

Alveolar bone is a unique osseous tissue due to the integration of the teeth and the proximity of dental plaque biofilms. Periodontal health and homeostasis are mediated by a balanced host immune response to polymicrobial oral biofilms. Shifts in the composition and quantity of microbes within dental plaque biofilms can drive a local proinflammatory immune response state in the epithelial and gingival barrier connective tissues. Infiltrating proinflammatory immune cells within the inflamed connective tissue secrete local factors that induce paracrine signaling to subjacent bone cells. Sustained chronic inflammation disrupts “coupled” osteoclast-osteoblast actions, which ultimately result in alveolar bone destruction.
This chapter will provide an overview of alveolar bone physiology and will highlight why the oral microbiota is a critical regulator of alveolar bone remodeling. The ecology of subgingival plaque biofilms will be reviewed considering our understanding that periodontitis is a polymicrobial disruption of host homeostasis. The pathogenesis of periodontal bone loss will be explained from both a historical and current perspective, providing the opportunity to revisit the role of fibrosis in alveolar bone destruction. The molecular basis of host-microbe interactions will be discussed in the context of the commensal oral flora in periodontal health and pathogenic shifts in the oral microbiota during periodontal disease states. The role of periodontal innate and adaptive immune cell interactions with bone cells will be reviewed based on our current understanding of osteoimmunological mechanisms influencing alveolar bone remodeling. Lastly, probiotic/prebiotic therapeutic interventions in the oral microbiota will be evaluated as a potential therapy to support alveolar bone homeostasis/ prevent periodontal bone loss.
1 |. ALVEOLAR BONE ANATOMY/PHYSIOLOGY
The alveolar process (commonly referred to as alveolar bone) is dependent on the development, eruption, and maintenance of the teeth.1,2 The primary functions of the alveolar bone are to protect the roots of the teeth and support masticatory function. Like nonoral skeletal tissues, the alveolar bone functions as a source of hematopoietic and mesenchymal stem cells and is a reservoir for calcium, phosphorus, and magnesium. Alveolar bone is responsive to calciotropic hormones, such as parathyroid hormone and calcitonin, which regulate serum calcium homeostasis. Alveolar bone metabolism is also influenced by endocrine signaling effects mediated by changes in sex hormones and circulating inflammatory factors.
Alveolar bone is composed of alveolar bone proper, supporting trabecular bone, and supporting cortical bone (ie, lingual and buccal cortical plates) (Figure 1). The alveolar bone proper is a 0.1–0.4 mm thick layer of bone that lines the alveolus (tooth socket) and supports the attachment of periodontal ligament Sharpey’s fibers.3–5 The outer surface of the alveolar bone proper is surrounded by supporting trabecular and cortical bone. The lingual/buccal cortical plates merge with the alveolar bone proper at the alveolar bone crest.4,6,7 Depending on the morphology of the roots and the buccal-lingual dimensions of the alveolar process, trabecular bone can be interposed between the alveolar bone proper and lingual/buccal cortical plates.4,6,7 The trabecular number, thickness, and organization of the alveolar bone are highly variable and do not appear to be influenced by age.5,8
FIGURE 1.
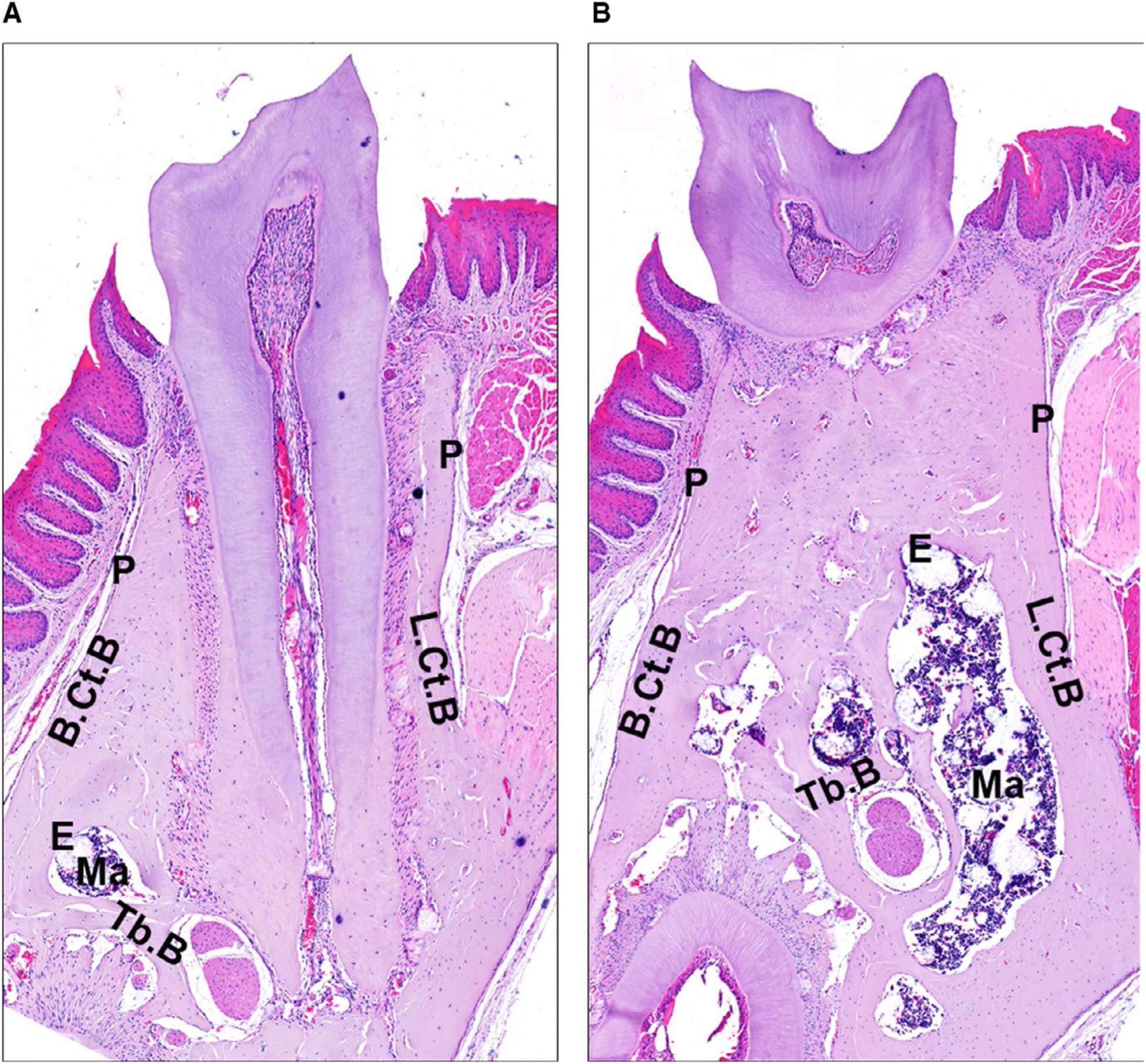
Murine alveolar bone anatomy. Twelve-week-old male C57BL6 mouse: serial frontal sections. A, Through the mesial root of the mandibular first molar. B, Through the furcation of the mandibular first molar. P, periosteum; E, endosteum; B.Ct.B, buccal cortical bone; L.Ct.B, lingual cortical bone; Ma, marrow; Tb.B, trabecular bone
Alveolar bone surfaces are lined by membranes (ie, periodontal ligament, periosteum, endosteum) that have a dense blood supply and serve as a robust source of progenitor cells. The periodontal ligament lines the inner surface of the alveolar bone proper; periosteum lines the outer surface of the lingual/buccal cortical plates; and endosteum lines the inner (endocortical) surface of the lingual/buccal cortical plates and interposed trabeculae. Recent seminal reports have applied cell lineage tracing techniques to discern the source of mesenchymal progenitor cells where bone-forming osteoblast cells originate. Orthopedic investigations of nonoral skeletal sites delineated that osteoblasts originate from mesenchymal progenitors derived from the bone marrow, endosteum, and periosteum.9–11 Orthopedic osseous wound healing studies further determined that the endosteum and periosteum are the primary sources of osteoblasts during osseous regeneration.9–11 Having direct implications for alveolar bone remodeling and regeneration, a timely report utilized cell lineage tracing and mineral double-labeling approaches to compare the periodontal ligament, periosteum, and endosteum as sources of osteogenic progenitors.12 This seminal study showed that bone-forming osteoblastic cells robustly originate from mesenchymal progenitors derived from the periodontal ligament, periosteum, and endosteum. Highlighting the importance of the periodontal ligament as a source of osteoblasts within the alveolar bone, progenitor cells originating from the periodontal ligament displayed a two to threefold higher mineral deposition rate than progenitor cells originating from the periosteum and endosteum.12
The alveolar bone medullary space, which is located between the endocortical surfaces of the cortical plates and around the interposed trabecular bone, is interspersed with hematopoietic marrow, adipocytes, and blood vessels.5,8 The adult bone marrow houses hematopoietic stem cells, which are self-renewing and multipotent progenitor cells that sustain erythropoiesis, myelopoiesis, and lymphopoiesis.13,14 Stem cell niches are tissue microenvironments that support the maintenance of stem cells and regulate their functions through the production of local factors.13,14 Current research supports the notion that hematopoietic stem cell niches in bone marrow are endosteal and/or perivascular in nature,13–16 and there is evidence that stromal-osteoblastic cells and osteoclastic cells produce local factors that influence the maintenance and modulate the function of hematopoietic stem cells.13–16 These hematopoietic stem cells and downstream progenitor cell populations play important roles in supporting tissue homeostasis and regeneration, both in health and disease.15,16 Future research is necessary to determine whether the hematopoietic stem cell niche housed within alveolar bone is similar to nonoral skeletal sites. Furthermore, periodontal disease has unknown effects on local hematopoietic stem cell niches.
2 |. BONE CELLS: OSTEOBLASTS, OSTEOCYTES, OSTEOCLASTS
Osteoblasts are bone-forming cells that are derived from the mesenchymal lineage (Figure 2). Osteoblast lineage cells include osteoblast precursors, osteoblasts, bone lining cells, and osteocytes.17–19 The transcription factors runt domain–containing transcription factor, osterix, and activating transcription factor 4 regulate osteoblast differentiation.17–19 Runt domain–containing transcription factor is required for the commitment of mesenchymal progenitors to the osteoblast lineage. Runt domain–containing transcription factor and downstream osterix are necessary for osteoblast differentiation and function. Activating transcription factor 4, which is independent of runt domain–containing transcription factor and osterix, is required for osteoblast differentiation and function. WNT, bone morphogenetic protein, fibroblast growth factor, and insulin-like growth factor signaling support osteoblast differentiation and function, whereas Notch signaling inhibits osteoblast differentiation.17–19 Osteoblasts secrete an extracellular matrix primarily composed of type I collagen fibers and noncollagenous proteins.20,21 The newly formed bone matrix is initially unmineralized, which is commonly referred to as osteoid. Secreted noncollagenous proteins, such as alkaline phosphatase, osteocalcin, and bone sialoprotein, support mineralization of the bone matrix.20,21 The mineralized inorganic phase of the bone matrix is hydroxyapatite, Ca10(PO4)6(OH)2.20,21 Osteoblasts eventually undergo apoptosis, become quiescent bone lining cells, or develop into osteocytes.
FIGURE 2.

Bone cells: osteoclasts, osteoblasts, and osteocytes. Twelve-week-old male C57BL6 mouse trabecular bone. Tartrate-resistant acid phosphatase–positive osteoclasts (red arrows) resorbing old bone, cuboidal bone forming osteoblasts (green arrows), and bone-embedded osteocytes (black arrows)
Osteocytes are terminally differentiated osteoblast lineage cells that are entombed within the mineralized bone matrix (Figure 2).20,22 While osteoblasts make up roughly 4%−6% of bone cells, osteocytes astonishingly account for more than 90% of bone cells.20,22 Osteocytes embedded within the bone matrix form dendritic processes. The osteocyte cell body is encased in an osseous space called the lacuna, and the dendritic processes travel through the bone matrix in small canals known as canaliculi. Osteocyte dendritic processes facilitate communication with other osteocytes, cells lining the bone surface, vascular spaces, and the bone marrow environment.22–24 Osteocytes play important roles in sensing mechanical loading forces on bone25 and are responsive to circulating endocrine signaling mediators.26 Osteocytes express biologic factors that critically regulate osteoblast activity, including dickkopf WNT signaling pathway inhibitor 1 and sclerostin. Dickkopf WNT signaling pathway inhibitor 1 is expressed broadly across cell types, whereas sclerostin is primarily synthesized by osteocytes. Both dickkopf WNT signaling pathway inhibitor 1 and sclerostin are negative regulators of the Wnt/β-catenin pathway in osteoblasts, which suppresses osteoblast-mediated bone formation.22–24
Osteoclasts are multinucleated bone resorbing cells that are derived from monocyte-macrophage precursor cells (Figure 2). Macrophage colony-stimulating factor27,28 and receptor activator of nuclear factor-kappa B ligand (RANKL)29–33 are signaling factors that are critical and necessary for osteoclastogenesis. The receptor activator of nuclear factor kappa B (RANK) is expressed on osteoclast precursors and osteoclasts. Macrophage colony-stimulating factor signaling at its cognate receptor, c-Fms, induces the expression of RANK34 and supports the proliferation and differentiation of osteoclast precursor cells.35 RANKL binding at its receptor on osteoclast precursors induces the expression of the transcription factor, NFATc1, which leads to osteoclast differentiation.36–38 RANKL:RANK binding on osteoclast cells mediates the expression of effector genes that drive osteoclast maturation, function, and survival.36–38 Dendritic cell–specific transmembrane protein39,40 and osteoclast stimulatory transmembrane protein41 are RANKL-induced fusion proteins critical for osteoclast maturation. RANKL-induced expression of the β3 integrin subunit is necessary for αVβ3-mediated binding to the bone matrix.36–38 Osteoclasts bound to the bone matrix form a sealing zone, where they secrete hydrogen ions and lytic enzymes that resorb the underlying osseous tissue. The secretion of the osteoclast proteolytic enzymes, tartrate-resistant acid phosphatase and the collagenase cathepsin K, are dependent on NFATc1-mediated RANKL signaling.36–38 RANKL-mediated osteoclastogenesis is critically regulated by its physiologic decoy receptor osteoprotegrin.42–45 Osteoprotegrin binding of RANKL inhibits RANKL signaling at the RANK receptor, which downregulates osteoclastogenesis. Appreciating that the RANKL:osteoprotegrin ratio determines the availability of free RANKL in the local environment, the RANKL:osteoprotegrin axis must be evaluated when assessing the potential of RANKL to drive osteoclastogenesis.
3 |. RANKL:OSTEOPROTEGRIN AXIS
Clinical and preclinical studies have discerned that dental plaque biofilms upregulate RANKL, downregulate osteoprotegrin, and increase the RANKL:osteoprotegrin ratio.46–48 Exogenous osteoprotegrin administration in rodent experimental periodontal disease models suppressed osteoclastogenesis and blunted periodontal bone loss,49,50 which demonstrates that the RANKL:osteoprotegrin axis critically regulates periodontitis-driven alveolar bone loss. The molecular underpinnings defining the cellular sources of RANKL and osteoprotegrin have been recently elucidated.
Diverse cells synthesize RANKL and osteoprotegrin under physiologic conditions, which supports basal osteoclastogenesis and homeostatic bone remodeling. Osteoblasts were initially perceived to be the primary source of RANKL30,51 and osteoprotegrin.52 A study in transgenic mice with inducible ablation of mature osteoblastic cells challenged the notion that osteoblast-derived RANKL regulates basal osteoclastogenesis.53 Osteoblast-ablated transgenic mice had impaired bone formation but lacked alterations in osteoclast-mediated bone resorption.53 Timely studies utilizing mesenchymal stem cell and osteocyte-specific conditional RANKL knockout models later discerned that RANKL derived from osteocytes, not osteoblasts, critically supports basal osteoclastogenesis and physiologic bone remodeling.54–56 T and B-cell–specific conditional RANKL knockout models did not demonstrate differences in bone mass, which implies that lymphocyte-derived RANKL does not regulate physiologic bone remodeling.57 Timely investigations in lymphocyte-specific knockout mice have revealed that B cells and T cells play important roles in the production of osteoprotegrin under physiologic conditions.58,59 B cell knockout mice have elevated bone resorption and reduced bone mass, which is secondary to deficient osteoprotegrin synthesis in the bone marrow environment.58 Furthermore, immunophenotyping in wild-type mice astonishingly revealed that 64% of marrow osteoprotegrin synthesis is attributed to the B cell lineage.58 T cells promote skeletal homeostasis through the expression of CD40 ligand, which indirectly regulates B cell osteoprotegrin synthesis. CD40 ligand activation of the CD40 costimulatory receptor induces B cell osteoprotegrin production,44,58 which supports basal osteoclastogenesis and physiologic bone remodeling.58,59
Dental plaque biofilms have been reported to upregulate RANKL and downregulate osteoprotegrin, resulting in an enhanced RANKL:osteoprotegrin ratio that drives osteoclastogenesis and catabolic alveolar bone loss.46,47 Whereas both supragingival and subgingival biofilms have the ability to upregulate the RANKL:osteoprotegrin ratio, subgingival biofilms profoundly skew the RANKL:osteoprotegrin axis.48 RANKL derived from diverse cell types critically regulates periodontitis-induced alveolar bone loss. Kawai et al initially reported that B and T lymphocytes are the primary source of RANKL in the bone resorptive lesion of periodontal disease.60 The report was based on immunofluorescent confocal microscopy analysis of CD3+RANKL+ T cells, CD20+RANKL+ B cells, and CD14+RANKL+ monocytes in healthy versus diseased clinical gingival tissue isolates. Notably, the study did not assess the impact of periodontitis on RANKL expression by osteoblasts, osteocytes, or periodontal ligament cells. Pacios et al later reported that osteoblast lineage cells play an essential role in periodontal bone loss through the activation of nuclear factor-kappa B.61 Transgenic mice with osteoblast lineage–specific depletion of nuclear factor-kappa B signaling were orally inoculated with Porphyromonas gingivalis and Fusobacterium nucleatum. Study outcomes revealing that wild-type mice had increased alveolar bone loss, osteoclastogenesis, and RANKL expression led the authors to postulate that osteoblast RANKL contributes to periodontal bone loss.61 Graves et al employed transgenic mice with osteocyte-specific deletion of RANKL to discern that osteocytes play an important role in periodontitis through expression of RANKL.62 Oral inoculation of transgenic mice with P. gingivalis and F. nucleatum showed that osteocyte RANKL critically regulates bacteria-driven osteoclastogenesis and periodontal bone loss. Most recently, Tsukasaki et al applied the murine ligature periodontitis model to transgenic mice with targeted deletion of RANKL in B cells, T cells, osteoblastic cells, and periodontal ligament cells.63 Study outcomes discerned that osteoblast RANKL and periodontal ligament RANKL most substantially contributed to periodontal bone loss. T-cell–derived RANKL induced periodontal bone loss to a lesser extent than osteoblast RANKL and periodontal ligament RANKL, whereas B-cell–derived RANKL had no effect. These studies support the notion that periodontitis-associated biofilms upregulate RANKL across diverse cell types to drive osteoclast-mediated alveolar bone loss. Future investigations are indicated to determine the role of cell-specific osteoprotegrin alterations in periodontitis-driven catabolic effects on alveolar bone.
4 |. BONE REMODELING
Bone remodeling (turnover) is the skeletal renewal process in which myeloid-lineage–derived osteoclastic cells resorb old bone matrix, and mesenchymal-lineage–derived osteoblastic cells subsequently form new bone matrix. Skeletal homeostasis occurs when osteoclast-osteoblast actions are balanced (“coupled”), and there is no net gain/loss of osseous tissue. Skeletal bone loss occurs when the actions of osteoclastic cells exceed those of osteoblastic cells, and there is a net loss of osseous tissue.64–67 Osteoclast-osteoblast–mediated bone remodeling processes are modulated by mechanical loading forces, paracrine/juxtracrine signaling interactions with nearby bone and immune cells, and endocrine signaling events induced by circulating hormones and immune factors.
Bone remodeling takes place in an asynchronous manner throughout the skeleton and occurs at distinct anatomic sites known as basic multicellular units.64 The basic multicellular unit consists of osteoclastic cells and osteoblastic cells that remodel (turnover) the bone through distinct and sequential phases: activation, resorption, reversal, formation, and termination.68 The healthy adult skeleton contains an estimated one million active basic multicellular units, which turn over approximately 10% of the entire skeleton per year.69,70 Trabecular bone is estimated to remodel at an average rate that is seven times more rapid than cortical bone,69,70 which highlights that trabecular bone may be more susceptible to inflammatory bone loss. Considering that the estimated lifespan of bone-resorbing osteoclasts is 2 weeks and bone-forming osteoblasts is 3 months in the basic multicellular unit,69,70 alterations in osteoblast activity can substantially impact skeletal homeostasis.
Saffar and coworkers71,72 performed seminal research in the golden hamster, which delineated that periodontal disease causes alveolar bone loss through the disruption (“uncoupling”) of balanced osteoclast-osteoblast–mediated bone remodeling. These reports showed that dietary plaque-induced periodontal inflammation critically altered the actions of osteoclasts and osteoblasts within the basic multicellular unit.71,72 Experimental periodontitis-induced alveolar bone destruction was driven by the following changes in physiologic bone remodeling: upregulated activation of bone cells initiating small foci of bone remodeling; increased osteoclast-mediated bone resorption; increased reversal (empty resorption lacunae lined by mononuclear cells), and decreased osteoblast-mediated bone formation.71,72 Importantly, these seminal studies clearly delineated that periodontitis-driven alveolar bone destruction is secondary to enhanced osteoclast-mediated bone resorption and suppressed osteoblast-mediated bone formation.
Early work by Irving et al73 lends support to the view that osteoblasts critically regulate periodontal bone loss. Weanling gnotobiotic rats were mono-infected with gram-positive bacteria: Actinomyces naeslundii, Actinomyces viscosus, or Streptococcus mutans. Osteoblast activity was evaluated via 3H proline labeling, and osteoclast activity was assessed by the acid phosphatase technique. Mono-infection with each of the three gram-positive bacteria induced periodontal disease as evidence by the plaque accumulation, apical migration of the junctional epithelium, and alveolar bone destruction. Notably, the alveolar bone destruction was attributed to cessation of active bone formation, not enhanced osteoclastogenesis. In order to validate that the aforementioned study outcomes were not due to animal species or gnotobiotic status, weanling conventional hamsters were subsequently superinfected with A. naeslundii. Periodontal bone loss occurred more slowly in the hamster model than in the rat model but was defined by severely impaired osteoblastogenesis.73
Irving et al also carried out a follow-up investigation to determine whether gram-negative bacteria cause periodontal bone destruction through actions on osteoblasts or osteoclasts.74 Weanling gnotobiotic rats were mono-infected with a gram-negative, anaerobic rod isolated from a case of human periodontitis. Periodontal disease was characterized by minimal plaque formation, apical migration of the junctional epithelium, and alveolar bone destruction. Alveolar bone destruction was attributed to severely suppressed active bone formation and substantially enhanced osteoclastogenesis. Irving et al speculated that the alveolar bone loss that occurred in the absence of gross plaque accumulation was likely attributed to endotoxin.74 The work by Irving and coworkers73 importantly reveals that both gram-positive and gram-negative bacterial biofilms have catabolic effects on alveolar bone, which are mediated in part through anti-osteoblastic actions.
Despite knowledge that periodontitis-driven bone loss is in part attributed to suppressed osteoblastogenesis,71,72 the osteoblastic cell lineage is, for the most part, overlooked by periodontal researchers. However, there is indirect evidence from recent studies that further highlights the importance of the osteoblastic cell lineage in the pathogenesis of periodontal bone loss (Figure 3). Exogenous intermittent administration of the first 34 amino acids of parathyroid hormone is a US Food and Drug Administration–approved anabolic drug for the treatment of osteoporosis. Exogenous intermittent parathyroid hormone 1–34 has anabolic effects on bone through stimulating osteoblast-mediated bone formation.18,75 Exogenous intermittent parathyroid hormone 1–34 administration in the rat ligature periodontitis model has been shown to prevent/protect against periodontitis-associated alveolar bone loss,76,77 and clinically has been shown to adjunctively enhance periodontal surgery outcomes.78 In January 2019, the Food and Drug Administration recommended approval of another anabolic drug, romosozumab, for the treatment of postmenopausal osteoporosis. Romosozumab is a monoclonal antibody that stimulates bone formation by binding to sclerostin, the osteocyte-derived protein that suppresses osteoblastogenesis.18,79 Interestingly, sclerostin-antibody administration has been shown to support alveolar bone regeneration in the rat ligature periodontitis.80,81
FIGURE 3.
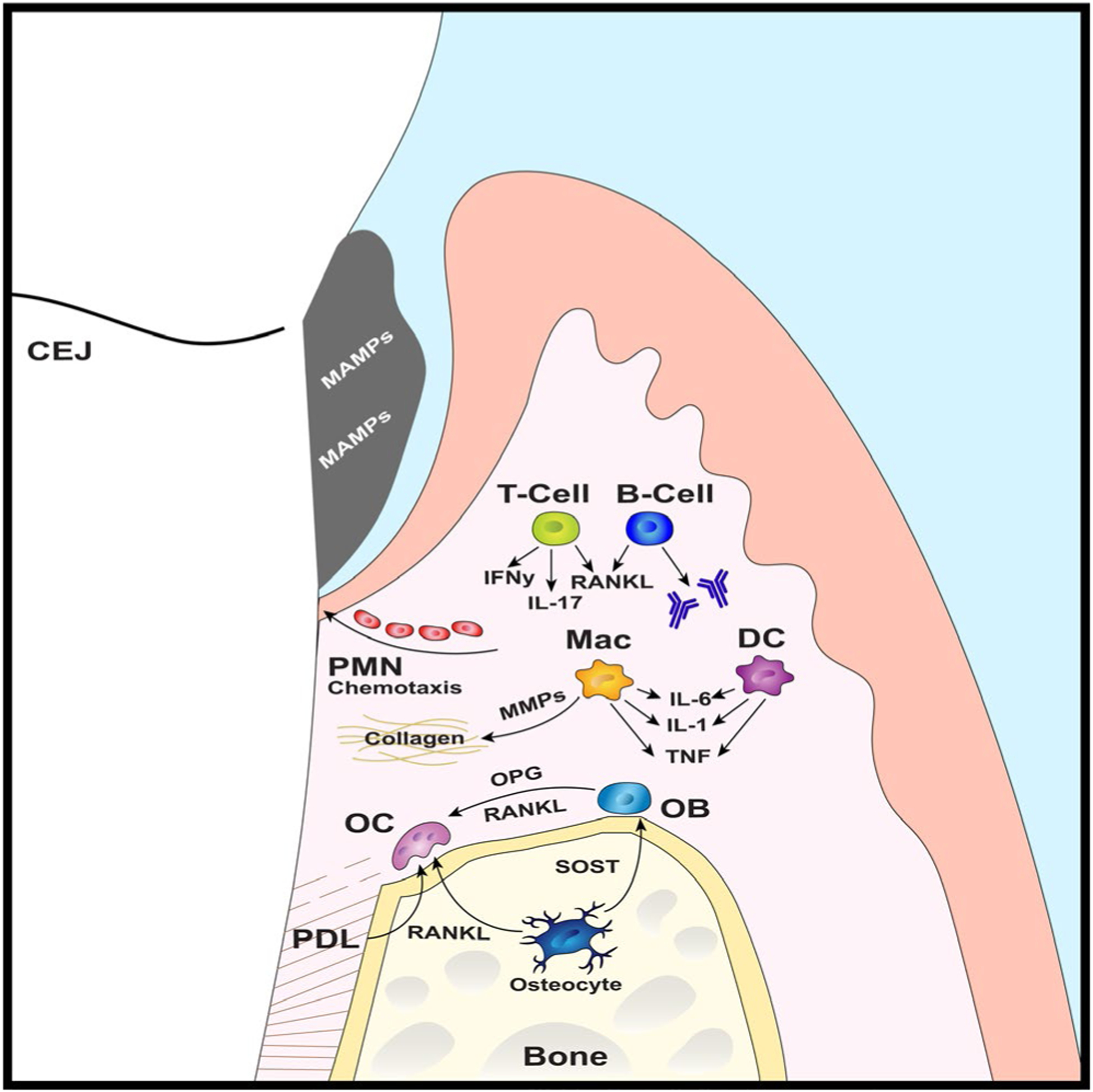
Periodontal immune microenvironment. Subgingival plaque biofilm derived microbe-associated molecular patterns stimulate the periodontal immune response. Neutrophils are recruited to the junctional epithelium via chemokine gradients. Innate immune cells, such as macrophages and dendritic cells, secrete matrix metalloproteinases and proinflammatory cytokines—that is, tumor necrosis factor (TNF), interleukin-1 (IL-1), interleukin-6 (IL-6)—which disrupt connective tissue homeostasis. Osteoblasts, osteocytes, periodontal ligament cells, and lymphocytes (ie, T cells, B cells) secrete receptor activator of nuclear factor-kappa B ligand (RANKL) and other proinflammatory factors, which support osteoclastogenesis and inhibit osteoblastogenesis. B cells synthesize antibodies that drive the humoral response. Osteocytes secrete sclerostin to suppress osteoblastogenesis. CEJ, cementoenamel junction; DC, dendritic cells; IFNy, interferon-gamma; Mac, macrophages; MAMPs, microbe-associated molecular patterns; MMP, matrix metalloproteinases; OB, osteoblasts; OC, osteoclasts; OPG, osteoprotegrin; PDL, periodontal ligament; PMN, neutrophils; SOST, sclerostin
5 |. ALVEOLAR BONE: A UNIQUE OSSEOUS TISSUE
Alveolar bone is a unique osseous tissue owing to its integration with the dentition and close proximity to oral biofilms. Bone remodeling in the alveolar bone complex has been reported to take place at a rate that is three to sixfold more robust than at nonoral skeletal sites.82–84 Mechanical loading studies estimate that functional strains are two to fourfold higher in alveolar bone than at nonoral skeletal sites,85–88 which implies that occlusal forces transmitted through the fibrous junction of the periodontal ligament contribute to the robust remodeling of alveolar bone. Periodontal research studies revealing that oral biofilms significantly modulate alveolar bone metabolism in periodontal health89 and disease71,72 highlight that oral biofilms critically regulate alveolar bone remodeling.
The host is colonized by diverse microorganisms at mucosal barrier surfaces exposed to the external environment. The collection of microbes colonizing distinct anatomic sites are referred to as microbiota communities.90–92 The oral cavity is a distinct anatomic barrier environment to colonizing microbes due to the presence of a transmucosal organ, the tooth, which supports vigorous biofilm formation and is integrated with alveolar bone. The spatial relationship of the oral microbiota to alveolar bone is unique, in that no other microbiota community (ie, gut, skin, urogenital, lung) colonizes an external body surface directly integrated with osseous tissue.
The dentogingival junction, which consists of the epithelial and connective tissue attachment to the tooth,93,94 protects the subjacent alveolar bone from microbes resident in the dental plaque biofilm.95,96 Whereas epithelial tissues typically act as an impermeable barrier to microbial biofilms colonizing external body surfaces, the junctional epithelial attachment at the tooth surface is highly permeable.95,97 The periodontal innate immune response defends against microbial invasion of the permeable junctional epithelium via the continuous transit of gingival crevicular fluid98,99 and leukocytes into the gingival sulcus.100–103 Notably, polymorphonuclear cells (neutrophils) transiting through the gingival vascular plexus to the junctional epithelium emigrate to the gingival sulcus, and they do not infiltrate or reside in the connective tissue extracellular matrix.95,96,104 Polymorphonuclear cells emigrating into the gingival sulcus form a barrier wall between the epithelium and plaque biofilm, and thus have been considered the first line of the periodontal immune defense.97,105,106 Dentogingival biofilms stimulate immune response effects in barrier epithelial and underlying connective tissues, which induce paracrine signaling that influences alveolar bone homeostasis.95,96,104,107
6 |. SUBGINGIVAL BIOFILM ECOSYSTEM: INTER ACTIONS WITH THE HOST
Advances in the understanding of periodontal biofilm ecosystems and the host immune response have led to evolving theories of periodontal disease pathogenesis. Early observations demonstrating that increased bacterial plaque accretions were associated with periodontal disease sites shaped the “nonspecific plaque hypothesis.” This hypothesis is based on the notion that the increased quantity of bacteria, irrespective of the presence of specific bacteria, stimulates periodontal tissue destruction.108,109 Advances in bacterial culturing techniques and the advent of whole-genome deoxyribonucleic acid probes enabled researchers to identify prominent bacteria in dental plaque biofilms from healthy versus diseased sites. Investigations of subgingival plaque biofilms from active periodontal disease sites led to the “specific plaque hypothesis.” This hypothesis dictates that infection by a specific putative periopathogenic bacterium (eg, P. gingivalis, Aggregatibacter actinomycetemcomitans) or a small cluster of interacting putative periopathogenic bacteria (eg, “red complex”: P. gingivalis, Tannerella forsythia, Treponema denticola) drives periodontitis-associated alveolar bone loss.109,110
Oral biofilms are dynamic polymicrobial communities that involve interspecies synergies and competition, which have implications for the periodontal immune response and alveolar bone homeostasis.111–113 Oral inoculation–induced experimental periodontitis investigations have been carried out to discern the role of single bacteria versus a consortium of bacteria in the etiology of periodontal bone loss. Mono versus polymicrobial oral infection models have been employed to evaluate the interactions of red complex (P. gingivalis, T. denticola, T. forsythia) periopathogenic bacteria in periodontitis-driven alveolar bone loss. F. nucleatum was also investigated based on evidence that it serves as a bridge bacterium between early and late-colonizing plaque biofilm species. Kesavalu et al114 subjected rats to P. gingivalis, T. denticola, or T. forsythia monomicrobial oral infections or combined polymicrobial oral infections with or without F. nucleatum. Rats that were infected with the polymicrobial consortium exhibited increased alveolar bone loss compared with rats infected with one of the microbes, which was independent of the presence of F. nucleatum.114 Polak et al orally infected mice with P. gingivalis or F. nucleatum alone or both bacteria together.115 Combined F. nucleatum/P. gingivalis infection relative to monoinfections induced more alveolar bone loss in vivo and increased tumor necrosis factor and interleukin-1beta levels in the subcutaneous chamber model. Orth et al116 orally inoculated mice with P. gingivalis or T. denticola alone or coinfected with P. gingivalis/T. denticola. Four doses of a co-inoculum at 1:1 ratio of P. gingivalis and T. denticola (5 × 108 total bacterial cells) was shown to induce the same level of bone loss as four doses of 1 × 1010 P. gingivalis cells. Co-inoculum induction of T cell proliferation and interferon-gamma–dominant responses relative to mono-inoculum treatments suggest that P. gingivalis and T. denticola act synergistically to stimulate the host immune response and drive periodontal bone loss. Settem et al subjected mice to monomicrobial infection with F. nucleatum or T. forsythia alone or coinfection with both bacteria.117 Coinfection of F. nucleatum/T. forsythia was reported to be more potent than monomicrobial infections in inducing nuclear factor kappa B activity and proinflammatory cytokine secretion (ie, interleukin-1beta, interleukin-6, tumor necrosis factor) in monocytic cells and primary murine macrophages. In vivo comparison demonstrated that coinfection relative to monomicrobial infections resulted in increased gingival inflammatory infiltrate, enhanced osteoclast cell numbers lining bone, and synergistic effects on alveolar bone loss.117 However, Yamazaki et al utilized periopathogenic bacteria (P. gingivalis, Filifactor alocis, and F. nucleatum) or healthy periodontal bacteria (A. naeslundii, Streptococcus mitis, and Veillonella rogosae) in germ-free mice to find no alveolar bone resorption in either group.118
Periodontitis is currently understood to be a polymicrobial disease that results from complex interactions among the commensal microbiota, host susceptibility, and environmental factors. Periodontal pathogenesis appears to be driven by an excessive proinflammatory immune response to shifts in the normal microbiota, which are exacerbated by disease-associated bacterial species. Investigations in the germ-free versus specific-pathogen-free mouse model have discerned that the oral commensal microbiota induces a low-grade proinflammatory immune response state that drives alveolar bone loss in health.89,119 We have recently shown that antibiotic perturbation of the commensal microbiota induces a supraphysiologic proinflammatory immune response state that has catabolic effects on the skeleton, both at oral (unpublished data) and nonoral120 sites. Hajishengallis et al provided evidence supporting the notion that proinflammatory alveolar bone loss is exacerbated by periopathogenic bacteria–induced shifts in the normal microbiota.119 The seminal murine experimental periodontitis study demonstrated that low-abundance P. gingivalis triggers changes to the amount and composition of the oral commensal microbiota, which drives proinflammatory periodontal bone loss.119
The application of powerful 16S ribosomal RNA gene amplification and sequencing techniques has enabled investigators to characterize the bacterial microbiome (genomic complement) of microbiota communities. The oral microbiota is a diverse microbial community made up of roughly 1000 bacterial species.121–123 454-Pyrosequencing of 16S ribosomal RNA gene amplicons in clinical subgingival plaque isolates has revealed that the subgingival microbiota consists of roughly 700 bacterial species.124 Advanced knowledge about the composition of subgingival bacterial biofilms in health and disease calls into question whether periodontitis is secondary to infection by empirically defined putative periopathogenic bacteria.
Griffen et al reported that subgingival plaque from chronic periodontitis versus healthy patients demonstrated an increased diversity, both in the number of species and the combination of richness and evenness reflected by the Shannon index.124 The bacterial species more prominent in health were not lost in disease but constituted a smaller fraction of the total community. The investigation confirmed associations of P. gingivalis, T. forsythia, and T. denticola with disease. However, the study revealed substantial numbers of additional species associated with disease that are not empirically defined putative periopathogenic bacteria. F. alocis, a bacterium not previously associated with periodontitis, was one of the top three associated species.124
Abusleme et al found that subgingival plaque from chronic periodontitis versus healthy patients demonstrated a higher bacterial load and richness.125 Empirically defined putative periopathogenic bacteria were present at low levels in health, which calls into question whether infection by specific microbes induces the host proinflammatory response that mediates periodontal tissue destruction. Based on findings that a higher community biomass was associated with disease, the supraphysiologic host immune response causing periodontal breakdown may be secondary to an overall greater bacterial challenge. Considering that periodontal disease was characterized by the emergence of newly abundant taxa concurrent with an increase in bacterial biomass,125 the report provides evidence supporting the postulate that the pathogenesis of periodontitis is secondary to ecological shifts in the subgingival microbiome.126
Our current understanding of periodontitis as a polymicrobial disruption of host homeostasis104,107 is centered on the bacterial component of the microbiota. However, the oral microbiota is comprised of diverse microbial species, including bacteria, fungi, viruses, archaea, and other eukaryotes.112,123,127 Recent advances in 18S ribosomal RNA and 28S ribosomal RNA genomic sequencing have begun to define the mycobiome (fungal component of the microbiome), which has led to research discerning the impact of the mycobiota (fungal component of the microbiota) on the host immune system.128,129 Candida albicans has recently been identified as the single, ubiquitous member of the microbiota that is central for the systemic induction of antifungal TH17 cell–mediated immunity.130,131 C. albicans is considered a commensal fungus with pathogenic potential that colonizes the gut, skin, and oral cavity at low levels during health. Bacher et al delineated that C. albicans is responsible for the systemic induction of human antifungal TH17 responses.130 Shao et al demonstrated that intestinal colonization of mice with C. albicans induces systemic expansion of fungal-specific TH17 CD4+ T cells and interleukin-17 responsiveness that protects against C. albicans invasive infection.131 Notably, C. albicans–induced TH17/interleukin-17–mediated immunity was shown to confer protection against invasive extracellular pathogens that extend beyond the fungal microorganisms, including Staphylococcus aureus.131
TH17 cell–mediated immunity has been shown to be essential for oral mucosal host defense against C. albicans–mediated oral candidiasis132 and periopathogenic bacteria–induced periodontal bone loss.133 However, the relationship between interkingdom microbial interactions and oral immunity is poorly understood. C. albicans and F. nucleatum species demonstrate strong coaggreagation,134,135 which has immunoregulatory effects on macrophage immune defense that appear to favor sustained colonization.136 The cooperation of C. albicans and F. nucleatum highlights the potential for interkingdom interactions in plaque biofilms, which may have implications for the pathogenesis of periodontitis-driven alveolar bone loss.
7 |. PATHOGENESIS OF ALVEOLAR BONE DESTRUCTION: RANGE OF EFFECTIVENESS
Subgingival plaque biofilms induce a supraphysiologic proinflammatory immune response state in the gingival connective tissue, leading to paracrine signaling that drives alveolar bone destruction. Page and Schroeder estimated that the “range of effectiveness” of subgingival plaque to induce alveolar bone destruction is about 2.5 mm.137 This postulate is supported by Garant and Cho’s theory that dental plaque–induced bone resorption factors in the connective tissue have an “effective radius of action,”138 and by Waerhaug’s work showing that the linear distance from the apical extension of the subgingival plaque to the alveolar crest ranged from 0.5 mm to 2.7 mm.139 Histologic investigations performed by Lindhe and Ericsson in the beagle dog ligature model140 and by Rowe and Bradley in clinical autopsy specimens141 have shown that the proximity of the inflammatory infiltrate to the alveolar bone correlates with the number of osteoclasts and resorption lacunae present.
Chronic pathophysiologic stimulation by dental plaque biofilms leads to the excessive production of proinflammatory mediators, which can ultimately have catabolic effects on balanced tissue remodeling processes. Subgingival plaque–induced supraphysiologic immune and inflammatory responses result in pathophysiologic levels of prostaglandins, proteases, matrix metalloproteinases, cytokines, and other host enzymes being released from epithelial cells, fibroblasts, polymorphonuclear leukocytes, monocytes, macrophages, osteoblasts, or other host cells.142–144 The chronic proinflammatory state dysregulates fibroblast-mediated remodeling of the gingival connective tissue and the periodontal ligament, which leads to reduced collagen content and compromised tissue integrity.142,143 Periodontal ligament collagen fibers irreversibly detach from the root surface, which results in the junctional epithelium extending apically. This facilitates further apical extension of subgingival biofilms (Figure 4). As the periodontal pocket deepens, the anaerobic microenvironment favors gram-negative periopathogenic bacteria, increasing the site’s risk for disease progression.137,145
FIGURE 4.

Localized periodontitis at maxillary right first molar. Apical migration of subgingival biofilm–mediated alveolar bone destruction at the distobuccal root
The inflammatory infiltrate spreads deeper within the periodontitis-afflicted connective tissue, driving proinflammatory paracrine signaling effects that dysregulate “coupled” osteoclast-osteoblast–mediated bone remodeling processes. Diverse cells within the local periodontal environment synthesize increased levels of RANKL and lower levels of osteoprotegrin, which skews the RANKL:osteoprotegrin axis to favor osteoclastic bone resorption.46,47 A multitude of cells in the local microenvironment synthesize proinflammatory cytokines (ie, tumor necrosis factor, interleukin-1beta, interleukin-6, interleukin-17) that further upregulate RANKL production and/or have synergistic effects on RANKL signaling, which exacerbates osteoclast bone resorptive processes (Figure 3).142–144 Furthermore, interleukin-1beta, interleukin-6, and tumor necrosis factor have potent anti-osteoblastic actions that suppress osteoblast differentiation and function (Figure 3).18,144 The subgingival plaque–induced proinflammatory host immune response state therefore drives periodontal bone destruction through enhanced osteoclast activity and/or suppressed osteoblastic activity.
8 |. PATHOGENESIS OF ALVEOLAR BONE DESTRUCTION: REVISITING FIBROSIS
Page and Schroeder146 laid the foundation for our understanding of cellular changes in epithelial and gingival connective tissue during periodontal health and disease. The investigation depended on histopathologic and ultrastructural analysis of periodontal tissues specimens. Despite not having advanced molecular-cellular assays available, the report was written from the perspective that the host immune response to colonizing oral biofilms drives the pathogenesis of periodontitis.146 The seminal work is still considered to be the most influential manuscript on periodontal disease pathogenesis due to current knowledge that the host immune response to plaque biofilms drives periodontal tissue destruction.
Page and Schroeder proposed that the pathogenesis of periodontitis occurred via a succession of stages (ie, initial lesion, early lesion, established lesion, advanced lesion), which resulted in pathologic changes to the structure and function of the periodontium.146 We will focus on the advanced lesion, since it is the only stage afflicted by appreciable bone loss. The advanced lesion is characterized by a dense infiltrate of lymphocytes, macrophages, and plasma cells that spreads deep through the gingival connective tissue, progressive loss of collagen subjacent to the pocket epithelium that continues to migrate apically/laterally, and destruction of the periodontal ligament attachment and supporting alveolar bone. Bone destruction initiates along the crest of the interdental septum, which results in fibrotic gingival thickening. Exposed marrow spaces become hypercellular, undergo fibrosis, and transform to dense fibrotic scar tissue.
Page and Schroeder posited that, “Generally, bone destruction begins along the crest of the interdental septum around the communicating blood vessels.146 As the marrow spaces are opened, both red and white marrow become hypercellular, undergo fibrosis and become transformed into scar like connective tissue.” Weinmann introduced the alveolar bone fibrosis theory, which was realized during a histologic investigation of the effects of chronic periodontitis in human periodontium specimens collected at autopsy.147 The author explained that the inflammatory infiltrate in the superficial connective tissue leads to resorption of the alveolar bone crest, an inflammatory reaction following along the blood vessels into the bone marrow spaces, and a transformation of the fatty marrow into fibrous marrow.147 Moskow and Polson performed an extensive histological investigation evaluating the extension of the inflammatory infiltrate in human periodontitis.148 The study included 350 autopsy and surgically retrieved segments of human jaws. The authors reported that the inflammatory infiltrate extends into the alveolar process and elicits a response. Penetrations of inflammatory cells into the marrow was commonly associated with fibrosis of the fatty marrow. The authors state, “Our observations are in agreement with those of Weinmann (1941), in that a major pathway of the inflammatory infiltrate in periodontitis is through the intra-alveolar vessels.”148
The clinical studies performed by Weinmann,147 Page and Schroeder146, and Moskow and Polson148 are, astonishingly, the only known published reports that have addressed the alveolar bone fibrosis phenomena. Personal communication with Roy Page provided valuable insight as to why the fibrosis mechanism of periodontal pathogenesis has been overlooked. Experimental periodontitis model limitations are largely responsible. Preclinical models do not recapitulate the long-term chronic proinflammatory immune response state induced by subgingival biofilms in chronic periodontitis. Small rodent animal models typically do not have naturally occurring periodontal disease. Furthermore, the standard experimental approaches employed to induce periodontitis (ie, periopathogen oral inoculation, ligature placement) have limited capacity to alter the indigenous subgingival microbiota and are too short in duration.
Fibrosis is the common pathologic outcome across many chronic inflammatory diseases. Extensive research efforts have begun to define the pathophysiology of fibrotic conditions. Reviewing this knowledge lends strong support to the postulate that chronic periodontitis–induced alveolar bone destruction occurs in part through fibrosis. General fibrosis principles that apply to all fibrotic diseases include the following:
Fibrosis is caused by unresolved chronic inflammation that dysregulates physiologic tissue remodeling and leads to aberrant fibrous connective tissue deposition.
Fibrosis ultimately results in loss of tissue architecture and progressive loss of organ function.149–151
In line with the first principle, chronic periodontitis has been shown to dysregulate balanced (“coupled”) osteoclast-osteoblast–mediated alveolar bone remodeling,71,72 leading to the deposition of fibrous scar-like tissue within the alveolar bone marrow compartment.146–148 In line with the second principle, chronic periodontitis ultimately results in loss of alveolar bone architecture and progressive loss of periodontal function, which is synonymous with loss of dentoalveolar support that can terminally result in tooth loss.
Cutting-edge microbiome research has recently revealed that distinct microbiota communities critically regulate the pathogenesis of fibrotic conditions afflicting the liver,152,153 lungs,154–156 and skin.157,158 Dysfunction of the gut epithelial barrier leads to increased translocation of gut bacterial by-products/metabolites into the portal venous circulation, which contributes to the development and progression of nonalcoholic fatty liver disease.152,153 Dysbiosis of the lung microbiota has been shown to exacerbate lung fibrosis via upregulating a profibrotic inflammatory cytokine network.156 Furthermore, the skin microbiota can have potent proinflammatory immunomodulatory actions that induce fibrous healing (scarring) of skin wounds.158 Knowledge that nonoral microbiota–host immune response effects contribute to fibrotic conditions further supports the notion that periodontitis-induced alveolar bone destruction occurs in part through fibrosis. Future research is critically needed to discern the role of fibrosis in the pathophysiology of clinical periodontal bone loss.
9 |. OSTEOIMMUNOLOGY: INTERACTION OF IMMUNE CELLS: BONE CELLS
The field of osteoimmunology has demonstrated that the immune cell interactions with bone cells regulate skeletal development and homeostasis, under physiologic and pathophysiologic conditions.58,159–161 Osteoimmunological studies have led to explorations of dissecting the crosstalk between bone cells and immune cells in the regulation of bone turnover.58,159–161 The commensal microbiota is a critical regulator of the host immune response and immunological education.162 Importantly, the commensal microbiota has recently been shown to have immunomodulatory actions that regulate osteoclast-osteoblast–mediated bone remodeling processes.89,163
Page and Schroeder provided a sound conceptual framework for the understanding of the impact of subgingival biofilms on cellular changes in the periodontium.146 Whereas Page and Schroeder focused on macrophages, lymphocytes, and plasma cells,146 the field of periodontal research has carried out elaborate investigations further defining the interactions of subgingival biofilms and specific adaptive and innate immune cells (Figure 5). The focus of this section is centered on the current understanding of oral microbiota-periodontal immune processes regulating alveolar bone homeostasis in health and disease.
FIGURE 5.
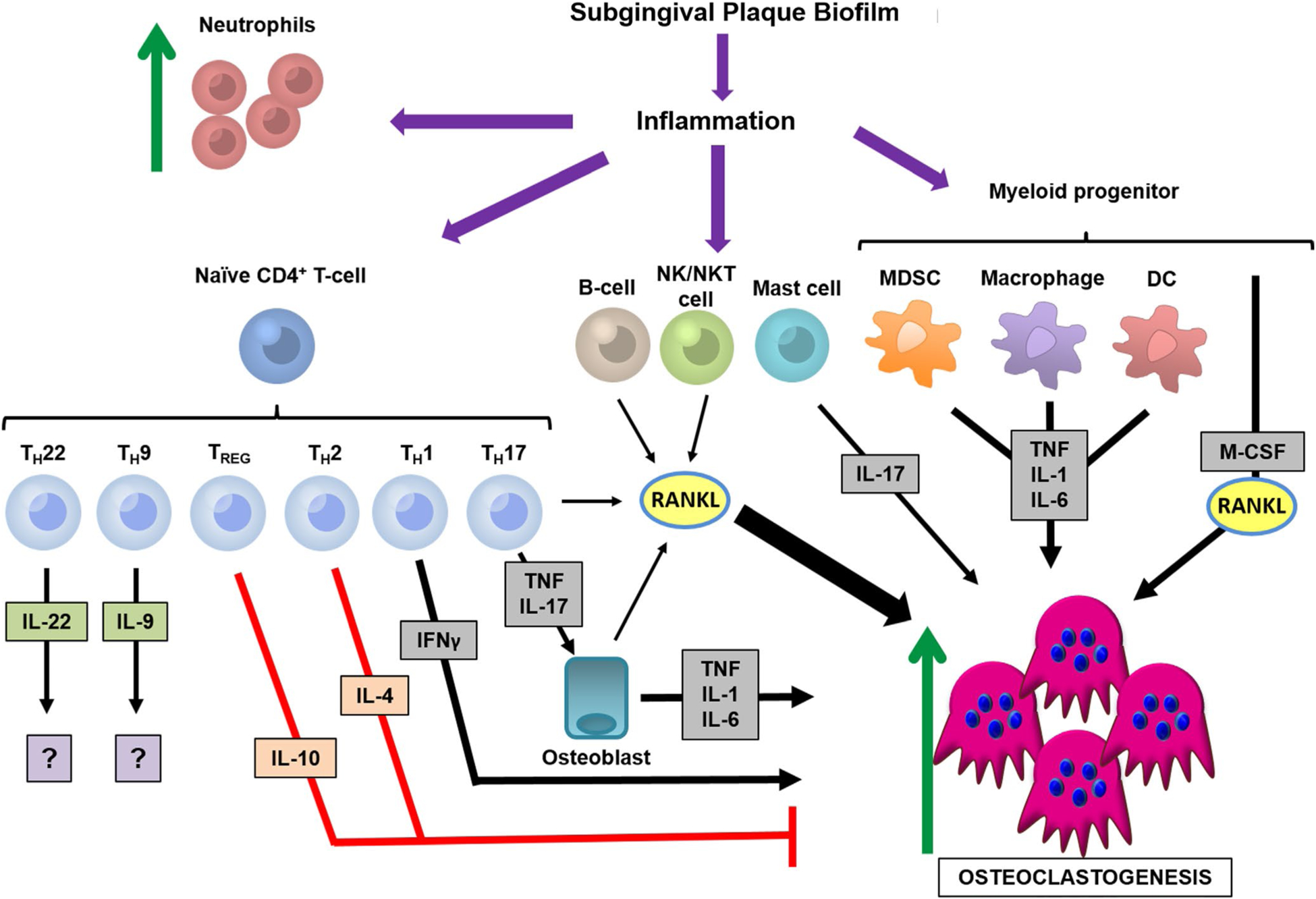
Periodontal osteoimmunology. The subgingival plaque biofilm induces a multitude of immune cells to mount the periodontal immune response. There is an increase in neutrophil chemotaxis, as well as differentiation of naive CD4+ T cells into several subsets. TH1 cells secrete interferon-gamma (IFNγ), which can have pro-osteoclastogenic properties. TH17 cells secrete interleukin-17 (IL-17) and tumor necrosis factor (TNF), which induce osteoblasts to synthesize proinflammatory/pro-osteoclastic factors. T regulatory cells and TH2 cells secrete interleukin-10 (IL-10) and interleukin-4 (IL-4), which have anti-osteoclastogenic effects. TH22 cells and TH9 cells produce interleukin-22 (IL-22) and interleukin-9 (IL-9), factors that currently have unclear roles in periodontal osteoimmunology. B cells, natural killer (NK) cells, and natural killer T (NKT) cells can secrete receptor activator of nuclear factor-kappa B ligand (RANKL), whereas mast cells can produce interleukin-17 (IL-17) to support osteoclastogenesis. Myeloid-lineage cells, such as myeloid-derived suppressor cells (MDSC), macrophages, and dendritic cells (DC), synthesize proinflammatory cytokines—that is, TNF, interleukin-1 (IL-1), interleukin-6 (IL-6)—that support osteoclastogenesis. M-CSF, macrophage colony-stimulating factor; TREG, T regulatory cells
9.1 |. CD8+/CD4+/gamma delta T cells
T cells are the critical players in adaptive immunity that regulate the function of antigen-presenting cells and B-cell–mediated humoral immunity.66 T cells recognize immunological epitopes through T-cell receptors and through antigens presented on major histocompatibility complex molecules from antigen-presenting cells. Unlike T cells that are specific for self-antigens expressed by thymic epithelial cells, commensal microbiota–specific T cells do not undergo negative selection in the thymus and are present in healthy individuals despite the constant stimulation of their cognate antigens.164 Furthermore, T cells are essential for productive, long-lasting immune responses and immunological memory.
CD8+ T cells function in monitoring shifts in peptide antigens presented on major histocompatibility complex class I molecules, which are expressed on all nucleated cells. CD8+ T cells also possess cytotoxic properties, where they can control other immune cells to avoid excessive immune activation and its pathological consequences.165 CD4+ T cells recognize both self and foreign peptides presented by the major histocompatibility complex class II pathway,166 allowing for a robust and lasting immune response and the establishment of effective immune memory.167 Gamma delta (γδ) T cells are an unconventional, less prominent T cell population that have important immune surveillance properties at epithelial and mucosal barriers,168 including the oral cavity.169,170 There are specific γδ T-cells subsets that are adapted to specific barrier sites. This cell population can be induced by the commensal microbiota and pathogens, influencing such properties as generation, effector functions, proliferation, and maintenance.168–170 Interestingly, oral mucosal homeostasis is orchestrates by mutual interplay between interleukin-17–producing γδ T cells and the microbiota.170,171
Under pathologic conditions, studies have clarified how T cells can influence bone remodeling through direct expression of pro-osteoclastic cytokines and indirect signaling to stromal-osteoblastic cells.30,172,173 Kong et al showed that T cells isolated from global RANKL knockout mice produced less interleukin-2 and interferon-gamma, which was attributed to a developmental defect of these T-cells and not a direct effect of RANKL on mature T cells.30 RANKL expression in T cells is induced by T-cell receptor engagement with an antigen receptor,172 which is dependent on T-cell receptor activation-induced calcium ion mobilization.173 T cells have been shown to critically regulate osteoclastogenesis through the expression of RANKL and other biologic mediators.66,174
Extensive periodontal research has been performed to discern the role of T cells in periodontitis-driven alveolar bone loss. Investigations from the late 1980s to 1990s often referred to the ratio of CD4:CD8 T cells as means to determine the extent of the immune response in periodontal lesions. Studies have reported a decreased CD4:CD8 ratio in periodontitis lesions compared with healthy sites or peripheral blood,175–178 although an increased CD4:CD8 ratio179 or no changes in CD4:CD8 have also been reported.180,181 Investigations characterizing T lymphocytes present in gingival tissues afflicted by periodontitis determined that T cells are primed to become memory T cells.182,183 During the progression of periodontal disease, subgingival plaque bacterial antigens are taken up by innate immune cells. These innate immune cells are believed to migrate to the cervical lymph nodes, where they present antigens to prompt the adaptive immune response. This allows for T cell activation leading to memory T cells, which have been found in the peripheral blood of chronic periodontitis patients versus healthy control subjects.184
Several studies used severe combined immunodeficient mice, which lack both T and B cells, to begin to delineate the role of T cells in periodontitis-induced alveolar bone loss. Teng et al transferred human peripheral blood leukocytes from periodontal disease patients into non-obese diabetic/severe combined immunodeficient mice and then orally challenged with A. actinomycetemcomitans.50 Outcomes from the investigation suggest that A. actinomycetemcomitans–driven periodontal bone loss is mediated in part by RANKL expression on T cells.50 Baker et al orally infected severe combined immunodeficient mice and immunocompetent control mice with P. gingivalis. Severe combined immunodeficient mice were more resistant to oral infection–induced periodontal bone loss, which implies that T cells and B cells significantly modulate P. gingivalis–driven alveolar bone destruction.185 Baker et al employed the P. gingivalis oral infection model in mice deficient in CD4+ T-cell signaling to major histocompatibility complex class II (Aβ-knockout mice) and mice lacking major histocompatibility complex class I signaling to CD8+ T cells and NK1+ T cells (B2m-knockout mice).186 Aβ-knockout mice (deficient in major histocompatibility complex class II–responsive CD4+ T cells) exhibited less alveolar bone loss than wild-type mice did, whereas B2m-knockout mice (deficient in major histocompatibility complex class I–responsive CD8+ T cells and NK1+ T cells) were not protected against P. gingivalis–induced alveolar bone loss.186 These outcomes support the notion that CD4+ T cells are important effectors of alveolar bone loss consequent to P. gingivalis oral infection.186
Though CD8+ T cells and γδ T cells may not be the primary inducer of periodontitis-driven alveolar bone loss, they likely play important roles modulating the host response to subgingival biofilms in periodontal disease. Importantly, CD8+ T cells suppress interferon-gamma–producing cells and favor humoral immune responses.187 In peripheral blood mononuclear cells isolated from chronic periodontitis patients, activated cytotoxic CD8+CD28+ T cells were significantly elevated in periodontitis patients compared with healthy controls.188 γδ T cells are present in epithelial and connective tissues in both healthy189–191 and chronically inflamed gingiva.189,192 Gemmell and Seymour reported that the proportion of γδ T cells was not significantly altered in gingival tissues’ isolates from patients afflicted by gingivitis and periodontitis.193 Tsukasaki et al evaluated changes in γδ T-cell frequency within gingival tissues of mice subjected to the ligature periodontitis model.63 Experimental periodontitis did not alter γδ T-cell frequency within gingival tissues,63 which is in line with previously reported clinical findings.193
Effector CD4+ T cell subsets, including TH1, TH2, T regulatory, TH17, TH9, and TH22, have begun to be dissected to understand their specific roles in osteoimmunology and periodontitis-driven alveolar bone loss.
9.2 |. TH1/TH2 cells
TH1 cells are CD4+ T-effector cells that target intracellular pathogens and are specifically defined by secretion of interleukin-2, interferon-gamma, and tumor necrosis factor.194,195 Interferon-gamma has been shown to have both pro and antiresorptive actions.196–201 Conversely, TH2 cells function in extracellular pathogen control and are specifically defined by interleukin-4, interleukin-5, and interleukin-13 cytokine secretion. Furthermore, TH2 cells potentiate humoral responses, supporting B cell proliferation and differentiation. TH1 and TH2 cells are mutually opposing and self-sustained, because interferon-gamma and interleukin-4 antagonize one another.194,195
TH1-derived interferon-gamma was initially reported to inhibit osteoclastogenesis, where interferon-gamma receptor–deficient mice had increased osteoclasts and enhanced bone loss.196 Interferon-gamma also has inhibitory actions on osteoclast precursors197 and tumor necrosis factor-α–induced bone marrow macrophage-derived osteoclasts.198 To the contrary, other studies reported that interferon-gamma enhances osteoclastogenesis.199–201 However, after much debate on whether interferon-gamma promotes or inhibits osteoclastogenesis, Gao et al determined that interferon-gamma dynamically regulates osteoclastogenesis.202 Interferon-gamma directly targets osteoclast precursors to inhibit osteoclast formation, and indirectly promotes osteoclast differentiation and maturation through antigen-dependent T cell activation, which induces T cells to secrete the pro-osteoclastic factors RANKL and tumor necrosis factor.202 Thus, interferon-gamma has direct anti-osteoclastogenic actions and indirect pro-osteoclastogenic actions, which are dependent upon physiologic conditions.
TH2 cytokines interleukin-4, interleukin-10, and interleukin-13 have been reported to have anti-osteoclastogenic functions.203–207 In RANKL-induced osteoclastogenesis, interleukin-4 inhibited NFATc1 expression by antagonizing nuclear factor kappa B signaling.203 Interleukin-10 also has inhibitor functions on NFATc1 expression and its nuclear translocation in osteoclastogenesis.204 Interleukin-4, interleukin-10, and interleukin-13 have been shown to upregulate osteoprotegrin and downregulate RANKL synthesis.205,206 Interleukin-10 also inhibits osteoclastogenesis by downregulating the production of pro-osteoclastogenic cytokines, such as tumor necrosis factor, interleukin-1beta, and interleukin-6.207
Reports have evaluated TH1 and TH2 characteristic cytokine expression in clinical periodontal tissues. Several studies purported more TH1 involvement over TH2 in chronic periodontitis.208,209 Investigations conveyed early onset periodontitis patients had increased interleukin-4 in gingival tissues.210–212 Others have postulated that TH2 cells are prominent in early onset and chronic periodontitis,213,214 and TH2 cytokines may be an exacerbating factor in progression from gingivitis to chronic periodontitis.211 Contradictory to these findings, several studies found that TH1 and TH2-related cytokines were comparable in vitro215 and between diseased and healthy periodontal tissues.216
Experimental periodontitis models have been utilized to define the role of TH1 and TH2 cells in inflammatory periodontal bone loss. An in vitro study found that P. gingivalis–specific T-cell lines produce both TH1 and TH2 cytokines.217 Another investigation found that P. gingivalis–specific clone T cells initially primed by cross-reactive F. nucleatum antigens were polarized to the TH2 subset, whereas T-cells stimulated with P. gingivalis alone maintained the profile of TH1 subset cells.218 Kawai et al aimed to evaluate the role of T cells, specifically using T-cell receptor/CD28–dependent TH1 or TH2 clones in rats.219 Gingival injection of A. actinomycetemcomitans antigen and lipopolysaccharide induced local bone resorption after the transfer of antigen-specific T-cell receptor/costimulatory B7–dependent TH1 clone cells but not after transfer of TH2 clone cells.219 Teng et al found that increased interferon-gamma was correlated with A. actinomycetemcomitans–specific RANKL+CD4+TH-cell–mediated alveolar bone loss during the progression of periodontal disease.220 Furthermore, Alayan et al reported that both TH1-cytokine (interleukin-12p40, interferon-gamma, tumor necrosis factor)–deficient mice and TH2-cytokine (interleukin-10, interleukin-4)–deficient mice exhibited significantly more alveolar bone loss than wild-type control mice.221 T. forsythia has been shown to drive alveolar bone loss through a TH2 bias.222 The aforementioned work demonstrates the complexity of subgingival plaque polymicrobial species in the modulation of TH1/TH2-mediated immunity in periodontitis-induced bone loss.
Historically, evaluating the TH1/TH2 cellular balance was considered an important outcome when assessing periodontal bone loss. The proposed theory was that TH1 cells mediate early stable lesions, whereas TH2 cells attempt to maintain homeostasis. However, as the disease progresses, TH2 cells become more prominent beyond the established lesion (reviewed by Gemmell and Seymour223). This notion is supported by the presence of elevated B cell activation and plasma cells that infiltrate inflamed tissue, where the TH2-derived cytokine interleukin-4 promotes B cell activation and proliferation. Because TH1 and TH2 influences on alveolar bone loss clinically and experimentally are not straightforward, this calls for ongoing studies to define the molecular underpinnings of each cell in periodontal disease progression.
9.3 |. T regulatory cells/TH17 cells
T regulatory cells maintain immune homeostasis by ameliorating inflammation through cytokine production of interleukin-10, interleukin-12, and transforming growth factor beta. In clinical periodontitis, T regulatory cells have been shown to be present during disease.224–227 T regulatory cell presence throughout progressive stages of periodontitis may imply a compensatory mechanism aiming to alleviate tissue destruction from the overreaction of the immune system. Yet, T regulatory cells have been shown to have high plasticity, where they can lose immunosuppressive function under chronic periodontal inflammation.228,229 Thus, it may be proposed that T regulatory cells may be present in early lesions to mitigate the adaptive immune response, but then, as the disease progresses, T regulatory cells are dominated by other proinflammatory cells and factors.
Kobayashi et al employed the murine P. gingivalis oral inoculation model to evaluate the induction of interleukin-10–producing CD4+ T cells in chronic periodontitis. The number of FoxP3+CD4+CD25+ T regulatory cells was increased in the experimental group during the late stage of infection, which correlated with elevated interleukin-10–producing CD4+ T cells.230 Garlet et al showed that A. actinomycetemcomitans oral inoculation in mice resulted in elevated CD4+CD25+ and CD4+FoxP3+ cells, characterizing the presence of T regulatory cells in the periodontal environment in a late stage after infection. T regulatory cell–associated cytokines, including interleukin-10 and transforming growth factor beta, were synthesized in kinetics that resemble T regulatory cell migration. Treatment with glucocorticoid-inducible tumor necrosis factor receptor (T regulatory cell function inhibitor) exacerbated inflammation and periodontal bone loss, which suggests the presence of T regulatory cells attenuates the severity of periodontitis.231 Furthermore, T regulatory cell migration and recruitment have been shown to be essential for ameliorating experimental periodontitis-induced alveolar bone loss, which may be dependent upon CCR4-CCL22 signaling.232,233 Other periodontal disease studies have proposed that T regulatory cells may be transdifferentiated into TH17 cells during intermediate stages of disease.63,229 Tsukasaki et al reported that murine experimental periodontitis-induced oral infection converted FoxP3+ T cells into TH17 cells. These bone-damaging exFoxP3 TH17 cells, which initially may suppress local infection and protect against pathogens, also contributed to alveolar bone resorption.63 Clinically, interleukin-17/FoxP3 double-positive cells were detected in periodontal lesions of chronic periodontitis tissues.229 In contrast, Parachuru et al reported no detection of interleukin-17/FoxP3 double-positive cells in active periodontal disease lesions.234
TH17 cells sustain homeostasis at mucosal barrier sites by regulating the microbiota, and they modulate autoimmune conditions, such as multiple sclerosis and rheumatoid arthritis.235 TH17 cells are induced from naive T cells via antigen presentation or through stimulation by proinflammatory cytokines, such as interleukin-1beta, interleukin-6, and interleukin-23.235 To the contrary, interferon-gamma and interleukin-4 can inhibit TH17 differentiation. TH17 cells predominantly secrete interleukin-17, as well as interleukin-21, interleukin-22 and tumor necrosis factor.235 Recent timely reports have highlighted the interplay between TH17 cells and the oral microbiota, which have been shown to have implications for alveolar bone homeostasis in health and disease.63,89,236,237 TH17 cells are of interest in bone remodeling due to interleukin-17 having potent pro-osteoclastic actions. Sato et al found that interleukin-17 has no effect on osteoclast precursor cell differentiation in the RANKL-macrophage colony–stimulating factor-osteoclast culture system.238 This implies that interleukin-17 promotes neighboring cells in the microenvironment to drive osteoclastogenesis, which is consistent with a report showing that interleukin-17 promotes osteoclastogenesis through osteoblastic cell–mediated RANKL induction.239
TH17 cells, , since their discovery, have been of great interest in periodontal disease and alveolar bone loss.240,241 Increased TH17 cells and interleukin-17 expression have been observed clinically and are elevated in isolates from periodontitis versus gingivitis patients.242–246 Interleukin-17 has also been reported to be increased in gingival isolates from active versus inactive periodontitis lesions.226,247,248 Interleukin-23, which can promote TH17 cell differentiation, has been reported to be increased in periodontitis lesions with elevated interleukin-17 expression.245,247 Dutzan et al designated TH17 cells as the primary cellular source of interleukin-17 in clinical periodontitis, where there was minimal interleukin-17 contribution from CD8+ T cells, γδ T cells, and non-T-cell lineages.249
Interleukin-17 receptor alpha–deficient mice (C57Bl/6 and Balb/c) inoculated with P. gingivalis have significantly more alveolar bone loss due to defects in the chemokine-neutrophil recruitment axis.133,250 Yu et al found this outcome to be sex dependent, with females having more severe periodontal bone loss, due to defective production of anti–P. gingivalis immunoglobulin G and the chemokines CXCL1 and CXCL2.250 Eskan et al showed that antibody neutralization of interleukin-17A blunted periodontal inflammation and alveolar bone loss in the P. gingivalis oral inoculation model.251 In vitro studies have delineated that specific serotypes of A. actinomycetemcomitans252 and P. gingivalis253 induce characteristic TH1/TH17 transcription factors and cytokines. A modified T. forsythia strain, with an altered O-glycan surface composition, has been reported to induce TH17-linked mobilization of neutrophils to the gingival tissues and suppress P. gingivalis–driven alveolar bone loss in mice.254,255 These reports highlight the complexity of the subgingival plaque polymicrobial ecosystem in the modulation of the TH17/interleukin-17 periodontal immune response.
Whereas early investigations evaluated the TH1/TH2 balance in periodontal disease, reports have evolved into describing the TH17/TH1 paradigm256 and TH17/T regulatory cell paradigm.257 The TH17/TH1 paradigm is centered on discerning which cell is more destructive, whereas the TH17/T regulatory cell paradigm is focused on T regulatory cell amelioration of inflammation and periodontal bone loss. Monasterio and coworkers recently employed the murine oral inoculation periodontitis model to show that the TH1/TH17 immune response and periodontal bone loss is strain dependent.258,259 Bi et al reported increased TH1/TH17 cells in periodontitis-afflicted gingival isolates, whereas TH2/T regulatory cells were upregulated in healthy gingiva.260 Serotype b of A. actinomycetemcomitans induced elevated T-bet+interferon-gamma+ and RAR-related orphan receptor gamma t+interleukin-17+ T cells in periodontal lesions, which were associated with increased RANKL and alveolar bone resorption.258 Capsular-defective mutant strains of P. gingivalis promoted less bone loss and decreased TH1/TH17 cells and related cytokines compared with the encapsulated W50 wild-type strain.259 Investigations focusing on the TH17/T regulatory cell paradigm have reported an upregulated TH17/T regulatory cell balance in the progression of ligature-induced periodontitis, both in the rat model261 and rhesus monkey model.262 Supporting these findings, Wang et al showed that vaccination with P. gingivalis ameliorates murine experimental periodontitis by offsetting the TH17/T regulatory cell imbalance.263 Recently, Cafferata et al reported alterations in the TH17/T regulatory cell axis but found that interleukin-35 treatment inhibited alveolar bone loss by modulating the TH17/T regulatory cell ratio with increased T regulatory cell–associated cytokines.264 Taken together, the balance of TH17 cells to TH1 cells and T regulatory cells may provide insight into defined TH17 cell parameters that correlate with periodontal disease progression.
9.4 |. TH9 and TH22 cells
Most recently, there have been further discoveries into defining additional CD4+ T-cell helper subsets, such as TH9 and TH22 cells. TH9 cells have been described in allergy and asthma, where they require transforming growth factor beta and interleukin-4 for their induction.265,266 TH9 cells are defined by the intracellular transcription factors GATA3 and interferon regulatory factor 4 and primarily secrete interleukin-9, driving immunoglobulin E/immunoglobulin G production.265,266 TH22 cells are involved in skin immunity and homeostasis, where they can primarily secrete interleukin-22 and tumor necrosis factor.267–269 TH17 and TH22 cells both can express the transcription factor RAR-related orphan receptor gamma t, but TH22 cells are also identified by aryl hydrocarbon receptor expression. Notably, TH22 cells synthesize interleukin-22, which is significant for host barrier defense against bacterial infections.267–269 It is currently unclear whether TH9 and TH22 cells are critical players in osteoimmunology and alveolar bone homeostasis. Of interest, several clinical research investigations have begun to elucidate the role of TH9 and TH22 cells in periodontal disease.270,271 Diaz-Zuniga et al evaluated the interleukin-9 and interleukin-22 cytokine levels in gingival crevicular fluid samples and biopsies and their association with periodontal disease severity.270 Increased levels of interleukin-22 and aryl hydrocarbon receptor were detected in patients with periodontitis compared with gingivitis and healthy individuals, and a significant correlation was reported between secreted interleukin-22 and clinical attachment level of the sampled periodontal pockets.270 Higher interleukin-9 levels were detected in gingivitis patients than in periodontitis and healthy counterparts.270 Osteoclasts stimulated with tissue homogenates obtained from periodontitis versus healthy patients demonstrated enhanced resorptive activity, which was dependent on the presence of interleukin-22.270 The same research group isolated dendritic cells and naive CD4+ T cells from healthy donors and stimulated with different serotypes of A. actinomycetemcomitans or purified lipopolysaccharide.271 A. actinomycetemcomitans serotype b increased levels of interleukin-22 and aryl hydrocarbon receptor in T lymphocytes.271 Further research is indicated to discern the role of the TH9 and TH22 helper T cell subsets in periodontal health and disease.
9.5 |. B cells
B cells derive from the hematopoietic lineage, where they are considered positive immune regulators due to antibody production. B cells are professional antigen-presenting cells as well as producers of cytokines that promote multiple humoral responses. Importantly, stromal-osteoblastic cells have been shown to support the commitment and differentiation of all stages of B cell development.272 Activated B cells express RANKL, which critically regulates skeletal homeostasis and disease.66,273 Interestingly, B cells can also inhibit osteoclast formation and viability through transforming growth factor beta secretion.274
The seminal report by Page and Schroeder described the plasma cells/B cells as a minor component of the inflammatory infiltrate detected within the early lesion of chronic periodontitis.146 Disease progression led to an increased presence of these cells in the affected connective tissue. The advanced lesion, which is the only stage afflicted by appreciable bone loss, was dominated by plasma cells/B cells.146 Subsequent studies have confirmed that plasma cells/B cells are the most prominent cellular constituent of the infiltrate in advanced lesions of chronic periodontitis.183,275–278 Interestingly, Jing et al identified CD138+CD38+ plasma cells as major contributors to the secretion of interleukin-35 and interleukin-37, which are postulated as anti-inflammatory cytokines in chronic periodontitis.279
Experimental animal models challenged by P. gingivalis280 and A. actinomycetemcomitans281 oral inoculation found that the serum antibody response preceded alveolar bone loss, and bacterial-responsive B cells were responsible for this bone loss. P. gingivalis–induced experimental periodontitis studies performed in mice deficient in immunoglobulin D (IgD)282 and immunoglobulin M (IgM)283,284, which have critical defects in B cell maturation, clearly demonstrate that B cells contribute to periodontal bone loss. Baker et al studied the contribution of B cell IgD to alveolar bone loss by carrying out P. gingivalis oral infection in IgD-deficient mice. P. gingivalis–specific antibody was lower and oral colonization was higher in IgD-deficient mice, yet alveolar bone loss was completely absent.282 Diminished proportions of CD4+ T cells and CD69+ activated B cells in IgD-deficient mice support the notion that IgD mediates alveolar bone resorption through antigen-specific coactivation of B cells and CD4+ T cells.282 Oliver-Bell et al reported that IgM-deficient mice were also protected from P. gingivalis–mediated periodontal bone loss, which was attributed to reduced RANKL-expressing B cells.283 Abe et al demonstrated that tumor necrosis factor ligand superfamily member 13 and B-lymphocyte stimulator are elevated in experimental murine periodontitis and clinical periodontitis.284 Tumor necrosis factor ligand superfamily member 13 and B-lymphocyte stimulator are tumor necrosis factor ligand family cytokines that are important for B cell proliferation, maturation, and survival. The upregulated expression of tumor necrosis factor ligand superfamily member 13 and B-lymphocyte stimulator correlated with increased numbers of B cells/plasma cells in gingival tissue isolates from both mice and humans.284 Ligature-induced periodontitis in IgM-deficient mice resulted in less alveolar bone loss compared with wild-type controls.284 Antibody neutralization of tumor necrosis factor ligand superfamily member 13 or B-lymphocyte stimulator lowered B cell numbers in the gingival tissue and inhibited alveolar bone loss in wild-type mice, which was not observed in IgM-deficient mice.284
Because activated B cells express RANKL,66,273 studies have attempted to elucidate the role of activated B cells in periodontal disease. Han et al immunized mice with A. actinomycetemcomitans and isolated activated B cells from immunized and nonimmunized mice.281 A. actinomycetemcomitans–specific activated B cells secreted greater RANKL levels in vitro than nonimmunized B cells did. A. actinomycetemcomitans–specific activated B-cell culture supernatants induced superior osteoclast outcomes in the RAW264.7 cell osteoclast culture system. Kanzaki et al evaluated RANKL expression on B cells and T cells within gingival tissue isolates from periodontitis patients, finding that RANKL expression was more profound on B cells than on T cells.285 Isolated B cells promoted osteoclast differentiation and function in the RAW264.7 cell osteoclast culture system.285 Demoersman et al showed that peripheral blood–activated B cells from chronic periodontitis patients versus healthy controls expressed more RANKL.286
Memory B cells can drive a proinflammatory infiltrate within active periodontal lesions through cytokine production, such as tumor necrosis factor or interleukin-6, or enhance synthesis of matrix metalloproteinases to influence tissue breakdown.277,287 Mahanonda et al demonstrated that memory B cells reside in the connective tissue near the junctional epithelium in normal gingiva,287 suggesting that memory B cells may be important in regulating periodontal health and homeostasis. Demoersman et al found memory B cells to be the predominant B cell subset in periodontitis patients compared with healthy controls.286 Experimental models of periodontitis performed by Han and coworkers288–290 have further defined the role of memory B cells in periodontal bone loss. In one report, rats were primed with systemic lipopolysaccharide injections and then memory B cells were isolated and adoptively transferred into rats that were subjected to lipopolysaccharide systemic challenge compared with vehicle control.288 Memory B cells were reported to enhance alveolar bone loss and osteoclast differentiation and maturation in lipopolysaccharide-challenged rats compared with control.288 In a rat ligature-induced periodontitis model, memory B cell numbers were suppressed but were found to express higher levels of RANKL than control did.290 Han et al further defined memory B cells in a study in which rats were primed with systemic injections of P. gingivalis.289 Memory B cells were then isolated and adoptively transferred into rats that were subjected to the ligature-induced periodontitis. Results from this study indicate that switched memory B cells (CD27+CD38−IgD−) expressed more RANKL, interleukin-6, and interleukin-1beta than naive, antibody-secreting, and unswitched memory B cells (CD27+CD38−IgD+) did.289 Switched memory B cells also induced greater periodontal bone loss and upregulated proinflammatory cytokines, TH1 cells, and TH17 cells in the gingiva and cervical lymph nodes.289 Though reports differ in outcomes concerning whether activated B cells or memory B cells express more RANKL, both B cell subsets have been shown to play important roles in regulating periodontal disease–driven alveolar bone loss.
Additional B cell subsets have been identified, including B1, B2, and regulatory B cells (also known as B10 cells). B1 and B2 cells are traditional B cells that can give rise to long-lived plasma cells and memory cells. Regulatory B cells are identified by the expression of interleukin-10.291 Ongoing studies have begun to identify these B cells subsets in periodontal health and disease. Donati et al evaluated gingival tissues from chronic periodontitis patients and found B1 and B2 cells constituted a larger proportion of cells than T cells and plasma cells.277 In contrast, Demoersman et al reported that B1 cells are diminished in peripheral blood of periodontitis patients compared with healthy patients.286 Timely reports have characterized regulatory B cells to play a critical role in suppressing inflammatory responses in vitro292,293 and ameliorating alveolar bone loss in vivo.293–295 Liu et al performed an in vitro study in which isolated splenic B cells were immunized with P. gingivalis and treated with CD40 ligand and CpG to induce B cell–derived toll-like receptor signaling.292 Immunized B cells secreted elevated interleukin-10 levels, compared with nonimmunized B cells.292 Based on these in vitro findings, further studies characterized regulatory B cells in rodent experimental periodontitis models. Hu et al used the rat ligature-induced periodontal disease model, with or without local CD40 ligand injections into the palatal gingiva.293 CD40 ligand elevated regulatory B cells, increased gingival Il10, decreased RANKL, and blunted alveolar bone loss.293 Local injection of CD40 ligand and CpG into the gingiva was studied by Yu et al in a murine ligature model294 and by Wang et al in a P. gingivalis-lipopolysaccharide systemic administration model.296 CD40 ligand and CpG–induced regulatory B cells alleviated periodontal bone loss.294,296 Zhao et al found that CD40 ligand and CpG induced regulatory B cells ameliorate ligature-induced periodontitis in a toll-like receptor-9–independent manner.295 Similarly, Shi et al adoptively transferred periodontopathogenic-specific regulatory B cells, which alleviated alveolar bone loss, increased interleukin-10 production, and reduced interleukin-17 and TH17 cells in the murine gingiva.297
Advancing knowledge about B cell subsets and their diverse roles in periodontal health and disease will provide opportunity for the development of therapeutic targets in the B cell lineage for the treatment of periodontitis.
9.6 |. Natural killer cells/natural killer T cells
Natural killer cells are cytolytic effector lymphocytes that are important in viral and bacterial infections and malignancies. Natural killer cells have several activating receptors, such as natural cytotoxicity receptors (NKp30, NKp44, and NKp46), of which only NKp46 has a mouse ortholog (NCR1). Peripheral natural killer cell homeostasis is mediated by interleukin-15, while natural killer cells can produce interferon-gamma as well as proinflammatory and anti-inflammatory cytokines, such as tumor necrosis factor and interleukin-10, respectively.298 Notably, natural killer–mediated interferon-gamma production modulates T cell responses in secondary lymphoid organs as well as impacts T cell and B cell immunity.298 Invariant natural killer T cells are defined by NK1.1 expression, which recognizes glycolipids:CD1d complexes expressed by antigen-presenting cells.299 Natural killer T cells also recognize pathogenic bacterial antigens and self-antigens.299 The difference between these cell types is that natural killer cells are large granular lymphocytes that mature in circulation, whereas natural killer T cells are a type of T cell that maturates in the thymus.298,299
Under pathologic conditions like rheumatoid arthritis, natural killer cells contribute to osteoclastogenesis by expressing soluble and surface-bound RANKL and macrophage colony-stimulating factor.300 In vitro studies have elucidated natural killer cell interactions with osteoclasts and osteoblasts.301,302 In the presence of interleukin-15, synovial fluid–derived natural killer cells were co-cultured with autologous monocytes, which differentiated monocytes into osteoclasts.301 Takeda et al isolated whole bone marrow, extracted either natural killer cells or T cells, and co-cultured with osteoblasts in the presence or absence of prostaglandin E2 and/or interleukin-15.302 Osteoblast numbers were decreased by dose-dependent interleukin-15 in whole marrow cultures and cultures lacking T cells.302 Elevated osteoblast numbers were observed in co-cultures lacking natural killer cells, which was attributed to interleukin-15 activation of natural killer cell–induced osteoblastic cell apoptosis.302
There are few studies determining the role of natural killer cells in the initiation and pathogenesis of periodontitis. Tsoumis et al reported a positive correlation between peripheral blood natural killer cell cytotoxicity and clinical periodontitis progression,303 and Fujita et al performed an immunohistochemical study that demonstrated natural killer cell accumulation in gingival tissue afflicted by periodontal disease.304 Chaushu et al orally inoculated P. gingivalis or F. nucleatum in mice deficient for the natural killer cell receptor NCR1. The NCR1 knockout mice, compared with wild-type mice, exhibited less alveolar bone loss when infected by P. gingivalis and were completely protected against F. nucleatum–induced alveolar loss.305 These studies imply that natural killer cells regulate the pathogenesis of periodontal disease and related alveolar bone loss.
Natural killer T cells have been shown to be more prominent in periodontitis lesions versus gingivitis lesions.306,307 Yet, Nowak et al reported natural killer T cells were not present in clinical chronic periodontal disease lesions.308 Baker et al orally inoculated P. gingivalis in β2m-knockout mice that are deficient in major histocompatibility complex class I–responsive CD8+ T cells and NK1+ T cells.186 The β2m-knockout mice had similar alveolar bone loss to wild-type mice, which implies that natural killer T cells do not modulate periodontitis-driven alveolar bone loss.186 Aoki-Nonaka et al orally inoculated P. gingivalis in CD1d knockout mice, which are deficient in responsive natural killer T cells.309 The CD1d knockout mice exhibited no alveolar bone loss, suggesting that natural killer T cells contribute to periodontal bone loss. The limited studies discerning the role of natural killer cells and natural killer T cells in periodontitis demonstrate the need for ongoing investigations.
9.7 |. Mast cells
Mast cells differentiate from the hematopoietic stem cell lineage and mature in peripheral connective tissues. Mast cells regulate mucosal epithelial barrier homeostasis by inducing both innate and adaptive immune cells through the production of proinflammatory mediators, such as tumor necrosis factor, nitric oxide, and matrix metalloproteinases.310 Mucosal tissues, such as the gut311 and oral cavity,312 contain mast cells that are continuously being exposed to foreign pathogens and allergens. Mast cells have been characterized in healthy gingiva,312,313 but they expand in inflamed tissues.314,315
Mast cell populations were observed to be increased in chronic periodontitis.316–318 Timely studies have elucidated the role of mast cells in periodontal disease progression, where the degree of degranulation correlates with disease severity.319–321 Interestingly, when mast cells were inhibited by lodoxamide ethyl in beagle dogs, alveolar bone loss was ameliorated.322 In contrast, Gemmell et al reported that tryptase+ mast cells were deceased in clinical periodontal lesions, suggesting there is not an increase in mast cell migration into diseased tissue.214 More recently, Huang et al reported higher toll-like receptor-4 expression on mast cells within gingival tissues of chronic periodontitis patients, suggesting that mast cell toll-like receptor-4 expression may mediate the disease process.323 Interleukin-31324 and interleukin-33325 have been investigated in relation to the role of mast cells in experimental periodontal disease models. Tada et al reported that P. gingivalis induces the production of interleukin-31 by human mast cells, which downregulated tight junction molecules to promote gingival epithelial barrier permeability.324 Koseoglu et al found that interleukin-33, which supports mast cell development, exhibited a positive correlation with the number of mast cells in a rat ligature periodontitis model.325 Upregulated interleukin-33 expression correlated with increased levels of RANKL and alveolar bone loss.325
Recognizing that mast cells have been linked with interleukin-17 production, Parachuru et al evaluated healthy, gingivitis, and chronic periodontitis tissues to discern the cellular source of interleukin-17.326 Interleukin-17+–labeled cells were neither CD4+ nor CD8+, but were tryptase+, a selective marker for mast cells.326 Furthermore, an experimental mouse model of periodontitis presented mast cells as contributors to P. gingivalis–induced alveolar bone loss by increasing tumor necrosis factor and interleukin-6 production.327 Though mast cells may be important in recognition and clearing of early infections, a constant inflammatory milieu may promote mast cells to contribute and exacerbate later stages of periodontal disease.
9.8 |. Neutrophils
Neutrophils are the first responders in primary host immune defense.328 When proinflammatory cytokines and chemokines are present, neutrophils utilize adhesion molecules to attach to endothelial cells within the blood vessels of gingival lamina propria.95,104,328,329 It has been estimated that about 30,000 neutrophils transit through the gingival tissue per minute,103 where this recruitment is necessary for periodontal tissue homeostasis.104 Individuals afflicted by neutrophil defects have a higher incidence of periodontal disease than healthy individuals do.330 Clinical neutrophil defects have facilitated characterizing the role of neutrophils and their function in the periodontal innate immune defense.104 In periodontal disease, neutrophils have enhanced survival, which may contribute to the chronic proinflammatory state that drives periodontal tissue destruction. Though neutrophils have been extensively characterized as an important immune cell in periodontitis (reviewed by Hajishengallis and Korostoff331), this cell population continues to be of interest owing to its close proximity to the oral microbiota and the modulation of other immune cells in periodontal health and disease.
9.9 |. Myeloid-derived suppressor cells
Myeloid-derived suppressor cells are an immature myeloid precursor cell population derived from bone marrow that regulate the innate and adaptive immune responses.332,333 Whereas myeloid-derived suppressor cells were initially studied in cancer, research advances have elucidated that myeloid-derived suppressor cells regulate immune responses and the pathogenesis of several inflammatory diseases, including rheumatoid arthritis, systemic lupus erythematosus, and periodontal disease.332–334 Under homeostatic conditions, myeloid-derived suppressor cells comprise about 30% of normal bone marrow cells.335
Chronic inflammation at peripheral sites drives recruitment of myeloid-derived suppressor cells, which promote ongoing tissue damage through several mechanisms. Primarily, myeloid-derived suppressor cells suppress T cell activation and proliferation,336 but also modulate the T regulatory cell population through major histocompatibility complex class II337 and inhibit natural killer cell functions.338,339 Furthermore, because myeloid-derived suppressor cells are an immature progenitor population, these cells can differentiate into other myeloid/monocyte lineage cells, like macrophages, plasmacytoid dendritic cells, inflammatory monocytes, and granulocytes, which is dependent upon proinflammatory factors in the local microenvironment.340,341 Interestingly, seminal reports have shown that myeloid-derived suppressor cells can be osteoclast progenitor cells in vivo and can be cultured into osteoclasts in vitro under inflammatory conditions.342–345 Cytokines such as tumor necrosis factor, interleukin-1beta, CCL3, and CCL4, which have pro-osteoclastic effects,346–348 support myeloid-derived suppressor cells recruitment to inflammatory sites.349,350 We recently published an initial report showing that perturbations in the microbiota elevate myeloid-derived suppressor cells in bone marrow.120 Antibiotic disruption of the commensal microbiota increased myeloid-derived suppressor cells in the bone marrow environment, which was linked to increased osteoclastogenesis through the major histocompatibility complex class II antigen presentation pathway.120
Though myeloid-derived suppressor cells have been well characterized in several chronic inflammatory conditions, little is known about their function in periodontal health and disease. Ezernitchi et al described the role of myeloid-derived suppressor cell–mediated immunosuppression in a murine model challenged by P. gingivalis injection, reporting that upregulated myeloid-derived suppressor cells suppressed T cell intracellular signaling.351 Arjunan et al drove monocytes down the myeloid-derived suppressor cell lineage in vitro, through stimulation of P. gingivalis.352 Both acute and chronic P. gingivalis oral infections in mice induced myeloid-derived suppressor cells, which suppressed cytotoxic T cells and upregulated FoxP3+ T cells.352 Su et al found that P. gingivalis oral infection expanded three subpopulations of myeloid-derived suppressor cells in mice: CD11b+Ly6G++Ly6C+, CD11b+Ly6G+Ly6C++, and CD11b+Ly6G+Ly6C+.334 Only the CD11b+Ly6G+Ly6C++-expressing cells suppressed T cell proliferation and were able to differentiate into osteoclasts in vitro.334 Recently, Cheng et al reported that periodontal inflammation supports the recruitment of distant metastatic cancer cells through myeloid-derived suppressor cells and related chemokines.353 Currently, there are no known studies attributing myeloid-derived suppressor cells’ immunosuppressive effects directly on alveolar bone homeostasis. The role of myeloid-derived suppressor cells and their immunosuppressive mechanisms influencing alveolar bone remodeling in periodontal health and disease remain to be defined.
9.10 |. Macrophages
Macrophages share a common progenitor cell population with myeloid-derived suppressor cells, monocytes, dendritic cells, and osteoclasts, and they are responsible for mediating inflammation, phagocytic activities, and immune surveillance via antigen presentation. These cells can polarize into proinflammatory-like (M1) or anti-inflammatory-like (M2) cells, dependent upon factors within the microenvironment.354 Defining the role of macrophages in osteoimmunology, bone marrow macrophages were identified in close proximity to osteoblasts, suggesting that they communicate with bone cells and support bone remodeling processes, specifically producing bone morphogenetic proteins to promote osteoblast maturation.355,356 Furthermore, macrophage depletion models support the notion that macrophages are critical osteoimmune cells that modulate both osteoblast and osteoclast processes.357,358 Alternatively, activated macrophages have been characterized as regulators of skeletal homeostasis, yet macrophage effects on bone remodeling in the alveolar bone complex are not as well understood.
Periodontal research has determined that macrophages play critical roles in periodontal tissue destruction (reviewed by Sima and Glogauer359). Recent investigations have focused on how macrophage polarization may be a vital target for monitoring disease periodontal progression. Studies imply that M1 macrophages are contributory in osteoclastogenesis with P. gingivalis infection in vitro360,361 and in vivo.360,362 M1 macrophages have been shown to induce interferon-gamma or interleukin-6 production and exacerbate alveolar bone loss.360,362 Huang et al hypothesized that P. gingivalis and A. actinomycetemcomitans would induce M1-type cells, whereas oral commensal bacteria would promote more of an M2-like response.361 M1 macrophage output from P. gingivalis challenge increased proinflammatory cytokines with less promotion of T cell recruitment, whereas A. actinomycetemcomitans challenge elevated both the proinflammatory cytokines and T-cell chemokines.361 In contrast to these findings, Yamaguchi et al found that M1 macrophages inhibited RANKL-induced osteoclastogenesis, which was interferon-gamma dependent.363
Macrophage skewing toward an M2 phenotype has also been investigated. Huang et al reported that M2 macrophages were induced by oral commensals, but not P. gingivalis or A. actinomycetemcomitans.361 Yu et al found M2 macrophages to be slightly enhanced in comparison with M1 macrophages in vitro and in vivo.360 Zhuang et al showed that induction of M2 macrophages prevented periodontal bone loss, utilizing both the P. gingivalis oral inoculation and ligature-induced experimental periodontitis models.364 Though considerable research efforts have been centered on macrophage polarization/skewing in periodontitis, the function of these cells has not been extensively investigated in periodontal health and homeostasis.
9.11 |. Dendritic cells
Like other professional antigen-presenting cells, such as macrophages and B cells, myeloid lineage–derived dendritic cells are necessary for thymic T cell education for immune tolerance and T cell interactions in lymphoid tissues in health and immune challenges.365 Dendritic cells function in immune surveillance, which is achieved through antigen presentation by major histocompatibility complex class I and class II complexes. These dynamic mechanisms are essential for innate-adaptive immune crosstalk presenting self/nonself-peptides to the adaptive immune system. Dendritic cells are critical in taking in nonself-antigen, migrating to secondary lymphoid tissues to present to T cells, promoting an adaptive immune response.
Dendritic cell subsets can be divided into plasmacytoid dendritic cells, Langerhans cells, and conventional dendritic cells. Plasmacytoid dendritic cells derive from lymphoid progenitors and are proinflammatory in nature due to production and secretion of type I interferons, interleukin-6, tumor necrosis factor, and proinflammatory chemokines.365 Plasmacytoid dendritic cells have also been associated with increased levels of CCL2, CCL5, and CXCL10 that may promote RANKL, thus leading to osteoclastogenesis induction.366,367 Conventional dendritic cells reside in connective and lymphoid tissues, where they express high levels of CD11c. These cells commonly present antigen and secrete multiple cytokines to stimulate specific T cell subsets, thus skewing T cells subsets for secretion of interferon-gamma, RANKL, and interleukin-17, promoting osteoclastogenesis.368 Immature conventional dendritic cells survey connective tissues for potential foreign threats, but, when activated, these cells will migrate and travel to nearby lymph nodes to present foreign antigens to T cells for an adaptive immune response.369 Langerhans cells are a distinct dendritic cell subset that express the C-type lectin receptor langerin (CD207). These cells reside in the skin epidermis and oral mucosal epithelium, where they function in immune surveillance by promoting either tolerance to self-antigen or an immune response to activate T cells.370–372 Langerhans cells are derived from pre-dendritic cells and monocytes through a possible transforming growth factor beta 1 signaling mechanism to promote mucosal homeostasis and prevent tissue damage.373
As previously stated, dendritic cells share a common progenitor with osteoclasts. Therefore, several reports have shown that dendritic cells can develop into osteoclasts.374,375 Murine bone marrow and splenic CD11c+ dendritic cells develop into TRAP+ cathepsin K+ functional osteoclasts in a RANKL/RANK-dependent manner, both in vitro375 and in vivo.368 These findings were supported by an investigation which showed that immature dendritic cells derived from spleen and marrow developed into osteoclasts in a macrophage colony-stimulating factor/RANKL–induced in vitro system, whereas mature conventional dendritic cells and plasmacytoid dendritic cells did not.374 Importantly, these reports suggest that immature dendritic cells can act as osteoclast precursor cells.
Because of the multifunctionality and common cellular lineage that dendritic cells share with other immune cells, dendritic cells have been investigated in experimental periodontitis models to understand dendritic cell–mediated disease initiation, progression, and support of osteoclastogenesis. A study investigating the role of Langerhans cells in oral mucosal immunity found that in vivo Langerhans cell depletion in P gingivalis-induced murine periodontitis led to increased T cell and B cell infiltration in periodontal tissues and exacerbated alveolar bone loss.376 However, a separate report stated that Langerhans cells were not necessary for P. gingivalis–induced alveolar bone loss.377 Furthermore, Bittner-Eddy et al reported that oral mucosal Langerhans cells promoted TH17 differentiation, and Langerhans cell ablation resulted in a TH1-driven response but T regulatory cells were unaffected.377 The authors speculated that Langerhans cell depletion led to suppression of TH17-mediated immunity, which was compensated by an upregulation of TH1-mediated immunity.377
Investigations have utilized experimental periodontal disease models to demonstrate that dendritic cells can drive proinflammatory cytokine production.378,379 Dendritic cell–mediated osteoclastogenesis has been shown to elevate alveolar bone loss, which is attributed to increased RANKL and numbers of osteoclasts in vitro and in vivo.380 Yet, P. gingivalis oral inoculation allowed for dendritic cells to generate TH2 responses381,382 and T regulatory cell induction through the secretion of interleukin-10 and transforming growth factor beta.383 Alnaeeli et al described dendritic cells as an essential immune cell in the pathogenesis of periodontitis.375 CD4+ T cells co-cultured with A. actinomycetemcomitans–primed dendritic cells differentiated into functional osteoclasts.375 Furthermore, adoptively transferred CD11c+ dendritic cell–derived osteoclasts induced bone loss in vivo.375 In a P. gingivalis–induced periodontitis model, silencing of cathepsin K led to reduced dendritic cell numbers in periodontal lesions, which was correlated with elevated TH17 cells.379 Xiao et al carried out a dendritic cell–lineage-specific deletion of FOXO1 in a P. gingivalis and F. nucleatum murine oral gavage model.384 FOXO1-deficient dendritic cells demonstrated impaired recruitment to the oral mucosal epithelium.384 FOXO1-deficient dendritic cells secreted less interleukin-12, which was compensated by elevated interleukin-1beta, interleukin-17, and RANKL.384 Future studies are indicated to further elucidate the specific roles dendritic cells play in mediating oral mucosal immunity and periodontal health and disease.
9.12 |. Highlights of osteoimmune interactions in periodontitis
The immune system plays a critical role in the maintenance of alveolar bone in health and disease. Innate immune cells, such as neutrophils, macrophages, and dendritic cells, have been classified as the early line of defense in periodontal disease. Recent research has highlighted the potential role of myeloid-derived suppressor cells, mast cells, and natural killer/natural killer T cells as candidate modulators of periodontitis. Historically, B cells and CD8+ and CD4+ T cells were identified as critical promoters in the pathogenesis of periodontal disease and alveolar bone destruction. Advances in characterizing B cell and T cell subsets have begun to elucidate the complexity of the adaptive immune response involved in periodontal bone loss. There has been an extensive focus on the TH17/interleukin-17 immune response in disease progression, and regulatory B cells have been introduced as protective immune mediators. Ongoing advances in the field of osteoimmunology are necessary to clarify the role of distinct immune cells in periodontal health and disease. This importantly will provide opportunities for therapeutic interventions in the treatment of periodontitis and prevention of alveolar bone destruction.
10 |. THERAPEUTIC INTERVENTIONS: PROBIOTICS AND PREBIOTICS
Probiotic/prebiotic therapies are noninvasive interventions intended to alter indigenous microbiota communities to improve health.385 Probiotic/prebiotic interventions in the gut microbiota modulate the host immune response, which has been shown to protect against skeletal deterioration driven by sex-steroid deficiency.386–388 There has been a recent surge in probiotic/prebiotic research for the prevention and treatment of periodontal disease. The review will focus on prebiotic/probiotic research that has evaluated alveolar bone outcomes (Table 1). Though there have been a multitude of clinical and preclinical studies, alveolar bone outcomes have predominantly been evaluated in experimental animal models.
TABLE 1.
Summary of preclinical studies testing probiotics on alveolar bone
| Probiotic strain | Species tested | Model | Probiotic duration | Outcomes | Reference |
|---|---|---|---|---|---|
| Lactobacillus rhamnosus GG | BALB/c mice | Porphyromonas gingivalis, Fusobacterium nucleatum oral gavage | 44 d | ↓ABL ↓Inflammation ↓# OCs |
389 |
| Lactobacillus rhamnosus GG | BALB/c mice | Porphyromonas gingivalis, Fusobacterium nucleatum oral gavage | 44 d | ↓ABL ↓Duodenum inflammation ↓Ileum IL-6 |
390 |
| Lactobacillus gasseri SBT2055 | BALB/c mice | Porphyromonas gingivalis oral gavage (days 21–35) | 35 d | ↓ABL ↓TNF, IL-6 ↓Gingival detachment |
391 |
| Bacillus subtilis | Wistar rats | Ligature (days 30–44) | 44 d | ↓ABL, RANKL/OPG ratio ↓Eosinophils |
392 |
| Bacillus subtilis | Wistar rats | Ligature (days 1–14) | 14 d (days 15–29) | ↓# OCs ↓ABL, attachment ↓L-1b, ↑IL-10 |
392 |
| Lactobacillus brevis CD2 | C57BL/6 mice | Ligature | 5 d | ↓ABL ↓TNF, Il-1b, Il-6, Il-17a ↓Anaerobic microbes, ↑aerobic microbes |
393 |
| Bifidobacterium animalis lactis HN019 | Wistar rats | Ligature | 0, 3, and 7 d | ↑OPG, β-defensins ↓Il-1b, Nfkb Altered oral flora ↓Tb.Sp |
394 |
| Lactobacillus paracasei NTU-101 | Wistar rats | Lipopolysaccharide induction | 4 wk | ↓ABL ↓TNF, Il-1b, Il-17a ↓Oral bacterial numbers |
395 |
| Bifidobacterium animalis lactis HN019 | Wistar rats | Ligature (days 1–14) | 14 d (days 15–29) | ↓ABL, attachment loss ↓# OCs, Il-1b, Cinc ↑Il-10, Tgfb ↑Anaerobic/aerobic ratio |
396 |
| Streptococcus sanguinis, Streptococcus salivarius, Streptococcus mitis | Beagle dogs | Natural-occurring periodontitis (12 wk) | Weeks 1, 2, and 4 | ↓ABL | 397 |
| Bacillus subtilis | Wistar rats | Ligature (days 30–44) | 44 d |
↑Attachment loss, ABL ↑Intestinal villous and crypt depth |
398 |
| Bacillus subtilis | Wistar rats | Ligature (days 30–44) | 44 d | ↓ABL, CTX-1; ↑OPG | 399 |
| Saccharomyces cerevisiae | Wistar rats | Ligature (7 d) | 96 h | ↓ABL 15 d post-ligature; ↓OCs, Tnf, Il-1b 30 d post-ligature | 400 |
| Weissella cibaria | ICR mice | Ligature (14 d) | 14 d | ↓ABL ↓Tnf, Il-1b, Il-6, Il-10 |
401 |
Note: Abbreviations: ABL, alveolar bone loss; Cinc, cytokine-induced neutrophil chemoattractant; CTX-1, C-terminal telopeptide of type I collagen; IL/Il, interleukin; Nfkb, nuclear factor-kappa B; OC, osteoclast; OPG, osteoprotegrin; RANKL, receptor activator of nuclear factor-kappa B ligand; Tb.Sp, trabecular spacing; Tgfb, transforming growth factor beta; TNF/TNF/Tnf, tumor necrosis factor.
Probiotics are viable microorganisms that provide benefits to the host.385 Lactobacilli and bifidobacteria are the most commonly used probiotic strains.385 Lactobacilli are indigenous bacteria colonizing the oral cavity and digestive tract, having critical roles in the prevention and treatment of gastrointestinal disorders.402 Bifidobacteria naturally occur in a range of ecological niches that are either directly or indirectly connected to the gastrointestinal tract, such as the human oral cavity.402 Several strains of probiotics have recently been reported to have beneficial outcomes in the prevention and treatment of periodontal disease. Nackaerts et al performed the first known investigation of probiotic effects on alveolar bone homeostasis. A probiotic combination of Streptococcus sanguinis, Streptococcus salivarius, and S. mitis, was administered three times over 12 weeks in dogs, where probiotic administration restricted naturally occurring alveolar bone loss.397 Other investigations have utilized ligature,392–394,396,398–401 perio-pathogen inoculation,389–391 or lipopolysaccharide administration395 to induce experimental periodontitis in rodent models to evaluate probiotic effects on the oral and gut microbiota, periodontal immune response, and alveolar bone homeostasis. Studies varied in methodology, where diverse probiotics were administered for different durations of time, prior to and/or during experimentally induced periodontitis (Table 1).389,391–396,398,399,400 Investigations utilizing a ligature model with a Bifidobacterium probiotic have reported decreased alveolar bone loss,392,396,398,399 reduced attachment loss,392,396,398 increased osteoprotegrin levels,394,399 suppressed proinflammatory cytokine levels,394,396 and alterations in anaerobic:aerobic oral microbiota composition.394,396 Maekawa and Hajishengallis employed a ligature-induced murine periodontitis model.393 Lactobacillus brevis CD2 was applied topically for 5 days after ligature application, which was found to reduce alveolar bone loss, decrease proinflammatory cytokines (tumor necrosis factor, interleukin-1beta, interleukin-6, interleukin-17), and increase aerobic bacteria while suppressing anaerobic bacteria in the oral cavity.393 Garcia et al used Saccharomyces cerevisiae to treat experimental periodontitis for 96 hours and the results were evaluated 7, 15, and 30 days following treatment.400 Probiotic treatment suppressed alveolar bone loss and upregulated interleukin-10 at 15 days, and it reduced osteoclast numbers and proinflammatory cytokine levels (tumor necrosis factor and interleukin-1beta) at 30 days.400 Kim et al utilized the probiotic Weissella cibaria and found decreased proinflammatory cytokines tumor necrosis factor, interleukin-1beta, and interleukin-6 in the gingiva and less alveolar bone loss.401 Two investigations utilized P. gingivalis and/or F. nucleatum to induce periodontitis in Balb/c mice to determine the effects of probiotics on alveolar bone.389,391 Gatej et al orally administered Lactobacillus rhamnosus GG 3 days prior to P. gingivalis and F. nucleatum infection and the treatment continued for 44 days.389 L. rhamnosus GG was reported to prevent alveolar bone loss, reduce inflammatory score, and lower numbers of osteoclasts.389 Kobayashi et al orally administered Lactobacillus gasseri SBT2055, which was found to reduce alveolar bone loss, clinical attachment loss, and inflammation in P. gingivalis–induced periodontitis.391 Liu et al applied the lipopolysaccharide-induced periodontitis model where rats were fed the probiotic Lactobacillus paracasei NTU-101.395 Dietary L. paracasei NTU-101 decreased alveolar bone loss, suppressed gingival Tnf, Il17a, and Il1b, and reduced the oral microbiota bacterial load.395 Interestingly, there was also an increase in anti–reactive oxygen species in the gingiva and serum of probiotic-treated animals compared with control.395 Notably, two studies employed probiotic use for the treatment of periodontitis and found benefits on intestinal outcomes: Messora et al reported greater intestinal villous and crypt depth,398 and Gatej et al found reduced inflammation in the duodenum and ileum with probiotic use.390 Clinical studies have employed probiotics either as a preventative measure to support periodontal health403–407 or as an intervention for the treatment of chronic periodontitis.408–412 However, clinical reports have neglected to quantify probiotic effects on alveolar bone outcomes.
Prebiotics are nondigestible food elements that benefit the host by promoting proliferation and/or function of selective gut microbes to have beneficial effects.385 Certain prebiotics can enhance resident commensal gut bacteria, such as lactobacilli and bifidobacteria, while also providing enhanced functions in deterring pathogenic bacteria colonization.413 The primary mechanism of action of prebiotics is assumed to be indirect; that is, facilitating the proliferation of beneficial components of the resident microbiota.413 Prebiotics have been shown to improve intestinal absorption and balance of calcium and magnesium,414,415 as well as improve femoral bone mineral density.416 There has been a recent increase in studies evaluating prebiotics and their effects on periodontal health and disease. An in vitro study that screened for several prebiotic candidates identified N-acetyl-d-mannosamine to promote biofilm formation of S. mitis and S. sanguinis without influencing pathogenic strains.417 De Oliveira Silva et al utilized beta-glucans as a prebiotic intervention in rat experimental periodontitis.418 Beta-glucans treatment reduced alveolar bone loss and gingival interleukin-10 expression.418 Levi et al utilized mannan oligosaccharide as a prebiotic in rat ligature-induced periodontitis.419 Mannan oligosaccharide administration was carried out for 30 days, followed by 14 days of ligature-induced periodontitis.419 Mannan oligosaccharide administration ameliorated alveolar bone loss, decreased interleukin-10, interferon-gamma, tumor necrosis factor, and interleukin-1beta, increased transforming growth factor beta, and elevated intestinal villous height and crypt depth.419
Future randomized, placebo-controlled, double-blind clinical trials are necessary to discern the potential for probiotics/prebiotics interventions in the microbiome to support alveolar bone homeostasis and prevent periodontal bone loss. Recognizing that probiotic/prebiotic manipulation of the gut microbiota has osteoimmunomodulatory effects that protect against nonoral skeletal deterioration,386–388 there is high potential for alveolar bone applications.
11 |. CONCLUSION
Alveolar bone is a unique osseous tissue owing to its incorporation with the dentition and proximity to oral biofilms. A balanced host immune response to polymicrobial oral biofilms is required for periodontal health and homeostasis. Further, the oral microbiota is a critical regulator of alveolar bone remodeling. However, shifts in the composition and quantity of microbes within dental plaque biofilms can drive a local proinflammatory immune response in oral barrier sites. Infiltrating proinflammatory immune cells within the inflamed connective tissue secrete local factors that induce paracrine signaling to subjacent bone cells. Prolonged chronic inflammation disrupts “coupled” osteoclast-osteoblast actions, which ultimately results in alveolar bone destruction. Though most studies focus on osteoclastogenesis, we highlight the importance of the osteoblastic cell lineage in the pathogenesis of periodontal bone loss, where the osteoblastic cell lineage, for the most part, is overlooked by periodontal researchers. Highlighted in this review is that knowledge that nonoral microbiota-host immune response effects contribute to fibrotic conditions supports the notion that periodontitis-induced alveolar bone destruction occurs in part through fibrosis. Future research is critically needed to discern the role of fibrosis in the pathophysiology of clinical periodontal bone loss. Furthermore, our current understanding of periodontal osteoimmunology influencing alveolar bone remodeling may play a vital role in alveolar osteoimmune mechanisms. However, with critical gaps in knowledge of several immune cells, this provides an opportunity for further development in periodontal bone osteoimmunology. Lastly, probiotic/prebiotic therapeutic interventions in the oral microbiota as a potential therapy may help to support alveolar bone homeostasis/ prevent periodontal bone loss. Future randomized, placebo-controlled, double-blind, clinical trials are needed to elucidate the noninvasive, therapeutic potential for probiotics/prebiotics interventions in the microbiome to promote alveolar bone homeostasis and prevent periodontal bone loss. Recognizing that probiotic/prebiotic shifts of the gut microbiota have osteoimmunomodulatory protective effects, there is high promise for alveolar bone applications.
ACKNOWLEDGMENTS
We thank Michael E. Chew for graphical design and support of the figures in this paper. The authors’ research is supported by NIH/NIDCR K08DE025337, NIH/NIDCR T32DE017551, and the American Society for Bone and Mineral Research Rising Star Award.
REFERENCES
- 1.Schroeder HE. Oral Structural Biology: Embryology, Structure, and Function of Normal Hard and Soft Tissues of the Oral Cavity and Temporomandibular Joints. New York, NY: G. Thieme Verlag; 1991. [Google Scholar]
- 2.Cho MI, Garant PR. Development and general structure of the periodontium. Periodontol 2000. 2000;24:9–27. [DOI] [PubMed] [Google Scholar]
- 3.Schroeder HE. The Periodontium. Handbook of Microscopic Anatomy. Vol. 5. Berlin, Germany: Springer; 1986. [Google Scholar]
- 4.Orban BJ, Sicher H. Orban’s Oral Histology and Embryology 5th edition Sicher H, ed. St Louis, MO: C.V. Mosby; 1962. [Google Scholar]
- 5.Severson JA, Moffett BC, Kokich V, Selipsky H. A histologic study of age changes in the adult human periodontal joint (ligament). J Periodontol. 1978;49:189–200. [DOI] [PubMed] [Google Scholar]
- 6.Hand AR, Frank ME. (eds) Fundamentals of Oral Histology and Physiology. Ames, IA: John Wiley & Sons Inc.; 2014. [Google Scholar]
- 7.Saffar JL, Lasfargues JJ, Cherruau M. Alveolar bone and the alveolar process: the socket that is never stable. Periodontol 2000. 1997;13:76–90. [DOI] [PubMed] [Google Scholar]
- 8.Parfitt GJ. An investigation of the normal variations in alveolar bone trabeculation. Oral Surg Oral Med Oral Pathol. 1962;15:1453–1463. [DOI] [PubMed] [Google Scholar]
- 9.Allen MR, Hock JM, Burr DB. Periosteum: biology, regulation, and response to osteoporosis therapies. Bone. 2004;35:1003–1012. [DOI] [PubMed] [Google Scholar]
- 10.Colnot C. Skeletal cell fate decisions within periosteum and bone marrow during bone regeneration. J Bone Miner Res. 2009;24:274–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bahney CS, Zondervan RL, Allison P, et al. Cellular biology of fracture healing. J Orthop Res. 2019;37(1):35–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ren Y, Han X, Ho SP, et al. Removal of SOST or blocking its product sclerostin rescues defects in the periodontitis mouse model. FASEB J. 2015;29:2702–2711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Morrison SJ, Scadden DT. The bone marrow niche for haematopoietic stem cells. Nature. 2014;505:327–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Crane GM, Jeffery E, Morrison SJ. Adult haematopoietic stem cell niches. Nat Rev Immunol. 2017;17:573–590. [DOI] [PubMed] [Google Scholar]
- 15.Calvi LM, Link DC. The hematopoietic stem cell niche in homeostasis and disease. Blood. 2015;126:2443–2451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mendelson A, Frenette PS. Hematopoietic stem cell niche maintenance during homeostasis and regeneration. Nat Med. 2014;20:833–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Long F. Building strong bones: molecular regulation of the osteoblast lineage. Nat Rev Mol Cell Biol. 2011;13:27–38. [DOI] [PubMed] [Google Scholar]
- 18.Redlich K, Smolen JS. Inflammatory bone loss: pathogenesis and therapeutic intervention. Nat Rev Drug Discov. 2012;11:234–250. [DOI] [PubMed] [Google Scholar]
- 19.Karner CM, Hilton MJ. Endochondral ossification. In: Bilezikian JP, Bouillon R, Clemens T, Compston J, Bauer DC, Ebeling PR, Engelke K, Goltzman D, Guise T, Jan de Beur SM, Jüppner H, Lyons K, McCauley L, McClung MR, Miller PD, Papapoulos SE, Roodman GD, Rosen CJ, Seeman E, Thakker RV, Whyte MP, Zaidi M, eds. Primer on the Metabolic Bone Diseases and Disorders of Mineral Metabolism. Hoboken, NJ: John Wiley & Sons Inc.; 2018:12–19. [Google Scholar]
- 20.Florencio-Silva R, Sasso GR, Sasso-Cerri E, Simoes MJ, Cerri PS. Biology of bone tissue: structure, function, and factors that influence bone cells. Biomed Res Int. 2015;2015:421746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bradley EW, Westendorf JJ, van Wijnen AJ, Dudakovic A. Osteoblasts: function, development, and regulation. In: Bilezikian JP, Bouillon R, Clemens T, Compston J, Bauer DC, Ebeling PR, Engelke K, Goltzman D, Guise T, Jan de Beur SM, Jüppner H, Lyons K, McCauley L, McClung MR, Miller PD, Papapoulos SE, Roodman GD, Rosen CJ, Seeman E, Thakker RV, Whyte MP, Zaidi M, eds. Primer on the Metabolic Bone Diseases and Disorders of Mineral Metabolism. Hoboken, NJ: John Wiley & Sons Inc; 2018:31–37. [Google Scholar]
- 22.Bonewald LF. Osteocytes. In: Bilezikian JP, Bouillon R, Clemens T, Compston J, Bauer DC, Ebeling PR, Engelke K, Goltzman D, Guise T, Jan de Beur SM, Jüppner H, Lyons K, McCauley L, McClung MR, Miller PD, Papapoulos SE, Roodman GD, Rosen CJ, Seeman E, Thakker RV, Whyte MP, Zaidi M, eds. Primer on the Metabolic Bone Diseases and Disorders of Mineral Metabolism. Hoboken, NJ: John Wiley & Sons Inc; 2018:38–45. [Google Scholar]
- 23.Bonewald LF. The amazing osteocyte. J Bone Miner Res. 2011;26:229–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bellido T. Osteocyte-driven bone remodeling. Calcif Tissue Int. 2014;94:25–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tatsumi S, Ishii K, Amizuka N, et al. Targeted ablation of osteocytes induces osteoporosis with defective mechanotransduction. Cell Metab. 2007;5:464–475. [DOI] [PubMed] [Google Scholar]
- 26.O’Brien CA, Plotkin LI, Galli C, et al. Control of bone mass and remodeling by PTH receptor signaling in osteocytes. PLoS One. 2008;3:e2942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wiktor-Jedrzejczak W, Bartocci A, Ferrante AW Jr, et al. Total absence of colony-stimulating factor 1 in the macrophage-deficient osteopetrotic (op/op) mouse. Proc Natl Acad Sci USA. 1990;87:4828–4832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yoshida H, Hayashi S, Kunisada T, et al. The murine mutation osteopetrosis is in the coding region of the macrophage colony stimulating factor gene. Nature. 1990;345:442–444. [DOI] [PubMed] [Google Scholar]
- 29.Lacey DL, Timms E, Tan HL, et al. Osteoprotegerin ligand is a cytokine that regulates osteoclast differentiation and activation. Cell. 1998;93:165–176. [DOI] [PubMed] [Google Scholar]
- 30.Kong YY, Yoshida H, Sarosi I, et al. OPGL is a key regulator of osteoclastogenesis, lymphocyte development and lymph-node organogenesis. Nature. 1999;397:315–323. [DOI] [PubMed] [Google Scholar]
- 31.Yasuda H, Shima N, Nakagawa N, et al. Osteoclast differentiation factor is a ligand for osteoprotegerin/osteoclastogenesis-inhibitory factor and is identical to TRANCE/RANKL. Proc Natl Acad Sci USA. 1998;95:3597–3602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li J, Sarosi I, Yan XQ, et al. RANK is the intrinsic hematopoietic cell surface receptor that controls osteoclastogenesis and regulation of bone mass and calcium metabolism. Proc Natl Acad Sci USA. 2000;97:1566–1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dougall WC, Glaccum M, Charrier K, et al. RANK is essential for osteoclast and lymph node development. Genes Dev. 1999;13:2412–2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Arai F, Miyamoto T, Ohneda O, et al. Commitment and differentiation of osteoclast precursor cells by the sequential expression of c-Fms and receptor activator of nuclear factor κB (RANK) receptors. J Exp Med. 1999;190:1741–1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tanaka S, Takahashi N, Udagawa N, et al. Macrophage colony-stimulating factor is indispensable for both proliferation and differentiation of osteoclast progenitors. J Clin Invest. 1993;91:257–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Boyce BF, Xing L. Functions of RANKL/RANK/OPG in bone modeling and remodeling. Arch Biochem Biophys. 2008;473:139–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Feng X, Teitelbaum SL. Osteoclasts: new insights. Bone Res. 2013;1:11–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nakashima T, Takayanagi H. New regulation mechanisms of osteoclast differentiation. Ann N Y Acad Sci. 2011;1240:E13–E18. [DOI] [PubMed] [Google Scholar]
- 39.Kukita T, Wada N, Kukita A, et al. RANKL-induced DC-STAMP is essential for osteoclastogenesis. J Exp Med. 2004;200:941–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yagi M, Miyamoto T, Sawatani Y, et al. DC-STAMP is essential for cell-cell fusion in osteoclasts and foreign body giant cells. J Exp Med. 2005;202:345–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yang M, Birnbaum MJ, MacKay CA, Mason-Savas A, Thompson B, Odgren PR. Osteoclast stimulatory transmembrane protein (OC-STAMP), a novel protein induced by RANKL that promotes osteoclast differentiation. J Cell Physiol. 2008;215:497–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Simonet WS, Lacey DL, Dunstan CR, et al. Osteoprotegerin: a novel secreted protein involved in the regulation of bone density. Cell. 1997;89:309–319. [DOI] [PubMed] [Google Scholar]
- 43.Bucay N, Sarosi I, Dunstan CR, et al. Osteoprotegerin-deficient mice develop early onset osteoporosis and arterial calcification. Genes Dev. 1998;12:1260–1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yun TJ, Chaudhary PM, Shu GL, et al. OPG/FDCR-1, a TNF receptor family member, is expressed in lymphoid cells and is up-regulated by ligating CD40. J Immunol. 1998;161:6113–6121. [PubMed] [Google Scholar]
- 45.Kajiya M, Giro G, Taubman MA, Han X, Mayer MP, Kawai T. Role of periodontal pathogenic bacteria in RANKL-mediated bone destruction in periodontal disease. J Oral Microbiol. 2010;2(1):5532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Belibasakis GN, Bostanci N. The RANKL-OPG system in clinical periodontology. J Clin Periodontol. 2012;39:239–248. [DOI] [PubMed] [Google Scholar]
- 47.Cochran DL. Inflammation and bone loss in periodontal disease. J Periodontol. 2008;79:1569–1576. [DOI] [PubMed] [Google Scholar]
- 48.Belibasakis GN, Meier A, Guggenheim B, Bostanci N. The RANKL-OPG system is differentially regulated by supragingival and subgingival biofilm supernatants. Cytokine. 2011;55:98–103. [DOI] [PubMed] [Google Scholar]
- 49.Jin Q, Cirelli JA, Park CH, et al. RANKL inhibition through osteoprotegerin blocks bone loss in experimental periodontitis. J Periodontol. 2007;78:1300–1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Teng YT, Nguyen H, Gao X, et al. Functional human T-cell immunity and osteoprotegerin ligand control alveolar bone destruction in periodontal infection. J Clin Invest. 2000;106:R59–R67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Udagawa N, Takahashi N, Jimi E, et al. Osteoblasts/stromal cells stimulate osteoclast activation through expression of osteoclast differentiation factor/RANKL but not macrophage colony-stimulating factor: receptor activator of NF-kappa B ligand. Bone. 1999;25:517–523. [DOI] [PubMed] [Google Scholar]
- 52.Udagawa N, Takahashi N, Yasuda H, et al. Osteoprotegerin produced by osteoblasts is an important regulator in osteoclast development and function. Endocrinology. 2000;141:3478–3484. [DOI] [PubMed] [Google Scholar]
- 53.Corral DA, Amling M, Priemel M, et al. Dissociation between bone resorption and bone formation in osteopenic transgenic mice. Proc Natl Acad Sci USA. 1998;95:13835–13840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Xiong J, Onal M, Jilka RL, Weinstein RS, Manolagas SC, O’Brien CA. Matrix-embedded cells control osteoclast formation. Nat Med. 2011;17:1235–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nakashima T, Hayashi M, Fukunaga T, et al. Evidence for osteocyte regulation of bone homeostasis through RANKL expression. Nat Med. 2011;17:1231–1234. [DOI] [PubMed] [Google Scholar]
- 56.Xiong J, Piemontese M, Onal M, et al. Osteocytes, not osteoblasts or lining cells, are the main source of the RANKL required for osteoclast formation in remodeling bone. PLoS One. 2015;10:e0138189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Onal M, Xiong J, Chen X, et al. Receptor activator of nuclear factor κB ligand (RANKL) protein expression by B lymphocytes contributes to ovariectomy-induced bone loss. J Biol Chem. 2012;287:29851–29860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Li Y, Toraldo G, Li A, et al. B cells and T cells are critical for the preservation of bone homeostasis and attainment of peak bone mass in vivo. Blood. 2007;109:3839–3848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Grcevic D, Lee SK, Marusic A, Lorenzo JA. Depletion of CD4 and CD8 T lymphocytes in mice in vivo enhances 1,25-dihydroxyvitamin D3-stimulated osteoclast-like cell formation in vitro by a mechanism that is dependent on prostaglandin synthesis. J Immunol. 2000;165:4231–4238. [DOI] [PubMed] [Google Scholar]
- 60.Kawai T, Matsuyama T, Hosokawa Y, et al. B and T lymphocytes are the primary sources of RANKL in the bone resorptive lesion of periodontal disease. Am J Pathol. 2006;169:987–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pacios S, Xiao W, Mattos M, et al. Osteoblast lineage cells play an essential role in periodontal bone loss through activation of nuclear factor-kappa B. Sci Rep. 2015;5:16694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Graves DT, Alshabab A, Albiero ML, et al. Osteocytes play an important role in experimental periodontitis in healthy and diabetic mice through expression of RANKL. J Clin Periodontol. 2018;45:285–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tsukasaki M, Komatsu N, Nagashima K, et al. Host defense against oral microbiota by bone-damaging T cells. Nat Commun. 2018;9:701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sims NA, Martin TJ. Coupling the activities of bone formation and resorption: a multitude of signals within the basic multicellular unit. Bonekey Rep. 2014;3:481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zaidi M. Skeletal remodeling in health and disease. Nat Med. 2007;13:791–801. [DOI] [PubMed] [Google Scholar]
- 66.Weitzmann MN, Ofotokun I. Physiological and pathophysiological bone turnover—role of the immune system. Nat Rev Endocrinol. 2016;12:518–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zaidi M, Yuen T, Sun L, Rosen CJ. Regulation of skeletal homeostasis. Endocr Rev. 2018;39:701–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Raggatt LJ, Partridge NC. Cellular and molecular mechanisms of bone remodeling. J Biol Chem. 2010;285:25103–25108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Parfitt AM. Osteonal and hemi-osteonal remodeling: the spatial and temporal framework for signal traffic in adult human bone. J Cell Biochem. 1994;55:273–286. [DOI] [PubMed] [Google Scholar]
- 70.Manolagas SC. Birth and death of bone cells: basic regulatory mechanisms and implications for the pathogenesis and treatment of osteoporosis. Endocr Rev. 2000;21:115–137. [DOI] [PubMed] [Google Scholar]
- 71.Baron R, Saffar JL. A quantitative study of bone remodeling during experimental periodontal disease in the golden hamster. J Periodontal Res. 1978;13:309–315. [DOI] [PubMed] [Google Scholar]
- 72.Makris GP, Saffar JL. Disturbances in bone remodeling during the progress of hamster periodontitis. A morphological and quantitative study. J Periodontal Res. 1985;20:411–420. [DOI] [PubMed] [Google Scholar]
- 73.Irving JT, Socransky SS, Heeley JD. Histological changes in experimental periodontal disease in gnotobiotic rats and conventional hamsters. J Periodontal Res. 1974;9:73–80. [DOI] [PubMed] [Google Scholar]
- 74.Irving JT, Newman MG, Socransky SS, Heely JD. Histological changes in experimental periodontal disease in rats mono-infected with a gram-negative organism. Arch Oral Biol. 1975;20:219–220. [DOI] [PubMed] [Google Scholar]
- 75.Silva BC, Bilezikian JP. Parathyroid hormone: anabolic and catabolic actions on the skeleton. Curr Opin Pharmacol. 2015;22:41–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Barros SP, Silva MA, Somerman MJ, Nociti FH Jr. Parathyroid hormone protects against periodontitis-associated bone loss. J Dent Res. 2003;82:791–795. [DOI] [PubMed] [Google Scholar]
- 77.Tokunaga K, Seto H, Ohba H, et al. Topical and intermittent application of parathyroid hormone recovers alveolar bone loss in rat experimental periodontitis. J Periodontal Res. 2011;46:655–662. [DOI] [PubMed] [Google Scholar]
- 78.Bashutski JD, Eber RM, Kinney JS, et al. Teriparatide and osseous regeneration in the oral cavity. N Engl J Med. 2010;363:2396–2405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lorentzon M. Treating osteoporosis to prevent fractures: current concepts and future developments. J Int Med. 2019;285:381–394. [DOI] [PubMed] [Google Scholar]
- 80.Taut AD, Jin Q, Chung JH, et al. Sclerostin antibody stimulates bone regeneration after experimental periodontitis. J Bone Miner Res. 2013;28:2347–2356. [DOI] [PubMed] [Google Scholar]
- 81.Chen H, Xu X, Liu M, et al. Sclerostin antibody treatment causes greater alveolar crest height and bone mass in an ovariectomized rat model of localized periodontitis. Bone. 2015;76:141–148. [DOI] [PubMed] [Google Scholar]
- 82.Huja SS, Fernandez SA, Hill KJ, Li Y. Remodeling dynamics in the alveolar process in skeletally mature dogs. Anat Rec A Discov Mol Cell Evol Biol. 2006;288:1243–1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Matsuura T, Tokutomi K, Sasaki M, Katafuchi M, Mizumachi E, Sato H. Distinct characteristics of mandibular bone collagen relative to long bone collagen: relevance to clinical dentistry. Biomed Res Int. 2014;2014:769414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wen D, Qing L, Harrison G, Golub E, Akintoye SO. Anatomic site variability in rat skeletal uptake and desorption of fluorescently labeled bisphosphonate. Oral Dis. 2011;17:427–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ehrlich PJ, Lanyon LE. Mechanical strain and bone cell function: a review. Osteoporos Int. 2002;13:688–700. [DOI] [PubMed] [Google Scholar]
- 86.Knoell AC. A mathematical model of an in vitro human mandible. J Biomech. 1977;10:159–166. [DOI] [PubMed] [Google Scholar]
- 87.Daegling DJ, Hylander WL. Occlusal forces and mandibular bone strain: is the primate jaw “overdesigned”? J Hum Evol. 1997;33:705–717. [DOI] [PubMed] [Google Scholar]
- 88.Mavropoulos A, Rizzoli R, Ammann P. Different responsiveness of alveolar and tibial bone to bone loss stimuli. J Bone Miner Res. 2007;22:403–410. [DOI] [PubMed] [Google Scholar]
- 89.Irie K, Novince CM, Darveau RP. Impact of the oral commensal flora on alveolar bone homeostasis. J Dent Res. 2014;93:801–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Institute of Medicine. Microbial Ecology in States of Health and Disease: Workshop Summary. Washington, DC: National Academies Press; 2014. [PubMed] [Google Scholar]
- 91.Belkaid Y, Naik S. Compartmentalized and systemic control of tissue immunity by commensals. Nat Immunol. 2013;14:646–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.The Human Microbiome Project Consortium. Structure, function, and diversity of the healthy human microbiome. Nature 2012;486:207–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Gargiulo AW, Wentz FM, Orban B. Dimensions and relations of the dentogingival junction in humans. J Periodontol. 1961;32:261–267. [Google Scholar]
- 94.Sicher H. Changing concepts of the supporting dental structures. Oral Surg Oral Med Oral Pathol. 1959;12:31–35. [DOI] [PubMed] [Google Scholar]
- 95.Bosshardt DD, Lang NP. The junctional epithelium: from health to disease. J Dent Res. 2005;84:9–20. [DOI] [PubMed] [Google Scholar]
- 96.Nanci A, Bosshardt DD. Structure of periodontal tissues in health and disease. Periodontol 2000. 2006;40:11–28. [DOI] [PubMed] [Google Scholar]
- 97.Schroeder HE, Listgarten MA. (1997) The gingival tissues: the architecture of periodontal protection. Periodontol 2000. 1997;13:91–120. [DOI] [PubMed] [Google Scholar]
- 98.Brill N, Krasse OB. The passage of tissue fluid into the clinically healthy gingival pocket. Acta Odontol Scand. 1958;16:233–245. [Google Scholar]
- 99.Brill N. The Gingival Pocket Fluid: Studies of its Occurrence, Composition, and Effect. Acta Odontologica Scandinavica, Vol. 20, Suppl 32. Copenhagen, Denmark: Royal Dental College; 1962;20(suppl):32. [Google Scholar]
- 100.Attström R. Presence of leukocytes in crevices of healthy and chronically inflamed gingivae. J Periodontal Res. 1970;5:42–47. [DOI] [PubMed] [Google Scholar]
- 101.Attström R, Egelberg J. Emigration of blood neutrophils and monocytes into the gingival crevices. J Periodontal Res. 1970;5:48–55. [DOI] [PubMed] [Google Scholar]
- 102.Attström R, Egelbert J. Presence of leukocytes within the gingival crevices during developing gingivitis in dogs. J Periodontal Res. 1971;6:110–114. [DOI] [PubMed] [Google Scholar]
- 103.Schiott CR, Loe H. The origin and variation in number of leukocytes in the human saliva. J Periodontal Res. 1970;5:36–41. [DOI] [PubMed] [Google Scholar]
- 104.Darveau RP. Periodontitis: a polymicrobial disruption of host homeostasis. Nat Rev Microbiol. 2010;8:481–490. [DOI] [PubMed] [Google Scholar]
- 105.Garant PR. Plaque-neutrophil interaction in monoinfected rats as visualized by transmission electron microscopy. J Periodontol. 1976;47:132–138. [DOI] [PubMed] [Google Scholar]
- 106.Attström R. The roles of gingival epithelium and phagocytosing leukocytes in gingival defence. J Clin Periodontol. 1975;2:25–32. [DOI] [PubMed] [Google Scholar]
- 107.Curtis MA, Zenobia C, Darveau RP. The relationship of the oral microbiotia to periodontal health and disease. Cell Host Microbe. 2011;10:302–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Theilade E. The non-specific theory in microbial etiology of inflammatory periodontal diseases. J Clin Periodontol. 1986;13:905–911. [DOI] [PubMed] [Google Scholar]
- 109.Berezow AB, Darveau RP. Microbial shift and periodontitis. Periodontol 2000. 2011;55(1):36–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Socransky SS, Haffajee AD, Cugini MA, Smith C, Kent RL Jr. Microbial complexes in subgingival plaque. J Clin Periodontol. 1998;25:134–144. [DOI] [PubMed] [Google Scholar]
- 111.Shirtliff ME, Peters BM, Jabra-Rizk MA. Cross-kingdom interactions: Candida albicans and bacteria. FEMS Microbiol Lett. 2009;299:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Baker JL, Bor B, Agnello M, Shi W, He X. Ecology of the oral microbiome: beyond bacteria. Trends Microbiol. 2017;25:362–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Lamont RJ, Koo H, Hajishengallis G. The oral microbiota: dynamic communities and host interactions. Nat Rev Microbiol. 2018;16:745–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Kesavalu L, Sathishkumar S, Bakthavatchalu V, et al. Rat model of polymicrobial infection, immunity, and alveolar bone resorption in periodontal disease. Infect Immun. 2007;75:1704–1712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Polak D, Wilensky A, Shapira L, et al. Mouse model of experimental periodontitis induced by Porphyromonas gingivalis/Fusobacterium nucleatum infection: bone loss and host response. J Clin Periodontol. 2009;36:406–410. [DOI] [PubMed] [Google Scholar]
- 116.Orth RK, O’Brien-Simpson NM, Dashper SG, Reynolds EC. Synergistic virulence of Porphyromonas gingivalis and Treponema denticola in a murine periodontitis model. Mol Oral Microbiol. 2011;26:229–240. [DOI] [PubMed] [Google Scholar]
- 117.Settem RP, El-Hassan AT, Honma K, Stafford GP, Sharma A Fusobacterium nucleatum and Tannerella forsythia induce synergistic alveolar bone loss in a mouse periodontitis model. Infect Immun. 2012;80:2436–2443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Yamazaki K, Sato K, Tsuzuno T, et al. Orally administered pathobionts and commensals have comparable and innocuous systemic effects on germ-free mice. Microb Pathog. 2020;140:103962. [DOI] [PubMed] [Google Scholar]
- 119.Hajishengallis G, Liang S, Payne MA, et al. Low-abundance biofilm species orchestrates inflammatory periodontal disease through the commensal microbiota and complement. Cell Host Microbe. 2011;10:497–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Hathaway-Schrader JD, Steinkamp HM, Chavez MB, et al. Antibiotic perturbation of gut microbiota dysregulates osteoimmune cross talk in postpubertal skeletal development. Am J Pathol. 2019;189:370–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Aas JA, Paster BJ, Stokes LN, Olsen I, Dewhirst FE. Defining the normal bacterial flora of the oral cavity. J Clin Microbiol. 2005;43:5721–5732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Dewhirst FE, Chen T, Izard J, et al. The human oral microbiome. J Bacteriol. 2010;192:5002–5017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Wade WG. The oral microbiome in health and disease. Pharmacol Res. 2013;69:137–143. [DOI] [PubMed] [Google Scholar]
- 124.Griffen AL, Beall CJ, Campbell JH, et al. Distinct and complex bacterial profiles in human periodontitis and health revealed by 16S pyrosequencing. ISME J. 2012;6:1176–1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Abusleme L, Dupuy AK, Dutzan N, et al. The subgingival microbiome in health and periodontitis and its relationship with community biomass and inflammation. ISME J. 2013;7:1016–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Marsh PD. Microbial ecology of dental plaque and its significance in health and disease. Adv Dent Res. 1994;8:263–271. [DOI] [PubMed] [Google Scholar]
- 127.Lloyd-Price J, Abu-Ali G, Huttenhower C. The healthy human microbiome. Genome Med. 2016;8:51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Underhill DM, Iliev ID. The mycobiota: interactions between commensal fungi and the host immune system. Nat Rev Immunol. 2014;14:405–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Limon JJ, Skalski JH, Underhill DM. Commensal fungi in health and disease. Cell Host Microbe. 2017;22:156–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Bacher P, Hohnstein T, Beerbaum E, et al. Human anti-fungal Th17 immunity and pathology rely on cross-reactivity against Candida albicans. Cell. 2019;176:1340–1355.e1315. [DOI] [PubMed] [Google Scholar]
- 131.Shao TY, Ang WXG, Jiang TT, et al. Commensal Candida albicans positively calibrates systemic Th17 immunological responses. Cell Host Microbe. 2019;25:404–417.e406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Conti HR, Shen F, Nayyar N, et al. Th17 cells and IL-17 receptor signaling are essential for mucosal host defense against oral candidiasis. J Exp Med. 2009;206:299–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Yu JJ, Ruddy MJ, Wong GC, et al. An essential role for IL-17 in preventing pathogen-initiated bone destruction: recruitment of neutrophils to inflamed bone requires IL-17 receptor–dependent signals. Blood. 2007;109:3794–3802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Grimaudo NJ, Nesbitt WE. Coaggregation of Candida albicans with oral Fusobacterium species. Oral Microbiol Immunol. 1997;12:168–173. [DOI] [PubMed] [Google Scholar]
- 135.Wu T, Cen L, Kaplan C, et al. Cellular components mediating coadherence of Candida albicans and Fusobacterium nucleatum. J Dent Res. 2015;94:1432–1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Bor B, Cen L, Agnello M, Shi W, He X. Morphological and physiological changes induced by contact-dependent interaction between Candida albicans and Fusobacterium nucleatum. Sci Rep. 2016;6:27956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Page RC, Schroeder HE. Periodontitis in Man and Other Animals: A Comparative Review. New York, NY: Karger; 1982. [Google Scholar]
- 138.Garant PR, Cho MI. Histopathogenesis of spontaneous periodontal disease in conventional rats. I. Histometric and histologic study. J Periodontal Res. 1979;14:297–309. [DOI] [PubMed] [Google Scholar]
- 139.Waerhaug J. The angular bone defect and its relationship to trauma from occlusion and downgrowth of subgingival plaque. J Clin Periodontol. 1979;6:61–82. [DOI] [PubMed] [Google Scholar]
- 140.Lindhe J, Ericsson I. Effect of ligature placement and dental plaque on periodontal tissue breakdown in the dog. J Periodontol. 1978;49:343–350. [DOI] [PubMed] [Google Scholar]
- 141.Rowe DJ, Bradley LS. Quantitative analyses of osteoclasts, bone loss and inflammation in human periodontal disease. J Periodontal Res. 1981;16:13–19. [DOI] [PubMed] [Google Scholar]
- 142.Liu YC, Lerner UH, Teng YT. Cytokine responses against periodontal infection: protective and destructive roles. Periodontol 2000. 2010;52:163–206. [DOI] [PubMed] [Google Scholar]
- 143.Garlet GP. Destructive and protective roles of cytokines in periodontitis: a re-appraisal from host defense and tissue destruction viewpoints. J Dent Res. 2010;89:1349–1363. [DOI] [PubMed] [Google Scholar]
- 144.Graves DT, Li J, Cochran DL. Inflammation and uncoupling as mechanisms of periodontal bone loss. J Dent Res. 2011;90:143–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Listgarten MA. Pathogenesis of periodontitis. J Clin Periodontol. 1986;13:418–430. [DOI] [PubMed] [Google Scholar]
- 146.Page RC, Schroeder HE. Pathogenesis of inflammatory periodontal disease. A summary of current work. Lab Invest. 1976;34:235–249. [PubMed] [Google Scholar]
- 147.Weinmann JP. Progress of gingival inflammation into the supporting structures of the teeth. J Periodontol. 1941;12:71–82. [Google Scholar]
- 148.Moskow BS, Polson AM. Histologic studies on the extension of the inflammatory infiltrate in human periodontitis. J Clin Periodontol. 1991;18:534–542. [DOI] [PubMed] [Google Scholar]
- 149.Thannickal VJ, Zhou Y, Gaggar A, Duncan SR. Fibrosis: ultimate and proximate causes. J Clin Invest. 2014;124:4673–4677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Wynn TA, Ramalingam TR. Mechanisms of fibrosis: therapeutic translation for fibrotic disease. Nat Med. 2012;18:1028–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Eming SA, Wynn TA, Martin P. Inflammation and metabolism in tissue repair and regeneration. Science. 2017;356:1026–1030. [DOI] [PubMed] [Google Scholar]
- 152.Tilg H, Cani PD, Mayer EA. Gut microbiome and liver diseases. Gut. 2016;65:2035–2044. [DOI] [PubMed] [Google Scholar]
- 153.Chu H, Duan Y, Yang L, Schnabl B. Small metabolites, possible big changes: a microbiota-centered view of non-alcoholic fatty liver disease. Gut. 2019;68:359–370. [DOI] [PubMed] [Google Scholar]
- 154.Weitnauer M, Mijosek V, Dalpke AH. Control of local immunity by airway epithelial cells. Mucosal Immunol. 2016;9:287–298. [DOI] [PubMed] [Google Scholar]
- 155.O’Dwyer DN, Dickson RP, Moore BB. The lung microbiome, immunity, and the pathogenesis of chronic lung disease. J Immunol. 2016;196:4839–4847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Yang D, Chen X, Wang J, et al. Dysregulated lung commensal bacteria drive interleukin-17B production to promote pulmonary fibrosis through their outer membrane vesicles. Immunity. 2019;50:692–706.e697. [DOI] [PubMed] [Google Scholar]
- 157.Chen YE, Fischbach MA, Belkaid Y. Skin microbiota-host interactions. Nature. 2018;553:427–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Canesso MC, Vieira AT, Castro TB, et al. Skin wound healing is accelerated and scarless in the absence of commensal microbiota. J Immunol. 2014;193:5171–5180. [DOI] [PubMed] [Google Scholar]
- 159.Lorenzo J, Horowitz M, Choi Y. Osteoimmunology: interactions of the bone and immune system. Endocr Rev. 2008;29:403–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Takayanagi H. Osteoimmunology and the effects of the immune system on bone. Nat Rev Rheumatol. 2009;5:667–676. [DOI] [PubMed] [Google Scholar]
- 161.Walsh MC, Takegahara N, Kim H, Choi Y. Updating osteoimmunology: regulation of bone cells by innate and adaptive immunity. Nat Rev Rheumatol. 2018;14:146–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Belkaid Y, Harrison OJ. Homeostatic immunity and the microbiota. Immunity. 2017;46:562–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Novince CM, Whittow CR, Aartun JD, et al. Commensal gut microbiota immunomodulatory actions in bone marrow and liver have catabolic effects on skeletal homeostasis in health. Sci Rep. 2017;7:5747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Klein L, Kyewski B, Allen PM, Hogquist KA. Positive and negative selection of the T cell repertoire: what thymocytes see (and don’t see). Nat Rev Immunol. 2014;14:377–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Bevan MJ. Helping the CD8+ T-cell response. Nat Rev Immunol. 2004;4:595–602. [DOI] [PubMed] [Google Scholar]
- 166.Reipert BM, Steinitz KN, van Helden PM, et al. Opportunities and limitations of mouse models humanized for HLA class II antigens. J Thromb Haemost. 2009;7(Suppl 1):92–97. [DOI] [PubMed] [Google Scholar]
- 167.Rocha N, Neefjes J. MHC class II molecules on the move for successful antigen presentation. EMBO J. 2008;27:1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Khairallah C, Dechanet-Merville J, Capone M. γδ T cell-mediated immunity to cytomegalovirus infection. Front Immunol. 2017;8:105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Krishnan S, Prise IE, Wemyss K, et al. Amphiregulin-producing γδ T cells are vital for safeguarding oral barrier immune homeostasis. Proc Natl Acad Sci USA. 2018;115:10738–10743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Wilharm A, Tabib Y, Nassar M, et al. Mutual interplay between IL-17-producing γδT cells and microbiota orchestrates oral mucosal homeostasis. Proc Natl Acad Sci USA. 2019;116:2652–2661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Fleming C, Cai Y, Sun X, et al. Microbiota-activated CD103(+) DCs stemming from microbiota adaptation specifically drive gammadeltaT17 proliferation and activation. Microbiome. 2017;5:46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Kong YY, Feige U, Sarosi I, et al. Activated T cells regulate bone loss and joint destruction in adjuvant arthritis through osteoprotegerin ligand. Nature. 1999;402:304–309. [DOI] [PubMed] [Google Scholar]
- 173.Wang R, Zhang L, Zhang X, et al. Regulation of activation-induced receptor activator of NF-κB ligand (RANKL) expression in T cells. Eur J Immunol. 2002;32:1090–1098. [DOI] [PubMed] [Google Scholar]
- 174.Pacifici R. T cells: critical bone regulators in health and disease. Bone. 2010;47:461–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Taubman MA, Stoufi ED, Ebersole JL, Smith DJ. Phenotypic studies of cells from periodontal disease tissues. J Periodontal Res. 1984;19:587–590. [DOI] [PubMed] [Google Scholar]
- 176.Cole KL, Seymour GJ, Powell RN. Phenotypic and functional analysis of T cells extracted from chronically inflamed human periodontal tissues. J Periodontol. 1987;58:569–573. [DOI] [PubMed] [Google Scholar]
- 177.Stoufi ED, Taubman MA, Ebersole JL, Smith DJ, Stashenko PP. Phenotypic analyses of mononuclear cells recovered from healthy and diseased human periodontal tissues. J Clin Immunol. 1987;7:235–245. [DOI] [PubMed] [Google Scholar]
- 178.Johannessen AC, Nilsen R, Kristoffersen T, Knudsen GE. Variation in the composition of gingival inflammatory cell infiltrates. J Clin Periodontol. 1990;17:298–305. [DOI] [PubMed] [Google Scholar]
- 179.Syrjanen S, Markkanen H, Syrjanen K. Inflammatory cells and their subsets in lesions of juvenile periodontitis. A family study. Acta Odontol Scand. 1984;42:285–292. [DOI] [PubMed] [Google Scholar]
- 180.Reinhardt RA, McDonald TL, Bolton RW, DuBois LM, Feely DE, Kaldahl WB. In situ activated T lymphocytes in active versus stable periodontal lesions. J Periodontal Res. 1988;23:295–302. [DOI] [PubMed] [Google Scholar]
- 181.Lappin DF, Koulouri O, Radvar M, Hodge P, Kinane DF. Relative proportions of mononuclear cell types in periodontal lesions analyzed by immunohistochemistry. J Clin Periodontol. 1999;26:183–189. [DOI] [PubMed] [Google Scholar]
- 182.Gemmell E, Feldner B, Seymour GJ. CD45RA and CD45RO positive CD4 cells in human peripheral blood and periodontal disease tissue before and after stimulation with periodontopathic bacteria. Oral Microbiol Immunol. 1992;7:84–88. [DOI] [PubMed] [Google Scholar]
- 183.Yamazaki K, Nakajima T, Aoyagi T, Hara K. Immunohistological analysis of memory T lymphocytes and activated B lymphocytes in tissues with periodontal disease. J Periodontal Res. 1993;28:324–334. [DOI] [PubMed] [Google Scholar]
- 184.Aoyagi T, Sugawara-Aoyagi M, Yamazaki K, Hara K. Interleukin 4 (IL-4) and IL-6-producing memory T-cells in peripheral blood and gingival tissue in periodontitis patients with high serum antibody titers to Porphyromonas gingivalis. Oral Microbiol Immunol. 1995;10:304–310. [DOI] [PubMed] [Google Scholar]
- 185.Baker PJ, Evans RT, Roopenian DC. Oral infection with Porphyromonas gingivalis and induced alveolar bone loss in immunocompetent and severe combined immunodeficient mice. Arch Oral Biol. 1994;39:1035–1040. [DOI] [PubMed] [Google Scholar]
- 186.Baker PJ, Dixon M, Evans RT, Dufour L, Johnson E, Roopenian DC. CD4+ T cells and the proinflammatory cytokines gamma interferon and interleukin-6 contribute to alveolar bone loss in mice. Infect Immun. 1999;67:2804–2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Wassenaar A, Reinhardus C, Abraham-Inpijn L, Kievits F. Type-1 and type-2 CD8+ T-cell subsets isolated from chronic adult periodontitis tissue differ in surface phenotype and biological functions. Immunology. 1996;87:113–118. [PMC free article] [PubMed] [Google Scholar]
- 188.Cifcibasi E, Ciblak M, Kiran B, et al. The role of activated cytotoxic T cells in etiopathogenesis of periodontal disease: does it harm or does it heal? Sci Rep. 2015;5:9262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Lundqvist C, Hammarstrom ML. T-cell receptor gamma delta-expressing intraepithelial lymphocytes are present in normal and chronically inflamed human gingiva. Immunology. 1993;79:38–45. [PMC free article] [PubMed] [Google Scholar]
- 190.Lundqvist C, Hammarstrom S, Hammarstrom ML. Intraepithelial lymphocytes expressing TCR gamma delta in human gingival tissues. Adv Exp Med Biol. 1995;371B:1097–1102. [PubMed] [Google Scholar]
- 191.Kawahara K, Fukunaga M, Takata T, Kawamura M, Morishita M, Iwamoto Y. Immunohistochemical study of gamma delta T cells in human gingival tissues. J Periodontol. 1995;66:775–779. [DOI] [PubMed] [Google Scholar]
- 192.Lundqvist C, Baranov V, Teglund S, Hammarstrom S, Hammarstrom ML. Cytokine profile and ultrastructure of intraepithelial gamma delta T cells in chronically inflamed human gingiva suggest a cytotoxic effector function. J Immunol. 1994;153:2302–2312. [PubMed] [Google Scholar]
- 193.Gemmell E, Seymour GJ. Gamma delta T lymphocytes in human periodontal disease tissue. J Periodontol. 1995;66:780–785. [DOI] [PubMed] [Google Scholar]
- 194.Kaiko GE, Horvat JC, Beagley KW, Hansbro PM. Immunological decision-making: how does the immune system decide to mount a helper T-cell response? Immunology. 2008;123:326–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Zhou L, Chong MM, Littman DR. Plasticity of CD4+ T cell lineage differentiation. Immunity. 2009;30:646–655. [DOI] [PubMed] [Google Scholar]
- 196.Takayanagi H, Ogasawara K, Hida S, et al. T-cell-mediated regulation of osteoclastogenesis by signalling cross-talk between RANKL and IFN-γ. Nature. 2000;408:600–605. [DOI] [PubMed] [Google Scholar]
- 197.Kohara H, Kitaura H, Fujimura Y, et al. IFN-γ directly inhibits TNF-α-induced osteoclastogenesis in vitro and in vivo and induces apoptosis mediated by Fas/Fas ligand interactions. Immunol Lett. 2011;137:53–61. [DOI] [PubMed] [Google Scholar]
- 198.Ji JD, Park-Min KH, Shen Z, et al. Inhibition of RANK expression and osteoclastogenesis by TLRs and IFN-γ in human osteoclast precursors. J Immunol. 2009;183:7223–7233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Fox SW, Chambers TJ. Interferon-gamma directly inhibits TRANCE-induced osteoclastogenesis. Biochem Biophys Res Commun. 2000;276:868–872. [DOI] [PubMed] [Google Scholar]
- 200.Kotake S, Nanke Y, Mogi M, et al. IFN-γ-producing human T cells directly induce osteoclastogenesis from human monocytes via the expression of RANKL. Eur J Immunol. 2005;35:3353–3363. [DOI] [PubMed] [Google Scholar]
- 201.Madyastha PR, Yang S, Ries WL, Key LL Jr. IFN-γ enhances oseoclast generation in cultures of peripheral blood from osteopetrotic patients and normalizes superoxide production. J Interferon Cytokine Res. 2000;20:645–652. [DOI] [PubMed] [Google Scholar]
- 202.Gao Y, Grassi F, Ryan MR, et al. IFN-γ stimulates osteoclast formation and bone loss in vivo via antigen-driven T cell activation. J Clin Invest. 2007;117:122–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Cheng J, Liu J, Shi Z, et al. Interleukin-4 inhibits RANKL-induced NFATc1 expression via STAT6: a novel mechanism mediating its blockade of osteoclastogenesis. J Cell Biochem. 2011;112:3385–3392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Evans KE, Fox SW. Interleukin-10 inhibits osteoclastogenesis by reducing NFATc1 expression and preventing its translocation to the nucleus. BMC Cell Biol. 2007;8:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Liu D, Yao S, Wise GE. Effect of interleukin-10 on gene expression of osteoclastogenic regulatory molecules in the rat dental follicle. Eur J Oral Sci. 2006;114:42–49. [DOI] [PubMed] [Google Scholar]
- 206.Palmqvist P, Lundberg P, Persson E, et al. Inhibition of hormone and cytokine-stimulated osteoclastogenesis and bone resorption by interleukin-4 and interleukin-13 is associated with increased osteoprotegerin and decreased RANKL and RANK in a STAT6-dependent pathway. J Biol Chem. 2006;281:2414–2429. [DOI] [PubMed] [Google Scholar]
- 207.Houri-Haddad Y, Soskolne WA, Halabi A, Shapira L. IL-10 gene transfer attenuates P. gingivalis-induced inflammation. J Dent Res. 2007;86:560–564. [DOI] [PubMed] [Google Scholar]
- 208.Takeichi O, Haber J, Kawai T, Smith DJ, Moro I, Taubman MA. Cytokine profiles of T-lymphocytes from gingival tissues with pathological pocketing. J Dent Res. 2000;79:1548–1555. [DOI] [PubMed] [Google Scholar]
- 209.Ukai T, Mori Y, Onoyama M, Hara Y. Immunohistological study of interferon-γ and interleukin-4-bearing cells in human periodontitis gingiva. Arch Oral Biol. 2001;46:901–908. [DOI] [PubMed] [Google Scholar]
- 210.Manhart SS, Reinhardt RA, Payne JB, et al. Gingival cell IL-2 and IL-4 in early-onset periodontitis. J Periodontol. 1994;65:807–813. [DOI] [PubMed] [Google Scholar]
- 211.Tokoro Y, Matsuki Y, Yamamoto T, Suzuki T, Hara K. Relevance of local Th2-type cytokine mRNA expression in immunocompetent infiltrates in inflamed gingival tissue to periodontal diseases. Clin Exp Immunol. 1997;107:166–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Bartova J, Kratka-Opatrna Z, Prochazkova J, et al. Th1 and Th2 cytokine profile in patients with early onset periodontitis and their healthy siblings. Mediators Inflamm. 2000;9:115–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Lappin DF, MacLeod CP, Kerr A, Mitchell T, Kinane DF. Anti-inflammatory cytokine IL-10 and T cell cytokine profile in periodontitis granulation tissue. Clin Exp Immunol. 2001;123:294–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Gemmell E, Carter CL, Seymour GJ. Mast cells in human periodontal disease. J Dent Res. 2004;83:384–387. [DOI] [PubMed] [Google Scholar]
- 215.Nakajima T, Yamazaki K, Cullinan MP, Gemmell E, Seymour GJ. T-cell antigen specificity in humans following stimulation with Porphyromonas gingivalis. Arch Oral Biol. 1999;44:1045–1053. [DOI] [PubMed] [Google Scholar]
- 216.Berglundh T, Liljenberg B, Lindhe J. Some cytokine profiles of T-helper cells in lesions of advanced periodontitis. J Clin Periodontol. 2002;29:705–709. [DOI] [PubMed] [Google Scholar]
- 217.Gemmell E, Carter CL, Grieco DA, Sugerman PB, Seymour GJ. P. gingivalis-specific T-cell lines produce Th1 and Th2 cytokines. J Dent Res. 2002;81:303–307. [DOI] [PubMed] [Google Scholar]
- 218.Choi JI, Borrello MA, Smith ES, Zauderer M. Polarization of Porphyromonas gingivalis-specific helper T-cell subsets by prior immunization with Fusobacterium nucleatum. Oral Microbiol Immunol. 2000;15:181–187. [DOI] [PubMed] [Google Scholar]
- 219.Kawai T, Eisen-Lev R, Seki M, Eastcott JW, Wilson ME, Taubman MA. Requirement of B7 costimulation for Th1-mediated inflammatory bone resorption in experimental periodontal disease. J Immunol. 2000;164:2102–2109. [DOI] [PubMed] [Google Scholar]
- 220.Teng YT, Mahamed D, Singh B. Gamma interferon positively modulates Actinobacillus actinomycetemcomitans–specific RANKL+ CD4+ Th-cell-mediated alveolar bone destruction in vivo. Infect Immun. 2005;73:3453–3461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Alayan J, Ivanovski S, Farah CS. Alveolar bone loss in T helper 1/T helper 2 cytokine-deficient mice. J Periodontal Res. 2007;42:97–103. [DOI] [PubMed] [Google Scholar]
- 222.Myneni SR, Settem RP, Connell TD, Keegan AD, Gaffen SL, Sharma A. TLR2 signaling and Th2 responses drive Tannerella forsythia–induced periodontal bone loss. J Immunol. 2011;187:501–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Gemmell E, Seymour GJ. Immunoregulatory control of Th1/Th2 cytokine profiles in periodontal disease. Periodontol 2000. 2004;35:21–41. [DOI] [PubMed] [Google Scholar]
- 224.Nakajima T, Ueki-Maruyama K, Oda T, et al. Regulatory T-cells infiltrate periodontal disease tissues. J Dent Res. 2005;84:639–643. [DOI] [PubMed] [Google Scholar]
- 225.Okui T, Ito H, Honda T, Amanuma R, Yoshie H, Yamazaki K. Characterization of CD4+ FOXP3+ T-cell clones established from chronic inflammatory lesions. Oral Microbiol Immunol. 2008;23:49–54. [DOI] [PubMed] [Google Scholar]
- 226.Dutzan N, Gamonal J, Silva A, Sanz M, Vernal R. Over-expression of forkhead box P3 and its association with receptor activator of nuclear factor-κ B ligand, interleukin (IL) 17, IL-10 and transforming growth factor-β during the progression of chronic periodontitis. J Clin Periodontol. 2009;36:396–403. [DOI] [PubMed] [Google Scholar]
- 227.Cardoso CR, Garlet GP, Moreira AP, Junior WM, Rossi MA, Silva JS. Characterization of CD4+CD25+ natural regulatory T cells in the inflammatory infiltrate of human chronic periodontitis. J Leukoc Biol. 2008;84:311–318. [DOI] [PubMed] [Google Scholar]
- 228.Ernst CW, Lee JE, Nakanishi T, et al. Diminished forkhead box P3/CD25 double-positive T regulatory cells are associated with the increased nuclear factor-κB ligand (RANKL+) T cells in bone resorption lesion of periodontal disease. Clin Exp Immunol. 2007;148:271–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Okui T, Aoki Y, Ito H, Honda T, Yamazaki K. The presence of IL-17+/FOXP3+ double-positive cells in periodontitis. J Dent Res. 2012;91:574–579. [DOI] [PubMed] [Google Scholar]
- 230.Kobayashi R, Kono T, Bolerjack BA, et al. Induction of IL-10-producing CD4+ T-cells in chronic periodontitis. J Dent Res. 2011;90:653–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Garlet GP, Cardoso CR, Mariano FS, et al. Regulatory T cells attenuate experimental periodontitis progression in mice. J Clin Periodontol. 2010;37:591–600. [DOI] [PubMed] [Google Scholar]
- 232.Glowacki AJ, Yoshizawa S, Jhunjhunwala S, et al. Prevention of inflammation-mediated bone loss in murine and canine periodontal disease via recruitment of regulatory lymphocytes. Proc Natl Acad Sci USA. 2013;110:18525–18530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Araujo-Pires AC, Vieira AE, Francisconi CF, et al. IL-4/CCL22/CCR4 axis controls regulatory T-cell migration that suppresses inflammatory bone loss in murine experimental periodontitis. J Bone Miner Res. 2015;30:412–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Parachuru VP, Coates DE, Milne TJ, Hussaini HM, Rich AM, Seymour GJ. Forkhead box P3-positive regulatory T-cells and interleukin 17-positive T-helper 17 cells in chronic inflammatory periodontal disease. J Periodontal Res. 2014;49:817–826. [DOI] [PubMed] [Google Scholar]
- 235.Stockinger B, Omenetti S. The dichotomous nature of T helper 17 cells. Nat Rev Immunol. 2017;17:535–544. [DOI] [PubMed] [Google Scholar]
- 236.Dutzan N, Abusleme L, Bridgeman H, et al. On-going mechanical damage from mastication drives homeostatic Th17 cell responses at the oral barrier. Immunity. 2017;46:133–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Dutzan N, Kajikawa T, Abusleme L, et al. A dysbiotic microbiome triggers TH17 cells to mediate oral mucosal immunopathology in mice and humans. Sci Transl Med. 2018;10(463):aat0797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Sato K, Suematsu A, Okamoto K, et al. Th17 functions as an osteoclastogenic helper T cell subset that links T cell activation and bone destruction. J Exp Med. 2006;203:2673–2682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Kotake S, Udagawa N, Takahashi N, et al. IL-17 in synovial fluids from patients with rheumatoid arthritis is a potent stimulator of osteoclastogenesis. J Clin Invest. 1999;103:1345–1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Harrington LE, Hatton RD, Mangan PR, et al. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol. 2005;6:1123–1132. [DOI] [PubMed] [Google Scholar]
- 241.Park H, Li Z, Yang XO, et al. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol. 2005;6:1133–1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Beklen A, Ainola M, Hukkanen M, Gurgan C, Sorsa T, Konttinen YT. MMPs, IL-1, and TNF are regulated by IL-17 in periodontitis. J Dent Res. 2007;86:347–351. [DOI] [PubMed] [Google Scholar]
- 243.Honda T, Aoki Y, Takahashi N, et al. Elevated expression of IL-17 and IL-12 genes in chronic inflammatory periodontal disease. Clin Chim Acta. 2008;395:137–141. [DOI] [PubMed] [Google Scholar]
- 244.Cardoso CR, Garlet GP, Crippa GE, et al. Evidence of the presence of T helper type 17 cells in chronic lesions of human periodontal disease. Oral Microbiol Immunol. 2009;24:1–6. [DOI] [PubMed] [Google Scholar]
- 245.Allam JP, Duan Y, Heinemann F, et al. IL-23-producing CD68+ macrophage-like cells predominate within an IL-17-polarized infiltrate in chronic periodontitis lesions. J Clin Periodontol. 2011;38:879–886. [DOI] [PubMed] [Google Scholar]
- 246.Adibrad M, Deyhimi P, Ganjalikhani Hakemi M, Behfarnia P, Shahabuei M, Rafiee L. Signs of the presence of Th17 cells in chronic periodontal disease. J Periodontal Res. 2012;47:525–531. [DOI] [PubMed] [Google Scholar]
- 247.Ohyama H, Kato-Kogoe N, Kuhara A, et al. The involvement of IL-23 and the Th17 pathway in periodontitis. J Dent Res. 2009;88:633–638. [DOI] [PubMed] [Google Scholar]
- 248.Sun L, Girnary M, Wang L, et al. IL-10 dampens an IL-17-mediated periodontitis-associated inflammatory network. J Immunol. 2020;204:2177–2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Dutzan N, Konkel JE, Greenwell-Wild T, Moutsopoulos NM. Characterization of the human immune cell network at the gingival barrier. Mucosal Immunol. 2016;9:1163–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Yu JJ, Ruddy MJ, Conti HR, Boonanantanasarn K, Gaffen SL. The interleukin-17 receptor plays a gender-dependent role in host protection against Porphyromonas gingivalis–induced periodontal bone loss. Infect Immun. 2008;76:4206–4213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Eskan MA, Jotwani R, Abe T, et al. The leukocyte integrin antagonist Del-1 inhibits IL-17-mediated inflammatory bone loss. Nat Immunol. 2012;13:465–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Diaz-Zuniga J, Melgar-Rodriguez S, Alvarez C, et al. T-lymphocyte phenotype and function triggered by Aggregatibacter actinomycetemcomitans is serotype-dependent. J Periodontal Res. 2015;50:824–835. [DOI] [PubMed] [Google Scholar]
- 253.Vernal R, Diaz-Zuniga J, Melgar-Rodriguez S, et al. Activation of RANKL-induced osteoclasts and memory T lymphocytes by Porphyromonas gingivalis is serotype dependant. J Clin Periodontol. 2014;41:451–459. [DOI] [PubMed] [Google Scholar]
- 254.Settem RP, Honma K, Nakajima T, et al. A bacterial glycan core linked to surface (S)-layer proteins modulates host immunity through Th17 suppression. Mucosal Immunol. 2013;6:415–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Settem RP, Honma K, Sharma A. Neutrophil mobilization by surface-glycan altered Th17-skewing bacteria mitigates periodontal pathogen persistence and associated alveolar bone loss. PLoS One. 2014;9:e108030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Gaffen SL, Hajishengallis G. A new inflammatory cytokine on the block: re-thinking periodontal disease and the Th1/Th2 paradigm in the context of Th17 cells and IL-17. J Dent Res. 2008;87:817–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Cafferata EA, Jerez A, Vernal R, Monasterio G, Pandis N, Faggion CM Jr. The therapeutic potential of regulatory T lymphocytes in periodontitis: a systematic review. J Periodontal Res. 2018. [DOI] [PubMed] [Google Scholar]
- 258.Monasterio G, Castillo F, Ibarra JP, et al. Alveolar bone resorption and Th1/Th17-associated immune response triggered during Aggregatibacter actinomycetemcomitans-induced experimental periodontitis are serotype-dependent. J Periodontol. 2018;89:1249–1261. [DOI] [PubMed] [Google Scholar]
- 259.Monasterio G, Fernandez B, Castillo F, et al. Capsular-defective Porphyromonas gingivalis mutant strains induce less alveolar bone resorption than W50 wild-type strain due to a decreased Th1/Th17 immune response and less osteoclast activity. J Periodontol. 2019;90(5):522–534. [DOI] [PubMed] [Google Scholar]
- 260.Bi CS, Sun LJ, Qu HL, et al. The relationship between T-helper cell polarization and the RANKL/OPG ratio in gingival tissues from chronic periodontitis patients. Clin Exp Dent Res. 2009;5:377–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Gao L, Zhao Y, Wang P, et al. Detection of Th17/Treg cells and related factors in gingival tissues and peripheral blood of rats with experimental periodontitis. Iran J Basic Med Sci. 2017;20:294–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Ebersole JL, Kirakodu S, Novak MJ, et al. Cytokine gene expression profiles during initiation, progression and resolution of periodontitis. J Clin Periodontol. 2014;41:853–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Wang L, Guan N, Jin Y, Lin X, Gao H. Subcutaneous vaccination with Porphyromonas gingivalis ameliorates periodontitis by modulating Th17/Treg imbalance in a murine model. Int Immunopharmacol. 2015;25:65–73. [DOI] [PubMed] [Google Scholar]
- 264.Cafferata EA, Terraza-Aguirre C, Barrera R, et al. Interleukin-35 inhibits alveolar bone resorption by modulating the Th17/Treg imbalance during periodontitis. J Clin Periodontol. 2020. [DOI] [PubMed] [Google Scholar]
- 265.Schmitt E, Klein M, Bopp T. Th9 cells, new players in adaptive immunity. Trends Immunol. 2014;35:61–68. [DOI] [PubMed] [Google Scholar]
- 266.Kaplan MH, Hufford MM, Olson MR. The development and in vivo function of T helper 9 cells. Nat Rev Immunol. 2015;15:295–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Eyerich S, Eyerich K, Pennino D, et al. Th22 cells represent a distinct human T cell subset involved in epidermal immunity and remodeling. J Clin Invest. 2009;119:3573–3585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Wolk K, Witte E, Witte K, Warszawska K, Sabat R. Biology of interleukin-22. Semin Immunopathol. 2010;32:17–31. [DOI] [PubMed] [Google Scholar]
- 269.Sabat R, Ouyang W, Wolk K. Therapeutic opportunities of the IL-22-IL-22R1 system. Nat Rev Drug Discov. 2014;13:21–38. [DOI] [PubMed] [Google Scholar]
- 270.Diaz-Zuniga J, Melgar-Rodriguez S, Rojas L, et al. Increased levels of the T-helper 22-associated cytokine (interleukin-22) and transcription factor (aryl hydrocarbon receptor) in patients with periodontitis are associated with osteoclast resorptive activity and severity of the disease. J Periodontal Res. 2017;52:893–902. [DOI] [PubMed] [Google Scholar]
- 271.Diaz-Zuniga J, Melgar-Rodriguez S, Monasterio G, et al. Differential human Th22-lymphocyte response triggered by Aggregatibacter actinomycetemcomitans serotypes. Arch Oral Biol. 2017;78:26–33. [DOI] [PubMed] [Google Scholar]
- 272.Horowitz MC, Fretz JA, Lorenzo JA. How B cells influence bone biology in health and disease. Bone. 2010;47:472–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Weitzmann MN. The role of Inflammatory cytokines, the RANKL/OPG axis, and the immunoskeletal interface in physiological bone turnover and osteoporosis. Scientifica. 2013;2013:125705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Weitzmann MN, Cenci S, Haug J, Brown C, DiPersio J, Pacifici R. B lymphocytes inhibit human osteoclastogenesis by secretion of TGFβ. J Cell Biochem. 2000;78:318–324. [DOI] [PubMed] [Google Scholar]
- 275.Seymour GJ, Greenspan JS. The phenotypic characterization of lymphocyte subpopulations in established human periodontal disease. J Periodontal Res. 1979;14:39–46. [DOI] [PubMed] [Google Scholar]
- 276.Reinhardt RA, Bolton RW, McDonald TL, DuBois LM, Kaldahl WB. In situ lymphocyte subpopulations from active versus stable periodontal sites. J Periodontol. 1988;59:656–670. [DOI] [PubMed] [Google Scholar]
- 277.Donati M, Liljenberg B, Zitzmann NU, Berglundh T. B-1a cells and plasma cells in periodontitis lesions. J Periodontal Res. 2009;44:683–688. [DOI] [PubMed] [Google Scholar]
- 278.Thorbert-Mros S, Larsson L, Berglundh T. Cellular composition of long-standing gingivitis and periodontitis lesions. J Periodontal Res. 2015;50:535–543. [DOI] [PubMed] [Google Scholar]
- 279.Jing L, Kim S, Sun L, et al. IL-37- and IL-35/IL-37-producing plasma cells in chronic periodontitis. J Dent Res. 2019;98:813–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Baker PJ, Carter S, Dixon M, Evans RT, Roopenian DC. Serum antibody response to oral infection precedes but does not prevent Porphyromonas gingivalis-induced alveolar bone loss in mice. Oral Microbiol Immunol. 1999;14:194–196. [DOI] [PubMed] [Google Scholar]
- 281.Han X, Kawai T, Eastcott JW, Taubman MA. Bacterial-responsive B lymphocytes induce periodontal bone resorption. J Immunol. 2006;176:625–631. [DOI] [PubMed] [Google Scholar]
- 282.Baker PJ, Boutaugh NR, Tiffany M, Roopenian DC. B cell IgD deletion prevents alveolar bone loss following murine oral infection. Interdiscip Perspect Infect Dis. 2009;2009:864359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Oliver-Bell J, Butcher JP, Malcolm J, et al. Periodontitis in the absence of B cells and specific anti-bacterial antibody. Mol Oral Microbiol. 2015;30:160–169. [DOI] [PubMed] [Google Scholar]
- 284.Abe T, AlSarhan M, Benakanakere MR, et al. The B cell–stimulatory cytokines BLyS and APRIL are elevated in human periodontitis and are required for B cell–dependent bone loss in experimental murine periodontitis. J Immunol. 2015;195:1427–1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Kanzaki H, Makihira S, Suzuki M, et al. Soluble RANKL cleaved from activated lymphocytes by TNF-α-converting enzyme contributes to osteoclastogenesis in periodontitis. J Immunol. 2016;197:3871–3883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Demoersman J, Pochard P, Framery C, et al. B cell subset distribution is altered in patients with severe periodontitis. PLoS One. 2018;13:e0192986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Mahanonda R, Champaiboon C, Subbalekha K, et al. Human memory B cells in healthy gingiva, gingivitis, and periodontitis. J Immunol. 2016;197:715–725. [DOI] [PubMed] [Google Scholar]
- 288.Han YK, Jin Y, Miao YB, Shi T, Lin XP. Improved RANKL production by memory B cells: a way for B cells promote alveolar bone destruction during periodontitis. Int Immunopharmacol. 2018;64:232–237. [DOI] [PubMed] [Google Scholar]
- 289.Han Y, Jin Y, Miao Y, Shi T, Lin X. Switched memory B cells promote alveolar bone damage during periodontitis: an adoptive transfer experiment. Int Immunopharmacol. 2018;62:147–154. [DOI] [PubMed] [Google Scholar]
- 290.Han Y, Jin Y, Miao Y, Shi T, Lin X. Improved RANKL expression and osteoclastogenesis induction of CD27+CD38− memory B cells: a link between B cells and alveolar bone damage in periodontitis. J Periodontal Res. 2019;54:73–80. [DOI] [PubMed] [Google Scholar]
- 291.Cherukuri A, Ding Q, Sharma A, Mohib K, Rothstein DM. Regulatory and effector B cells: a new path toward biomarkers and therapeutic targets to improve transplant outcomes? Clin Lab Med. 2019;39:15–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Liu Z, Hu Y, Yu P, et al. Toll-like receptor agonists Porphyromonas gingivalis LPS and CpG differentially regulate IL-10 competency and frequencies of mouse B10 cells. J Appl Oral Sci. 2017;25:90–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Hu Y, Yu P, Yu X, Hu X, Kawai T, Han X. IL-21/anti-Tim1/CD40 ligand promotes B10 activity in vitro and alleviates bone loss in experimental periodontitis in vivo. Biochim Biophys Acta Mol Basis Dis. 2017;1863:2149–2157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Yu P, Hu Y, Liu Z, et al. Local induction of B cell interleukin-10 competency alleviates inflammation and bone loss in ligature-induced experimental periodontitis in mice. Infect Immun. 2017;85(1):e00645–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Zhao Q, Hu Y, Deng S, et al. Cytidine-phosphate-guanosine oligodeoxynucleotides in combination with CD40 ligand decrease periodontal inflammation and alveolar bone loss in a TLR9-independent manner. J Appl Oral Sci. 2018;26:e20170451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Wang Y, Yu X, Lin J, et al. B10 cells alleviate periodontal bone loss in experimental periodontitis. Infect Immun. 2017;85(9):e00335–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Shi T, Jin Y, Miao Y, Wang Y, Zhou Y, Lin X. IL-10 secreting B cells regulate periodontal immune response during periodontitis. Odontology. 2020;108:350–357. [DOI] [PubMed] [Google Scholar]
- 298.Hammer Q, Ruckert T, Romagnani C. Natural killer cell specificity for viral infections. Nat Immunol. 2018;19:800–808. [DOI] [PubMed] [Google Scholar]
- 299.Lawson V. Turned on by danger: activation of CD1d-restricted invariant natural killer T cells. Immunology. 2012;137:20–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Pridgeon C, Lennon GP, Pazmany L, Thompson RN, Christmas SE, Moots RJ. Natural killer cells in the synovial fluid of rheumatoid arthritis patients exhibit a CD56bright, CD94bright, CD158negative phenotype. Rheumatology. 2003;42:870–878. [DOI] [PubMed] [Google Scholar]
- 301.Soderstrom K, Stein E, Colmenero P, et al. Natural killer cells trigger osteoclastogenesis and bone destruction in arthritis. Proc Natl Acad Sci USA. 2010;107:13028–13033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Takeda H, Kikuchi T, Soboku K, et al. Effect of IL-15 and natural killer cells on osteoclasts and osteoblasts in a mouse coculture. Inflammation. 2014;37:657–669. [DOI] [PubMed] [Google Scholar]
- 303.Tsoumis GS, Singh G, Dolby AE. Human antibody-dependent cellular cytotoxicity and natural killer cytotoxicity in periodontal disease. A preliminary report. J Periodontal Res. 1985;20:122–130. [DOI] [PubMed] [Google Scholar]
- 304.Fujita S, Takahashi H, Okabe H, Ozaki Y, Hara Y, Kato I. Distribution of natural killer cells in periodontal diseases: an immunohistochemical study. J Periodontol. 1992;63:686–689. [DOI] [PubMed] [Google Scholar]
- 305.Chaushu S, Wilensky A, Gur C, et al. Direct recognition of Fusobacterium nucleatum by the NK cell natural cytotoxicity receptor NKp46 aggravates periodontal disease. PLoS Pathog. 2012;8:e1002601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.Yamazaki K, Ohsawa Y, Yoshie H. Elevated proportion of natural killer T cells in periodontitis lesions: a common feature of chronic inflammatory diseases. Am J Pathol. 2001;158:1391–1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307.Amanuma R, Nakajima T, Yoshie H, Yamazaki K. Increased infiltration of CD1d and natural killer T cells in periodontal disease tissues. J Periodontal Res. 2006;41:73–79. [DOI] [PubMed] [Google Scholar]
- 308.Nowak M, Kramer B, Haupt M, et al. Activation of invariant NK T cells in periodontitis lesions. J Immunol. 2013;190:2282–2291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309.Aoki-Nonaka Y, Nakajima T, Miyauchi S, et al. Natural killer T cells mediate alveolar bone resorption and a systemic inflammatory response in response to oral infection of mice with Porphyromonas gingivalis. J Periodontal Res. 2014;49:69–76. [DOI] [PubMed] [Google Scholar]
- 310.Bidri M, Ktorza S, Vouldoukis I, et al. Nitric oxide pathway is induced by FcεRI and up-regulated by stem cell factor in mouse mast cells. Eur J Immunol. 1997;27:2907–2913. [DOI] [PubMed] [Google Scholar]
- 311.Kunii J, Takahashi K, Kasakura K, et al. Commensal bacteria promote migration of mast cells into the intestine. Immunobiology. 2011;216:692–697. [DOI] [PubMed] [Google Scholar]
- 312.Carranza FA Jr, Cabrini RL. Mast cells in human gingiva. Oral Surg Oral Med Oral Pathol. 1955;8:1093–1099. [DOI] [PubMed] [Google Scholar]
- 313.Zachrisson BU. Mast cells of the human gingiva. I. Investigations concerning the preservation and demonstration of mast cells in the gingival area. Odontol Revy. 1968;19:1–22. [PubMed] [Google Scholar]
- 314.Zachrisson BU. Mast cells of the human gingiva. 3. Histochemical demonstration of immature mast cells in chronically inflamed tissue. J Periodontal Res. 1968;3:136–145. [DOI] [PubMed] [Google Scholar]
- 315.Zachrisson BU. Mast cells of the human gingiva. 2. Metachromatic cells at low pH in healthy and inflamed tissue. J Periodontal Res. 1967;2:87–105. [DOI] [PubMed] [Google Scholar]
- 316.Gunhan M, Bostanci H, Gunhan O, Demiriz M. Mast cells in periodontal disease. Ann Dent. 1991;50:25–29. [PubMed] [Google Scholar]
- 317.Ramadan AE, Younan AE, Goubran EZ, Nour Z. Mast cell population in marginal periodontitis. Alex Dent J. 1976;1:46–59. [PubMed] [Google Scholar]
- 318.Batista AC, Rodini CO, Lara VS. Quantification of mast cells in different stages of human periodontal disease. Oral Dis. 2005;11:249–254. [DOI] [PubMed] [Google Scholar]
- 319.Huang S, Lu F, Chen Y, Huang B, Liu M. Mast cell degranulation in human periodontitis. J Periodontol. 2013;84:248–255. [DOI] [PubMed] [Google Scholar]
- 320.Huang S, Lu F, Li J, et al. Quantification of tryptase-TIM-3 double-positive mast cells in human chronic periodontitis. Arch Oral Biol. 2014;59:654–661. [DOI] [PubMed] [Google Scholar]
- 321.Agrawal R, Gupta J, Gupta KK, Kumar V. Correlation of mast cells in different stages of human periodontal diseases: pilot study. J Oral Maxillofac Pathol. 2016;20:91–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322.Jeffcoat MK, Williams RC, Johnson HG, Wechter WJ, Goldhaber P. Treatment of periodontal disease in beagles with lodoxamide ethyl, an inhibitor of mast cell release. J Periodontal Res. 1985;20:532–541. [DOI] [PubMed] [Google Scholar]
- 323.Huang B, Dai Q, Huang SG. Expression of toll-like receptor 4 on mast cells in gingival tissues of human chronic periodontitis. Mol Med Rep. 2018;17:6731–6735. [DOI] [PubMed] [Google Scholar]
- 324.Tada H, Nishioka T, Takase A, Numazaki K, Bando K, Matsushita K. Porphyromonas gingivalis induces the production of interleukin-31 by human mast cells, resulting in dysfunction of the gingival epithelial barrier. Cell Microbiol. 2018;e12972. [DOI] [PubMed] [Google Scholar]
- 325.Koseoglu S, Hatipoglu M, Saglam M, Enhos S, Esen HH. Interleukin-33 could play an important role in the pathogenesis of periodontitis. J Periodontal Res. 2015;50:525–534. [DOI] [PubMed] [Google Scholar]
- 326.Parachuru VPB, Coates DE, Milne TJ, Rich AM, Seymour GJ. FoxP3+ regulatory T cells, interleukin 17 and mast cells in chronic inflammatory periodontal disease. J Periodontal Res. 2018;53:622–635. [DOI] [PubMed] [Google Scholar]
- 327.Malcolm J, Millington O, Millhouse E, et al. Mast cells contribute to Porphyromonas gingivalis–induced bone loss. J Dent Res. 2016;95:704–710. [DOI] [PubMed] [Google Scholar]
- 328.Bisson C, Massin F, Lefevre PA, Thilly N, Miller N, Gibot S. Increased gingival crevicular fluid levels of soluble triggering receptor expressed on myeloid cells (sTREM) 1 in severe periodontitis. J Clin Periodontol. 2012;39:1141–1148. [DOI] [PubMed] [Google Scholar]
- 329.Tonetti MS, Imboden MA, Lang NP. Neutrophil migration into the gingival sulcus is associated with transepithelial gradients of interleukin-8 and ICAM-1. J Periodontol. 1998;69:1139–1147. [DOI] [PubMed] [Google Scholar]
- 330.Meyle J, Gonzales JR. Influences of systemic diseases on periodontitis in children and adolescents. Periodontol 2000. 2001;26:92–112. [DOI] [PubMed] [Google Scholar]
- 331.Hajishengallis G, Korostoff JM. (2017) Revisiting the Page & Schroeder model: the good, the bad and the unknowns in the periodontal host response 40 years later. Periodontol 2000. 2017;75:116–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 332.Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009;9:162–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 333.Veglia F, Perego M, Gabrilovich D. Myeloid-derived suppressor cells coming of age. Nat Immunol. 2018;19:108–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 334.Su L, Xu Q, Zhang P, Michalek SM, Katz J. Phenotype and function of myeloid-derived suppressor cells induced by Porphyromonas gingivalis infection. Infect Immun. 2017;85(8):e00213–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 335.Zhao E, Xu H, Wang L, et al. Bone marrow and the control of immunity. Cell Mol Immunol. 2012;9:11–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 336.Dugast AS, Haudebourg T, Coulon F, et al. Myeloid-derived suppressor cells accumulate in kidney allograft tolerance and specifically suppress effector T cell expansion. J Immunol. 2008;180:7898–7906. [DOI] [PubMed] [Google Scholar]
- 337.Watanabe S, Deguchi K, Zheng R, et al. Tumor-induced CD11b+Gr-1+ myeloid cells suppress T cell sensitization in tumor-draining lymph nodes. J Immunol. 2008;181:3291–3300. [DOI] [PubMed] [Google Scholar]
- 338.Li H, Han Y, Guo Q, Zhang M, Cao X. Cancer-expanded myeloid-derived suppressor cells induce anergy of NK cells through membrane-bound TGF-β1. J Immunol. 2009;182:240–249. [DOI] [PubMed] [Google Scholar]
- 339.Stiff A, Trikha P, Mundy-Bosse B, et al. Nitric oxide production by myeloid-derived suppressor cells plays a role in impairing Fc receptor-mediated natural killer cell function. Clin Cancer Res 2018;24(5)1891–1904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 340.Song X, Krelin Y, Dvorkin T, et al. CD11b+/Gr-1+ immature myeloid cells mediate suppression of T cells in mice bearing tumors of IL-1β-secreting cells. J Immunol. 2005;175:8200–8208. [DOI] [PubMed] [Google Scholar]
- 341.Kennedy DE, Knight KL. Inhibition of B Lymphopoiesis by adipocytes and IL-1-producing myeloid-derived suppressor cells. J Immunol. 2015;195:2666–2674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 342.Charles JF, Hsu LY, Niemi EC, Weiss A, Aliprantis AO, Nakamura MC. Inflammatory arthritis increases mouse osteoclast precursors with myeloid suppressor function. J Clin Invest. 2012;122:4592–4605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 343.Zhuang J, Zhang J, Lwin ST, et al. Osteoclasts in multiple myeloma are derived from Gr-1+CD11b+myeloid-derived suppressor cells. PLoS One. 2012;7:e48871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 344.Sawant A, Deshane J, Jules J, et al. Myeloid-derived suppressor cells function as novel osteoclast progenitors enhancing bone loss in breast cancer. Cancer Res. 2013;73:672–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 345.Zhang H, Wang S, Huang Y, et al. Myeloid-derived suppressor cells are proinflammatory and regulate collagen-induced arthritis through manipulating Th17 cell differentiation. Clin Immunol. 2015;157:175–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 346.Han JH, Choi SJ, Kurihara N, Koide M, Oba Y, Roodman GD. Macrophage inflammatory protein-1α is an osteoclastogenic factor in myeloma that is independent of receptor activator of nuclear factor κB ligand. Blood. 2001;97:3349–3353. [DOI] [PubMed] [Google Scholar]
- 347.Watanabe T, Kukita T, Kukita A, et al. Direct stimulation of osteoclastogenesis by MIP-1α: evidence obtained from studies using RAW264 cell clone highly responsive to RANKL. J Endocrinol. 2004;180:193–201. [DOI] [PubMed] [Google Scholar]
- 348.Yu X, Huang Y, Collin-Osdoby P, Osdoby P. CCR1 chemokines promote the chemotactic recruitment, RANKL development, and motility of osteoclasts and are induced by inflammatory cytokines in osteoblasts. J Bone Miner Res. 2004;19:2065–2077. [DOI] [PubMed] [Google Scholar]
- 349.Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol. 2012;12:253–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 350.Wang Z, Liu Y, Zhang Y, Shang Y, Gao Q. MDSC-decreasing chemotherapy increases the efficacy of cytokine-induced killer cell immunotherapy in metastatic renal cell carcinoma and pancreatic cancer. Oncotarget. 2016;7:4760–4769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 351.Ezernitchi AV, Vaknin I, Cohen-Daniel L, et al. TCR ζ down-regulation under chronic inflammation is mediated by myeloid suppressor cells differentially distributed between various lymphatic organs. J Immunol. 2006;177:4763–4772. [DOI] [PubMed] [Google Scholar]
- 352.Arjunan P, Meghil MM, Pi W, et al. Oral pathobiont activates anti-apoptotic pathway, promoting both immune suppression and oncogenic cell proliferation. Sci Rep. 2018;8:16607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 353.Cheng R, Billet S, Liu C, et al. Periodontal inflammation recruits distant metastatic breast cancer cells by increasing myeloid-derived suppressor cells. Oncogene. 2020;39:1543–1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 354.Michalski MN, McCauley LK. Macrophages and skeletal health. Pharmacol Ther. 2017;174:43–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 355.Chang MK, Raggatt LJ, Alexander KA, et al. Osteal tissue macrophages are intercalated throughout human and mouse bone lining tissues and regulate osteoblast function in vitro and in vivo. J Immunol. 2008;181:1232–1244. [DOI] [PubMed] [Google Scholar]
- 356.Nicolaidou V, Wong MM, Redpath AN, et al. Monocytes induce STAT3 activation in human mesenchymal stem cells to promote osteoblast formation. PLoS One. 2012;7:e39871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 357.Burnett SH, Beus BJ, Avdiushko R, Qualls J, Kaplan AM, Cohen DA. Development of peritoneal adhesions in macrophage depleted mice. J Surg Res. 2006;131:296–301. [DOI] [PubMed] [Google Scholar]
- 358.Clausen BE, Burkhardt C, Reith W, Renkawitz R, Forster I. Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res. 1999;8:265–277. [DOI] [PubMed] [Google Scholar]
- 359.Sima C, Glogauer M. Macrophage subsets and osteoimmunology: tuning of the immunological recognition and effector systems that maintain alveolar bone. Periodontol 2000. 2013;63:80–101. [DOI] [PubMed] [Google Scholar]
- 360.Yu T, Zhao L, Huang X, et al. Enhanced activity of the macrophage M1/M2 phenotypes and phenotypic switch to M1 in periodontal infection. J Periodontol. 2016;87:1092–1102. [DOI] [PubMed] [Google Scholar]
- 361.Huang CB, Alimova Y, Ebersole JL. Macrophage polarization in response to oral commensals and pathogens. Pathog Dis. 2016;74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 362.Lam RS, O’Brien-Simpson NM, Lenzo JC, et al. Macrophage depletion abates Porphyromonas gingivalis–induced alveolar bone resorption in mice. J Immunol. 2014;193:2349–2362. [DOI] [PubMed] [Google Scholar]
- 363.Yamaguchi T, Movila A, Kataoka S, et al. Proinflammatory M1 macrophages inhibit RANKL-induced osteoclastogenesis. Infect Immun. 2016;84:2802–2812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 364.Zhuang Z, Yoshizawa-Smith S, Glowacki A, et al. Induction of M2 macrophages prevents bone loss in murine periodontitis models. J Dent Res. 2019;98(2):200–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 365.Song L, Dong G, Guo L, Graves DT. The function of dendritic cells in modulating the host response. Mol Oral Microbiol. 2018;33:13–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 366.Chauhan D, Singh AV, Brahmandam M, et al. Functional interaction of plasmacytoid dendritic cells with multiple myeloma cells: a therapeutic target. Cancer Cell. 2009;16:309–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 367.Sawant A, Ponnazhagan S. Role of plasmacytoid dendritic cells in breast cancer bone dissemination. Oncoimmunology. 2013;2:e22983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 368.Alnaeeli M, Teng YT. Dendritic cells differentiate into osteoclasts in bone marrow microenvironment in vivo. Blood. 2009;113:264–265; author reply 265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 369.Heath WR, Carbone FR. Dendritic cell subsets in primary and secondary T cell responses at body surfaces. Nat Immunol. 2009;10:1237–1244. [DOI] [PubMed] [Google Scholar]
- 370.Kaplan DH. In vivo function of Langerhans cells and dermal dendritic cells. Trends Immunol. 2010;31:446–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 371.Wilensky A, Segev H, Mizraji G, et al. Dendritic cells and their role in periodontal disease. Oral Dis. 2014;20:119–126. [DOI] [PubMed] [Google Scholar]
- 372.Allam JP, Peng WM, Appel T, et al. Toll-like receptor 4 ligation enforces tolerogenic properties of oral mucosal Langerhans cells. J Allergy Clin Immunol. 2008;121:368–374.e361. [DOI] [PubMed] [Google Scholar]
- 373.Capucha T, Koren N, Nassar M, et al. Sequential BMP7/TGF-β1 signaling and microbiota instruct mucosal Langerhans cell differentiation. J Exp Med. 2018;215:481–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 374.Speziani C, Rivollier A, Gallois A, et al. Murine dendritic cell transdifferentiation into osteoclasts is differentially regulated by innate and adaptive cytokines. Eur J Immunol. 2007;37:747–757. [DOI] [PubMed] [Google Scholar]
- 375.Alnaeeli M, Penninger JM, Teng YT. Immune interactions with CD4+ T cells promote the development of functional osteoclasts from murine CD11c+ dendritic cells. J Immunol. 2006;177:3314–3326. [DOI] [PubMed] [Google Scholar]
- 376.Arizon M, Nudel I, Segev H, et al. Langerhans cells down-regulate inflammation-driven alveolar bone loss. Proc Natl Acad Sci USA. 2012;109:7043–7048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 377.Bittner-Eddy PD, Fischer LA, Kaplan DH, Thieu K, Costalonga M. Mucosal Langerhans cells promote differentiation of Th17 cells in a murine model of periodontitis but are not required for Porphyromonas gingivalis–driven alveolar bone destruction. J Immunol. 2016;197:1435–1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 378.Mizraji G, Nassar M, Segev H, et al. Porphyromonas gingivalis promotes unrestrained type I interferon production by dysregulating TAM signaling via MYD88 degradation. Cell Rep. 2017;18:419–431. [DOI] [PubMed] [Google Scholar]
- 379.Chen W, Gao B, Hao L, et al. The silencing of cathepsin K used in gene therapy for periodontal disease reveals the role of cathepsin K in chronic infection and inflammation. J Periodontal Res. 2016;51:647–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 380.Zhang X, Alnaeeli M, Singh B, Teng YT. Involvement of SOCS3 in regulation of CD11c+ dendritic cell-derived osteoclastogenesis and severe alveolar bone loss. Infect Immun. 2009;77:2000–2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 381.Su H, Yan X, Dong Z, Chen W, Lin ZT, Hu QG. Differential roles of Porphyromonas gingivalis lipopolysaccharide and Escherichia coli lipopolysaccharide in maturation and antigen-presenting functions of dentritic cells. Eur Rev Med Pharmacol Sci. 2015;19:2482–2492. [PubMed] [Google Scholar]
- 382.Mahanonda R, Pothiraksanon P, Sa-Ard-Iam N, et al. The effects of Porphyromonas gingivalis LPS and Actinobacillus actinomycetemcomitans LPS on human dendritic cells in vitro, and in a mouse model in vivo. Asian Pac J Allergy Immunol. 2006;24:223–228. [PubMed] [Google Scholar]
- 383.Tyagi RK, Miles B, Parmar R, et al. Human IDO-competent, long-lived immunoregulatory dendritic cells induced by intracellular pathogen, and their fate in humanized mice. Sci Rep. 2017;7:41083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 384.Xiao W, Dong G, Pacios S, et al. FOXO1 deletion reduces dendritic cell function and enhances susceptibility to periodontitis. Am J Pathol. 2015;185:1085–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 385.Cerdo T, Garcia-Santos JA, Bermúdez MC, Campoy C. The role of probiotics and prebiotics in the prevention and treatment of obesity. Nutrients. 2019;11:635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 386.Li JY, Chassaing B, Tyagi AM, et al. Sex steroid deficiency–associated bone loss is microbiota dependent and prevented by probiotics. J Clin Invest. 2016;126:2049–2063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 387.Nilsson AG, Sundh D, Backhed F, Lorentzon M. Lactobacillus reuteri reduces bone loss in older women with low bone mineral density: a randomized, placebo-controlled, double-blind, clinical trial. J Intern Med. 2018;284:307–317. [DOI] [PubMed] [Google Scholar]
- 388.Takimoto T, Hatanaka M, Hoshino T, et al. Effect of Bacillus subtilis C-3102 on bone mineral density in healthy postmenopausal Japanese women: a randomized, placebo-controlled, double-blind clinical trial. Biosci Microbiota Food Health. 2018;37:87–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 389.Gatej SM, Marino V, Bright R, et al. Probiotic Lactobacillus rhamnosus GG prevents alveolar bone loss in a mouse model of experimental periodontitis. J Clin Periodontol. 2018;45:204–212. [DOI] [PubMed] [Google Scholar]
- 390.Gatej SM, Bright R, Weyrich LS, et al. Probiotic Lactobacillus rhamnosus GG protects against P. gingivalis and F. nucleatum gut dysbiosis. J Int Acad Periodontol. 2020;22:18–27. [PubMed] [Google Scholar]
- 391.Kobayashi R, Kobayashi T, Sakai F, Hosoya T, Yamamoto M, Kurita-Ochiai T. Oral administration of Lactobacillus gasseri SBT2055 is effective in preventing Porphyromonas gingivalis-accelerated periodontal disease. Sci Rep. 2017;7:545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 392.Messora MR, Pereira LJ, Foureaux R, et al. Favourable effects of Bacillus subtilis and Bacillus licheniformis on experimental periodontitis in rats. Arch Oral Biol. 2016;66:108–119. [DOI] [PubMed] [Google Scholar]
- 393.Maekawa T, Hajishengallis G. Topical treatment with probiotic Lactobacillus brevis CD2 inhibits experimental periodontal inflammation and bone loss. J Periodontal Res. 2014;49:785–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 394.Oliveira LF, Salvador SL, Silva PH, et al. Benefits of Bifidobacterium animalis subsp. lactis probiotic in experimental periodontitis. J Periodontol. 2017;88:197–208. [DOI] [PubMed] [Google Scholar]
- 395.Liu TH, Tsai TY, Pan TM. Effects of an ethanol extract from Lactobacillus paracasei subsp. paracasei NTU 101 fermented skimmed milk on lipopolysaccharide-induced periodontal inflammation in rats. Food Funct. 2018;9:4916–4925. [DOI] [PubMed] [Google Scholar]
- 396.Ricoldi MST, Furlaneto FAC, Oliveira LFF, et al. Effects of the probiotic Bifidobacterium animalis subsp. lactis on the non-surgical treatment of periodontitis. A histomorphometric, microtomographic and immunohistochemical study in rats. PLoS One. 2017;12:e0179946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 397.Nackaerts O, Jacobs R, Quirynen M, Rober M, Sun Y, Teughels W. Replacement therapy for periodontitis: pilot radiographic evaluation in a dog model. J Clin Periodontol. 2008;35:1048–1052. [DOI] [PubMed] [Google Scholar]
- 398.Messora MR, Oliveira LF, Foureaux RC, et al. Probiotic therapy reduces periodontal tissue destruction and improves the intestinal morphology in rats with ligature-induced periodontitis. J Periodontol. 2013;84:1818–1826. [DOI] [PubMed] [Google Scholar]
- 399.Foureaux Rde C, Messora MR, de Oliveira LF, et al. Effects of probiotic therapy on metabolic and inflammatory parameters of rats with ligature-induced periodontitis associated with restraint stress. J Periodontol. 2014;85:975–983. [DOI] [PubMed] [Google Scholar]
- 400.Garcia VG, Knoll LR, Longo M, et al. Effect of the probiotic Saccharomyces cerevisiae on ligature-induced periodontitis in rats. J Periodontal Res. 2016;51:26–37. [DOI] [PubMed] [Google Scholar]
- 401.Kim JW, Jung BH, Lee JH, et al. Effect of Weissella cibaria on the reduction of periodontal tissue destruction in mice. J Periodontol. 2020. [DOI] [PubMed] [Google Scholar]
- 402.O’Callaghan A, van Sinderen D. Bifidobacteria and their role as members of the human gut microbiota. Front Microbiol. 2016;7:925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 403.Kuru BE, Laleman I, Yalnizoglu T, Kuru L, Teughels W. The influence of a Bifidobacterium animalis probiotic on gingival health: a randomized controlled clinical trial. J Periodontol. 2017;88:1115–1123. [DOI] [PubMed] [Google Scholar]
- 404.Toiviainen A, Jalasvuori H, Lahti E, et al. Impact of orally administered lozenges with Lactobacillus rhamnosus GG and Bifidobacterium animalis subsp. lactis BB-12 on the number of salivary mutans streptococci, amount of plaque, gingival inflammation and the oral microbiome in healthy adults. Clin Oral Investig. 2015;19:77–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 405.Jasberg H, Tervahartiala T, Sorsa T, Soderling E, Haukioja A. Probiotic intervention influences the salivary levels of matrix metalloproteinase (MMP)-9 and tissue inhibitor of metalloproteinases (TIMP)-1 in healthy adults. Arch Oral Biol. 2018;85:58–63. [DOI] [PubMed] [Google Scholar]
- 406.Mayanagi G, Kimura M, Nakaya S, et al. Probiotic effects of orally administered Lactobacillus salivarius WB21-containing tablets on periodontopathic bacteria: a double-blinded, placebo-controlled, randomized clinical trial. J Clin Periodontol. 2009;36:506–513. [DOI] [PubMed] [Google Scholar]
- 407.Shimauchi H, Mayanagi G, Nakaya S, et al. Improvement of periodontal condition by probiotics with Lactobacillus salivarius WB21: a randomized, double-blind, placebo-controlled study. J Clin Periodontol. 2008;35:897–905. [DOI] [PubMed] [Google Scholar]
- 408.Vivekananda MR, Vandana KL, Bhat KG. Effect of the probiotic Lactobacilli reuteri (Prodentis) in the management of periodontal disease: a preliminary randomized clinical trial. J Oral Microbiol. 2010;2(1):Article ID 5344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 409.Tekce M, Ince G, Gursoy H, et al. Clinical and microbiological effects of probiotic lozenges in the treatment of chronic periodontitis: a 1-year follow-up study. J Clin Periodontol. 2015;42:363–372. [DOI] [PubMed] [Google Scholar]
- 410.Ince G, Gursoy H, Ipci SD, Cakar G, Emekli-Alturfan E, Yilmaz S. Clinical and biochemical evaluation of lozenges containing Lactobacillus reuteri as an adjunct to non-surgical periodontal therapy in chronic periodontitis. J Periodontol. 2015;86:746–754. [DOI] [PubMed] [Google Scholar]
- 411.Morales A, Carvajal P, Silva N, et al. Clinical effects of Lactobacillus rhamnosus in non-surgical treatment of chronic periodontitis: a randomized placebo-controlled trial with 1-year follow-up. J Periodontol. 2016;87:944–952. [DOI] [PubMed] [Google Scholar]
- 412.Morales A, Gandolfo A, Bravo J, et al. Microbiological and clinical effects of probiotics and antibiotics on nonsurgical treatment of chronic periodontitis: a randomized placebo- controlled trial with 9-month follow-up. J Appl Oral Sci. 2018;26:e20170075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 413.Gibson GR, McCartney AL, Rastall RA. Prebiotics and resistance to gastrointestinal infections. Br J Nutr. 2005;93(Suppl 1):S31–34. [DOI] [PubMed] [Google Scholar]
- 414.Younes H, Coudray C, Bellanger J, Demigne C, Rayssiguier Y, Remesy C. Effects of two fermentable carbohydrates (inulin and resistant starch) and their combination on calcium and magnesium balance in rats. Br J Nutr. 2001;86:479–485. [DOI] [PubMed] [Google Scholar]
- 415.Coudray C, Bellanger J, Vermorel M, et al. Two polyol, low digestible carbohydrates improve the apparent absorption of magnesium but not of calcium in healthy young men. J Nutr. 2003;133:90–93. [DOI] [PubMed] [Google Scholar]
- 416.Kruger MC, Brown KE, Collett G, Layton L, Schollum LM. The effect of fructooligosaccharides with various degrees of polymerization on calcium bioavailability in the growing rat. Exp Biol Med. 2003;228:683–688. [DOI] [PubMed] [Google Scholar]
- 417.Slomka V, Hernandez-Sanabria E, Herrero ER, et al. Nutritional stimulation of commensal oral bacteria suppresses pathogens: the prebiotic concept. J Clin Periodontol. 2017;44:344–352. [DOI] [PubMed] [Google Scholar]
- 418.De Oliveira Silva V, Lobato RV, Andrade EF, et al. Effects of β-glucans ingestion on alveolar bone loss, intestinal morphology, systemic inflammatory profile, and pancreatic β-cell function in rats with periodontitis and diabetes. Nutrients 2017;9(9):Article no 1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 419.Levi Y, Novais GS, Dias RB, et al. Effects of the prebiotic mannan oligosaccharide on the experimental periodontitis in rats. J Clin Periodontol. 2018;45:1078–1089. [DOI] [PubMed] [Google Scholar]