

Abstract
Deoxyribonucleic acid replication is one of the most crucial tasks taking place in the cell, and it has to be precisely regulated. This process is initiated in the replication origins (ORIs), and thus it is essential to identify such sites for a deeper understanding of the cellular processes and functions related to the regulation of gene expression. Considering the important tasks performed by ORIs, several experimental and computational approaches have been developed in the prediction of such sites. However, existing computational predictors for ORIs have certain curbs, such as building only single-feature encoding models, limited systematic feature engineering efforts and failure to validate model robustness. Hence, we developed a novel species-specific yeast predictor called yORIpred that accurately identify ORIs in the yeast genomes. To develop yORIpred, we first constructed optimal 40 baseline models by exploring eight different sequence-based encodings and five different machine learning classifiers. Subsequently, the predicted probability of 40 models was considered as the novel feature vector and carried out iterative feature learning approach independently using five different classifiers. Our systematic analysis revealed that the feature representation learned by the support vector machine algorithm (yORIpred) could well discriminate the distribution characteristics between ORIs and non-ORIs when compared with the other four algorithms. Comprehensive benchmarking experiments showed that yORIpred achieved superior and stable performance when compared with the existing predictors on the same training datasets. Furthermore, independent evaluation showcased the best and accurate performance of yORIpred thus underscoring the significance of iterative feature representation. To facilitate the users in obtaining their desired results without undergoing any mathematical, statistical or computational hassles, we developed a web server for the yORIpred predictor, which is available at: http://thegleelab.org/yORIpred.
Keywords: iterative feature representation, support vector machine, replication origin, machine learning
Introduction
Deoxyribonucleic acid (DNA) is the main carrier of genetic information for life. Before cell division, biomolecules and organelles including the duplication of DNA molecules need to be copied for appropriate distribution among the new daughter cells. This process of DNA duplication happening in the dividing cell is called DNA replication or semi-conservative replication [1]. DNA replication occurs in the cytoplasm and nucleus for prokaryotes and eukaryotes, respectively. However, the basic process of DNA replication remains the same. The replication origin serves as the site for genome replication, and hence it has to be tightly regulated [2]. Bacterial genome has only one replication origin [3] in contrast to the presence of multiple replication origins (ORIs) in the eukaryotic genome [4]. Since the duplication of DNA is initiated in the ORIs, it is necessary to identify such sites for better understanding the transmission of genetic information.
Several in vitro and in vivo experimental techniques, such as chromatin immunoprecipitation (ChIP), ChIP-sequencing, DNase I footprinting technique, electrophoretic mobility shift assays, gel retardation assay, isothermal titration calorimetry, replication initiation point mapping and surface plasmon resonance have been developed to identify DNA ORIs [5]. Lee and Bell [6] reported that the origin recognition complex (ORC) that binds ORIs could be accurately detected [7]. The ORIs of Saccharomyces cerevisiae possess two characteristic sequence patterns [8]: (1) ORIs have highly specific sequence patterns, where ORC identifies the T-rich 17-bp ARS motif and its consensus sequence that may interact with its neighboring B1/B2/B3 elements [9] and (2) ORIs’ nucleosome exclusion [10]. However, due to their time- and cost-ineffectiveness, computational tools based on various skew types were established. High-throughput sequencing-based marker frequency analysis was developed to analyze replication characteristics and to map ORIs in both prokaryotes [11] and eukaryotes [12]. Ori-Finder [13] and Ori-Finder 2 [14] apply Z-curve method and comparative genomics analysis to identify ORCs in bacterial and archaeal genomes. Other computational tools include CG software [15] and GraphDNA [16]. The limitation of using such computational tools is that these predictors were built only based on positive samples containing information about ORIs.
However, in the recent decade, this limitation has been overcome by the introduction of various computational methods, which have utilized both positive and negative samples to train the replication origin predictors and to attenuate the occurrence of false positives and false negatives. Chen et al. [17] predicted a method to identify ORIs based on DNA structural properties, whereas Li et al. [18] utilized pseudo k-tuple nucleotide compositions (PseKNC) in predicting replication origin sites in S. cerevisiae. Subsequently, Zhang et al. [19] and Xiao et al. [20] integrated dinucleotide physicochemical features and position-specific features, respectively, with general pseudo amino acid composition [21] and enhanced their respective predictors’ performance accuracies. Later, Liu et al. [22] developed a predictor called ‘iRO-3wPseKNC’ to predict ORIs in four yeast species using three-window-based PseKNC. Subsequently, the same group implemented different lengths of ORIs and Guanine/Cytosine (GC) asymmetry bias and developed a prediction algorithm called ‘iRO-PsekGCC’ for two yeast species [23]. Two research groups developed their respective predictors to predict ORIs in S. cerevisiae using the same dataset but different approaches [24, 25]. Recently, Dao et al. [26] proposed a computational platform to identify replication origin from eukaryotes, where they applied feature selection technique on hybrid features (a combination of Kmer and binary encoding) and identified optimal feature set, which improved prediction accuracies on multiple species. Among the existing methods, four methods are applicable to yeast species. Of those, two methods (iRO-3wPseKNC and iRO-PsekGCC) were developed using diverse ORI lengths, and the remaining two methods were developed using fixed ORI lengths. In this work, our primary objective was to develop species-specific prediction models from the dataset containing diverse ORI lengths. Despite the advantages of the existing methods (iRO-3wPseKNC and iRO-PsekGCC) and performance improvements [22, 23], certain limitations cannot be ruled out: (i) existing methods employed only a single encoding and classifier; (ii) only limited feature engineering efforts have been made to explore the efficiency of different feature types and classifiers for better classification, as the existing tools focused on extracting only a few feature encodings and (iii) failure to evaluate the model transferability.
By addressing the above-mentioned issues, we developed a novel species-specific predictor called yORIpred, which accurately predict ORIs from four yeast species including S. cerevisiae, Kluyveromyces lactis, Pichia pastoris and Schizosaccharomyces pombe. To develop yORIpred, firstly, 40 baseline models were generated by exploring five different classification algorithms [random forest (RF), gradient boosting (GB), artificial neural network (ANN), support vector machine (SVM) and extremely randomized tree (ERT)] and eight diverse sequence encoding schemes [Kmer composition, composition of k-spaced nucleic acid pairs (CKSNAP), electron-ion interaction pseudopotentials (EIIP), dinucleotide physicochemical properties (DPCP), PseKNC, pseudo dinucleotide composition (PseDNC), series correlation pseudo trinucleotide composition (SCPseTNC) and trinucleotide physicochemical properties (TPCP)] to provide comprehensive feature information for model training. Notably, a two-step feature selection approach was employed during the baseline model generation. Secondly, the predicted probability of ORIs from 40 baseline models was considered as novel features, where iterative feature learning approach was applied to learn probabilistic features in order to promote the feature representation capability in a highly supervised iterative manner. Our systematic analysis revealed that feature representation learned by SVM has a high discrimination distribution characteristic between ORIs and non-non-ORIs when compared with the remaining four classifiers. yORIpred showed promising results in both cross-validation (CV) analysis and independent evaluation, thus indicating that the iterative feature representation is solely responsible for its accurate and robust prediction. We anticipate that our proposed predictor may help to identify novel ORIs and useful in elucidating their functional mechanisms.
Methods
Dataset construction
Recently, Liu et al. [22] constructed training datasets with the varying length for four yeast species (S. cerevisiae, K. lactis P. pastoris and S. pombe) based on DeOri database [14]. Of those, S. cerevisiae contains 341 ORI sequences and 343 non-ORI sequences; S. pombe contains 339 ORI sequences and 336 non-ORI sequences; P. pastoris contains 306 ORI sequences and 303 non-ORI sequences and K. lactis contains 148 ORI sequences and 148 non-ORI sequences. Since DeOri database was not updated, Liu et al. dataset was considered in this study for the following reasons: (i) they applied several filtering schemes and constructed a reliable training dataset; (ii) none of the sequences for each species-specific dataset possesses greater than 80% sequence identity with other sequences and (iii) employing such datasets for the development of model enables a fair comparison between the existing methods and our proposed method.
To evaluate our prediction model, we constructed an independent dataset for the S. cerevisiae species by utilizing a recently reported ORI dataset (Ori-Finder 3) [27]. Generally, random sequences have been used as non-ORIs, but we employed sequences other than ORI functions reported for S. cerevisiae. Specifically, we considered recombination hot/cold spot sequences downloaded from iRSpot-Pse6NC2.0 [28] and considered them as non-ORIs. Subsequently, samples have been excluded that share >75% sequence identity with the training dataset, which resulted in 67 ORIs and 837 non-ORIs.
Feature descriptors
Generally, DNA sequences with variable length should be converted into fixed length of numerical vectors by means of feature extraction [29]. Here, we employed eight different feature descriptors and evaluated their contribution in classifying ORIs from non-ORI sequences, when provided as an input to different machine learning (ML) classifiers. In brief, each descriptor is described as follows:
Kmer
Kmer represents the normalized occurrence frequencies of k neighboring base pairs in the DNA sequence [28, 30, 31]. For instance, Kmer (k = 2) descriptor can be calculated as:
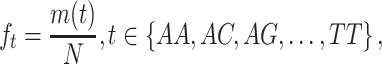 |
(1) |
where m(t) represents the total number of the Kmer type t and N denotes the sequence length. Here k is set as 2, 3, 4 and 5 and combined together that resulted as 1360=(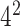 +
+ 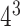 +
+ 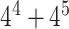 )-dimensional (1360D) feature vector.
)-dimensional (1360D) feature vector.
CKSNAP
CKSNAP converts a DNA sequence into a numerical feature vector by computing the occurrence frequency of all possible k-spaced nucleotide pairs (KNP) along the sequence. For instance, in the sequence ‘AXXTXXXG’, ‘AT’ and ‘TG’, respectively, represent two-spaced and three-spaced nucleotide pairs. The frequency of KNP can be defined as:
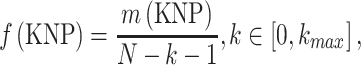 |
(2) |
where m(KNP) represents the number of KNP along the sequence, and (N-k-l) represents the number of KNP along a sequence with length N. We kept kmax = 5 that generated 96D feature vector.
EIIP
EIIP represents the distribution of free-electron energies along the DNA sequence. The EIIP values of nucleotides A, T, G and C are 0.1260, 0.1335, 0.0806 and 0.1340, respectively. Utilizing these values, EIIP generated 64D feature vector for a given DNA sequence (S) that can be computed as follows:
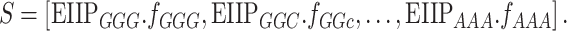 |
(3) |
The various combination of trinucleotides is shown as subscripts in Equation (3); EIIPxyz = EIIPx + EIIPy + EIIPz, expresses the EIIP values of one of the trinucleotides (xyz), where x, y, z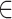 {G, T, C, A}; and fxyz denotes normalized trinucleotide frequency.
{G, T, C, A}; and fxyz denotes normalized trinucleotide frequency.
DPCP
We employed 15 PCPs: PCP1, F-rise; PCP2, F-tilt; PCP3, F-slide; PCP4, F-twist; PCP5, F-roll; PCP6, roll; PCP7, F-shift; PCP8, twist; PCP9, tilt; PCP10, shift; PCP11, slide; PCP12, rise; PCP13, enthalpy; PCP14, energy; and PCP15, entropy. DPCP is computed as follows:
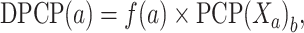 |
(4) |
X a is the value of bth (b = 1,2,…,15) dinucleotide PCP. Ultimately, DPCP provides a 240D vector.
TPCP
We employed 11 PCPs: PCP1, bendability (consensus); PCP2, bendability (DNase); PCP3, nucleosome positioning; PCP4, trinucleotide GC content; PCP5, consensus (rigid); PCP6, consensus (roll); PCP7, DNase I (rigid); PCP8, nucleosome (rigid); PCP9, molecular weight (daltons); PCP10, DNase I; and PCP11, nucleosome. Notably, the PCP values of both trinucleotides and dinucleotides have been provided in our previous study [32, 33]. These encodings have been widely applied in computational biology [34–36].
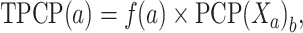 |
(5) |
X m is the value of bth (b = 1,2,…,11) trinucleotide PCP. Eventually, TPCP provides 704D vector.
PseDNC
PseDNC integrates both local and global sequence-order information of DNA sequences, whose feature vector for a given DNA sequence is defined as:
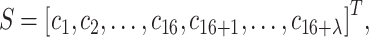 |
(6) |
where
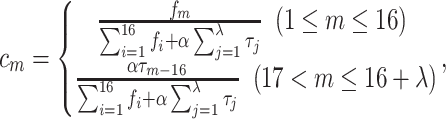 |
(7) |
where 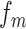 represents the normalized different dinucleotides frequency,
represents the normalized different dinucleotides frequency, 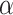 is the weight factor,
is the weight factor, 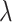 represents the total number of pseudo components and
represents the total number of pseudo components and 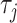 represents the j-tier correlation factor that reflects the sequence-order correlation between all the j-th adjacent dinucleotides along a DNA sequence.
represents the j-tier correlation factor that reflects the sequence-order correlation between all the j-th adjacent dinucleotides along a DNA sequence. 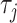 is defined as:
is defined as:
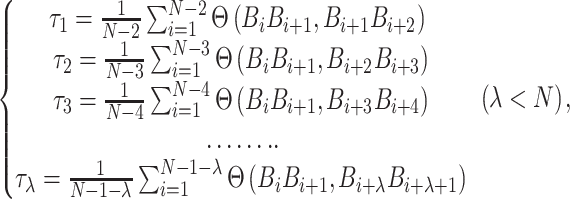 |
(8) |
where 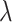 is an integer and
is an integer and 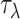 represents
represents 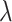 -tier correlation factor, whose correlation function is given as:
-tier correlation factor, whose correlation function is given as:
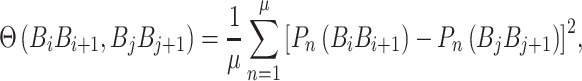 |
(9) |
where 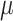 is fourteen DNA local structural properties (Tilt, Roll, Rise, Shift, Slide, Twist, GC content, Adenine content, Thymine content, Stacking energy, Bending stiffness, Electron_interaction, Enthalpy and Entropy). The standardized values of 14 structural properties taken from previous work [24].
is fourteen DNA local structural properties (Tilt, Roll, Rise, Shift, Slide, Twist, GC content, Adenine content, Thymine content, Stacking energy, Bending stiffness, Electron_interaction, Enthalpy and Entropy). The standardized values of 14 structural properties taken from previous work [24]. 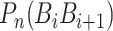 is the value of the nth DNA local property for the dinucleotide (
is the value of the nth DNA local property for the dinucleotide (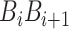 ) at position i and
) at position i and 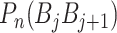 represents the corresponding value for the dinucleotide (
represents the corresponding value for the dinucleotide (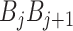 ) at position j. They are calculated as follows:
) at position j. They are calculated as follows:
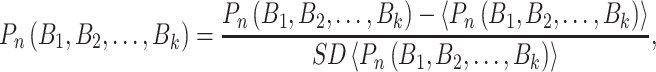 |
(10) |
where <> and SD represent the mean and standard deviation. Finally, PseDNC with the following parameters λ = 2 and 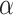 = 1.0, generated a total of 18D feature vector.
= 1.0, generated a total of 18D feature vector.
PseKNC
PseKNC is an extended version of PseDNC that incorporates k-tuple nucleotide composition [37, 38]. PseKNC feature vector for a given DNA sequence is expressed as follows:
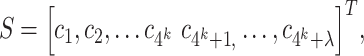 |
(11) |
where
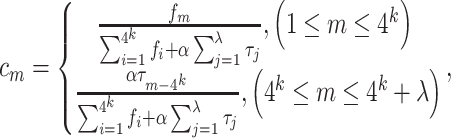 |
(12) |
where 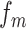 represents the normalized occurence frequency of the ith k-tuple nucleotide,
represents the normalized occurence frequency of the ith k-tuple nucleotide, 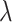 represents the total number of counted ranks or tiers of the correlations along the nucleotide sequence;
represents the total number of counted ranks or tiers of the correlations along the nucleotide sequence; 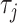 is defined as:
is defined as:
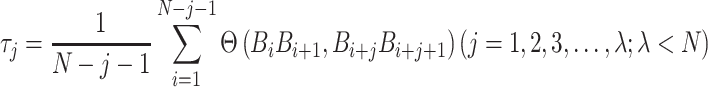 |
(13) |
The correlation function is given as follows:
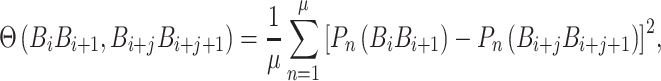 |
(14) |
where 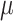 is fourteen DNA local structural properties as mentioned in PseDNC.
is fourteen DNA local structural properties as mentioned in PseDNC. 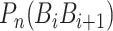 is the value of the nth DNA local property for the dinucleotide (
is the value of the nth DNA local property for the dinucleotide (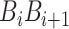 ) at position i and
) at position i and 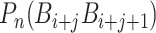 represents the corresponding value for the dinucleotide (
represents the corresponding value for the dinucleotide (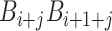 ) at position i + j. Eventually, PseDNC with the following parameters Kmer = 5, λ = 5 and
) at position i + j. Eventually, PseDNC with the following parameters Kmer = 5, λ = 5 and 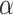 = 0.8, generated a total of 1029D feature vector.
= 0.8, generated a total of 1029D feature vector.
SCPseTNC
In SCPseTNC approach, trinucleotide physiochemical indices are incorporated to generate the representations of DNA sequences. It is computed as follows:
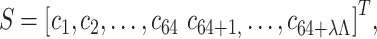 |
(15) |
where
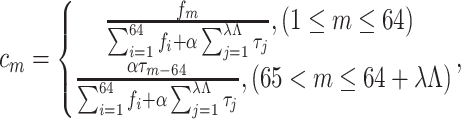 |
(16) |
where 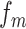 and
and 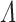 represent the normalized trinucleotide frequency and number of physicochemical indices, respectively.
represent the normalized trinucleotide frequency and number of physicochemical indices, respectively. 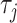 is defined as:
is defined as:
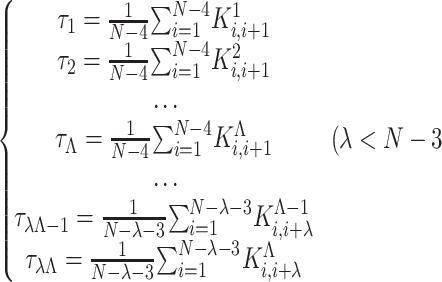 ) (17)
) (17)
The correlation function is given by:
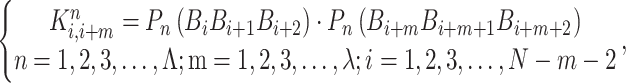 |
(18) |
where 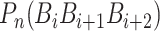 is the numerical value of the nth (n = 1,2,3,…,
is the numerical value of the nth (n = 1,2,3,…,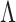 ) physicochemical index of the trinucleotide
) physicochemical index of the trinucleotide 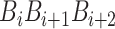 at position i and
at position i and 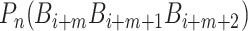 represents the corresponding value of the trinucleotide
represents the corresponding value of the trinucleotide 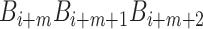 at position i + m. Ultimately, SCPseDNC with the following parameters λ = 2 and
at position i + m. Ultimately, SCPseDNC with the following parameters λ = 2 and 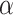 = 0.5, generated a total of 86D feature vector.
= 0.5, generated a total of 86D feature vector.
ML classifiers
Five different classifiers were utilized namely, SVM, ANN, RF, GB and ERT. Since SVM contribution is significant in yORIpred implementation, a brief description of SVM and its utilization is detailed below.
SVM
SVM is one of the powerful ML algorithms that have been widely applied in various prediction problems [39–45]. The aim of SVM is to transform the input features into a high-dimensional space and find the optimal hyperplane that can maximize the distance between ORIs and non-ORIs [46]. Two SVM parameters, namely kernel parameter 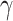 and penalty parameter C, have to be optimized during the training to achieve the best performance. We employed a grid search approach to optimize these parameters with the following search range: 2–15
and penalty parameter C, have to be optimized during the training to achieve the best performance. We employed a grid search approach to optimize these parameters with the following search range: 2–15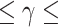 2−5 with a step size of −2 and 2–5
2−5 with a step size of −2 and 2–5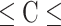 215 with a step size of 2. It is worth mentioning that our preliminary analysis showed that radial basis function achieved superior performance when compared with the other three kernel functions (Sigmoid, Polynomial and Linear). A brief description of the remaining classifier has been provided in previous studies [47–49], whose respective classifier tuning hyperparameters search range is provided in Table S1. Notably, 10-fold CV was employed for tuning ML parameters on training dataset and examined their performances.
215 with a step size of 2. It is worth mentioning that our preliminary analysis showed that radial basis function achieved superior performance when compared with the other three kernel functions (Sigmoid, Polynomial and Linear). A brief description of the remaining classifier has been provided in previous studies [47–49], whose respective classifier tuning hyperparameters search range is provided in Table S1. Notably, 10-fold CV was employed for tuning ML parameters on training dataset and examined their performances.
Iterative feature representation learning
We employed iterative feature representation learning to develop a robust prediction model. This approach has been widely applied in computational biology [32, 50]. Figure 1 shows the iterative feature learning framework that involves three sub-steps: (1) feature optimization; (2) probabilistic feature generation and (3) iterative feature generation, which are as follows:
Figure 1 .
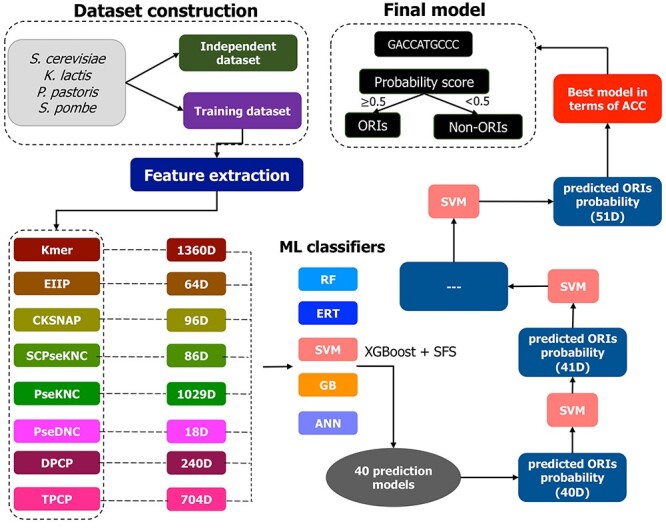
An overview of the species-specific yORIpred framework for predicting ORIs from yeast species. It involves the following steps: (i) the construction of the training and independent datasets, (ii) feature extraction by employing eight feature representation algorithms and (iii) iterative feature representation scheme that has three sub-steps. Finally, each sequence is assigned a score ranging from 0.0 to 1.0 by yORIpred. If the score > 0.5, it indicates that the sequence is predicted to be ORIs; otherwise, it is non-ORIs.
Feature optimization
Generally, original feature dimension may contain irrelevant information, which may lead to poor performance of prediction model and low model robustness [51–53]. Feature selection protocol is one of the important steps in developing prediction model to reduce or remove redundancy for improving the model performance and reducing the computational time. To include more relevant information from each descriptor, feature optimization was applied on seven feature encodings (Kmer, CKSNAP, PseKNC, SCPseKNC, EIIP, DPCP and TPCP) and excluded PseDNC (18D) due to its smaller feature dimension. A systematic two-step feature selection approach was applied to identify the optimal feature set from the original feature dimension, where the first step is to rank features followed by the sequential forward search (SFS). Generally, extreme GB (XGBoost) has been known as a prediction algorithm [54]. Besides prediction, it can effectively rank features according to the importance score, and it has been widely applied in various problems to identify the relevant feature set [55, 56]. Here, we employed XGBoost for ranking features and sorted them in descending order based on the score. Subsequently, SFS was applied to identify the optimal feature subset from each encoding subsets. In SFS, k (k = 2) features were added each time to five different ML classifiers and evaluated their performance using 10-fold CV. Ultimately, a feature set that achieved a superior performance in terms of accuracy (ACC) is considered as the optimal model for each classifier.
Generation of probabilistic features
From step 1, we obtained 40 optimal models (8 encodings 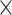 5 classifiers), whose predicted probability of ORIs were concatenated and considered as a 40D novel feature vector.
5 classifiers), whose predicted probability of ORIs were concatenated and considered as a 40D novel feature vector.
Iterative feature generation
Inspired by the layer-wise way of learning features in deep neural networks [57], we employed a similar strategy to develop a final prediction model. Here, we employed five different classifiers individually for iterative approach. The procedure is as follows: In the first run of the iterative strategy, 40D probabilistic feature vector obtained from step 2 inputted to ML classifier (e.g. SVM) and developed their corresponding model using 10-fold CV. The predicted probability of ORIs from the optimal model was considered as a new feature vector, which was combined with the previous 40D vector and obtained 41D feature vector. We repeat this process in an iterative manner and obtained 1D predicted probability of ORIs by training multidimensional features at each successive step, and then fusing both output and input characteristics as a input feature to the subsequent iteration process. This iterative approach will be terminated after 11 rounds to avoid over-fitting.
Feature fusion approach
The feature fusion approach is quite popular in bioinformatics and computational biology [58]. In addition to the iterative feature representation, we employed a feature fusion approach and evaluated whether this approach improves the prediction performance. Briefly, eight different sequence-based encoding vectors of Kmer, CKSNAP, PseKNC, PseDNC, SCPseKNC, EIIP, DPCP and TPCP were merged as follows:
 |
(19) |
FF is the sequential fusion that yielded 3597D features. Notably, fused features may contain irrelevant or mutual information that directly impacts performance. Therefore, we applied the two-step feature selection approach (as mentioned above) on FF by employing five different classifiers independently and developed their corresponding optimal model.
Evaluation metrics
To quantify the performance of developed models and evaluate among them, we employed five commonly used evaluation metrics [59–61]: sensitivity (Sn), specificity (Sp), ACC, Matthews correlation coefficient (MCC), balanced accuracy (BACC). The definition of each metric is expressed as:
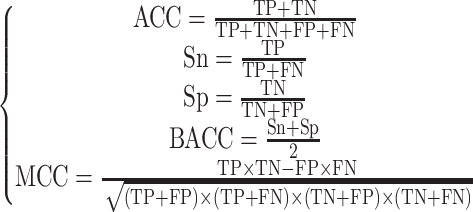 |
(20) |
where TP is the number of ORIs correctly predicted as ORIs, TN is the number of non-ORIs correctly predicted as non-ORIs, FP is the number of ORIs incorrectly predicted as non-ORIs and FN is the number of non-ORIs incorrectly predicted as ORIs, respectively. In addition to the above metrics, we employed receiver operating characteristic (ROC) curves that plot Sn as a function of (1 − Sp) with different decision thresholds. Subsequently, we computed area under the curve from ROC curve. Furthermore, a two-tailed test was employed to compute the statistical differences between two ROC curves [62].
Results and Discussion
Assessing the contribution of different feature descriptors and classifiers
We employed five different classifiers (ANN, RF, GB, ERT and SVM) and eight different feature encodings (Kmer, CKSNAP, DPCP, PseDNC, TPCP, PseKNC, SCPseKNC and EIIP) to assess their contribution in ORIs prediction for each yeast species. Conventionally, each encoding was inputted to five different classifiers and developed their respective prediction model using 10-fold CV. In total, 40 prediction models (8 encodings  5 ML classifiers) were developed for each species, whose performances are shown in Figure 2 and Tables S2–S5. Figure 2 shows that each classifier performance fluctuates for different feature encodings, where it shares similar MCC with the other classifiers, sometimes slightly superior MCC and marginally lower MCC. For instance, SVM-based performance in P. pastoris is outstanding using CKSNAP, Kmer and TPCP, similar to MCC with the different classifiers using DPCP, EIIP and SCPseTNC, while slightly deteriorating using PseDNC and PseKNC (Figure 2B). Parallelly, a similar phenomenon was observed in other three species (K. lactis, S. cerevisiae and S. pombe). It is worth mentioning that we did not observe any standout classifier that consistently performed well regardless of using eight different feature encodings.
5 ML classifiers) were developed for each species, whose performances are shown in Figure 2 and Tables S2–S5. Figure 2 shows that each classifier performance fluctuates for different feature encodings, where it shares similar MCC with the other classifiers, sometimes slightly superior MCC and marginally lower MCC. For instance, SVM-based performance in P. pastoris is outstanding using CKSNAP, Kmer and TPCP, similar to MCC with the different classifiers using DPCP, EIIP and SCPseTNC, while slightly deteriorating using PseDNC and PseKNC (Figure 2B). Parallelly, a similar phenomenon was observed in other three species (K. lactis, S. cerevisiae and S. pombe). It is worth mentioning that we did not observe any standout classifier that consistently performed well regardless of using eight different feature encodings.
Figure 2 .
Performance comparison of different classifiers trained using eight different feature types based on 10-fold cross-validation for four yeast species. (A) K. lactis, (B) P. pastoris, (C) S. cerevisiae and (D) S. pombe. Kmer, Kmer composition; CKSNAP, composition of k-spaced nucleic acid pairs; EIIP, electron-ion interaction pseudopotentials; DPCP, dinucleotide physicochemical properties; PseKNC, pseudo k-tuple composition; PseDNC, pseudo dinucleotide composition; SCPseTNC, series correlation pseudo trinucleotide composition; TPCP, trinucleotide physicochemical properties.
To obtain an overview performance of each classifier, we computed average performances of eight encodings as shown in Figure S1. Results indicate that RF, ERT and GB achieved similar performances with MCC in the range of 0.690–0.700, superior to SVM and ANN in K. lactis. In the case of S. cerevisiae, RF, ERT and SVM achieved similar performances with MCC in the range of 0.414–0.418, which is better than GB and ANN. Furthermore, SVM showed superior performance in the remaining two datasets. Overall, the above analysis indicates that the three classifiers’ (RF, ERT and SVM) performances are similar regardless of the datasets, while slightly superior to ANN and GB.
Feature optimization results via two-step feature selection
As described above, we obtained 40 baseline models and considered only 35 models for feature optimization, and excluded five models (five classifiers based on PseDNC) because of the lower feature dimension (18D). Firstly, we used XGBoost to predict the feature importance score for seven encodings separately and selected the non-zero importance score features. Figure 3A shows that feature encoding possesses less than 100 original dimension (CKSNAP, EIIP and SCPseTNC) and holds 86–100% non-zero feature importance score regardless of the datasets. Interestingly, features with higher-dimensions (Kmer, DPCP, PseKNC and TPCP) contained significantly reduced features containing non-zero feature values. Subsequently, we ranked these non-zero features according to their score and carried out SFS to identify the optimal feature set.
Figure 3 .
Effect of feature selection and optimization. (A) The percentage of features identified to be important by XGBoost for seven different encodings using four species-specific datasets. (B) Percentage of improvement in terms of accuracy compared between optimal feature set models and control. We considered only feature selection improved models and excluded deteriorated models from seven encodings. The number of improved models is mentioned in the figure.
Table S2-S5 shows the optimal performance of 35 models and their optimal feature set dimension for K. lactis, P. pastoris, S. cerevisiae and S. pombe. We observed that our feature selection approach improves the majority of the baseline models’ prediction performances compared with their control. At the same time, it also deteriorates some baseline model performances. For instance, the optimal feature sets of three encodings (CKSNAP, TPCP, SCPseTNC) performances for SVM is lower than their control in P. pastoris (Table S3). The deterioration of the model performance due to the following reasons: (i) all features in the original dimension is equally essential for model performance, mainly encodings with lower dimension and (ii) XGBoost excluded many features by assigning zero from the higher-dimensional feature that may include relevant feature information. Instead of detailing each optimal feature set based model on different datasets, we computed the average ACC for baseline models only whose performance improved by feature selection approach and compared with the respective control. Figure 3B shows that all five classifiers’ average improvement is similar to the range of 2.29–2.78% for the P. pastoris dataset. Notably, we observed a similar phenomenon for the S. pombe dataset. In the case of K. lactis, SVM improvement is immense, followed by RF and remaining three classifiers. In the case of S. cerevisiae, except ANN, the remaining four classifiers achieved similar enhancements. Overall, feature selection improved 31/35, 29/35, 30/35 and 27/35 models, respectively, for P. pastoris, K. lactis, S. cerevisiae and S. pombe. These improved models obtained from feature selection, PseDNC-based five models excluded from feature optimization, and models with original features for their deterioration during feature selection considered for the subsequently analysis.
Impact of different classifiers during iterative feature learning process
We obtained the predicted probability of ORI from 40 baseline models and considered the novel features fed to five different classifiers, and independently performed an iterative feature learning process. To intuitively examine the results, we plotted ACC change as incrementing the feature dimension at each iteration step (Figure 4). The result shows that RF, ERT, GB and ANN have a common trend for each species; performance remains in equilibrium (4A, C, E and G). However, SVM has a general tendency for two species (K. lactis and P. pastoris), ACC peaked at the beginning and converged to the steady-state (4A and C). In the case of S. cerevisiae, the ACC curve of SVM increased initially, peaked and assembled to a steady-state. Next, we compared the best models’ performances for three species (K. lactis, P. pastoris and S. cerevisiae). Figure 4B, D and F shows that SVM significantly outperformed the other classifiers in all five metrics. In the case of S. pombe, all classifiers achieved maximum ACC either in the first or second iteration, whose performances are similar (Figure 4H). Due to the excellent and consistent performance of SVM-based model in our iterative approach, we selected this as the final model for each species and named it as yORIpred.
Figure 4 .
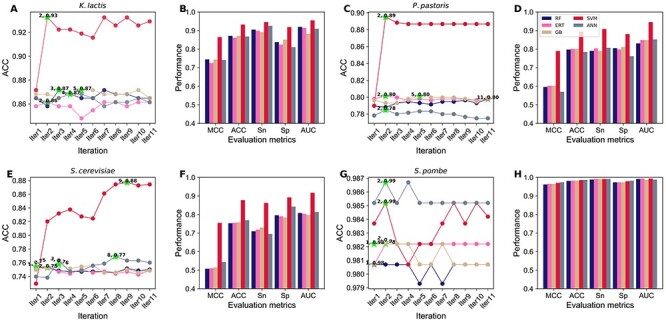
Performance comparisons of five different classifiers during iterative feature learning process. (A) Performance-based on K. lactis and the best performance is shown in green asterisk, and the comparison of the best model from five classifiers is shown in (B). (C) Performance-based on P. pastoris and the comparison of the best model from five classifiers is shown in (D). (E) Performance-based on S. cerevisiae and the comparison of the best model from five classifiers is shown in (F). (G) Performance-based on S. pombe and the comparison of the best model from five classifiers is shown in (H).
SVM-based iterative feature representation learning strategy significantly improved the performance when compared with their counterparts. However, it is unclear whether simply the classifier choice played a key role or some other hidden factors. Hence, we carried out two different analyses for three species (K. lactis, P. pastoris and S. cerevisiae) to understand this event: (1) SVM was used to assess the performance based on other classifiers’ probabilistic features at each iteration. SVM performance topology was identified to be similar to the four classifiers (data not shown), indicating features generated by four classifiers (using iterative approach) showed a moderate discriminative capability between ORIs and non-ORIs. (2) SVM-based probabilistic features were inputted into four classifiers and evaluated their performance at each iteration. Figure 5 shows that all four classifiers ACC graph topology is identical to SVM regardless of the species, thus indicating that SVM-based generated predicted probability values at each iteration step possess a very high discriminative capability between ORIs and non-ORIs. As a result, not only SVM but also other methods performed exceptionally well. Overall, the above analysis shows that the classifier’s choice (SVM) to generate probabilistic features (during the iterative approach) plays a significant role in yORIpred-improved performance.
Figure 5 .
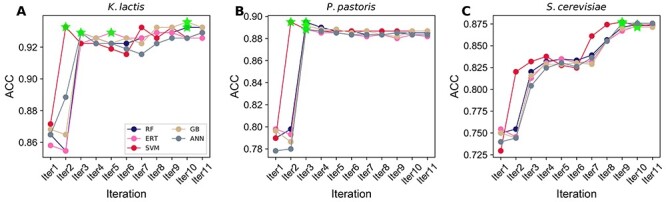
Performance comparison of SVM with the other four classifiers using the SVM generated predicted probability features. A classifier that reached the maximum accuracy is shown in green asterisk. (A) K. lactis, (B) P. pastoris and (C) S. cerevisiae.
Comparison of yORIpred performance with the feature fusion approach
Recently, several studies demonstrated the advantage of feature fusion approaches in several prediction problems [56, 63]. Hence, we applied the feature fusion method to investigate whether it can enhance the prediction performance compared with the iterative feature learning approach. Precisely, all eight different encodings combined linearly and generated a 3597D feature vector. For each species, firstly, we ranked features with non-zero scores identified by XGBoost and subsequently applied SFS by employing five different classifiers (as mentioned above) and identified the corresponding optimal model. Figure 6 shows the performance of five different classifiers for each species. Interestingly, three classifiers’ (RF, ERT and GB) ACC curve topology are similar to each other regardless of the species. However, ANN and SVM have a different ACC curve topology. The maximum ACC achieved by the optimal feature set is shown in a green asterisk. Next, we compared the optimal models obtained from feature fusion with yORIpred, and the result is shown in Table 1, where the feature fusion model is named with the prefix FF. The result shows that yORIpred performance is consistently better than feature fusion models. To get a statistical significance between yORIpred and other methods, we compared two AUC values and computed P-value using a two-tailed test [62]. By applying a P-value threshold of 0.05, yORIpred significantly outperformed all feature fusion models for two species (P. pastoris and S. cerevisiae). In the case of K. lactis, yORIpred performed better than FF-RF, FF-ERT and FF-GB and significantly outperformed the other two models. Furthermore, yORIpred performance is similar to other FF models in S. pombe. Altogether, the above comparative analysis highlights that the iterative learning approach is the sole reason for the yORIpred-improved performance.
Figure 6 .
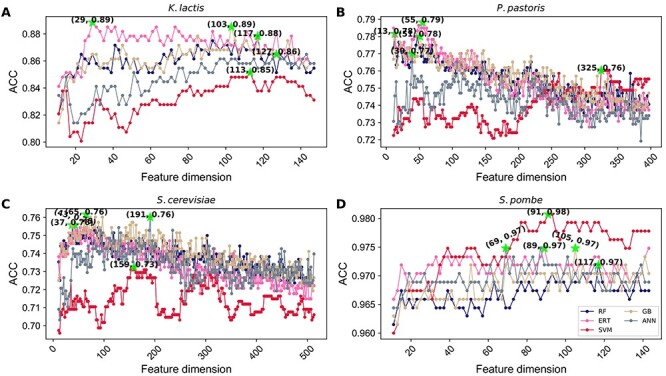
Selection of the optimal feature set based models for five different classifiers from the feature fusion. The relationship curve of prediction accuracy and dimension of feature subset. The curve in this figure reflects the change of predictor accuracy with the varying feature subset dimension. The selected model for each classifier is mentioned in the green asterisk. (A) K. lactis, (B) P. pastoris and (C) S. cerevisiae and (D) S. pombe.
Table 1.
Performance comparison of yORIpred with the predictors based on feature fusion approach
| Species | Method | MCC | ACC | Sn | Sp | AUC | P-value |
|---|---|---|---|---|---|---|---|
| K. lactis | yORIpred | 0.865 | 0.932 | 0.946 | 0.919 | 0.955 | — |
| FF-RF | 0.757 | 0.878 | 0.899 | 0.858 | 0.937 | 0.353 | |
| FF-ERT | 0.779 | 0.889 | 0.919 | 0.858 | 0.939 | 0.404 | |
| FF-GB | 0.770 | 0.885 | 0.885 | 0.885 | 0.932 | 0.246 | |
| FF-ANN | 0.730 | 0.865 | 0.878 | 0.851 | 0.897 | 0.010 | |
| FF-SVM | 0.703 | 0.851 | 0.872 | 0.831 | 0.913 | 0.049 | |
| P. pastoris | yORIpred | 0.790 | 0.895 | 0.909 | 0.881 | 0.946 | — |
| FF-RF | 0.563 | 0.782 | 0.781 | 0.782 | 0.828 | <0.0001 | |
| FF-ERT | 0.577 | 0.788 | 0.778 | 0.799 | 0.833 | <0.0001 | |
| FF-GB | 0.560 | 0.780 | 0.778 | 0.782 | 0.818 | <0.0001 | |
| FF-ANN | 0.540 | 0.770 | 0.775 | 0.766 | 0.820 | <0.0001 | |
| FF-SVM | 0.521 | 0.760 | 0.752 | 0.769 | 0.799 | <0.0001 | |
| S. cerevisiae | yORIpred | 0.755 | 0.877 | 0.862 | 0.892 | 0.917 | — |
| FF-SVM | 0.513 | 0.756 | 0.719 | 0.793 | 0.812 | <0.0001 | |
| FF-RF | 0.513 | 0.756 | 0.724 | 0.787 | 0.812 | <0.0001 | |
| FF-ERT | 0.524 | 0.762 | 0.745 | 0.778 | 0.809 | <0.0001 | |
| FF-GB | 0.521 | 0.760 | 0.736 | 0.784 | 0.802 | <0.0001 | |
| FF-ANN | 0.465 | 0.733 | 0.707 | 0.758 | 0.788 | <0.0001 | |
| S. pombe | yORIpred | 0.970 | 0.985 | 0.991 | 0.979 | 0.994 | — |
| FF-SVM | 0.944 | 0.972 | 0.988 | 0.955 | 0.989 | 0.322 | |
| FF-RF | 0.950 | 0.975 | 0.994 | 0.955 | 0.994 | 1.0 | |
| FF-ERT | 0.950 | 0.975 | 0.988 | 0.961 | 0.982 | 0.045 | |
| FF-GB | 0.950 | 0.975 | 0.988 | 0.961 | 0.984 | 0.082 | |
| FF-ANN | 0.962 | 0.981 | 0.994 | 0.967 | 0.994 | 1.0 |
The first and second columns, respectively, represent species and its corresponding prediction method. A P-value <0.05 was considered to indicate a statistically significant difference between yORIpred and the selected method (shown in italic).
Performance comparison of yORIpred with the existing predictors on the same training dataset
We compared the performance of yORIpred with the existing methods, namely iRO-3wPseKNC and iRO-PsekGCC, where both methods were developed using the same training datasets and, respectively, contained four and two species-specific prediction models. Generally, comparing the CV or independent performances using the same dataset is a more objective approach that avoids bias. Table 2 shows the performance comparison results between yORIpred and the existing predictors revealing a better predictive performance by yORIpred than other predictors in terms of MCC, ACC, Sn, Sp and AUC. Specifically, the MCC achieved by yORIpred was 16.2% higher than iRO-3wPseKNC for K. lactis; 31.0–37.0% higher than two existing methods for P. pastoris; 22.5–29.6% higher than two current methods for S. cerevisiae and 4.0% higher than iRO-3wPseKNC for S. pombe.
Table 2.
Performance comparison of yORIpred with the existing methods on the same training dataset
| Species | Method | MCC | ACC | Sn | Sp | AUC | P-value |
|---|---|---|---|---|---|---|---|
| K. lactis | yORIpred | 0.865 | 0.932 | 0.946 | 0.919 | 0.955 | — |
| iRO-3wPseKNC | 0.703 | 0.851 | 0.858 | 0.845 | 0.901 | 0.0158 | |
| P. pastoris | yORIpred | 0.790 | 0.895 | 0.909 | 0.881 | 0.946 | — |
| iRO-3wPseKNC | 0.422 | 0.711 | 0.699 | 0.723 | 0.796 | <0.001 | |
| iRO-PseKGCC | 0.484 | 0.742 | 0.745 | 0.739 | 0.800 | <0.001 | |
| S. cerevisiae | yORIpred | 0.755 | 0.877 | 0.862 | 0.892 | 0.917 | — |
| iRO-3wPseKNC | 0.459 | 0.730 | 0.707 | 0.752 | 0.808 | <0.001 | |
| iRO-PseKGCC | 0.530 | 0.765 | 0.739 | 0.781 | 0.813 | <0.001 | |
| S. pombe | yORIpred | 0.970 | 0.985 | 0.991 | 0.979 | 0.994 | — |
| iRO-3wPseKNC | 0.929 | 0.965 | 0.979 | 0.949 | 0.986 | 0.144 |
The first and second columns, respectively, represent species and its corresponding prediction method. A P-value <0.05 was considered to indicate a statistically significant difference between yORIpred and the selected method (shown in italic).
By applying a statistical cut-off of 0.05, yORIpred significantly outperformed iRO-3wPseKNC for three species, namely K. lactis, P. pastoris and S. cerevisiae and slightly better than iRO-3wPseKNC for S. pombe. Furthermore, yORIpred significantly outperformed iRO-PseKGCC for both species. It should be noted that the same RF classifier with different feature encodings employed in the existing methods. As a result, it could not capture the pattern that discriminates well between ORIs and non-ORIs. However, our approach overcomes the limitations of the existing methods and the feature fusion approach employed in this study and showed excellent performance. The possible reason for yORIpred superior performance includes: (i) integrating different classifiers and feature encoding schemes and generating probabilistic features; (ii) systematic evaluation of different classifiers during iterative feature learning and selecting SVM and (iii) probability features generated by SVM shows a very high discriminative capability of ORIs from non-ORIs.
Performance validation on the independent dataset
We compared the predictive performance of yORIpred against the existing methods using the independent dataset for S. cerevisiae, which includes 67 ORIs and 837 non-ORIs. Since the dataset is imbalanced, comparison using ACC is not straightforward; hence, we used BACC to rank the methods as shown in Table 3. The comparison results revealed that yORIpred achieved the best performance with MCC and BACC of 0.583 and 0.879. Specifically, MCC performed by yORIpred was 6.4% higher than the second-best method, iRO-3wPseKNC. Among the existing methods, iRO-PseKGCC performance was low in our evaluation. Overall, yORIpred not only performed well on the training dataset but also replicated its CV performances during independent evaluation, thus highlighting its stability and robustness in ORIs prediction.
Table 3.
Performance comparison of various methods on S. cerevisiae independent dataset
| Method | MCC | BACC | Sn | Sp | AUC |
|---|---|---|---|---|---|
| yORIpred | 0.583 | 0.879 | 0.836 | 0.922 | 0.915 |
| iRO-3wPseKNC | 0.519 | 0.838 | 0.761 | 0.915 | NA |
| iRO-PseKGCC | −0.013 | 0.490 | 0.746 | 0.233 | NA |
The first column represents the method employed for the evaluation. iRO-3wPseKNC and iRO-PseKGCC did not give predicted probability values during our evaluation. Hence, we cannot provide an AUC value. It is mentioned as not available (NA).
Web server development
As an implementation of the developed yORIpred approach, we have made available an online web server version of yORIpred at http://thegleelab.org/yORIpred. The web server is equipped with 16 cores, 64 GB memory and a 2 TB hard disk. To utilize the yORIpred web server, users should select particular species from the homepage and input DNA sequences in the textbox or upload sequences in Fast Adaptive Shrinkage Threshold Algorithm (FASTA) format via the file-selection dialog box. The prediction results are provided in table format with detailed information about the serial number, FASTA ID, predicted class (ORIs or non-ORIs) and predicted probability of ORIs. Notably, the probability score of ORIs is in the range from 0 to 1, where a probability score close to 1 means the result is likely to be an ORI, and a score close to 0 means the result is unlikely to be an ORI. More detailed instructions for using the yORIpred web server can be found at the web server’s README page.
Limitations and future work
Despite the performance of yORIpred for predicting species-specific ORIs from yeast species, it has the following limitations: (i) The training dataset of yORIpred is relatively limited (especially for S. pombe) due to the lack of experimentally characterized ORIs. In the future, when more ORI sequences become available, additional data should be collected for the development of a more robust model and enable reliable whole-genome ORIs annotation; (ii) The existing methods employed random sequences as non-ORIs during their model construction. As a result, the prediction performance might be affected when the user provides other functional DNA sequences (promoter and coding sequence). In the future, DNA sequences other than ORI functions should be considered along with the random sequences as non-ORIs during model development, which will be helpful to eliminate the false-positive results; and (iii) yORIpred predictive capability relies on multiple ML classifiers trained on numerous sequence-derived features. Generally, the machine-learning models’ performance is directly proportional to the informative features extracted from the training dataset. In this regard, it is challenging to find a novel sequence-based encoding scheme to improve model performance. With more data available in the future, we plan to apply several classical approaches reported recently [63–67] and identify the most appropriate method.
Conclusions
Here, we proposed a novel species-specific yORIpred predictor for accurate identification of ORI sites from yeast species. To establish an accurate and efficient prediction model, we developed 40 baseline models by exploring various feature encodings and classical ML classifiers. Subsequently, a 40D probabilistic feature vectors generated from the baseline models were fed into SVM and an iterative feature representation learning scheme was applied to create more informative features. Our empirical studies based on CV and independent evaluation demonstrated the effectiveness of yORIpred species-specific models by outperforming the existing methods, iRO-3wPseKNC and iRO-PsekGCC. Furthermore, we demonstrated the superiority of yORIpred compared with the feature fusion approach. The improved performance of yORIpred is mainly due to the following aspects: (i) integrating different classifiers and feature encoding schemes to generate probabilistic features, (ii) identifying an appropriate SVM classifier during iterative feature learning and (iii) probabilistic features obtained through the iterative approach has a very high discrimination capacity of ORIs from non-ORIs. A user-friendly web server has been established that allows the prediction of ORIs from the given genomic sequences, which will significantly enhance its impact in driving genome biology. We anticipate that yORIpred could be a powerful tool for accurate and high-throughput ORIs prediction from DNA sequences. Moreover, the current approach could be extended to other DNA sequence-based prediction problems, such as post replication modification sites, recombination hot-spot and enhancer predictions.
Key Points
In this study, we present yORIpred, a powerful bioinformatics tool for the accurate prediction of species-specific yeast ORIs.
yORIpred utilized eight different feature encodings schemes to encode the sequences and integrated with iterative feature representation algorithm to build the stable predictor.
Benchmarking comparison shows that yORIpred significantly outperforms the state-of-the-art predictors.
A user-friendly web server is available (http://thegleelab.org/yORIpred) to facilitate online high-throughput prediction of ORIs.
Supplementary Material
Funding
This work is supported by the Basic Science Research Program through the National Research Foundation of Korea funded by the Ministry of Science and ICT [2018R1D1A1B07049572, 2016M3C7A1904392, 2019R1I1A1A01062260, 2020R1A4A4079722, 2020M3E5D9080661].
Balachandran Manavalan is a research professor at the Department of Physiology, Ajou University School of Medicine, Republic of Korea. He is also an associate member of KIAS, Republic of Korea. His main research interests include prediction of protein structures, machine learning, data mining, computational biology and functional genomics.
Shaherin Basith is a research assistant professor in the Department of Physiology, Ajou University School of Medicine, Republic of Korea. Her main area of research focuses on exploring the structure–function relationships of proteins using state-of-the-art molecular modeling tools, phylogenetic analysis and biomolecular simulations. She is also actively involved in the application of machine learning tools for peptide and small molecule drug discovery.
Tae Hwan Shin is a research fellow in the Department of Physiology, Ajou University School of Medicine, Republic of Korea. His main research interests include nanotoxicology, integration of triple omics (transcriptomics, metabolomics and proteomics), Parkinson’s disease, human mesenchymal stem cells and machine learning.
Gwang Lee is a professor in the Department of Physiology, Ajou University School of Medicine, Republic of Korea. His main research interests include the integration of triple omics (transcriptomics, metabolomics and proteomics) for nanotoxicity studies, machine learning, neurodegenerative diseases and biomolecular simulations targeting therapeutically important proteins or enzymes.
Contributor Information
Balachandran Manavalan, Department of Physiology, Ajou University School of Medicine, Republic of Korea.
Shaherin Basith, Department of Physiology, Ajou University School of Medicine, Republic of Korea.
Tae Hwan Shin, Department of Physiology, Ajou University School of Medicine, Republic of Korea.
Gwang Lee, Department of Physiology, Ajou University School of Medicine, Republic of Korea.
Data Availability
Training and independent datasets used in this study could be freely downloaded using the below link: http://thegleelab.org/yORIpred/yORIData.html.
Conflict of Interest
The authors declare that they have no competing interests.
References
- 1. Watson JD, Crick FH. Molecular structure of nucleic acids; a structure for deoxyribose nucleic acid. Nature 1953;171:737–8. [DOI] [PubMed] [Google Scholar]
- 2. Mott ML, Berger JM. DNA replication initiation: mechanisms and regulation in bacteria. Nat Rev Microbiol 2007;5:343–54. [DOI] [PubMed] [Google Scholar]
- 3. Skarstad K, Katayama T. Regulating DNA replication in bacteria. Cold Spring Harb Perspect Biol 2013;5:a012922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bogenschutz NL, Rodriguez J, Tsukiyama T. Initiation of DNA replication from non-canonical sites on an origin-depleted chromosome. PLoS One 2014;9:e114545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Song C, Zhang S, Huang H. Choosing a suitable method for the identification of replication origins in microbial genomes. Front Microbiol 2015;6:1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lee DG, Bell SP. Architecture of the yeast origin recognition complex bound to origins of DNA replication. Mol Cell Biol 1997;17:7159–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lou C, Zhao J, Shi R, et al. sefOri: selecting the best-engineered sequence features to predict DNA replication origins. Bioinformatics 2020;36:49–55. [DOI] [PubMed] [Google Scholar]
- 8. Liachko I, Bhaskar A, Lee C, et al. A comprehensive genome-wide map of autonomously replicating sequences in a naive genome. PLoS Genet 2010;6:e1000946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Biswas SB, Khopde SM, Biswas-Fiss EE. Control of ATP-dependent binding of Saccharomyces cerevisiae origin recognition complex to autonomously replicating DNA sequences. Cell Cycle 2005;4:494–500. [DOI] [PubMed] [Google Scholar]
- 10. Nieduszynski CA, Knox Y, Donaldson AD. Genome-wide identification of replication origins in yeast by comparative genomics. Genes Dev 2006;20:1874–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Khodursky AB, Peter BJ, Cozzarelli NR, et al. DNA microarray analysis of gene expression in response to physiological and genetic changes that affect tryptophan metabolism in Escherichia coli. Proc Natl Acad Sci 2000;97:12170–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Raghuraman M, Winzeler EA, Collingwood D, et al. Replication dynamics of the yeast genome. Science 2001;294:115–21. [DOI] [PubMed] [Google Scholar]
- 13. Gao F, Zhang CT. Ori-finder: a web-based system for finding oriCs in unannotated bacterial genomes. BMC Bioinformatics 2008;9:79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Luo H, Zhang CT, Gao F. Ori-finder 2, an integrated tool to predict replication origins in the archaeal genomes. Front Microbiol 2014;5:482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Roten CA, Gamba P, Barblan JL, et al. Comparative genometrics (CG): a database dedicated to biometric comparisons of whole genomes. Nucleic Acids Res 2002;30:142–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Thomas JM, Horspool D, Brown G, et al. GraphDNA: a java program for graphical display of DNA composition analyses. BMC Bioinformatics 2007;8:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chen W, Feng P, Lin H. Prediction of replication origins by calculating DNA structural properties. FEBS Lett 2012;586:934–8. [DOI] [PubMed] [Google Scholar]
- 18. Wen-Chao Li E-ZD, Ding H, Chen W, et al. iORI-PseKNC: a predictor for identifying origin of replication with pseudo k-tuple nucleotide composition. Chemom Intel Lab Syst 2015;141:100–6. [Google Scholar]
- 19. Zhang CJ, Tang H, Li WC, et al. iOri-human: identify human origin of replication by incorporating dinucleotide physicochemical properties into pseudo nucleotide composition. Oncotarget 2016;7:69783–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Xiao X, Ye HX, Liu Z, et al. iROS-gPseKNC: predicting replication origin sites in DNA by incorporating dinucleotide position-specific propensity into general pseudo nucleotide composition. Oncotarget 2016;7:34180–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chou KC. Some remarks on protein attribute prediction and pseudo amino acid composition. J Theor Biol 2011;273:236–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Liu B, Weng F, Huang DS, et al. iRO-3wPseKNC: identify DNA replication origins by three-window-based PseKNC. Bioinformatics 2018;34:3086–93. [DOI] [PubMed] [Google Scholar]
- 23. Liu B, Chen S, Yan K, et al. iRO-PsekGCC: identify DNA replication origins based on pseudo k-tuple GC composition. Front Genet 2019;10:842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Dao FY, Lv H, Wang F, et al. Identify origin of replication in Saccharomyces cerevisiae using two-step feature selection technique. Bioinformatics 2019;35:2075–83. [DOI] [PubMed] [Google Scholar]
- 25. Do DT, Le NQK. Using extreme gradient boosting to identify origin of replication in Saccharomyces cerevisiae via hybrid features. Genomics 2020;112:2445–51. [DOI] [PubMed] [Google Scholar]
- 26. Dao FY, Lv H, Zulfiqar H, et al. A computational platform to identify origins of replication sites in eukaryotes. Brief Bioinform 2020. doi: 10.1093/bib/bbaa017. [DOI] [PubMed] [Google Scholar]
- 27. Wang D, Lai FL, Gao F. Ori-finder 3: a web server for genome-wide prediction of replication origins in Saccharomyces cerevisiae. Brief Bioinform 2020. doi: 10.1093/bib/bbaa182. [DOI] [PubMed] [Google Scholar]
- 28. Yang H, Yang W, Dao FY, et al. A comparison and assessment of computational method for identifying recombination hotspots in Saccharomyces cerevisiae. Brief Bioinform 2019. doi: 10.1093/bib/bbz123. [DOI] [PubMed] [Google Scholar]
- 29. Du P, Wang X, Xu C, et al. PseAAC-builder: a cross-platform stand-alone program for generating various special Chou's pseudo-amino acid compositions. Anal Biochem 2012;425:117–9. [DOI] [PubMed] [Google Scholar]
- 30. Zhang ZY, Yang YH, Ding H, et al. Design powerful predictor for mRNA subcellular location prediction in Homo sapiens. Brief Bioinform 2020. doi: 10.1093/bib/bbz177. [DOI] [PubMed] [Google Scholar]
- 31. Lv H, Dao F-Y, Zhang D, et al. iDNA-MS: an integrated computational tool for detecting DNA modification sites in multiple genomes. iScience 2020;23(4):100991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Manavalan B, Basith S, Shin TH, et al. 4mCpred-EL: an ensemble learning framework for identification of DNA N4-Methylcytosine sites in the mouse genome. Cell 2019;8:1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Manavalan B, Basith S, Shin TH, et al. Meta-4mCpred: a sequence-based meta-predictor for accurate DNA 4mC site prediction using effective feature representation. Molecular Therapy-Nucleic Acids 2019;16:733–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hasan MM, Manavalan B, Khatun MS, et al. i4mC-ROSE, a bioinformatics tool for the identification of DNA N4-methylcytosine sites in the Rosaceae genome. Int J Biol Macromol 2019;157:752–8. [DOI] [PubMed] [Google Scholar]
- 35. Hasan MM, Manavalan B, Shoombuatong W, et al. i6mA-fuse: improved and robust prediction of DNA 6 mA sites in the Rosaceae genome by fusing multiple feature representation. Plant Mol Biol 2020;103(1-2):225–34. [DOI] [PubMed] [Google Scholar]
- 36. Liu K, Chen W. iMRM: a platform for simultaneously identifying multiple kinds of RNA modifications. Bioinformatics 2020;36(11):3336–42. [DOI] [PubMed] [Google Scholar]
- 37. Lai HY, Zhang ZY, Su ZD, et al. iProEP: a computational predictor for predicting promoter. Mol Ther Nucleic Acids 2019;17:337–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Feng CQ, Zhang ZY, Zhu XJ, et al. iTerm-PseKNC: a sequence-based tool for predicting bacterial transcriptional terminators. Bioinformatics 2019;35:1469–77. [DOI] [PubMed] [Google Scholar]
- 39. Hasan MM, Schaduangrat N, Basith S, et al. HLPpred-fuse: improved and robust prediction of hemolytic peptide and its activity by fusing multiple feature representation. Bioinformatics 2020;36(11):3350–6. [DOI] [PubMed] [Google Scholar]
- 40. Yang W, Zhu XJ, Huang J, et al. A brief survey of machine learning methods in protein sub-Golgi localization. Current Bioinformatics 2019;14:234–40. [Google Scholar]
- 41. Tan JX, Li SH, Zhang ZM, et al. Identification of hormone binding proteins based on machine learning methods. Math Biosci Eng 2019;16:2466–80. [DOI] [PubMed] [Google Scholar]
- 42. Charoenkwan P, Shoombuatong W, Lee H-C, et al. SCMCRYS: predicting protein crystallization using an ensemble scoring card method with estimating propensity scores of P-collocated amino acid pairs. PloS one 2013;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Schaduangrat N, Nantasenamat C, Prachayasittikul V, et al. ACPred: a computational tool for the prediction and analysis of anticancer peptides. Molecules 2019;24:1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Schaduangrat N, Nantasenamat C, Prachayasittikul V, et al. Meta-iAVP: a sequence-based meta-predictor for improving the prediction of antiviral peptides using effective feature representation. Int J Mol Sci 2019;20:5743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Shoombuatong W, Schaduangrat N, Pratiwi R, et al. THPep: a machine learning-based approach for predicting tumor homing peptides. Comput Biol Chem 2019;80:441–51. [DOI] [PubMed] [Google Scholar]
- 46. Chang C-C, Lin C-J. LIBSVM: a library for support vector machines. ACM TIST 2011;2:1–27. [Google Scholar]
- 47. Basith S, Manavalan B, Shin TH, et al. SDM6A: a web-based integrative machine-learning framework for predicting 6mA sites in the Rice genome. Mol Ther Nucleic Acids 2019;18:131–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wang X, Li C, Li F, et al. SIMLIN: a bioinformatics tool for prediction of S-sulphenylation in the human proteome based on multi-stage ensemble-learning models. BMC Bioinformatics 2019;20:602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Hasan MM, Basith S, Khatun MS, et al. Meta-i6mA: an interspecies predictor for identifying DNA N6-methyladenine sites of plant genomes by exploiting informative features in an integrative machine-learning framework. Brief Bioinform 2020. doi: 10.1093/bib/bbaa202. [DOI] [PubMed] [Google Scholar]
- 50. Xu Y, Zhao X, Liu Set al. LncPred-IEL: a long non-coding RNA prediction method using iterative ensemble learning. In: 2019 IEEE International Conference on Bioinformatics and Biomedicine (BIBM). 2019, p. 555–62. IEEE. [Google Scholar]
- 51. Basith S, Manavalan B, Shin TH, et al. iGHBP: computational identification of growth hormone binding proteins from sequences using extremely randomised tree. Comput Struct Biotechnol J 2018;16:412–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Manavalan B, Basith S, Shin TH, et al. AtbPpred: a robust sequence-based prediction of anti-tubercular peptides using extremely randomized trees. Comput Struct Biotechnol J 2019;17:972–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Charoenkwan P, Schaduangrat N, Nantasenamat C, et al. iQSP: a sequence-based tool for the prediction and analysis of quorum sensing peptides via Chou's 5-steps rule and informative physicochemical properties. Int J Mol Sci 2019;21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Yu B, Qiu W, Chen C, et al. SubMito-XGBoost: predicting protein submitochondrial localization by fusing multiple feature information and eXtreme gradient boosting. Bioinformatics 2020;36:1074–81. [DOI] [PubMed] [Google Scholar]
- 55. Jia C, Bi Y, Chen J, et al. PASSION: an ensemble neural network approach for identifying the binding sites of RBPs on circRNAs. Bioinformatics 2020;36(15):4276–82. [DOI] [PubMed] [Google Scholar]
- 56. Elbasir A, Mall R, Kunji K, et al. BCrystal: an interpretable sequence-based protein crystallization predictor. Bioinformatics 2020;36:1429–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Zhou Z-H, Feng J. Deep forest: towards an alternative to deep neural networks, arXiv preprint arXiv:1702.08835. 2017.
- 58. Cai J, Wang D, Chen R, et al. A bioinformatics tool for the prediction of DNA N6-Methyladenine modifications based on feature fusion and optimization protocol. Front Bioeng Biotechnol 2020;8:502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Basith S, Manavalan B, Hwan Shin T, et al. Machine intelligence in peptide therapeutics: a next-generation tool for rapid disease screening. Med Res Rev 2020;40(4):1276–1314. [DOI] [PubMed] [Google Scholar]
- 60. Su R, Hu J, Zou Q, et al. Empirical comparison and analysis of web-based cell-penetrating peptide prediction tools. Brief Bioinform 2019;21(2):408–420. [DOI] [PubMed] [Google Scholar]
- 61. Khanal J, Tayara H, Chong KT. Identifying enhancers and their strength by the integration of word embedding and convolution neural network. IEEE Access 2020;8:58369–76. [Google Scholar]
- 62. Hanley JA, McNeil BJ. The meaning and use of the area under a receiver operating characteristic (ROC) curve. Radiology 1982;143:29–36. [DOI] [PubMed] [Google Scholar]
- 63. Li F, Chen J, Ge Z, et al. Computational prediction and interpretation of both general and specific types of promoters in Escherichia coli by exploiting a stacked ensemble-learning framework. Brief Bioinform 2020. doi: 10.1093/bib/bbaa049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Xie R, Li J, Wang J, et al. DeepVF: a deep learning-based hybrid framework for identifying virulence factors using the stacking strategy. Brief Bioinform 2020. doi: 10.1093/bib/bbaa125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Song J, Wang Y, Li F, et al. iProt-sub: a comprehensive package for accurately mapping and predicting protease-specific substrates and cleavage sites. Brief Bioinform 2019;20:638–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Liu Q, Chen J, Wang Y, et al. DeepTorrent: a deep learning-based approach for predicting DNA N4-methylcytosine sites. Brief Bioinform 2020. doi: 10.1093/bib/bbaa124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Wei L, He W, Malik A, et al. Computational prediction and interpretation of cell-specific replication origin sites from multiple eukaryotes by exploiting stacking framework. Brief Bioinform 2020. doi: 10.1093/bib/bbaa275. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Training and independent datasets used in this study could be freely downloaded using the below link: http://thegleelab.org/yORIpred/yORIData.html.