

Abstract
There is a great interest in increasing proteins’ stability to widen their usability in numerous biomedical and biotechnological applications. However, native proteins cannot usually withstand the harsh industrial environment, since they are evolved to function under mild conditions. Ancestral sequence reconstruction is a well-established method for deducing the evolutionary history of genes. Besides its applicability to discover the most probable evolutionary ancestors of the modern proteins, ancestral sequence reconstruction has proven to be a useful approach for the design of highly stable proteins. Recently, several computational tools were developed, which make the ancestral reconstruction algorithms accessible to the community, while leaving the most crucial steps of the preparation of the input data on users’ side. FireProtASR aims to overcome this obstacle by constructing a fully automated workflow, allowing even the unexperienced users to obtain ancestral sequences based on a sequence query as the only input. FireProtASR is complemented with an interactive, easy-to-use web interface and is freely available at https://loschmidt.chemi.muni.cz/fireprotasr/.
Keywords: ancestral sequence reconstruction, ancestral enzymes, evolution, phylogeny-based analysis, protein stability
Introduction
Proteins are widely used in numerous biomedical and biotechnological applications. Native proteins have mainly evolved under mild intracellular conditions [1]. Therefore, their applicability is often limited in the harsh industrial environments characterized by inhospitable temperature, extreme pH, high pressure or the presence of organic co-solvents. As a result, there is a continuous interest in increasing protein stability. New approaches in the field of protein engineering, such as fluorescence-activated cell sorting and microfluidics, have widened the throughput of directed evolution experiments. However, saturation mutagenesis of all positions and systematic re-combinations of many single-point mutations of the protein of interest is often out of reach.
In the past decades, various computational methods were designed to unburden costly and laborious experimental work by narrowing down the search space for potential stabilizing mutations. Most of those methods can be assigned to one of the three categories: (i) machine learning, (ii) force-field-based predictions and (iii) molecular evolution. Each category has its advantages and shortcomings [2]. Machine-learning methods are able to unearth hidden features and dependencies overreaching the current state of expert knowledge, while still struggling with the insufficient size, quality and diversity of the experimental data, essential for training and validation of statistically significant models. Force-field-based approaches are a robust solution for the prediction of protein stability; however, they rely on the high-resolution protein structures that are available for only a small fraction of the known proteins. Evolution-based approaches do not suffer from these limitations due to the rapid growth of the sequence databases. However, this continuous growth widens the search space and increases noise in the data, requiring laborious and time-demanding manual corrections from the side of the user with expert knowledge of the system of interest. Inexperienced user may not therefore utilize evolution-based methods effectively to obtain accurate and reliable results.
The two most widely used evolution-based methods for stability engineering are ancestral sequence reconstruction (ASR) and consensus design. Both methods start with the multiple-sequence alignment (MSA) of the set of relevant homolog sequences. Consensus design relies on the simple analysis of the conservation of the amino acids on the individual positions in the sequence alignment. As a result, it cannot account for the coevolution of the residues located in the sites responsible for the protein’s activity [3] and is utilized mostly as a part of the hybrid workflows [4, 5]. In comparison, ASR goes much further by also considering evolutionary information depicted by the phylogenetic tree. This inclusion of the evolutionary distances inscribed into the phylogenetic tree is mostly negligent at the positions with low Shannon entropy; however, the discrepancies grow stronger with noisy MSA [6]. ASR is a probabilistic method that explores the deep evolutionary history of homolog sequences to reassemble protein’s evolutionary trajectory [7]. ASR is able to unearth sequences of the long-extinct genes and organisms from which the current ones evolved and is, therefore, an invaluable tool in the field of evolutionary biology [8, 9]. ASR has also been shown to be a very effective strategy not only for thermostability engineering [10, 11], but also for improving other protein’s characteristics such as specificity [12], activity, or expression [13]. Furthermore, ASR was previously proven to be an effective strategy for the stabilization of prokaryotic proteins [10, 11], as well as for the improvement of significantly more complex eukaryotic proteins such as cytochrome P450 [14, 15]. Two main algorithms, maximum-likelihood [16, 17] (ML) and Bayesian inference [18] (BI) were designed to infer ancestral sequence from MSA and phylogenetic tree. Many tools were built over the years to make those algorithms accessible to the community. However, the requirement of the MSA of carefully selected homologs and the rooted phylogenetic tree are still huge limiting steps for the general use of ASR method by the non-expert users.
FireProtASR addresses those limitations by introducing one-stop-shop solution for the ancestral sequence reconstruction. It covers all steps of ancestral inference including search for homolog sequences, selection of the biologically relevant subset of the sequences, construction of the multiple-sequence alignment, construction and rooting of the phylogenetic tree and finally the ancestral inference with the use of ML. Our computational workflow is fully automated and removes the need for extensive expert knowledge of the system of interest as well as employed bioinformatics tools. Furthermore, a novel algorithm based on the localized weighted back-to-consensus analysis was utilized to resolve an issue of the ancestral gaps reconstruction. Assembled workflow and developed web server were thoroughly validated using: (i) in-house laboratory experiments, (ii) detailed comparison with three previously published studies and (iii) a large number of proteins representing structurally and functionally different families. FireProtASR does not require installation and settings of any software packages as the method is implemented in the interactive web interface freely available at: https://loschmidt.chemi.muni.cz/fireprotasr/.
Methods
Workflow description
The basic workflow of the FireProtASR method is outlined in Figure 1. To infer ancestral sequences representing all ancestral nodes of the evolutionary tree in a fully automated way, a set of biologically relevant homologous sequences must be collected from genomic databases and reduced to a suitable size (Phase 1). With the initial set of homologous sequences in hand, several state-of-the-art methods are utilized to construct a multiple-sequence alignment and a phylogenetic tree, which are then used to support the inference of ancestral nodes and reconstruction of ancestral gaps (Phase 2). The FireProtASR workflow requires no user intervention beyond providing a query sequence and (in the case of enzymes) selecting catalytic residues used to identify a biologically relevant set of homologous sequences. However, it is also possible to start a calculation with a user-defined initial set of homologous sequences, MSA, or even a phylogenetic tree instead of a single sequence, thus skipping the first phase of the calculation.
Figure 1 .
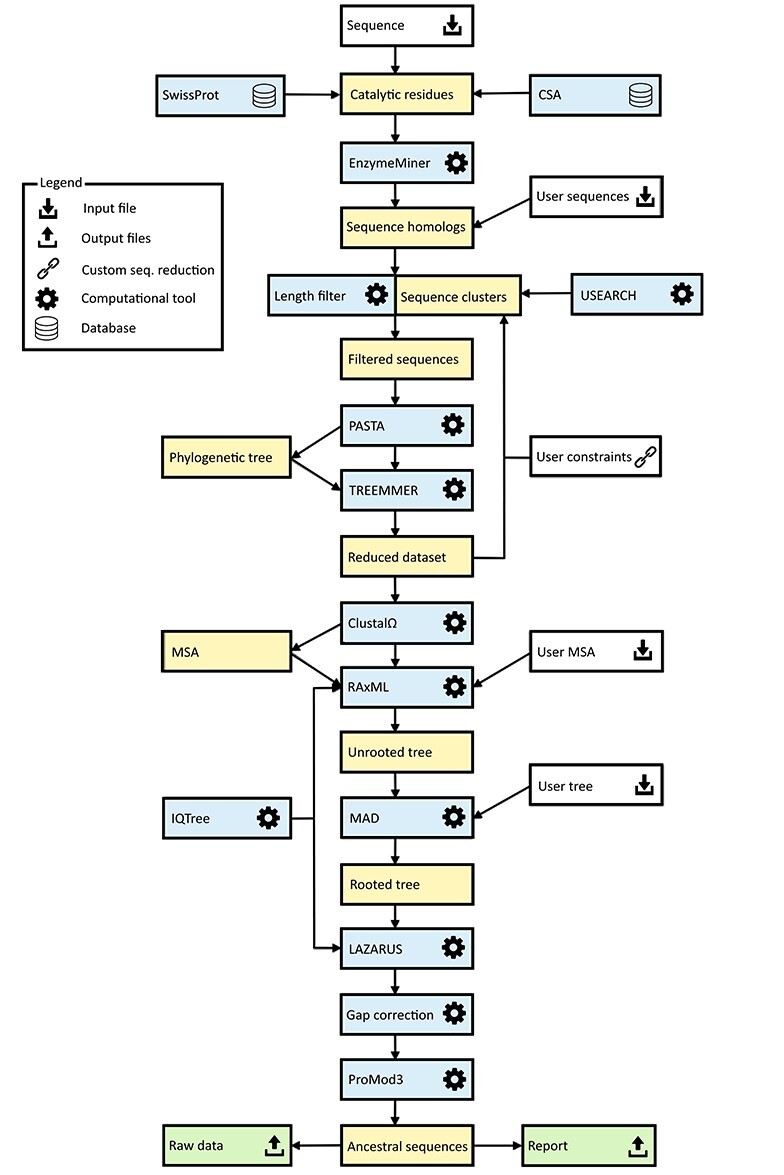
Workflow diagram for the FireProtASR method. The workflow has two phases: (1) collection of the initial set of homologous sequences and (2) ancestral sequence reconstruction. Colour coding: yellow denotes intermediate results and blue denotes computational tools. Grey and green denote inputs and outputs of the calculations, respectively.
Phase 1: collection of the initial set of homologous sequences
The query sequence of the target protein in plain text or FASTA format is the only input required from the side of the user. Once the query sequence has been uploaded to the server and checked for validity, searches for the catalytic residues are performed automatically using SwissProt [19] and the Catalytic Site Atlas [20]. The user can also specify the catalytic residues by themselves if no/incorrect catalytic residues are found. Once the catalytic residues and query sequence have been specified, an in-house tool called EnzymeMiner [21] is used to collect an initial set of homologous sequences. EnzymeMiner first performs two rounds of PSI-BLAST [22] against the NCBI nr database [23] and then filters out all sequences lacking the designated catalytic residues, thereby ensuring the biological relevance of the remaining homologs. EnzymeMiner searches can yield up to tens of thousands of homologous sequences for large families. If no catalytic residues were selected or provided by the user, BLAST [24] will be used instead of EnzymeMiner, to obtain an initial set of homologous sequences with potentially lower quality.
Next, the FireProtASR reduces the set of homologous sequences to the required number, which is set to 150 sequences by default. Several filters are applied during this process. First, all homologs with sequence lengths 20% higher or lower than that of the query sequence are excluded from the initial set. This sequence length normalization is done to remove potential outliers that could lead to a construction of a noisy MSA with many gaps. Second, all homologs whose sequence identity to the query falls outside a certain range are removed from the initial set. By default, the upper and lower similarity limits are set to 90 and 30%, respectively. This step ensures that the phylogenetic tree is unbiased towards the query sequence while removing distant homologs that would degrade the quality of the sequence alignment. Third, USEARCH [25] is used to cluster the remaining sequences with 90% sequence identity, and a single sequence is randomly selected from each cluster.
Applying these filters produces a diverse set containing hundreds to thousands of homologous sequences. An initial phylogenetic tree is quickly constructed with the PASTA software suite [26], using MAFFT [27] and the swift neighbour-joining algorithm implemented in FastTree 2.0 [28]. The resulting phylogenetic tree is then forwarded to Treemmer [29], which iteratively prunes leaves from the input tree until a specific number of leaves remains, while minimizing the loss of genetic diversity. The pruned tree is then displayed to the user via the interactive user interface, allowing the user to choose to exclude selected branches or even whole subtrees of the phylogenetic tree from further calculations.
Phase 2: ancestral sequence reconstruction
In the second phase, the ancestral sequences are inferred from the initial set of up to 150 homologs approved by the user. To begin with, a new MSA is constructed from the reduced set of homologous sequences. For this task, ClustalΩ [30] is utilized by default, but other methods will be available in upcoming versions of FireProtASR. For inference of the final phylogenetic tree, the best-fitting evolutionary matrix must be selected. This is done using one of the modules of the IQTREE package [31]. Alternatively, if the user prefers a specific evolutionary matrix for the biological system of interest, the appropriate model and all the relevant modifiers can be specified manually when setting up the calculation.
The evolutionary model and its parameter settings along with the MSA are then forwarded into RAxML [17], which is used to construct a robust phylogenetic tree. By default, fifty bootstraps are performed at the start of the maximum-likelihood search; since no outgroup is provided, the resulting phylogenetic tree is unrooted. Automated outgroup sequence selection is not straightforward, especially for prokaryotic proteins due to the high frequency of horizontal gene transfers. Rooting of the tree is therefore performed using a minimal ancestor deviation algorithm, which was shown to achieve comparable levels of accuracy to outgroup rooting in trees describing the evolution of eukaryotes, and to surpass both outgroup and midpoint rooting in the case of prokaryotes [32].
The MSA constructed with ClustalΩ, the selected evolutionary model, and the rooted phylogenetic tree from RAxML are used as inputs for the Lazarus method [33], which is implemented using the PAML software package [16]. The Lazarus method was re-implemented for FireProtASR to enable calculations to be performed without specifying outgroup. Consequently, ancestral sequences of all ancestral nodes are parsed from their posterior probabilities and provided to users in separate files in FASTA format. Additionally, BLASTp [24] is used to search for a template in the PDB database [34], and a model structure of the query sequence is constructed by homology modelling using the ProMod3 program [35]. This model is shown in the web interface to allow users to visualize the differences between the query sequence and the selected ancestor.
Finally, due to the large number of undesirable ancestral gaps inserted into ancestral sequences by Lazarus, a novel algorithm for ancestral gap reconstruction was designed for use in FireProtASR. This algorithm is based on the principle of localized weighted back-to-consensus because consensus analysis has proven to be an effective approach for increasing proteins’ thermal stability [36–38]. To begin with, each terminal node of the phylogenetic tree is assigned a binary vector of length equal to the length of the corresponding sequence in the MSA. Each position in this vector is assigned a value of −1 or 1, indicating the presence of a gap or standard amino acid, respectively, at the corresponding position of the relevant sequence. On moving from the terminals towards the root of the tree, the probability of a gap in ancestral node An at position i is calculated as 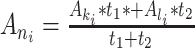 , where Ak, Al are the child nodes of An and t1, t2 are the evolutionary distances between An and its child nodes. Taking t3 to be the evolutionary distance between An and its parental node, its value can be updated based on the values of t1 and t2 as follows:
, where Ak, Al are the child nodes of An and t1, t2 are the evolutionary distances between An and its child nodes. Taking t3 to be the evolutionary distance between An and its parental node, its value can be updated based on the values of t1 and t2 as follows: 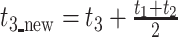 . This new value is computed before proceeding with the calculation for the parental node; its use increases the relative impact of well-branched subtrees and therefore limits the impact of lone sequences and small subtrees compared to that of well-represented ones. Finally, ancestral sequences are reconstructed based on the scores in the corresponding vector. Positions with values lower than 0 are assigned as gaps, and the remaining amino acids are selected based on their posterior probabilities as estimated by Lazarus. The nature of inconclusive positions with scores in the interval <−0.1, 0.1> is determined based on the frequencies of gaps in the global alignment and the state of the parental node. To include the ancestral gap, frequencies of gaps in the global alignment should reach over 60%, or over 40% if the ancestral gap is present in the parental node sequence. The model case for a single position in the sequence alignment is shown in Figure 2.
. This new value is computed before proceeding with the calculation for the parental node; its use increases the relative impact of well-branched subtrees and therefore limits the impact of lone sequences and small subtrees compared to that of well-represented ones. Finally, ancestral sequences are reconstructed based on the scores in the corresponding vector. Positions with values lower than 0 are assigned as gaps, and the remaining amino acids are selected based on their posterior probabilities as estimated by Lazarus. The nature of inconclusive positions with scores in the interval <−0.1, 0.1> is determined based on the frequencies of gaps in the global alignment and the state of the parental node. To include the ancestral gap, frequencies of gaps in the global alignment should reach over 60%, or over 40% if the ancestral gap is present in the parental node sequence. The model case for a single position in the sequence alignment is shown in Figure 2.
Figure 2 .
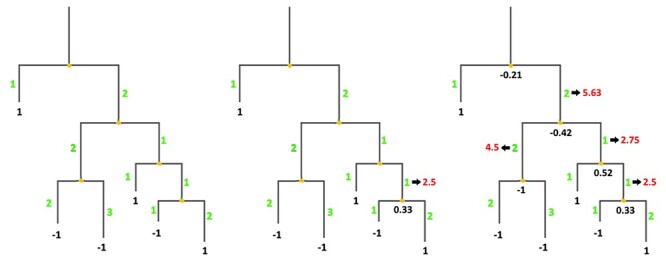
Ancestral gaps reconstruction algorithm. Green colour denotes the initial branch lengths of the phylogenetic tree. Black numbers indicate the values of the vectors of the terminal and the ancestral sequences at the given position in the multiple sequence alignment. Red values show the modified branch lengths that are updated after the calculation of the underlying ancestral node.
Experimental validation
The workflow was experimentally validated using haloalkane dehalogenases as a model enzyme. This enzyme was selected as a typical representative of the α/β superfamily, counting over 100 000 proteins. The sequence of the haloalkane dehalogenase DhaA (UniProt ID P0A3G2) was used as the sole input for the calculation. Six different ancestral sequences were selected and experimentally characterized.
Chemicals and growth media
1-bromobutane and LB medium were purchased from Sigma-Aldrich Co. (St. Louis, MO, USA). IPTG was purchased from Duchefa Biochemie B.V. (Haarlem, The Netherlands). All chemicals used in this work were of analytical grade.
Expression in Escherichia coli BL21 (DE3)
Escherichia coli Dh5α cells were obtained from Invitrogen and Escherichia coli BL21 (DE3) from New England Biolabs. The genes for the ancestral dehalogenases were synthesized and subcloned into the expression vector pET21b. The generated plasmids were transformed into chemo-competent E.coli BL21 (DE3) cells. Obtained colonies were used to prepare precultures by inoculation into 10 ml of LB medium (with 100 μg/ml ampicillin) followed by overnight incubation at 37°C and 180 rpm. For expression of each variant, 1 l of LB medium supplemented with 100 μg/ml ampicillin was inoculated with 5 mL of the appropriate pre-culture (1/200). The flasks were incubated at 37°C and 180 rpm until OD600 0.6–0.8 was reached, then incubated at 20°C for 30 min. β-D-1-thiogalactopyranoside (IPTG, 0.2 mM) was then added for induction, and the culture was incubated at 20°C and 180 rpm overnight. Finally, the culture was harvested by centrifugation at 4500 × g, 4°C for 15 min, after which the cell pellets were frozen at −80°C until further use.
Protein purification
The cell pellets were suspended in 50 ml of equilibration buffer (20 mM phosphate buffer pH 7.5 containing 0.5 M NaCl and 10 mM imidazole) and disrupted by sonication with a Hielscher UP200S ultrasonic processor (Hielscher, Germany) four times for 4 min each. Disrupted cells were centrifuged at 13 000 × g and 4°C for 1 h (Laborzentrifugen, Germany). The crude extract was then collected, filtered and loaded onto a Ni-NTA Superflow Cartridge (Qiagen, Germany) in equilibration buffer. Unbound and weakly bound proteins were washed out using increasing imidazole concentrations. The target enzyme was eluted with purification buffer containing 300 mM of imidazole. The eluted protein was dialyzed three times overnight against 50 mM of phosphate buffer (pH 7.5), after which its purity was checked by SDS–polyacrylamide gel electrophoresis (SDS–PAGE). About, 15% polyacrylamide gels were stained with Instant Blue (Fluka, Switzerland). Protein concentrations were determined by NanoDrop (Sigma-Aldrich, USA). The enzymes were lyophilized using a vacuum pump system for long-term storage.
Circular dichroism (CD) spectroscopy
CD spectra were recorded at 20°C using a spectropolarimeter Chirascan (Applied Photophysics, United Kingdom). Data were collected from 190 to 260 nm, at 100 nm/min with a 1-s response time and 1-nm bandwidth using a 0.1-cm quartz cuvette. Each spectrum shown is the average of five individual scans and was corrected for absorbance caused by the buffer. Collected CD data were expressed in terms of the mean residue ellipticity (ΘMRE), which was calculated using the equation:
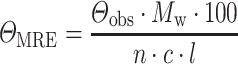 |
where Θobs is the observed ellipticity in degrees, Mw is the protein molecular weight, n is number of residues, l is the cell path length, c is the protein concentration (0.2 mg/ml) and the factor 100 originates from the conversion of the molecular weight to mg/dmol.
Thermal denaturation
Thermal unfolding was followed by monitoring the ellipticity at 224 nm over the temperature range of 20–94°C, with a resolution of 0.1°C at a heating rate of 1°C/min. Recorded thermal denaturation curves were roughly normalized to represent signal changes between approximately 1 and 0 and fitted to sigmoidal curves using Origin 6.1 (OriginLab Corporation, USA). The melting temperature (Tm) was evaluated as the midpoint of the normalized thermal transition.
Enzymatic haloalkane dehalogenase activity
Dehalogenation activity was assayed using the colorimetric method of Iwasaki et al. [49]. The release of halide ions was analyzed spectrophotometrically at 460 nm using an Eon microplate reader (BioTek, USA) after reaction with mercuric thiocyanate and ferric ammonium sulfate. The reactions were performed at 37°C in 25-ml Reacti Flasks closed with Mininert Valves. The reaction mixtures consisted of 10 ml 100 mM glycine buffer (pH 8.6) and 10 μl of the substrate 1-bromobutane. Reactions were initiated by adding the enzyme to a final concentration of 0.01 (DhaA 172Loc), 0.0065 (DhaA 172Glob), 0.0052 (DhaA 230Glob), 0.028 (DhaA 238Loc) or 0.014 mg/ml (DhaA 238Glob). Reactions were monitored by withdrawing 1 ml of samples from the reaction mixture after 0, 5, 10, 15, 20 and 30 min. The samples were immediately mixed with 0.1 ml of 35% nitric acid to stop the reaction. Dehalogenation activities were quantified as rates of product formation over time. Each activity was measured in three independent replicates.
Enzymatic luciferase activity
Luminescence activity measurements were performed with a FLUOstar OPTIMA Microplate reader (BMG Labtech, Germany) using coelenterazine as the substrate at 37°C. A 25 μl of sample of purified enzyme at a concentration of about 1 mg/ml was placed into a microtiter plate well. After baseline collection for 10 s, the luminescence reaction was initiated by adding 225 μl of 8.8 μM coelenterazine in reaction buffer (100 mM potassium phosphate buffer, pH 7.5). Luminescence was recorded for 72.5 s, and each sample was measured in at least three independent experiments. The areas of the resulting luminescence intensity peaks in relative luminescence units (RLU) were converted into values in units of RLU/mg/s.
Results
Web server input
The only required input to the web server is a query sequence of the target protein in plain text or FASTA format. Alternatively, one can upload a FASTA file containing an initial set of sequence homologs or a multiple sequence alignment (MSA). Rooted and unrooted phylogenetic trees in the standard Newick format can also be provided. When performing calculations in basic mode, only the table containing the essential residues is available to the user. Essential residues are identified automatically by searching in SwissProt [19] and mCSA [20]. However, the initial selection can be changed by the user. The default values and settings of individual computational tools are optimized to provide reliable results for most systems. Operating in advanced mode expands the list of modifiable parameters to include those related to: (i) the thresholds of the homolog identity filters and sequence clustering, (ii) selection of the evolutionary model and (iii) construction of the phylogenetic tree. Advanced mode allows experts to fine-tune the calculation’s parameters based on the studied biological system, which may be useful when dealing with particularly small or large protein families.
Selection and reduction
Upon submission, a unique identifier is assigned to each job to track the calculation. The ‘calculation browser’ informs the user about the status of the individual steps in the ancestral sequence reconstruction workflow. Once the first phase of the job is finished, the initial phylogenetic tree is displayed to the user using a strongly updated adaptation of PhyloTree library (Figure 3A) [39], together with the table of removed sequences (Figure 3B). By clicking on the individual leaves of the phylogenetic tree, the user can exclude selected sequences from future calculations. Furthermore, whole subtrees can be removed by choosing this option in the menu of the selected ancestral node. The MSA of the homologous sequences can be also visualized by switching to the multiple sequence alignment tab. This mode is intended for the expert users with the greater knowledge of the system of interest as it allows for the removal of the noise and outliers from the initial set of homolog sequences. If the expert mode is utilized, it is recommended to exclude the sequences that do not share the function similar to the query protein or that cause a significant disturbance in the MSA.
Figure 3 .
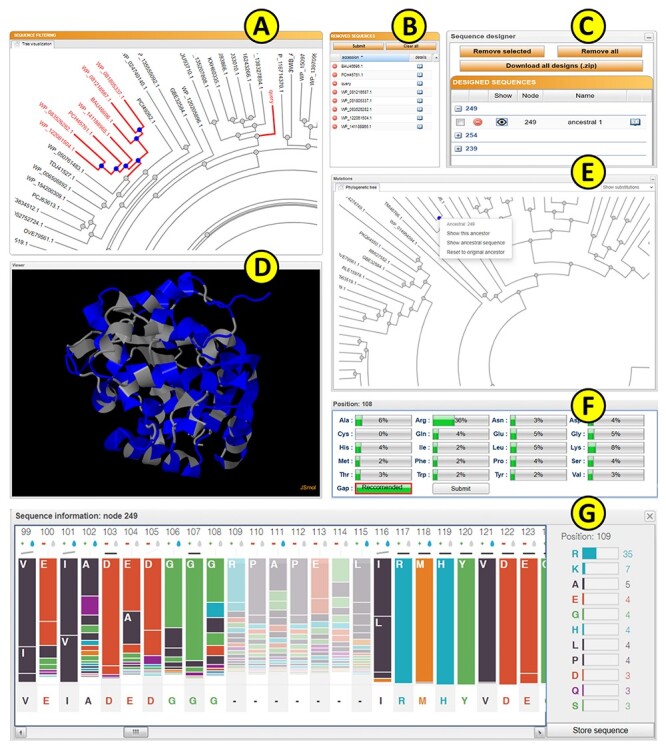
The FireProtASR graphical user interface showing results obtained for the haloalkane dehalogenase DhaA (UniProt ID P0A3G2, PDB ID 4E46). (A) The sequence-filtering panel allows users to exclude selected branches from the calculation. (B) The reduction table shows the list of removed sequences. (C) The sequence designer allows users to download and edit ancestral sequences. (D) The JSmol viewer provides interactive protein visualization. (E) The mutations panel contains all designed ancestral sequences in the ancestral tree. (F) The edit window enables amino acid substitutions at individual positions. (G) The sequence information window shows detailed information on selected ancestral sequences.
Web server output
The calculation’s progress can be tracked in the ‘calculation browser’ similarly to the selection step. Once finished, users can either download the results in the zipped archive directly from the calculation page or navigate to the ‘Result page’ for further analysis. The ‘Result page’ is organized into several panels allowing users to interactively visualize and design ancestral enzymes.
Protein visualization
The homology model of the query protein predicted by ProMod3 is interactively visualized in the web browser using the JSmol applet [40] (Figure 3D). Users can switch between different visualization styles such as backbone, wireframe or cartoon and change the quality of the visualized structure. It is also possible to visualize the differences between the query and the selected ancestral sequence on the modelled protein structure: substitutions and deletions are shown in blue and red, respectively, while insertions are indicated by regions between red and yellow residues.
Ancestral tree panel
The ‘ancestral panel’ shows the final phylogenetic tree constructed by RAxML [17] along with further information about the precalculated ancestral sequences (Figure 3E). By selecting any of the ancestral nodes, it is possible to either (i) visualize the differences between a wild-type protein and the selected ancestor node on the protein structure or (ii) open a new window providing an overview of the posterior probabilities for individual amino acids in the sequence of the selected ancestor (Figure 3G). Posterior probabilities are shown in the bar-styled sequence logo together with the percentages for each considered amino acid, and each bar is expanded with information about the charge and hydrophobicity of the most probable amino acids. The bar representation was in part derived from the SequenceLogo library [41]. The user can edit the ancestral sequence and store it as a new user-defined ancestor (Figure 3F). This option is useful for the experts with more in-depth knowledge of the system of interest and allows to force some specific mutations, e.g., the mutations with the previously known effect on proteins stability, into the constructed ancestral sequence. It can also be used to bring some biological insight into the positions with noisy posterior probabilities. Furthermore, the ancestral sequences’ MSA can be visualized in the multiple sequence alignment tab for further analysis.
Sequence designer
The ‘Sequence designer’ panel allows users to manage and edit user-defined ancestral sequences. Additionally, new sequences can be created by modifying existing custom ancestors (Figure 3C). Differences between the query sequence and custom ancestors can also be visualized on the protein structure in this panel. All prepared designs can be downloaded in one zipped archive together with the original ancestors and the structure prepared by homology modelling.
Web server experimental validation
In one of our previous studies, we have presented experimental characterizations of six inferred ancestral proteins from haloalkane dehalogenase subfamily II [10]. Relative to their contemporary counterparts, these ancestral proteins exhibited higher thermal stability (by 8–24°C), improved yields and broadened substrate specificity. Those ancestral sequences were reconstructed by clustering an initial set of homologous sequences that was reduced by inspection in the sequence-editing program BioEdit [42]. A multiple sequence alignment was then manually curated using a structure-guided alignment of eight proteins from HLD-II and poorly conserved regions were removed from the alignment. The topology of the phylogenetic tree was optimized by subtree pruning and re-grafting, and the tree’s root was established using outgroup selected on the basis of expert judgement. Finally, the ancestral sequences and positioning of gaps were refined by manual inspection.
As part of the validation of FireProtASR, we tried to replicate these results by using the sequence of haloalkane dehalogenase DhaA (UniProt ID P0A3G2) as the only input query. All steps of the calculation, including homologous sequence selection, multiple sequence alignment construction, phylogenetic rooting and ancestral reconstruction were carried out automatically. Three pairs of ancestral sequences were selected, each pair containing one ‘global’ and one ‘local’ ancestral node (Figure 4A). Global ancestor (Glob) represents ancestral sequence obtained directly from the fully automated workflow, while local ancestor (Loc) was constructed by carrying out FireProtASR workflow for a second time using only the sequences included in the subtree beneath the selected ancestral node. Local ancestor therefore represents a root of a phylogenetic tree constructed from only the sequences most relevant to the selected ancestral node. Node 238 (Figure 4D) is an ancestor of only five leaves and was selected because of its close proximity to luciferase and dehalogenase, providing a fair comparison to the previously published ancestors. Similar comparison can be also achieved with node 172 (Figure 4B), having several stable dehalogenases in its progeny. Finally, node 230 (Figure 4C) was highlighted as a more distant ancestor of both luciferase and dehalogenase. No pruning, curation or re-grafting was performed in the process. Selected ancestral sequences were then subjected to the experimental validation. MSA of the query protein, selected ancestors, and the sequence provided by executing back-to-consensus analysis is attached in Figure 4E.
Figure 4 .
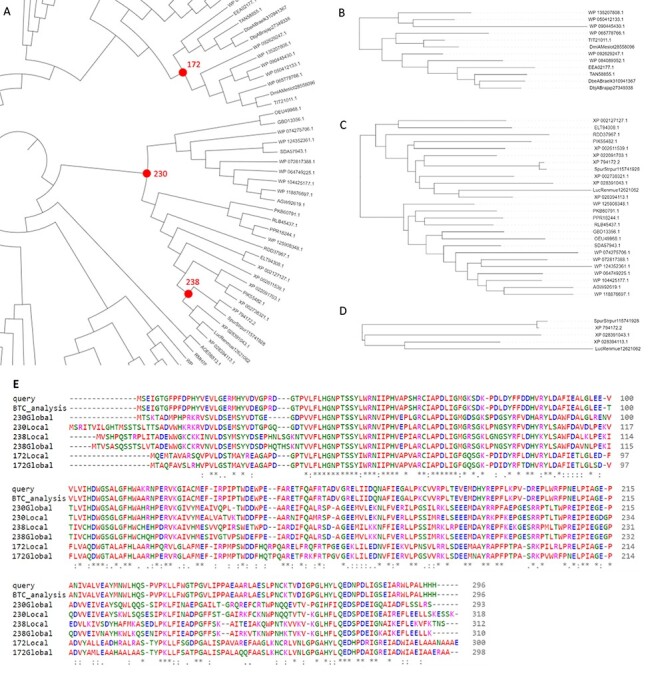
Results provided by the FireProtASR workflow using haloalkane dehalogenase DhaA as an input query. (A) Phylogenetic tree of the HLD-II constructed by the FireProtASR strategy with indicated three global ancestors reconstructed within this study. (B) Phylogenetic tree for the local ancestor of the ancestral node 172. (C) Phylogenetic tree for the local ancestor of ancestral node 230. (D) Phylogenetic tree for the local ancestor of ancestral node 238. (E) Multiple sequence alignment comparing the query sequence with the suggested ancestral sequences and the result of the back-to-consensus analysis.
Although the selected sequences have high implied sequence similarity (92–97%) with the inferred ancestors, experimental validation showed that the ancestors’ thermal stability was 20–26°C higher than that of wild-type DhaA (Table 1). The ancestral proteins also exhibited high expressibility, solubility, yields and catalytic activity. Moreover, inference based on both haloalkane dehalogenases and luciferases led to the discovery of the very interesting enzyme ancHLD-Rluc, which exhibits dual dehalogenase and monooxygenase activity. This experimental validation provides direct experimental evidence of the good functionality and reliability of the fully automated version of FireProtASR.
Table 1.
Characteristics of reconstructed and experimentally characterized ancestral haloalkane dehalogenases
| Protein code | Expression (% of total protein) | Solubility (%) | Yield (mg/l) | T m (°C) | HLD act. (μmol/mg·s) | LUC act. (RLU/mg·s) |
|---|---|---|---|---|---|---|
| DhaA wt | 17 | 83.1 | 91.1 | 50.56 ± 2.4 | 0.032 ± 0.0059 | n.a. |
| DhaA 172Loc | 23 | 85.5 | 74.9 | 71.60 ± 0.7 | 0.038 ± 0.0002 | 1.41 ± 0.26 |
| DhaA 172Glob | 21 | 65.2 | 88.2 | 70.04 ± 1.5 | 0.061 ± 0.0045 | n.a. |
| DhaA 230Loc | 20 | n.d. | n.d. | n.d. | n.d. | n.d. |
| DhaA 230Glob | 23 | 84.8 | 108.5 | 72.14 ± 0.4 | 0.061 ± 0.0118 | n.a. |
| DhaA 238Loc | 23 | 63.2 | 74.9 | 70.36 ± 0.6 | 0.014 ± 0.0021 | 353.5 ± 14.58 |
| DhaA 238Glob | 19 | 83.3 | 94.4 | 76.19 ± 0.2 | 0.030 ± 0.0012 | 3.18 ± 0.33 |
Notes: DhaA, haloalkane dehalogenase from Rhodococcus rhodochrous NCIMB 13064; wt, wild type; Loc, ancestral protein inferred from local alignment; Glob, ancestral protein inferred from global alignment; Tm, melting temperature; HLD act., haloalkane dehalogenases activity; LUC act., luciferase activity; n.d., not determined due to poor solubility of this protein; n.a., not active under tested conditions.
Additionally, results obtained using FireProtASR were thoroughly and quantitatively compared to three previously published experimental studies. For this purpose, Euclidean distance [43], and the Subtree prune and regraft distance [44] were calculated to compare the trees obtained from the FireProtASR and published literature. The two trees were also graphically compared using the Jaccard index utilizing ColorBrewer [45] scheme. Detailed comparison of all three experimental studies with the results produced by FireProtASR server is attached in Supplementary Data 1–3, available online at https://academic.oup.com/bib. Finally, the robustness and reliability of the FireProtASR server was tested using 60 diverse proteins from various protein families (see Supplementary Data 4 available online at https://academic.oup.com/bib).
Discussion
ASR has been shown to be a very effective strategy for the protein thermostability engineering and as such was implemented in various computational tools using maximum-likelihood (FastML [46], RaxML [17], Ancestors [47]) or Bayesian inference (HandAlign [48], MrBayes [18]) methods. However, a significant limitation of those methods is that they require complex input data to be uploaded by the users. Those requirements are reaching from a simple set of homolog sequences to the MSA or even rooted phylogenetic tree, leaving the most crucial and laborious parts of the calculation in the hands of the users. Non-expert users without the deep knowledge of the bioinformatics tools and the system of interest are therefore hindered from the successful use of the ASR method.
FireProtASR is a web server that aims to provide users with one-stop-shop solution for the ancestral sequence reconstruction. FireProtASR requires minimal input from the users, and the whole calculation can be processed from a single protein sequence, set of homolog sequences, MSA and phylogenetic tree. All steps of the calculation, including the search for biologically relevant homolog sequences, dataset reduction and the ancestral reconstruction are automated. Moreover, a novel algorithm based on localized weighted back-to-consensus analysis is implemented to resolve an issue with ancestral gap reconstruction. FireProtASR web server is also complemented by an easy-to-use web interface that allows users to interactively analyze sequences of the individual ancestral nodes together with the ability to design their own ancestral sequences based on the posterior probabilities of the existing nodes.
The robustness and reliability of the results produced by the FireProtASR workflow was evaluated by experimental characterization of six ancestral sequences of haloalkane dehalogenase from HLD-II subfamily. With the exception of the local variant of the ancestral node 230, all designed ancestral sequences are soluble and also retain high expressibility and yields on the levels comparable to the DhaA wild type. However, the thermal stability has increased by over 20°C and global variants 172 and 230 have also increased the HLD activity by two-fold. Increase in HLD activity cannot be observed in the constructed local variants that utilize smaller subsets of homolog sequences, and thus only a limited amount of evolutionary information. This would encourage the usage of the global variants for the design of highly stable and active proteins. However, more focused view using a localized variants of the ancestral nodes can provide some useful results as can be observed in the local variant of the node 238 that shows both dehalogenase and monooxygenase activity. High thermal stabilization was also achieved in those variants.
Finally, the results provided by the FireProtASR web server are consistent with the designs presented in the published literature as the fully automatized designs obtained by FireProtASR method maintain high sequence similarity (>90%) with the manually designed and curated ancestors. Finally, the comprehensive analysis of approximately 60 different proteins from various protein families have proven the robustness and reliability of the presented method.
The full automation of the FireProtASR method eliminates the need to select, install and evaluate individual tools, optimize their parameters and interpret intermediate results. Together with its general applicability for a wide range of protein families, FireProtASR makes the procedure of ancestral reconstruction accessible to the users without any prior expertise in bioinformatics, and the intuitive web interface allows for a further analysis utilizing both sequence and structural information.
Key Points
FireProtASR is a web service for a fully automated design of stable proteins using ancestral sequence reconstruction and is accompanied by an interactive and easy-to-use interface.
FireProtASR allows users to utilize ancestral reconstruction without prior knowledge of the necessary bioinformatics tools and the biological system.
The robustness and reliability of the FireProtASR method were thoroughly tested by both laboratory experiments and by comparing predictions with the results published in scientific literature.
Laboratory characterization of the ancestral designs showed up to 26°C improvement in thermostability and some of the proteins poses even dual catalytic activity.
Supplementary Material
Milos Musil is a bioinformatician at Loschmidt Laboratories, Masaryk University. He carries out his doctoral thesis at the Brno University of Technology, designing and implementing bioinformatics tools for the automatized design of stable proteins.
Rayyan Tariq Khan is a doctoral candidate at Loschmidt Laboratories, Masaryk University. His work is focused on ancestral sequence reconstruction, experimental evolution and design of bioinformatics tools.
Andy Beier is doing his postdoc in protein engineering at the Loschmidt Laboratories, Masaryk University. His main responsibilities are the mutagenesis, production and detailed biochemical and biophysical characterization of enzymes and the development of an ultra-high-throughput assay for dehalogenases.
Jan Stourac is a bioinformatician at Loschmidt Laboratories, Masaryk University. He carries out his doctoral thesis at the Faculty of Informatics, Masaryk University, focusing on the design and implementation of the bioinformatics tools for the analysis of protein tunnels.
Hannes Konegger is a former MSCA fellow, examined the biochemical basis of evolutionary- and structure-based protein engineering methods. He recently turned his field of interest towards microbial ecology and applied bioenergetics.
Jiri Damborsky is a professor of Biochemistry at Masaryk University and group leader at International Clinical Research Center at St. Ann’s Teaching Hospital. He is interested in development of software tools for computational enzyme design.
David Bednar is a team leader at Loschmidt Laboratories, Masaryk University. His team is focused on molecular modelling, bioinformatics and development of software for protein engineering.
Contributor Information
Milos Musil, Loschmidt Laboratories, Masaryk University.
Rayyan Tariq Khan, Loschmidt Laboratories, Masaryk University.
Andy Beier, Loschmidt Laboratories, Masaryk University.
Jan Stourac, Loschmidt Laboratories, Masaryk University.
Hannes Konegger, MSCA.
Jiri Damborsky, International Clinical Research Center at St. Ann’s Teaching Hospital.
David Bednar, Loschmidt Laboratories, Masaryk University.
Data availability
All data validating the robustness and accuracy of our service are available in the Supplementary materials 1-4. Web service and tutorials are freely available at https://loschmidt.chemi.muni.cz/fireprotasr/.
Funding
Czech Ministry of Education (CZ.02.1.01/0.0/0.0/17_043/0009632, 857560, CZ.02.1.01/0.0/0.0/16_026/0008451); the Czech Grant Agency (20-15915Y); the Technology Agency of Czech Republic (TH02010219); Brno University of Technology (FIT-S-20-6293); and the European Commission (720776, 814418, 722610) and Marie Curie@MUNI (CZ.02.2.69/0.0/0.0/19_074/0012727). Computational resources were supplied by the project ‘e-Infrastruktura CZ’ (LM2018140) and ELIXIR (LM2015047). This project has received funding from the European Union’s Horizon 2020 research and Innovation programme. The article reflects the author’s view and the Agency is not responsible for any use that may be made of the information it contains.
References
- 1. Modarres HP, Mofrad MR, Sanati-Nezhad A. Protein thermostability engineering. RSC Adv 2016;6:115252–70. [Google Scholar]
- 2. Musil M, Konegger H, Hon J, et al. . Computational design of stable and soluble biocatalysts. ACS Catal 2019;9:1033–54. [Google Scholar]
- 3. Hendrikse NM, Charpentier G, Nordling E, et al. . Ancestral diterpene cyclases show increased thermostability and substrate acceptance. FEBS J 2018;285:4660–73. [DOI] [PubMed] [Google Scholar]
- 4. Musil M, Stourac J, Bendl J, et al. . FireProt: web server for automated design of thermostable proteins. Nucleic Acids Res 2017;45:W393–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Goldenzweig A, Goldsmith M, Hill SE, et al. . Automated structure- and sequence-based design of proteins for high bacterial expression and stability. Mol Cell 2016;63:337–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Risso VA, Gavira JA, Gaucher EA, et al. . Phenotypic comparisons of consensus variants versus laboratory resurrections of Precambrian proteins. Proteins Struct Funct Bioinforma 2014;82:887–96. [DOI] [PubMed] [Google Scholar]
- 7. Hochberg GKA, Thornton JW. Reconstructing ancient proteins to understand the causes of structure and function. Annu Rev Biophys 2017;46:247–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bickelmann C, Morrow JM, Du J, et al. . The molecular origin and evolution of dim-light vision in mammals. Evolution 2015;69:2995–3003. [DOI] [PubMed] [Google Scholar]
- 9. Hobbs JK, Prentice EJ, Groussin M, et al. . Reconstructed ancestral enzymes impose a fitness cost upon modern bacteria despite exhibiting favourable biochemical properties. J Mol Evol 2015;81:110–20. [DOI] [PubMed] [Google Scholar]
- 10. Babkova P, Sebestova E, Brezovsky J, et al. . Ancestral haloalkane dehalogenases show robustness and unique substrate specificity. Chembiochem 2017;18:1448–56. [DOI] [PubMed] [Google Scholar]
- 11. Risso VA, Gavira JA, Sanchez-Ruiz JM. Thermostable and promiscuous Precambrian proteins. Environ Microbiol 2014;16:1485–9. [DOI] [PubMed] [Google Scholar]
- 12. Wheeler LC, Lim SA, Marqusee S, et al. . The thermostability and specificity of ancient proteins. Curr Opin Struct Biol 2016;38:37–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zakas PM, Brown HC, Knight K, et al. . Enhancing the pharmaceutical properties of protein drugs by ancestral sequence reconstruction. Nat Biotechnol 2017;35:35–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bart AG, Harris KL, Gillam EMJ, et al. . Structure of an ancestral mammalian family 1B1 cytochrome P450 with increased thermostability. J Biol Chem 2020;295:5640–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gumulya Y, Baek J-M, Wun S-J, et al. . Engineering highly functional thermostable proteins using ancestral sequence reconstruction. Nat Catal 2018;1:878–88. [Google Scholar]
- 16. Yang Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol 2007;24:1586–91. [DOI] [PubMed] [Google Scholar]
- 17. Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinforma Oxf Engl 2014;30:1312–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ronquist F, Teslenko M, Mark P, et al. . MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol 2012;61:539–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Boeckmann B, Bairoch A, Apweiler R, et al. . The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res 2003;31:365–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ribeiro AJM, Holiday GL. Mechanism and catalytic site atlas (M-CSA): a database of enzyme reaction mechanisms and active sites. Nucleic Acids Res 2018;46:618–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hon J, Borko S, Stourac J, et al. . EnzymeMiner: automated mining of soluble enzymes with diverse structures, catalytic properties and stabilities. Nucleic Acids Res 2020;48:W104–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Altschul SF, Madden TL, Schaffer AA, et al. . PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 1997;17:3389–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. NCBI Resource Coordinators . Database resources of the National Center for Biotechnology Information. Nucleic Acids Res 2016;44:D7–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Camacho C, Coulouris G, Avagyan V, et al. . BLAST+: architecture and applications. BMC Bioinforma 2009;10:421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinforma Oxf Engl 2010;26:2460–1. [DOI] [PubMed] [Google Scholar]
- 26. Mirarab S, Nguyen N, Guo S, et al. . PASTA: ultra-large multiple sequence alignment for nucleotide and amino-acid sequences. J Comput Biol 2015;22:377–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol 2013;30:772–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Price MN, Dehal PS, Ap A. FastTree 2—approximately maximum-likelihood trees for large alignments. PLoS One 2010;5(3):e9490. doi: 10.1371/journal.pone.0009490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Menardo F, Loiseau C, Brites D, et al. . Treemmer: a tool to reduce large phylogenetic datasets with minimal loss of diversity. BMC Bioinformatics 2018;19:164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sievers F, Wilm A, Dineen D, et al. . Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal omega. Mol Syst Biol 2011;7:539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Nguyen L-T, Schmidt HA, Haeseler A, et al. . IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol 2015;32:268–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Tria FDK, Landan G, Dagan T. Phylogenetic rooting using minimal ancestor deviation. Nat Ecol Evol 2017;1:193. [DOI] [PubMed] [Google Scholar]
- 33. Hanson-Smith V, Kolaczkowski B, Thornton JW. Robustness of ancestral sequence reconstruction to phylogenetic uncertainty. Mol Biol Evol 2010;27:1988–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sussman JL, Lin D, Jiang J, et al. . Protein data Bank (PDB): database of three-dimensional structural information of biological macromolecules. Acta Crystallogr D Biol Crystallogr 1998;54:1078–84. [DOI] [PubMed] [Google Scholar]
- 35. Biasini M, Schmidt T, Bienert S, et al. . OpenStructure: an integrated software framework for computational structural biology. Acta Crystallogr D Biol Crystallogr 2013;69:701–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Amin N, Liu AD, Ramer S, et al. . Construction of stabilized proteins by combinatorial consensus mutagenesis. Protein Eng Des Sel 2004;17:787–93. [DOI] [PubMed] [Google Scholar]
- 37. Lehmann M, Loch C, Middendorf A, et al. . The consensus concept for thermostability engineering of proteins: further proof of concept. Protein Eng Des Sel 2002;15:403–11. [DOI] [PubMed] [Google Scholar]
- 38. Sullivan BJ, Nguyen T, Durani V, et al. . Stabilizing proteins from sequence statistics: the interplay of conservation and correlation in triosephosphate isomerase stability. J Mol Biol 2012;420:384–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Shank SD, Weaver S, Kosakovsky Pond SL. Phylotree.Js—a JavaScript library for application development and interactive data visualization in phylogenetics. BMC Bioinformatics 2018;19:276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hanson RM, Prilusky J, Renjian Z, et al. . JSmol and the next-generation web-based representation of 3D molecular structure as applied to Proteopedia. Isr J Chem 2013;53:207–16. [Google Scholar]
- 41. Maguire E, Rocca-Serra P, Sansone S-A, et al. . Redesigning the sequence logo with glyph-based approaches to aid interpretation. In: Proceedings of EuroVis 2014 Short Paper, IEEE Visualization and Graphics Technical Committee (IEEE VGTC) 2014. [Google Scholar]
- 42. Kirmani S. A user friendly approach for design and economic analysis of standalone SPV system. Smart Grid Renew Energy 2015;06:67–74. [Google Scholar]
- 43. Vienne DM, Aguileta G, Ollier S. Euclidean nature of phylogenetic distance matrices. Syst Biol 2011;60:826–32. [DOI] [PubMed] [Google Scholar]
- 44. Bordewich M, Semple C. On the computational complexity of the rooted subtree prune and Regraft distance. Ann Comb 2005;8:409–23. [Google Scholar]
- 45. Harrower M, Brewer CA. ColorBrewer.Org: an online tool for selecting colour schemes for maps. Cartogr J 2003;40:27–37. [Google Scholar]
- 46. Ashkenazy H, Penn O, Doron-Faigenboim A, et al. . FastML: a web server for probabilistic reconstruction of ancestral sequences. Nucleic Acids Res 2012;40:W580–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Diallo AB, Makarenkov V, Blanchette M. Ancestors 1.0: a web server for ancestral sequence reconstruction. Bioinforma Oxf Engl 2010;26:130–1. [DOI] [PubMed] [Google Scholar]
- 48. Westesson O, Barquist L, Holmes I. HandAlign: Bayesian multiple sequence alignment, phylogeny and ancestral reconstruction. Bioinforma Oxf Engl 2012;28:1170–1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Iwasaki I, Utsumi S, Ozawa T. New colorimetric determination of chloride using mercuric thiocyanate and ferric ion. Bulletin of the Chemical Society of Japan 1952;25(3):226. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data validating the robustness and accuracy of our service are available in the Supplementary materials 1-4. Web service and tutorials are freely available at https://loschmidt.chemi.muni.cz/fireprotasr/.