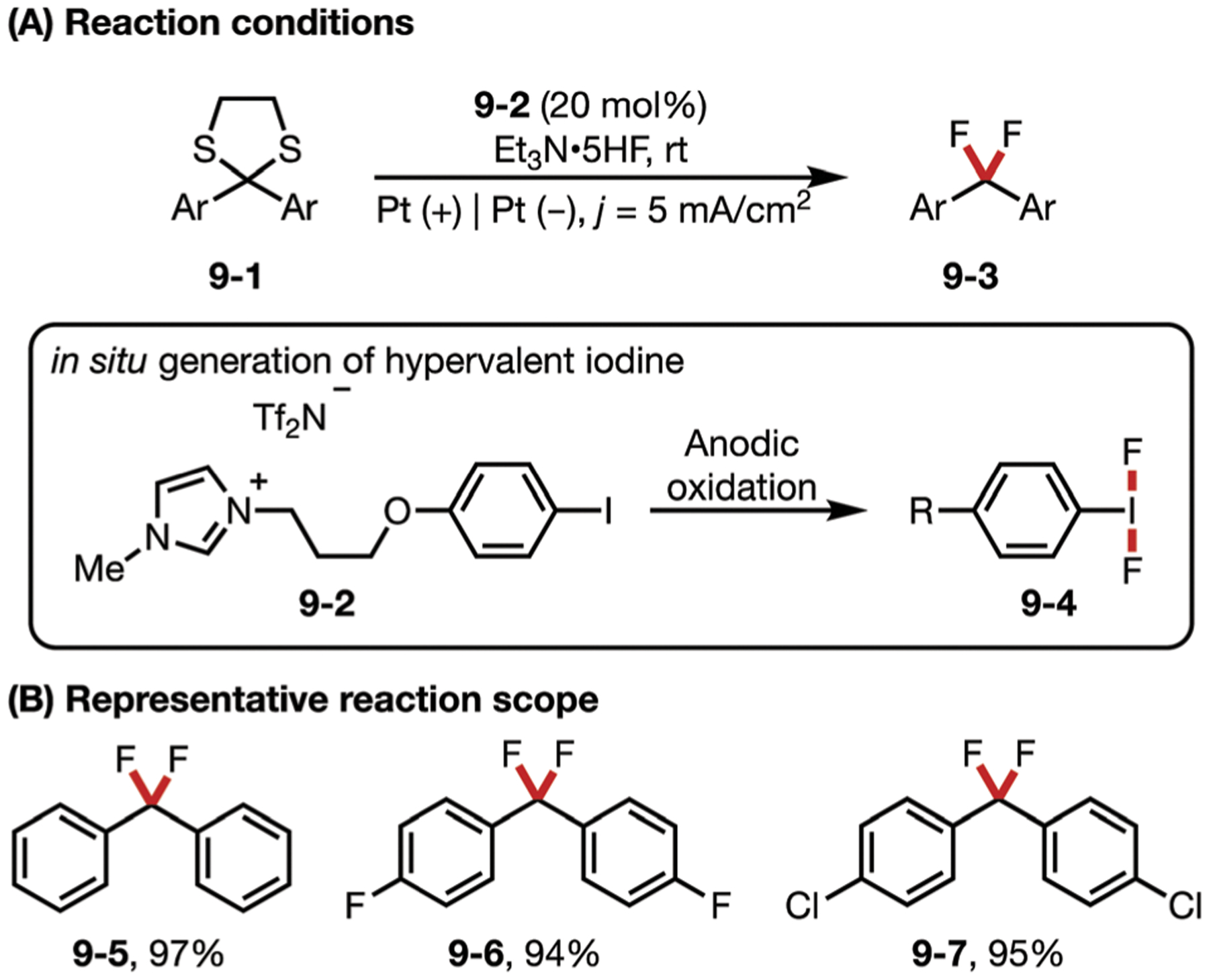
(A and B) Hypervalent iodine-mediated fluorodesulfurization of dithiolanes.
2.1.5. Oxidation of nitrogenated functional groups.
The cleavage of weak nitrogen–halogen bonds is the basis for the development of a wide range of reactions. Two possible mechanisms for this reactivity have been proposed. In an ionic mechanism, upon formation of the iodinated amine intermediate, a simple beta-H+ elimination step promoted by base will generate a C=N group to complete the mediated dehydrogenation.58–60 In radical mechanisms, an N-centered radical is generated from the halogenated amine by modes of single-electron activation such as photoirradiation (see Section 7: Electrophotocatalysis).61,62 As an example of the ionic scenario, in 2014, Little and co-workers demonstrated the use of anodically generated electrophilic iodine species to promote the formation of benzoxazoles from 2-aminobenzoxazolines via key intermediate 10–452 (Fig. 10).
Fig. 10.

(A and B) Iodide-mediated synthesis of 2-aminobenzoxazoles.
2.2. Nitrate mediators
The nitrate radical was first identified in the atmosphere during the late 1800s and has been shown to play an important role in atmospheric chemistry. It can be formed by reaction of NO2 with ozone or through the decomposition of N2O5.63 Nitrate radicals can react with aldehydes via an HAT process, resulting in acyl radicals and nitric acid. Nitrate radicals can also add to alkenes, to furnish organic nitrate functional groups.
2.2.1. Alcohol oxidation.
Nitrate radicals can also be generated electrochemically via anodic oxidation of nitrate anions. Despite this interesting reactivities, this species has been rarely used in synthetic electrochemistry. One of these examples was reported in 2012 by Raja and co-workers, where an electrochemical oxidation of alcohols to aldehydes or ketones was mediated by nitrate radical64,65 (Fig. 11). This reaction is thought to proceed by an HAT event, whereby a nitrate radical removes a hydrogen from the carbinolic position, followed by a second oxidation proceeding through direct anodic oxidation of the carbon centered radical or a second oxidation mediated by nitrate radical.
Fig. 11.
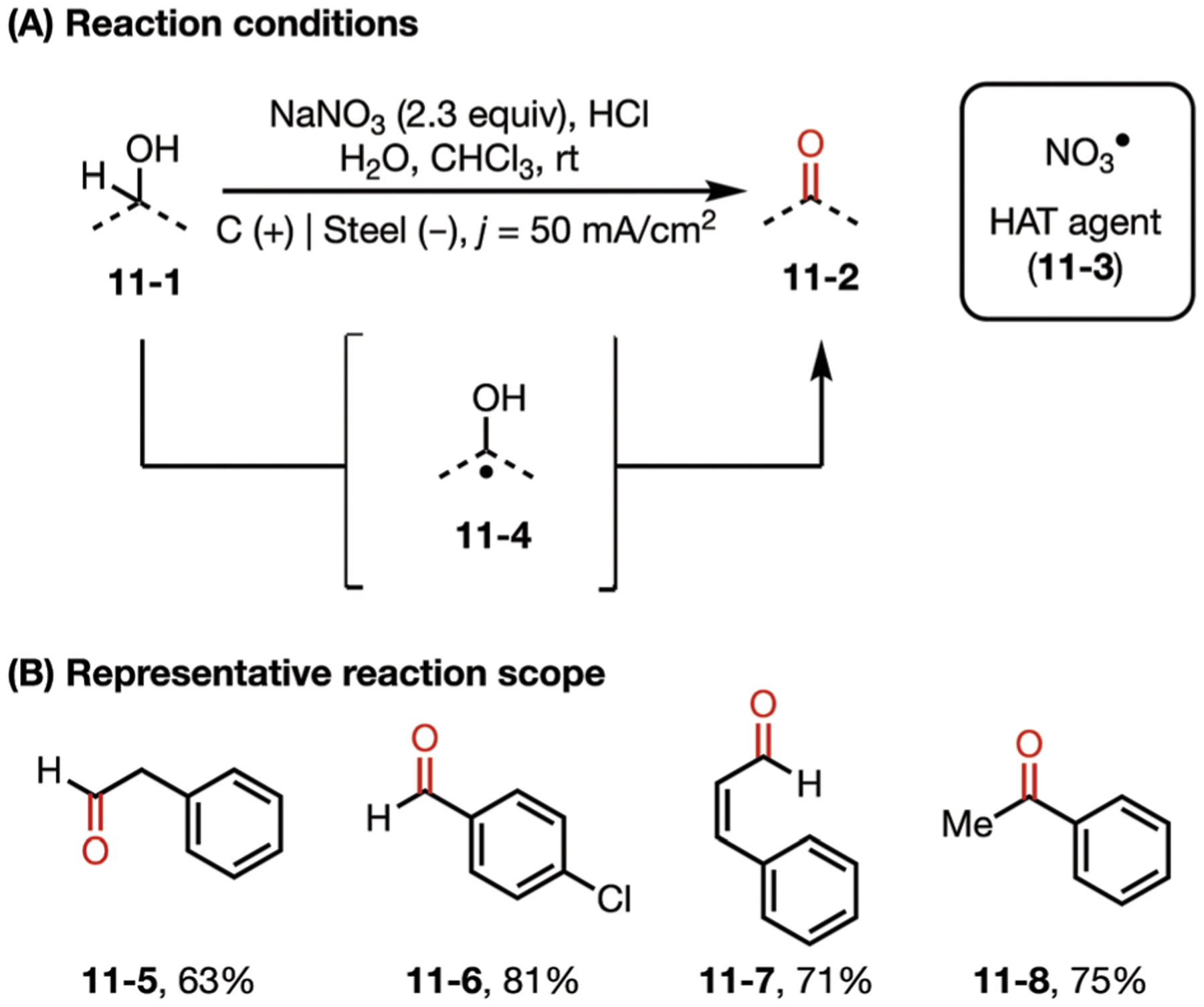
(A and B) Nitrate-mediated oxidation of alcohols.
2.2.2. Fluorination of unactivated C(sp3)–H bonds.
In 2019, Baran and co-workers employed nitrate radicals to achieve fluorination of unactivated C(sp3)–H bonds66 (Fig. 12). The proposed mechanism starts with a HAT process mediated by a nitrate radical, which leads to a radical chain process that is sustained by the nitrogen-centered radical cation 12–4 derived from Selectfluor (12–3), the fluorinating agent. This methodology provided good yields for substrates containing ketones, esters, carboxylic acids, and imides.
Fig. 12.

(A–C) Nitrate-mediated C(sp3)–H fluorination.
2.3. Nitroxyl mediators
Stable nitroxyl radicals have been reported since the 1960s67–71 and have since attracted the attention of synthetic chemists. These radicals can be oxidized electrochemically to oxoammonium cations, which have been extensively studied for the oxidation of alcohols and amines. In 2015, Minteer, Sigman, and co-workers combined experimental and computational techniques to group nitroxyl mediators into four distinct classes, matching high or low oxidation potentials of the mediator with high or low catalytic activity for alcohol oxidation (Fig. 13).72 The top left quadrant of the graph originated from this study contains nitroxyls with high activity that can be generated at low potential, which is desirable for energy applications. On the other extreme, the bottom right corner contains poorly active mediators that require high oxidation potentials, which are useful as EPR labels. Curiously, TEMPO (labeled as number 1 in Fig. 13), the most frequently employed nitroxyl catalyst and EPR label, is not the best option for either applications, and its common use can be understood due to its relative ease of access. In 2018, Stahl, Rafiee, and co-workers extensively reviewed the use of nitroxyl radicals in electrocatalytic reactions.73
Fig. 13.

Minteer and Sigman’s model for nitroxyl mediators. This figure has been reproduced from ref. 72 with permission from American Chemical Society, copyright 2015.
2.3.1. Alcohol oxidation.
During the 2000’s, the research groups of Tanaka,74–76 Onomura,77 and Frontana-Uribe78 reported cooperative electrochemical systems involving bromide and nitroxyl radicals, which could generate oxoammonium cations capable of oxidizing primary and secondary alcohols to the corresponding carbonyl compounds. Tanaka79 and Brown80 also reported direct oxidation of TEMPO to induce oxidation of alcohols to carboxylic acids, or aldehydes, and ketones.
During the last decade, Stahl and co-workers have explored the oxidation of alcohols mediated by anodically generated oxoammonium cations.81–84 Stahl observed that ACT (4-acetamido-TEMPO) (E = 0.65 V vs. Ag/AgCl) is more electrocatalytic active than AZADO (2-azaadamantane-N-oxyl) and ABNO (9-azabicyclo[3.3.1]nonane N-oxyl) (EAZADO = 0.45 V, EABNO = 0.48 V, vs. Ag/AgCl) which are less sterically hindered.81 The higher activity of ACT is attributed to the fact that its corresponding oxoammonium cation is generated at a higher potential. A higher pH environment further contributes to the increase of turnover frequency of the electrocatalyst. This finding was explored for oxidation of alcohols and aldehydes into the corresponding carboxylic acids82,83 (Fig. 14).
Fig. 14.

(A and B) Nitroxyl-mediated oxidation of alcohols and aldehydes to carboxylic acids.
Identification of electrocatalytic systems that operate at low potentials and exhibit high turnover frequencies is highly relevant to the energy-conversion field. In 2016, Stahl and co-workers demonstrated an elegant example with a cooperative electrochemical system, composed of CuII(bpy) and TEMPO, for oxidation of alcohols85 (Fig. 15). This system combines CuII’s ability to promote single-electron oxidations with the electron-proton acceptor property of nitroxyl radicals. Since high energy intermediates are avoided in this catalytic cycle, this reaction can be realized with an anodic potential 500 mV lower than a system using only TEMPO, which would involve oxoammonium cations (−0.14 V and +0.36 V, vs. Fc0/Fc+). This cooperative system also proved to be advantageous in terms of kinetics, with a kobs (11.6 s−1) nearly 5 times higher than the corresponding kobs (2.3 s−1) for the TEMPO-only system.
Fig. 15.

Cooperative copper-nitroxyl system for oxidation of alcohols to aldehydes.
2.3.2. Oxidation of amines and carbamates.
The electrochemical generation of oxoammonium cations has been applied to oxidation of amines and carbamates, yielding amides and imides, respectively. While Little86 used a cooperative system formed by bromide/TEMPO, Kashiwagi87 and Stahl88 employed modified versions of TEMPO to accomplish this type of transformation (Fig. 16). The proposed mechanism first involves a two-electron-one-proton transfer to generate a stabilized iminium species 16–3 which is subsequently trapped by water to form a hemiaminal 16–4. In a second step, the hemiaminal 16–4 is subjected to a two-electron-one-proton transfer, forming the carbonyl group.
Fig. 16.

(A–C) Nitroxyl-mediated oxidation of carbamates.
2.3.3. Generation of nitrogen-centered radicals.
New reactivities of nitroxyl radicals and the corresponding oxoammonium cations have emerged recently. In 2014, Xu and co-workers reported the formation of nitrogen-centered radicals via the oxidation of amides by TEMPO+ under basic conditions89 (Fig. 17). Mechanistic investigation suggests that this process might involve a single-electron transfer from a nitrogen-centered anion to an oxoammonium cation. The nitrogenated radical was employed in a subsequent intramolecular reaction with an olefin to produce bicyclic products. In this reaction, TEMPO is used in stoichiometric amounts, since it is also employed as a radical trap.
Fig. 17.

(A and B) Radical cyclization triggered by electrochemical generated nitrogen centered radicals.
The formation of nitroxyl radicals was also explored by Lin and co-workers. During the investigation of an electrochemical alkene azidooxygenation,90 Lin postulated the anodically generation of a metastable charge-transfer complex (CTC) arising from the combination of azide anion with TEMPO+. This structure was confirmed by X-ray crystallography in 2021.91 When CTC is in solution, an equilibrium is established with formation of small amounts of azidyl radical, which can add to an alkene, generating a carbon centered radical that can be trapped by TEMPO.
During the development of this azidooxygenation of alkenes, minor amounts of vicinal diazides were frequently identified as side-products. In order to favor the formation of the diazides, Lin designed a new sterically hindered nitroxyl catalyst (CHAMPO, Fig. 18).92 Once the carbon-centered radical is generated, CHAMPO would be too sterically hindered to form a C–O bond, and a second azidyl group could be transferred, possibly via an inner-sphere mechanism.
Fig. 18.

(A–C) Nitroxyl-mediated diazidation of alkenes.
2.4. Phthalimide N-oxyl radical (PINO) and quinuclidine radical cation as HAT agents
The phthalimide N-oxyl radical (PINO) has been used in radical reactions since 1977, beginning with a seminal report by Grochowski and co-workers.93,94 The majority of reactions employing PINO take advantage of the high BDE of N-hydroxyphthalimide’s O–H bond (NHPI). Pedulli and co-workers95 measured a BDE equal to 88.1 kcal mol−1, which is similar to the C–H bonds of allylic (88.8 kcal mol−1) and benzylic (89.7 kcal mol−1) positions.96
During the 1980s, Masui and co-workers pioneered the electrochemical generation of PINO from catalytic amounts of NHPI, and employed this radical for a wide range of oxidative reactions, including the oxidation of alcohols to carbonyl compounds,97,98 conversion of acetals and ethers to esters,96,99,100 generation of imides from amides,101 and early examples of allylic and benzylic oxidations.96,102 Kinetic studies of the oxidation of benzhydrol to benzophenone mediated by electrochemically-generated PINO revealed a large primary KIE (kH/kD = 10.6 at 25 °C), indicating that a HAT process should be the rate-determining step.103
2.4.1. Allylic oxidation.
Oxidation of allylic C–H bonds offers a convenient and often selective approach for the functionalization of complex molecules104 and thus has been widely employed in total synthesis of natural products. In 2016, Baran and co-workers studied this transformation taking Masui’s pioneering work as a foundation to develop an optimized protocol. Baran incorporated two major modifications in their system105 (Fig. 19). Firstly, tBuOOH replaced O2 as terminal oxidant, which contributed to a substantial improvement in reaction conversion and reproducibility. Secondly, after the evaluation of different redox mediators, tetrachloro-N-hydroxyphthalimide (Cl4NHPI) was identified as an optimal choice. Cyclic voltammetry analysis showed that the N-oxyl radical derived from Cl4NHPI should be higher in energy and more reactive than the PINO radical, which facilitates the HAT step. Following the formation of an allylic peroxide intermediate, this peroxide was subsequently converted into the enone product. Control experiments showed that this final step can proceed without electrochemical assistance. This method was also employed in the oxidation of a wide range of terpenes and olefins.
Fig. 19.

(A–C) N-hydroxy phthalimide-mediated allylic oxidation.
2.4.2. Benzylic oxidations.
In 2017, Stahl and co-workers studied a cooperative catalytic system employing cobalt(II) acetate and NHPI under aerobic conditions to promote benzylic oxidation.106 During the development of this chemical method, a few substrates showed poor reactivity, and the authors hypothesized that the oxidation product would chelate cobalt, inhibiting the catalytic cycle. To overcome this limitation, the authors explored an electrochemical route for the generation of the PINO radical without cobalt, leading to improved yields.
In a subsequent work, Stahl and co-workers utilized anodically generated PINO to produce benzyl radicals, which were trapped by iodine, yielding benzylic iodides107 (Fig. 20). The choice of base was shown to be decisive in regulating the outcome of this reaction; when the sterically hindered 2,6-lutidine is used as base, the benzylic iodides can be generated efficiently. However, the use of pyridine as base leads to the unproductive formation of benzyl-pyridinium salts. Competition between hydrogen evolution and the reduction of iodine to iodide at the cathode required the use of a divided cell for these experiments, keeping all iodine in the anodic chamber.
Fig. 20.

(A and B) N-hydroxy phthalimide-mediated benzylic iodination.
2.4.3. Oxidation of unactivated C(sp3)–H bonds.
The oxygenation of unactivated C(sp3)–H bonds is a challenging transformation in organic synthesis and is typically approached through the use of highly reactive species. Curci and co-workers have studied the utilization of dioxiranes to promote these challenging oxidations,108 the most common members of this class of compounds, DMDO (dimethyldioxirane) and TFDO (methyltrifluoromethyldioxirane), are not commercially available due to their tendency to easily decompose. In 2007, White and co-workers reported the use of a new iron-oxo complex to achieve selective oxidation in complex architectures.109 These developments are transformative in the selective oxidation of complex molecules, especially in medicinal chemistry. Nevertheless, the application of many existing chemical methods in large-scale synthesis can still be challenging considering the cost of the required oxidants (e.g., trifluoroacetone) and catalysts.
In 2017, Baran and co-workers used PINO radicals and N-oxyl radicals derived from Cl4NHPI to promote a HAT process on unactivated C(sp3)–H bonds.110 However, this approach was unsuccessful, likely due to the inert nature of such C–H bonds. During an evaluation of different mediators, a stoichiometric amount of quinuclidine was identified as a promising option. Quinuclidine can be anodically oxidized to the corresponding radical cation (21–5), which is reactive enough to abstract hydrogen from electron-rich C(sp3)–H bonds, forming alkyl radicals that can be later trapped by O2. Use of this methodology culminated in the total synthesis of (+)-2-oxo-yahazunone (21–13; Fig. 21) by employing sclareolide (21–11) as a substrate. In 2019, Magauer and co-workers explored the electrochemical generation of the radical cation of quinuclidine to synthesize the natural product mitrephorone A (21–15).111
Fig. 21.

(A–C) Quinuclidine-mediated C(sp3)–H oxidation.
2.5. Dehydrogenation mediated by DDQ
DDQ (2,3-dichloro-5,6-dicyano-1,4-benzoquinone) is a useful oxidant used to mediate the dehydrogenation of different functional groups. DDQ produces a hydroquinone (DDHQ) as a by-product, which can be anodically oxidized back to DDQ, allowing for a catalytic process (Fig. 22). This approach has been explored for the oxidation of alcohols to aldehydes,112 secondary amines to imines,113 and for the synthesis of benzoxazoles.114
Fig. 22.
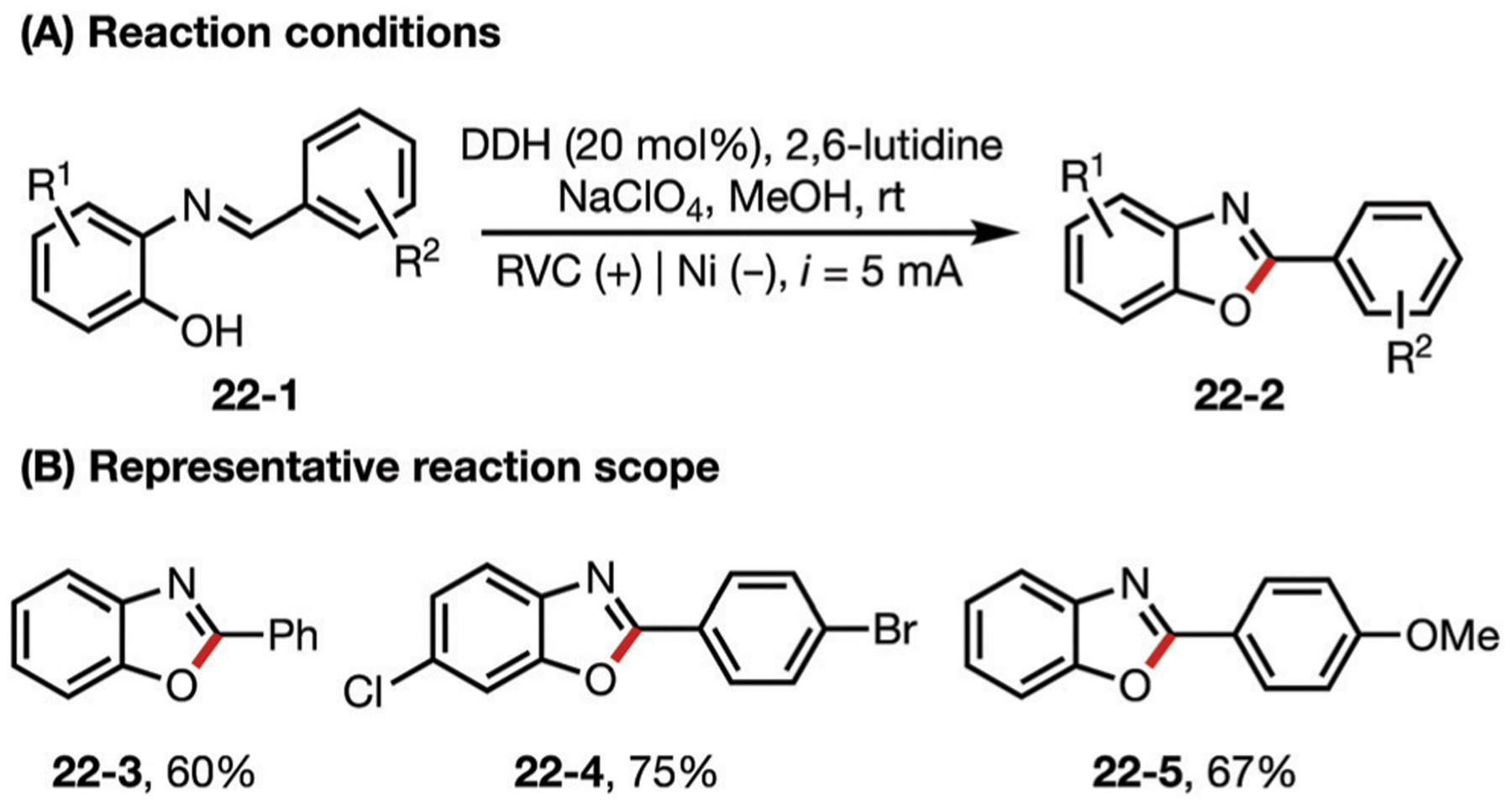
(A and B) DDQ-mediated generation of benzoxazoles.
2.6. Nitrogen centered radical cation as electron transfer agents
In the 1960s, Nelson and co-workers demonstrated that anodic oxidation of triphenylamine (23–1) led to a rapid coupling to form tetraphenylbenzidine (23–4), which would undergo subsequent oxidation to form radical cation and dication species (Fig. 23). Notably, the addition of a para-substituent on the three aromatic rings (23–6) would alter the reactivity and allow the formation of a very stable nitrogen centered radical cation (23–7), which was observed by a reversible profile on cyclic voltammetry analysis.115,116 Introduction of electron-donating or electron-withdrawing groups on the para position of the aryl rings allows easy modulation of the oxidation potential. These mediators have been employed in a wide range of transformations to promote electron transfer events, such as the oxidation of sulfides,117,118 stilbenes,119 and strained bicycles.120,121
Fig. 23.
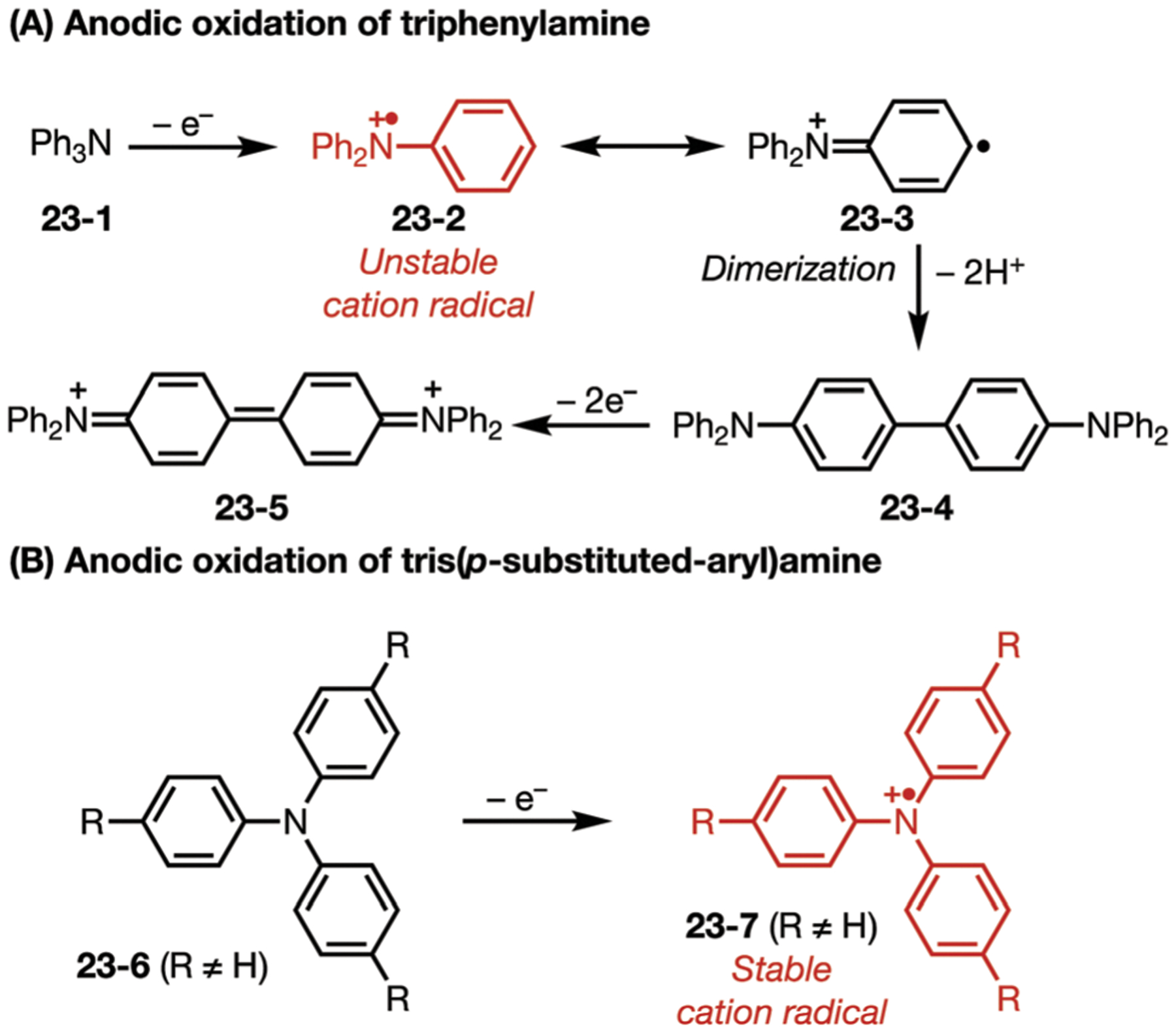
(A and B) Different stability profiles of triarylamine cation-radicals.
2.6.1. Oxidation of electron-rich aromatics.
In 2015, Little and co-workers explored the use of the cation-radical of tris-(p-bromophenyl)amine to oxidize benzylic alcohols to aldehydes and benzylic ethers to benzoate esters, among other transformations (Fig. 24).122 The use of a triarylamine allowed the authors to perform the oxidation at a much lower potential than the oxidation potential of the substrate, which enables higher yields and broader functional group compatibility. In this work, a polymeric ionic liquid was employed as electrolyte, which could be recovered and reused, simplifying product isolation and minimizing waste generation.
Fig. 24.

(A and B) Triarylamine-mediated oxidation of alcohols and ethers.
Inspired by the useful properties of radical cations derived from triarylamines, Zeng, Little, and co-workers developed analogue redox mediators using a triarylimidazole (TAI) scaf-fold that can be easily synthesized.123,124 The radical cation derived from TAI was able to oxidize benzylic alcohols and p-methoxytoluene to the corresponding benzaldehydes, benzylic ethers to benzoate esters, and chalcone epoxides (25–1) to radical cations (25–4) that can further engage in a Friedel–Crafts mechanism (Fig. 25).125
Fig. 25.

(A and B) Triarylimidazole-mediated oxidation of chalcone epoxides mediated by TAI cation-radical.
Little and co-workers also explored a fused-ring form of TAI mediator (2-aryl-1-methylphenanthro[9,10-d]imidazole, see 26–2), which yields a more stable radical cation than original TAIs. The higher stability of this radical cation was observed by cyclic voltammetry analysis: when the scan rate was decreased from 100 mV s−1 to 10 mV s−1 (a longer analysis time), the fused mediator 26–2 still presented a quasi-reversible profile, indicating a longer lifetime of the corresponding radical cation as compared to the radical cation derived from mediator 26–1126 (Fig. 26). Another analogue redox mediator, N-aryl-carbazole, was studied by Ma and co-workers, and the corresponding N-centered radical cation was also suggested as the key reactive species in oxidative transformations.127
Fig. 26.

Cyclic voltammogram of TAI 26–1 and its cyclized analogue 26–2. This figure has been adapted from ref. 126 with permission from American Chemical Society, copyright 2014.
2.6.2. Oxidation of anions into radicals.
In 2018, Xu and co-workers developed a radical cascade to produce imidazo-fused N-heterocycles128 (Fig. 27). Their approach relies on the generation of a nitrogen-centered radical by oxidation of the corresponding anion, which has an oxidation potential (Ep/2 = 0.66 V vs. SCE) significantly lower than common triarylamines (TBPA, Ep/2 = 1.08 V vs. SCE). This difference in potentials indicates that the substrate would likely undergo a direct oxidation, and that the triarylamine radical cation would play a minor role in the mechanism. Since triarylamines gave low yields (11–39%) of the desired product, the authors turned to tetraarylhydrazine mediators which proved more efficient. Tetra-(4-tert-butylphenyl)hydrazine showed an oxidation potential (Ep/2 = 0.68 V vs. SCE) lower than that of triarylamines, and could generate the desired bicyclic product in 89% yield. This was the first application of this mediator in synthetic electrochemistry.
Fig. 27.

(A and B) Tetraarylhydrazine-mediated synthesis of imidazo-fused N-heterocycles.
Xu and co-workers also explored mediated oxidation of enolates derived from dimethyl malonate and methyl acetoacetate (Fig. 28), which have low oxidation potential (enolate of dimethylmalonate, Ep/2 = 0.31 V vs. SCE).129 In order to achieve a mediated process, a phenothiazine (28–4; Ep/2 = 0.52 V vs. SCE) was employed, which has a lower oxidation potential than triarylamines, despite architectural similarities. The concentration of enolate 28–5 was kept small, as it was generated continuously by deprotonation of the dicarbonyl compound with cathodically generated hydroxide, which is an important feature to guarantee a mediated process. The resultant carbon-centered radical 28–6 was employed on the construction of substituted pyrrolidines and tetrahydropyridines.
Fig. 28.

(A–C) Phenothiazine-mediated synthesis of substituted pyrrolidines.
Transition metal complex as mediators
The combination of transition metal catalysis and electrochemistry offers the advantages of higher atom efficiency, milder reaction conditions, and safer protocols in place of using stoichiometric chemical oxidants to induce catalyst turnover. Furthermore, using electrochemistry to activate metal catalysts or key reactive intermediates derived from these catalysts has been shown to promote new reactivities that were previously challenging or unknown.
Metalla-electrochemical reactions fall into two broad categories. First, transition-metal complexes can serve as direct mediators to transfer electrons between electrodes and target substrates. Specifically, transition metal-bound substrate adducts exhibit lower oxidation potentials, and the oxidized open-shell species possess characteristics of persistent radicals, thus rendering the group transfer process to the substrates more selective and efficient. This type of metalla-electrochemical methods has given rise to numerous new transformations that were impossible or challenging to accomplish otherwise. A more detailed review on these methods can be found in a recent account.130
Second, electrochemistry supports traditional transition-metal catalyzed C–H activations, leading to a variety of transformations forging carbon–carbon and carbon–halogen bonds. In a transition-metal-catalyzed C–H activation, anodic oxidation can typically play two roles: to enable regeneration of the active transition metal catalyst (Fig. 29A) and to oxidize the transition metal catalyst intermediate to a high-valent species to induce subsequent reductive elimination (Fig. 29B). Detailed discussion on transition-metal catalyzed C–activation reactions can be found in recent reviews.131 An important advantage of metalla-electrochemistry is that C–H activation reactions employ electricity as the oxidizing equivalent, thus avoiding the use of external chemical oxidants and generation of wasteful by-products.
Fig. 29.

(A and B) Modes of electrochemical transition metal-catalyzed C–H functionalization.
Traditionally, anodic C–H activation has been dominated by precious transition metal catalysts based on palladium, ruthenium, iridium, and rhodium; more recently, however, 3d earth-abundant metals such as nickel and cobalt have seen increasing use in electrochemical C–H activation. In the following section we will discuss recent advances in metalla-electrochemical reactions grouped by different metal mediators.
2.7. Manganese
Manganese is a particularly attractive mediator in electrocatalytic systems due to its abundance, low toxicity, and low cost. Traditionally, manganese complexes have been employed in alkene and C–H functionalizations to form C–N, C–O, and C–halogen bonds via radical pathways.132 However, it was not until recently that redox-active manganese complexes matured as competent mediators for electrochemically-driven transformations. Mechanistic investigation of these catalytic cycles suggest that a MnII/MnIII redox pair is operating in many of these transformations.
2.7.1. Olefin functionalization.
In 1998, Snider reported the diazidation of alkenes and glycals using 3 equivalents of Mn(OAc)3·H2O.133 Snider proposed that the diazidation of alkene occurs via rapid azide transfer from MnIII-azide to the alkene and subsequently to the incipient alkyl radical intermediate. This pioneering discovery has provided inspiration for several recent developments in electrocatalysis.
In 2017, the Lin lab developed an electrochemical diazidation of alkenes using MnII salts as efficient and selective electrocatalysts.134 The resulting vicinal diazides can be easily reduced to form diamines, which are prevalent motifs in a variety of bioactive natural products, pharmaceuticals, and molecular catalysts. Unlike Snider’s reaction system, the MnII salt was added as precatalyst, and was then oxidized under electrochemical conditions to generate a MnIII–N3 intermediate (Fig. 30B). This oxidized intermediate serves as a latent azidyl radical source that delivers a first azide equivalent to the alkene (30–1), followed by addition of a second azide equivalent to the resultant alkyl radical (30–3) to furnish the diazide product 30–2. This work also features significantly milder reaction conditions than those reported by Snider. The Lin group later applied this strategy to the dichlorination of alkenes (Fig. 30D).135
Fig. 30.

(A–D) Manganese-catalyzed electrocatalytic diazidation and dichlorination of alkenes.
To extend the utility of the strategy used for this electrochemical diazidation protocol, the Lin group developed an additional electrochemical method for the heterodifunctionalization of alkenes based on anodically coupled electrolysis (ACE) strategy.136 The implementation of ACE strategy relies on the appropriate pairing of radical precursors (X1)− and (X2)− with a suitable metal catalyst. Two parallel anodic oxidations will take place: one radical precursor would undergo single-electron oxidation to generate a transient free radical (X1)• while the other precursor (X2)− would undergo catalyst-assisted anodic oxidation to form a (X2)• equivalent in the form of a metal-bound adduct. This adduct could then transfer the functional group of interest to the alkene substrate. The persistent radical effect would dictate the order in which radicals (X1)• and (X2)• add to the substrate, thereby inducing a highly chemo- and regioselective strategy for the heterodifunctionalization of alkenes. In the reaction reported by Lin, two different radical intermediates including a transient CF3 radical and a MnIII–Cl adduct were generated simultaneously via ACE from Langlois’s reagent and MgCl2 in the presence of MnII precatalyst. As shown in Fig. 31, the anodically generated CF3 radical adds to the alkene, which is followed by transfer of chlorine from MnIII–Cl to deliver the chlorotrifluoromethylated product 31–2. The use of ACE as a mechanistic design principle has also been utilized for the chlorophosphinoylation (Fig. 31C),137 and haloalkylation of alkenes (Fig. 31D).138
Fig. 31.

(A–D) Manganese-catalyzed electrocatalytic heterofunctionalization of alkenes.
The Lin group applied their strategy for the electrochemical heterodifunctionalization of alkenes to the radical cyclization of 1,6-enynes.139 In this reaction, the Langlois reagent (CF3SO2Na) and MgCl2 served as functional group donors and MnII served as the electrocatalyst. As depicted in Fig. 32, the 1,6-enyne substrate undergoes consecutive trifluoromethylation, radical ene–yne cyclization, and chlorination to furnish the substituted cyclized product (Fig. 32). The E/Z selectivity observed in the product was proposed to be sterically controlled. To explore the validity of this proposal, addition of bulky ligand (e.g. 2,2,-bipyridine) yielded near-complete selectivity for a single product isomer.
Fig. 32.

(A–C) Synthesis of chlorotrifluoromethylated pyrrolidines via electrocatalytic radical ene–yne cyclization.
The Mo group developed an electrochemical method to make trifluoromethylated oxindoles and related heterocycles.140 Langlois’ reagent is used as the CF3 source. Upon anodic oxidation, this reagent releases a CF3 radical which adds to the substrate to generate a tertiary alkyl radical and triggers cyclization onto the aromatic ring (Fig. 33). This protocol has been employed to forge complex ring systems through cascade cyclization (Fig. 33C).
Fig. 33.

(A–C) Manganese-mediated electrochemical trifluoromethylation for the synthesis of azaheterocycles.
The Lei group has also used the MnII/MnIII redox pair to enable electrochemically driven radical cyclization reactions.141 In this work, the authors reported an electrochemically-driven, Mn-catalyzed coupling reaction between N-substituted 2-arylbenzoimidazoles (34–1) and alkylboronic acids (Fig. 34). EPR and CV data led them to propose a mechanism including the direct anodic oxidation of alkylboronic acids to alkyl radicals. Subsequent addition of these alkyl radicals to substrate 34–1 was proposed to be facilitated by a MnIII species, ultimately leading to the formation of cyclized product 34–2.
Fig. 34.
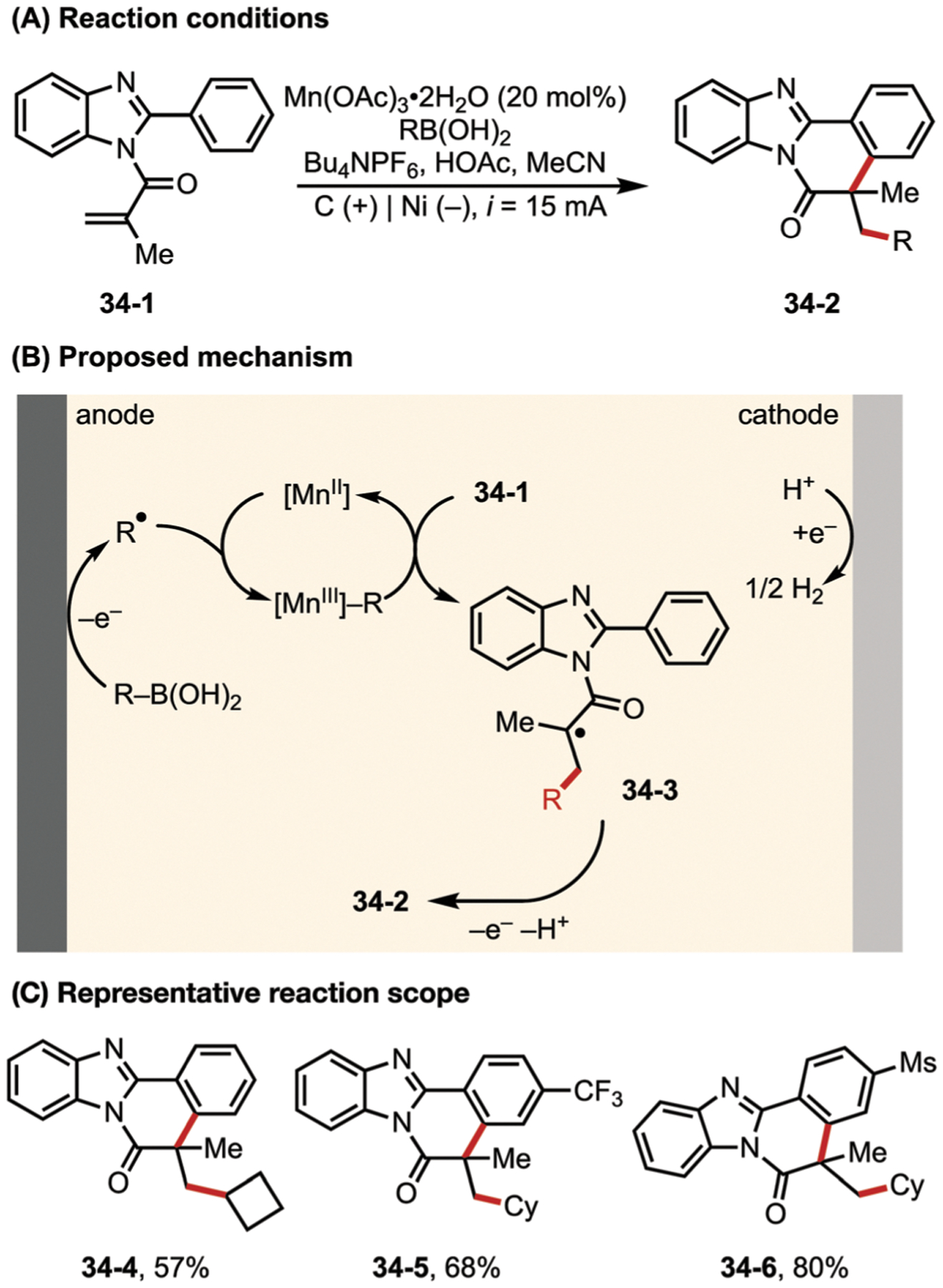
(A–C) Manganese-catalyzed electrochemical cyclization reaction of N-substituted 2-arylbenzoimidazoles with alkylboronic acids.
2.7.2. C(sp3)–H functionalization.
In addition to alkene functionalization, manganese electrochemical mediators have been utilized in C(sp3)–H functionalization. An interesting example from the Lei group described an electrochemical azidation of C(sp3)–H bonds facilitated by visible light catalysis (Fig. 35).142 In this work, DDQ was utilized as a HAT reagent to activate inert tertiary C–H bonds, which is then trapped by the azide-transfer complex MnIII–N3 formed in situ on the anode. Mechanistic studies suggested that both excited photocatalyst DDQ and the anodically generated azidyl radicals are capable of HAT to form alkyl or benzyl radicals. This reaction technically belongs to the electrophotocatalysis section (see Section 7) but is discussed here owing to its similarity in catalytic mechanism to other examples in this section.
Fig. 35.

(A–C) Manganese-catalyzed oxidative azidation of C(sp3)–H bonds under electrophotocatalytic conditions.
In 2021, the Ackermann group reported an electrochemical manganese porphyrin/salen complex-catalyzed azidation of unactivated secondary, tertiary, and benzylic C–H bonds.143 In this work, mechanistic studies suggest that a MnIII/MnIV redox pair is in operation.
A similar mechanism was proposed in another example by Morrill and co-workers that used a MnII catalyst to mediate an electrochemical synthesis of β- and γ-chlorinated ketones (Fig. 36).144 In this system, anodically generated MnIII–Cl is responsible for the activation of a cyclopropanol or cyclobutanol substrate (36–1) to afford alkoxy radical 36–3, which undergoes C–C bond homolysis to generate a primary alkyl radical 36–4. The release of ring strain is thought to be the driving force for cleavage of the C–C bond. In an alternative pathway, a MnIII-alkoxide resulting from ligand exchange between MnIII–Cl and 36–1 may undergo reversible β-scission to form 36–4. Trapping of this transient primary carbon-centered radical with the persistent MnIII–Cl species forms a new C−Cl bond.
Fig. 36.

(A and B) Manganese-catalyzed electrochemical deconstructive chlorination of cycloalkanols via alkoxy radicals.
2.7.3. Manganese-catalyzed alcohol oxidation.
Mn-Based mediators have also been shown to mediate alcohol oxidation reactions. Huang and co-workers developed an electrochemical Mn-catalyzed cyclization reaction to construct quinazolinones from aminobenzamides and alcohols in an undivided cell (Fig. 37).145 The authors proposed that the MnIII species generated on the anode can oxidize the alcohol to benzaldehyde, which then condenses with aminobenzamides to be further transformed into the cyclized product 37–3.
Fig. 37.

(A–C) Manganese-catalyzed electrochemical synthesis of quinazolinones.
2.8. Iron
The relatively low oxidation potential of ferrocene (E1/2(Cp2Fe) = 0.34 V vs. SCE in 9 : 1 MeCN : H2O) makes it a selective redox catalyst that bears a broad functional group tolerance.146 Ferrocene has been utilized in numerous transformations to activate N–H bonds to generate highly reactive amidyl radicals. Additionally, other iron complexes have found applications in electrochemical C–H activation reactions.
2.8.1. Radical cyclization.
In 2018, the Xu group reported a ferrocene-mediated electrochemical alkyne functionalization to access fluorinated dibenzazepines147 using CF2HSO2NHNHBoc as a CF2H radical precursor (Fig. 38). The electrolytic process commences with the anodic oxidation of Cp2Fe to Cp2Fe+. At the same time, methanol is reduced on the cathode to generate H2 and MeO−. The deprotonation and subsequent oxidation of CF2HSO2NHNHBoc (38–2) generates intermediate 38–5, which further loses an electron and a proton to afford diazene 38–6. The following decomposition of 38–6 releases a CF2H radical which can react with an alkyne group to furnish vinyl radical 38–8. This vinyl radical then cyclizes onto phenyl rings to afford a 7-member ring structure 38–9. Lastly, oxidative rearomatization of the cyclized intermediate affords the dibenzazepine product 38–3.
Fig. 38.

(A–C) Electrochemical difluoromethylarylation of alkynes.
In another work by the Xu group, Lewis acids were used to acidify the tertiary C–H bond on malonate amides, which was then deprotonated by electrochemically generated methoxide (Fig. 39). Oxidation of the resulting anion 39–4 by a Fc/Fc+ couple then generates the alkyl radical intermediate 39–5, which subsequently undergoes cyclization to deliver product 39–6.148
Fig. 39.
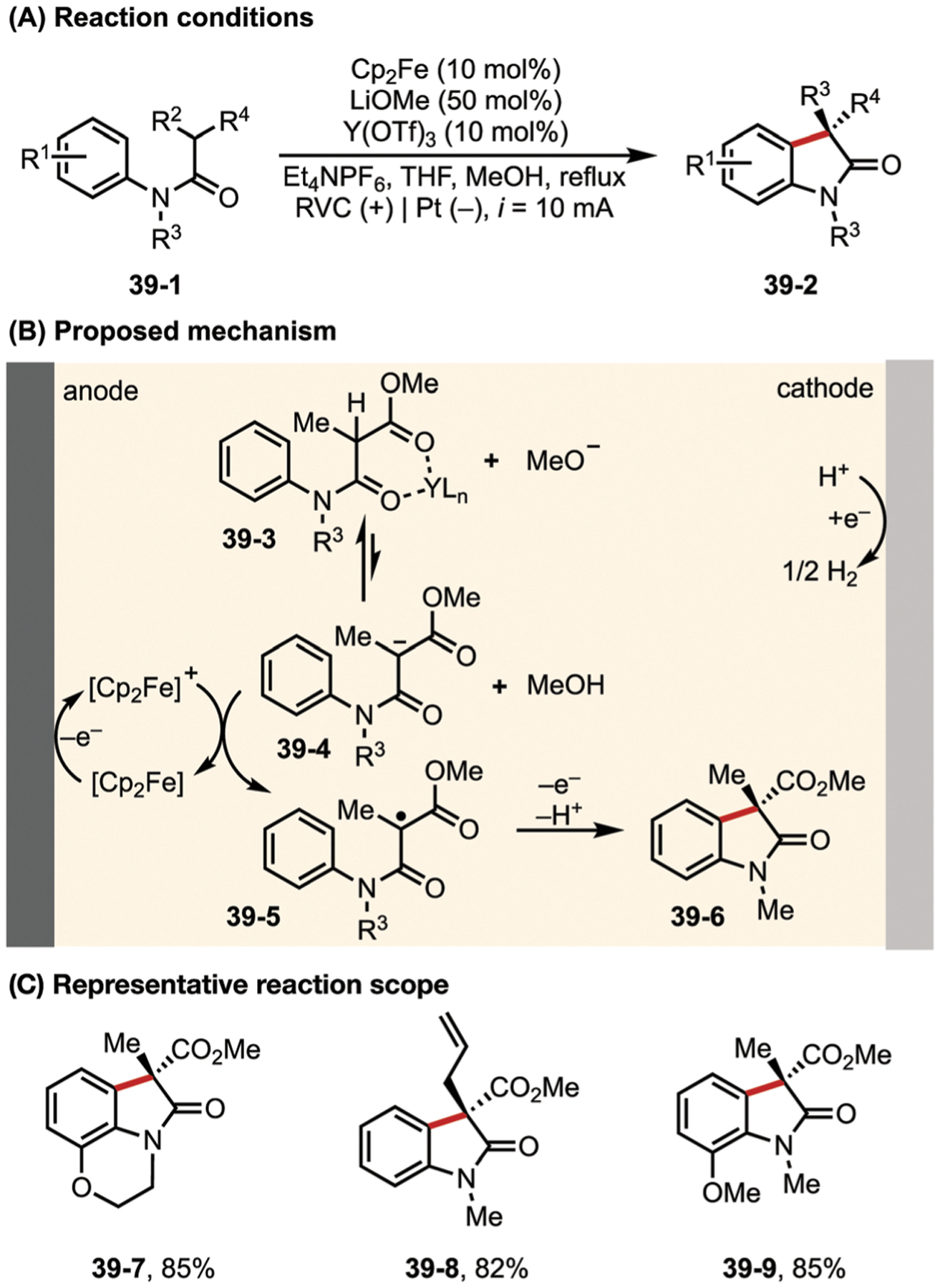
(A–C) Electrochemical dehydrogenative cyclization of 1,3-di-carbonyls.
The Xu group employed a similar strategy for radical cascade cyclization reaction to access functionalized 7-member carbo-cyclic compounds (Fig. 40).149 The anodically generated amidyl radical by means of Cp2Fe+ catalysis undergoes stepwise 5-exo-trig and 7-endo-trig cyclization followed by HAT from 1,4-cyclo-hexadiene (1,4-CHD) to deliver product 40–2. It was concluded that the trans disposition of the alkyl radical and the alkene in the intermediate 40–4 is critical to inducing an 7-endo-trig cyclization pathway instead of the 6-exo-trig cyclization pathway in the second radical addition event.150 The typically favored 6-exo-trig cyclization is likely inhibited by the ring strain from the resultant trans-fused bicyclic ring system.
Fig. 40.
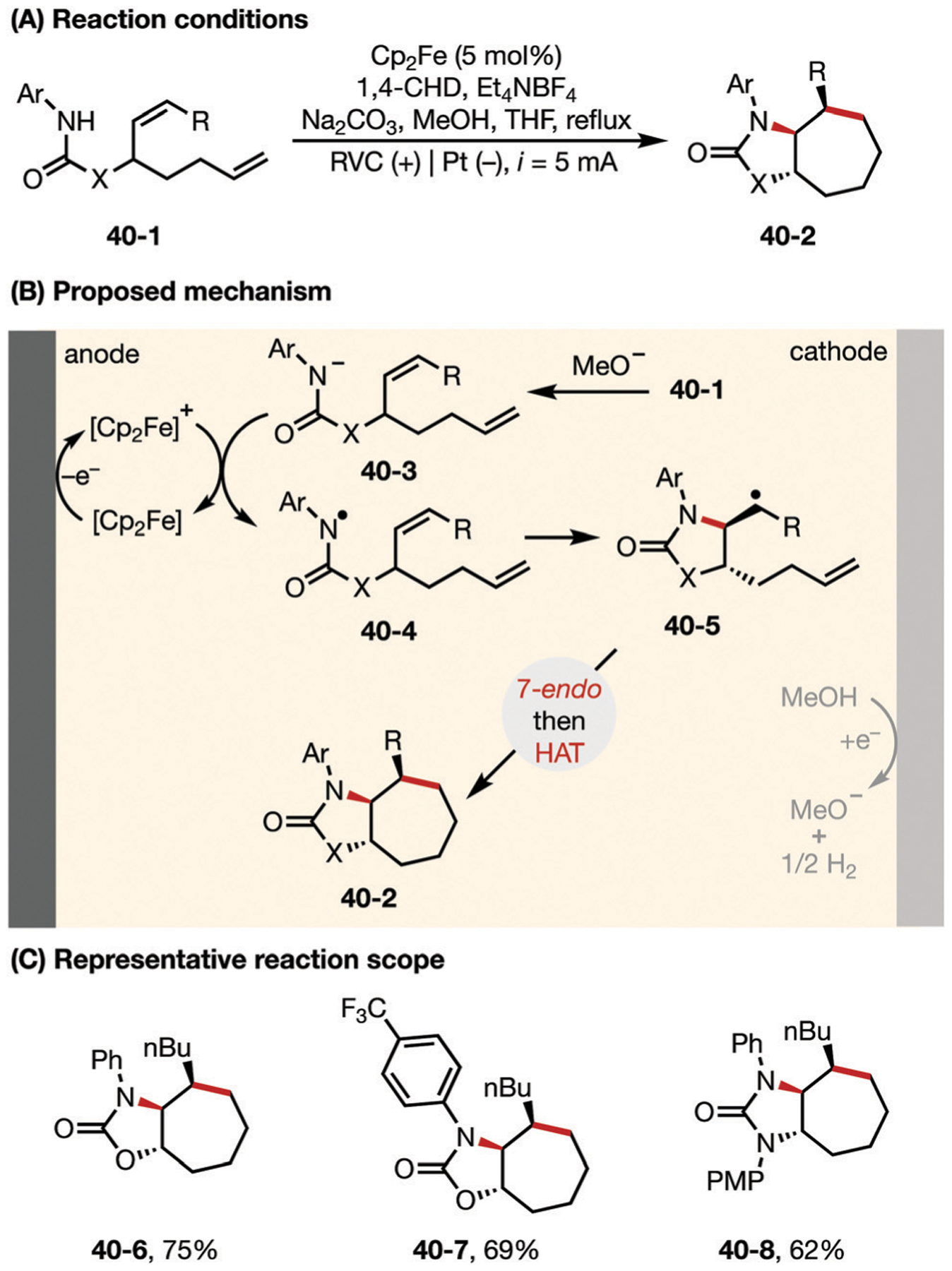
(A–C) Electrochemical synthesis of 7-membered carbocycles through cascade 7-endo-trig radical cyclization.
2.8.2. Aromatic C–H functionalization.
Fe(acac)3 has also been used as a catalyst in electrochemical C–H activation. The Ackermann group described a ferraelectrocatalytic C–H arylation in 2019.151 As shown in Fig. 41, this transformation is thought to proceed through a C–H cleavage (via a sigma bond metathesis pathway or through a base-assisted internal electrophilic substitution). After this initial event, the resultant FeII intermediate goes through an anodic single-electron-transfer (SET) and transmetallation to generate five-membered FeIII intermediate 41–4, which then reductively eliminates to release the arylated product. The iron catalyst is then regenerated on the anode.
Fig. 41.

(A and B) Electrochemistry-driven iron-catalyzed C–H arylation.
2.8.3. Various amination reactions.
In a similar report in 2018, Luo and co-workers demonstrated an Fe-catalyzed electrochemical cycloaddition of diaza-oxyallyl cations to synthesize diamines.152 Like some previous examples in this section, this reaction also featured cathodic reduction of methanol paired with the anodic oxidation of ferrocene (Fig. 42). On the anodic side, the oxidized ferrocenium serves as a mediator to oxidize the deprotonated N,N-dibenzyloxyurea (42–4). As such, diaza-oxyallyl cation 42–5 is generated in an efficient fashion and reacts with a variety of heteroarenes to form value-added diamine products.
Fig. 42.
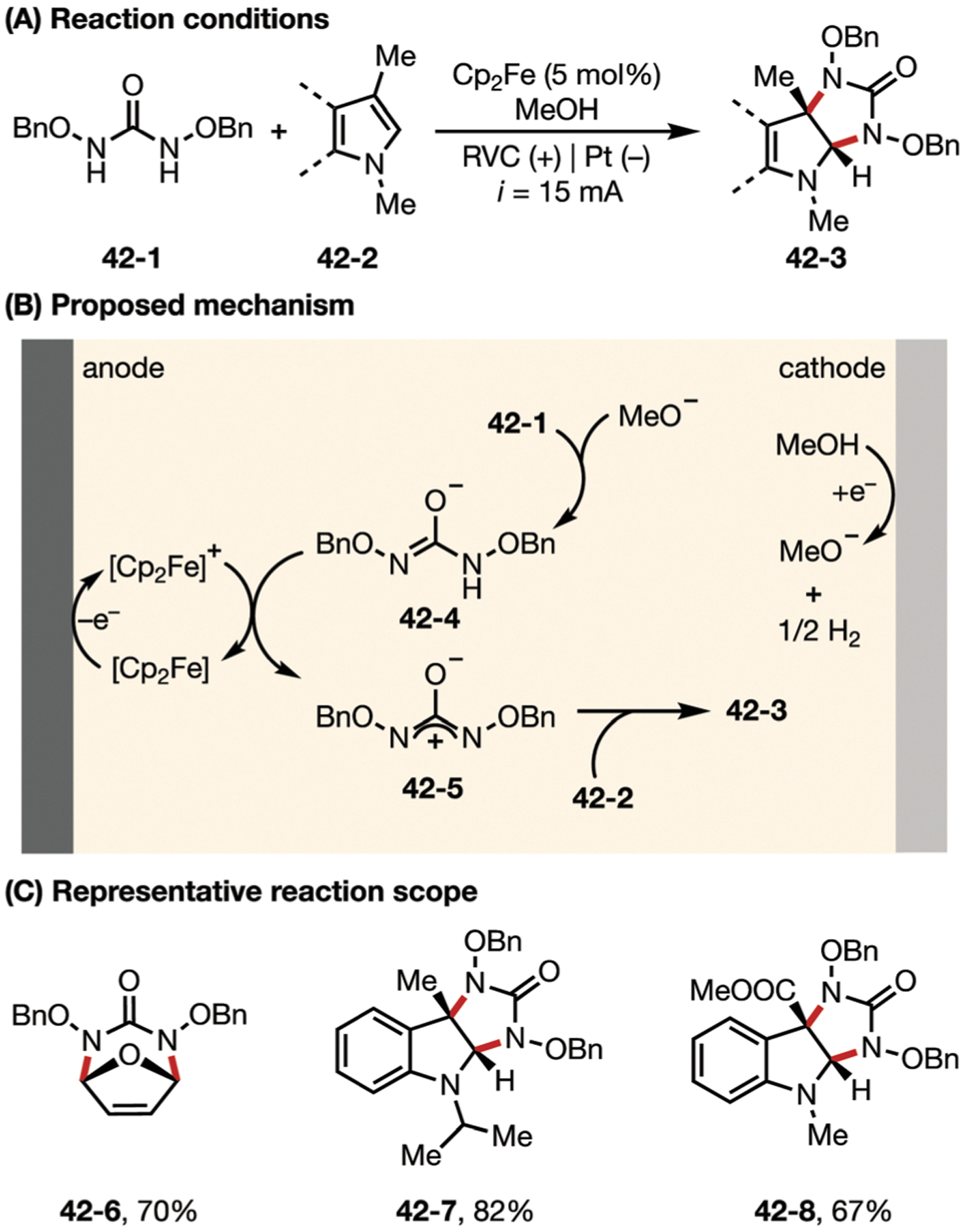
(A–C) Electro-oxidative generation of diaza-oxy-allyl cation toward the synthesis of diamine.
Furthermore, Banerjee and co-workers reported a synthesis of benzimidazolone and oxazinones from aryl benzyloxyureas using a ferrocene catalyst and paired electrolysis.153 Here, water was reduced cathodically and the resultant hydroxide ion acts as the active base to remove the proton on urea 43–1 (Fig. 43). The resultant species then undergoes Fc+-mediated oxidation to form zwitterionic nitrenium intermediate 43–3. Loss of a nitrene leaving group gives rise to an isocyanate intermediate 43–4, which undergoes 5-exo cyclization to give the desired product.
Fig. 43.

(A and B) Electrochemical access to benzimidazolone and quinazolinone via in situ generation of isocyanates.
2.9. Copper
2.9.1. Cyanation of alkenes.
In 2019, the Lin group reported a Cu-catalyzed enantioselective electrocatalytic cyanophosphinoylation of vinylarenes that made use of ACE strategy (Fig. 44). A secondary phosphine is oxidized in a Cu-mediated process to form a P-centered transient radical 44–4 and meanwhile CuII–CN is oxidized to CuIII–CN. This pair of open-shell intermediates will then react with the alkene sequentially following the persistent radical effect to give product 44–2. The bisoxazoline (BOX) ligands along with CN− play a key role in preventing the cathodic reduction of copper ions, which will result in Cu metal plating out.149 The high enantioselectivity of this transformation was found to be related to the structure of the ester groups on the sBOX ligand.
Fig. 44.

(A–C) Enantioselective electrocatalytic cyanophosphorylation of vinylarenes.
In 2020, the Lin group reported a dual electrocatalytic approach for the asymmetric hydrocyanation of conjugated alkenes (Fig. 45).154 Anodic oxidation generates a CoIII–H complex and a CuIII–CN complex in a concerted fashion. The CoIII–H species enables hydrogen-atom transfer to the alkene substrate, and CuIII–CN delivers a cyanide radical equivalent to the resultant alkyl radical. The enantioselectivity of the hydrocyanation reaction was again induced by the use of chiral Cu(sBOX) catalysts. Electrochemistry turns over both catalysts so that an external oxidant is not necessary. Notably, performing the same hydrocyanation reaction using traditional chemical oxidants without electrical input has not proven successful to give comparable efficiencies or enantioselectivities to the electrocatalytic protocol. This reaction is broadly applicable to the functionalization of conjugated alkenes, including alkenylarenes, dienes, enynes, and allenes. Computational studies using DFT unravelled the mechanism of enantioinduction facilitated by the Cu(sBOX) catalyst: the pendant ester group played a key part in enhancing the enantioselectivity of this reaction via a C–H⋯π interaction with the aryl group of the alkene substrate as illustrated in transition state 45–7.
Fig. 45.

(A–C) Dual electrocatalysis enables enantioselective hydrocyanation of conjugated alkenes.
2.9.2. C–H functionalization.
Ackermann and co-workers developed a Cu catalyzed C–H functionalization of benzamides to access isoindolones (Fig. 46).155 The CuI catalyst was oxidized on the anode the first time to CuII, which complexes with substrate 46–1, and a second oxidation occurs to generate a CuIII species (46–6) which is responsible for C–H activation. Thereafter, metalation of the terminal alkyne via ligand exchange and subsequent reductive elimination delivers the C–H alkynylated arene 46–10, which readily undergoes base-promoted cyclization to form isoindolone 46–3.
Fig. 46.
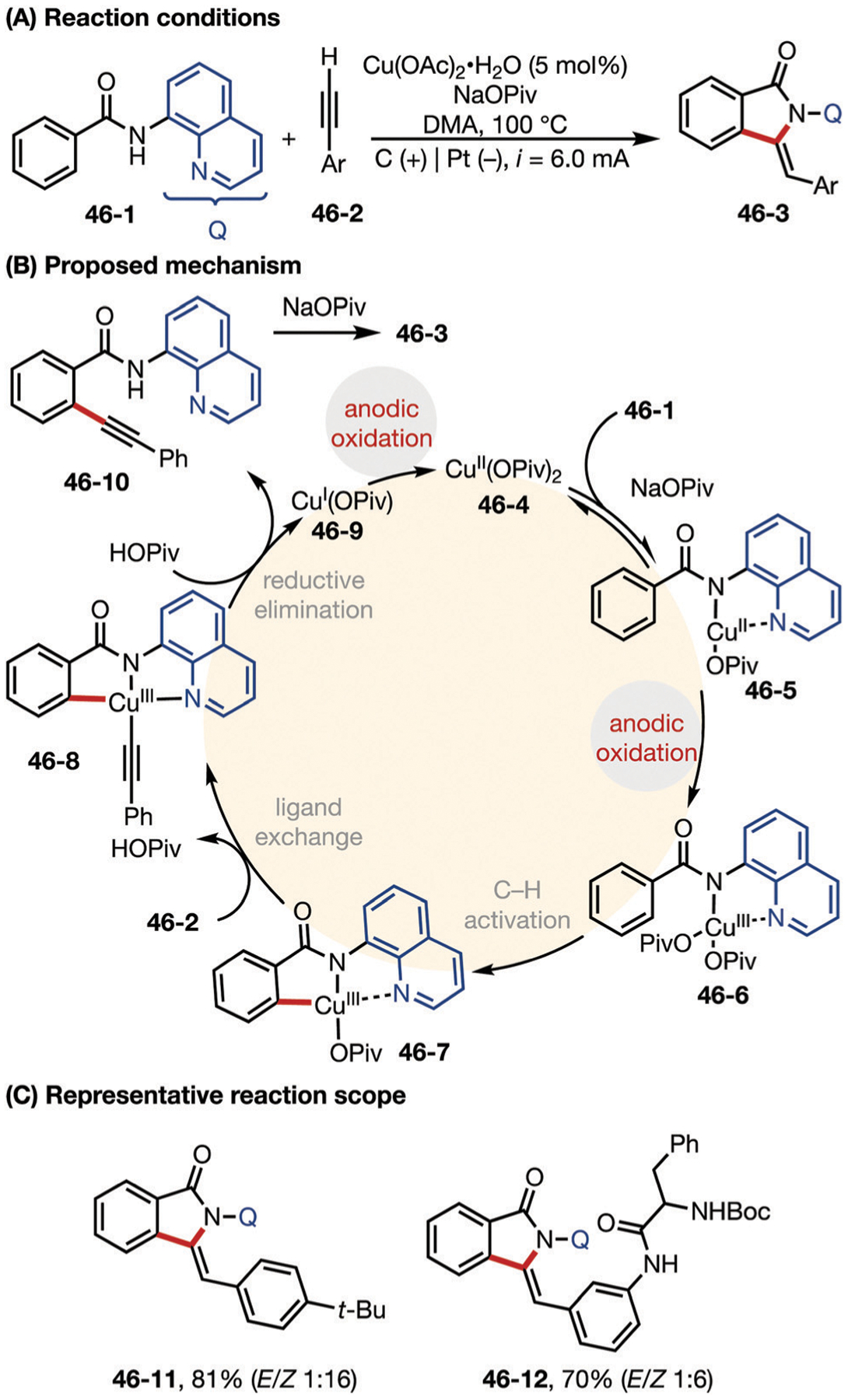
(A–C) Copper-catalyzed alkyne annulation.
In 2020, the Mei group reported a Cu/TEMPO co-catalyzed asymmetric Shono-type electrooxidation reaction. TEMPO acts as the redox mediator while copper with bisoxazoline ligands induce high enantioselectivities to afford C1-alkynylated tetra-hydroisoquinolines (Fig. 47).156
Fig. 47.
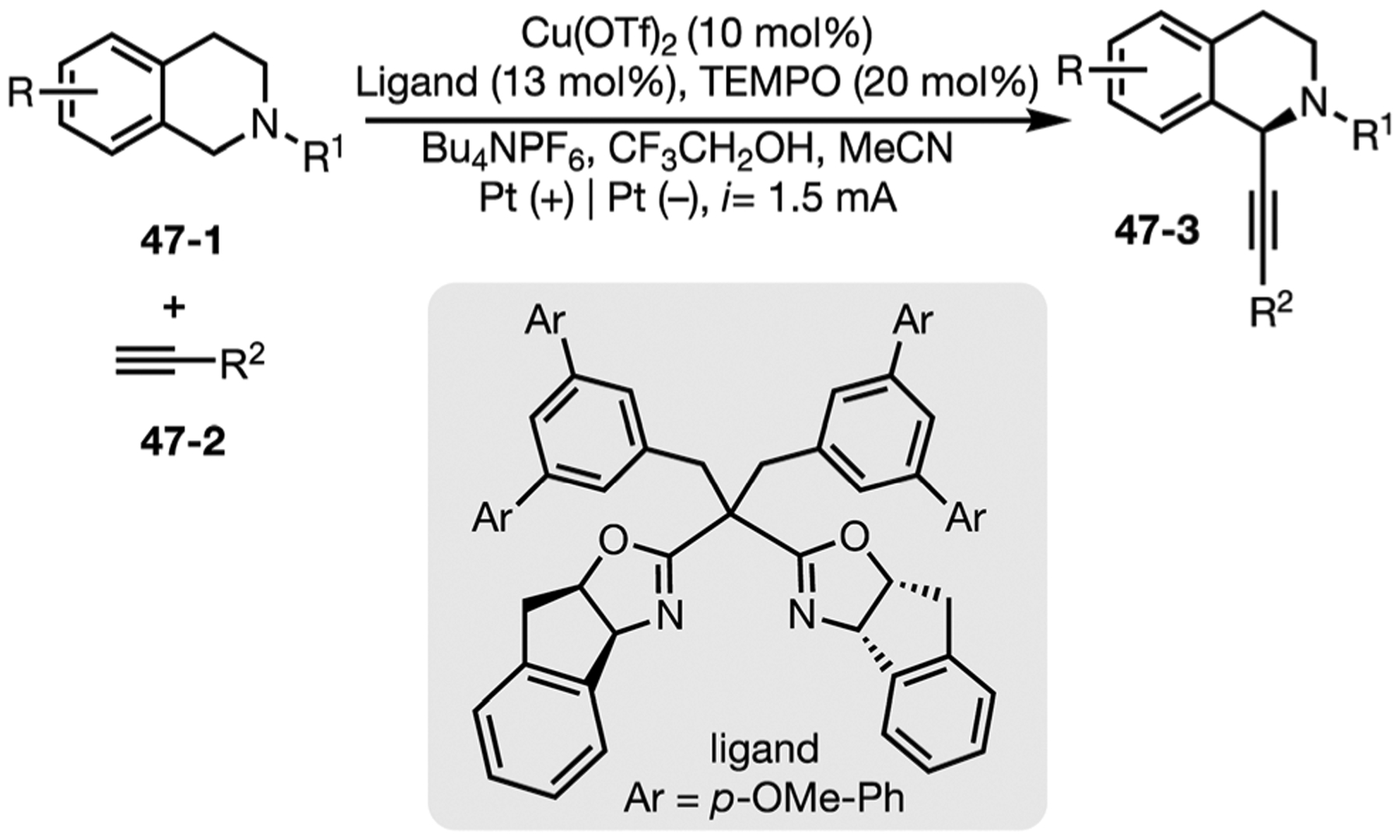
Cu(II)/TEMPO-coatalyzed enantioselective C(sp3)–H alkynylation of tertiary cyclic amines.
Mei’s group also demonstrated a Cu-catalyzed electrochemical C–H amination of arenes (Fig. 48).157 Another redox mediator used in this system is n-Bu4NI, which is directly recycled on the anode. Kinetic studies indicated that the electron transfer event between the iodine radical and copper complex is the rate-determining step. The authors proposed that the catalytic cycle starts with the coordination of substrates 48–1 and 48–2 to CuII catalyst to generate intermediate 48–5, and this complex is then oxidized by an iodine radical to form a CuIII species 48–6. Complex 48–6 then undergoes metal-to-ligand electron transfer to form radical cation 48–7. Subsequent intramolecular amine transfer to the radical cation and ligand exchange affords product 48–3 and generate a CuI species, which is oxidized on the anode to restore the catalyst to its active form. In a related report, Nicholls and co-workers disclosed a Cu(OAc)2 catalyzed directed C–H amination of benzamides with morpholine.158
Fig. 48.
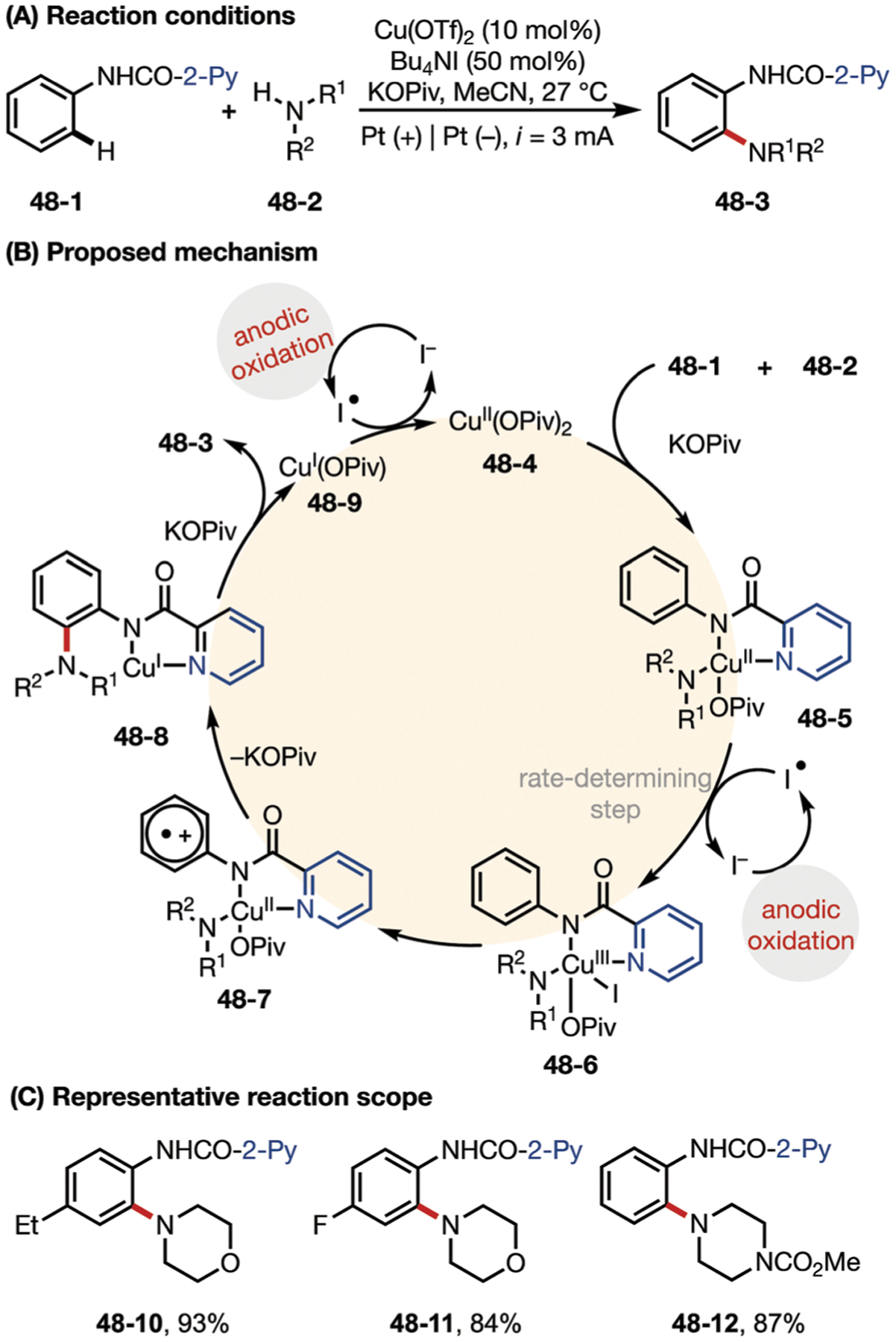
(A–C) Copper-catalyzed electrochemical C–H amination of arenes with secondary amines.
2.9.3. Allylic amination.
The Hu group described an electrochemical method to accomplish a formal aza-Wacker cyclization of internal alkenes.159 In this protocol, as shown in Fig. 49, electrochemical oxidation generates amidyl radical 49–4 which subsequently cyclizes to generate alkyl radical 49–5. This radical intermediate is then trapped by the Cu catalytic cycle, and an elimination assisted by base yields the alkene product. After releasing the product, the resultant CuI catalyst is oxidized at the electrode to CuII to re-enter the catalytic cycle. A wide range of 5-membered N-heterocycles including oxazolidinone and pyrrolidinone-containing functionalizable alkenes can be prepared under mild conditions.
Fig. 49.

(A–C) Formal aza-Wacker cyclization by tandem electrochemical oxidation and copper catalysis.
2.10. Palladium
In 2007, Amatore, Jutand, and co-workers reported an electrochemical PdII-catalyzed C–H bond olefination reaction of N-acetyl-anilines.160 In this reaction, benzoquinone acts as a sub-stoichiometric redox mediator, which is electrochemically recycled.
Pd-Catalyzed electrocatalytic reactions have also enabled the strategic formation of C–O bonds. In 2013, the Budnikova group reported the electrochemical Pd-catalyzed C–H oxygenation of 2-phenylpyridine using perfluoroalkyl carboxylic acids (Fig. 50A).161 Cyclic voltammograms and NMR data suggested that this C–H functionalization reaction proceed through monometallic PdIII intermediates in acetonitrile or through bimetallic fluorinated PdII intermediates in dichloromethane.
Fig. 50.

(A) Electrochemical palladium-catalyzed ortho-oxygenation of 2-phenylpyridines with perfluorocarboxylic acids. (B) Electrochemical palladium-catalyzed phosphonation of pyridines.
The same group later developed an electrooxidative Pd-catalyzed phosphonation reaction of 2-phenylpyridine (Fig. 50B).162 The authors postulated and subsequently isolated a key intermediate, binuclear phosphonate bipalladium [(PhPy)Pd(EtO)2P(O)]2, whose structure was confirmed by X-ray analysis. This species could be electrochemically oxidized to a PdIII or PdIV reactive intermediate that undergoes reductive elimination to afford the phosphonated product.
Electrochemical Pd-catalyzed transformations have also been applied in carbon–halogen bond formation. In 2017, the Kakiuchi group reported a palladium-catalyzed ortho-selective chlorination of N-quinolinylbenzamide derivatives under anodic oxidation conditions (Fig. 51).163
Fig. 51.

Electrochemical palladium-catalyzed chlorination of N-quinolinylbenzamide derivatives.
The breadth of Pd electrocatalysis is further showcased in C–C bond formation. In 2017, the Mei group reported Pd-catalyzed C(sp2)–H functionalization of oximes using organoboron reagents or benzoyl acetic acids, providing ortho-methylated or ortho-acylated products, respectively.164 The proposed reaction mechanism starts with C–H activation to generate palladacycle 52–5. It is proposed that MeBF3K can be directly oxidized to afford a methyl radical, which is then trapped by palladacycle 52–5. Alternatively, 52–5 can undergo direct transmetallation with MeBF3K followed by anodic oxidation to form a PdIII or PdIV intermediate 52–6. This high-valent intermediate readily undergoes reductive elimination to deliver the methylated product and regenerate the PdII catalyst (Fig. 52).
Fig. 52.

(A and B) Palladium-catalyzed electrooxidative C–C bond formation of oximes.
In 2017, Surendranath and co-workers developed an electrochemical strategy to oxidize methane with PdSO4 as the mediator.165 Mechanistic studies support an electrochemical oxidation of PdII catalyst to a putative bimetallic PdIII,III2 intermediate, which rapidly functionalizes methane to generate CH3OSO3H and CH3SO3H via concurrent faradaic and non-faradaic pathways, respectively. Both CH3OSO3H and CH3SO3H can be thermally and hydrolytically converted to methanol (Fig. 53).
Fig. 53.

Electrochemical palladium-catalyzed methane monofunctionalization.
The Lei group reported a Pd-catalyzed electrochemical aminocarbonylation of alkynes under atmospheric pressure of CO in an undivided cell (Fig. 54).166 Based on cyclic voltammetry, kinetic analysis, and XAFS spectroscopy data, the authors proposed that the catalytic cycle proceeds through a Pd0/PdII redox pair. The reaction starts with amine substrate 54–2 binding to PdII catalyst 54–4, followed by CO insertion to generate intermediate 54–6, which reacts with alkyne 54–1 to give the palladium acetylide 54–7. This species then undergoes reductive elimination to release 2-ynamide 54–3. The resultant Pd0 subsequently gets oxidized on the anode to regenerate the catalytic cycle. Diverse primary and secondary amines with various functional groups are viable substrates for this reactivity.
Fig. 54.

(A–C) Electrochemical oxidative aminocarbonylation of terminal alkynes.
In 2017, the Mei group reported a PdII-catalyzed C(sp3)–H oxygenation via anodic oxidation. According to the proposed mechanism, electrochemical oxidation of divalent palladacycle 55–4 to a PdIII or PdIV intermediate (55–5) occurs to induce the subsequent reductive elimination (Fig. 55).167 Kinetic isotope effect (KIE) experiments suggested that the C–H activation of the substrate is the rate-limiting step (kH/kD = 3.7). The acylated product can re-enter the catalytic cycle to undergo an additional C–H activation, eventually resulting in di- or tri-acetoxylation products. The Sanford group has also applied electrochemistry in Pd-catalyzed electrochemical acetoxylations of C(sp2)–H and C(sp3)–H bonds.168
Fig. 55.

(A–C) Electrochemical palladium-catalyzed C(sp3)–H bond acetoxylation.
2.11. Ruthenium
A common practice for inducing C–H activation is to utilize nitrogen-based directing groups. In 2018, the Ackermann group reported a Ru-catalyzed electrocatalytic C–H activation with aromatic carbamates and phenols (Fig. 56).169 Based on mechanistic studies, the authors proposed that the catalytic cycle starts with the coordination of aromatic carbamates accompanied by C–H activation to generate ruthena(II)cycle intermediate 56–5. Subsequent coordination of an alkyne followed by migratory insertion affords the seven-membered ruthena(II)cycle 56–7. This species then undergoes reductive elimination to deliver the product and Ru0 intermediate 56–8. The anodic oxidation of intermediate 56–8 back to the active RuII species 56–4 completes the catalytic cycle.
Fig. 56.

(A–C) Electrochemical ruthenium-catalyzed alkyne annulations of arylcarbamates.
The Ackermann group later reported a Ru-catalyzed C–H activation to enable annulations between alkynes and benzoic acids.170 The proposed mechanism is similar to that of the previous example. In 2021, the group published an electrochemical three-component assembly of isoquinolines catalyzed by a RuII catalyst, shown in Fig. 57.171
Fig. 57.

(A and B) Ruthenaelectro-catalyzed three-component alkyne annulation.
In 2018, the Xu group described a Ru-catalyzed electrochemical dehydrogenative annulation reaction of aniline derivatives and alkynes for the synthesis of indoles (Fig. 58).172 Similar to Ackermann’s work, an electric current is used to oxidize Ru0 precatalyst 58–9 to its active form. Xu proposed that NaOAc converts precatalyst [RuCl2(p-cymene)]2 into ruthenium diacetate complex 58–4. Coordination with the pyrimidine directing group promotes reversible C–H activation to form the six-membered ruthenacycle 58–6. Subsequently, intermediate 58–6 goes through ligand exchange, and migratory insertion of the bound alkyne into the Ru–C bond triggers the metal center to dissociate from the pyrimidyl nitrogen and instead complex with the amino group of the aniline moiety to obtain the six-membered ruthenacycle in the resultant intermediate 58–8. Eventually, reductive elimination delivers the product.
Fig. 58.
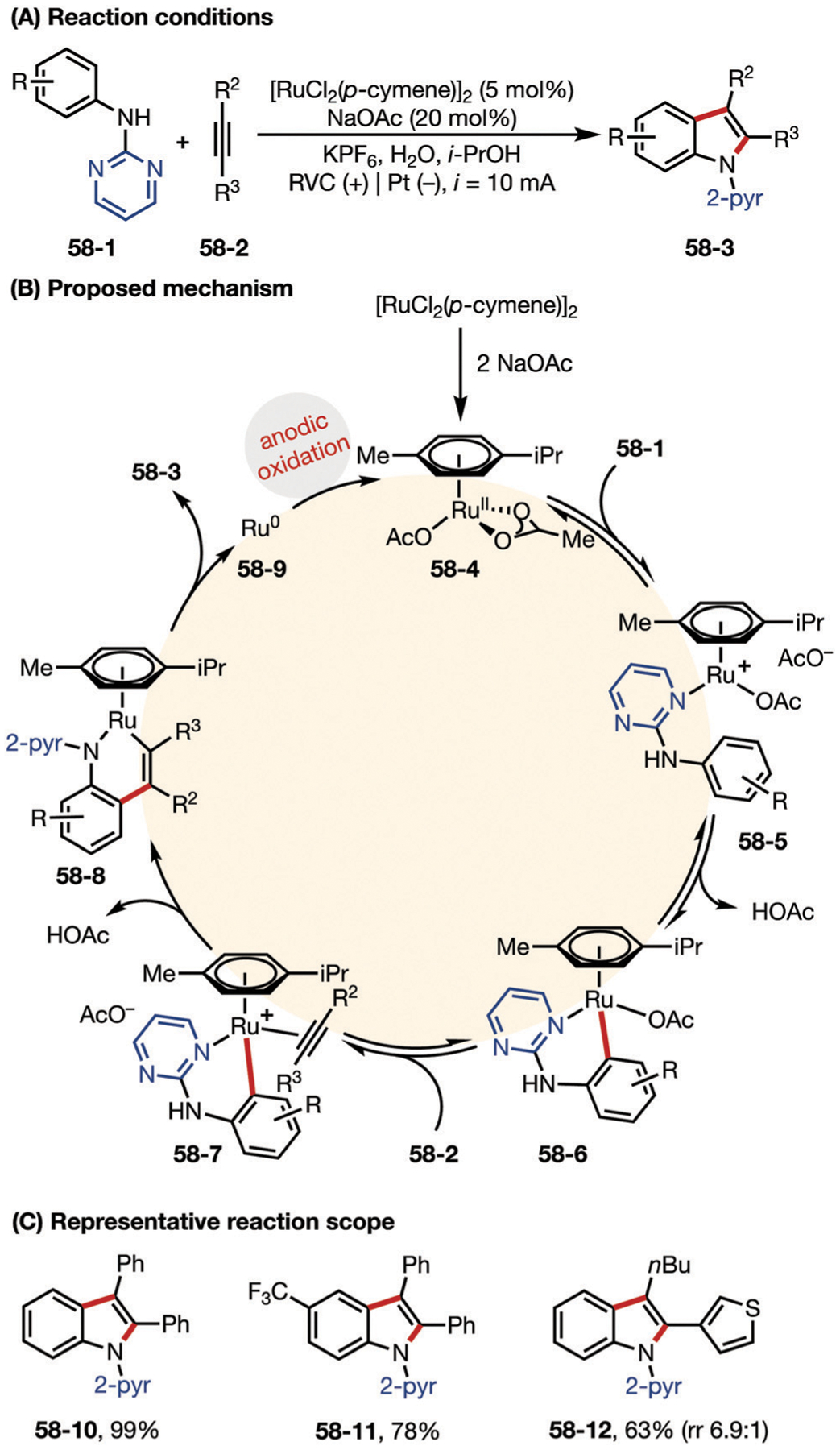
(A–C) Ruthenium-catalyzed Electrochemical dehydrogenative alkyne annulation.
2.12. Iridium
In 2018, the Ackermann group developed an Ir/benzoquinone co-catalyzed electrochemical C–H alkenylation of benzoic acids via ortho-coordination.173 Mechanistic studies in Fig. 59 suggested a facile C–H cleavage and the subsequent formation of IrIII cycle 59–7, which undergoes β-H elimination and reductive elimination to form IrI intermediate 59–8. Benzoquinone-mediated anodic oxidation then restores the IrI species to the active catalyst 59–4.
Fig. 59.
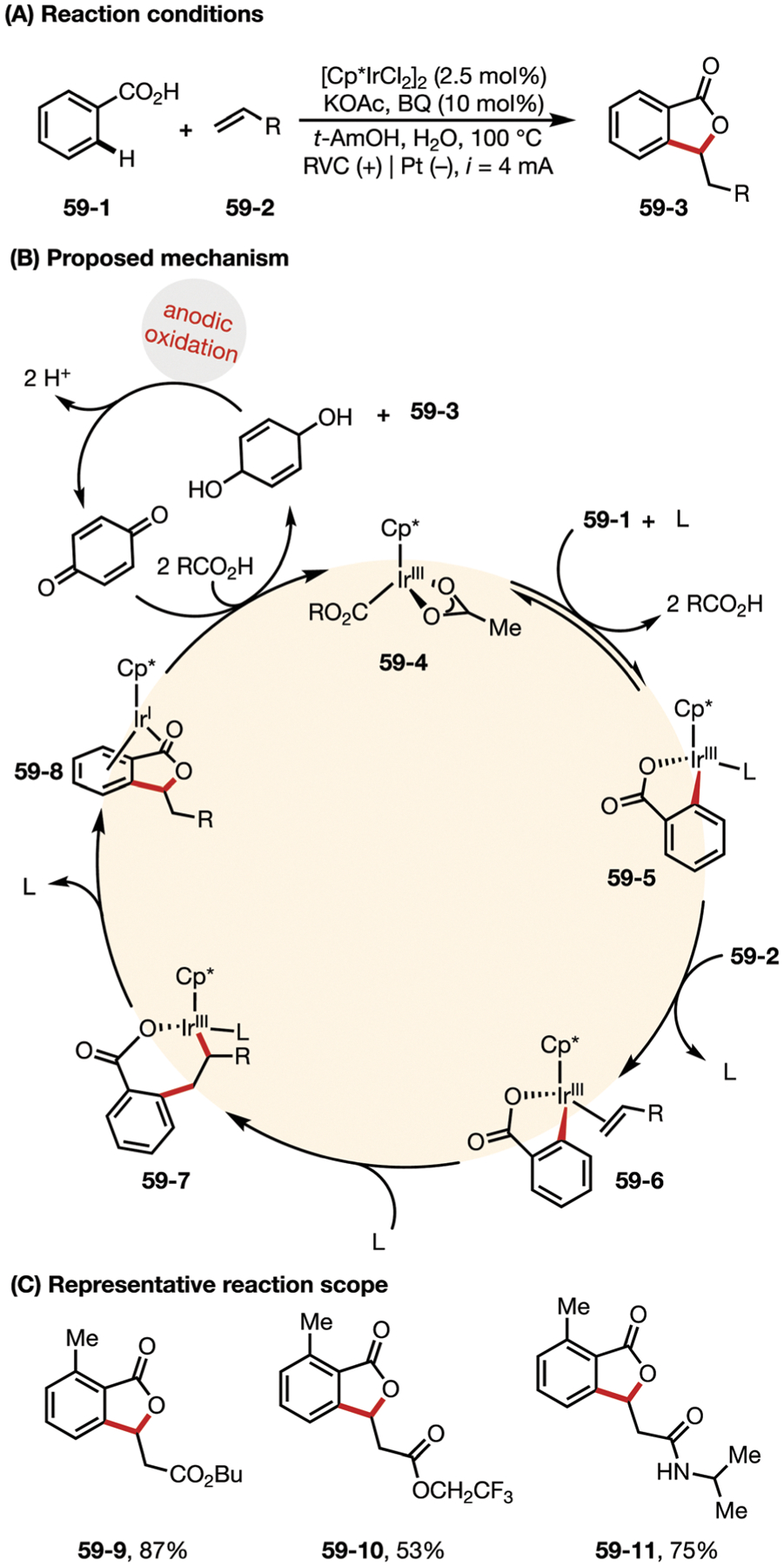
(A–C) Cooperative iridium-catalyzed electrooxidative C–H alkenylations.
In 2019, Mei and co-workers demonstrated that the synergistic combination of iridium catalysis and electrochemistry enables vinylic C–H activation of acrylic acids for alkyne annulation, producing α-pyrones in an undivided cell (Fig. 60).174 Without electricity, traditional chemical oxidants such as Ag0 or CuII did not give significant amounts of the desired product.
Fig. 60.

(A–C) Electrochemistry-enabled Ir-Catalyzed vinylic C–H Functionalization for alkyne annulation.
2.13. Cobalt
Cobalt electrocatalysis is a particularly powerful platform for a variety of C–H transformations and has been applied for the construction of C−O, C−N, and C–C bonds.
The Ackermann group has pioneered the development of cobalt-catalyzed metalla-electrochemical C–H activations.175 In 2018, the group reported a Co-catalyzed electrochemical C–H/N–H annulation of alkynes at room temperature.176 The proposed mechanism is shown in Fig. 61, the catalytic cycle starts with the anodic oxidation of CoI or CoII to an active CoIII carboxylate species 61–4. Thereafter, this species engages in carboxylate-assisted C–H activation of benzamides to generate intermediate 61–5, which then binds to phenyl alkyne to provide CoIII intermediate 61–6. Lastly, reductive elimination affords the desired product and form the putative CoI species 61–7. The catalytically active CoIII carboxylate complex is regenerated by anodic oxidation, thereby avoiding the use of sacrificial stoichiometric oxidants.
Fig. 61.
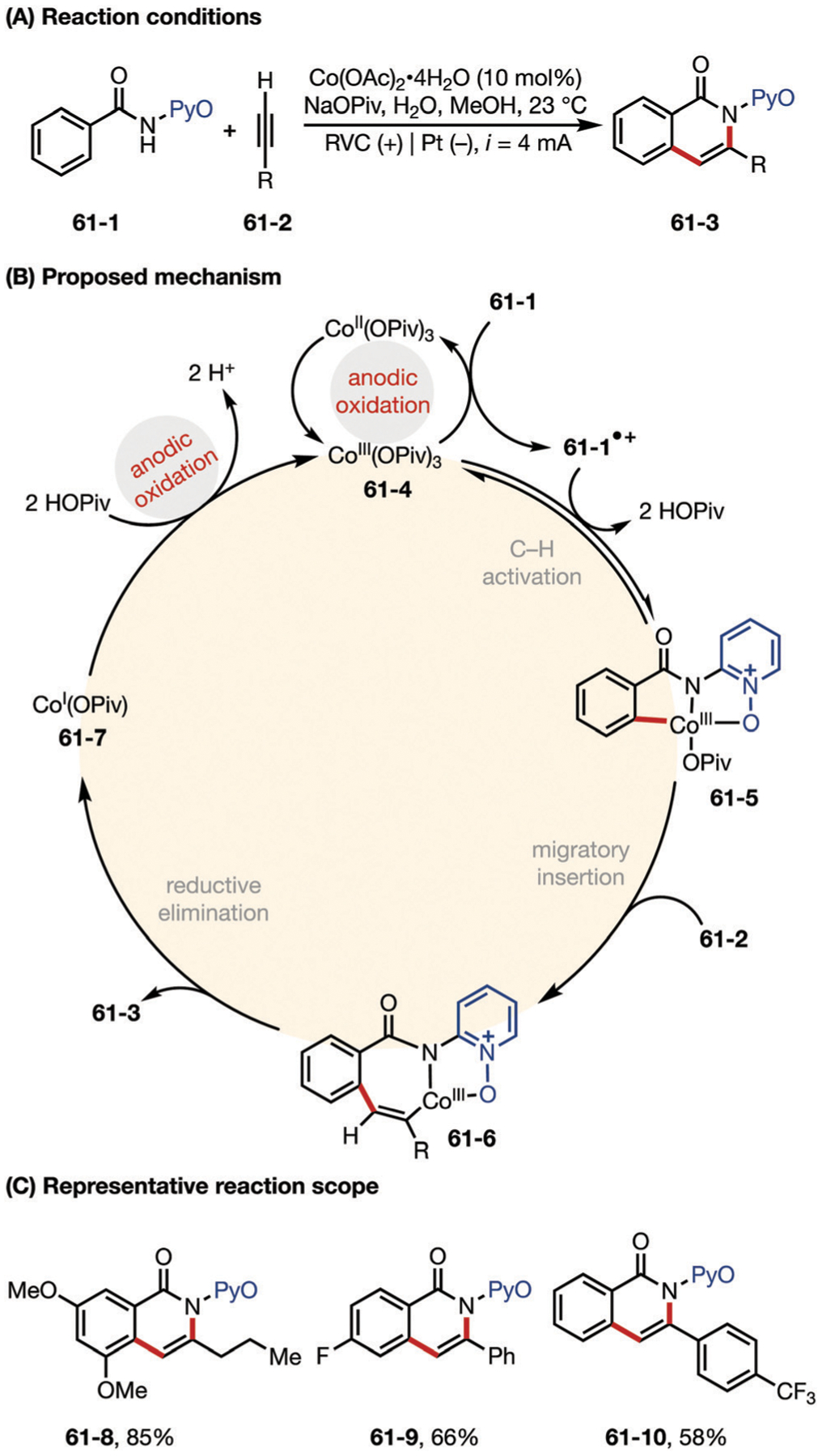
(A–C) Electrochemistry-enabled cobalt-catalyzed C–H/N–H Activation. PyO = 2-pyridyl N-oxide.
In 2020, the same group reported a cobalt-catalyzed electrochemical C–H allylation of benzamides with electronically unactivated alkenes.177 A similar mechanism involving CoII/III intermediates is proposed.
The Lei group has described several Co-catalyzed electrochemical C–H activation protocols.178 In 2018, Lei published a cobalt-catalyzed C–H amination of arenes to access synthetically useful arylamines (Fig. 62).179 Later, the authors reported a Co-catalyzed electrochemical [4+2] annulation of sulfonamides with alkynes to make a variety of sultams (Fig. 63).180 In this work, it was proposed that the [4+2] annulation reaction possibly proceeds through the following pathway: CoII species first coordinates with substrate 63–1 to generate CoII complex 63–4 and then undergoes single electron oxidation to access the CoIII complex 63–5. It is noteworthy that oxygen is critical for the reaction to proceed, and the authors hypothesize that H2O2 generated from O2 reduction could also oxidize CoII complex 63–4 to CoIII complex 63–5. Next, C–H activation followed by phenylacetylene insertion to generate intermediate 63–7, which undergoes reductive elimination to form the annulation product and CoI species. The generated CoI species is oxidized at anode to regenerate CoII. At the same time, O2 is reduced at the cathode to generate the superoxide radical anion, which then reacts with the solvent mixture to produce H2O2.
Fig. 62.

Cobalt-catalyzed electrooxidative C–H amination of arenes with alkylamines.
Fig. 63.

(A–C) Electrochemistry-enabled cobalt-catalyzed [4+2] annulation for the synthesis of sultams.
In 2019, the Yu group described a Co-catalyzed electrochemical oxidative C–H/N–H intramolecular annulation with isocyanides to synthesize iminoisoindolinone derivatives (Fig. 64).181 Anodic oxidation serves a similar role as in the previous examples.
Fig. 64.
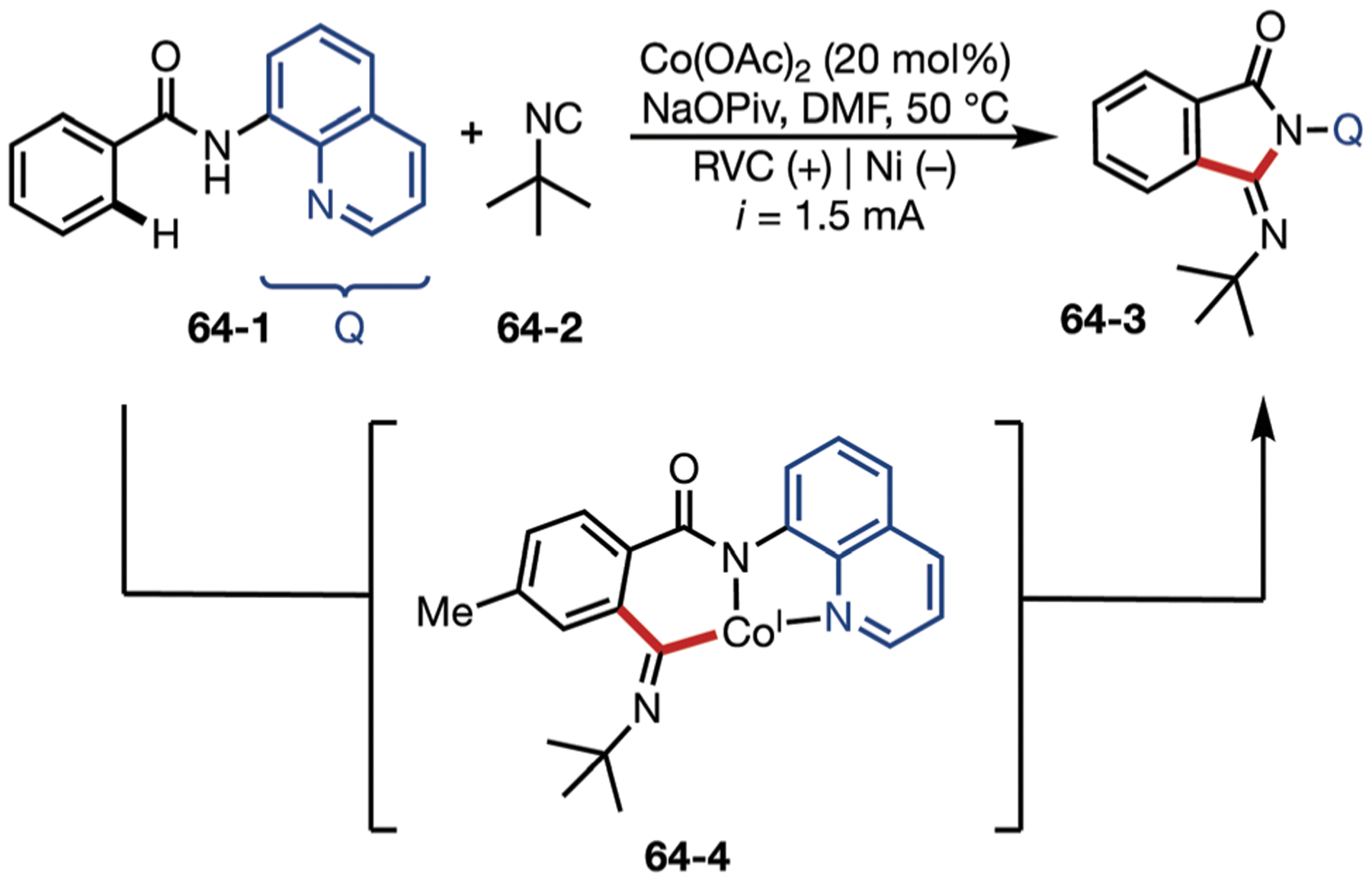
Electrochemical oxidative C–H/N–H intramolecular annulation with isocyanides for iminoisoindolinone synthesis.
2.14. Nickel
In an early work, the Budnikova group developed a Ni- or Pd-catalyzed electrochemical ortho-fluoroalkylation of 2-phenylpyridine.182 Based on the cyclic voltammogram of [Ni(bpy)3]2+ catalyst, the authors proposed that the C–H functionalization process goes through a NiII/NiIII redox pair.
In 2019, the Ackermann group reported Ni-catalyzed electrochemical alkoxylations of benzamides and secondary alcohols.183 Notably, this reaction has not been shown possible with other transition metal catalysts prior to this work. Anodic oxidation is used to induce reductive elimination of a high valent NiIII or NiIV complex. In order to rationalize the C–O bond formation process, the well-defined NiIII complex 65–4 was synthesized and fully characterized, including X-ray diffraction analysis. Cyclic voltam-metric studies of 65–4 revealed an oxidation feature at a potential of 0.50 V vs. Fc0/+, suggesting the formation of a higher valent complex 65–5 (Fig. 65). DFT calculations indicate the redox non-innocence of the ligand during oxidation; thus the oxidation is best described as a ligand-centered process. Reductive elimination of this species gave the final product.
Fig. 65.

Nickellaelectro-catalyzed C–H alkoxylation with secondary alcohols.
2.15. Rhodium
In 2018, the Ackermann group reported a Rh-catalyzed electrochemical C–H alkenylation of benzoic acids mediated by a rhodium catalyst.184 As shown in Fig. 66, the authors proposed a catalytic cycle that proceeds via an ortho C–H activation of benzoic acid 66–1 followed by migratory insertion of alkene 66–2. Intermediate 66–7 subsequently undergoes β-hydride elimination and reductive elimination. Finally, anodic oxidation of RhI intermediate 66–8 regenerates the catalytically active complex 66–4 while generating the desired product 66–3. Based on competition experiments with CF3-substituted and CH3-substituted benzoic acids and isotope labelling experiments, it is believed that the C–H activation proceeds through a base-assisted internal electrophilic substitution mechanism.
Fig. 66.

(A–C) Electrooxidative rhodium-catalyzed C–H/C–H activation for dehydrogenative alkenylation.
The Ackermann group also published an electrochemical arene alkenylation reaction catalyzed by a Rh complex. Like the previous example, this catalytic cycle also involves a RhI/RhIII redox pair. The proposed mechanism includes a key C–C bond activation step that leads to the formation of ketone by-product 67–4 (Fig. 67).185
Fig. 67.
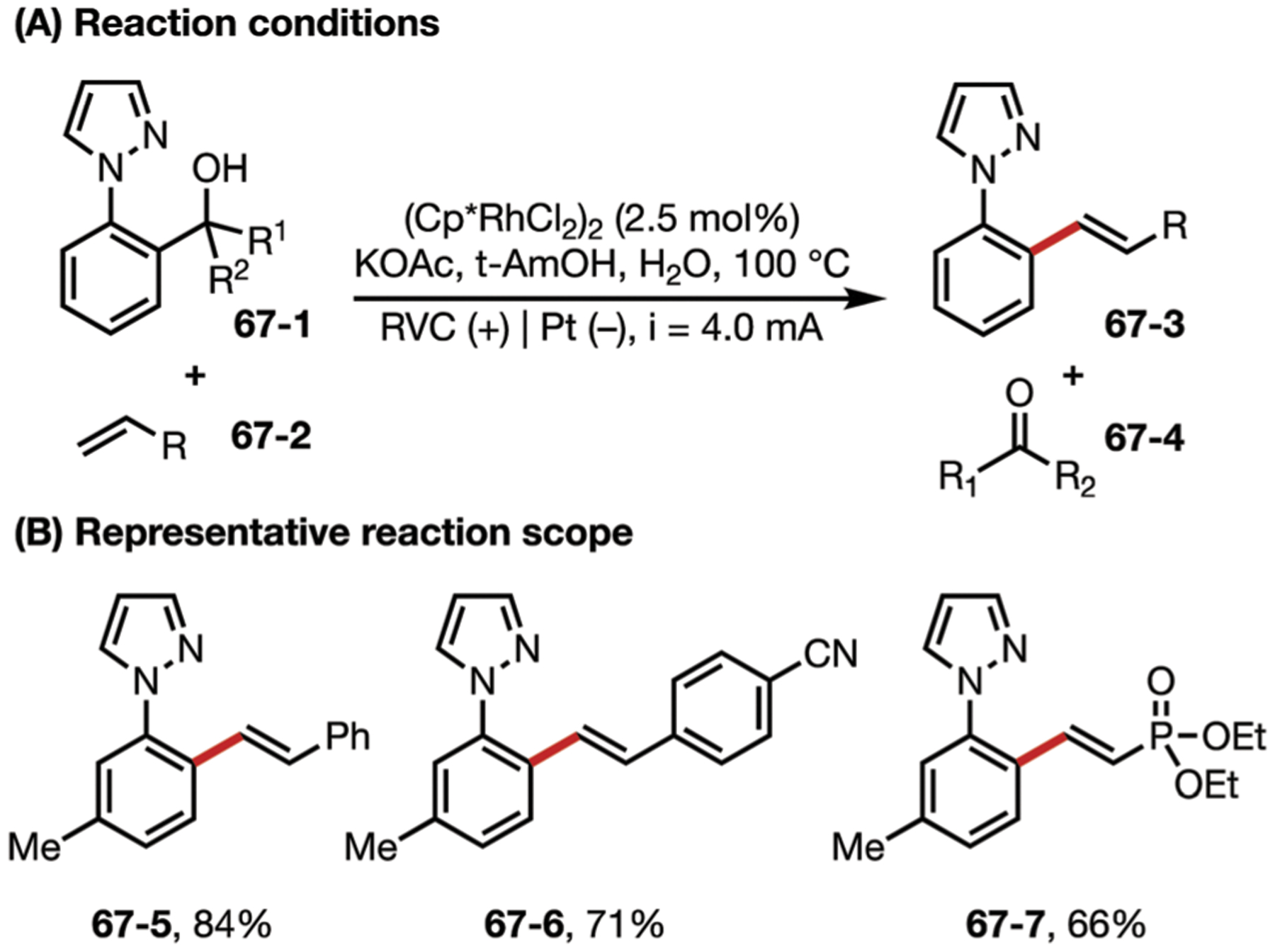
(A and B) Electrochemical C–C bond activation C–C bond alkenylation by rhodium(III) catalysis.
The Xu group reported a Rh-catalyzed electrocatalytic C–H phosphorylation reaction in 2019.186 This method enabled the synthesis of triarylphosphine oxides from diarylphosphine oxides. In this work, electricity is used to oxidize RhIII intermediate 68–6 to facilitate reductive elimination towards C–P bond formation (Fig. 68). However, the authors stated that the exact oxidation state of intermediate 68–7 prior to reductive elimination remains unknown. Pyrazole, pyridazine, purines, and pyrrolo-pyrimidine directing groups were shown to be compatible with the C–H activation step.
Fig. 68.

(A–C) Rhodium(III)-catalyzed aryl C–H phosphorylation enabled by anodic oxidation induced reductive elimination.
3. Cathodic reduction
Compared to electrooxidative methodologies, the utility of electroreductive chemistry remains relatively underexplored. In recent years, electrochemistry has emerged as a powerful and efficient method for achieving challenging transformations at deeply reducing potentials.187–190 In contrast to traditional reductive reactions which use heterogeneous reducing metals or homogenous organic compounds as stoichiometric reductants, electroreductive chemistry provides several advantages including highly efficient electron transfer, tunable reducing potential, and good scalability. Nevertheless, these approaches rely on the direct reduction of reactants on the cathode surface, which may result in electrode passivation leading to lower selectivity. As such, reductive electrocatalysis has attracted increasing attention in synthetic chemistry for its ability to decrease reaction overpotential and regulate downstream chemical pathways. Currently, nickel complexes are the most predominant catalysts used in electroreductive chemistry. However, a variety of 3d block metals such as cobalt and zinc as well as organic compounds have also been demonstrated as efficient reductive mediators. Electroreductive transformations are usually coupled with the oxidation of sacrificial anodes, such as Mg, Zn or Fe, or readily oxidizable additives such as triethyl amine (Et3N). During electrolysis, the anodic oxidation of these sacrificial reductants generates metal salts or amine radical cations, which are typically innocuous. In addition, sacrificial reductants may also prevent the undesired oxidation of substrates or reactive intermediates when the electrolysis is carried out in an undivided cell.
3.1. Nickel
In this section, cathodic reduction utilizing nickel complexes is discussed. Electrochemistry advantageously supports the in situ generation of often sensitive Ni species, resulting in more practical conditions for Ni-catalyzed transformations compared to traditional methods. Consequently, a wide variety of protocols have been reported in this direction including cross-electrophile coupling (XEC), alkene difunctionalization, C–H activation, as well as C–N, C–S, and C–P bond formation.
3.1.1. Homocoupling of aryl halides.
Among the earliest examples of electrochemically promoted homocoupling of electrophiles was Ban’s work using cathodically generated Ni0 species 69–3 (Fig. 69A).191 When used stoichiometrically, this nickel species was shown to facilitate the homocoupling of bromobenzene, which had previously been accomplished via chemical reductants such as Zn or AlEt3. This seminal work led to a number of further developments in Ni-catalyzed electrochemical coupling reactions.192–200
Fig. 69.
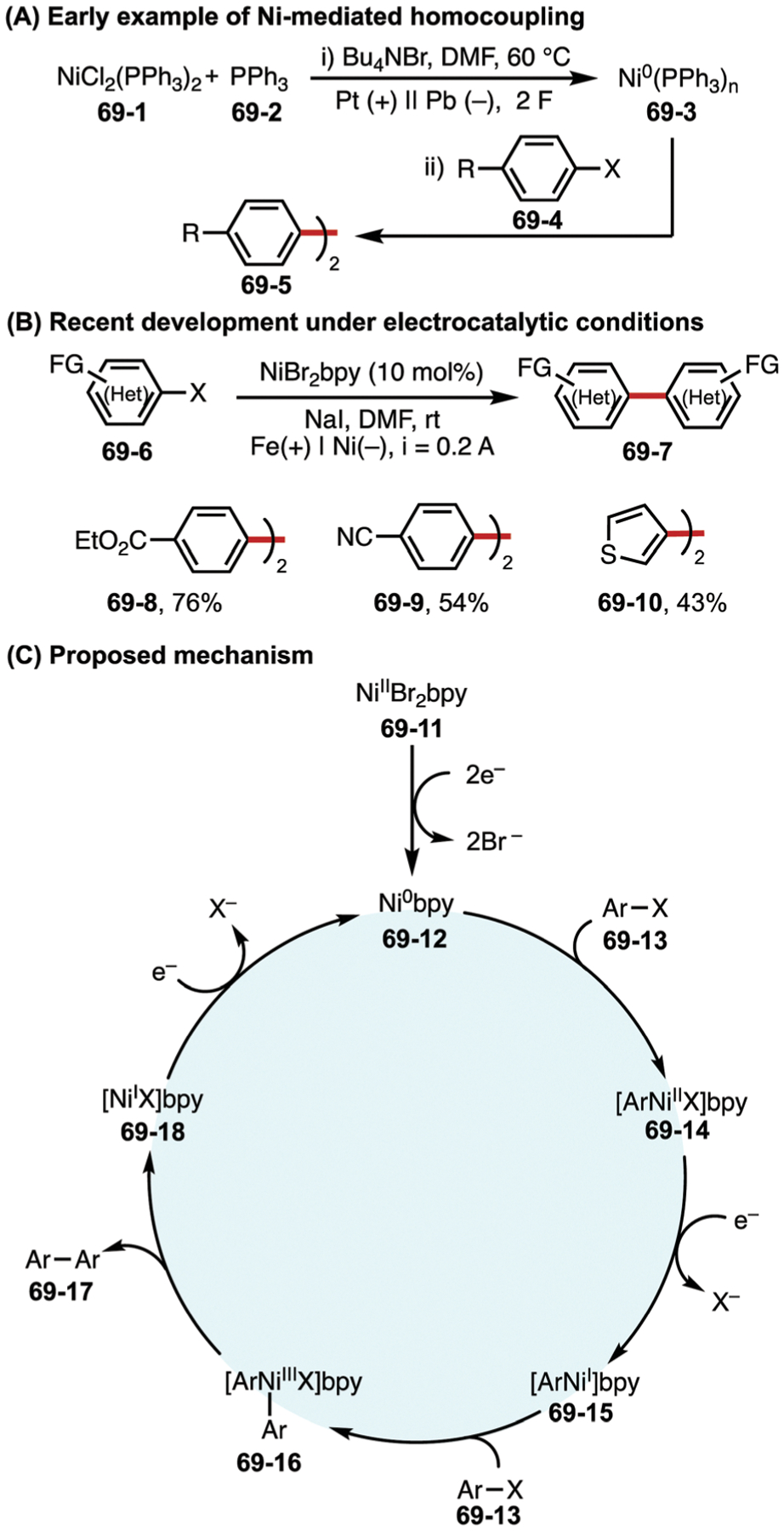
(A–C) Nickel-catalyzed electrochemical homocoupling reaction.
In 2018, the Léonel group reported an electrocatalytic homocoupling of aryl and heteroaryl halides mediated by a nickel bipyridine catalyst (Fig. 69B).201 In the proposed catalytic cycle, nickel pre-catalyst 69–11 is first reduced to Ni0 complex 69–12 via cathodic reduction. Oxidative addition of this intermediate to the C–X bond of the aryl halide 69–13 then furnishes intermediate 69–14. Cathodic single-electron reduction of this intermediate, followed by oxidative addition to a second aryl halide equivalent, furnishes NiIII complex 69–16. A reductive elimination step provides the desired coupling product 69–17. This step also generates NiI species 69–18 which is further reduced electrochemically to regenerate Ni0 complex 69–12 (Fig. 69C). The authors achieved the desired homocoupling in good yields and demonstrated functional group tolerance of esters, nitriles, and thiophenes.
In 2020, the Mei group reported a Ni-catalyzed enantioselective homocoupling of aryl bromides using chiral pyridi-neoxazoline ligands, providing biaryls in good yields and enantioselectivities using an undivided cell (Fig. 70A).202 This transformation provides protected BINOL derivatives that could not be easily generated via traditional reductants (such as Mn dust). The reaction also tolerates a variety of functional groups including boronic esters and aryl chlorides (Fig. 70B).
Fig. 70.

(A and B) Nickel-catalyzed electrochemical enantioselective homocoupling reaction.
3.1.2. C(sp2)–C(sp2) XEC.
Early examples of Ni-catalyzed XEC were disclosed by Périchon and co-workers.203,204 In these reports, an N-heteroaryl halide and a simple aryl halide can be cross coupled in good yields. The authors attributed the cross-coupling selectivity to the intrinsic reactivity of the nickel catalyst employed: (1) Ni0 undergoes oxidative addition to a simple aryl halide faster than an N-heteroaryl halide, giving rise to NiII aryl, and (2) the NiI aryl complex upon a single-electron reduction is more reactive towards oxidative addition to an N-heteroaryl halide than to another equivalent of the simple aryl halide. Depending on the electronic property of the substrates and the identity of the leaving group, homocoupling products can sometimes also be observed. Sengmany and co-workers also extended the scope of this XEC to reactions between phenyl halides and chloropyrimidines or chloropyridazines.205–208 The corresponding products resemble fragments of pharmaceutical and bioactive compounds (Fig. 71).
Fig. 71.

Nickel-catalyzed electrochemical cross coupling reaction. X = Br or Cl.
In 2019, Sevov reported a Mizoroki–Heck coupling with aryl halides and alkenes, which was achieved by combining electrochemistry and nickel catalysis in lieu of the traditional Pd catalyst (Fig. 72A).209 To overcome the slow kinetics that is characteristic of NiII aryl complexes in the Heck reaction, the authors employed cyclohexenone (72–4) as an additive to transform the redox inert NiII aryl complex 72–8 to a redox active NiII species 72–9. This intermediate thus undergoes single-electron reduction on the cathode and subsequent migratory insertion into the corresponding alkenes followed by β-H elimination to give coupling product 72–14. Good selectivity towards the linear products over the branched products was observed. A wide range of simple alkenes containing functional groups such as silyl, hydroxy, and sulfone are compatible in this system (Fig. 72C).
Fig. 72.

(A–C) Nickel-catalyzed electrochemical Mizoroki–Heck coupling reaction.
3.1.3. C(sp2)–C(sp3) XEC.
Nickel electrocatalysis has also been demonstrated to promote C(sp2)–C(sp3) coupling with α-haloketones (73–1), α-haloesters (73–2), or allyl acetate (73–3) as a coupling partner (Fig. 73).210–213 Early developments in this area are discussed in a review by Périchon.214
Fig. 73.

Nickel-catalyzed electrochemical cross coupling of aryl, heteroaryl or vinyl halides with activated alkyl chlorides.
In 2017, Hansen and co-workers further extended the scope of this reactivity to cross coupling between unactivated alkyl bromides and aryl bromides (Fig. 74A).215 The group reported that electrochemistry offered improved reproducibility over traditional metal reductants for Ni-catalyzed cross coupling reactions. Inspired by the mechanism that Weix proposed for reducing metal-promoted cross coupling,216 the authors suggested a reaction pathway that is distinct from Perichón’s original hypothesis.207 In this mechanism, the Ni(alkyl)(aryl) intermediate (74–6) formed prior to reductive elimination is generated in a radical recombination process instead of traditional oxidative addition of NiI to the alkyl halide. Specifically, NiI intermediate 74–8 reacts with alkyl halide 74–2 via halogen abstraction to form an alkyl radical 74–9, which then adds to NiII aryl species 74–5 to furnish NiIII intermediate 74–6. The rest of the catalytic cycle is analogous to the previous proposal (Fig. 74B). The same group also reported a modified protocol using MeCN as a more process-friendly solvent instead of DMA and a secondary amine as the sacrificial oxidant instead of a sacrificial anode to avoid the production of stoichiometric metal salts (e.g., ZnX2, MnX2 or NiX2).217
Fig. 74.
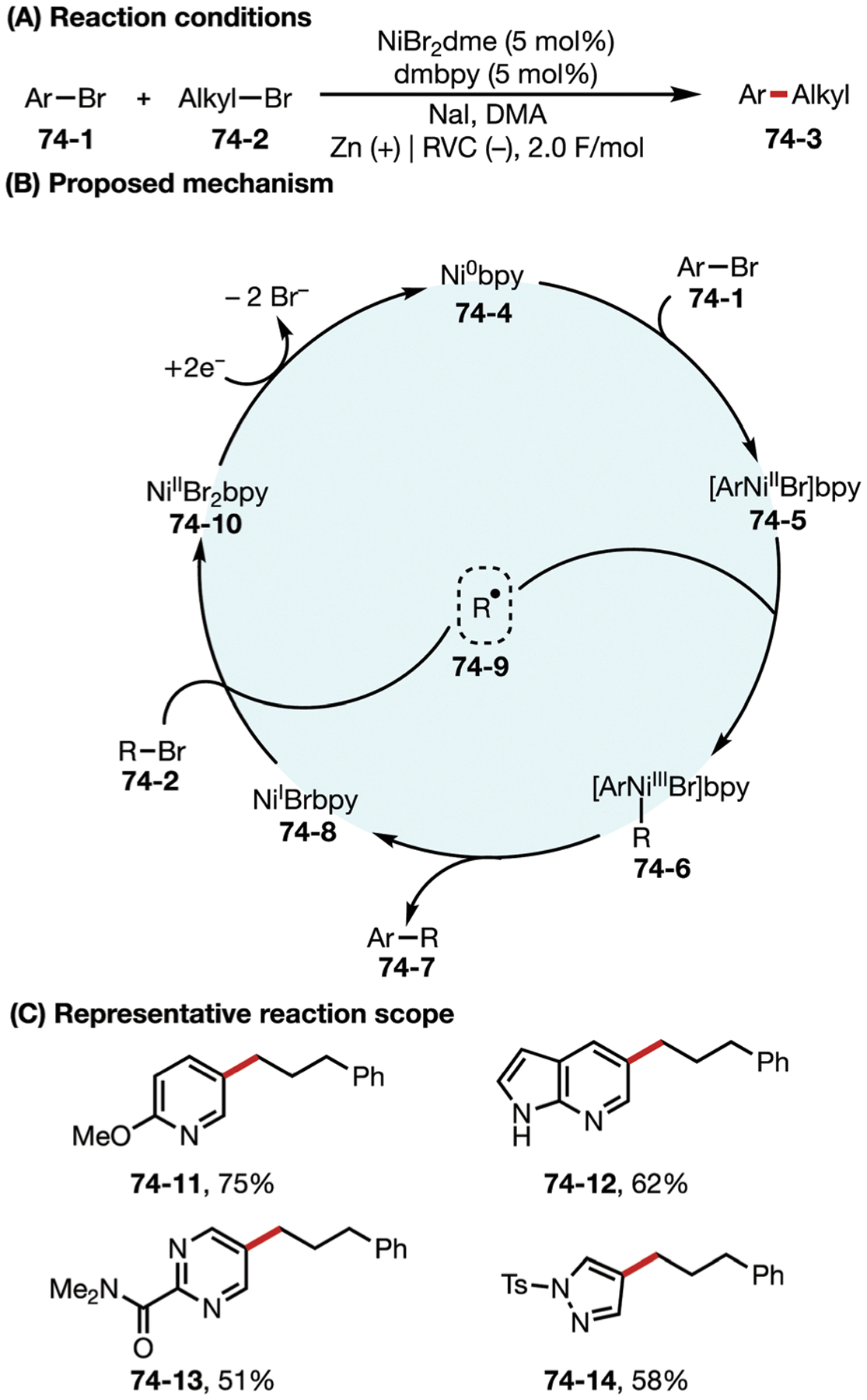
(A–C) Nickel-catalyzed electrochemical cross-electrophile coupling reactions of unactivated alkyl halides.
In 2018, Bio and co-workers reported nickel-catalyzed electroreductive coupling using N-hydroxyphthalimide esters (75–1) as coupling agents (Fig. 75A).218 The cathodic reduction potential of the redox active ester 75–1 is 1.2 V (vs. Ag/AgCl), which matches the reduction potential of the NiI complex 75–9, thus enabling the cathodic reduction of both species to take place simultaneously in a cathodically coupled electrolysis (CaCE) towards the desired reactivity (Fig. 75B). The C(sp3) radical 75–5 generated from 75–1 reduction is intercepted by a nickel catalyst 75–7, which arises from the oxidative addition of electrogenerated Ni0 75–6 to aryl halide 75–2. The resultant NiIII species 75–8 undergoes reductive elimination to form the C(sp2)–C(sp3) coupled product 75–3. Notably, electron-rich tertiary amines are used as sacrificial reductants, as they can easily undergo anodic oxidation to complete the full electrochemical reaction. The authors also carried out this reaction in a continuous flow system, showing that reaction yield and chemo-selectivity can be optimized through systematic tuning of flow rate and current density. In 2019, Loren’s group reported a one-pot strategy for the same transformation involving premixing of alkyl carboxylates and N-hydroxyphthalimide tetramethyluronium hexafluorophosphate (PITU) to form the corresponding N-hydroxyphthalimide esters prior to electrolysis.219
Fig. 75.

(A–C) Nickel-catalyzed electrochemical cathodically coupled electrolysis.
In 2020, the Mei group and the Rueping group independently reported Ni-catalyzed electroreductive XEC via a chain-walking mechanism (Fig. 76A).220,221 These two groups used terminal alkyl bromides 76–1 as the C(sp3) electrophiles. Upon formation of NiI(alkyl)(aryl) intermediate 76–3, a series of β-H elimination and migratory insertion processes took place to complete “chain walking,” eventually giving rise to thermo-dynamically stable benzyl nickel species 76–4. Subsequent reductive elimination furnished the products 76–2. This system was initially susceptible to several unproductive pathways such as early reductive elimination of NiII complex 76–3 homocoupling between two aryl halides, and generation of alkenes via β-H elimination. However, both groups were able to tackle these problems through the use of a bipyridine ligand which drastically changed the catalyst’s CV profile (as shown in Mei’s work) and smoothly facilitated the chain-walking mechanism.
Fig. 76.

Nickel-catalyzed chain-walking cross-electrophile coupling reaction.
In 2020, the Sevov group introduced a redox shuttle (77–5) strategy in a Ni-catalyzed electroreductive XEC reaction that serves to mitigate overreduction and degradation of the catalyst (Fig. 77A).222 The authors found that the difference between the cathodic potential required for XEC coupling and the potential needed to induce catalyst degradation, which leads to protonation and homocoupling, is minimal. Taking advantage of the redox shuttles developed by the energy-storage community, the reaction yield improved substantially (from 43% to 97% for bromobiphenyl and from 5% to 89% for bromobenzoate) (Fig. 77B). The reaction tolerates sulfones, nitriles, amides, esters, and heteroarenes such as pyridines and quinolines. The authors later found that maintaining a proper concentration of these redox shuttles is crucial for achieving both high reactivity and overcharge protection, as high concentration of 77–5 inhibits product formation whereas low concentration cannot sufficiently suppress catalyst overreduction.
Fig. 77.
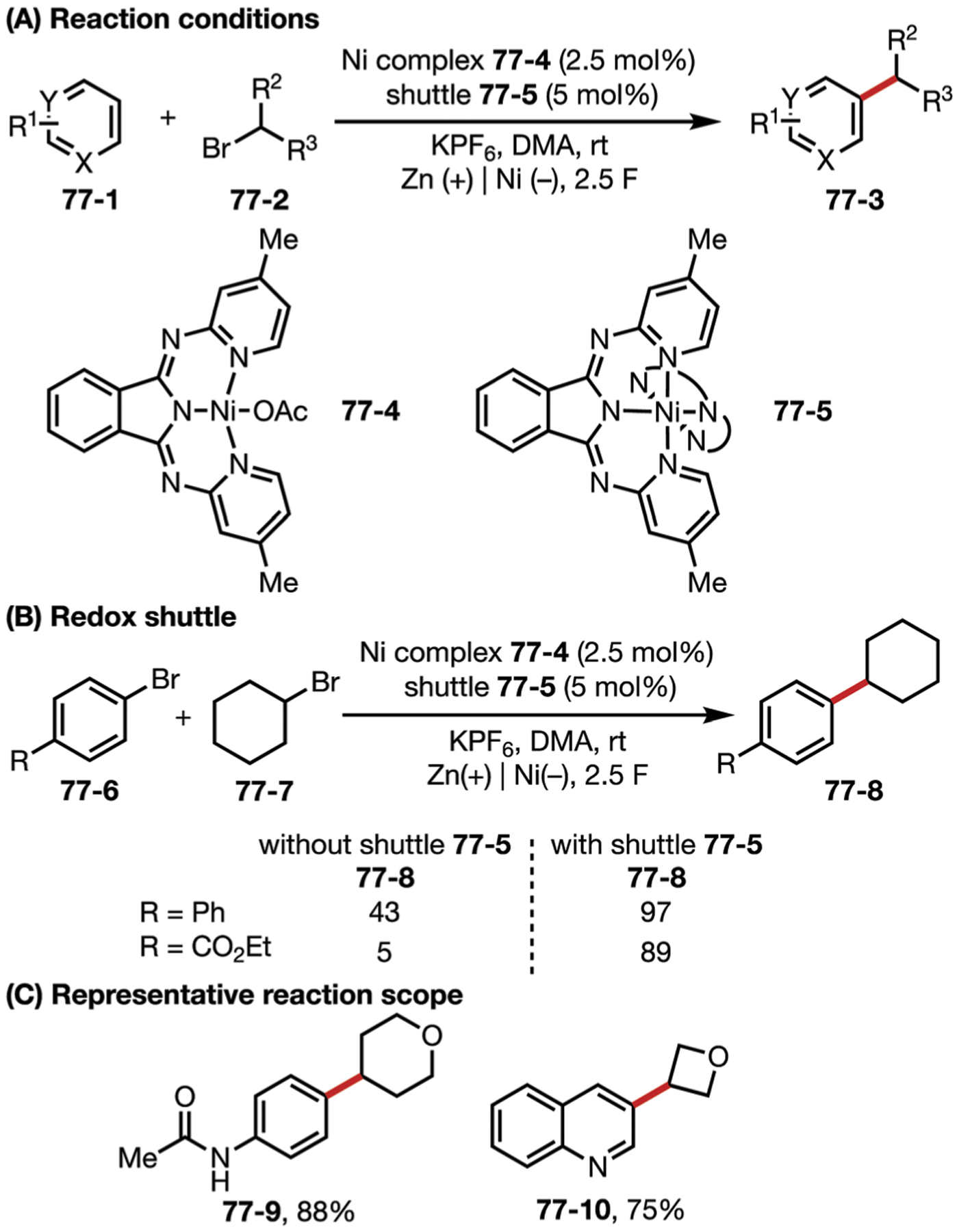
(A–C) Nickel-catalyzed electroreductive electrophile cross coupling reaction via redox shuttles.
In 2019, Reisman and co-workers reported an enantioselective variant of Ni catalyzed C(sp3)–C(sp2) XEC (Fig. 78A), where the asymmetric electroreductive coupling between alkenyl bromides (78–1) and benzyl chlorides (78–2) was explored.223 The high enantioselectivity of this transformation was achieved using an aminoindanol-derived bis(oxazoline) ligand.
Fig. 78.

(A and B) Nickel-catalyzed electrochemical enantioselective cross coupling reaction.
3.1.4. Alkene/alkyne functionalization.
An early example of Nickel-catalyzed electroreductive alkene/alkyne functionalization was achieved by the Périchon group in 1995 using aryl bromides and ethyl acrylate.224 The pioneering work led to a variety of intramolecular variations of this reactivity.225–227 These developments served to expand the scope of the starting materials to alkynes228,229 and other Michael acceptors,230,231 as well as to perfluorinated organohalides.232 In 2005, Little et al. employed a Ni(salen) catalyst to achieve the electroreductive cyclization of an aldehyde or ketone onto a pendant Michael acceptor (Fig. 79).233
Fig. 79.

Nickel-catalyzed electroreductive alkene fuctionalization.
In 2020, the Findlater group reported electroreductive pyridylation of electron-deficient alkenes, utilizing para-cyanopyridine as the heterocycle donor (Fig. 80A).234 Through CV experiments, the authors proposed two possible pathways, both of which include the reduction of the Michael acceptor to the corresponding radical anion. Subsequently, either radical cross coupling with a cathodically generated cyanopyridyl radical anion or radical addition to Ni-activated cyanopyridine results in C–C bond formation.
Fig. 80.

(A and B) Electroreductive pyridylation of electron-deficient alkenes. The anodic reaction is not discussed by the authors.
3.1.5. C–N bond formation.
In 2017, the Baran group reported the electrochemical nickel-catalyzed coupling between aryl halides and amines (Fig. 81A).235 This work features mild conditions, is amenable to large scale preparation, and exhibits complementary substrate suitability to established methods facilitated by transition metal catalysis or photochemistry. Baran, Minteer, and Neurock subsequently carried out mechanistic studies and demonstrated additional applications of this reaction in the modification of medicinally relevant compounds.236 The reaction is proposed to begin with cathodic reduction of the nickel precatalyst to form 81–4, followed by oxidative addition of 81–4 to aryl bromide 81–5. Upon formation of 81–6, ligand exchange between bromide and free amine was proposed to be inefficient. Consequently, cathodic reduction take place to form 81–7 as a stable intermediate. With assistance from a base (1,8-diazabicyclo[5.4.0]undec-7-ene, DBU), ligand exchange occurs to yield amine-coordinated 81–8. Subsequent anodic oxidation and reductive elimination provide the desired product and turn over the Ni catalyst (Fig. 81B). The authors applied this reaction towards the derivatization of nucleosides and oligopeptides (Fig. 81C). This reaction technically adopts a paired electrolysis mechanism (see Section 4) but is discussed here owing to its similarity in catalytic mechanism to other examples in this section.
Fig. 81.
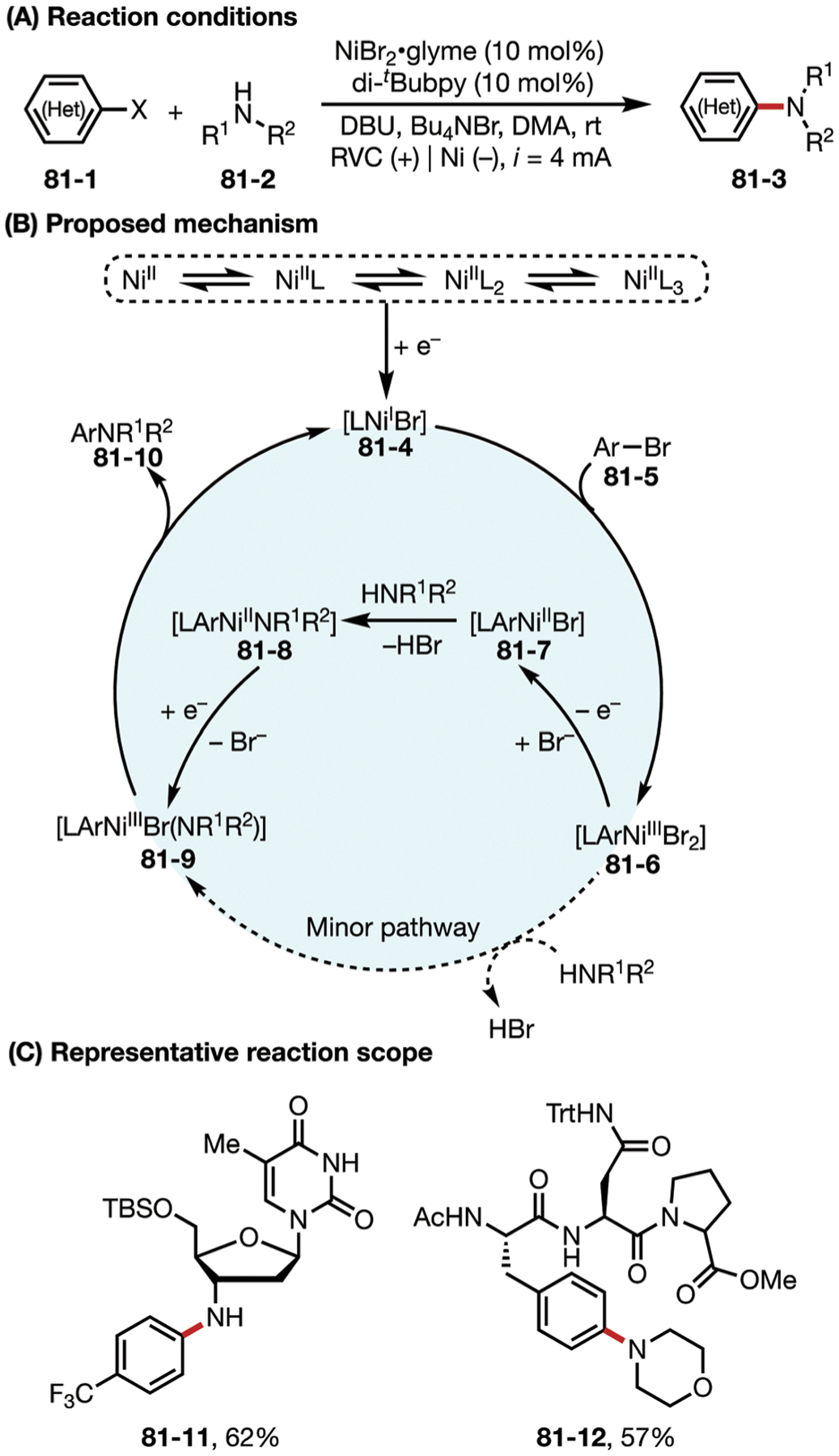
(A–C) Electrochemical nickel-catalyzed coupling between aryl halides and amines.
3.1.6. C–S and C–P bond formation.
The Mei group reported a Ni-catalyzed thiolation of aryl bromides and chlorides via electrocatalysis (Fig. 82A).237 The work features mild and base-free reaction conditions and exhibits good tolerance with heterocycles (Fig. 82C). The authors proposed a mechanism that is distinct from that of the aforementioned amination. Specifically, electrogenerated Ni0 82–5 is hypothesized to add to aryl halide 82–6, and the resultant NiII intermediate 82–7 then undergoes ligand exchange with thiol 82–11 to form product 82–9. However, the participation of an electrogenerated thiyl radical 82–12 as a possible pathway has not been definitively ruled out (Fig. 82B).
Fig. 82.

(A–C) Electrochemical nickel-catalyzed thiolation of aryl halides and heteroaryl halides.
Meanwhile, the Pan group reported a similar transformation using aryl iodides as the coupling partner (Fig. 83).238 In this work, a mechanism involving the addition of a thiyl radical to Ni is proposed, which was based on CV data and control experiments. In 2020, the Messaoudi group further expanded the scope of thiol coupling partners to various anomeric glycosyl thiols, achieving coupling with aryl, alkenyl, and alkynyl bromides (Fig. 83).239
Fig. 83.
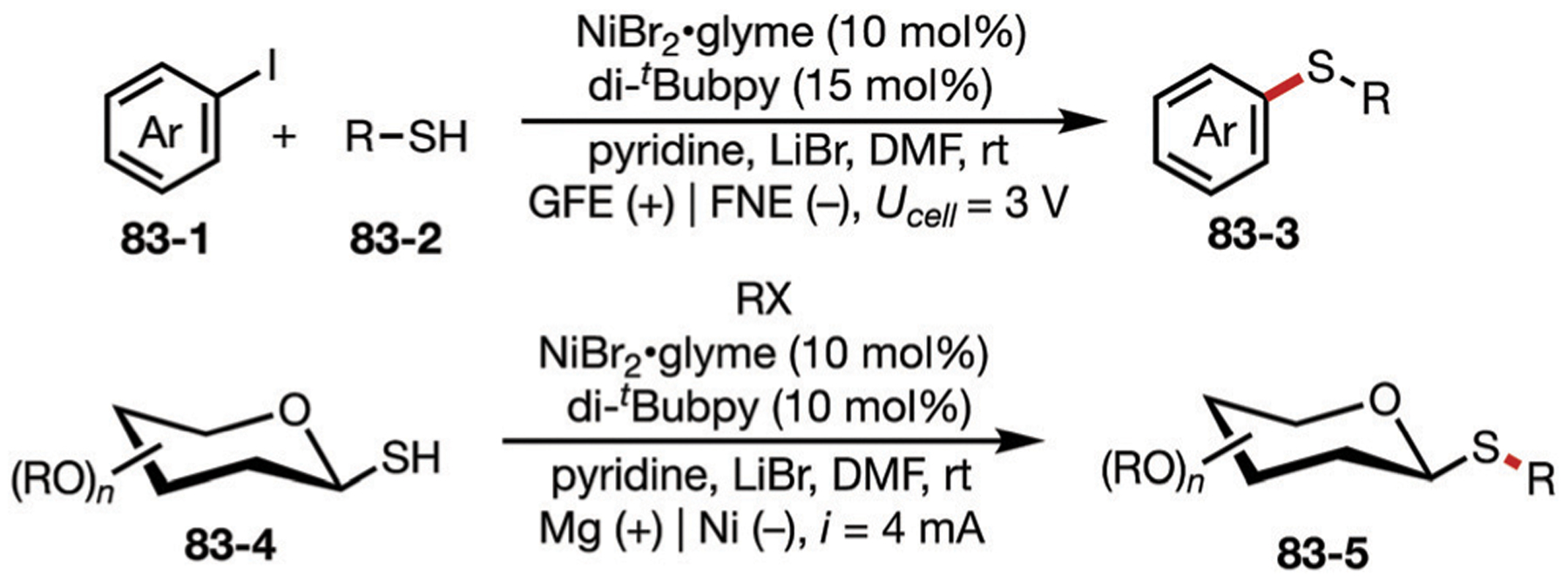
Electrochemical nickel-catalyzed carbon–sulfur bond formation.
Analogously, Ni has been shown to catalyze C–P bond formation using organophosphites as the nucleophile. An electrochemical version of this reaction was reported in 2018 by the Léonel (Fig. 84A).240 Several dialkylphosphites and trimethyl phosphites proved suitable coupling partners, providing the corresponding aryl phosphonates in good efficiency. The proposed mechanism is analogous to the thiolation reaction described previously (Fig. 84B).
Fig. 84.

(A and B) Electrochemical nickel-catalyzed phosphorination of aryl halides and heteroaryl halides.
The Xiang group reported a similar system with a scope that extended to nitriles, esters, ketones, pyrimidines, thiophenes, and benzo[d]thiazoles without the use of a sacrificial anode (Fig. 85A).241 A different mechanism is proposed for this transformation in which dialkyl phosphite 85–17 undergoes proton-transfer/electron-transfer on the anode to generate P-centered radical 85–18. Meanwhile, Ni species 85–13 is formed from cathodic reduction of the catalyst precursor and oxidative addition with the aryl halide. The radical addition of 85–18 to 85–13 precedes reductive elimination to deliver the product 85–16 and NiI 85–15, the latter of which is reduced on the cathode to regenerate Ni0 85–11 (Fig. 85B). This reaction technically adopts a paired electrolysis mechanism (see Section 4) but is discussed here owing to its similarity in catalytic mechanism to other examples in this section.
Fig. 85.

(A–C) Electrochemical nickel-catalyzed carbon–phosphine bond formation.
The Léonel group later published a follow-up work on electrochemical nickel-catalyzed C–P formation and further extended the scope of P-donors.242 The Rueping group also reported a similar transformation as well as a C–Se coupling protocol.243
3.1.7. C–H functionalization.
In 2019, the Zeng group developed an electrochemically enabled, nickel-catalyzed reductive decarboxylative coupling reaction between N-hydroxyphthalimide (NHP) esters (86–2) and quinoxalinones (86–1) (Fig. 86A).244 The proposed mechanism starts with reduction of NiII catalyst 86–2 to NiI species 86–1 at the cathode, followed by a single-electron transfer from NiI to NHP ester 86–2 and subsequent decarboxylation to give cyclohexyl radical 86–6 (Fig. 86B). The authors also proposed that direct reduction of 86–2 at the cathode can provide the same radical species 86–6. The cyclohexyl radical then adds to substrate 86–1 in a Minisci-type pathway to generate radical cation 86–8, and this process is proposed to be facilitated by Ni coordination to (86–7). A series of follow-up events ultimately lead to the formation of product 86–3. Meanwhile, sacrificial reductant Et3N is oxidized at the anode to give a triethylamine radical cation.
Fig. 86.

(A and B) Electrochemically enabled, NiCl2-catalyzed reductive decarboxylative coupling of N-hydroxyphthalimide (NHP) esters with quinoxalinones.
In 2020, the Ackermann group reported a nickel-catalyzed electrochemical C–H activation using primary and secondary alkyl halides via a directing group strategy245 (Fig. 87). This reaction mechanism is analogous to similar C–H activation reactions reported by the same group using other transition metals (see Section 2.14) but relies on the cathodic reduction of Ni to generate the active catalyst and maintain the catalytic cycle.
Fig. 87.
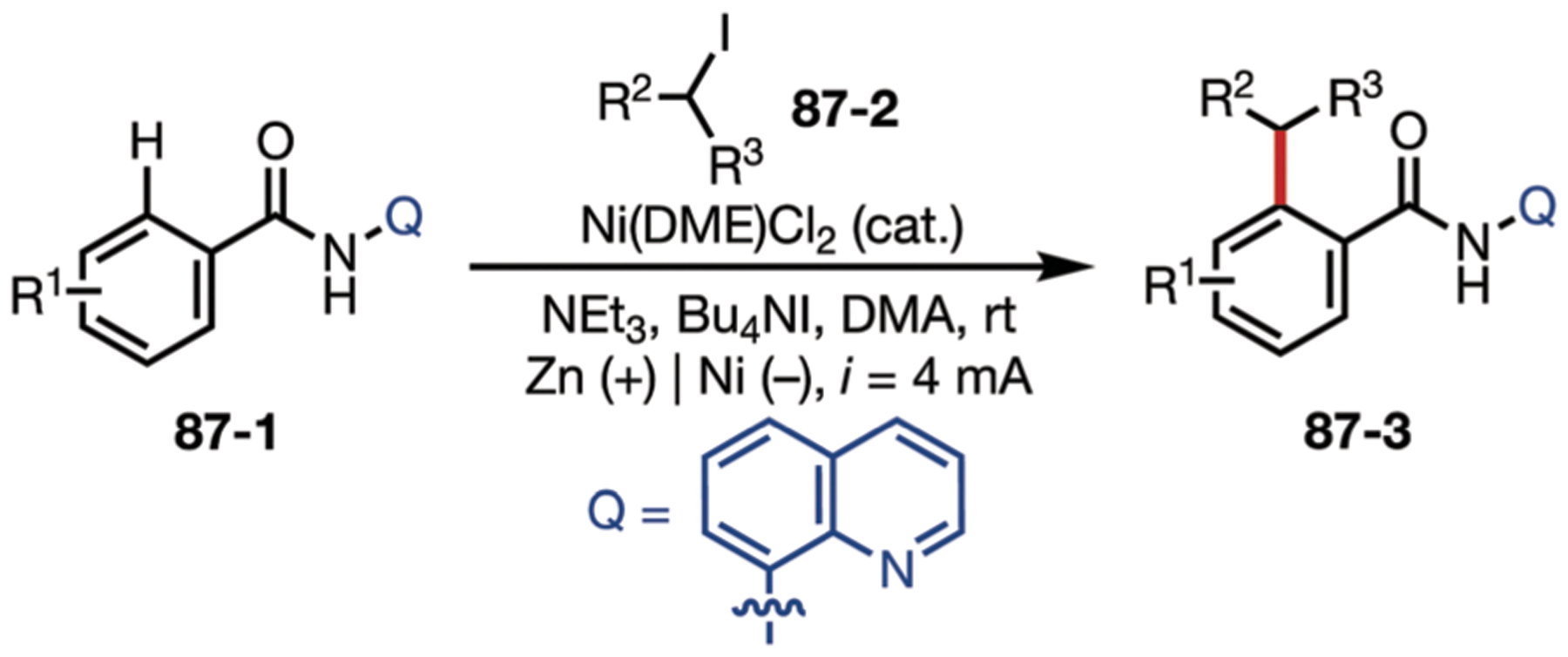
Electrochemical C–H activation of benzamides.
3.1.8. Miscellaneous transformations.
Nickel-catalyzed reductive electrochemical decarboxylation was first reported by the Dunach group in 2000.246 In a later work by the same group, this strategy was extended to deprotect carbamates and allyl β-keto esters (Fig. 88A).247 Mechanistically, the reaction proceeds through the electrochemical generation of 88–6, which inerts to the C–O bond of 88–7 to form a π-allyl NiII complex 88–8 (Fig. 88B). After decarboxylation and deallylation, 88–2 is formed.248
Fig. 88.

(A and B) Nickel-catalyzed reductive electrochemical decarboxylation.
3.2. Cobalt
3.2.1. XEC.
In the early 2000s, Gosmini, Nedéléc, and co-workers demonstrated a series of cobalt-catalyzed electroreductive XEC of aryl halides (Fig. 89).249,250 In these reactions, a CoI species is proposed to be the active catalyst, which is generated via electroreduction of CoII chloride and reacts with aryl halides (89–1, 89–2, 89–4) to initiate the reaction. Several functionalities, such as esters, trifluoromethyl, and ether groups, have been explored in this scheme and exhibit good reactivity. These early studies have paved the way for a variety of new developments in the area of Co electroreductive chemistry.
Fig. 89.

Cobalt-catalyzed aryl halides coupling: early contributions from Gosmini, Nedéléc and co-workers.
3.2.2. Alkene hydroalkylation.
Nedéléc and co-worker also reported a CoII(salen) mediated electroreductive coupling of alkyl bromides with methyl vinyl ketone. In this reaction, the reduced CoI intermediate (90–4) is transformed to CoII (90–5) or CoIII (90–6) via oxidative addition, which mediates the alkyl addition to alkene 90–2 in either radical-polar crossover (Fig. 90, pathway a) or anionic pathways (Fig. 90, pathway b).251 Bedioui and co-workers further studied this system, expanding the scope to several alkyl bromides and activated olefins, including acrylate and acrylonitriles.252
Fig. 90.
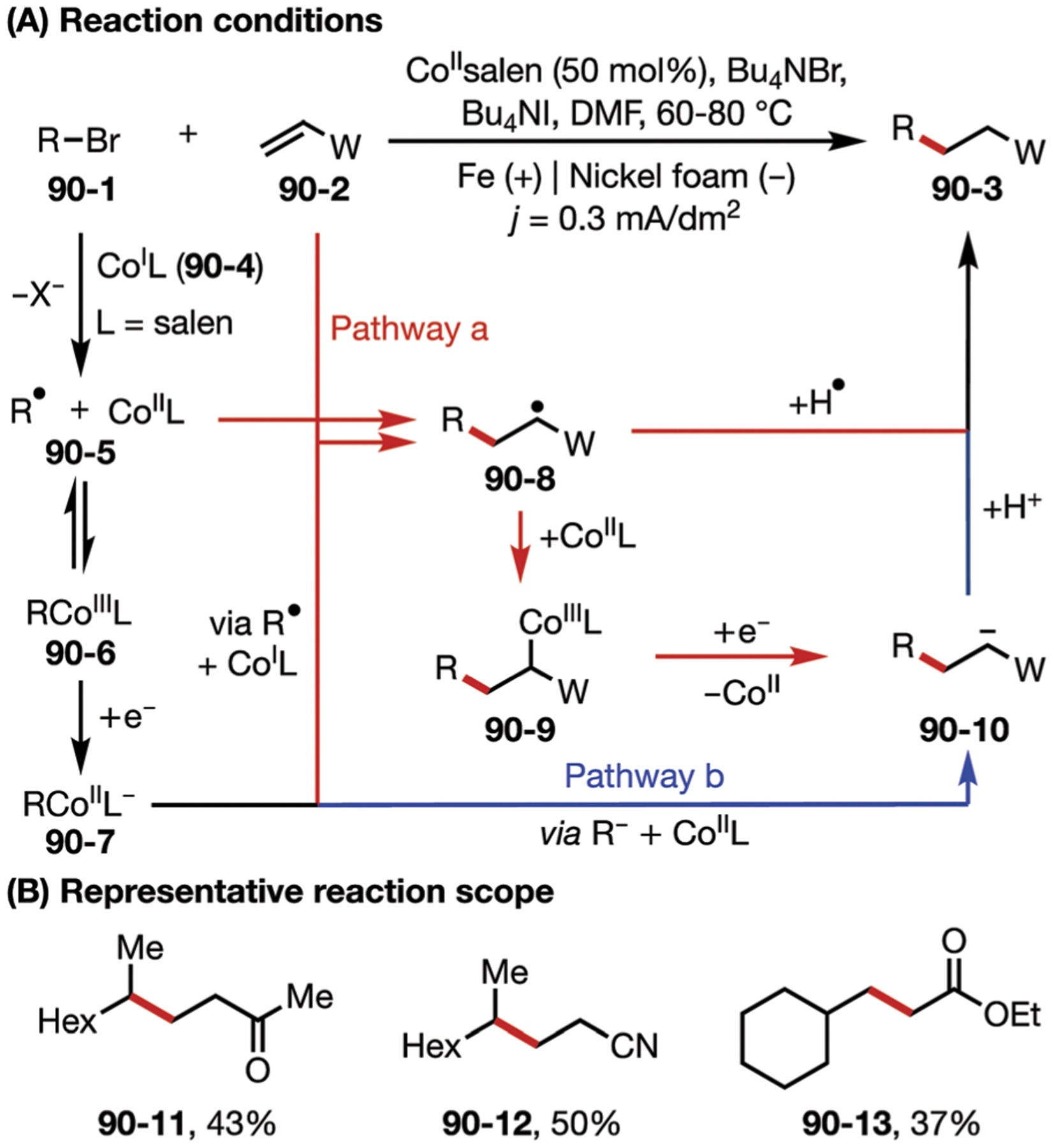
(A and B) Co(salen)-catalyzed alkylation. W = electron-withdrawing group (EWG).
3.2.3. Reductive dehalogenation.
Cobalt and its organometallic complexes have recently emerged as versatile redox mediators in electrocatalysis.253 In 2013, the Atobe group reported a reductive debromination of open-chain and cyclic dibromoalkanes with a CoII(salen) electrocatalyst (Fig. 91).254 In this context, CoII(salen) is reduced to CoI(salen) and abstracts a bromine atom from substrate 91–3 to generate carbon-centred radical 91–4. Upon reduction with a second equivalent of CoI(salen), carbanion 91–5 undergoes protonation to complete the debromination to form 91–6. This series of events is repeated to provide fully debrominated product 91–7. In the case with a vicinal dibromide substrate, β-elimination takes place from intermediate 91–5 to yield an alkene product (e.g., 91–10; Fig. 91C).
Fig. 91.

(A–C) Co(salen)-catalyzed debromination. GC = glassy carbon. DSA = Dimensionally Stable Anodes, titanium mesh with metal oxides (RuO2, IrO2 or PtO2). The anodic reaction was not discussed.
3.2.4. Reductive carboxylation.
Lu and co-workers reported an asymmetric electrocarboxylation of 1-phenylethyl chloride with a chiral Co(salen) catalyst (Fig. 92).255 In the first step, the active CoI(salen) species is generated via cathodic reduction of CoII(salen) and reacts with benzyl chloride 92–1 via oxidative addition, thus forming CoIII intermediate 92–3. This intermediate undergoes a second single-electron reduction to form CoII species 92–4. Upon subsequent nucleophilic addition to CO2 directed by the chiral catalyst enantioenriched carboxylate 92–5 is formed with the regeneration of CoII(salen).
Fig. 92.

(A and B) Co(salen)-catalyzed enantioselective carboxylation.
In 2020, the Ackerman group demonstrated a cross-electrophile coupling of cinnamoyl chlorides and CO2 in a Co-mediated electroreductive pathway (Fig. 93).256 In this system, CoII acetate is cathodically reduced to CoI species 93–4, which undergoes oxidative addition to cinnamoyl chloride 93–1 to form π-allyl CoIII complex 93–5. This intermediate then undergoes rearrangement to give η1-allyl CoIII intermediates 93–6 and 93–7, which are then converted to CoI complexes 93–8 and 93–9, respectively, via two-electron reduction. These low valent Co species then react with CO2 to deliver carboxylate products 93–10 and 93–11 and liberate the active CoI catalyst. Both electron-withdrawing and electron-donating aryl substituents are well-tolerated in this system, providing a useful method for the synthesis of vinylacetic acids.
Fig. 93.

(A–C) Cobalt-catalyzed allylic carboxylation.
3.2.5. Other Co-mediated transformations.
Cobalt-containing vitamin B12 has also been widely used as a mediator in electroreductive reactions.257 In a report from Rusling and co-workers, vitamin B12 was employed to achieve cyclopropenation of styrene with dichloromethane. In this reaction, vitamin B12 is reduced to CoI, which then catalyzes the generation of carbon-centered radical 94–4 (Fig. 94).258 This radical intermediate adds to styrene to generate benzylic radical 94–5, which is reduced to carbanion 94–6 before intramolecular cyclization to form a cyclopropane ring.
Fig. 94.

Vitamin B12-catalyzed cyclopropanation of styrene.
Hisaeda used a similar vitamin B12-mediated protocol to synthesize esters, amides, and 1,2-dichlorostilbenes from tri-chlorotoluenes by means of generation of 1,1-dichlorobenzyl radicals (95–6; Fig. 95).259 Here, electrogenerated CoI undergoes oxidative addition to a C−Cl bond of substrate 95–1, forming CoIII intermediate 95–4. Direct homolysis of the CoIII–C bond in 95–4 (Fig. 95, solid arrow) or single-electron reduction followed by mesolytic cleavage of the CoIII–C bond in 95–5 (Fig. 95, dashed arrow) give 95–6. Upon further reaction with oxygen to generate an acyl chloride intermediate and subsequent reaction with a nucleophile (e.g., an alcohol or an amine), the desired ester 95–2 or amide 95–3 can be obtained.
Fig. 95.

(A and B) Vitamin B12-catalyzed ester/amide synthesis.
Peters and co-workers designed a cobaltocene mediator to facilitate concerted proton-electron transfers (CPET) under electroreductive conditions (Fig. 96).260 CPET enables milder conditions for redox transformations by lowering the reaction energy barrier compared with conventional stepwise electron transfer-proton transfer processes. The system developed by Peters employs an aniline-substituted cobaltocene mediator, which promotes CPET reactivity to drive pinacol coupling while suppressing the competing hydrogen evolution reaction. Mechanistic studies revealed that protonated CoII catalyst 96–1 reacts with acetophenone via CPET to give a neutral α-radical 96–3 and CoIII 96–5. Radical dimerization of 96–3 affords pinacol 96–4 as the final product in 83%. Meanwhile, 96–5 is protonated to form the dimethylanilinium Brønsted acid 96–6, which undergoes cathodic reduction at a potential of −1.30 V vs. Fc+/Fc0 to regenerate CoII catalyst 96–1. This approach provides a potential solution to improving functionality tolerance for electrolysis that otherwise requires highly biased potentials.
Fig. 96.
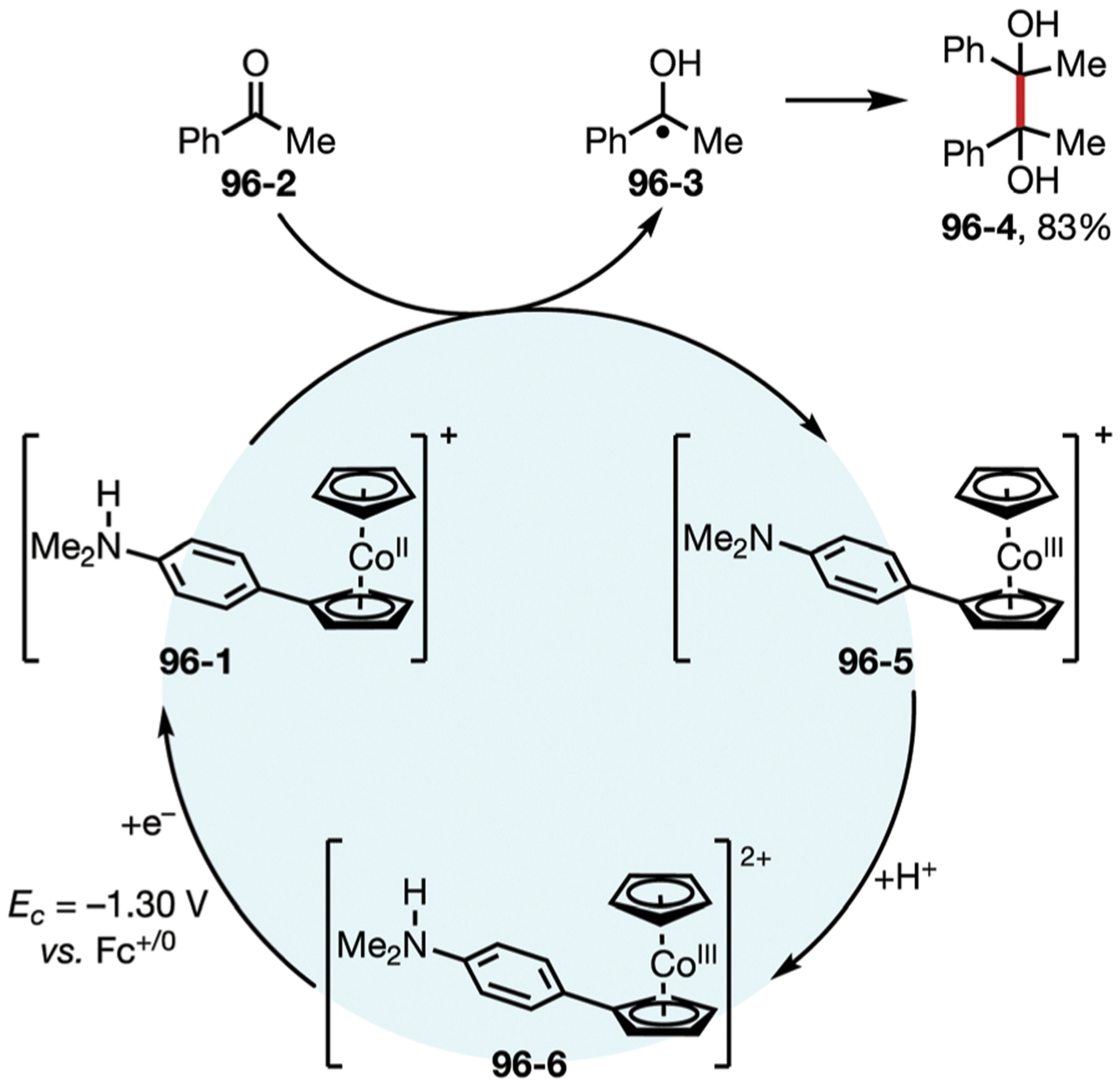
Cobalt-catalyzed reductive ketone dimerization via CPET. Boron-doped diamond (BDD) electrode is used as cathode and glassy carbon is used as counter electrode.
3.3. Titanium
An early example of titanocene-catalyzed electroreductive dehalogenation of organic halides was disclosed by Magdesieva and co-workers (Fig. 97).261 After the electroreductive generation of active TiIII species 97–6, halogen atom abstract takes place to transform the benzyl halide or pseudohalide substrate (97–1) to a benzyl radical prior to formation of product 97–2. In 2011, this reactivity was extended to an electroreductive epoxide ring opening reaction by the same group using a 30% loading of titanocene dichloride as the mediator (Fig. 97).262 In this context, TiIII catalyst 97–6 induces homolytic ring opening of epoxide 97–3 to form carbon-centered radical 97–7. Hydrogen atom transfer from 1,4-cyclohexadiene (1,4-CHD) to 97–7 followed by transmetallation of the resultant 97–8 with tri-methylsilyl chloride affords the product 97–4.
Fig. 97.
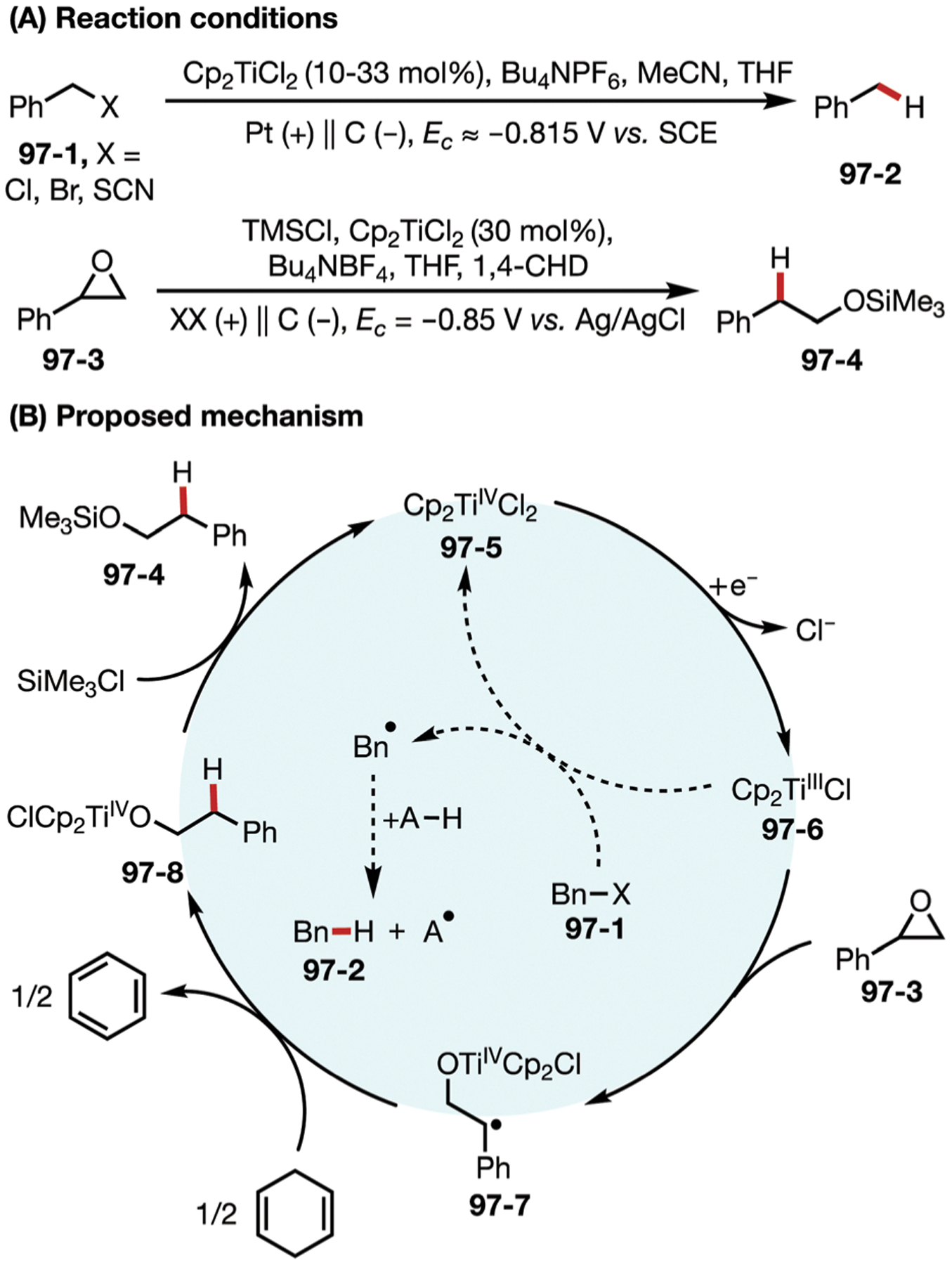
(A and B) Titanocene-catalyzed dehalogenation and epoxide opening. The anode [XX(+)] in the epoxide ring-opening reaction was not disclosed.
In addition, Lisitsyn and co-workers achieved amination of arenes via electrochemical reduction of hydroxylamine using a TiIV mediator (Fig. 98).263 Mechanistic investigation of this transformation revealed that the initial cathodic reduction of TiIV sulfate generates active TiIII, which reacts with hydroxylamine to form TiIV-bound aminyl radical 98–3. 98–3 is in equilibrium with ammonia radical cation (98–4). The authors proposed that either of these intermediates can react with an arene substrate (98–1), giving rise to the aniline product (98–2). Anisole,264 benzene,265 and chloroaniline266 were shown to be suitable substrates for this method.
Fig. 98.

(A and B) Titanium-catalyzed amination of arenes.
In 2018, Gansäuer and co-workers achieved the radical arylation of epoxides using a pre-generated TiIII catalyst via electrochemistry. This approach avoids the use of heterogenous metal reductant and generates indoline products in high efficiency (Fig. 99).267 Thiourea is found to facilitate halide abstraction from reduced titanocene to generate the active Cp2TiCl catalyst.
Fig. 99.

Titanium-catalyzed epoxide ring-opening arylation.
3.4. Zinc
Zinc salts are frequently used as Lewis acids, but if these salts are reduced to Zn0, they can be used to mediate reductive transformations. For example, Huang and co-workers have demonstrated a series of electrochemical allylation reactions of carbonyl species in aqueous solution via a Zn-mediated approach (Fig. 100).268 The sacrificial zinc anode is oxidized and releases Zn2+ into the reaction medium. Upon forming a complex with ammonia, ZnII is reduced on the cathode to give active Zn0 particles, which react with allyl bromides (100–2) to afford allyl zinc reagent 100–4. 100–4 then undergoes nucleophilic addition to aldehyde 100–1 to generate 100–5, followed by hydrolysis to give the products 100–3. Subsequent studies improved the atom efficiency of this reaction by introducing Pt as the anode in a divided cell to avoid consumption of stoichiometric amounts of Zn on a sacrificial anode.269 The authors propose that in this case, oxidation of water happens on the Pt anode, which results in the formation of protons that can diffuse to the cathode chamber to participate in the reaction.
Fig. 100.
(A–C) Zinc-mediated allylation of aldehydes.
In 2014, Chakraborty, Roy and co-workers demonstrated the same transformation using a transparent conductive oxide (TCO) glass cathode and a Zn anode with additional catalytic Zn2+ in the solution (Fig. 101).270 It was found that ZnII was reduced to Zn0 on the cathode surface as nanowires, which promote the reaction. In addition to aldehydes, N-tosylimines can also undergo allylation in good yields.
Fig. 101.

(A and B) Zinc catalyzed allylation of aldehydes and sulfonimines. TCO glass = transparent conductive oxide glass, indium tin oxide.
3.5. Palladium
In 1985, Torii and co-workers reported an early example of an electroreductive Pd-catalyzed homocoupling of aryl halides (Fig. 102A).271 In 2005, the Moeller group developed a Heck-type reaction assisted by Pd and electrochemistry (Fig. 102B).272 Both reactions are proposed to be promoted by cathodically generated Pd0. These early developments demonstrated the possibility of reductive Pd electrocatalysis.
Fig. 102.

(A) Palladium catalysed homocoupling of aryl halides; (B and C) palladium catalysed Heck type reaction.
In 2017, Huang and co-workers reported an electrochemical Pd-mediated allylic alkylation reaction in an undivided cell system (Fig. 103).273 A variety of alkyl iodides and benzyl bromides were converted to the desired product in high yields. Aryl halide, ester, carboxylic acid and alcohol functionalities were well tolerated.
Fig. 103.

(A and B) Palladium-catalyzed allylic halide coupling.
In 2018, Mei and co-workers reported a Pd-catalyzed electroreductive carboxylation of allyl ethers (Fig. 104).274 The oxidative addition of electrogenerated Pd0 (104–3) to the allyl electrophile 104–1 generates a cationic π-allyl PdII complex (104–4). Complex 104–4 is in equilibrium with a terminal η1-allyl PdII species, and this energetically favorable isomer is further reduced to Pd0 complex 104–6. This species reacts with electrophilic CO2 to deliver the product 104–7 and regenerate Pd0. Notably, enantioselective carboxylation was also achieved when a chiral phosphine ligand, (R)-MeO-BIPHEP, is used.
Fig. 104.

(A–C) Palladium-catalyzed carboxylation of allyl ethers. DPPPH = 1,2-bis(diphenylphosphino)benzene; (R)-MeO-BIPHEP = (R)-(+)-2,2″-bis(diphenylphosphino)-6,6″-dimethoxy-1,1″-biphenyl.
3.6. Samarium
SmI2, also known as Kagan’s reagent, has been shown to be an outstanding single electron reductant in synthetic organic chemistry.275 However, SmI2 is a potent reductant that is air sensitive and can be difficult handle or store. Thus, in situ generation of SmI2 via electroreduction constitutes an attractive approach to achieve the same types of transformations.276–278 For example, Mellah and co-workers introduced Sm as the sacrificial anode to electrochemically generate Sm2+ in situ (Fig. 105A),279 which reacts with iodide in the medium to form Kagan’s reagent. This system was shown to promote the pinacol coupling of carbonyl compounds. This approach can also be applied to allylation, benzylideneaniline coupling, and Reformatsky-type reactions.280 In these systems, the authors showed that catalytic SmII can be regenerated via cathodic reduction of SmIII on a Sm cathode (Fig. 105B–D).
Fig. 105.

(A–D) Samarium-mediated coupling reactions.
In 2017, the same group reported a Sm-mediated electroreduction of nitroarenes to azoarenes (Fig. 106).281 In the first step, a pre-electrolysis is performed (Fig. 106, red conditions) to oxidize a Sm anode, releasing SmII into solution until a 20 mol% loading with respect to the organic substrate is attained. Thereafter, the polarity of the electrolysis is reversed (Fig. 106, blue conditions), and Sm now acts as the cathode. Nitrobenzene (106–1) is then reduced by SmII to generate nitrosobenzene 106–3. 106–3 then undergoes a one-electron reduction by SmII to form nitroxyl radical anions which quickly dimerize to intermediate 106–4. Upon reduction of 106–4 with SmII, azobenzene product (106–2) was formed in high yields. The reaction has also been used to achieve cross coupling of two different nitroarenes by adjusting the relative stoichiometry of the substrates. Thus, when a 1 : 3 molar ratio of two nitrobenzenes are used, over 99% selectivity can be achieved.
Fig. 106.

(A and B) Samarium-catalyzed reductive coupling of nitroarenes.
3.7. Tin
Since 1980s, tin has been enlisted in the electroreductive allylation of carbonyl compounds. The reaction was performed by the Torii group in a divided cell with two Pt electrodes and catalyzed by a 10 mol% loading of tin (Fig. 107).282 It is proposed that low valent tin species 107–4 reacts with two equivalents of allyl bromide to generate active diallyltin dibromide intermediate 107–5 which reacts with a ketone or aldehyde (107–1) to form the product 107–3. Upon ligand exchange with methanol, monomethoxide tin intermediate 107–6 is formed and reduced cathodically to 107–4, completing the cycle.
Fig. 107.
(A and B) Tin-catalyzed allylation of ketones/aldehydes.
Subsequently, Wang, Zhang, and co-workers demonstrated a Sn-mediated aldehyde allylation in aqueous media with two graphite electrodes.283 The tin catalyst can be recycled by extraction and show little decrease of reactivity even after five cycles of reuse. This reaction has also been merged with anodic alcohol oxidation in a divided cell system to achieve parallel paired electrolysis.284
In a follow-up development, the same group developed a paired electrolysis system to synthesize secondary homoallylic alcohols.285,286 In this case, alcohol substrate (108–1) is anodically oxidized to the aldehydes or ketones (108–4), which diffuse to the cathode to undergo Sn0-mediated allylation with allyl bromide (Fig. 108). Both benzyl and unactivated alcohols are suitable substrates and a variety of functionalities are tolerated.
Fig. 108.
(A–C) Tin-catalyzed ketone allylation in one pot.
In addition to these allylation reactions, the Zhang group also demonstrated the synthesis of oximes from alcohols and KNO3 through paired electrolysis (Fig. 109).287 Here, alcohol substrate 109–1 was oxidized to an aldehyde or ketone (109–3) on the anode, which then combined with the hydroxylamine generated via cathodic reduction of KNO3 to form the desired product 109–2.
Fig. 109.

(A–C) Tin-catalyzed synthesis of oximes.
Organic mediators
3.8. Arenes
Arenes are well-known for their ability to stabilize radical and charged species. This feature renders arenes suitable as mediators in electrocatalytic transformations. An early example was reported by Ban and co-workers, wherein anthracene mediates the reductive removal of arenesulfonyl groups of sulfonamides.288 Here, anthracene serves as a redox shuttle to transfer electrons to the sulfonamide substrate. In 2010, the Senboku group further optimized this reaction system by using a Mg anode/Pt cathode system to eliminate the use of hazardous mercury electrodes in the original system (Fig. 110).289 A variety of arene mediators were screened, and the targeted detosylation was achieved in high yields with good functional group tolerance when naphthalene was used.
Fig. 110.

(A and B) Naphthalene-mediated detosylation.
Another example of arene-mediated electroreduction is the dehalogenative functionalization of aryl halides via aryl radical formation. In early contributions by Tokuda and Kurono, phenanthrene was employed to promote the reduction of aryl halides.290,291 In 2013, Suga and co-workers introduced fluorene derivatives as mediators and achieved the reduction of aryl chlorides (111–1) in high efficiency (Fig. 111A).292 This protocol can also be incorporated in radical cyclization reactions (Fig. 111B).293
Fig. 111.
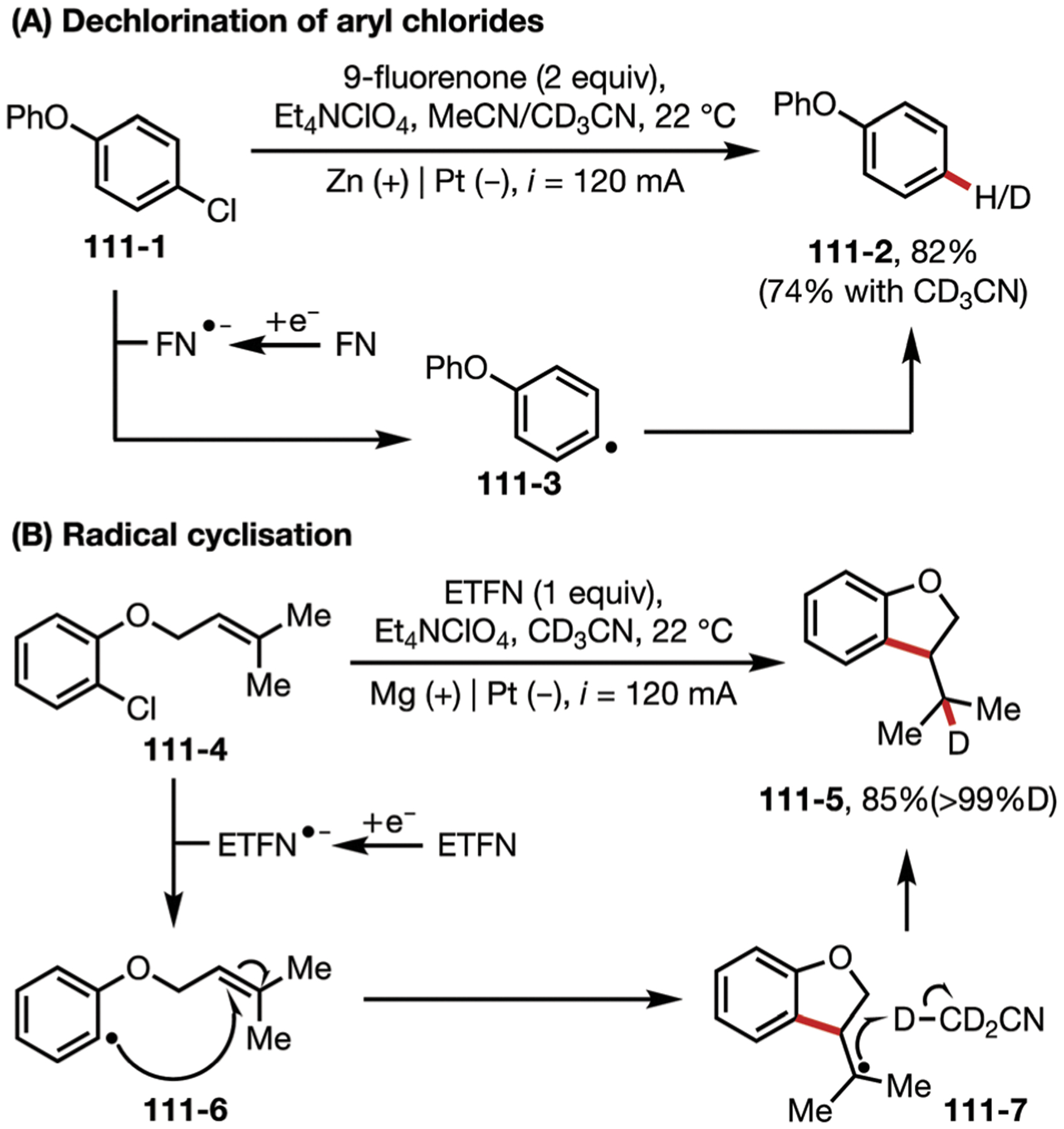
Fluorene mediated: (A) dechlorination, FN = 9-fluorenone; (B) radical cyclization of aryl halides, ETFN = 9,9-diethyl-9H-fluorene.
Perylene diimide derivatives (PDIs) are an intriguing class of organic dyes which have been widely used as semiconductors and photocatalysts.294 In 2016, Wan and co-workers reported an electroreductive arylation of pyrroles through a PDI-catalyzed pathway (Fig. 112).295 Several aryl halides (Cl, Br, I) with electron-withdrawing substituents and protected or unprotected pyrroles were converted to the coupled products in good yields. Mechanistic insights suggest that the PDI radical anion 112–4 acts as the key mediator of this reaction. Similar to previous examples of arene mediated reduction, PDI accepts one electron from the cathode to form 112–4, which then reduces aryl halide 112–1 to the corresponding aryl radical. Subsequent addition of this radical intermediate to pyrrole 112–2 constructs the biaryl C–C bond and eventually led to the formation the desired product 112–3 (the mechanism after radical addition to 112–2 is not discussed).
Fig. 112.

(A and B) PDI-catalyzed C–H arylation of pyrroles.
3.9. Benzoate-catalyzed carboxylation
A benzoate-mediated electrochemical reaction was developed by Senboku and co-workers to achieve reductive radical cyclization of aryl halides with alkenes followed by tandem carboxylation (Fig. 113).296 Cyclic voltammetry studies revealed a reversible peak of 4-tert-butylbenzoate at −2.9 V vs. Ag/Ag+, which is higher than that of the aryl bromide substrate (−3.2 V), thus supporting the benzoate-mediated pathway for substrate reduction. It is proposed that 4-tert-butylbenzoate (113–4) undergoes a one-electron reduction to radical anion 113–3 which reduces aryl bromide 113–1 to aryl radical 113–5. Following an intramolecular radical addition to the pendant alkene moiety of the substrate, the resultant carbon-centered radical 113–6 was reduced to carbanion 113–7 and then captured by CO2 to complete the reaction. When an alkyne was used as the substrate, dicarboxylic acid was formed (e.g., 113–9).297 This protocol can also be applied to vinyl bromide substrates (e.g., 113–10)298 and grants access to a variety of value-added heterocyclic carboxylic acids.
Fig. 113.

(A and B) Benzoate-catalyzed radical cyclization carboxylation.
3.10. Carborane/fullerene-catalyzed debromination
Fullerenes and carboranes are valuable materials in the fields of polymer synthesis,299 supramolecular chemistry,300 and solar cells.301 In the context of organic synthesis, fullerene compounds can undergo a variety of transformations such as the Bingel302 and Prato303 reactions, and carboranes have also been engaged in interesting reactions such as borylation and metal coordination.304 In 2011, Fuchigami and co-workers demonstrated the reduction of 1,2-dibromo-1,2-diphenylethane in a divided cell system via an o-carborane mediated pathway (Fig. 114).305 Here, o-carborane 114–1 underwent consecutive cathodic reduction to generate first radical anion 114–2 and then dianion species 114–3. Electron transfer between 114–3 and vicinal dibromide substrate 114–4 proceeds rapidly, generating trans-stilbene 114–5 and catalyst 114–2. The stilbene product can be easily separated from reaction mixture by hexanes extraction in 95% yield with only 1 mol% catalyst loading. A similar system was also reported by the same group, which used C60 as a mediator to cathodically reduce vic-dihalides to the corresponding alkenes following a similar mechanism.306
Fig. 114.

Carborane-catalyzed debromination. The anodic reaction was not discussed.
3.11. Viologen/Pd-catalyzed dimerization of aryl halides
Viologens are well-suited for applications in electrocatalysis. Not only do viologens exhibit high electrochemical reversibility, but they can be modification at the pyridyl groups to achieve tunable redox potentials.307 The Tanaka group used viologens as catalytic reductants to promote a Pd-catalyzed homocoupling of aryl halides (Fig. 115).308,309 Importantly, mechanistic studies revealed that dicationic salt (V2+) is initially reduced to neutral V0 which reduces PdII precursor to Pd0 (115–3). V0 could be regenerated via cathodic reduction of the resultant V+, and a sacrificial Zn anode is used to circumvent unwanted Pd oxidation. Using this dual catalytic system, a variety of aryl halides with electron-withdrawing and neutral substituents can be efficiently converted to biaryls, whereas diminished reactivity is observed for electron-donating substituents. Pyridinium and diquat derivatives are also proven to be effective mediators for this reaction.310
Fig. 115.

(A–C) Palladium/viologen catalyzed aryl halides coupling.
4. Paired electrolysis
Paired electrolysis has attracted substantial attention in recent years for its ability to couple two desirable electrochemical reactions simultaneously within the same reaction system. This efficient and atom-economical technique has been applied to industrial synthesis311 and energy conversion processes.312 Despite recent advances, the strategic application of paired electrolysis remains relatively rare in the context of organic synthesis. Sequential and convergent paired electrolysis, wherein both cathode and anode are employed to promote the same transformation, can often lead to ingenious reaction design and new solutions to synthetic problems. However, challenges can arise from the competition between interelectrode mass transport of electrogenerated reactive intermediates and their undesired degradation pathways. Moreover, in many cases balancing the rates of cathodic and anodic events can be crucial to the reaction selectivity. In light of these challenges, recent developments in paired electrolysis have made use of electrochemical mediators to enhance the reaction efficiency and selectivity. In previously sections, we have discussed several examples wherein a paired electrolysis scenario is proposed to contribute to the overall transformation. In this section, we will primarily focus on recently developments in which paired electrolysis is strategically implemented to solve synthetic problems.
4.1. Nickel catalysis
In 2020, the Hu group reported a combination of 4,4′-dimethoxy-2–2′-bipyridine (L1) and (DME)NiBr2 catalyst, THF/CH3CN co-solvent, fluorine-doped tin oxide (FTO) coated glass anode and carbon fibre cathode to achieve the direct arylation of benzylic C–H bonds.313 The reaction tolerates a variety of functional groups including ketones, esters, sulfones, and amides. The reaction is initiated by the oxidation of a toluene derivative 116–1 at the anode to form the benzylic radical 116–7 (Fig. 116). Meanwhile, catalytic precursor LNiIBr (116–10) undergoes cathodic reduction and oxidative addition with ArBr 116–2 to generate NiII species 116–8. This intermediate is then intercepted by radical 116–7 to form NiIII complex 116–9. The subsequent reductive elimination gives the desired product 116–3.
Fig. 116.
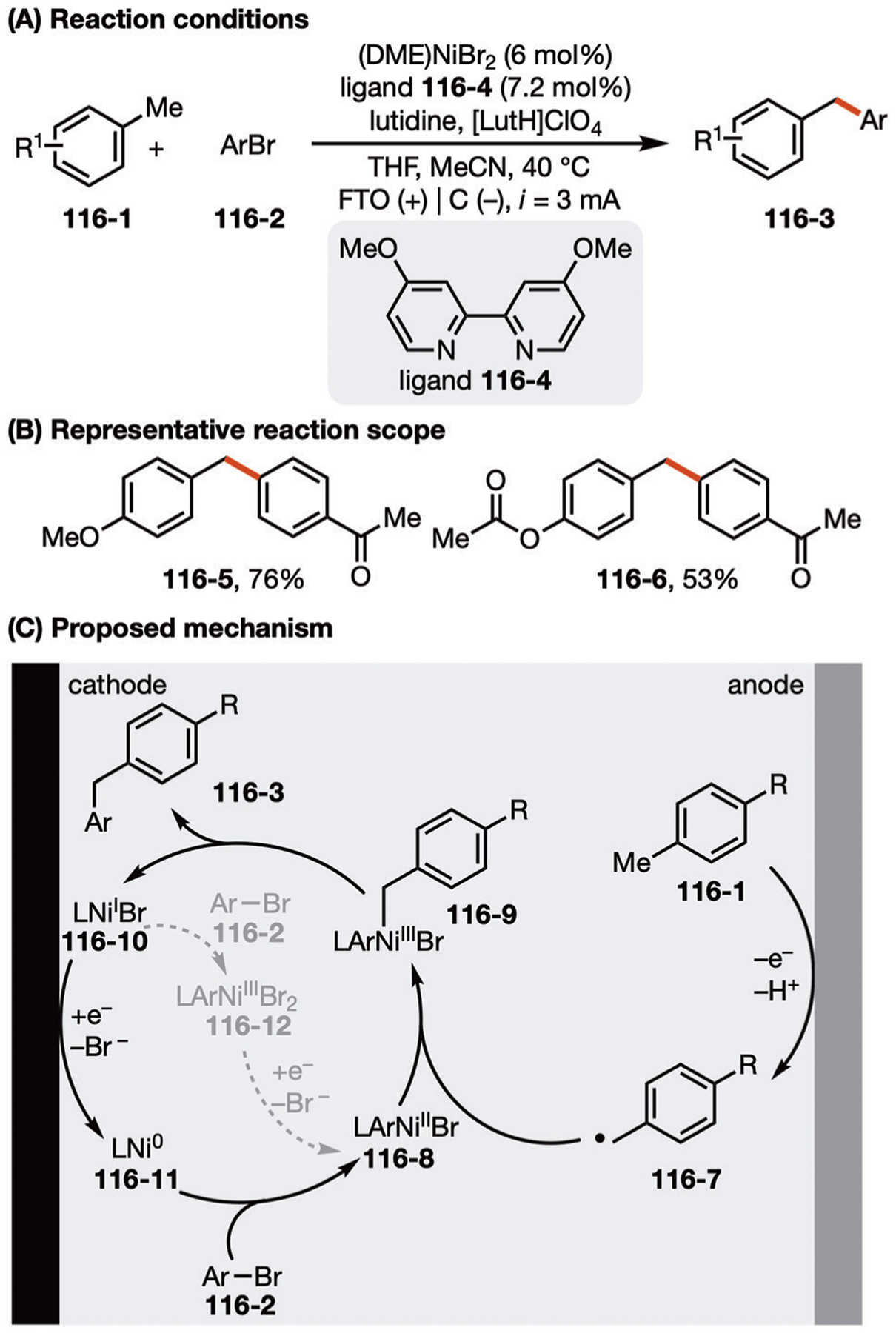
(A–C) Electrochemical nickel-catalyzed direct arylation of benzylic C–H bonds.
In the same year, Buchwald, Jensen, and co-workers designed a microfluidic redox-neutral electrochemical cell and developed a series of reactions including α-amino C–H arylation, deboronative arylation, allylic C–H arylation, Minisci-type radical addition to heteroarenes, and Ni-catalyzed C–O coupling (Fig. 117).314 The authors strategically implemented a flow cell with very narrow and variable interelectrode distance, which allowed them to overcome the intrinsic challenge of mass transport of reactive intermediates in convergent paired electrolysis.
Fig. 117.
Ni-Catalyzed C–O cross coupling via microfluidic cell.
In 2021, Liu and co-workers reported the use of paired electrolysis to achieve redox-neutral cross coupling reaction via nickel catalysis (Fig. 118).315 The combination of anodic oxidation of potassium benzyl trifluoroborate and cathodic reduction of organonickel intermediates enables this transformation to proceed with a 93% yield for the model substrates. Control experiments also suggest that electricity is essential to the reactivity despite the redox-neutral nature of the transformation. The proposed mechanism differs from Hu’s system in terms of the activation of Ni catalyst by the cathode (Fig. 118). The reaction is initiated by cathodic reduction of NiII precursor to NiI complex 118–9, which is followed by oxidative addition of 118–9 to 118–2 to form NiIII complex 118–10. This intermediate undergoes a second cathodic reduction to form 118–7. At this point, anodically generated alkyl radical 118–6 adds to 118–7 followed by reductive elimination to furnish desired product 118–4 and turn over the NiI catalyst 118–9. This transformation is also compatible with esters, nitriles, aldehydes, alkenes, amides, and N-heteroarenes.
Fig. 118.
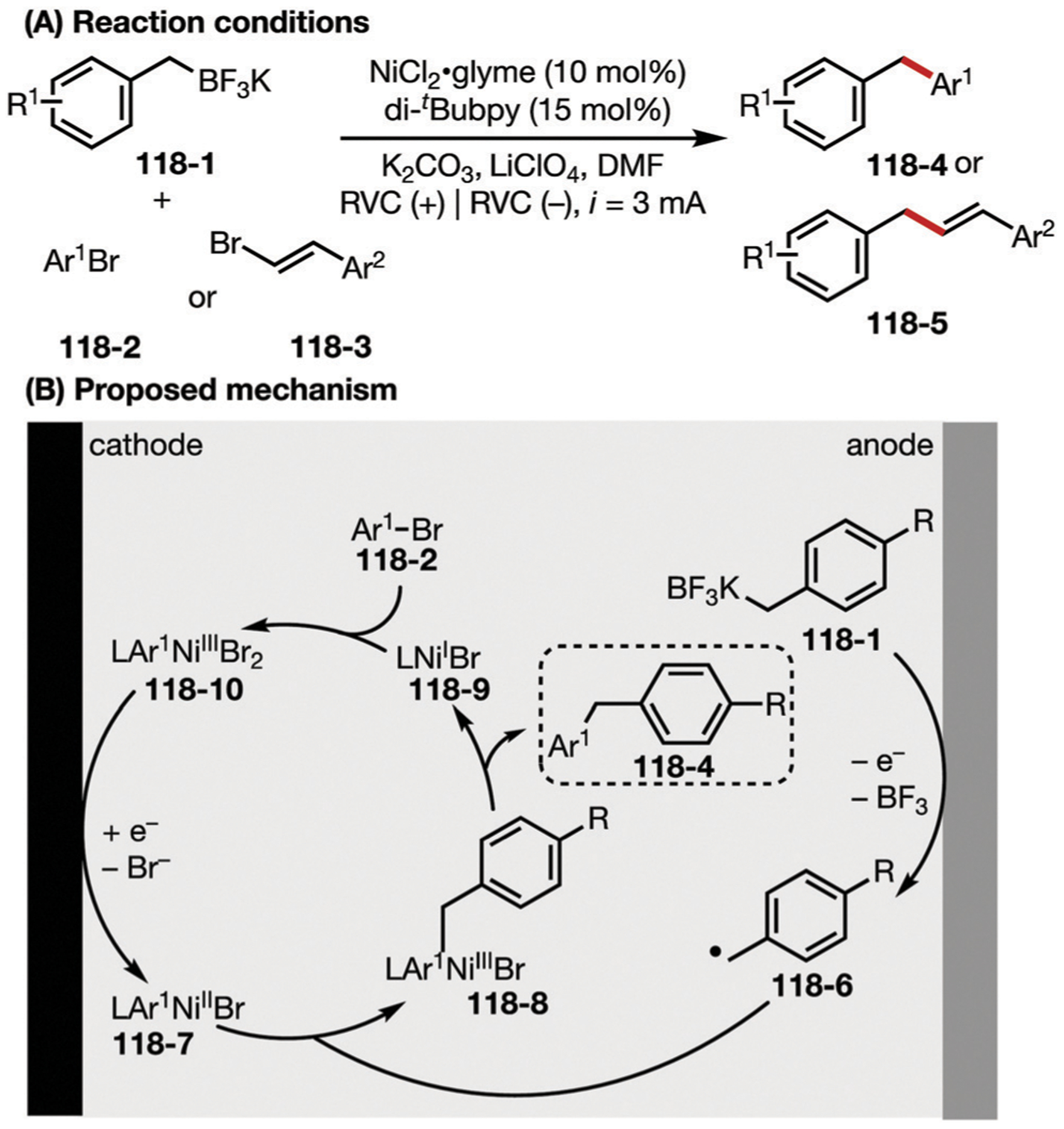
(A and B) Nickel-catalyzed electrochemical cross-coupling reactions of benzyl trifluoroborate and organic halides.
In 2021, the Jiao group reported nickel-catalyzed oxygenation of sulfides and phosphines using water as the oxygen source.316 The authors proposed that anodically generated O2 and cathodically generated Ni0 react to form NiII-peroxo species, which delivers an oxygen atom to the substrate such as sulfides or triaryl phosphines. Notably, this transformation tolerates several other oxidatively sensitive functional groups such as aldehydes and pyridines (Fig. 119).
Fig. 119.

(A and B) Electrochemically induced nickel catalysis for oxygenation reactions with water.
4.2. Other transformations
The Nonaka group demonstrated an early work on paired electrolytic synthesis of epoxides and dibromides in parallel (Fig. 120).317 In this system, a divided cell is used with a dimensionally stable anode (DSA) and graphite cathode. In the anodic chamber, bromide anions were oxidized to form bromine which then reacts with alkenes to form dibromides; in the cathodic chamber, oxygen gas undergoes reduction to peroxides which then oxidize vanadyl(IV) acetylacetonate (120–1) to vanadyl(V) (120–2), which catalyzes alkene epoxidation. Several electron deficient olefins were demonstrated to be suitable substrates and the current efficiency of the reaction could reach 132%.
Fig. 120.

Paired electrolysis of aldehyde condensation and carbon dioxide reduction.
In the recent years, the Moeller group also employed a divided cell design to pair anodic cerium-catalyzed condensation reaction and cathodic rhenium-catalyzed carbon dioxide reduction (Fig. 121).318 Synthetically useful benzimidazole (121–2) and carbon monoxide were generated in the anodic and cathodic chambers respectively with high faradaic efficiency and in good yields.
Fig. 121.

Paired electrolysis of epoxide and dibromide synthesis.
Berlinguette and co-workers paired alcohol oxidation and Pd-catalyzed alkyne hydrogenation in a three-compartment cell system (Fig. 122).319 Notably, the use of a Pd foam electrode enabled the two electrochemical process to be performed under two distinct conditions (aqueous and organic solvents) and high selectivity was achieved for both transformations. This protocol has also been used in the deuteration of unsaturated bonds.320
Fig. 122.

Paired electrolysis of alcohol oxidation with Pd-mediated hydrogenation of alkynes.
In 2018, Xu and co-workers reported a TEMPO-catalyzed electrochemical C–N bond formation to synthesize polycyclic N-heteroarenes (Fig. 123).321 In the first step, TEMPO was oxidized anodically to give TEMPO+ which reacts with the oxime substrate (123–1) to afford iminoxyl radical intermediate (123–4). Upon radical cyclization to 123–5 and aromatization by one electron oxidation with TMEPO+, N-oxide intermediate (123–2) was attained. Interestingly, the selectivity can be controlled by using different counter electrodes. When Pt is used as the cathode, hydrogenation become predominant and 123–2 is the final product. By introducing Pb cathode, which is known to suppress hydrogenation, cathodic cleavage of N–O bond is favored and give N-heteroarenes (123–3) as desired product.
Fig. 123.
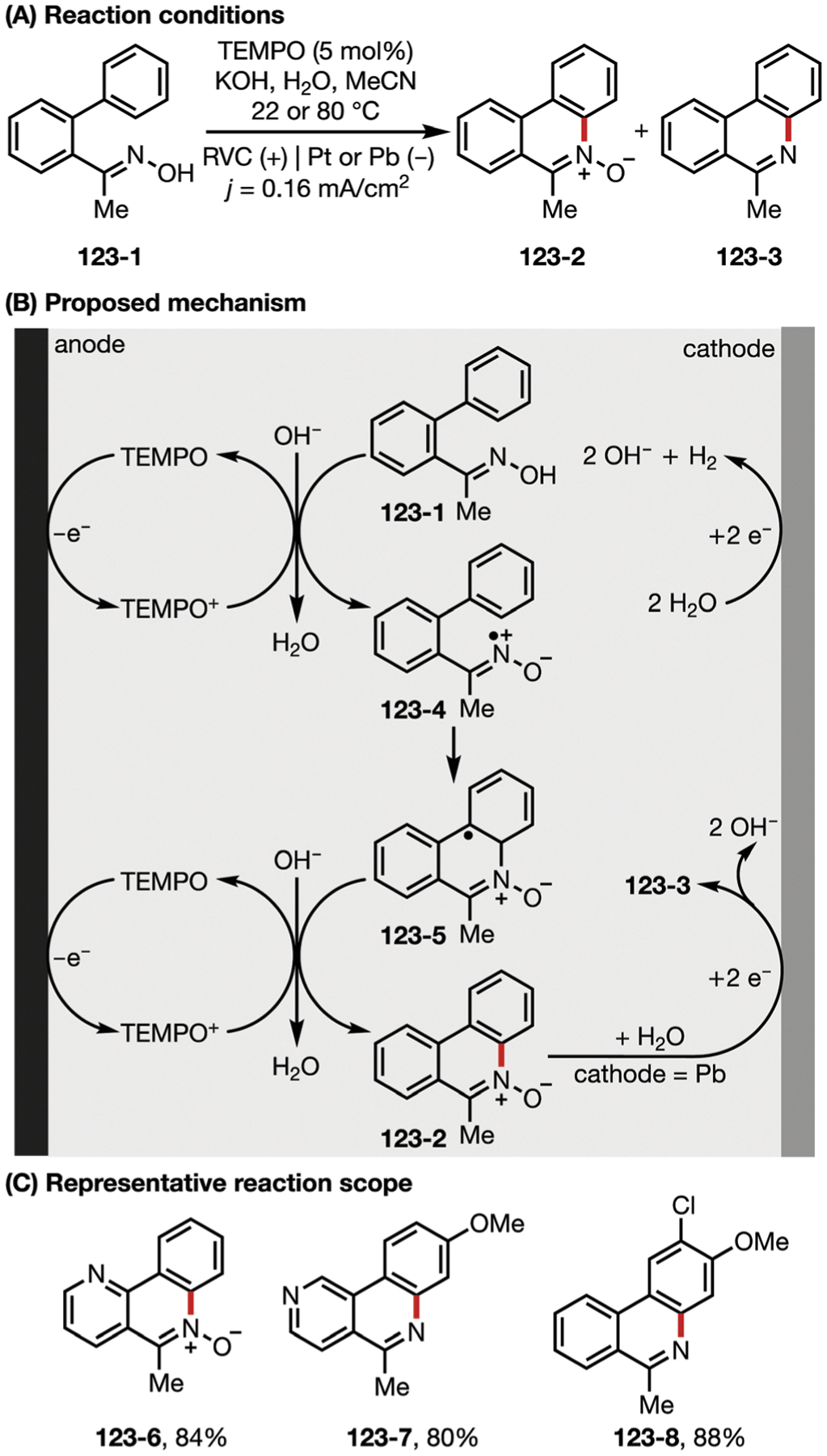
(A–C) TEMPO-catalyzed aryl C-N bond construction.
5. Heterogeneous electrocatalysis
Electrodes are capable of intimately interacting with substrates during electron transfer events and can even exhibit catalytic properties themselves. Since the surface of an electrode does not fall under the same classification as the small-molecule mediators discussed in the previous sections, the role of the electrode in facilitating a specific transformation can be challenging to determine. When an electrode is used as a heterogenous catalyst, the catalytic nature of the electrode encompasses interactions of substrates and intermediates with the surface of electrode (adsorption and desorption), such as electrostatic forces, π-interactions, and chemical bonds.322
Such catalytic properties of electrodes have been considerably less explored in organic synthesis. In 2020, the editorial team of ACS Catalysis defined several features which define electrode as an electrocatalyst, such as improved TOF or product selectivity relative to alternative electrodes.10 Some of the examples discussed in this section compare the different properties of carbon and platinum electrodes, while other examples highlight the use of electrodes modified with nanoparticles. This section does not intend to provide an exhaustive summary of developments in heterogenous electrocatalysis, but rather aims to bring attention of the community to this emerging area in the context of synthetic organic chemistry.
5.1. Carbon versus platinum electrodes
The differences between carbon and platinum anodes have been explored for oxidation of carboxylates (Fig. 124).323,324 The initial step in this transformation is a single electron oxidation followed by decarboxylation. When platinum was used as anode, the resulting radical can desorb from the surface of the electrode and engage in a dimerization reaction. However, when carbon was used as anode, the intermediate radical is more strongly adsorbed, putatively through interaction with paramagnetic centers in the electrode, and a second electron can be removed generating a carbocation, which desorbs and reacts with nucleophiles present in the reaction.
Fig. 124.

Electrochemical decarboxylation of phenylacetic acid with different anode materials.
These characteristics were employed by Moeller and co-workers during an anodic intramolecular cyclization of an enol-ether with an allyl-silane (Fig. 125).325 When platinum was used as anode, a small amount of a vinyl-silane was identified, which indicated the intermediacy of a β-silyl radical. Changing the anode to RVC (reticulated vitreous carbon) allowed for this radical intermediate to be oxidized to a carbocation before desorption from the electrode surface, improving the yield of the desired product.
Fig. 125.

Electrochemical cyclization with different anode materials.
In 2021, Waldvogel and co-workers discussed how different carbon-based anodes can provide surprisingly disparate outcomes for the same reaction.326 This arises from differences in morphology, the sp2/sp3 ratio of carbon, and manufacturing process for these materials. A remarkable recent example highlights the efficacy of different carbon anodes to promote radical or cationic mechanisms.327 Han and co-workers demonstrated that upon anodic oxidation of hydroxamic acids, the resultant oxygen-centered radical can add to a styrene double bond, resulting in the formation of a benzylic radical (Fig. 126). If RVC is used as anode, this benzylic radical is oxidized to a carbocation, which engages in a Friedel–Crafts reaction. However, if carbon felt is used as anode the benzylic radical supposedly can desorb from the surface of the electrode and react with an N-oxyl radical, generating a bis-oxygenated product.
Fig. 126.

(A–C) Electrochemical difunctionalization of alkenes with hydroxamic acid using different anode materials.
5.2. Modified electrodes
In 2019, Manthiram and co-workers explored the use of a carbon electrode coated with manganese oxide nanoparticles to promote epoxidation of alkenes (Fig. 127).328 The authors propose the presence of a MnIV–oxo species as the epoxidation promoter. After the oxygen transfer step, the resulting MnII species would be converted back to MnIV after two anodic oxidation events. Remarkably, the oxygen transferred to the epoxide comes only from water, which is also responsible for providing protons for hydrogen evolution reaction occurring at the cathode.
Fig. 127.
Electrochemical epoxidation of alkenes with nanoparticles coated anode.
On the reductive side of modified catalytic electrodes, Sun, Williard, Yu, and co-workers reported a cross-electrophile coupling promoted by a cathode coated with copper and palladium nanoparticles (CuPd NP; Fig. 128).329 The systems containing these nanoparticles proved to be more selective than a Pd foil cathode or purely Pd nanoparticles. On the other hand, nanoparticles with a large excess of copper proved ineffective for the target transformation, highlighting the importance of the Pd/Cu ratio on the surface of electrode. The optimal CuPd NP could be recycled up to 5 times without loss in yield, and the morphology of the catalyst did not change. Although the mechanism of this transformation is not fully elucidated at this time, the authors propose an initial reduction of alkyl iodide, which is partially supported by the observation of dimer formation.
Fig. 128.

(A and B) Electrochemical cross-electrophile coupling with nanoparticles coated cathode.
The possibility of recycling the catalyst with a simple washing procedure is an outstanding property of heterogeneous catalysts. The mechanism understanding usually requires investigation of the catalyst surface, employing a range of techniques, such as transmission electron microscopy (TEM), X-ray powder diffraction (XRD) and X-ray photoelectron spectroscopy (XPS). The field will certainly benefit from the more frequently collaborative interactions between materials scientists and organic chemists, and we foresee the development of new heterogeneous electrocatalysts for the construction of functional organic molecules as a growing area of research.
6. Bioelectrochemistry
Bioelectrocatalysis is an interdisciplinary field that aims to combine the merits of both biocatalysis and electrocatalysis for chemical synthesis. Bioelectrocatalytic methods utilize redox-active enzymes or electroactive microbial cells as catalysts to enable redox reactions. The advantages of bioelectrocatalysis include high activity, high selectivity, high atom efficiency, and mild reaction conditions. The past few years have seen an increasing number of applications of bioelectrocatalysis in the production of clean biofuels, degradable materials, and value-added chemicals.330a
Common bioelectrocatalysts include isolated oxidoreductases and electroactive microbial cells. Isolated oxidoreductases are frequently used in biosynthesis, and rapid advancements in protein engineering strategies such as directed evolution have allowed chemists to further optimize the reactivity of enzymes towards the desired organic transformations. Living electroactive microbials can also perform electron transfer and are used in bioelectrochemical systems. For microbial cell-based biocatalysis, diverse metabolic pathways enable microbial cells to catalyze many chemical reactions and produce a variety of products.
There are two general operating mechanisms of electron transfer between electrodes and bioelectrocatalysts: direct electron transfer (DET) and mediated electron transfer (MET). The following discussion of bioelectrocatalysis will focus on the applications of bioelectrocatalysis in the synthesis of fine chemicals and chiral organic products. More extensive reviews of bioelectrocatalysis are published recently.330
6.1. Direct electron transfer
DET processes involve the direct transfer of electrons from the electrode to the enzyme active site.330a For most oxidoreductases, these active sites are buried deep within the protein structures, thereby leading to slow electron transfer rate.331 Additional challenges arise due to the difficulties associates with maintaining stability of the enzymes when they are within close proximity to the electrode surface. In light of these considerations, research efforts have focused on developing docking motifs to modify enzymes to promote efficient electron transfer. Some examples of docking motifs include nanoparticles that act as electron relay agents,332 His-tag on the C or N-terminus of the enzyme to increase its binding affinity to the modified electrode surface,333 and pyrene-modified linear poly(ethylenimine) (LPEI) that can immobilize oxidoreductases by cross-linking the enzyme and the multiwalled carbon nanotubes at the carbon electrode.334 For example, in 2003, Willner and co-workers developed a strategy to enhance DET by reconstitution of apo-glucose oxidase with gold nanocrystals functionalized with a FAD coenzyme (Fig. 129).332 The bioelectrocatalysis rate of the resulting “artificial” protein was enhanced significantly, as the gold nanoparticles served to efficiently wire the enzyme redox centers to the electrode.
Fig. 129.

Nanowiring of redox enzymes by a gold nanoparticle. a = 1 or 2, b = 0 or 1. FAD = flavin adenine dinucleotide, GOx = glucose oxidase. The author did not specify the role of the glassy carbon cathode.
6.2. Mediated electron transfer
6.2.1. Mediated electron transfer of free enzymes.
Mediated electron transfer processes make use of small, diffusive redox mediators or redox polymers to serve as electron shuttles between the enzyme active site and the electrode surface. Some commonly used redox mediators include viologens, quinones, dyes, tetrathiafulvalene, and metal complexes such as ferrocenes.330a These mediators have been coupled with a variety of oxidoreductases to synthesize fine chemicals.
Electricity can be utilized to regenerate expensive cofactor NADH in enzyme-catalyzed reactions. In 2019, the Minteer group developed a bioelectrocatalytic N2 fixation system using methyl viologen as the redox mediator (Fig. 130).335 The N2 fixation step is catalyzed by nitrogenase, and the resultant ammonia is further transformed into alanine (130–4) by l-alanine dehydrogenase with the consumption of oxalic acid (130–3) and NADH. Methyl viologen also facilitates the regeneration of cofactor NADH catalyzed by diaphorase. The alanine intermediate is utilized by ω-transaminase as amino-group donor to catalyze the amination of ketone substrate 130–1 to produce chiral amines 130–2 with up to 99% ee.
Fig. 130.

(A and B) Bioelectrocatalytic N2 fixation: from N2 to chiral amine intermediates. The platinum anode generates protons and O2 from water.
Neutral red is another common mediator and has been applied in the reduction of CO2 to formic acid.336 This reduction was catalyzed by formate dehydrogenase (FDH) which was immobilized on a graphite-based cathode.
Redox polymers can also be used to facilitate efficient electron transfer. In 2019, the Minteer group demonstrated that diaphorase immobilized in the three-dimensional network of cobaltocene-modified poly(allylamine) redox polymers (DH/Cc-PAA) can efficiently regenerate NADH, which is used by alcohol dehydrognenase to reduce aldehydes into alcohols (Fig. 131).337 The polymer immobilization strategy allows the enzyme and redox mediators to be retained on the electrode surface after a reaction is completed, enabling these catalysts to be recycled and reused. The high efficiency, catalytic stability, and recyclability (91% of the maximum activity after five experimental cycles) renders this system among the most promising means of NADH regeneration developed heretofore.
Fig. 131.

NADH regeneration by a redox polymer-immobilized enzymatic system. DH = diaphorase, ADH = alcohol dehydrogenase. The platinum-wire anode generates O2 from water.
On the basis of this pioneering work, in 2020, the Minteer group reported a biphasic bioelectrocatalytic system to synthesize chiral β-hydroxy nitriles from ethyl 4-chloroacetate using the DH/Cc-PAA bioelectrode (Fig. 132).338 Powered by electrogenerated NADH, ADH catalyzed the asymmetric reduction of 132–1 to generate ethyl (S)-4-chloro-3-hydroxy-butanoate 132–2 in high enantioselectivity. Next, 132–2 was converted to 132–3 through nucleophilic substitution catalyzed by HHDH. The biphasic system ensures that the ester substrate, intermediate and product pre-dominantly reside in the organic phase, thus suppressing their hydrolysis in the aqueous phase. At the same time, the biphasic system minimizes the inhibition of HHDH activity by the substrate 132–1, enabling the two-step enzymatic synthesis to proceed smoothly.
Fig. 132.

Biphasic bioelectrocatalytic synthesis of chiral β-hydroxy nitriles. DH = diaphorase, ADH = alcohol dehydrogenase HHDH = halohydrin dehalogenase. A platinum mesh serves as the anode. The authors do not specify the anodic reaction.
With advancements in protein engineering technologies, the catalytic properties of oxidoreductases can be tuned and enhanced to meet the requirements of synthetic transformations. Directed evolution, for example, has been used to modify the redox potentials of certain enzymes to increase the power output of biofuel cells.339
6.2.2. Mediated electron transfer of electroactive microbial cells.
Electroactive microbial cells can also act as bioelectrocatalysts. However, because the cell membrane layers of microbes are typically not conductive, extracellular electron transfer rates are slow. Recent research efforts have been directed towards overcoming this challenge using membrane-transmissible mediators.340 Another strategy to overcome this challenge involves the combination of electrochemical technology with metabolic engineering of cells. A creative strategy to construct an engineered strain is the introduction of an electron pathway to establish electrochemical communication between the bacterial cells and electrode. Sturm-Richter and co-workers developed a protocol to heterologously express c-type cytochromes CymA, MtrA and STC from S. oneidensis in an E. coli cell to construct an electron transport chain (Fig. 133).341 The presence of an electron transport chain accelerates the electron transfer efficiency by 183%. Based on this work, Mayr and co-workers integrated an NADPH-dependent alcohol dehydrogenase from L. brevis into this engineered E. coli platform to perform the asymmetric reduction of acetophenone to (R)-1-phenylethanol 133–2.342 Methyl viologen acts as the redox mediator that facilitates the electron transfer in the periplasm.
Fig. 133.

Resting E. coli as a chassis for microbial electrosynthesis: production of chiral alcohols. MtrA, STC and CymA: proteins of the electron transfer pathway in Shewanella oneidensis. MR-1, LbADH: An alcohol dehydrogenase from Lactobacillus brevis. The graphite anode generates O2 from water.
7. Electrophotocatalysis
Electrophotochemistry is a strategy that combines electrochemistry’s ability to generate reactive intermediates with photochemistry’s ability to access excited state intermediates. The synergistic combination has been applied to creatively solve challenging problems in organic synthesis. In 1970, Moutet and Reverdy first proposed electrophotochemistry as a concept in the context of photoexcitation of electrochemically generated phenothiazine radical cations.343 In a similar pioneering work in 1985, Rusling demonstrated that the reduction of 4-chlorobiphenyl could be promoted by a super reductant in the form of photoexcited, electrochemically generated anthracene radical anion.344 In the following section, we discuss recent developments in electrophotocatalysis wherein redox-active catalytic species participate in both electrochemical and photochemical activation events to achieve desired transformations. These examples are grouped into two major categories depending on whether the electrochemical steps are coupled to or decoupled from photochemical steps. For additional information outside of what is discussed in this section, we refer the reader to recent reviews or primary research articles on electrophotocatalysis.345,346
7.1. Replacing external oxidant or reductant
Electrophotocatalysis is an excellent strategy for accessing highly reactive intermediates in a selective and controlled fashion. A typical electrophotocatalytic process entails photochemical and electrochemical steps that function in tandem to activate a single catalytic species, thus giving rise to a highly reactive photoexcited catalyst or radical species without using a terminal oxidant or reductant.
In 2019, Lin et al. employed electophotocatalysis for the oxidation of alcohols using riboflavin tetraacetate (RFT) and thiourea catalysts (Fig. 134).347 In the catalytic cycle, the RFT catalyst is first photoexcited to RFT* and subsequently reacts with thiourea 134–3 to generate semiquinone form RFT•-H and thiyl radical 134–4. This thiyl radical then undergoes hydrogen atom transfer (HAT) with the alcohol substrate (134–1) followed by reaction of resultant intermediate 134–5 with RFT•-H to complete both catalytic cycles and yield the carbonyl product (134–2). Finally, RFT-H2 is turned over at the anode, completing the catalytic cycle. Notably, the need for a sacrificial oxidant (i.e., O2) is replaced by anodic oxidation, which avoids the oxidant-induced degradation of thiourea cocatalyst and lead to critical expansion of the scope of flavin-catalyzed alcohol oxidation to include challenging unactivated aliphatic alcohols.
Fig. 134.

(A–C) Electrophotochemical oxidation of alcohols mediated by riboflavin tetraacetate.
The generation of alkyl radicals from alkanes is challenging due to the high BDE of unactivated C(sp3)–H bonds (98.5 kcal mol−1 for methylene groups).96 In 2020, Xu and co-workers addressed this challenge via the electrophotochemical generation of chlorine radicals,348 which can abstract unactivated hydrogen atoms and generate HCl (BDE = 102 kcal mol−1). The resultant alkyl radicals then engage in a Minisci-type reaction to construct C2- and C4-alkylated quinolines and pyridines (Fig. 135). This method also used chlorine radicals to perform HAT of activated C–H bonds at benzylic positions and positions α to heteroatoms. In separate works, the Xu group has also explored Minisci-type reactivity through electrophotochemically generated radicals from carboxylic acids349 and potassium trifluoroborate salts.350
Fig. 135.

(A–C) Electrophotochemical alkylation of heteroarenes mediated by chloride.
In 2021, Ackermann group used an acridinium-based photocatalyst Mes-Acr+ to achieve the electrophotocatalytic C–H trifluoromethylation of arenes (Fig. 136).351 Anodic oxidation and subsequent irradiation of the Mes-Acr+ generates its oxidized excited state Mes-Acr+*. Single electron transfer between Mes-Acr+* and the sulfinate anion generates acridinyl radical and CF3•, the latter of which is trapped by the arene substrate, leading to final product 136–2. The reduced form of photocatalyst Mes-Acr• is turned over anodically without the use of a chemical oxidant.
Fig. 136.

(A and B) Electrophotochemical trifluoromethylation of arenes.
In 2021, Ravelli group adopted an electrophotocatalytic approach to achieve dehydrogenative functionalization of benzothiazoles with simple alkanes.352 Electrochemistry is utilized to turnover photocatalyst tetra-n-butylammonium decatungstate (TBADT), which undergoes photo-induced HAT with simple alkanes (e.g., 137–2) to generate alkyl radical intermediates (TBADT is designated as W in Fig. 137).
Fig. 137.
Electrophotochemical alkylation of benzothiazole.
7.2. Accessing extremely oxidizing or reducing potentials
Another prominent application of electrophotocatalysis is the generation of super oxidants and reductants by means of tandem electro- and photochemical activation.
In 2019, Lambert and co-workers employed a trisaminocyclopropenium cation (TAC+) as an electrophotocatalyst to oxidize benzene toward C–H amination with azoles (Fig. 138).353 In this work, TAC+ is first oxidized on the electrode to the radical dication TAC2+. This oxidation is then followed by visible light photoexcitation to generate the highly oxidizing species TAC2+* (E ≈ 3.3 V). TAC2+* is capable of oxidizing simple arenes to their corresponding radical cations, which are then trapped by an azole to deliver the coupled product. In 2020, this strategy was also applied to the TAC+-catalyzed oxidative C–H functionalization of ethers and DDQ-catalyzed nucleophilic aromatic substitution.354,355
Fig. 138.
(A–C) Electrophotochemical coupling of arenes with azoles mediated by trisaminocyclopropenium cation.
In 2021, Lambert’s group reported the diamination of alkanes using the same electrophotocatalytic strategy (Fig. 139).356 Alkane substrates with a benzylic C–H bond (139–1) can be converted into 3,4-dihydroimidazoles (139–2), which can then be transformed to valuable vicinal diamines in a subsequent chemical reaction without purification of the intermediates. The author proposed that a key step involves the oxidation of substrates by the highly oxidizing radical dication TAC2+.
Fig. 139.
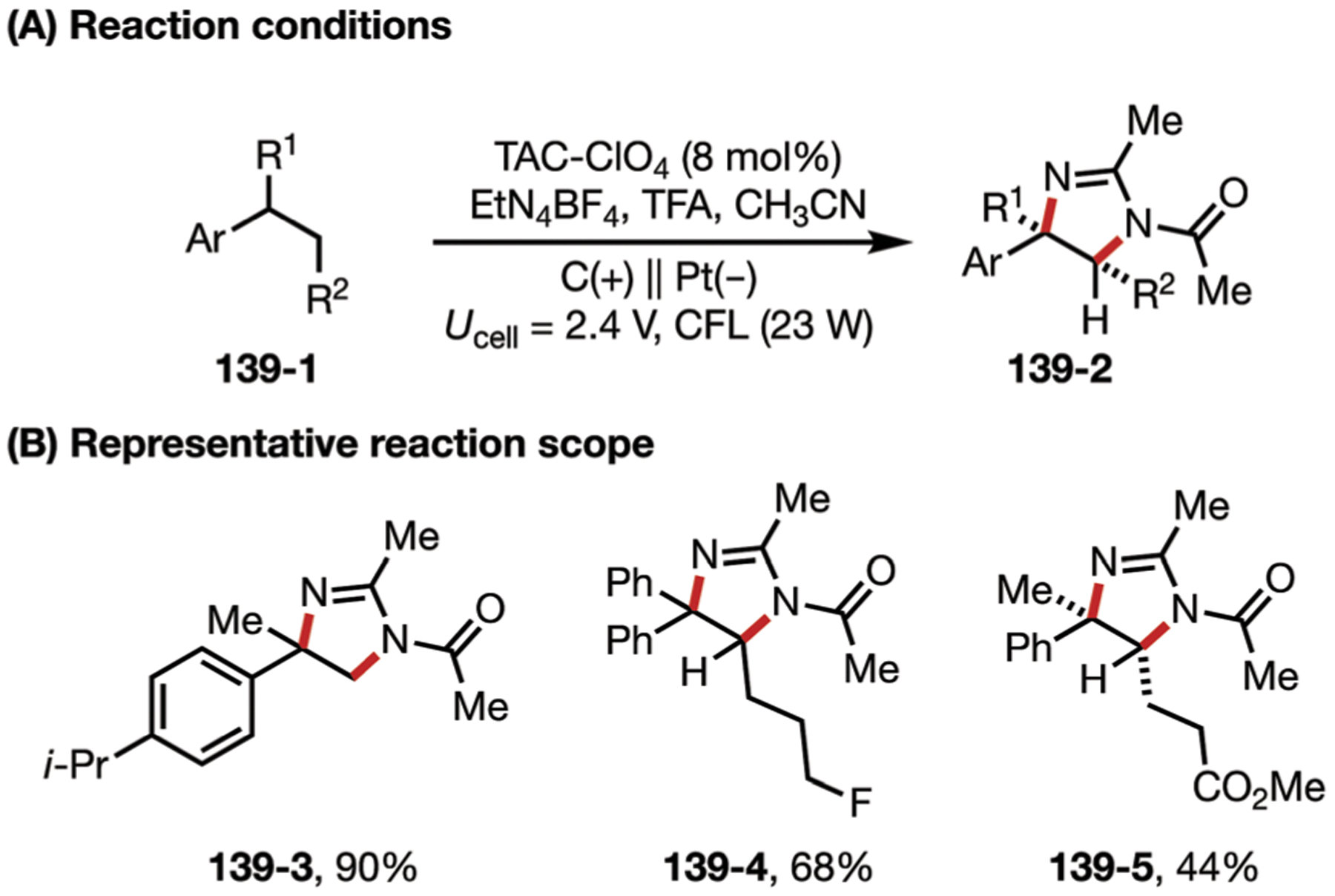
Electrophotocatalytic deamination of vicinal C–H bonds.
In the same year, Barham and co-workers demonstrated that electrophotocatalyst (trisbiaryl)aminium radical cation (e.g., TdCBPA radical cation) can oxidatively activate C–H bonds of arenes (Fig. 140).357 The authors proposed that the pre-complexation between the catalyst and the arene substrate via π-stacking is key to achieving this challenging transformation.
Fig. 140.

Electrophotocatalytic azolation of arenes. TdCBPA = tris(2′,4;-dicyano-[1,1′-biphenyl]-4-yl)amine.
In 2020, Lambert and Lin extended this strategy to access highly reducing electrophotocatalysts.358 Specifically, dicyanoanthracene (DCA) was used as an electrophotocatalyst to achieve the functionalization of aryl halides (Fig. 141). DCA is first reduced on the cathode prior to photoexcitation to furnish a highly reducing excited-state radical anion 141–5 (Ered ≈ −3.2 V vs. SCE) with a sufficient lifetime to undergo subsequent electron transfer (~13.5 ns).359 This species is potent enough to reduce electron-rich aromatic substrates such as 1-chloro-4-methoxybenzene (Ered = −2.9 V vs. SCE)360 to the corresponding radical anion, which further proceeds to deliver the product 141–9 in good yields.
Fig. 141.

(A–C) Electrophotochemical reduction of aryl halides mediated by dicyanoanthracene.
Concurrently, Wickens reported a similar electrophotocatalytic strategy employing naphthalene-based imide (NpMI) to promote reduction of aryl halides (Fig. 142).361 The photoexcited radical anion of NpMI was estimated to have a reducing potential similar to Na (−2.9 V vs. SCE) and Li (−3.3 V vs. SCE). Cathodic reduction of the substrate and subsequent fragmentation of the incipient radical anion generates an aryl radical, which can undergo phosphorylation and heteroarylation upon radical trapping.
Fig. 142.

(A and B) Electrophotochemical reduction of aryl halides mediated by naphthalene-based imide.
These examples showcase the highly biased redox potentials enabled by electrophotocatalysis. Recent developments in this area continue to illustrate the capacity of electrophotocatalysis to enable organic transformations at these extreme potentials, thereby expanding the toolbox of synthetic chemists.
7.3. Decoupled electrophotocatalysis
Unlike typical electrophotocatalytic systems described in this section, electrochemistry and photoexcitation can also be simultaneously harnessed to activate two discrete reactive species in nonconsecutive steps of the same reaction pathway toward the desired transformation. This type of systems is referred as decoupled electrophotocatalysis.362
In 2019, Stahl and co-workers demonstrated the use of anodically generated iodine for the intramolecular C–H amination (Fig. 143).363 The proposed mechanism involves the generation of a nitrogen-centered radical from visible light promoted homolytic cleavage of a weak N–I bond in key intermediate 143–3, which is formed from substrate 143–1 and electrogenerated I2. A 1,5-hydrogen atom transfer process leads to the activation of an inert C–H bond in a regioselective fashion, which is followed by capture of the resultant C-centered radical 143–5 with I2 to form a C–I bond (143–6). Subsequently, an intramolecular SN2 reaction constructs a C–N bond to furnish cyclized N-tosyl amine 143–2. This overall sequence was employed for the preparation of substituted pyrrolidines, oxazolines, and vicinal amino alcohols.
Fig. 143.
(A–C) Electrophotochemical synthesis of pyrrolidines mediated by iodide. CFL = compact fluorescent lamp.
8. Concluding remarks and future directions
The past ten years have brought significant growth and attention towards applications of electrocatalysis in organic synthesis. Such advancements have been prompted by increased recognition of electrochemistry’s ability to harness electrical current as an “imaginary” reagent to facilitate a range of single-electron oxidation and reduction events. The developments highlighted in this review demonstrate how this ability is further augmented by the introduction of electrocatalysts which serve to increase the selectivity and efficiency of electrochemically powered methodologies. Electrocatalysis has proven to be a viable pathway for accessing highly reactive radical intermediates from abundant starting materials, as well as for regulating their downstream reactivity to enable complex transformations in a single step. To date, electrocatalysis has expanded the scope of many staple reactivities such as C–C and C–X bond formation, alkene functionalization, and C–H activation, often under mild conditions.
Even with these advancements, we expect that electrocatalysis will continue to expand into new chemical spaces to address pressing challenges of efficiency and sustainability encountered in synthesis. From the perspective of this review, we see several emerging research directions amidst the renaissance of synthetic electrocatalysis, listed as follows:
Application of electrocatalytic methods for the late-stage modification of complex bioactive molecules and bioorthogonal functionalization of biomacromolecules.
Exploration of electroreductive strategies which allow access to deeply reducing potentials needed for challenging reductive transformations.
Development of paired electrolysis systems enabling highly efficient transformations through simultaneous and synergistic use of anodic and cathodic redox events.
Expansion of electrocatalysis into interdisciplinary areas such as electrophotocatalysis and bioelectrocatalysis to leverage the unique abilities of these systems.
Application of heterogenous electrocatalysis and employment of modern characterization techniques to understand and rationally design catalytic electrodes.
Altogether, electrocatalysis has developed into a powerful enabling technology in the field of organic synthesis. The diversity of new synthetic transformations enabled by electrochemistry can be attributed to the myriad optimization parameters of electrochemical systems (applied potential, current, electrode material, electrolyte, cell type, etc.), all of which can be used to intimately and selectively influence chemical reactivity. We anticipate that future developments in the field of electrocatalysis will power the discovery of new reaction strategies needed to solve key challenges in contemporary organic synthesis.
Acknowledgements
Financial support was provided by NIGMS (R01GM130928). S. L. thank the Research Corporation of Science Advancements for a Cottrell Scholar Award. L. F. T. N. thank The Brazilian National Council for Scientific and Technological Development for a CNPq Postdoctoral Fellowship.
Biographies

Luiz F. T. Novaes
Luiz Novaes received his BS (2013) and MS (2015) degrees in Chemistry from University of Campinas under the supervision of Prof. Ronaldo Pilli, working on total synthesis of natural poly-ketides. During his PhD studies (2015–2019), Luiz worked on total synthesis of the natural meroterpene actinoranone and the sesquiterpene pleocarpanone under the guidance of Prof. Julio Pastre (University of Campinas). In September of 2019, he joined the Lin group at Cornell University as a postdoctoral scholar, and has focused on developing electrochemical methodologies to enable the late stage functionalization of complex molecules.

Jinjian Liu
Jinjian Liu received her BS (2017) degree in Chemistry from The College of William and Mary under the supervision of Prof. William. R. McNamara, working on transition-metal catalyzed hydrogenation reactions. She joined Prof. Song Lin’s research group at Cornell in 2018, since then, she has worked on electrophotochemical C–H functionalization of inert substrates. Her current research focuses on the development of electrochemical methodologies for enantioselective functionalization of complex molecules.

Yifan Shen
Yifan Shen is currently a doctoral candidate of Organic Chemistry at Cornell University, under the supervision of Prof. Song Lin. He received his BS degree in Chemistry from Shanghai Jiao Tong University in 2017, working on transition metal catalyzed oxidation reaction. His research interests include electrocatalysis and radical relay chemistry.

Lingxiang Lu
Lingxiang Lu received his BS degree (Hons) in Chemistry from Wuhan University under the supervision of Prof. Hexiang Deng and Fusheng Ke, working on metal–organic frameworks and lithium sulphur batteries. In 2017, he joined Prof. Song Lin’s research group at Cornell for PhD studies on electrosynthesis. He is currently working on electroreductive transformations to construct value-added compounds.

Jonathan M. Meinhardt
Jonathan Meinhardt is an undergraduate student at Cornell University working in the research group of Professor Song Lin. Since joining the Lin group in 2019, Jon has worked on projects which developed and utilized structure–property relationships in the contexts of asymmetric catalysis and the design of energetic materials. In March of 2021, he received a Barry M. Goldwater Scholarship based on his efforts in these research areas. His current research interests involve the development of new strategies for site-selective C–H activation.

Song Lin
Song Lin is an Assistant Professor of Chemistry and Howard Milstein Faculty Fellow at Cornell University. He obtained BS at Peking University in 2008 and PhD at Harvard University, working under the guidance of Prof. Eric Jacobsen. After postdoctoral training with Prof. Christopher Chang at UC Berkeley, he joined Cornell University as a faculty member in 2016. Prof. Lin has served on the Early Career Advisory Board of ACS Catalysis and Chemistry – A European Journal. The Lin Laboratory is interested in developing new catalytic strategies for organic reaction discovery with a particular emphasis on electrochemistry and radical catalysis.
Footnotes
Conflicts of interest
There are no conflicts to declare.
Notes and references
- 1.Lund H, J. Electrochem. Soc, 2002, 149, S21–S33. [Google Scholar]
- 2.Waldvogel SR and Janza B, Angew. Chem., Int. Ed, 2014, 53, 7122–7123. [DOI] [PubMed] [Google Scholar]
- 3.McClymont KS, Wang F-Y, Minakar A and Baran PS, J. Am. Chem. Soc, 2020, 142, 8608–8613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Evans DH, Chem. Rev, 2008, 108, 2113–2144. [DOI] [PubMed] [Google Scholar]
- 5.Moeller KD, Chem. Rev, 2018, 118, 4817–4833. [DOI] [PubMed] [Google Scholar]
- 6.Franckle R and Little RD, Chem. Soc. Rev, 2014, 43, 2492–2521. [DOI] [PubMed] [Google Scholar]
- 7.Lee K, Elgrishi N, Kandemir B and Dempsey JL., Nat. Rev. Chem, 2017, 1, 0039. [Google Scholar]
- 8.Jackson MN and Surendranath Y, Acc. Chem. Res, 2019, 52, 3432–3441. [DOI] [PubMed] [Google Scholar]
- 9.Saveant JM, Chem. Rev, 2008, 108, 2348–2378. [DOI] [PubMed] [Google Scholar]
- 10.Dey A, Brent Gunnoe T and Stamenkovic VR, ACS Catal, 2020, 10, 13156–13158. [Google Scholar]
- 11. We note that the IUPAC definition of “electrocatalysis” states that “[e]lectrocatalysis is an electrochemical reaction that takes advantage of a catalyst.” We interpret this as inclusive of both molecular and surface catalysts, which is consistent with the definition of electrocatalysis used in this review. See: https://iupac.org/materialschemistryedu/energy/photocatalysis/.
- 12.Kärkäs MD, Verho O, Johnston EV and Åkermark B, Chem. Rev, 2014, 114, 11863–12001. [DOI] [PubMed] [Google Scholar]
- 13.Shao M, Chang Q, Dodelet J-P and Chenitz R, Chem. Rev, 2016, 116, 3594–3657. [DOI] [PubMed] [Google Scholar]
- 14.Appel AM, Bercaw JE, Bocarsly AB, Dobbek H, Dubois DL, Dupuis M, Ferry JG, Fujita E, Hille R, Kenis PJA, Kerfeld CA, Morris RH, Peden CHF, Portis AR, Ragsdale SW, Rauchfuss TB, Reek JNH, Seefeldt LC, Thauer RK and Waldrop GL, Chem. Rev, 2013, 113, 6621–6658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Horn EJ, Rosen BR and Baran PS, ACS Cent. Sci, 2016, 2, 302–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Siu JC, Fu N and Lin S, Acc. Chem. Res, 2020, 53, 547–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hilt G, ChemElectroChem, 2020, 7, 395–405. [Google Scholar]
- 18.Schotten C, Nicholls TP, Bourne RA, Kapur N, Nguyen BN and Willans CE, Green Chem, 2020, 22, 3358–3375. [Google Scholar]
- 19.Kingston C, Palkowitz MD, Takahira Y, Vantourout JC, Peters BK, Kawamata Y and Baran PS, Acc. Chem. Res, 2020, 53, 72–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Steves JE and Stahl SS, J. Am. Chem. Soc, 2013, 135, 15742–15745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhao J, Nanjo T, de Lucca EC Jr and White MC, Nat. Chem, 2019, 11, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bryan MC, Dunn PJ, Entwistle D, Gallou F, Koenig SG, Hayler JD, Hickey MR, Hughes S, Kopach ME, Moine G, Richardson P, Roschangar F, Steven A and Weiberth FJ, Green Chem, 2018, 20, 5082–5103. [Google Scholar]
- 23.Medici A, Pedrini P, Battisti A, Fantin G, Fogagnolo M and Guerrini A, Steroids, 2001, 66, 63–69. [DOI] [PubMed] [Google Scholar]
- 24.Maki T, Iikawa S, Mogami G, Harasawa H, Matsumura Y and Onomura O, Chem. – Eur. J, 2009, 15, 5364–5370. [DOI] [PubMed] [Google Scholar]
- 25.Liang S, Zeng C-C, Luo X-G, Ren F-Z, Tian H-Y, Sun B-G and Little RD, Green Chem, 2016, 18, 2222–2230. [Google Scholar]
- 26.Doobary S, Sedikides AT, Caldora HP, Poole DL and Lennox AJJ, Angew. Chem., Int. Ed, 2020, 59, 1155–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vereshchagin AN, Elinson MN, Dorofeeva EO, Demchuk DV, Bushmarinov IS, Goloveshkin AS and Nikishin GI, Tetrahedron, 2013, 69, 5234–5241. [Google Scholar]
- 28.Vereshchagin AN, Elinson MN, Dorofeeva EO, Nasybullin RF, Bushmarinovb IS, Goloveshkinb AS and Egorov MP, Electrochim. Acta, 2015, 165, 116–121. [Google Scholar]
- 29.Okimoto M, Yamamori H, Ohashi K, Hoshi M and Yoshida T, Synlett, 2013, 1568–1572. [Google Scholar]
- 30.Elinson MN, Dorofeeva EO, Vereshchagin AN, Nasybullin RF and Egorov MP, Catal. Sci. Technol, 2015, 5, 2384–2387. [Google Scholar]
- 31.Chen L, Barton LM, Vantourout JC, Xu Y, Chu C, Johnson EC, Sabatini JJ and Baran PS, Org. Process Res. Dev, DOI: 10.1021/acs.oprd.0c00270. [DOI] [Google Scholar]
- 32.Okimoto M, Yamamori H, Ohashi K, Nishikawa S, Hoshi M and Yoshida T, Synlett, 2012, 2544–2548. [Google Scholar]
- 33.Minato D, Mizuta S, Kuriyama M, Matsumura Y and Onomura O, Tetrahedron, 2009, 65, 9742–9748. [Google Scholar]
- 34.Yao C, Wang Y, Li T, Yu C, Li L and Wang C, Tetrahedron, 2013, 69, 10593–10597. [Google Scholar]
- 35.Elinson MN, Dorofeev AS, Feducovich SK, Belyakov PA and Nikishin GI, Eur. J. Org. Chem, 2007, 3023–3027. [Google Scholar]
- 36.Gao H, Zha Z, Zhang Z, Ma H and Wang Z, Chem. Commun, 2014, 50, 5034–5036. [DOI] [PubMed] [Google Scholar]
- 37.Liang S, Zeng C-C, Tian H-Y, Sun B-G, Luo X-G and Ren F-Z, J. Org. Chem, 2016, 81, 11565–11573. [DOI] [PubMed] [Google Scholar]
- 38.Pan X-J, Gao J and Yuan G-Q, Tetrahedron, 2015, 71, 5525–5530. [Google Scholar]
- 39.Kang L-S, Luo M-H, Lam CM, Hu L-M, Little RD and Zeng C-C, Green Chem, 2016, 18, 3767–3774. [Google Scholar]
- 40.Zhang Z, Su J, Zha Z and Wang Z, Chem. – Eur. J, 2013, 19, 17711–17714. [DOI] [PubMed] [Google Scholar]
- 41.Zhang Z, Su J, Zha Z and Wang Z, Chem. Commun, 2013, 49, 8982–8984. [DOI] [PubMed] [Google Scholar]
- 42.Xu K, Zhang Z, Qian P, Zha Z and Wang Z, Chem. Commun, 2015, 51, 11108–11111. [DOI] [PubMed] [Google Scholar]
- 43.Qian P, Su J-H, Wang Y, Bi M, Zha Z and Wang Z, J. Org. Lett, 2017, 82, 6434–6440. [DOI] [PubMed] [Google Scholar]
- 44.Elinson MN, Feducovich SK, Dorofeev AS, Vereshchagin AN and Nikishin GI, Tetrahedron, 2000, 56, 9999–10003. [Google Scholar]
- 45.Elinson MN, Feducovich SK, Dmitriev DE, Dorofeev AS, Vereshchagin AN and Nikishin GI, Tetrahedron Lett, 2001, 42, 5557–5559. [Google Scholar]
- 46.Elinson MN, Dorofeev AS, Feducovich SK, Nasybullin RF, Litvin EF, Kopyshev Mi. V. and Nikishin GI, Tetrahedron, 2006, 62, 8021–8028. [Google Scholar]
- 47.Gao W-J, Lam CM, Sun B-G, Little RD and Zeng C-C, Tetrahedron, 2017, 73, 2447–2454. [Google Scholar]
- 48.Khan FN, Jayakumar R and Pillai CN, J. Mol. Catal. A: Chem, 2003, 195, 139–145. [Google Scholar]
- 49.Bäumer U.-St. and Schäfer HJ, Electrochim. Acta, 2003, 48, 489–495. [Google Scholar]
- 50.Arndt S, Weis D, Donsbach K and Waldvogel SR, Angew. Chem., Int. Ed, 2020, 59, 8036–8041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Honjo E, Kutsumura N, Ishikawa Y and Nishiyama Sh., Tetrahedron, 2008, 64, 9495–9506. [Google Scholar]
- 52.Kitada S, Takahashi M, Yamaguchi Y, Okada Y and Chiba K, Org. Lett, 2012, 14, 5960–5963. [DOI] [PubMed] [Google Scholar]
- 53.Wen J, Shi W, Zhang F, Liu D, Tang S, Wang H, Lin X-M and Lei A, Org. Lett, 2017, 19, 3131–3134. [DOI] [PubMed] [Google Scholar]
- 54.Jiang Y-Y, Liang S, Zeng C-C, Hu L-M and Sun B-G, Green Chem, 2016, 18, 6311–6319. [Google Scholar]
- 55.Zhang C, Chen Y and Yuan G, Chin. J. Chem, 2016, 34, 1277–1282. [Google Scholar]
- 56.Sawamura T, Kuribayashi S, Inagi S and Fuchigami T, Org. Lett, 2010, 12, 644–646. [DOI] [PubMed] [Google Scholar]
- 57.Sawamura T, Kuribayashi S, Inagi S and Fuchigami T, Adv. Synth. Catal, 2010, 352, 2757–2760. [Google Scholar]
- 58.Gao W-J, Li W-C, Zeng C-C, Tian H-Y, Hu L-M and Little RD, J. Org. Chem, 2014, 79, 9613–9618. [DOI] [PubMed] [Google Scholar]
- 59.Qu Q, Gao X, Gao J and Yuan G, Sci. China: Chem, 2015, 58, 747–750. [Google Scholar]
- 60.Baba D and Fuchigami T, Tetrahedron Lett, 2003, 44, 3133–3136. [Google Scholar]
- 61.Chen J, Yan W-Q, Lam CM, Zeng C-C, Hu L-M and Little RD, Org. Lett, 2015, 17, 986–989. [DOI] [PubMed] [Google Scholar]
- 62.Tang S, Gao X and Lei A, Chem. Commun, 2017, 53, 3354–3356. [DOI] [PubMed] [Google Scholar]
- 63.Wayne RP, Barnes I, Biggs P, Burrows JP, Canosa-Mas CE, Hjorth J, Le Bras G, Moortgat GK, Perner D, Poulet G, Restelli G and Sidebottom H, Atmos. Environ., Part A, 1991, 25, 1–203. [Google Scholar]
- 64.Christopher C, Lawrence S, Bosco AJ, Xavier N and Raja S, Catal. Sci. Technol, 2012, 2, 824–827. [Google Scholar]
- 65.Christopher C, Lawrence S, Kulandainathan MA, Kulangiappar K, Raja ME, Xavier N and Raja S, Tetrahedron Lett, 2012, 53, 2802–2804. [Google Scholar]
- 66.Takahira Y, Chen M, Kawamata Y, Mykhailiuk P, Nakamura H, Peters BK, Reisberg SH, Li C, Chen L, Hoshikawa T, Shibuguchi T and Baran PS, Synlett, 2019, 1178–1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lebedev OL and Kazarnovskii SN, Zhur. Obshch. Khim, 1960, 30, 1631–1635. [Google Scholar]
- 68.Forrester AR and Thomson RH, Nature, 1964, 203, 74–75. [Google Scholar]
- 69.Metodiewa D, Skolimowski J and Karolczak S, Preparation of ACT (originally named as tempace): IUBMB Life, 1996, 40, 1211–1219. [DOI] [PubMed] [Google Scholar]
- 70.Shibuya M, Tomizawa M, Suzuki I and Iwabuchi Y, Preparation of AZADO: J. Am. Chem. Soc, 2006, 128, 8412–8413. [DOI] [PubMed] [Google Scholar]
- 71.Shibuya M, Tomizawa M, Sasano Y and Iwabuchi Y, Preparation of ABNO: J. Org. Chem, 2009, 74, 4619–4622. [DOI] [PubMed] [Google Scholar]
- 72.Hickey DP, Schiedler DA, Matanovic I, Doan PV, Atanassov P, Minteer SD and Sigman MS, J. Am. Chem. Soc, 2015, 137, 16179–16186. [DOI] [PubMed] [Google Scholar]
- 73.Nutting JE, Rafiee M and Stahl SS, Chem. Rev, 2018, 118, 4834–4885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kuroboshi M, Yoshihisa H, Cortona MN, Kawakami Y, Gao Z and Tanaka H, Tetrahedron Lett, 2000, 41, 8131–8135. [Google Scholar]
- 75.Tanaka H, Kawakami Y, Goto K and Kuroboshi M, Tetrahedron Lett, 2001, 42, 445–448. [Google Scholar]
- 76.Kubota J, Shimizu Y, Mitsudo K and Tanaka H, Tetrahedron Lett, 2005, 46, 8975–8979. [Google Scholar]
- 77.Demizu Y, Shiigi H, Oda T, Matsumura Y and Onomura O, Tetrahedron Lett, 2008, 49, 48–52. [Google Scholar]
- 78.Palma A, Cárdenas J and Frontana-Uribe BA, Green Chem, 2009, 11, 283–293. [Google Scholar]
- 79.Kuroboshi M, Yoshida T, Oshitani J, Goto K and Tanaka H, Tetrahedron, 2009, 65, 7177–7185. [Google Scholar]
- 80.Hill-Cousins JT, Kuleshova J, Green RA, Birkin PR, Pletcher D, Underwood TJ, Leach SG and Brown RCD, ChemSusChem, 2012, 5, 326–331. [DOI] [PubMed] [Google Scholar]
- 81.Rafiee M, Miles KC and Stahl SS, J. Am. Chem. Soc, 2015, 137, 14751–14757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Rafiee M, Konz ZM, Graaf MD, Koolman HF and Stahl SS, ACS Catal, 2018, 8, 6738–6744. [Google Scholar]
- 83.Rafiee M, Alherech M, Karlen SD and Stahl SS, J. Am. Chem. Soc, 2019, 141, 15266–15276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Goes SL, Mayer MN, Nutting JE, Hoober-Burkhardt LE, Stahl SS and Rafiee M, J. Chem. Educ, 2021, 98, 600–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Badalyan A and Stahl SS, Nature, 2016, 535, 406–410. [DOI] [PubMed] [Google Scholar]
- 86.Li C, Zeng C-C, Hu L-M, Yang F-L, Yoo SJ and Little RD, Electrochim. Acta, 2013, 114, 560–566. [Google Scholar]
- 87.Kashiwagi Y and Anzai J.-i., Chem. Pharm. Bull, 2001, 49, 324–326. [DOI] [PubMed] [Google Scholar]
- 88.Wang F, Rafiee M and Stahl SS, Angew. Chem., Int. Ed, 2018, 57, 6686–6690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Xu F, Zhu L, Zhu S, Yan X and Xu H-C, Chem. – Eur. J, 2014, 20, 12740–12744. [DOI] [PubMed] [Google Scholar]
- 90.Siu JC, Sauer GS, Saha A, Macey RL, Fu N, Chauviré T, Lancaster KM and Lin S, J. Am. Chem. Soc, 2018, 140, 12511–12520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Nelson HM, Siu JC, Saha A, Cascio D, MacMillan SN, Wu S-B, Lu C, Rodríguez JA, Houk KN and Lin S, Org. Lett, 2021, 23, 454–458. [DOI] [PubMed] [Google Scholar]
- 92.Siu JC, Parry JB and Lin S, J. Am. Chem. Soc, 2019, 141, 2825–2831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Grochowski E, Bolesławska T and Jurczak J, Synthesis, 1977, 718–720. [Google Scholar]
- 94.Recupero F and Punta C, Chem. Rev, 2007, 107, 3800–3842. [DOI] [PubMed] [Google Scholar]
- 95.Amorati R, Lucarini M, Mugnaini V, Pedulli GF, Minisci F, Recupero F, Fontana F, Astolfi P and Greci L, J. Org. Chem, 2003, 68, 1747–1754. [DOI] [PubMed] [Google Scholar]
- 96.Blanksby SJ and Ellison GB, Acc. Chem. Res, 2003, 36, 255–263. [DOI] [PubMed] [Google Scholar]
- 97.Masui M, Ueshima T and Ozaki S, J. Chem. Soc., Chem. Commun, 1983, 479–480. [Google Scholar]
- 98.Masui M, Hara S, Ueshima T, Kawaguchi T and Ozaki S, Chem. Pharm. Bull, 1983, 31, 4209–4211. [Google Scholar]
- 99.Masui M, Kawaguchi T and Ozaki S, J. Chem. Soc., Chem. Commun, 1985, 1484–1485. [Google Scholar]
- 100.Masui M, Kawaguchi T, Yoshida S and Ozaki S, Chem. Pharm. Bull, 1986, 34, 1837–1839. [Google Scholar]
- 101.Masui M, Hara S and Ozaki S, Chem. Pharm. Bull, 1986, 34, 975–979. [Google Scholar]
- 102.Masui M, Hosomi K, Tsuchida K and Ozaki S, Chem. Pharm. Bull, 1985, 33, 4798–4802. [Google Scholar]
- 103.Ueda C, Noyama M, Ohmori H and Masui M, Chem. Pharm. Bull, 1987, 35, 1372–1377. [Google Scholar]
- 104.Nakamura A and Nakada M, Synthesis, 2013, 1421–1451. [Google Scholar]
- 105.Horn EJ, Rosen BR, Chen Y, Tang J, Chen K, Eastgate MD and Baran PS, Nature, 2016, 533, 77–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Hruszkewycz DP, Miles KC, Thielb OR and Stahl SS, Chem. Sci, 2017, 8, 1282–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Rafiee M, Wang F, Hruszkewycz DP and Stahl SS, J. Am. Chem. Soc, 2018, 140, 22–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Curci R, D’Accolti L and Fusco C, Acc. Chem. Res, 2006, 39, 1–9. [DOI] [PubMed] [Google Scholar]
- 109.Chen MS and White MC, Science, 2007, 318, 783–787. [DOI] [PubMed] [Google Scholar]
- 110.Kawamata Y, Yan M, Liu Z, Bao D-H, Chen J, Starr JT and Baran PS, J. Am. Chem. Soc, 2017, 139, 7448–7451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Wein LA, Wurst K, Angyal P, Weisheit L and Magauer T, J. Am. Chem. Soc, 2019, 141, 19589–19593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Utley JHP and Rozenberg GG, J. Appl. Electrochem, 2003, 33, 525–532. [Google Scholar]
- 113.Luca OR, Wang T, Konezny SJ, Batista VS and Crabtree RH, New J. Chem, 2011, 35, 998–999. [Google Scholar]
- 114.Kang L-S, Xiao H.-l., Zeng C-C, Hu L-M and Little RD, J. Electroanal. Chem, 2016, 767, 13–17. [Google Scholar]
- 115.Seo ET, Nelson RF, Fritsch JM, Marcoux LS, Leedy DW and Adams RN, J. Am. Chem. Soc, 1966, 88, 3498–3503. [Google Scholar]
- 116.Nelson RF and Adams RN, J. Am. Chem. Soc, 1968, 90, 3925–3930. [Google Scholar]
- 117.Fuchigami T, Tetsu M, Tajima T and Ishii H, Synlett, 2001, 1269–1271. [Google Scholar]
- 118.Drouin L, Compton RG and Fairbanks AJ, J. Phys. Org. Chem, 2008, 21, 516–522. [Google Scholar]
- 119.Wu X, Davis AP and Fry AJ, Org. Lett, 2007, 9, 5633–5636. [DOI] [PubMed] [Google Scholar]
- 120.Park YS, Wang SC, Tantillo DJ and Little RD, J. Org. Chem, 2007, 72, 4351–4357. [DOI] [PubMed] [Google Scholar]
- 121.Park YS and Little RD, J. Org. Chem, 2008, 73, 6807–6815. [DOI] [PubMed] [Google Scholar]
- 122.Yoo SJ, Li L-J, Zeng C-C and Little RD, Angew. Chem., Int. Ed, 2015, 54, 3744–3747. [DOI] [PubMed] [Google Scholar]
- 123.Zeng C.-c., Zhang N.-t., Lam CM and Little RD, Org. Lett, 2012, 14, 1314–1317. [DOI] [PubMed] [Google Scholar]
- 124.Lu N.-n., Yoo SJ, Li L-J, Zeng C-C and Little RD, Electrochim. Acta, 2014, 142, 254–260. [Google Scholar]
- 125.Lu N.-n., Zhang N.-t., Zeng C-C, Hu L-M, Yoo SJ and Little RD, J. Org. Chem, 2015, 80, 781–789. [DOI] [PubMed] [Google Scholar]
- 126.Francke R and Little RD, J. Am. Chem. Soc, 2014, 136, 427–435. [DOI] [PubMed] [Google Scholar]
- 127.Zhu Y, Zhang J, Chen Z, Zhang A and Ma C, Chinese J. Catal, 2016, 37, 533–538. [Google Scholar]
- 128.Hou Z-W, Mao Z-Y, Melcamu YY, Lu X and Xu H-C, Angew. Chem., Int. Ed, 2018, 57, 1636–1639. [DOI] [PubMed] [Google Scholar]
- 129.Wu Z-J, Li S-R and Xu H-C, Angew. Chem., Int. Ed, 2018, 57, 14070–14074. [DOI] [PubMed] [Google Scholar]
- 130.Siu JC, Fu N and Lin S, Acc. Chem. Res, 2020, 53, 547–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.(a) Ma C, Fang P and Mei T-S, ACS Catal, 2018, 8, 7179–7189; [Google Scholar]; (b) Liu W and Ackermann L, ACS Catal, 2016, 6, 3743–3752; [Google Scholar]; (c) Ackermann L, Acc. Chem. Res, 2020, 53(1), 84–104. [DOI] [PubMed] [Google Scholar]
- 132.(a) Liu W and Ackermann L, ACS Catal, 2016, 6, 3743–3752; [Google Scholar]; (b) Palucki M, Finney NS, Pospisil PJ, Güler ML, Ishida T and Jacobsen EN, J. Am. Chem. Soc, 1998, 120, 948–954; [Google Scholar]; (c) Huang X and Groves JT, Chem. Rev, 2018, 118, 2491–2553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Snider BB and Lin H, Synth. Commun, 1998, 28, 1913–1922. [Google Scholar]
- 134.Fu N, Sauer GS, Saha A, Loo A and Lin S, Science, 2017, 357, 575–579. [DOI] [PubMed] [Google Scholar]
- 135.Fu N, Sauer GS and Lin S, J. Am. Chem. Soc, 2017, 139, 15548–15553. [DOI] [PubMed] [Google Scholar]
- 136.Ye K, Pombar G, Fu N, Sauer GS, Keresztes I and Lin S, J. Am. Chem. Soc, 2018, 140, 2438–2441. [DOI] [PubMed] [Google Scholar]
- 137.Lu L, Fu N and Lin S, Synlett, 2019, 1199–1203. [Google Scholar]
- 138.Fu N, Shen Y, Allen AR, Song L, Ozaki A and Lin S, ACS Catal, 2019, 9, 746–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Ye K-Y, Song Z, Sauer GS, Harenberg JH, Fu N and Lin S, Chem. – Eur. J, 2018, 24, 12274–12279. [DOI] [PubMed] [Google Scholar]
- 140.Zhang Z, Zhang L, Cao Y, Li F, Bai G, Liu G, Yang Y and Mo F, Org. Lett, 2019, 21, 762–766. [DOI] [PubMed] [Google Scholar]
- 141.Yuan Y, Zheng Y, Xu B, Liao J, Bu F, Wang S, Hu J-G and Lei A, ACS Catal, 2020, 10, 6676–6681. [Google Scholar]
- 142.Niu L, Jiang C, Liang Y, Liu D, Bu F, Shi R, Chen H, Chowdhury AD and Lei A, J. Am. Chem. Soc, 2020, 142, 17693–17702. [DOI] [PubMed] [Google Scholar]
- 143.Meyer TH, Samanta RC, Del Vecchio A and Ackermann L, Chem. Sci, 2021, 12, 2890–2897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Allen BDW, Hareram MD, Seastram AC, McBride T, Wirth T, Browne DL and Morrill LC, Org. Lett, 2019, 21, 9241–9246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Lin D, Lai Y and Huang J, ChemElectroChem, 2019, 6, 4188–4193. [Google Scholar]
- 146.Xiong P and Xu HC, Acc. Chem. Res, 2019, 52, 3339–3350. [DOI] [PubMed] [Google Scholar]
- 147.Xiong P, Xu H-H, Song J and Xu H-C, J. Am. Chem. Soc, 2018, 140, 2460–2464. [DOI] [PubMed] [Google Scholar]
- 148.Wu Z-J, Li S-R, Long H and Xu H-C, Chem. Commun, 2018, 54, 4601–4604. [DOI] [PubMed] [Google Scholar]
- 149.Long H, Song J and Xu H-C, Org. Chem. Front, 2018, 5, 3129–3132. [Google Scholar]
- 150.Beckwith ALJ and Schiesser CH, Tetrahedron, 1985, 41, 3925–3941. [Google Scholar]
- 151.Zhu C, Stangier M, Oliveira JCA, Massignan L and Ackermann L, Chem. – Eur. J, 2019, 25, 16382–16389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Li LJ and Luo SZ, Org. Lett, 2018, 20, 1324–1327. [DOI] [PubMed] [Google Scholar]
- 153.Saha D, Taily IM, Naik S and Banerjee P, Chem. Commun, 2021, 57, 631–634. [DOI] [PubMed] [Google Scholar]
- 154.Song L, Fu N, Ernst BG, Lee WH, Frederick MO, DiStasio RA and Lin S, Nat. Chem, 2020, 12, 747–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Tian C, Dhawa U, Scheremetjew A and Ackermann L, ACS Catal, 2019, 9, 7690–7696. [Google Scholar]
- 156.Gao P, Weng X, Wang Z, Zheng C, Sun B, Chen Z, You S and Mei T, Angew. Chem, 2020, 132, 15366–15371. [DOI] [PubMed] [Google Scholar]
- 157.Yang QL, Wang XY, Lu JY, Zhang LP, Fang P and Mei TS, J. Am. Chem. Soc, 2018, 140, 11487–11494. [DOI] [PubMed] [Google Scholar]
- 158.Kathiravan S, Suriyanarayanan S and Nicholls IA, Org. Lett, 2019, 21, 1968–1972. [DOI] [PubMed] [Google Scholar]
- 159.Yi X and Hu X, Angew. Chem., Int. Ed, 2019, 58, 4700–4704. [DOI] [PubMed] [Google Scholar]
- 160.Amatore C, Cammoun C and Jutand A, Eur. J. Org. Chem, 2008, 4567–4570. [Google Scholar]
- 161.Dudkina YB, Mikhaylov DY, Gryaznova TV, Tufatullin AI, Kataeva ON, Vicic DA and Budnikova YH, Organometallics, 2013, 32, 4785–4792. [Google Scholar]
- 162.Grayaznova TV, Dudkina YB, Islamov DR, Kataeva ON, Sinyashin OG, Vicic DA and Budnikova YH, J. Organomet. Chem, 2015, 785, 68–71. [Google Scholar]
- 163.Konishi M, Tsuchida K, Sano K, Kochi T and Kakiuchi F, J. Org. Chem, 2017, 82, 8716–8724. [DOI] [PubMed] [Google Scholar]
- 164.Ma C, Zhao C-Q, Li Y-Q, Zhang L-P, Xu X-T, Zhang K and Mei T-S, Chem. Commun, 2017, 53, 12189–12192. [DOI] [PubMed] [Google Scholar]
- 165.O’Reilly ME, Kim RS, Oh S and Surendranath Y, ACS Cent. Sci, 2017, 3, 1174–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Zeng L, Li H, Hu J, Zhang D, Hu J, Peng P, Wang S, Shi R, Peng J, Pao C-W, Chen J-L, Lee J-F, Zhang H, Chen Y-H and Lei A, Nat. Catal, 2020, 3, 438–445. [Google Scholar]
- 167.Yang Q-L, Li Y-Q, Ma C, Fang P, Zhang X-J and Mei T-S, J. Am. Chem. Soc, 2017, 139, 3293–3298. [DOI] [PubMed] [Google Scholar]
- 168.Shrestha A, Lee M, Dunn AL and Sanford MS, Org. Lett, 2018, 20, 204–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Mei R, Koeller J and Ackermann L, Chem. Commun, 2018, 54, 12879–12882. [DOI] [PubMed] [Google Scholar]
- 170.Qiu Y, Tian C, Massignan L, Rogge T and Ackermann L, Angew. Chem., Int. Ed, 2018, 57, 5818–5822. [DOI] [PubMed] [Google Scholar]
- 171.Tan X, Hou X, Rogge T and Ackermann L, Angew. Chem., Int. Ed, 2021, 60, 4619–4624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Xu F, Li Y-J, Huang C and Xu H-C, ACS Catal, 2018, 8, 3820–3824. [Google Scholar]
- 173.Qiu Y, Stangier M, Meyer TH, Oliveira JCA and Ackermann L, Angew. Chem., Int. Ed, 2018, 57, 14179–14183. [DOI] [PubMed] [Google Scholar]
- 174.Yang Q-L, Xing Y-K, Wang X-Y, Ma H-X, Weng X-J, Yang X, Guo H-M and Mei T-S, J. Am. Chem. Soc, 2019, 141, 18970–18976. [DOI] [PubMed] [Google Scholar]
- 175.(a) Tian C, Massignan L, Meyer TH and Ackermann L, Angew. Chem., Int. Ed, 2018, 57, 2383–2387; [DOI] [PubMed] [Google Scholar]; (b) Mei R, Sauermann N, Oliveira JCA and Ackermann L, J. Am. Chem. Soc, 2018, 140(25), 7913–7921. [DOI] [PubMed] [Google Scholar]
- 176.Tian C, Massignan L, Meyer TH and Ackermann L, Angew. Chem., Int. Ed, 2018, 57, 2383–2387. [DOI] [PubMed] [Google Scholar]
- 177.Dhawa U, Tian C, Li W and Ackermann L, ACS Catal, 2020, 10, 6457–6462. [Google Scholar]
- 178.(a) Tang S, Wang D, Liu Y, Zeng L and Lei A, Nat. Commun, 2018, 9, 1–7; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Gao X, Wang P, Zeng L, Tang S and Lei A, J. Am. Chem. Soc, 2018, 140, 4195–4199. [DOI] [PubMed] [Google Scholar]
- 179.Gao X, Wang P, Zeng L, Tang S and Lei A, J. Am. Chem. Soc, 2018, 140, 4195–4199. [DOI] [PubMed] [Google Scholar]
- 180.Cao Y, Yuan Y, Lin Y, Jiang X, Weng Y, Wang T, Bu F, Zeng L and Lei A, Green Chem, 2020, 22, 1548–1552. [Google Scholar]
- 181.Chen J, Jin L, Zhou J, Jiang X and Yu C, Tetrahedron Lett, 2019, 60, 2054–2058. [Google Scholar]
- 182.Dudkina YB, Mikhaylov DY, Gryaznova TV, Sinyashin OG, Vicic DA and Budnikova YH, Eur. J. Org. Chem, 2012, 2114–2117. [Google Scholar]
- 183.Zhang S, Struwe J, Hu L and Ackermann L, Angew. Chem., Int. Ed, 2020, 59, 3178–3183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Qiu Y, Kong W-J, Struwe J, Sauermann N, Rogge T, Scheremetjew A and Ackermann L, Angew. Chem., Int. Ed, 2018, 57, 5828–5832. [DOI] [PubMed] [Google Scholar]
- 185.Qiu Y, Scheremetjew A and Ackermann L, J. Am. Chem. Soc, 2019, 141, 2731–2738. [DOI] [PubMed] [Google Scholar]
- 186.Wu Z, Su F, Lin W, Song J, Wen T, Zhang H and Xu H, Angew. Chem., Int. Ed, 2019, 58, 16770–16774. [DOI] [PubMed] [Google Scholar]
- 187.Peters BK, Rodriguez KX, Reisberg SH, Beil SB, Hickey DP, Kawamata Y, Collins M, Starr J, Chen L, Udyavara S, Klunder K, Gorey TJ, Anderson SL, Neurock M, Minteer SD and Baran PS, Science, 2019, 363, 838–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Manabe S, Wong CM and Sevov CS, J. Am. Chem. Soc, 2020, 142, 3024–3031. [DOI] [PubMed] [Google Scholar]
- 189.Lu L, Siu JC, Lai Y and Lin S, J. Am. Chem. Soc, 2020, 142, 21272–21278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Zhang W and Lin S, J. Am. Chem. Soc, 2020, 142, 20661–20670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Mori M, Hashimoto Y and Ban Y, Tetrahedron Lett, 1980, 21, 631–634. [Google Scholar]
- 192.Schiavon G, Bontempelli G and Corain B, J. Chem. Soc., Dalton Trans, 1981, 1074–1081. [Google Scholar]
- 193.Rollin Y, Troupel M, Tuck DG and Perichon J, J. Organomet. Chem, 1986, 303, 131–137. [Google Scholar]
- 194.Chaussard J, Folest J-C, Nedelec J-Y, Périchon J, Sibille S and Troupel M, Synthesis, 1990, 369–381. [Google Scholar]
- 195.Yamamoto T and Saito N, Macromol. Chem. Phys, 1996, 197, 165–175. [Google Scholar]
- 196.Nédélec J-Y, Périchon J and Troupel M, Top. Curr. Chem, 1997, 185, 141–173. [Google Scholar]
- 197.Cassol TM, Demnitz FWJ, Navarro M and De Neves EA, Tetrahedron Lett, 2000, 41, 8203–8206. [Google Scholar]
- 198.de França KWR, Navarro M,Léonel É, Durandetti M and Nédélec J-Y, J. Org. Chem, 2002, 67, 1838–1842. [DOI] [PubMed] [Google Scholar]
- 199.Oliveira JL, Silva MJ, Florencio T, Urgin K, Sengmany S, Léonel E, Nédélec J-Y and Navarro M, Tetrahedron, 2012, 68, 2383–2390. [Google Scholar]
- 200.Oliveira JL, Le Gall E, Sengmany S, Léonel E, Dubot P, Cénédèse P and Navarro M, Electrochim. Acta, 2015, 173, 465. [Google Scholar]
- 201.Rahil R, Sengmany S, Le Gall E and Léonel E, Synthesis, 2018, 146–154. [Google Scholar]
- 202.Qiu H, Shuai B, Wang Y-Z, Liu D, Chen Y-G, Gao P-S, Ma H-X, Chen S and Mei T-S, J. Am. Chem. Soc, 2020, 142, 9872–9878. [DOI] [PubMed] [Google Scholar]
- 203.Gosmini C, Lasry S, Nedelec J-Y and Périchon J, Tetrahedron, 1998, 54, 1289–1298. [Google Scholar]
- 204.Gosmini C, Nédélec J-Y and Périchon J, Tetrahedron Lett, 2000, 41, 201–203. [Google Scholar]
- 205.Sengmany S, Léonel E, Polissaint F, Nédélec J-Y, Pipelier M, Thobie-Gautier C and Dubreuil D, J. Org. Chem, 2007, 72, 5631–5636. [DOI] [PubMed] [Google Scholar]
- 206.Sengmany S, Le Gall E and Léonel E, Molecules, 2011, 16, 5550–5560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Sengmany S, Vitu-Thiebaud A, Le Gall E, Condon S, Léonel E, Thobie-Gautier C, Pipelier M, Lebreton J and Dubreuil D, J. Org. Chem, 2013, 78, 370–379. [DOI] [PubMed] [Google Scholar]
- 208.Urgin K, Barhdadi R, Condon S, Léonel E, Pipelier M, Blot V, Thobie-Gautier C and Dubreuil D, Electrochim. Acta, 2010, 55, 4495–4500. [Google Scholar]
- 209.Walker BR and Sevov CS, ACS Catal, 2019, 9, 7197–7203. [Google Scholar]
- 210.Conan A, Sibille S, D’Incan E and Périchon J, J. Chem. Soc., Chem. Commun, 1990, 1, 48–49. [Google Scholar]
- 211.Durandetti M, Sibille S, Nédélec J-Y and Périchon J, Synth. Commun, 1994, 24, 145–151. [Google Scholar]
- 212.Durandetti M, Nédélec JY and Périchon J, J. Org. Chem, 1996, 61, 1748–1755. [DOI] [PubMed] [Google Scholar]
- 213.Durandetti M, Périchon J and Nédélec JY, J. Org. Chem, 1997, 62, 7914–7915. [DOI] [PubMed] [Google Scholar]
- 214.Durandetti M and Périchon J, Synthesis, 2004, 3079–3083. [Google Scholar]
- 215.Perkins RJ, Pedro JD and Hansen EC, Org. Lett, 2017, 19, 3755–3758. [DOI] [PubMed] [Google Scholar]
- 216.Biswas S and Weix DJ, J. Am. Chem. Soc, 2013, 135, 16192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Perkins RJ, Hughes JA, Weix DJ and Hansen EC, Org. Process Res. Dev, 2019, 23, 1746–1751. [Google Scholar]
- 218.Li H, Breen CP, Seo H, Jamison TF, Fang Y-Q and Bio MM, Org. Lett, 2018, 20, 1338–1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Koyanagi T, Herath A, Chong A, Ratnikov M, Valiere A, Chang J, Molteni V and Loren J, Org. Lett, 2019, 21, 816–820. [DOI] [PubMed] [Google Scholar]
- 220.Kumar GS, Peshkov A, Brzozowska A, Nikolaienko P, Zhu C and Rueping M, Angew. Chem., Int. Ed, 2020, 59, 6513–6519. [DOI] [PubMed] [Google Scholar]
- 221.Jiao KJ, Liu D, Ma HX, Qiu H, Fang P and Mei TS, Angew. Chem., Int. Ed, 2020, 59, 6520–6524. [DOI] [PubMed] [Google Scholar]
- 222.Truesdell BL, Hamby TB and Sevov CS, J. Am. Chem. Soc, 2020, 142, 5884–5893. [DOI] [PubMed] [Google Scholar]
- 223.DeLano TJ and Reisman SE, ACS Catal, 2019, 9, 6751–6754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Condon-Gueugnot S, Léonel E, Nédélec J-Y and Périchon J, J. Org. Chem, 1995, 60, 7684–7686. [Google Scholar]
- 225.de Medonca Cavalcanti JC, Goulart MOF, Léonel E and Nédélec J-Y, Tetrahedron Lett, 2002, 43, 6343–6345. [Google Scholar]
- 226.Pelletier J, Olivero S and Dunach E, Synth. Commun, 2004, 34, 3343–3348. [Google Scholar]
- 227.Dunach E, Esteves AP, Medeiros MJ, dos Santos Neves CS and Olivero S, C. R. Chim, 2009, 12, 889–894. [Google Scholar]
- 228.Esteves AP, Goken DM, Klein LJ, Lemos MA, Medeiros MJ and Peters DG, J. Org. Chem, 2003, 68, 1024–1029. [DOI] [PubMed] [Google Scholar]
- 229.Ischay MA, Mubarak MS and Peters DG, J. Org. Chem, 2006, 71, 623–628. [DOI] [PubMed] [Google Scholar]
- 230.Condon S, Dupré D, Falgayrac G and Nédélec J-Y, Eur. J. Org. Chem, 2002, 105–111. [Google Scholar]
- 231.Condon S, Dupré D, Lachaise I and Nédélec J-Y, Synthesis, 2002, 1752–1758. [Google Scholar]
- 232.Mikhaylov D, Gryaznova T, Dudkina Y, Khrizanphorov M, Latypov S, Kataeva O, Vicic DA, Sinyashina OG and Budnikova Y, Dalton Trans, 2012, 41, 165–172. [DOI] [PubMed] [Google Scholar]
- 233.Miranda JA, Wade CJ and Little RD, J. Org. Chem, 2005, 70(20), 8017–8026. [DOI] [PubMed] [Google Scholar]
- 234.Zhang S, Li L, Li X, Zhang J, Xu K, Li G and Findlater M, Org. Lett, 2020, 22, 3570–3575. [DOI] [PubMed] [Google Scholar]
- 235.Li C, Kawamata Y, Nakamura H, Vantourout JC, Liu Z, Hou Q, Bao D, Starr JT, Chen J, Yan M and Baran PS, Angew. Chem., Int. Ed, 2017, 56, 13088–13093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Kawamata Y, Vantourout JC, Hickey DP, Bai P, Chen L, Hou Q, Qiao W, Barman K, Edwards MA, Garrido-Castro AF, deGruyter JN, Nakamura H, Knouse K, Qin C, Clay KJ, Bao D, Li C, Starr JT, Garcia-Irizarry C, Sach N, White HS, Neurock M, Minteer SD and Baran PS, J. Am. Chem. Soc, 2019, 141, 6392–6402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Liu D, Ma H-X, Fang P and Mei T-S, Angew. Chem., Int. Ed, 2019, 58, 5033–5037. [DOI] [PubMed] [Google Scholar]
- 238.Wang Y, Deng L, Wang X, Wu Z, Wang Y and Pan Y, ACS Catal, 2019, 9, 1630–1634. [Google Scholar]
- 239.Zhu M, Alami M and Messaoudi S, Chem. Commun, 2020, 56, 4464–4467. [DOI] [PubMed] [Google Scholar]
- 240.Sengmany S, Ollivier A, Le Gall E and Léonel E, Org. Biomol. Chem, 2018, 16, 4495–4500. [DOI] [PubMed] [Google Scholar]
- 241.Bai Y, Liu N, Wang S, Wang S, Ning S, Shi L, Cui L, Zhang Z and Xiang J, Org. Lett, 2019, 21, 6835–6838. [DOI] [PubMed] [Google Scholar]
- 242.Daili F, Ouarti A, Pinaud M, Kribii I, Sengmany S, Gall EL and Léonel E, Eur. J. Org. Chem, 2020, 3452–3455. [Google Scholar]
- 243.Zhu C, Yue H, Nikolaienko P and Rueping M, CCS Chem, 2020, 2, 179–190. [Google Scholar]
- 244.Lian F, Xu K, Meng W, Zhang H, Tan Z and Zeng C, Chem. Commun, 2019, 55, 14685–14688. [DOI] [PubMed] [Google Scholar]
- 245.Samanta RC, Struwe J and Ackermann L, Angew. Chem., Int. Ed, 2020, 59, 14154–14159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Franco D and Dunach E, Tetrahedron Lett, 2000, 41, 7333–7336. [Google Scholar]
- 247.Franco D, Riahi A, Henin F, Muzart J and Dunach E, Eur. J. Org. Chem, 2002, 2257–2259. [Google Scholar]
- 248.Yamamoto T, Ishizu J and Yamamoto A, J. Am. Chem. Soc, 1981, 103, 6863–6869. [Google Scholar]
- 249.Gomes P, Fillon H, Gosmini C, Labbé E and Périchon J, Tetrahedron, 2002, 58, 8417–8424. [Google Scholar]
- 250.Le Gall E, Gosmini C, Nédélec J-Y and Périchon J, Tetrahedron Lett, 2001, 42, 267–269. [Google Scholar]
- 251.Cannes C, Bedioui F, Gueugnot SC, Nédélec JY and Devynck J, New J. Chem, 1999, 23, 489–494. [Google Scholar]
- 252.Condon S, Céline C and Bedioui F, J. Chem, 2019, 1–6. [Google Scholar]
- 253.Gandeepan P, Finger LH, Meyer TH and Ackermann L, Chem. Soc. Rev, 2020, 49, 4254–4272. [DOI] [PubMed] [Google Scholar]
- 254.Shen Y, Inagi S, Atobe M and Fuchigami T, Res. Chem. Intermed, 2013, 39, 89–99. [Google Scholar]
- 255.Chen BL, Zhu HW, Xiao Y, Sun QL, Wang H and Lu JX, Electrochem. Commun, 2014, 42, 55–59. [Google Scholar]
- 256.Ang NWJ, Oliveira JCA and Ackermann L, Angew. Chem., Int. Ed, 2020, 59, 12842–12847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Shimakoshi H and Hisaeda Y, Curr. Opin. Electrochem, 2018, 8, 24–30. [Google Scholar]
- 258.Njue CK, Nuthakki B, Vaze A, Bobbitt JM and Rusling JF, Electrochem. Commun, 2001, 3, 733–736. [Google Scholar]
- 259.Shimakoshi H, Luo Z, Inaba T and Hisaeda Y, Dalton Trans, 2016, 45, 10173–10180. [DOI] [PubMed] [Google Scholar]
- 260.Chalkley MJ, Garrido-Barros P and Peters JC, Science, 2020, 369, 850–854. [DOI] [PubMed] [Google Scholar]
- 261.Magdesieva TV, Graczyk M, Vallat A, Nikitin OM, Demyanov PI, Butin KP and Vorotyntsev MA, Electrochim. Acta, 2006, 52, 1265–1280. [Google Scholar]
- 262.Nikitin OM and Magdesieva TV, Mendeleev Commun, 2011, 21, 194–195. [Google Scholar]
- 263.Lisitsyn YA and Grigor’eva LV, Russ. J. Electrochem, 2009, 45, 132–138. [Google Scholar]
- 264.Lisitsyn YA and Sukhov AV, Russ. J. Electrochem, 2011, 47, 1180–1185. [Google Scholar]
- 265.Lisitsyn YA and Sukhov AV, Russ. J. Electrochem, 2015, 51, 1092–1095. [Google Scholar]
- 266.Lisitsyn YA and Sukhov AV, Russ. J. Electrochem, 2020, 56, 426–430. [Google Scholar]
- 267.Liedtke T, Spannring P, Riccardi L and Gansäuer A, Angew. Chem., Int. Ed, 2018, 57, 5006–5010. [DOI] [PubMed] [Google Scholar]
- 268.Huang J-M and Dong Y, Chem. Commun, 2009, 3943–3945. [DOI] [PubMed] [Google Scholar]
- 269.Huang JM and Ren HR, Chem. Commun, 2010, 46, 2286–2288. [DOI] [PubMed] [Google Scholar]
- 270.Sinha AK, Mondal B, Kundu M, Chakraborty B and Roy UK, Org. Chem. Front, 2014, 1, 1270–1275. [Google Scholar]
- 271.Torii S, Tanaka H and Morisaki K, Tetrahedron Lett, 1985, 26, 1655–1658. [Google Scholar]
- 272.Tian J and Moeller KD, Org. Lett, 2005, 7, 5381–5383. [DOI] [PubMed] [Google Scholar]
- 273.Lai Y-L and Huang J-M, Org. Lett, 2017, 19, 2022–2025. [DOI] [PubMed] [Google Scholar]
- 274.Jiao KJ, Li ZM, Xu XT, Zhang LP, Li YQ, Zhang K and Mei TS, Org. Chem. Front, 2018, 5, 2244–2248. [Google Scholar]
- 275.Krief A and Laval AM, Chem. Rev, 1999, 99, 745–778. [DOI] [PubMed] [Google Scholar]
- 276.Léonard E, Duñach E and Périchon J, J. Chem. Soc., Chem. Commun, 1989, 5, 276–277. [Google Scholar]
- 277.Hebri H, Duñach E, Heintz M, Troupel M and Périchon J, Synlett, 1991, 901–902. [Google Scholar]
- 278.Parrish JD and Little RL, Tetrahedron Lett, 2001, 42, 7767–7770. [Google Scholar]
- 279.Sahloul K, Sun L, Requet A, Chahine Y and Mellah M, Chem. – Eur. J, 2012, 18, 11205–11209. [DOI] [PubMed] [Google Scholar]
- 280.Sun L, Sahloul K and Mellah M, ACS Catal, 2013, 3, 2568–2573. [Google Scholar]
- 281.Zhang YF and Mellah M, ACS Catal, 2017, 7, 8480. [Google Scholar]
- 282.Uneyama K, Matsuda H and Torii S, Tetrahedron Lett, 1984, 25, 6017–6020. [Google Scholar]
- 283.Zha Z, Hui A, Zhou Y, Miao Q, Wang Z and Zhang H, Org. Lett, 2005, 7, 1903–1905. [DOI] [PubMed] [Google Scholar]
- 284.Zhang L, Zha Z, Wang Z and Fu S, Tetrahedron Lett, 2010, 51, 1426–1429. [Google Scholar]
- 285.Zhang L, Zha Z, Zhang Z, Li Y and Wang Z, Chem. Commun, 2010, 46, 7196–7198. [DOI] [PubMed] [Google Scholar]
- 286.Zhang L, Zha ZG and Wang ZY, Synlett, 2010, 1915–1918. [Google Scholar]
- 287.Zhang L, Chen H, Zha ZG and Wang ZY, Chem. Commun, 2012, 48, 6574. [DOI] [PubMed] [Google Scholar]
- 288.Oda K, Ohnuma T and Ban Y, J. Org. Chem, 1984, 49, 953–959. [Google Scholar]
- 289.Senboku H, Nakahara K, Fukuhara T and Hara S, Tetrahedron Lett, 2010, 51, 435–438. [Google Scholar]
- 290.Kurono N, Honda E, Komatsu F, Orito K and Tokuda M, Chem. Lett, 2003, 32, 720–721. [Google Scholar]
- 291.Kurono N, Honda E, Komatsu F, Orito K and Tokuda M, Tetrahedron, 2004, 60, 1791–1801. [Google Scholar]
- 292.Mitsudo K, Okada T, Shimohara S, Mandai H and Suga S, Electrochemistry, 2013, 81, 362–364. [Google Scholar]
- 293.Mitsudo K, Nakagawa Y, Mizukawa JI, Tanaka H, Akaba R, Okada T and Suga S, Electrochim. Acta, 2012, 82, 444–449. [Google Scholar]
- 294.Li C and Wonneberger H, Adv. Mater, 2012, 24, 613–636. [DOI] [PubMed] [Google Scholar]
- 295.Sun G, Ren S, Zhu X, Huang M and Wan Y, Org. Lett, 2016, 18, 544–547. [DOI] [PubMed] [Google Scholar]
- 296.Senboku H, Michinishi JY and Hara S, Synlett, 2011, 1567–1572. [Google Scholar]
- 297.Katayama A, Senboku H and Hara S, Tetrahedron, 2016, 72, 4626–4636. [Google Scholar]
- 298.Katayama A and Senboku H, ChemElectroChem, 2016, 3, 2052–2057. [Google Scholar]
- 299.Grimes RN, Dalton Trans, 2015, 44, 5939–5956. [DOI] [PubMed] [Google Scholar]
- 300.Diederich F and GómezLópez M, Chem. Soc. Rev, 1999, 28, 263–277. [Google Scholar]
- 301.Yu G, Gao J, Hummelen JC, Wudl F and Heeger AJ, Science, 1995, 270, 1789–1791. [Google Scholar]
- 302.Bingel K, Chem. Ber, 1993, 126, 1957–1959. [Google Scholar]
- 303.Maggini M, Scorrano G and Prato M, J. Am. Chem. Soc, 1993, 115, 9798–9799. [Google Scholar]
- 304.Spokoyny AM, Pure Appl. Chem, 2013, 85, 903–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305.Hosoi K, Inagi S, Kubo T and Fuchigami T, Chem. Commun, 2011, 47, 8632–8634. [DOI] [PubMed] [Google Scholar]
- 306.Fuchigami T, Kasuga M and Konno A, J. Electroanal. Chem, 1996, 411, 115–119. [Google Scholar]
- 307.Porter WW III, Vaid TP and Rheingold AL, J. Am. Chem. Soc, 2005, 127, 16559–16566. [DOI] [PubMed] [Google Scholar]
- 308.Kuroboshi M, Kobayashi R, Nakagawa T and Tanaka H, Synlett, 2009, 85–88. [Google Scholar]
- 309.Kuroboshi M, Shiba T and Tanaka H, Tetrahedron Lett, 2013, 54, 3666–3668. [Google Scholar]
- 310.Tanaka H, Kuroboshi M, Kataoka R and Suzuki R, Electrochemistry, 2013, 81, 356–358. [Google Scholar]
- 311.Steckhan E, Arns T, Heineman WR, Hilt G, Hoormann D, Jorissen J, Kröner L, Lewall B and Putter H, Chemosphere, 2001, 43, 63–73. [DOI] [PubMed] [Google Scholar]
- 312.Zhang B, Qu X, Qu J, Chen X, Xie H, Xing P, Wang D and Yin H, Green Chem, 2020, 22, 8633–8641. [Google Scholar]
- 313.Zhang L and Hu X, Chem. Sci, 2020, 11, 10786–10791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314.Mo Y, Lu Z, Rughoobur G, Patil P, Gershenfeld N, Akinwande AI, Buchwald SL and Jensen KF, Science, 2020, 368, 1352–1357. [DOI] [PubMed] [Google Scholar]
- 315.Luo J, Hu B, Wu W, Hu M and Liu TL, Angew. Chem., Int. Ed, 2021, 60, 2–12. [DOI] [PubMed] [Google Scholar]
- 316.Liang Y, Shi S-H, Jin R, Qiu X, Wei J, Tan H, Jiang X, Shi X, Song S and Jiao N, Nat. Catal, 2021, 4, 116–123. [Google Scholar]
- 317.Shen Y, Atobe M, Li W and Nonaka T, Electrochim. Acta, 2003, 48, 1041–1046. [Google Scholar]
- 318.Llorente MJ, Nguyen BH, Kubiak CP and Moeller KD, J. Am. Chem. Soc, 2016, 138, 15110–15113. [DOI] [PubMed] [Google Scholar]
- 319.Sherbo RS, Delima RS, Chiykowski VA, MacLeod BP and Berlinguette CP, Nat. Catal, 2018, 1, 501–507. [Google Scholar]
- 320.Kurimoto A, Sherbo RS, Cao Y, Loo NW and Berlinguette CP, Nat. Catal, 2020, 3, 719–726. [Google Scholar]
- 321.Zhao H-B, Xu P, Song J and Xu H-C, Angew. Chem., Int. Ed, 2018, 57, 15153–15156. [DOI] [PubMed] [Google Scholar]
- 322.Heard DM and Lennox AJJ, Angew. Chem., Int. Ed, 2020, 59, 18866–18884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323.Ross SD and Finkelstein M, J. Org. Chem, 1969, 34, 2923–2927. [Google Scholar]
- 324.Brennan MPJ and Brettle R, J. Chem. Soc., Perkin Trans 1, 1973, 257–261. [Google Scholar]
- 325.Hudson CM, Marzabadi MR, Moeller KD and New DG, J. Am. Chem. Soc, 1991, 113, 7372–7385. [Google Scholar]
- 326.Beil S, Pollok D and Waldvogel SR, Angew. Chem., Int. Ed, DOI: 10.1002/anie.202014544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327.Wei B-Y, Xie D-T, Lai S-Q, Jiang Y, Fu H, Wei D and Han B, Angew. Chem., Int. Ed, 2021, 60, 3182–3188. [DOI] [PubMed] [Google Scholar]
- 328.Jin K, Maalouf JH, Lazouski N, Corbin N, Yang D and Manthiram K, J. Am. Chem. Soc, 2019, 141, 6413–6418. [DOI] [PubMed] [Google Scholar]
- 329.Yin Z, Pang H, Guo X, Lin H, Muzzio M, Shen M, Wei K, Yu C, Williard P and Sun S, Angew. Chem., Int. Ed, 2020, 132, 16067–16070. [DOI] [PubMed] [Google Scholar]
- 330.For examples of reviews on bioelectrocatalysis, see:; (a) Chen H, Simoska O, Lim K, Grattieri M, Yuan M, Dong F, Lee YS, Beaver K, Weliwatte S, Gaffney EM and Minteer SD, Chem. Rev, 2020, 120, 12903–12993; [DOI] [PubMed] [Google Scholar]; (b) Chen H, Dong FY and Minteer SD, Nat. Catal, 2020, 3, 225–244; [Google Scholar]; (c) Xiao X, Xia H, Wu R, Bai L, Yan L, Magner E, Cosnier S, Lojou E, Zhu Z and Liu A, Chem. Rev, 2019, 119, 9509–9558. [DOI] [PubMed] [Google Scholar]
- 331.Masa J and Schuhmann W, Nano Energy, 2016, 29, 466–475. [Google Scholar]
- 332.Xiao Y, Patolsky F, Katz E, Hainfeld JF and Willner I, Science, 2003, 299, 1877–1881. [DOI] [PubMed] [Google Scholar]
- 333.Koto A, Taniya S, Sakamoto H, Satomura T, Sakuraba H, Ohshima T and Suye S, Biosens. Bioelectron, 2014, 5, 1. [Google Scholar]
- 334.Hickey DP, Lim K, Cai R, Patterson AR, Yuan M, Sahin S, Abdellaoui S and Minteer SD, Chem. Sci, 2018, 9, 5172–5177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 335.Chen H, Cai R, Patel J, Dong F, Chen H and Minteer SD, J. Am. Chem. Soc, 2019, 141, 4963–4971. [DOI] [PubMed] [Google Scholar]
- 336.Seelajaroen H, Haberbauer M, Hemmelmair C, Aljabour A, Dumitru LM, Hassel AW and Sariciftci NS, ChemBioChem, 2019, 20, 1196–1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 337.Yuan MW, Kummer MJ, Milton RD, Quah T and Minteer SD, ACS Catal, 2019, 9, 5486–5495. [Google Scholar]
- 338.Dong F, Chen H, Malapit CA, Prater MB, Li M, Yuan M, Lim K and Minteer SD, J. Am. Chem. Soc, 2020, 142, 8374–8382. [DOI] [PubMed] [Google Scholar]
- 339.Zhang L, Cui H, Zou Z, Garakani TM, Novoa-Henriquez C, Jooyeh B and Schwaneberg U, Angew. Chem, 2019, 131, 4610–4613. [DOI] [PubMed] [Google Scholar]
- 340.Liu C, Sakimoto KK, Colon BC, Silver PA and Nocera DG, Proc. Natl. Acad. Sci. U. S. A, 2017, 114, 6450–6455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 341.Sturm-Richter K, Golitsch F, Sturm G, Kipf E, Dittrich A, Beblawy S, Kerzenmacher S and Gescher J, Bioresour. Technol, 2015, 186, 89–96. [DOI] [PubMed] [Google Scholar]
- 342.Mayr JC, Grosch JH, Hartmann L, Rosa LFM, Spiess AC and Harnisch F, ChemSusChem, 2019, 12, 1631–1634. [DOI] [PubMed] [Google Scholar]
- 343.Moutet JC and Reverdy G, Tetrahedron Lett, 1979, 20, 2389–2392. [Google Scholar]
- 344.Shukla SS and Rusling JF, J. Phys. Chem, 1985, 89, 3353–3358. [Google Scholar]
- 345.Yu Y, Guo P, Zhong JS, Yuan Y and Ye KY, Org. Chem. Front, 2019, 7, 131–135. [Google Scholar]
- 346.Barham JP and König B, Angew. Chem., Int. Ed, 2020, 59, 11732–11747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 347.Zhang W, Carpenter KL and Lin S, Angew. Chem., Int. Ed, 2020, 59, 409–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 348.Xu P, Chen P-Y and Xu H-C, Angew. Chem., Int. Ed, 2020, 59, 14275–14280. [DOI] [PubMed] [Google Scholar]
- 349.Lai X-L, Shu X-M, Song J and Xu H-C, Angew. Chem., Int. Ed, 2020, 59, 10626–10632. [DOI] [PubMed] [Google Scholar]
- 350.Yan H, Hou Z-W and Xu H-C, Angew. Chem., Int. Ed, 2019, 58, 4592–4595. [DOI] [PubMed] [Google Scholar]
- 351.Qiu Y, Scheremetjew A, Finger LH and Ackermann L, Chem. – Eur. J, 2020, 26, 3241–3246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 352.Capaldo L, Quadri LL, Merli D and Ravelli D, Chem. Commun, 2021, 57, 4424–4427. [DOI] [PubMed] [Google Scholar]
- 353.Huang H, Strater ZM, Rauch M, Shee J, Sisto TJ, Nuckolls C and Lambert TH, Angew. Chem., Int. Ed, 2019, 58, 13318–13322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 354.Huang H and Lambert TH, Angew. Chem., Int. Ed, 2020, 59, 658–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 355.Huang H and Lambert TH, Angew. Chem., Int. Ed, 2021, 60, 11163–11167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 356.Shen T and Lambert TH, Science, 2021, 371, 620–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 357.Wu S, Žurauskas J, Domański M, Hitzfeld PS, Butera V, Scott DJ, Rehbein J, Kumar A, Thyrhaug E, Hauer J and Barham JP, Org. Chem. Front, 2021, 8, 1132–1142. [Google Scholar]
- 358.Kim H, Kim H, Lambert TH and Lin S, J. Am. Chem. Soc, 2020, 142, 2087–2092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 359.Eriksen J, Lund H, Nyvad AI, Yamato T, Mitchell RH, Dingle TW, Williams RV and Mahedevan R, Acta Chem. Scand, 1983, 37, 459–466. [Google Scholar]
- 360.Choi GJ, Zhu Q, Miller DC, Gu CJ and Knowles RR, Nature, 2016, 539, 268–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 361.Cowper NGW, Chernowsky CP, Williams OP and Wickens ZK, J. Am. Chem. Soc, 2020, 142, 2093–2099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 362.Liu J, Lu L, Wood D and Lin S, ACS Cent. Sci, 2020, 6, 1317–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 363.Wang F and Stahl SS, Angew. Chem., Int. Ed, 2019, 58, 6385–6390. [DOI] [PMC free article] [PubMed] [Google Scholar]