

Abstract
Current variant calling (VC) approaches have been designed to leverage populations of long-range haplotypes and were benchmarked using populations of European descent, whereas most genetic diversity is found in non-European such as Africa populations. Working with these genetically diverse populations, VC tools may produce false positive and false negative results, which may produce misleading conclusions in prioritization of mutations, clinical relevancy and actionability of genes. The most prominent question is which tool or pipeline has a high rate of sensitivity and precision when analysing African data with either low or high sequence coverage, given the high genetic diversity and heterogeneity of this data. Here, a total of 100 synthetic Whole Genome Sequencing (WGS) samples, mimicking the genetics profile of African and European subjects for different specific coverage levels (high/low), have been generated to assess the performance of nine different VC tools on these contrasting datasets. The performances of these tools were assessed in false positive and false negative call rates by comparing the simulated golden variants to the variants identified by each VC tool. Combining our results on sensitivity and positive predictive value (PPV), VarDict [PPV = 0.999 and Matthews correlation coefficient (MCC) = 0.832] and BCFtools (PPV = 0.999 and MCC = 0.813) perform best when using African population data on high and low coverage data. Overall, current VC tools produce high false positive and false negative rates when analysing African compared with European data. This highlights the need for development of VC approaches with high sensitivity and precision tailored for populations characterized by high genetic variations and low linkage disequilibrium.
Keywords: DNA sequence, next-generation sequence, simulation, variant calling, genomics
Introduction
Over the past decade, the whole human genome DNA sequencing revolution has improved our understanding of the human genome and impacted modern biological science [1]. This revolution has resulted in a reduction in DNA sequencing costs and parallelism of throughput, thanks to next-generation sequencing (NGS) [2]. Furthermore, this revolution has led to improved understanding of disease aetiology, diagnosis management, treatment and planning for both communicable and non-communicable diseases. One of the essential steps in the downstream analysis of NGS data is variant calling (VC), which is the process of identifying variants that are different from the chosen reference sample. These variants may have different impacts on the molecular biology of the system and subsequently diseases, so it is essential to identify the true variant by using a precise VC tool. This is considered a challenge as there are many VC tools with different underlying algorithms, such as heuristic and statistical approaches, which subsequently produce different results, which are almost never perfect [3, 4].
Several previous studies have evaluated VC tools on populations of European descent. To the best of our knowledge, none of these studies have evaluated the performance of VC tools using data from African populations. Most of these studies showed that the tools were able to detect true variants with high accuracy and specificity, even when VC tools performance was assessed using data with differing characteristics such as whole genome, whole exome, somatic variants or germline variants and with other parameters. Furthermore, many authors compared their tools using data with different depths of read coverage, as this affects the detection of variants from NGS data, and found that the higher the read depth coverage, the more confident the base calls, and hence these calls were more distinguishable from sequencing errors [5]. Subsequently, many studies use high coverage read sequence depth as it improves the accuracy of VC, whereas others consider variant filtering to be a suggested step as it could improve the specificity and sensitivity and reduce false positive (FP) rate [6]. Numerous have benchmarked VC tools using the set of NA12878 Genome in a Bottle high-confidence GRCh37 variants as a gold standard reference set [1, 7, 8], whereas other studies used simulated data as a gold standard. Further studies are summarized in Supplementary Tables S1 and S2.
Despite the advancements of NGS and the downstream analysis tools of genomic data, the downstream analysis process is still an important bottleneck [3, 9–12]. Harbouring the highest genetic diversity, African populations are disproportionately underrepresented in public databases and reference panels [13, 14]. As mentioned earlier, current VC approaches (Supplementary Tables S1 and S2) have been designed to leverage populations of long-range haplotypes and were benchmarked using populations of European descent [13, 15, 16], even though most genetic diversity, with high rates of heterogeneity, is found in non-European populations such as African populations. These VC tools may produce FP and false negative (FN) results, which can lead to misleading conclusions in prioritization of mutations, clinical relevancy and actionability of variants. In addition, because non-European, particularly African genomes, harbour the highest genetic diversity alongside a low level of linkage disequilibrium compared with other populations [17–20], differences in genetic characteristics as mentioned above can significantly affect the performance of not only the variant discovery tools, but also downstream bioinformatics analysis tools. Another concern with VC in populations of non-European descent is the use of inappropriate reference samples, which leads to an increased rate of FP and FN single nucleotide polymorphisms (SNPs).
Here, we leverage a total of 100 synthetic African and European WGS samples that mimic the genetic profiles of African and European subjects for different sample coverage levels (high and low) to benchmark the nine state-of-the-art VC tools (Supplementary Table S2), namely, VarScan2 [21], Samtools [22], genomic analysis toolkit (GATK)-HaplotypeCaller (GATK-HC) [23], SNVer [24], BCFtools [21], FreeBayes [25], LoFreq [26], Platypus [27] and VarDict [28]. The literature gathering and review were conducted by using PubMed and the University of Cape Town library (PRIMO), mainly for the period between 2008 and 2020; VC tools were chosen if the underlying method paper has at least two citations.
VC: overview and brief classification variant
Calling tools are designed to identify specific types of variants. These include germline variants, somatic variants, copy number variants (CNV) and structural variants (SV). Consequently, all VC methods fall into four categories: germline callers, somatic callers, CNV identifiers and SV identifiers [3, 29]. These programs have different underlying processes to optimize the identification of these different types of variants. Here, we focus on the identification of whole genome germline variants and hence germline callers as these are the variants that result in general population level differences, between European and non-European individuals. Within germline variant callers, there are two approaches employed by these variants calling programs: heuristic and statistical approach [30].
The heuristic approach is the original method of VC employed by tools such as VarScan2 [1]. This method uses filtering and quality cut-off values to identify an initial set of genotypes from which SNPs are inferred. This approach is sometimes preferred as there are no potential violable assumptions made about the data. A shortcoming, however, is that without a high degree of sequencing depth, the heuristic method is prone to under-calling SNPs at heterozygous genome sites. Consequently, for accurate VC, high sequencing depth is required and thus is computationally more complex and demanding [30]. This method is also unable to utilize information on the quality of individual reads and therefore provides no measure of uncertainty for each predicted variant. As the called SNPs are based on identified genotypes, if the identified genotype is incorrect, the called variants will also be incorrect [31].
The alternate approach is the statistical method, which solves many of the problems associated with the heuristic approach [30] and provides a measure of uncertainty for each called variant, thus increasing the accuracy of the output data. The statistical method is based on the likelihood of observing a specific outcome, given all prior known information. Bayes’ formula is used to determine the posterior probability Pr (G│X) of the genotype identified (G), given the data received. The highest posterior probabilities are chosen and likelihood ratios of this form a measure of confidence in the output. This method also allows easy incorporation of prior data such as known population scale-allele frequencies.
We have chosen nine different germline VC tools: VarDict [28], FreeBayes [25], GATK-HC [32], Platypus [27], SNVer [23], VarScan2 [1], LoFreq [26], Samtools and BCFtools [21, 33]. These are the most cited based on literature from PubMed, well maintained and accept standard alignment input formats [34]. These programs have varied approaches to variant identification and as a result produce different variant calls [34]. Consequently, the only way to identify which approach is best suited to population-specific genomic data is to test and compare the methods.
VarScan2 [1] has been designed to use a heuristic and statistical approach to identify variants and may also be used to identify somatic mutations [34] (Supplementary Table S2). The Samtools package consists of two different variants calling tools such as Samtools and BCFtools. Samtools and BCFtools both have Bayesian underlying processes and do not require any genotyping assumptions to call variants [21, 33]. The key difference between these tools is that BCFtools performs VC with a multiallelic calling model, whereas Samtools uses a consensus calling model (Supplementary Table S2). SNVer uses a binomial–binomial model to test the significance of observed allele frequencies against the sequencing error rates [23] (Supplementary Table S2). GATK-HC also uses a Bayesian approach to identify variants; however, this is under the assumption of uniform copy numbers [31] and does not involve genotype calling to inform variant identification [32]. GATK-HC incorporates ‘technical covariates, known sites of variation, genotypes for individuals, Linkage Disequilibrium, family and population structure’ into its VC approach to separate true variants from machine artefacts. As GATK has been developed as a toolkit, it also offers the ability for local realignment and base quality score recalibration to eliminate FP variants (Supplementary Table S2) [32].
LoFreq uses a Poisson–binomial distribution [34] to model sequencer platform that runs specific error rates and as result can call rare variants [26] (Supplementary Table S2). FreeBayes is haplotype-based VC tool that uses a Bayesian statistical framework that can model multiallelic loci in a set of individuals with non-uniform copy numbers [25] (Supplementary Table S2). Platypus is also a multisample haplotype-based variant caller [27]. Platypus integrates various approaches into one to perform VC such as mapping-based assembly and reference-free assembly approaches, which are incorporated into a Bayesian framework to perform VC (Supplementary Table S2). VarDict is the newest of the tools chosen for comparison, and uses two types of local realignment to improve estimated allele frequencies and is also able to call complex combinations of variants simultaneously [28].
Generating synthetic African and European WGS datasets
All African and European samples were extracted from 1000 Genomes (The 1000 Genomes Project Consortium, 2015) variant call file (VCF) (Figure 1A). To better capture samples across all African and European populations, stratified sampling approach was adopted using Plink [35] to sample 50 individuals from African samples and 50 individuals from European samples. NEAT-genReads [36] was used to simulate WGS data using parameters as shown in Table 1. NEAT-genReads has various submodules, one being GenMutModel that allows the generation of mutation models that reflect mutations present in input data. Another tool in this package is GenReads, which is used to generate the simulated reads. We used the GenMutModel module on the human leukocyte antigen region (chromosome 6) to characterize African and European mutation models. For the African samples, the 2933807–40173830 regions on chromosome 6 were used, and for the European samples, the 29733807–40312825 regions on chromosome 6 were used. These regions differ slightly as these models were generated from VCF files, and as expected, the SNPs present in African and European samples are not identical. FastQ files for forward and reverse reads, along with golden Burrows-Wheeler Aligner (BAM) and golden VCF files were generated using the GenReads module with the hg19 human reference sequence. We performed the simulation procedure twice, first without the mutation model and a second time using a mutation rate of 0.1 to mimic the standard Illumina sequencing error rate [37]. During the first simulation run, high coverage samples had a coverage ranging between 60 and 85, whereas low coverage samples had a coverage ranging between 10 and 50. During the second round of simulation, we produced only low coverage data. Two population-specific simulated datasets at two-depth coverage (high and low), each set representing African (AFR) and European (EUR) populations, have been generated. These were further classified into four datasets, each having 25 WGS samples of AFR high, AFR low, EUR high and EUR low coverage.
Figure 1 .

Overview of the variant calling analysis pipeline.
Table 1.
Data generated by NEAT-genReads and used to analyse the performance of variant calling tools
| African population | European population | |||
|---|---|---|---|---|
| Coverage depth | High | Low | High | Low |
| Sample numbers | 25 | 25 | 25 | 25 |
| Sequencer | Illumina | |||
| Sequencing error rate | 0.1 | |||
| Total samples | 100 | |||
Quality check, read alignment of synthetic African and European WGS datasets
The output from NEAT-genReads results in three different forms: (1) forward and reverse FastQ file, (2) golden BAM and (3) golden VCF. We used the forward and reverse FastQ files for further analysis. We performed quality control using FastQC. FastQC reports were aggregated using MultiQC [38], based on each simulated AFR high (Supplementary Figure S1), AFR low (Supplementary Figure S2), EUR high (Supplementary Figure S3) and EUR low FastQ (Supplementary Figure S4). All simulated 100 WGS samples were of good quality. Figure 1B illustrates the VC analysis pipeline.
The forward and reverse FastQ files were aligned to the latest human reference genome hg38/GRCh38 using the Burrows–Wheeler Aligner Memory (BWA-MEM) [33], the result was saved in Sequence Alignment/Map (SAM) format.
We replaced the reading group in the SAM file by using Picard [32], as the simulation tool generated one `@RG' tag for all the generated data, which is not accepted by GATK and other VC tools, as the reading group must be unique for each sample. Second, the SAM files were sorted and resulted in generated BAM files by using Picard (SortSam). Finally, BAM files were indexed using Picard (BuildBamIndex). As some VC tools can be sensitive to post-alignment quality control and of course since the data used are simulated, we opted not to conduct further post-alignment quality control such as mark duplication and realignment around indels.
Evaluating VC and performance measures and analysis
The resulting BAM files from the previous section were used as an input for each of the nine VC tools listed in the Supplementary Table S2. All 100 BAM samples from simulated AFR high, AFR low, EUR high and EUR low were used (Table 1). We performed population joint-call of all the samples at autosomal chromosomes for Samtools, BCFtools, FreeBayes, SNVer, GATK, Platypus and VarScan2, except for LoFreq and VarDict. All VC tools were used with the respective suggested standard parameters to allow equal chances of accurate variant identification. However, if the tool supports frequency-based calls such as VarDict and FreeBayes, we set the rate to 0.01 as the minimum allele frequency. Supplementary Figure S5 illustrates the intersections of the variant positions produced by Samtools, BCFtools, SNVer, GATK, Platypus, VarScan, LoFreq and VarDict on African/European based on high/low coverage data, supporting the hypothesis that existing VC tools do not predict and discover same number of variants. This supports the prior suggestions from scholars [18, 36, 38] to use a consensus or a combination of two to more VC tools for validating and achieving high-confident variants discovery.
All resulting VCFs from each VC tool were compared against golden VCF produced from NEAT-genReads. VC tool performance was assessed by extracting variant positions from the VCF files produced and comparing this to the true expected VCF (produced by NEAT). The calls were compared and subsequently were identified as either true positive (TP), FP, FN or true negative calls. Accordingly, we identified the number of positions called by each tool as well as the number of FP and FN positions called. Using these values, we calculated the percentage of FP positions identified and the percentage of FN positions identified. These values were calculated as
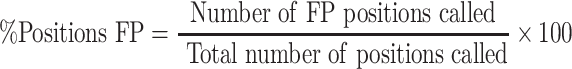 |
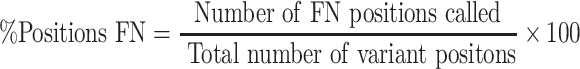 |
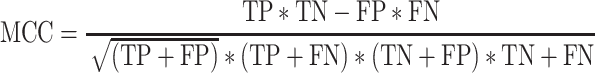 |
Matthews correlation coefficient (MCC) lies between −1 and +1, score of +1 is a perfect model and −1 is a poor model. The total number of variable sites present in the golden VCF samples gave the total number of variant positions. Furthermore, we calculated the sensitivity (Recall), precision [positive predictive value (PPV)] and F-score. Although the Ti/Tv (transition-to-transversion ratio) was obtained from BCFtools-stat, the Intervene tool [33] was used to visualize the golden VCF. As shown in Table 2, we were able to calculate MCC performance metrics.
Table 2.
Summary of the performance metrics of the nine variant calling tools evaluated from simulated data representing African and European populations
| African population | ||||||
|---|---|---|---|---|---|---|
| Coverage | Variant caller | Recall | PPV* | F-score | Ti/Tv | MCC |
| High | VarScan | 0.2945 | 0.991 | 0.454 | 1.67 | 0.791 |
| Samtools | 0.572 | 0.994 | 0.727 | 1.73 | 0.800 | |
| GATK-HC | 0.876 | 0.748 | 0.807 | 1.70 | 0.808 | |
| SNVer | 0.637 | 0.827 | 0.720 | 1.67 | 0.797 | |
| BCFtools | 0.053 | 0.999 | 0.100 | 1.73 | 0.813 | |
| FreeBayes | 0.371 | 0.871 | 0.761 | 1.72 | 0.795 | |
| LoFreq | 0.858 | 0.983 | 0.917 | 1.76 | 0.798 | |
| Platypus | 0.100 | 0.998 | 0.182 | 2.80 | 0.802 | |
| VarDict | 0.005 | 0.999 | 0.010 | 1.67 | 0.832 | |
| Low | VarScan | 0.268 | 0.869 | 0.409 | 1.68 | 0.789 |
| Samtools | 0.601 | 0.997 | 0.750 | 1.73 | 0.791 | |
| GATK-HC | 0.809 | 0.788 | 0.799 | 1.71 | 0.791 | |
| SNVer | 0.761 | 0.800 | 0.780 | 1.66 | 0.777 | |
| BCFtools | 0.037 | 0.999 | 0.072 | 1.73 | 0.813 | |
| FreeBayes | ||||||
| LoFreq | 0.781 | 0.999 | 0.877 | 1.76 | 0.791 | |
| Platypus | 0.058 | 0.575 | 0.105 | 2.43 | 0.801 | |
| VarDict | 0.004 | 0.999 | 0.009 | 1.66 | 0.831 | |
| European population | ||||||
| Coverage | Variant caller | Recall | PPV* | F-score | Ti/Tv | |
| High | VarScan | 0.236 | 0.867 | 0.371 | 1.54 | 0.791 |
| Samtools | 0.584 | 0.997 | 0.737 | 1.59 | 0.831 | |
| GATK-HC | 0.890 | 0.729 | 0.801 | 1.56 | 0.841 | |
| SNVer | 0.608 | 0.830 | 0.702 | 1.54 | 0.810 | |
| BCFtools | 0.030 | 0.999 | 0.059 | 1.59 | 0.847 | |
| FreeBayes | 0.351 | 0.771 | 0.712 | 1.56 | 0.791 | |
| LoFreq | 0.818 | 0.999 | 0.900 | 1.63 | 0.858 | |
| Platypus | 0.107 | 0.999 | 0.194 | 2.39 | 0.801 | |
| VarDict | 0.004 | 0.999 | 0.009 | 1.55 | 0.820 | |
| Low | VarScan | 0.283 | 0.873 | 0.428 | 1.55 | 0.790 |
| Samtools | 0.190 | 0.999 | 0.319 | 1.60 | 0.830 | |
| GATK-HC | 0.405 | 0.861 | 0.551 | 1.56 | 0.832 | |
| SNVer | 0.832 | 0.757 | 0.793 | 1.54 | 0.801 | |
| BCFtools | 0.410 | 0.997 | 0.581 | 1.59 | 0.832 | |
| FreeBayes | 0.361 | 0.821 | 0.751 | 1.58 | 0.790 | |
| LoFreq | 0.787 | 0.999 | 0.881 | 1.63 | 0.857 | |
| Platypus | 0.096 | 0.999 | 0.175 | 2.23 | 0.792 | |
| VarDict | 0.004 | 0.999 | 0.009 | 1.54 | 0.812 | |
*Positive predictive value (PPV); Matthews correlation coefficient (MCC)
Assessing high coverage sequences
The African golden-high coverage VCF contains a total of 1, 634, 027, 480 SNPs within all 25 samples, GATK-HC and LoFreq have the highest TP rates across all VC tools compared with sensitivity (0.87 and 0.85), respectively. GATK produced results with the highest sensitivity accompanying a low PPV (PPV = 0.7). All VC tools resulted in good precision; however, VarDict achieved the highest with a very low FP rate (PPV = 0.999 and MCC = 0.832), followed by BCFtools (PPV = 0.999 and MCC = 0.813). VarDict was computationally demanding and tested the limits of our computational capacities, which led to some compromises with respect to the parameters set for this tool. This may have resulted in poorer performance, as optimal parameters may not have been specified. VarDict requires a post-processing step, which involves the use of R and Perl, R, this process greatly slowed down the analysis of the results from the VarDict tool. The resulting performance of VarDict was good with respect to the TP rates as almost all the variants that were detected are considered to be TPs. The total number of variants of African-high coverage that are called from VarDict was 8, 462, 987, and the TPs number was 8, 462, 928 (PPV = 0.999 and MCC = 0.832). VarDict is therefore suggested to be suitable for African targeted sequence data.
Finally, F-score, which measures the overall performance, indicates that LoFreq had the highest performance among all (F-score = 0.91 and MCC = 0.858), followed by GATK-HC (F-score = 0.80 and MCC = 0.841) from European simulated data. The European golden-high coverage VCF contains 1, 671, 991, 580 true SNPs, for these samples, GATK had the highest FP rate (sensitivity = 0.89) followed by LoFreq (sensitivity = 0.81) and high F-score (F-score = 0.90), all tools produced results with good PPV.
Assessing low coverage sequences
The African golden-low coverage VCF contains 1, 719, 357, 177 SNPs, and VarDict had the lowest FP rate accompanied by a high precision (PPV = 0.999 and MCC = 0.831); however, BCFtools performed much better (PPV = 0.999 and MCC = 0.791). Results of low coverage from European based on LoFreq produced the highest F-score (F-score = 0.87 and MCC = 0.857). GATK-HC had the highest TP rate among all the tools. Comparing the European golden-low coverage versus inferred VCF, SNVer had the highest TP rate among all tools (sensitivity = 0.82), and LoFreq had the highest F-score of 0.88. All tools resulted in a good PPV using European low coverage data/Tv ratio is a measure of the likelihood of FP calls. For WGS data, this value should be ~2.0–2.1, with lower values implying FP calls [32]. The ratio was adequate from the output of all VC tools; however, this ratio was the highest within results from Platypus for both European and African data for both high and low coverage samples. VarDict and BCFtools appear to generate fewer FP variant calls and appear to perform the best with African datasets.
As expected, the result shows a higher number of calls for high coverage than low coverage data (Table 2 and Figure 2). The overall performances of each of the VC tools are rather similar using either the European or African population data. GATK-HC, LoFreq, SNVer and Samtools have the lowest FN rates among all tools, whereas VarDict, BCFtools, LoFreq and Samtools have the lowest FP rates among all VC tools compared. The tools that produced the highest TP rates were GATK-HC, LoFreq, SNVer and Samtools.
Figure 2 .

Relationship between positive predictive value (PPV) and sensitivity of variant calling tools on African and European genomic data of different coverages. VarScan2 (pink), Samtools (sky blue), GATK-HaplotypeCaller (red), SNver (dark blue), BCFtools (yellow), LoFreq (purple), Platypus (marron) and VarDict (green).
Discussion
The development of NGS has increased the scope for generation and analysis of human genomes and promoted the use of sequence data for personalized medicine. This progress in NGS has resulted in the development of downstream analysis tools and pipelines to handle such data. For example, many VC tools have been developed, which raises the question: which tool is most appropriate when dealing with a complex and diverse genome such as the African genome? We have compared nine VC tools (VarScan2, Samtools, GATK-HC, SNVer, BCFtools, FreeBayes, LoFreq, Platypus and VarDict) on simulated data representing two populations (African and European) at varied coverages (high and low) as four different sets of datasets. We assessed these tools based on sensitivity and precision and, most importantly, the F-score and MCC to measure the overall performance; low sensitivity and precision result in a low F-score—indicating a poor performance.
An increased specificity may result in the loss of TP calls, whereas a prioritized sensitivity will result in increased FP data. Depending on the desired output for a study, sensitivity or specificity must be favoured as there is a trade-off between these two. The total average of FPs for the African population at the different coverages levels (= 98508519, 31) was a bit higher than the European population (= 91271677, 25); hence, the ability of VC tools to detect true variants is higher when dealing with European genomic data, which supports our hypothesis that many VC tools are more suitable for analysing European genomic data than African genomic data.
Among the nine VC tools we compared, LoFreq in low/high coverage from European data simulation showed remarkably good results. The MCC from the analysis of all data scenarios (European: high and low, African: high and low coverage samples) was the highest for LoFreq compared with all tools tested along with a good sensitivity (Recall) and precision (PPV), especially from the African-high coverage data case. Figure 2 illustrates the relationship between PPV and sensitivity for the different tools and datasets for the different specified coverage levels. LoFreq (seen in purple) shows good performance as it maintained an overall high sensitivity and high PPV in all scenarios.
The differences in performance between BCFtools and Samtools are notable (Table 2). BCFtools performed better than Samtools when calling variants from African samples. This suggests that the multiallelic model that BCFtools employed to perform VC allows it to make variant calls with greater accuracy when analysing African genomic data, compared with the consensus calling method employed by Samtools. When calling variants from both the African and European genomic data, GATK-HC produced an extremely large number of FP base calls. We may expect this as there is a trade-off between specificity and sensitivity, and the increased sensitivity leads to greater rate of FP variant calls. This may indicate that the Bayesian approach for VC employed by GATK, with the assumption of uniform copy numbers, is not well suited to data with characteristics corresponding to that of African genomic data.
The overall performance from each VC tool was lower when analysing African genomic data, compared with the analysis of European data. This, along with the higher average percentage of FP positions identified by VC tools when calling on African data, supports our belief that VC tools are not designed to appropriately deal with the characteristics of African genomic data. Similarly, the average percentage of FN positions was larger for VC using African data. We also saw a strong agreement among the called variants between VC tools when using European data. This supports our hypothesis that VC tools have been designed to identify variants within European data. However, it is worth to note that the performance of several VC tools used in the course of this present evaluation might be sensitive to simulated datasets we generated that basically mimic the standard Illumina sequencing error rate [37, 38], and as well as the aligner tool applied on these simulated data [8]. For example, GATK is best for Illumina data [39] and Samtools is best for Ion Proton data [40]. Thus, the development of a VC tool that considers other characteristics of genomic data and is not biased towards the analysis of any particular data types is imperative. Our findings should help to inform their choice of VC tool (Supplementary Tables S1 and S2) to use when working with African genomic data, though the choice of optimal VC tool may also depend on a specific goal. The tool with the lowest percentage of FP positions was LoFreq, and therefore, if specificity is the end goal, we recommend this tool. However, if sensitivity were more important, the optimal tool choice would be BCFtools.
Concluding remarks
In summary, higher sequence depths help VC tools to accurately call true variants; this confirms and supports previous findings [34]. Considering both sensitivity and PPV, LoFreq outperformed all VC tools. It can accurately call complex and large variants with high TP rates and low FP rates, even given a very low coverage. These results suggest that LoFreq would be an appropriate option when VC WGS from African populations, whereas VarDict can also be considered for the analysis of targeted sequences, particularly for African data. Given the large amount of non-overlapping variants identified by the VC tools compared here, and the differing number of variants discovered by these tools, it is reasonable to consider using multiple VC tools to allow cross-validation of variants discovered for improved accuracy and reliability of results. Overall, this paper not only shed light on the performance of current VC tools (Supplementary Table S2), but also describes why and how the choice of VC tool is important. We highlight gaps in benchmarking VC tools in diverse populations and we believe this review could foster development of non-identical tools or maintenance of existing tools to circumvent the issue of replicating tools and reduce FNs and FPs in diverse populations. As genomic research is growing rapidly, VC tools will remain an integral part of the field. Given this, we recommend that future VC tools should be flexible enough to allow the user to choose any type of variants to identify during VC, and should incorporate all the advantages of the best VC tools into one and subsequently cater for advances in sequence generation by reducing FN and FP variants identified.
Key Points
VC is an important aspect of genomic studies as accurate discovery of polymorphism information through VC can be used to inform clinical decisions and that the choice of VC tool is critical.
Evaluation of 10 state-of-the-art VC tools demonstrated that LoFreq can currently be a great option in calling WGS from African populations and that VarDict can be considered for targeted sequence in African populations.
Future VC tool should be flexible enough to allow the user to choose any type of VC, incorporate all the advantages of current best VC tools into one and cater for advances in sequence generation in reducing FN and FP variants.
Supplementary Material
Acknowledgement
The authors are grateful to H3ABioNet consortium members for helpful discussions and Centre for High-Performance Computing (CHPC) (https://www.chpc.ac.za/) facility for providing a computing platform.
Shatha Alosaimi is an MSc in Human Genetics at the Division of Human Genetics, University of Cape Town.
Noelle van Biljon has Honours in Bioinformatics at Computational Biology Division at University of Cape Town.
Denis Awany is a PhD candidate in Human Genetics at University of Cape Town.
Prisca Thami is a PhD candidate at Division of Human Genetics, University of Cape Town.
Joel Defo is a PhD student at the Division of Human Genetics, University of Cape Town. Jacquiline Mugo is a PhD candidate at Computational Biology Division at University of Cape Town.
Christian D. Bope is a PhD in Computational Biology and Associate Professor at Department of Mathematics and Computer Science, Faculty of Sciences, the University of Kinshasa, DRC.
Gaston K. Mazandu is a PhD in Bioinformatics and Senior Lecturer Professor at the Division of Human Genetics, Department of Pathology, University of Cape Town.
Nicola J. Mulder is a PhD in Medical Microbiology and Professor and Head of the Computational Biology Division at University of Cape Town (UCT) and PI of H3ABioNet.
Emile R. Chimusa is a PhD in Bioinformatics and Associate Professor at the Division of Human Genetics, Department of Pathology, University of Cape Town.
Contributor Information
Shatha Alosaimi, Faculty of Health Sciences, Division of Human Genetics, Department of Pathology, University of Cape Town, Cape Town, South Africa.
Noëlle van Biljon, Department of Statistical Sciences, University of Cape Town, Cape Town, South Africa.
Denis Awany, Faculty of Health Sciences, Division of Human Genetics, Department of Pathology, University of Cape Town, Cape Town, South Africa.
Prisca K Thami, Faculty of Health Sciences, Division of Human Genetics, Department of Pathology, University of Cape Town, Cape Town, South Africa.
Joel Defo, Faculty of Health Sciences, Division of Human Genetics, Department of Pathology, University of Cape Town, Cape Town, South Africa.
Jacquiline W Mugo, Faculty of Health Sciences, Division of Computational Biology, Department of Biomedical Sciences, University of Cape Town, Cape Town, South Africa.
Christian D Bope, Faculty of Sciences, Department of Mathematics and Computer Science, University of Kinshasa, Kinshasa, DRC.
Gaston K Mazandu, Faculty of Health Sciences, Division of Human Genetics, Department of Pathology, University of Cape Town, Cape Town, South Africa; Faculty of Health Sciences, Division of Computational Biology, Department of Biomedical Sciences, University of Cape Town, Cape Town, South Africa.
Nicola J Mulder, Faculty of Health Sciences, Division of Computational Biology, Department of Biomedical Sciences, University of Cape Town, Cape Town, South Africa; Institute of Infectious Disease and Molecular Medicine, University of Cape Town, Anzio Road, Observatory, Cape Town 7925, South Africa.
Emile R Chimusa, Faculty of Health Sciences, Division of Human Genetics, Department of Pathology, University of Cape Town, Cape Town, South Africa; Institute of Infectious Disease and Molecular Medicine, University of Cape Town, Anzio Road, Observatory, Cape Town 7925, South Africa.
Funding
This work was funded in part by DAAD, the German Academic Exchange Programme, under Reference number 91653117 and 91628092; National Institutes of Health Common Fund under grant number U24HG006941 (H3ABioNet); the sub-Saharan African Network for TB/HIV Research Excellence (SANTHE), a DELTAS Africa Initiative (grant #DEL-15-006). The DELTAS Africa Initiative is an independent funding scheme of the African Academy of Sciences (AAS) Alliance for Accelerating Excellence in Science in Africa and supported by the New Partnership for Africa’s Development Planning and Coordinating Agency (NEPAD Agency) with funding from the Wellcome Trust (grant #107752/Z/15/Z) and the UK government. The views expressed in this publication are those of the authors and not necessarily those of AAS, NEPAD Agency, Wellcome Trust or the UK government. The content of this publication is solely the responsibility of the authors and does not necessarily represent the official views of the funders.
References
- 1. Koboldt DC, Zhang Q, Larson DE, et al. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res 2012;22:568–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Shen T, Pajoro-Van de Stadt SH, Yeat NC, et al. Clinical applications of next generation sequencing in cancer: from panels, to exomes, to genomes. Front Genet 2015;6:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Pabinger S, Dander A, Fischer M, et al. A survey of tools for variant analysis of next-generation genome sequencing data. Brief Bioinform 2012;15(2):256–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bao R, Huang L, Ndrade J, et al. Review of current methods, applications, and data management for the bioinformatics analysis of whole exome sequencing. Cancer Inform 2014;13(2):67–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Spencer DH, Tyagi M, Vallania F, et al. Performance of common analysis methods for detecting low-frequency single nucleotide variants in targeted next-generation sequence data. J Mol Diagn 2014;16:75–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Liu X, Han S, Wang Z, et al. Variant callers for next-generation sequencing data: a comparison study. PLoS One 2013;8(9):e75619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Laurie S, Fernandez-Callejo M, Marco-Sola S, et al. From wet-lab to variations: concordance and speed of bioinformatics pipelines for whole genome and whole exome sequencing. Hum Mutat 2016;37:1263–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kumaran M, Subramanian U, Devarajan B. Performance assessment of variant calling pipelines using human whole exome sequencing and simulated data. BMC Bioinformatics 2019;20(1):1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hentzsche JD, Robinson WA, Tan AC. A survey of computational tools to analyze and interpret whole exome sequencing data. Int J Genomics 2016;2016:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Xu C. A review of somatic single nucleotide variant calling algorithms for next-generation sequencing data. Comput Struct Biotechnol J 2018;16:15–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Xu H, Di Carlo J, Satya R, et al. Comparison of somatic mutation calling methods in amplicon and whole exome sequence data. BMC Genomics 2014;15:244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Campbell MC, Tishkoff SA. African genetic diversity: implications for human demographic history, modern human origins, and complex disease mapping. Annu Rev Genomics Hum Genet 2008;9(1):403–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Popejoy AB, Fullerton SM. Genomics is failing on diversity. Nature 2016;538:161–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lek M, Karczewski KJ, Minikel EV, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016;536:285–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Martin AR, Teferra S, Moller M, et al. The critical needs and challenges for genetic architecture studies in Africa. Curr Opin Genet Dev 2018;53:113–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sirugo G, Hennig BJ, Adeyemo AA, et al. Genetic studies of African populations: an overview on disease susceptibility and response to vaccines and therapeutics. Hum Genet 2008;123:557–98. [DOI] [PubMed] [Google Scholar]
- 17. Rotimi CN, Bentley AR, Doumatey AP, et al. The genomic landscape of African populations in health and disease. Hum Mol Genet 2017;26(2):225–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cheng AY, Teo YY, Ong RTH. Assessing single nucleotide variant detection and genotype calling on whole-genome sequenced individuals. Bioinformatics 2014;30(12):1707–13. [DOI] [PubMed] [Google Scholar]
- 19. Bope CD, Chimusa ER, Nembaware V, et al. Dissecting in silico mutation prediction of variants in African genomes challenges and perspectives. Front Genet 2019;10(601). 10.3389/fgene.2019.00601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Retshabile G, Mlotshwa BC, Williams L, et al. Whole-exome sequencing reveals uncaptured variation and distinct ancestry in the southern African population of Botswana. Am J Hum Genet 2018;102:731–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Li H, Handsaker B, Wysoker A, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009;25:2078–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. McKenna A, Hanna M, Banks E, et al. The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 2010;20: 1297–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wei Z, Wang W, Hu P, et al. SNVer: a statistical tool for variant calling in analysis of pooled or individual next-generation sequencing data. Nucleic Acids Res 2011;39:e132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Li H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 2011;27:2987–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Garrison E, Marth G. Haplotype-based variant detection from short-read sequencing. arXiv 2012; preprint, ArXiv:1207.3907(q-bio.GN). [Google Scholar]
- 26. Wilm A, Aw PP, Bertrand D, et al. LoFreq: a sequence-quality aware, ultra-sensitive variant caller for uncovering cell-population heterogeneity from high-throughput sequencing datasets. Nucleic Acids Res 2012;40:11189–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Rimmer A, Phan H, Mathieson I, et al. Integrating mapping-, assembly- and haplotype-based approaches for calling variants in clinical sequencing applications. Nat Genet 2014;46:912–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lai Z, Markovets A, Ahdesmaki M, et al. VarDict: a novel and versatile variant caller for next-generation sequencing in cancer research. Nucleic Acids Res 2016;44:e108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Luo R, Wong C-L, Wong Y-S, et al. Clair: exploring the limit of using a deep neural network on pileup data for germline variant calling. Nat Mach Intell 2020;2:220–27. [Google Scholar]
- 30. Mielczarek M, Szyda J. Review of alignment and SNP calling algorithms for next-generation sequencing data. J Appl Genet 2016;57(1):71–9. [DOI] [PubMed] [Google Scholar]
- 31. DePristo MA, Banks E, Poplin R, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 2011;43(5):491–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009;25(14): 1754–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sandmann S, Graaf AO, Karimi M, et al. Evaluating variant calling tools for non-matched next-generation sequencing data. Sci Rep 2017;7:43169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Purcell S, Neale B, Todd-Brown K, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 2007;81:559–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chen YT, Cong J, Lei J, et al.. A novel high-throughput acceleration engine for read alignment. In 2015 IEEE 23rd Annual International Symposium on Field-Programmable Custom Computing Machines. IEEE. 2005; pp. 199–202. [Google Scholar]
- 36. Glenn TC. Field guide to next-generation DNA sequencers. Mol Ecol Resour 2011;11(5):759–69. [DOI] [PubMed] [Google Scholar]
- 37. Ewels P, Magnusson M, Lundin S, et al. MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics 2016;32(19):3047–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Alosaimi S, Bandiang A, Biljon N, et al. A broad survey of DNA sequence data simulation tools. Brief Funct Genomics 2020;19(1):49–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Yi M, Zhao Y, Jia L, et al. Performance comparison of SNV detection tools with Illumina exome sequencing data - an assessment using both family pedigree information and sample-matched SNV array data. Nucleic Acids Res 2014;42:101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hwang S, Kim E, Lee I, et al. Systematic comparison of variant calling pipelines using gold standard personal exome variants. Sci Rep 2015;5:17875. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.