

Abstract
This report describes the development of a nickel-catalyzed decarbonylative reaction for the synthesis of fluoroalkyl thioethers (RFSR) from the corresponding thioesters. Readily available, inexpensive, and stable fluoroalkyl carboxylic acids (RFCO2H) serve as the fluoroalkyl (RF) source in this transformation. Stoichiometric organometallic studies reveal that RF–S bond-forming reductive elimination is a challenging step in the catalytic cycle. This led to the identification of diphenylphosphinoferrocene as the optimal ligand for this transformation. Ultimately, this method was applied to the construction of diverse fluoroalkyl thioethers (RFSR), with R = both aryl and alkyl.
Keywords: Nickel-catalysis, decarbonylation, fluoroalkyl carboxylic acids, thioether synthesis, fluoroalkylation
Graphical Abstract
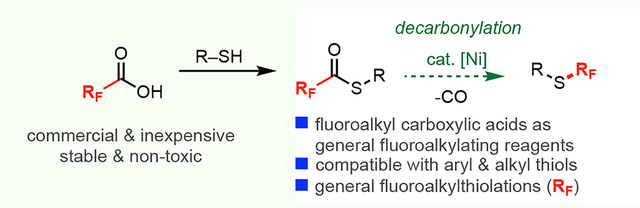
Fluoroalkyl thioethers (RFSR) have emerged as increasingly common motifs in bioactive molecules due to their unique physiochemical properties.1 As shown in Figure 1A, thioethers bearing diverse fluoroalkyl substituents (for example, CF2H, CFH2, and CH2CF3) appear in lead structures relevant to both medicinal and agricultural chemistry.2,3 The most common synthetic routes to RFSR involve either the electrophilic fluoroalkylation of thiols (Figure 1B, i)3–5 or the coupling of aryl/alkyl electrophiles with [M]–SRF nucleophiles (Figure 1B, ii).3,6–8 Both approaches have significant limitations with respect to the breadth of RF substituents that can be introduced, since very few of the necessary RF-containing electrophiles/nucleophiles are commercially available.6,7 Furthermore, many of these methods require other toxic, unstable, or expensive reagents.3,4,6,7 Overall, more general synthetic approaches to fluoroalkyl thioethers are of high interest, and the use of readily available fluoroalkyl carboxylic acids as RF precursors would be particularly enabling in this context.
Figure 1.
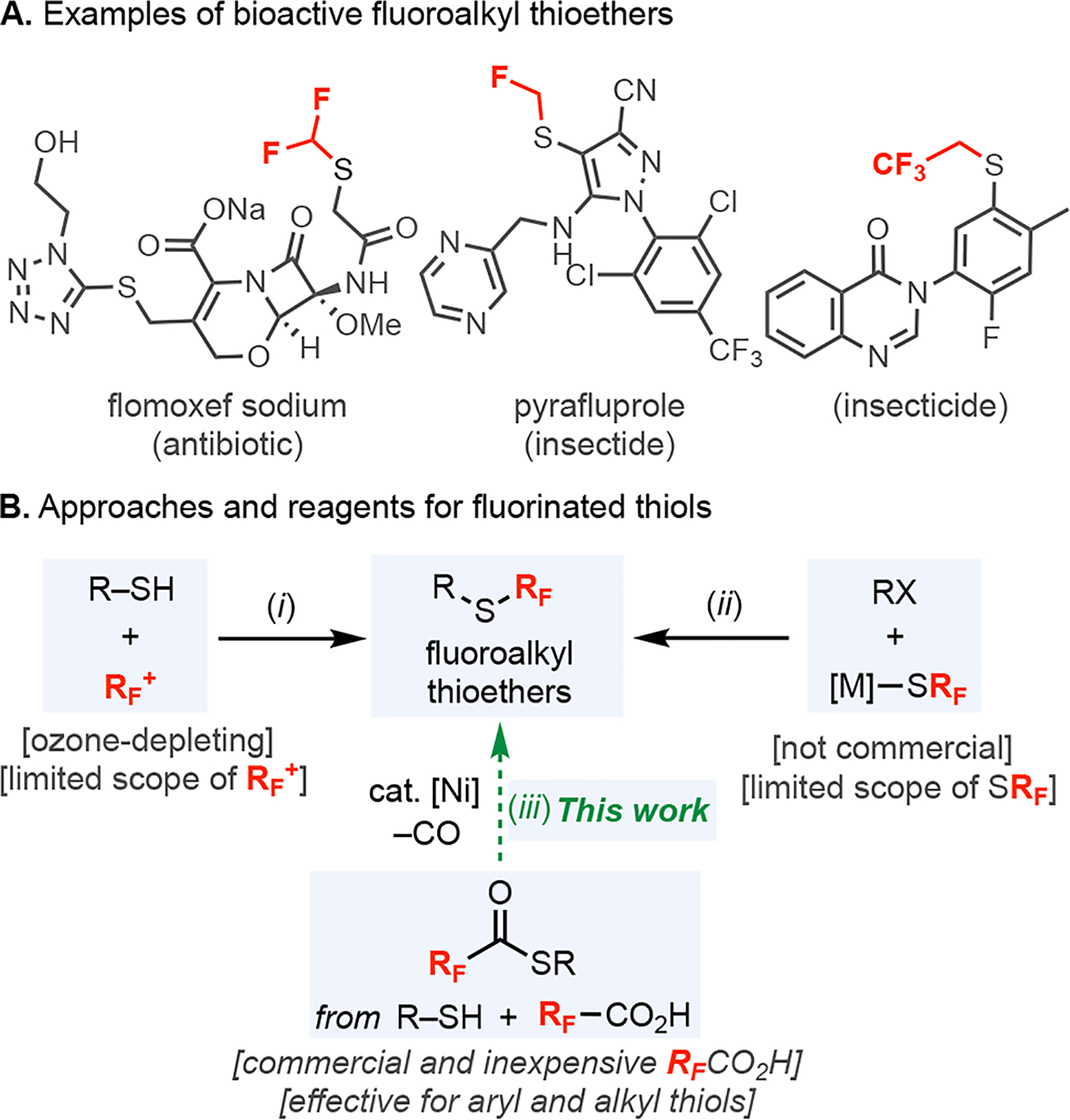
(A) Representative examples of bioactive molecules containing fluoroalkyl thioethers (RFSR). (B) Existing synthetic approaches to RFSR (i, ii) and our approach (iii).
This report describes the development of a Ni-catalyzed reaction for constructing fluoroalkyl thioethers from the corresponding thioesters (Figure 1B, iii). Our approach leverages fluoroalkyl carboxylic acids as inexpensive, stable, and commercially-available RF precursors.9–12 As such, it enables the construction of a variety of different fluoroalkyl thioethers from a single thiol starting material.
Recent studies on Ni-catalyzed coupling reactions of carboxylic acid derivatives13,14 led us to propose this decarbonylative route to fluoroalkyl thioethers. Recent reports from our group13c and others15–17 have demonstrated that Ni(0) phosphine complexes catalyze decarbonylative C–S coupling reactions of (hetero)aryl thioesters to afford (hetero)aryl thioether products (for example, see Figure 2A). We hypothesized that an analogous pathway, using fluoroalkyl thioesters as starting materials, could offer a route to RFSR products. The proposed catalytic cycle (Figure 2B), involves initial oxidative addition of the fluoroalkyl thioester at a Ni(0) catalyst to form the acyl Ni(II)-intermediate, I (step i). Carbonyl de-insertion then generates the Ni(II)(fluoroalkyl)(thiolate) intermediate II (step ii). Finally, II undergoes C–S bond-forming reductive elimination (step iii) to yield the target fluoroalkyl thioether product and regenerate the Ni(0) catalyst.
Figure 2.
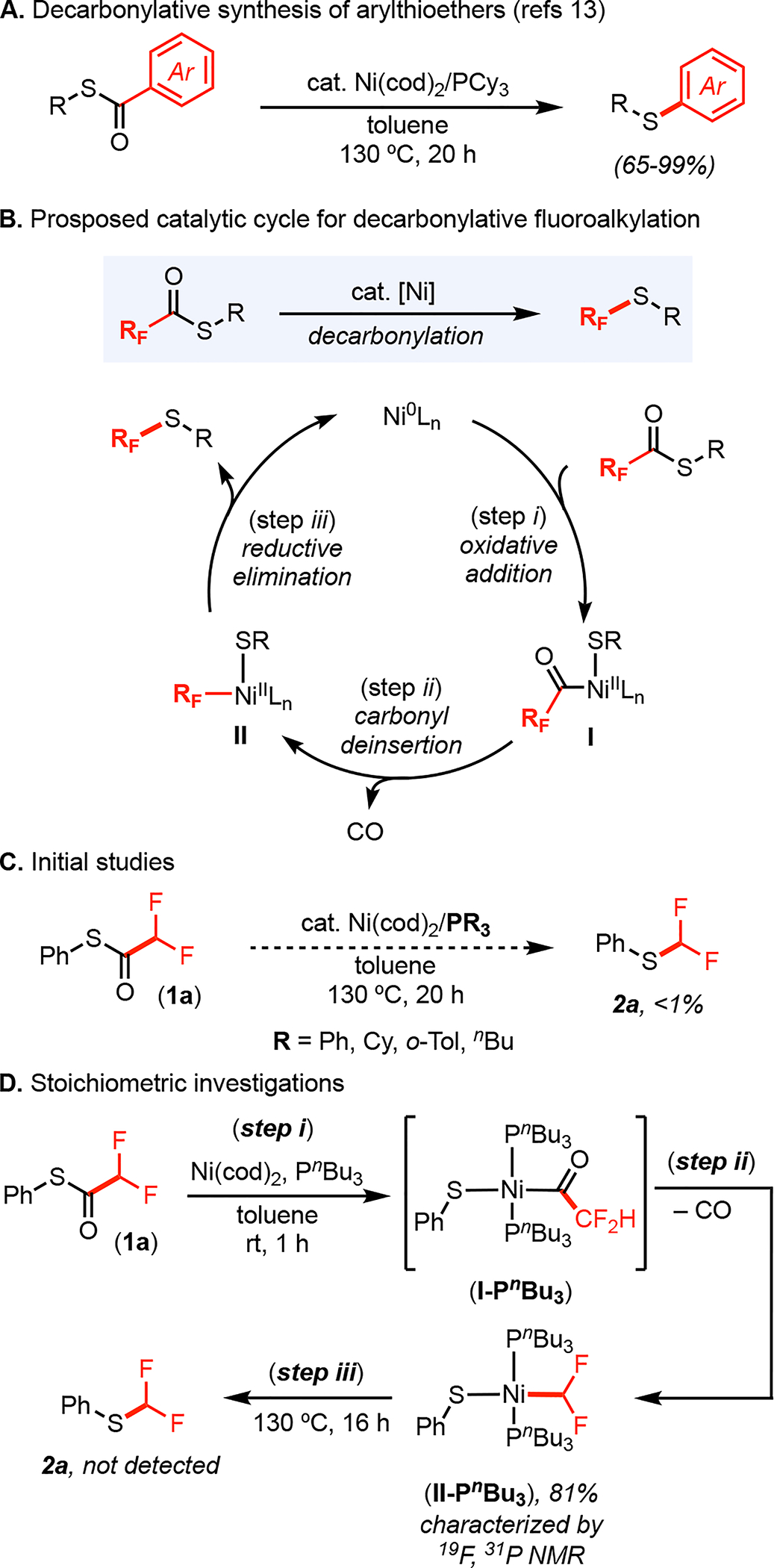
(A) Example of precedent for decarbonylative thioetherification. (B) Proposed catalytic cycle. (C) Initial catalysis studies. (D) Stoichiometric studies with PnBu3 as ligand.
We initiated these investigations by targeting the conversion of difluoromethyl thioester 1a to thioether 2a (Figure 2C). We focused on catalysts based on a combination of Ni(cod)2 and monodentate phosphine ligands (PR3), which were previously employed for the transformation in Figure 2A.13c,15–17 However, only traces (<1%) of product 2a were detected using PPh3, P(o-Tol)3, PCy3, or PBu3 (Figure 2C). In all of these systems, the majority of the mass balance was the unreacted starting material 1a.
We next conducted stoichiometric studies to identify the problematic step(s) in this sequence. The treatment of a toluene solution of Ni(cod)2/PnBu3 with 1 equiv of 1a resulted in the formation of (PnBu3)2Ni(SPh)(CF2H) (II-PnBu3) within 1 h at ambient temperature (Figure 2D). Complex II-PnBu3 was characterized in situ via 19F and 31P NMR spectroscopy, which show resonances indicative of a trans configuration, with three-bond coupling between the CF2H and PnBu3 ligands (JPF = 26.5 Hz). The formation of II-PnBu3 implicates the feasibility of two key steps of the catalytic cycle: oxidative addition (step i) and carbonyl de-insertion (step ii). However, when in situ-generated II-PnBu3 was heated at 130 °C for 2 h, none of the thioether product 2a was formed (step iii). Instead, the resonances associated with II-PnBu3 slowly decayed, without the observation of identifiable organic products. This suggests that F2HC–S bond-forming reductive elimination is challenging in this system and that alternative ligands are required to enable this step.
Literature reports have shown that 1,1’-bis(diphenylphosphino)ferrocene (dppf) is particularly effective for promoting challenging reductive elimination reactions.18 As such, we next conducted an analogous stoichiometric experiment with Ni(cod)2/dppf. As shown in Figure 3A, the treatment of a toluene solution of Ni(cod)2/dppf with 1 equiv of 1a resulted in 70% consumption of 1a within 1 h at 50 °C. This was accompanied by the formation of 2a (in 12% yield) along with broad signals in the 19F NMR spectrum. Based on previous reports,18a these broad signals are indicative of fluxional (dppf)NiII intermediates. Subsequent heating at 130 °C for 1 h resulted in S–CF2H bond-formation to generate 2a in 90% yield by 19F NMR spectroscopy (Figure 3A).19 Dppf was next examined as a ligand for the catalytic transformation of 1a to 2a. As shown in Figure 3B, the combination of 10 mol % Ni(cod)2 and 12 mol % dppf afforded 2a in 58% yield over 20 h at 130 °C in toluene. Further optimization of the reaction solvent and time resulted in nearly quantitative yield over 4 h in THF (Figure 3B).20
Figure 3.
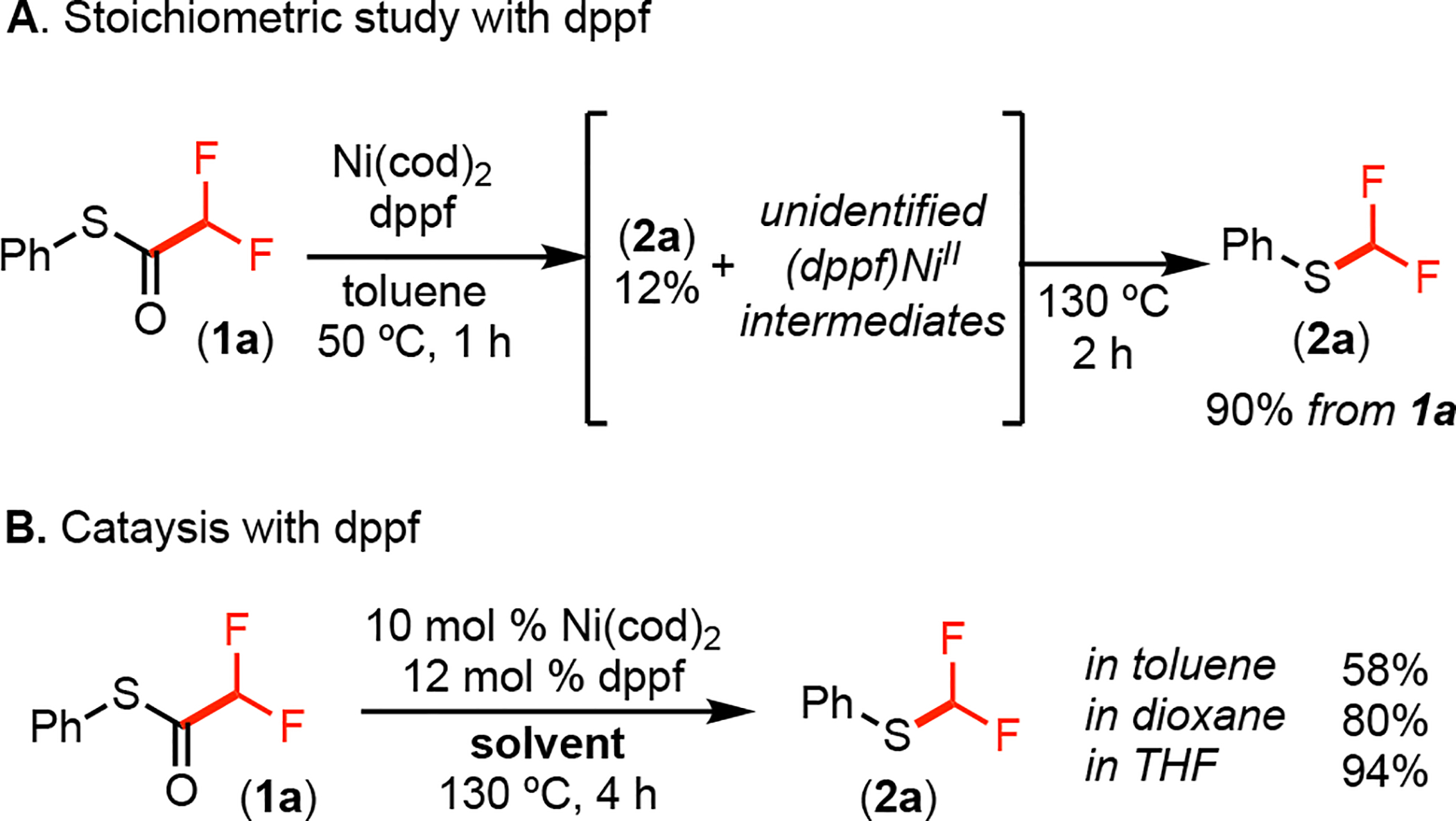
(A) Stoichiometric and (B) catalytic studies with dppf.
The scope of this transformation was first explored with respect to the substitution on sulfur (Figure 4). The difluoromethyl thioester substrates 1a-w were prepared via the reaction of RSH with difluoroacetic anhydride. These were typically obtained in quantitative yield without the need for purification by column chromatography. Aryl thioesters bearing electron-donating and -neutral substituents (1b-f) afforded good yields of the difluoromethyl thioether products (Figure 4A). Substituents such as ethers, amines, and amides were compatible. Aryl thioesters bearing electron withdrawing groups resulted in lower yields (see products 2h-2l), with the exception of 4-fluorothiophenol derivative, 2g. In these systems, the major side products were diarylthioethers, which are likely formed via competing activation of the aryl–S bond of the product by the Ni(0) catalyst.21 This transformation showed modest sensitivity to sterics on the aryl ring, and substrates containing either one or two electron-donating ortho-substituents afforded 2m-2o in moderate to good yields.
Figure 4.
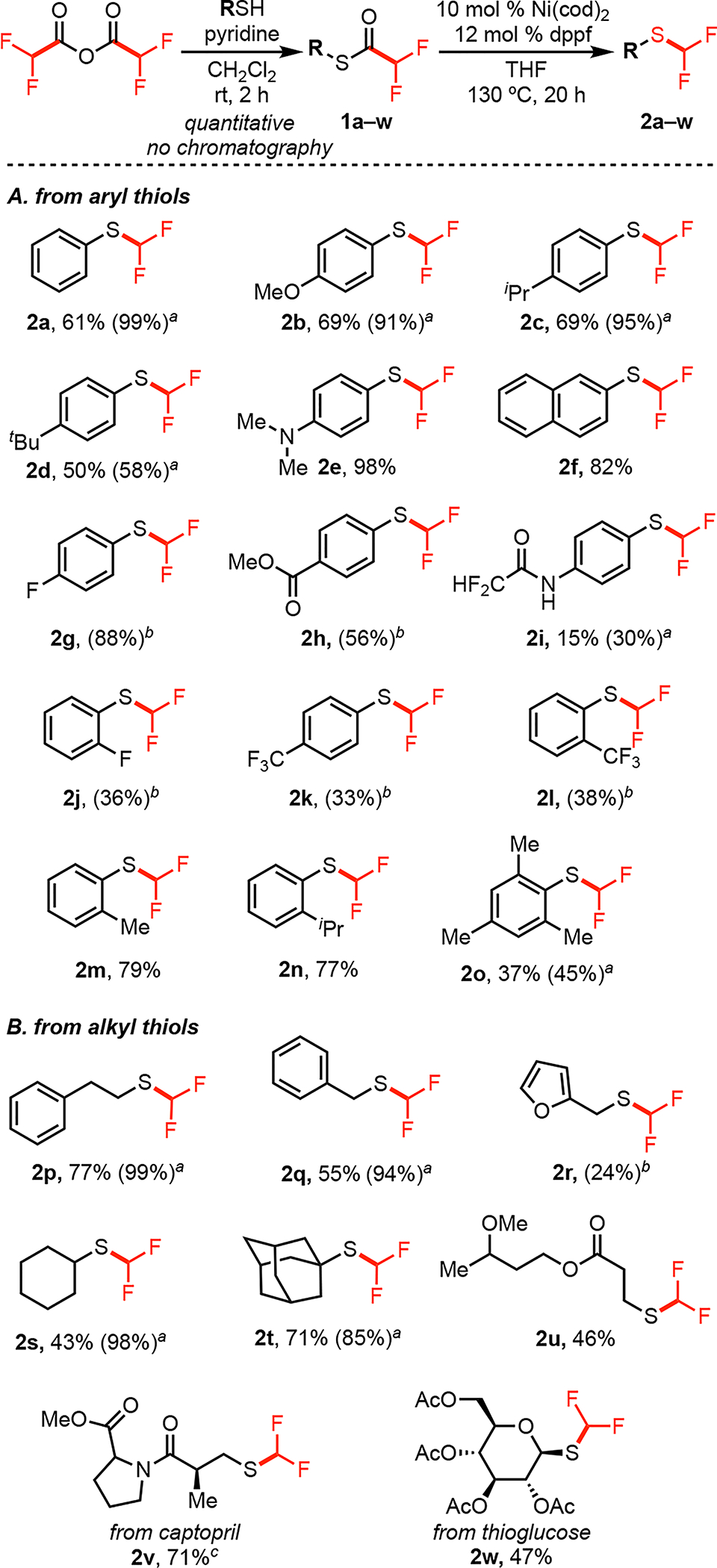
Scope of (A) aryl and (B) alkyl thioethers. a% conversion of 1 to 2 as determined by 19F NMR spectroscopy. bY-ield determined by 19F NMR spectroscopy with 4–fluorotoluene as internal standard. cCatalyst loading was increased to 15 mol% Ni(cod)2, 18 mol% dppf.
Primary, secondary and tertiary alkyl thiols were also effective substrates for this transformation (for example 2p, 2s, and 2t in Figure 4B). Thiol-containing biologically active compounds such as captopril (2v) and thioglucose (2w) underwent conversion to the corresponding difluoromethyl thioethers in good yields. In these systems, unreacted starting material accounted for the remaining mass balance when the yields were modest. Importantly, the catalytic cycle does not require an exogenous base. This limits racemization of substrates like 2v during catalysis.
Finally, we used this approach to synthesize a series of different fluoroalkyl thioethers. As shown in Figure 5, the substrates for this transformation were synthesized from commercially available RFCO2H and thiols. Catalytic decarbonylation then provided the partially fluorinated thioether products 2x-2ab in good to excellent yields. Importantly, these products are challenging to access using most existing approaches (Figure 1B), due to the inaccessibility of the required fluoroalkylating reagents. One current limitation of this approach is that fluorinated derivatives (e.g., SCF3, SCF2CF3) afford none of the desired fluoroalkyl thioether product.22 A stoichiometric study of the CF3 system showed the formation of Ni–CF3 intermediates; however, no thioether product was detected upon heating these species. This result suggests that the S–RF reductive elimination step remains a challenge in these systems.23
Figure 5.
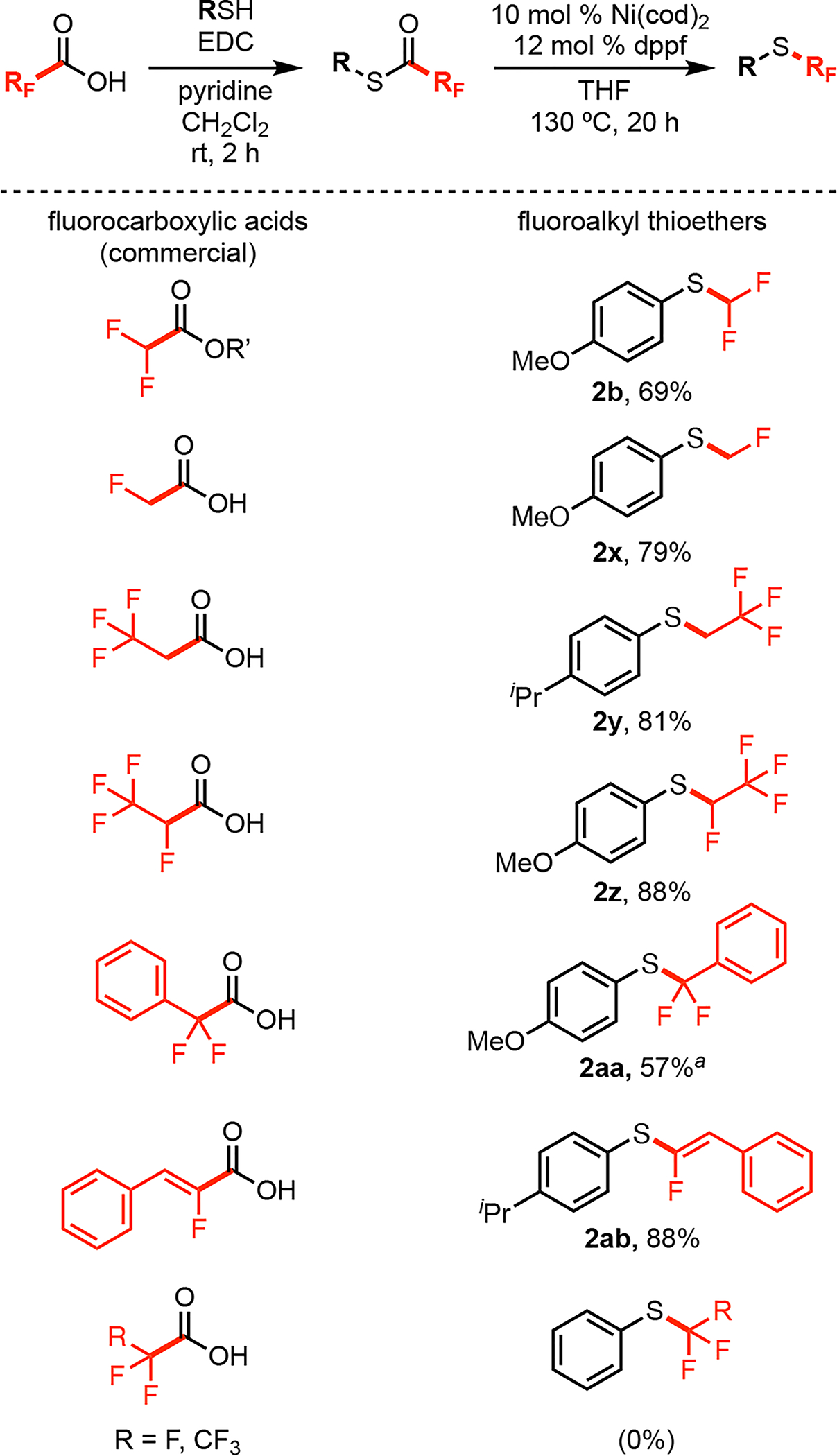
Scope of fluoroalkyl groups derived from commercial RFCO2H. Isolated yields. See the SI for details. aCatalyst loading was increased to 20 mol% Ni(cod)2 and ligand was Xantphos (24 mol %).
In summary, a nickel-catalyzed decarbonylative coupling reaction was developed to convert fluoroalkyl thioesters to the analogous thioethers. This method leverages readily available fluorocarboxylic acids as commercial and stable fluoroalkyl sources to install these functional groups, which are increasingly prevalent in biologically active molecules.
Supplementary Material
ACKNOWLEDGMENT
We acknowledge financial support from NIH NIGMS (GM073836 and GM136332) and the Danish National Research Foundation (Carbon Dioxide Activation Center; CADIAC) for support.
Footnotes
Supporting Information
Supporting Information is available free of charge on the ACS Publications website.
Experimental details, characterization data, and NMR spectra of compounds (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).(a) Purser S; Moore PR; Swallow S; Gouverneur V Fluorine in medicinal chemistry. Chem. Soc. Rev 2008, 37, 320–330. [DOI] [PubMed] [Google Scholar]; (b) Hagmann WK The Many Roles for Fluorine in Medicinal Chemistry. J. Med. Chem 2008, 51, 4359–4369. [DOI] [PubMed] [Google Scholar]; (c) Meanwell NA Synopsis of Some Recent Tactical Applications of Bioisoteres in Drug Design. J. Med. Chem 2011, 54, 2529–2591. [DOI] [PubMed] [Google Scholar]; (d) Leroux F; Jeschke P; Schlosser M α-Fluorinated Ethers, Thioethers, and Amines: Anomerically Biased species. Chem. Rev 2005, 105, 827–856. [DOI] [PubMed] [Google Scholar]; (e) Zafrani Y; Yeffet D; Sod-Moriah G; Berliner A; Amier D; Marciano D; Gershonov E; Saphier S Difluoromethyl Bioiostere: Examining the “Lipophilic Hydrogen Bond Donor” Concept. J. Med. Chem 2017, 60, 797–804. [DOI] [PubMed] [Google Scholar]
- (2).(a) Röschenthaler GV; Kazakova O Chemistry of α-Fluorinated Ethers- and Thioethers-Containing Heterocycles. In Fluorine in Heterocyclic Chemistry Nenajdenko V Eds.; Springer: Moscow, 2014. [Google Scholar]; (b) Kaiser F; Grob S; Langewald J; Narine A Insecticidal Active Mixtures Comprising Arylquinazolinone Compounds WO 2013/030262, August 13, 2012.
- (3).Xiong H; Pannecoucke X; Besset T Recent Advances in the Synthesis of SCF2H- and SCF2FG- Containing Molecules. Chem. Eur. J 2016, 16734–16749. [DOI] [PubMed] [Google Scholar]
- (4).(a) Hine J; Porter JJ Methylene Derivatives as Intermediates in Polar Reactions. J. Am. Chem. Soc 1957, 79, 5493–5496. [Google Scholar]; (b) Langlois BR Improvement of the Synthesis of Aryl Difluoromethyl Ethers and Thioethers by Using a Solid–Liquid Phase–Transfer Technique. J. Fluorine Chem 1988, 41, 247–261. [Google Scholar]; (c) Mehta VP; Greaney MF S-, N-, and Se-Difluoromethylation Using Sodium Chlorodifluoroacetate. Org. Lett 2013, 15, 5036–5039. [DOI] [PubMed] [Google Scholar]
- (5).(a) Zhang W; Wang F; Hu J N-Tosyl-S-Difluoromethyl-S-phenylsulfoximine: A New Difluoromethylation Reagent for S-, N- and C-Nucleophiles. Org. Lett 2009, 11, 2109–2112. [DOI] [PubMed] [Google Scholar]; (b) Zafrani Y; Sod-Moriah G; Segall Y Diethyl Bromodifluoromethylphosphonate: A Highly Efficient and Environmentally Benign Difluorocarbene Precursor. Tetrahedron 2009, 65, 5278–5283. [Google Scholar]; (c) Fier PS; Hartwig JF Synthesis of Difluoromethyl Ethers with Difluoromethyltriflate. Angew. Chem. Int. Ed 2013, 52, 2092–2095. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Thomoson CS; Dolbier WR Jr. Use of Fluoroform as a Source of Difluorocarbene in the Synthesis of Difluoromethoxy- and Difluorothiomethoxyarenes. J. Org. Chem 2013, 78, 8904–8908. [DOI] [PubMed] [Google Scholar]; (e) Deng X-Y; Lin J-H; Zheng J; Xiao J-C Difluoromethylation and gem-Difluorocyclopropenation with Difluorocarbene Generated by Decarboxylation. Chem. Commun 2015, 51, 8805–8808. [DOI] [PubMed] [Google Scholar]; (f) Orsi DL; Easley BJ; Lick AM; Altman RA Base Catalysis Enables Access to α, α-Difluoroalkylthioethers. Org. Lett 2017, 19, 1570–1573. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Straathof NJW; Tegelbeckers BJP; Hessel V; Wang X; Noël T A Mild and Fast Photocatalyic Trifluoromethylation of Thiols in Batch and Continuous-Flow. Chem. Sci 2014, 5, 4768–4773. [Google Scholar]; (h) Bottecchia C; Wei X-J; Kuijpers KPL; Hessel V; Noël T Visible Light-Induced Trifluoromethylation and Pefluoroalkylation of Cysteine Residues in Batch and Continuous Flow. J. Org. Chem 2016. 81, 7301–7307. [DOI] [PubMed] [Google Scholar]
- (6).(a) Arimori S; Matsubara O; Takada M; Shiro M; Shibata N Difluoromethylsulfonyl Hypervalent Iodonium Ylides for Electrophilic Difluoromethylthiolation Reactions Under Copper Catalysis. R. Soc. Open Sci 2016, 3, 160102. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ran Y; Lin Q-Y; Xu X-H; Qing F-L Radical Difluoromethylation of Thiols with Difluoromethylphosphonium Triflate Under Photoredox Catalysis. J. Org. Chem 2017, 82, 7373–7378. [DOI] [PubMed] [Google Scholar]; (c) Yang J; Jiang M; Jin Y; Yang H; Fu H Visible-light Photoredox Difluoromethylation of Phenols and Thiophenols with Commercially Available Difluorobromoacetic Acid. Org. Lett 2017, 19, 2758–2761. [DOI] [PubMed] [Google Scholar]
- (7).(a) Wu J; Gu Y; Leng X; Shen Q Copper-Promoted Sandmeyer Difluoromethylthiolation of Aryl and Heteroaryl Diazonium Salts. Angew. Chem. Int. Ed 2015, 54, 7648–7652. [DOI] [PubMed] [Google Scholar]; (b) Zhu D; Gu Y; Lu L; Shen Q N-Difluoromethylthiophthalimide: A Shelf-Stable, Electrophilic Reagent for Difluoromethylthiolation. J. Am. Chem 2015, 137, 10547–10553. [DOI] [PubMed] [Google Scholar]
- (8).Chen S; Zheng M; Liao X; Weng Z Copper-Catalyzed 2,2,2-Trifluoroethylthiolation of Aryl Halides. J. Org. Chem 2016, 81, 7993–8000. [DOI] [PubMed] [Google Scholar]
- (9). Fluorinated carboxylic derivatives have been used for some decarboxylative trifluoromethylation and difluoromethylation reactions; however, these systems are typically limited to explorations of Ar–CF3 and Ar–CF2H coupling (see refs 10 and 11). To our knowledge, fluoroalkyl carboxylic acid derivatives have not been utilized to access fluoroalkyl thioethers.
- (10).For examples of the use of RFCO2H and its derivatives as RF sources, see:; (a) Beatty JW; Douglas JJ; Cole KP; Stephenson CRJA Scalable and Operationally Simple Radical Trifluoromethylation. Nat. Commun 2015, 6, 7919–7925. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kyohide M; Etsuko T; Midori A; Kiyosi K A Convenient Trifluoromethylation of Aromatic Halides with Sodium Trifluoroacetate. Chem. Lett 1981, 10, 1719–1720. [Google Scholar]; (c) Chen M; Buchwald SL Rapid and Efficient Trifluoromethylation of Aromatic and Heteroaromatic Compounds Using Potassium Trifluoroacetate Enabled by a Flow System. Angew. Chem. Int. Ed 2013, 52, 11628–11631. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Ambler BR; Zhu L; Altman RA Copper-Catalyzed Synthesis of Trifluoroethylarenes from Benzylic Bromodifluoroacetates. J. Org. Chem 2015, 80, 8449–8457. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Ambler BR; Santosh P; Altman RA Ligand-Controlled Regioselective Copper-Catalyzed Trifluoromethlyation to Generate (Trifluoromethyl)allenes. Org. Lett 2015, 17, 2506–2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).For examples of the use of difluoroacetic acid or its derivatives for the construction of ArCF2H, see:; (a) Sun AC; McClain EJ; Beatty JW; Stephenson CRJ Visible Light-Mediated Decarboxylative Alkylation of Pharmaceutically Relevant Heterocycles. Org. Lett 2018, 20, 3487–3490. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Tung TT; Christensen SB; Nielsen J Diflluoroacetic Acid as a New Reagent for Direct C–H Difluoromethylation of Heteroaromatic Compounds. Chem. Eur. J 2017, 23, 18125–18128. [DOI] [PubMed] [Google Scholar]
- (12).For other examples of fluoroalkyl carboxylic acid derivatives being used as a fluoroalkyl sources, see:; (a) Yang M-H; Orsi DL; Altman RA Ligand-Controlled Regiodivergent Palladium-Catalyzed Decarboxylative Allylation Reaction to Access, α,α-Difluoroketones. Angew. Chem. Int. Ed 2015, 54, 2361–2365. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Yang M-H; Hunt JR; Sharifi N; Altman RA Palladium Catalysis Enables Benzylation of, α,α-Difluoroketone Enolates. Angew. Chem. Int. Ed 2016, 55, 9080–9083. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Ambler BR; Yang M-H; Altman RA Metal-Catalyzed Decarboxylative Fluoroalkylation Reactions. Synlett 2016, 27, 2747–2755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).(a) Maleckis A; Sanford MS Synthesis of Fluoroalkyl Palladium and Nickel Complexes via Decarbonylation of Acylmetal Species. Organometallics 2014, 33, 3831–3839. [Google Scholar]; (b) Malapit CA; Ichiishi N; Sanford MS Pd-Catalyzed Decarbonylative Cross-Couplings of Aroyl Chlorides. Org. Lett 2017, 19, 4142–4145. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Ichiishi N; Malapit CA; Wozniak L; Sanford MS Palladium- and Nickel-Catalyzed Decarbonylative C–S Coupling to Convert Thioesters to Thioethers. Org. Lett 2018, 20, 44–47. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Malapit CA; Bour JR; Brigham CE; Sanford MS Base-Free Nickel-Catalysed Decarbonylative Suzuki-Miyaura Coupling of Acid Fluorides. Nature 2018, 563, 100–104. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Malapit CA; Bour JR; Laursen SR; Sanford MS Mechanism and Scope of Nickel-Catalyzed Decarbonylative Borylation of Carboxylic Acid Fluorides. J. Am. Chem. Soc 2019, 141, 17322–17330. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Malapit CA; Borrell M; Milbauer MW; Brigham CE; Sanford MS Nickel-Catalyzed Decarbonylative Amination of Carboxylic Acid Esters. J. Am. Chem. Soc 2020, 142, 5918–5923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).For selected recent reviews on decarbonylative coupling reactions, see:; (a) Ogiwara Y; Sakai N Acyl Fluorides in Late Transition-Metal Catalysis. Angew. Chem. Int. Ed 2020, 59, 574–594. [DOI] [PubMed] [Google Scholar]; (b) Blanchard N; Bizet V Acid Fluorides in Transition-Metal Catalysis: A Good Balance Between Stability and Reactivity. Angew. Chem. Int. Ed 2019, 58, 6814–6187. [DOI] [PubMed] [Google Scholar]; (c) Guo L; Rueping M Decarbonylative Cross-Couplings: Nickel Catalyzed Functional Group Interconversion Strategies for the Construction of Complex Organic Molecules. Acc. Chem. Res 2018, 51, 1185–1195. [DOI] [PubMed] [Google Scholar]; (d) Meng G; Szostak M N-Acyl-Glutarimides: Privileged Scaffolds in Amide N–C Bond Cross-Coupling. Eur. J. Org. Chem 2018, 2352–2365. [Google Scholar]; (e) Takise R; Muto K; Yamaguchi J Cross-Coupling of Aromatic Esters and Amides. Chem. Soc. Rev 2017, 46, 5864–5888. [DOI] [PubMed] [Google Scholar]; (f) Wang Z; Wang X; Nishihara Y Nickel or Palladium-Catalyzed Decarbonylative Tranformations of Carboxylic Acid Derivatives. Chem Asian J 2020, 15, 1234–1247. [DOI] [PubMed] [Google Scholar]; (g) Pan F; Boursalian GB; Ritter T Palladium-Catalyzed Decarbonylative Difluoromethylation of Acid Chlorides at Room Temperature. Angew. Chem. Int. Ed 2018, 57, 16871–16876. [DOI] [PubMed] [Google Scholar]; (h) Nakatani S; Ito Y; Sakurai S; Kodama T; Tobisu M Nickel-Catalyzed Decarbonylation of Acylsilanes. J. Org. Chem 2020, 85, 7588–7594. [DOI] [PubMed] [Google Scholar]
- (15).(a) Liu C; Szostak M Decarbonylative Thioetherification by Nickel Catalyst Using Air- and Moisture-Stable Nickel Precatalysts. Chem. Commun 2018, 53, 2130–2133. [DOI] [PubMed] [Google Scholar]; (b) Lee SC; Liao HH; Chatupheeraphat A; Rueping M Nickel-Catalyzed C–S Bond Formation via Decarbonylative Thioetherification of Esters, Amides and Intramolecular Recombination Fragment Coupling of Thioesters. Chem. Eur. J 2018, 24, 3608–3612. [DOI] [PubMed] [Google Scholar]; (c) Ishitobi K; Isshiki R; Asahara KK; Lim C; Muto K; Yamaguchi J Decarbonylative Aryl Thioether Synthesis by Ni Catalysis. Chem. Lett 2018, 47, 756–759. [Google Scholar]; (d) Zheng Z-J; Jiang C; Shao P-C; Liu W-F; Zhao T-T; Xu P-F; Wei, Hao. Controlled Ni-Catalyzed Mono- and Double- Decarbonylations of α-Ketothioesters. Chem. Commun 2019, 55, 1907–1910. [DOI] [PubMed] [Google Scholar]; (e) Zhou J-Y; Tao S-W; Liu R-Q; Zhu Y-M Forging C–S Bonds Through Nickel-Catalyzed Aryl Anhydrides with Thiophenols: Decarbonylation or Decarbonylation Accompanied by Decarboxylation. J. Org. Chem 2019, 84, 11891–11901. [DOI] [PubMed] [Google Scholar]
- (16).For decarboxylative fluoroalkylthiolation, see:; (a) Li M; Petersen JL; Hoover JM Silver-Mediated Oxidative Decarboxylative Trifluoromethylthiolation of Coumarin-3-carboxylic Acids. Org. Lett 2017, 19, 638–641. [DOI] [PubMed] [Google Scholar]; (b) Cheng Z-F; Tao T-T; Feng Y-S; Tang W-K; Xu J; Dai J-J; Xu H-J Cu(II)-Mediated Decarboxylative Trifluoromethylthiolation of α,β-Unsaturated Carboxylic Acids. J. Org. Chem 2018, 83, 499–504. [DOI] [PubMed] [Google Scholar]
- (17).For related intramolecular decarbonylative coupling for the construction of other carbon-heteroatom bonds, see:; (a) Takise R; Isshiki R; Muto K; Itami K; Yamaguchi J Decarbonylative Diaryl Ether Synthesis by Pd and Ni Catalysis. J. Am. Chem. Soc 2017, 139, 3340–3343. [DOI] [PubMed] [Google Scholar]; (b) Mao R; Bera S; Cheseaux A; Hu X Deoxygenative Trifluoromethylation of Carboxylic Acids. Chem. Sci 2019, 10, 9555–9559. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Guo L; Rueping M Transition-Metal-Catalyzed Decarbonylative Coupling Reactions: Concepts, Classifications, and Applications. Chem. Eur. J 2018, 24, 7794–7809. [DOI] [PubMed] [Google Scholar]
- (18).(a) Ferguson DM; Bour JR; Canty AJ; Kampf JW; Sanford MS Aryl–CF3 Coupling from Phosphinoferrocene-Li-gated Palladium(II) Complexes. Organometallics 2019, 38, 519–526. [Google Scholar]; (b) Xu L; Vicic DA Direct Difluoromethylation of Aryl Halides via Base Metal Catalysis at Room Temperature. J. Am. Chem. Soc 2016, 138, 2536–2539. [DOI] [PubMed] [Google Scholar]; (c) Aikawa K; Serizawa H; Ishii K; Mikami K Palladium Catalyzed Negishi Cross-Coupling of Aryl Halides with (Difluoromethyl)zinc Reagent. Org. Lett 2016, 18, 3690–3693. [DOI] [PubMed] [Google Scholar]; (d) Mann G; Baranano D; Hartwig JF; Rheingold AL; Guzei IA Carbon–Sulfur Bond-Forming Reductive Elimination Involving sp-, sp2-, and sp3-Hybridized Carbon. Mechanism, Steric Effects, and Electronic Effect on Sulfide Formation. J. Am. Chem. Soc 1998, 120, 36, 9205–9219. [Google Scholar]
- (19). Decarbonylative conversion of 1a to 2a could be carried out in high yields with both stoichiometric and catalytic loadings of Ni(cod)2 and dppf. Attempts to characterize the organometallic intermediate(s) in situ by NMR spectroscopy were impeded by their fluxionality (see SI for details).
- (20). Several other large bite angle bidentate phosphines, including Xantphos and DPEphos, afforded significant yields in this transformation. See SI for more details on reaction optimization.
- (21).(a) Osakada K; Maeda M; Nakamura Y; Yamamoto T; Yamamoto A Reversible Oxidative Addition and Reductive Elimination of Diaryl Sulphide Involving C–S Bond Cleavage and Formation: Exchange of Two Aryl Groups in Aryl(Arylthiolato)Nickel Complexes Having Tertiary Phosphine Ligands. J. Chem. Soc., Chem. Commun 1986, 6, 442–443. [Google Scholar]; (b) Barbero N; Martin R Ligand-Free Ni-Catalyzed Reductive Cleavage of Inert Carbon–Sulfur Bonds. Org. Lett 2012, 14, 796–799. [DOI] [PubMed] [Google Scholar]; (c) Wang L; He W; Yu Z Transition-Metal Mediated Carbon–Sulfur Bond Activation and Transformations. Chem. Soc. Rev 2013, 42, 599–621. [DOI] [PubMed] [Google Scholar]; (d) Ma Y; Cammarata J; Cornella J Ni-Catalyzed Reductive Liebeskin–Srogl Alkylation of Heterocycles. J. Am. Chem. Soc 2019, 141, 1918–1922. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Delcaillau T; Bismuto A; Lian Z; Morandi B Nickel-Catalyzed Inter- and Intra-Molecular Aryl Thioether Metathesis by Reversible Arylation. Angew. Chem. Int. Ed 2020, 59, 2110–2114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22). See SI for catalyst screening of the decarbonylative trifluoromethylation of thiophenol.
- (23). Stoichiometric studies of the reaction between Ni(cod)2/dppf and (trifluoromethyl)thioesters showed the formation of Ni–CF3 intermediates by 19F NMR spectroscopy (see SI), suggesting that oxidative addition and carbonyl de-insertion are taking place. However, no S–CF3 coupling was observed by 19F NMR spectroscopy or GCMS when these intermediates were heated.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.