

Abstract
Anti-cancer peptides (ACPs) are known as potential therapeutics for cancer. Due to their unique ability to target cancer cells without affecting healthy cells directly, they have been extensively studied. Many peptide-based drugs are currently evaluated in the preclinical and clinical trials. Accurate identification of ACPs has received considerable attention in recent years; as such, a number of machine learning-based methods for in silico identification of ACPs have been developed. These methods promote the research on the mechanism of ACPs therapeutics against cancer to some extent. There is a vast difference in these methods in terms of their training/testing datasets, machine learning algorithms, feature encoding schemes, feature selection methods and evaluation strategies used. Therefore, it is desirable to summarize the advantages and disadvantages of the existing methods, provide useful insights and suggestions for the development and improvement of novel computational tools to characterize and identify ACPs. With this in mind, we firstly comprehensively investigate 16 state-of-the-art predictors for ACPs in terms of their core algorithms, feature encoding schemes, performance evaluation metrics and webserver/software usability. Then, comprehensive performance assessment is conducted to evaluate the robustness and scalability of the existing predictors using a well-prepared benchmark dataset. We provide potential strategies for the model performance improvement. Moreover, we propose a novel ensemble learning framework, termed ACPredStackL, for the accurate identification of ACPs. ACPredStackL is developed based on the stacking ensemble strategy combined with SVM, Naïve Bayesian, lightGBM and KNN. Empirical benchmarking experiments against the state-of-the-art methods demonstrate that ACPredStackL achieves a comparative performance for predicting ACPs. The webserver and source code of ACPredStackL is freely available at http://bigdata.biocie.cn/ACPredStackL/ and https://github.com/liangxiaoq/ACPredStackL, respectively.
Keywords: anti-cancer peptides, bioinformatics, prediction, sequence analysis, ensemble learning, performance assessment
Introduction
Cancer is a leading cause of death worldwide and has been a significant public health concern [1]. Conventional chemotherapy is one of the primary treatments for cancer. However, it is expensive and can cause adverse effects on healthy cells [2]. Moreover, highly adaptable cancer cells often develop drug resistance [3]. Further, drug resistance poses significant challenges for chemotherapy and development of anti-tumour drugs [4]. Thus, it is urgent to find more effective strategies for cancer treatment. Anti-cancer peptides (ACPs), a particular type of antimicrobial peptides (AMPs), are known as small peptides that usually have less than 50 amino acids [5]. It has been reported that ACPs can directly target cancer cells without affecting healthy cells [6]. Moreover, compared with conventional therapeutics, peptides have other attractive advantages as summarized in [5], such as high specificity, low intrinsic toxicity, high tissue penetration and ease of modifications. Therefore, ACPs have been extensively studied as an alternative approach for cancer treatment in recent years [6].
Several public databases, such as CancerPPD [5], DADP [7], CAMP [8] and APD [9], collect more and more experimentally validated ACPs, providing rich resources for knowledge-based discovery of novel ACPs. Although experimental techniques facilitate the discovery of ACPs, these techniques are costly, labour-intensive and are not practically suitable for the discovery of ACPs in a cost-effective manner. Computational methods that are capable of identifying ACPs accurately can complement with experimental techniques and greatly facilitate the high-throughput characterization of ACPs.
To date, a variety of computational methods have been developed for the identification of ACPs. Table 1 summarizes the existing computational methods and covers a wide range of aspects, including the applied machine learning (ML) algorithms, extracted biological features, evaluation strategies and webserver/software availability. In these studies, the ACPs identification task is formulated as a binary classification problem. There are three major steps in these studies: the first step is to collect experimentally validated ACPs as positive samples and non-ACPs as negative samples to construct benchmark datasets. At the second step, a variety of peptide sequence-based features are calculated and extracted for model training. Finally, the extracted feature sets are used as the input to feed into machine learning algorithms to train the prediction models and predict whether a query peptide is ACP or not.
Table 1.
A comprehensive list of the existing methods and tools for prediction of anti-cancer peptides
| Group | Methods/tools | Year | Model | Features | Evaluation strategy | Webserver/software availability |
|---|---|---|---|---|---|---|
| Conventional ML-based method | AntiCP [15] | 2013 | SVM | DPC, AAC, BP | IT, 5-fold-CV, 10-fold-CV | Yes |
| Hajisharifi et al. [2] | 2014 | SVM | PAAC, LAK | 5-fold-CV | No | |
| ACPP [16] | 2015 | SVM | PRM | IT, 10-fold-CV | Yes | |
| iACP [17] | 2016 | SVM | G-DPC | 5-fold-CV, JT | Yes | |
| Li and Wang [18] | 2016 | SVM | AAC, acACS, RAAC | 5-fold-CV, JT | No | |
| MLACP [12] | 2017 | RF, SVM | AAC, DPC, ATC, PCP | 10-fold-CV, LOOCV | No | |
| SAP [19] | 2018 | SVM | G-DPC | 10-fold-CV | No | |
| ACPred-FL [20] | 2018 | SVM | BP, OLP, TOB, CTD, AAC, G-DPC, ASDC | 10-fold-CV, IT | not accessible | |
| TargetACP [21] | 2018 | SVM | PPSSM, SAAC, CPSR | IT, JT | No | |
| ACPred [22] | 2019 | SVM | AAC, DPC, PCP, PAAC, APAAC | 5-fold-CV, JT | Yes | |
| mACPpred [23] | 2019 | SVM | AAC, DPC, CTD, QSO, AAI, BP, CJT | IT, 10-fold-CV | Yes | |
| PEPred-Suite [10] | 2019 | RF | AAC, ASDC, CTD, 188D, GGAP, BIT20, BIT21, OLP, IT | IT, 10-fold-CV | Yes | |
| ACPred-Fuse [11] | 2019 | RF | 29 features including AAC, GAAC, GDPC, GGAP, ASDC, TPC, GTPC, et al. | IT, 10-fold-CV | Yes | |
| AntiCP 2.0 [24] | 2020 | SVM, RF, KNN, ANN, ETree, Ridge | AAC, DPC, TC, BP | 5-fold-CV | Yes | |
| Ensemble-based method | iACP-GAEnsC [13] | 2017 | Ensemble | P-G-DPC, APAAC, RAAA | JT | No |
| DL-based method | ACP-DL [14] | 2019 | DL | BP, K-mer-SM | 5-fold-CV | No |
Model Abbreviations. RF: random forest, SVM: support vector machine, DL: deep learning, KNN: k-nearest neighbours, ETree: extra trees, ANN: artificial neural network.
Evaluation strategy Abbreviations. k-fold-CV: k-fold cross-validation, IT: Independent Test, JT: Jackknife test, LOOCV: Leave-one-out cross-validation.
Features abbreviations. DPC: dipeptide composition; AAC: amino acid composition; BP: binary profiles; PAAC: pseudo-amino acid composition; LAK: local alignment kernel; PRM: protein relatedness measure; g-DPC: g-gap dipeptide compositions; acACS: auto covariance of the average chemical shift; RAAC: reduced amino acid composition; ATC: atomic composition; PCP: physicochemical properties; OLP: overlapping property; TOB: twenty-one-bit; CTD: composition–transition–distribution; ASDC: adaptive skip dipeptide composition; PPSSM: pseudo position-specific scoring matrix; SAAC: split amino acid composition; CPSR: composite protein sequence representation; APAAC: amphiphilic pseudo-amino acid composition; QSO: quasi-sequence-order; AAI: amino acid index; CJT: conjoint triad; GGAP: gap dipeptide composition; GAAC: grouped amino acid composition; GDPC: grouped dipeptide composition; TPC: tripeptide composition; GTPC: grouped tripeptide composition; P-G-DPC: pseudo g-gap dipeptide composition; RAAA: reduced amino acid alphabet; K-mer-SM: k-mer sparse matrix; 188D: 188-dimensional feature; BIT20: twenty-bit features; BIT21: twenty-one-bit features; IT: information theory; TC: terminus composition.
We categorize these methods in Table 1 into three groups according to the types of machine learning methods employed: (i) The first group is the single ML-based methods, and they are developed based on conventional ML algorithms, such as support vector machine (SVM) and random forest (RF). As can be seen from Table 1, SVM is the most popular algorithm. In all, 10 out of 14 methods are developed based on SVM. Besides, there are two methods PEPred-Suite [10] and ACPred-Fuse [11] are developed based on RF. In addition, MLACP [12] is developed using both SVM and RF, and AntiCP 2.0 is implemented using SVM, RF, KNN, ETree, ANN and ridge, respectively; (ii) the second group is the ensemble learning-based method, and there is only one predictor, termed iACP-GAEnsC [13], in this group. iACP-GAEnsC is developed based on five different base-classifiers, including SVM, Probabilistic Neutral network (PNN), RF, Generalize regression neural network (GRNN) and K-nearest neighbour (KNN). The final ensemble model is constructed based on these base-classifiers by using genetic algorithm and voting strategy, (iii) the third group is the deep learning (DL)-based method. There is also only one predictor, named ACP-DL [14], in this group. ACP-DL utilizes the k-mer sparse matrix and a binary profile to encode the input sequences into a 2D matrix, and then feeds the encoded matrix into the long short-term memory (LSTM) neural network to train the prediction model.
All three groups of methods reviewed above need to extract and calculate a variety of peptide sequence or sequence-derived features for model training. These sequence features and sequence-based features are also summarized in Table 1. Amino acid composition (AAC), dipeptide composition (DPC), Composition-Transition-Distribution (CTD) and Binary Profiles (BP) features are among the most widely employed types of features in these methods. Different methods employ various features that favour the ML-based models to predict ACPs. Apparently, the employed features used by each method are essential for the method to identify ACPs. Accordingly, it would be important to understand the feature importance and the impact of individual features on the output of the prediction model. Five evaluation strategies including 10-fold cross-validation, 5-fold cross-validation, independent test, leave-one-out cross-validation and jackknife test were employed by the existing methods. All these evaluation strategies belong to cross-validation [25]. Among these, jackknife test, also called leave-one-out cross-validation, can be conducted to obtain a consistent outcome for a predictor [26, 27] on the same benchmark dataset. For k-fold cross-validation, when dividing a benchmark dataset, there are a large number of possible divisions even for a very simple dataset [25, 27]. Thus, a same predictor would yield different results when selecting different divisions for a same benchmark dataset [27]. Independent test is often used to evaluate the generalization ability of the model. Experimentally verified ACPs are currently very limited. With the increasing availability of ACPs in the future, newly identified ACPs may be independent on current ACPs. Thus, they can be regarded as more reliable independent datasets to evaluate the performance of existing predictors. Certainly, with more ACPs being incorporated into the training dataset, the performance of existing predictors is expected to be further improved. Out of 16 predictors, 9 have been implemented as online webservers; however, some webservers are not developed and made available. It is recommended that more webservers or stand-alone software should be made publicly accessible to facilitate users’ research efforts.
These ML-based methods greatly facilitate the identification of ACPs and promote the research progress in the field. However, several issues still remain in most of these methods that need to be addressed. First, the currently available ACPs datasets are relatively small, which are not suitable for training a robust deep learning-based prediction model [28]. Therefore, the performance of ACP-DL, which is the only existing deep learning-based method for ACPs identification, is substantially limited by the size of the dataset available [14]. Second, the single machine learning-based methods only apply single algorithms to train the model, and the performance is not robust in some cases. From the viewpoint of classification, individual classifiers are often limited and only focus on certain aspects of the classification. As a consequence, the performance of individual classifier-based methods of ACPs identification is often limited in terms of their classification capability and performance. In this context, ensemble learning can combine multiple different classifiers to harness the merits of all assembled models to improve the predictive performance. It has been demonstrated that ensemble learning usually outperforms individual classifiers [29, 30]. Ensemble learning has been successfully used in many bioinformatics studies, such as DNA-binding proteins identification [31, 32], non-classical secreted protein prediction [33], promoter prediction [34] and ncRNA-protein interaction prediction [35]. While there is only one ensemble learning-based method currently available, called iACP-GAEnsC [13], for ACPs identification, more ensemble learning frameworks should be considered and leveraged to develop more accurate and robust predictors in the future. Three, all existing methods are ‘Black-box’ models, and as such, it is difficult to interpret and understand why these models make the predictions. In this regard, cutting-edge model interpretation algorithms can be applied to interpret the machine learning-based methods, which can help us better understand why the predictors make such predictions. Finally, all existing methods are developed and evaluated using different benchmark and independent test datasets. Table 2 summarizes the datasets used for the development and assessment of the existing methods. There is a lack of a benchmark model to assess and evaluate the existing models comprehensively. This benchmark model can serve as a baseline to assist model comparison and facilitate users to choose the appropriate method to use.
Table 2.
A summary of the training and independent test datasets used in the existing methods
| Datasetsa | Training datasets | Independent test datasets | CD-HIT threshold | Dataset availability | ||
|---|---|---|---|---|---|---|
| Number of ACPs | Number of non-ACPs | Number of ACPs | Number of non-ACPs | |||
| Tyagi2013 [15] | 225 | 225 | 50 | 50 | – | Yes |
| Hajisharifi2014 [2] | 138 | 206 | No | No | 0.9 | Yes |
| Vijayakumar2015 [16] | 217 | 3979 | 40 | 40 | 1.0 | Yes |
| Chen2016 [17] | 138 | 206 | 150 | 150 | 0.9 | Yes |
| Li2016 [18] | 138 | 206 | 150 | 150 | 0.9 | Yes |
| Akbar2017 [13] | 138 | 206 | No | No | 0.9 | Yes |
| Manavalan2017b [12] | 187 | 398 | 422 + 126 | 422 + 205 | 0.9 | No |
| Xu2018 [19] | 138 | 206 | No | No | 0.9 | Yes |
| Wei2018 [20] | 250 | 250 | 82 | 82 | 0.9 | Yes |
| Kabir2018 [21] | 138 | 206 | 150 | 150 | 0.9 | Yes |
| Schaduangrat2019 [22] | 138 | 205 | No | No | 0.9 | Yes |
| Boopathi2019 [23] | 266 | 266 | 157 | 157 | 0.8 | Yes |
| Yi2019 [14] | 376 | 364 | 129 | 111 | 0.9 | Yes |
| Rao2019 [11] | 250 | 250 | 82 | 2628 | 0.8 | Yes |
| Agrawal2020 [24] | 689 | 689 | 172 | 172 | - | Yes |
| Basith2020c [36] | - | - | 246 | 1733 | 0.9, 0.6 | Yes |
aDatasets are named using the second name of the first author plus the publication year in the corresponding literature.
b Manavalan2017 contains two independent test datasets. One contains 422 ACPs and 422 non-ACPs, and the other contains 126 ACPs and 126 non-ACPs, respectively.
cIn Basith2020, positive and negative samples with >90% and > 60% sequence identity with training data sets used in existing methods were removed.
Next we propose a new stacking ensemble learning-based approach, termed ACPredStackL (Anti-Cancer peptide Predictor based on Stacking Learning), for the improved identification of ACPs. The stacking strategy can consider some different and weak classifiers, learn them in parallel and integrate them by training a meta-model to make the prediction based on different weak models’ predictions. The proposed ACPredStackL framework consists of two-level classifiers. The first-level classifier of ACPredStackL trains four different weak learners, including KNN, Naïve Bayesian (NB), LightGBM and SVM. Then the combined prediction outcomes of the first-level classifier are fed into a logistics regression (LR) algorithm to train the meta-model (i.e., the second-level learner). The predicted outcomes of the LR model will be used as the final prediction results. We validate and compare the predictive performance of ACPredStackL and other state-of-the-art methods based on the datasets reviewed in Table 2. Moreover, we further develop an online webserver of ACPredStack to facilitate community-wide efforts in analyzing and identifying ACPs, which is freely available at http://bigdata.biocie.cn/ACPredStackL/.
Materials and methods
Datasets
We review all the datasets used for developing the currently available ACP predictors. The details of these datasets are presented in Table 2. Among these datasets, Tyagi2013 [15] and Hajisharifi2014 [2] are two fundamental datasets. Tyagi2013 was collected from a well-known antimicrobial database (APD2) [9], antimicrobial peptides (CAMP) [8] and database of anuran defence peptides (DADP) [7]. Hajisharifi2014 was collected from APD2. The other datasets were mostly derived from these two datasets. For instance, the training datasets in Chen2016 [17], Li2016 [18], Akbar2017 [13], Xu2018 [19], Kabir2018 [21] and Schaduangrat2019 [22] are the same as Hajisharifi2014. The training datasets and ACPs in the independent datasets used in Wei2018 [20] and Rao2019 [11] are the same datasets derived from Tyagi2013, Chen2016 and the ACP database CancerPPD [5]. Yi2019 integrated Wei2018 and Chen2016 into a larger dataset. Agrawal2020 was obtained from Yi2019, Vijayakumar2015, Wei2018, Tyagi2013, Chen2016 and CancerPPD [5].
In this study, we applied the Boopathi2019 dataset as the benchmark dataset to evaluate the performance of ACPredStackL. There are three main reasons why we used Boopathi2019: (i) Boopathi2019 contained a larger number of samples than the other datasets. Figure 1 illustrates the relationships between Boopathi2019 and the other datasets. As can be seen, the training datasets of Boopathi2019 integrated three datasets, Tyagi2013, Wei2018 and Chen2016. The independent testing datasets of Boopathi2019 were collected from four datasets, including DADP [7], DBAASP [37], DRAMP [38] and LAMP [39]. As a result, Boopathi2019 covers the majority of the datasets used in other predictors; (ii) Boopathi2019 excludes those sequences that have more than 50 amino acid residues that may form the outliers during the prediction model training, (iii) The training datasets of Boopathi2019 applied a lower CD-HIT [40] threshold of 0.8 (Table 2) to exclude more redundant sequences than the other datasets and avoid over-estimation of the model performance.
Figure 1 .
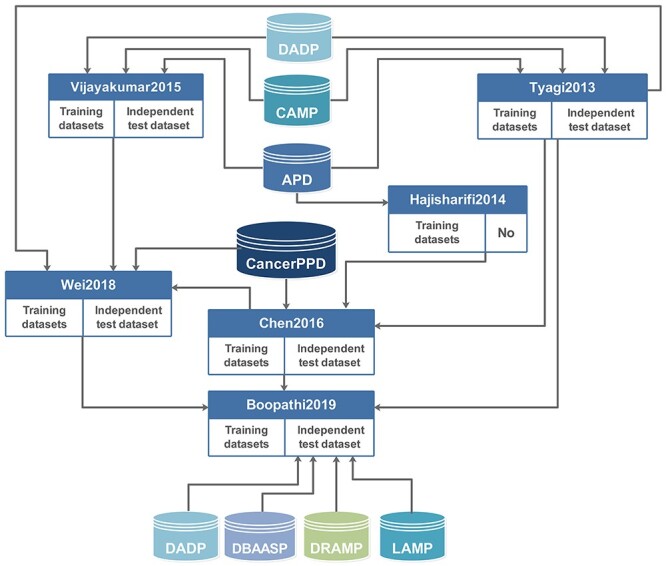
Relationships among the various datasets related to the Boopathi2019 dataset.
Feature extraction
In this study, we employed five different sequence encoding schemes provided in the iFeature software package [41] to encode the protein sequences, which included amino acid composition (AAC), pseudo-amino acid composition (PAAC), Composition, Transition and Distribution (CTD), composition of k-spaced amino acid pairs (CKSAAP), and quasi-sequence-order descriptors (QSOrder). These five sequence encoding schemes are introduced in the following subsections.
AAC
AAC represents the occurrence frequencies of 20 amino acids in a protein sequence. It has been previously used as a feature descriptor in several ACPs predictors [10–12, 15, 18, 20, 22]. For the amino acid aa and a protein sequence s, the occurrence frequency of aa in s is 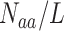 , where
, where 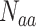 is the occurrence time of aa in s and
is the occurrence time of aa in s and 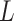 is the length of s. As a result, we obtain the occurrence frequencies of 20 amino acids in s, and then represent s as a 20-dimensional vector using the AAC encoding scheme.
is the length of s. As a result, we obtain the occurrence frequencies of 20 amino acids in s, and then represent s as a 20-dimensional vector using the AAC encoding scheme.
PAAC
The conventional amino acid composition only takes into account the occurrence frequencies of 20 amino acids in a protein sequence. However, in this way it loses the sequence-order information of the protein chain. PAAC incorporates the sequence-order effect and the frequency of 20 amino acids into a composite encoding scheme [42]. In PAAC, a protein sequence is encoded as a (20 + λ)-dimensional vector, where the first 20 components of the vector represent the frequency of 20 amino acids, and the last λ components represent the sequence-order information. Since PAAC is originally proposed, it has proven to be an effective feature encoding scheme and has been widely applied to improve the prediction performance in several domains [43]. It has been employed as a useful feature descriptor in ACPs predictors such as Hajisharifi et al. [2] and ACPred [22]. Calculation of PAAC features of the input protein sequences is performed using the PseAAC webserver [43] and the iFeature package [41]. In this work, we set λ = 4. As a result, a peptide sequence is encoded as a 24-dimensional vector.
The CTD feature
CTD employs seven different physicochemical properties to represent a protein or peptide sequence. For each property, twenty amino acids are partitioned into three groups. CTD uses three descriptors including composition (C), transition (T) and distribution (D) to describe the amino acid distribution of each property in each group along a protein sequence [44]. In our work, we only used the descriptor D to encode a peptide sequence, as D was found to lead to a better predictive performance than C and T descriptors in our preliminary analysis. The descriptor D has been used for prediction of ACPs in ACPred-FL [20], PEPred-Suite [10], mACPpred [23] and ACPred-Fuse [11]. For each property, D uses five descriptor values to denote the fractions of the entire sequence for each of the three groups. Thus, there are 15 descriptor values for each property. Refer to [44] for more details about calculation of the D predictor. Among the seven physicochemical properties, hydrophobicity is represented by seven descriptors in the iFeature package [41]. There are a total of 13 properties consisting of seven types of hydrophobicity and six other properties. Thus, in this work, a peptide sequence is encoded as a vector with 13 × 15 = 195 descriptor values.
CKSAAP
CKSAAP is known as amino acid pair spectrum encoding and has been used for prediction of ACPs in ACPred-Fuse [11]. It transforms a protein sequence into a numerical vector by calculating the occurrence frequency of all possible k-spaced amino acid pairs along the protein sequence. A k-spaced amino acid pair denotes that there are k spaces between any two amino acids in the sequence. For example, in the sequence ‘AXXGXXXT’, ‘AG’ is a 2-spaced amino acid pair, whereas ‘GT’ is a 3-spaced amino acid pair. Let 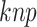 denote the k-spaced amino acid pair, then the frequency of
denote the k-spaced amino acid pair, then the frequency of 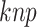 can be defined as follows:
can be defined as follows:
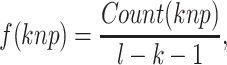 |
(1) |
where 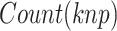 represents the count of
represents the count of 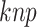 along the protein sequence, while l is the length of the protein sequence.
along the protein sequence, while l is the length of the protein sequence. 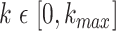 denotes the space between amino acid pairs. Thus,
denotes the space between amino acid pairs. Thus, 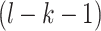 denotes the number of all
denotes the number of all 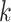 -spaced amino acid pairs along a protein sequence with the length
-spaced amino acid pairs along a protein sequence with the length 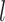 . As a result, the protein sequence is encoded as a 20
. As a result, the protein sequence is encoded as a 20 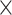 20
20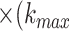 +1
+1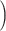 dimensional vector. In this work, we set
dimensional vector. In this work, we set 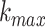 = 4.
= 4.
QSOrder
QSOrder describes the amino acid distribution based on the distance between any two amino acids located in two different positions along a protein or peptide sequence [44]. It has been used for prediction of ACPs in mACPpred [23]. For a protein or peptide sequence P with L amino acids, the sequence-order-coupling is defined as:
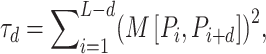 |
(2) |
where the matrix 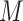 represents the Schneider-Wrede physicochemical distance matrix [45] or the Grantham chemical distance matrix [46], and
represents the Schneider-Wrede physicochemical distance matrix [45] or the Grantham chemical distance matrix [46], and 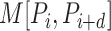 denotes the distance between the two amino acids at positions
denotes the distance between the two amino acids at positions 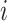 and
and 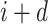 along the sequence P. In this work, we set
along the sequence P. In this work, we set 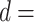 1, 2, 3, 4 and 5, respectively. For each type of amino acid aa, two QSOrder descriptors are defined as:
1, 2, 3, 4 and 5, respectively. For each type of amino acid aa, two QSOrder descriptors are defined as:
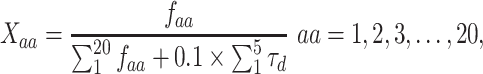 |
(3) |
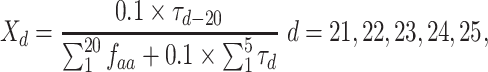 |
(4) |
where  denotes the normalized occurrence of aa in P. Therefore, a peptide sequence is encoded as a 25-dimensional vector using one of the two amino acid distance matrices. There are a total of 50 QSOrder features encoded for a peptide sequence.
denotes the normalized occurrence of aa in P. Therefore, a peptide sequence is encoded as a 25-dimensional vector using one of the two amino acid distance matrices. There are a total of 50 QSOrder features encoded for a peptide sequence.
Framework of ACPredStackL
We summarize the stacking ensemble learning-based framework of ACPredStackL in Figure 2. The three major steps in the overall workflow are respectively described below.
Figure 2 .
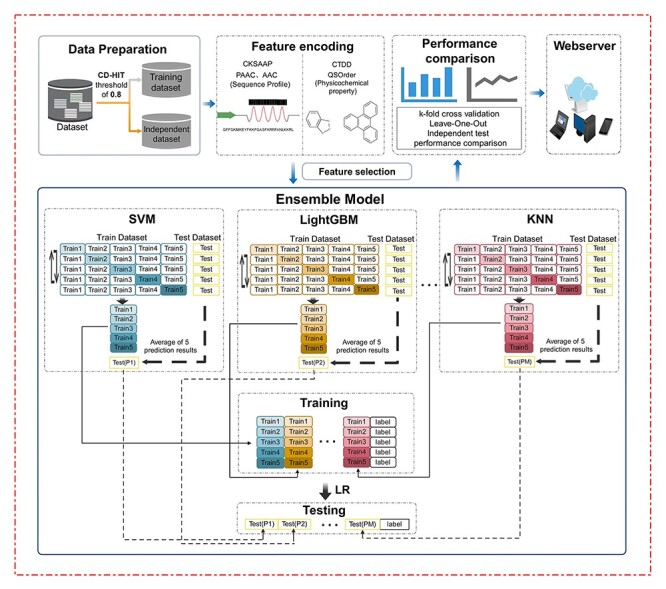
The overall framework of ACPredStack. Its development involves several major stages, including data preparation, feature encoding, ensemble modelling, performance comparison and webserver implementation.
Step 1. Feature representation
The sequences in the input datasets are encoded based on AAC, PAAC, CTD, CKSAAP and QSOrder encoding schemes. The dimensions of features generated by these five encoding schemes are 20, 24, 195, 2000 and 50, respectively. Each peptide sequence in the input datasets is therefore converted into a 2289-dimensional feature vector. Thus, the benchmark training and test datasets used in this study are represented as a 266 × 2289 matrix and a 157 × 2289 matrix, respectively. After that, we employed the ZScore algorithm using the iFeature package [41] to normalize the feature values.
Step 2. Feature selection
Each input sequence is encoded as a high dimensional feature vector (with a total dimension of 2289). High-dimensional features may contain redundant or noisy features that often lead to the decreased predictive performance of the trained model. We employ a two-step feature selection strategy [47–50] to select the most informative features from the original features. The procedure is described as follows. In the first step, the F-score algorithm from the iLearn package [51] is used to rank the features according to their ability to distinguish ACPs from non-ACPs. At the second step, we apply a sequential forward search procedure (FSP) [52] to select the suboptimum subset of features with stronger identification ability of ACPs. Let s and r denote the suboptimum subset and the ranking feature list, respectively, s is initialized with an empty set. FSP first moves the best feature from r to s, and then move d most informative features from r to s, and use 10-fold cross-validation to evaluate the classification performance of features in s. This procedure continues until r becomes empty. The selected subset s generated after such multiple iterations is used as the final suboptimum feature set that achieves the best classification performance. In this study, to improve the feature search efficiency, we first set a larger d = 50 to roughly select the top t features as an initial suboptimum feature subset. Then, we set a smaller d = 5 to further select an optimum feature subset from the features within the range of top [t-100, t + 100] in r. The final optimum s consisted of the top 600 features.
Step 3. Stacking ensemble learning
There exist three different ensemble learning strategies, including boosting, bagging and stacking [29, 53]. In this study, ACPredStackL applied the stacking strategy to integrate KNN, NB, LightGBM and SVM as the base-classifiers (i.e., first-level) and logistic regression as the meta-classifier (i.e., second-level). At the first level, we constructed three SVM classifiers with the RBF kernel by tuning the two hyperparameters C and γ. We named the classifier with C = 2.0 and γ = 0.00049 as SVMa, C = 1.0 and γ = 1 as SVMb, C = 8.0 and γ = 0.00049 as SVMc, respectively. As a result, ACPredStackL integrated six base-classifiers in the first-level learner. For ease of description, let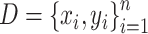 denote the training datasets and
denote the training datasets and 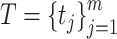 represent the test dataset, respectively. Here,
represent the test dataset, respectively. Here, 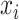 denotes the feature vector of a peptide sequence,
denotes the feature vector of a peptide sequence, 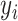 = 1 if
= 1 if 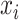 is an ACP and
is an ACP and 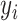 = 0 otherwise. The corresponding
= 0 otherwise. The corresponding 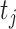 is a peptide sequence that will be predicted as an ACP or non-ACP. The workflow of our stacking ensemble strategy is as follows.
is a peptide sequence that will be predicted as an ACP or non-ACP. The workflow of our stacking ensemble strategy is as follows.
Firstly, the first-level classifiers transform the input datasets into new datasets for the second-level classifier. To avoid overfitting [53], we implemented our stacking model using the stacking cross-validation algorithm provided in the ‘mlxtend’ package [54] to prepare the inputs for the second-level classifier. In our model, 5-fold cross-validation was employed to split into five equal size subsets:
into five equal size subsets: 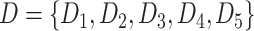 . For each
. For each 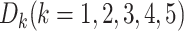 , we learn six base classifiers
, we learn six base classifiers 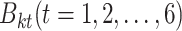 from the remaining 4-fold subsets
from the remaining 4-fold subsets 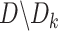 . For each
. For each 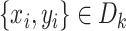 , we obtain a new sample
, we obtain a new sample 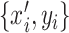 , where
, where 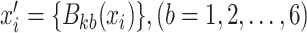 , and then transform
, and then transform 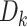 into
into 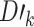 that uses six predicted probability values as new features. In the same way, for each
that uses six predicted probability values as new features. In the same way, for each 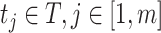 , a feature vector consisting of six predicted probability values
, a feature vector consisting of six predicted probability values 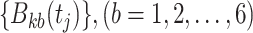 is constructed as a new sample, and then
is constructed as a new sample, and then 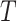 is transformed into a new test dataset
is transformed into a new test dataset 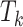 that is represented as a
that is represented as a 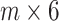 matrix, here
matrix, here 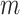 is the number of test samples. After five rounds of iterations, we construct a new training dataset
is the number of test samples. After five rounds of iterations, we construct a new training dataset 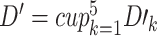 , and five test datasets
, and five test datasets 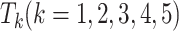 . Then, we integrate five
. Then, we integrate five 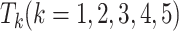 to obtain a final test dataset
to obtain a final test dataset 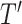 that is represented as a
that is represented as a 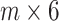 matrix.
matrix. 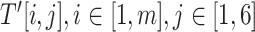 , denotes the cell value of the i-th row and the j-th column of
, denotes the cell value of the i-th row and the j-th column of 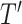 , which is defined as
, which is defined as 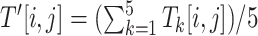 .
.
Secondly, the new training dataset 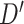 and test dataset
and test dataset 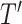 are provided as the input datasets of the second-level classifier. Then a logistic regression classifier is trained based on
are provided as the input datasets of the second-level classifier. Then a logistic regression classifier is trained based on  , and 10-fold cross-validation and jackknife test are conducted to evaluate the trained stacking ensemble model.
, and 10-fold cross-validation and jackknife test are conducted to evaluate the trained stacking ensemble model.  is used to further evaluate the generalization ability of the stacking ensemble model.
is used to further evaluate the generalization ability of the stacking ensemble model.
Performance evaluation
To quantify the performance of ACPredStackL and compare with that of other methods, we used five common performance evaluation metrics [11, 33, 55–65], including sensitivity (Sn), specificity (Sp), precision, accuracy (Acc) and Matthew’s Correlation Coefficient (MCC), which are defined as follows:
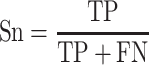 |
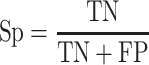 |
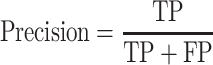 |
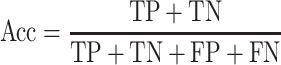 |
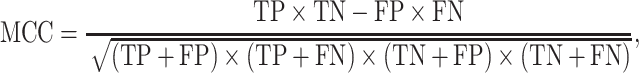 |
(5) |
where 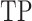 ,
, 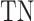 ,
, 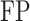 and
and 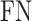 denote the numbers of true positives, true negatives, false positives and false negatives, respectively. In addition, we also plot the receiver-operating characteristic (ROC) curves based on the output of ACPredStackL, and accordingly calculated the Area Under the Curve (AUC) values.
denote the numbers of true positives, true negatives, false positives and false negatives, respectively. In addition, we also plot the receiver-operating characteristic (ROC) curves based on the output of ACPredStackL, and accordingly calculated the Area Under the Curve (AUC) values.
We conducted a series of benchmarking tests including 5-fold cross-validation, 10-fold cross-validation, jackknife cross-validation and independent test to validate and compare the predictive performance of ACPredStackL and other existing predictors.
Results and discussion
In this study, we evaluate and compare the performance of ACPredStackL with other computational methods on several benchmark datasets. We also identify the most important features for the model prediction and interpret prediction outputs of ACPredStackL.
Performance evaluation on benchmark datasets
In this section, we first examine the ability of individual sequence features for identification of ACPs. Five features with the best performance are selected and then used as the input to ACPredStackL to evaluate the feature representation ability of our model. Subsequently, 10-fold cross-validation and independent test are conducted to further evaluate the performance of ACPredStackL.
Performance evaluation of different features
Firstly, we evaluated and examined the ability of different sequence features in distinguishing ACPs from non-ACPs. LightGBM was employed to evaluate the performance of 23 selected features derived from sequences [41]. The detailed results of 10-fold cross-validation test are provided in Supplementary Table S1. The results demonstrate that five types of features, including PAAC, AAC, CTDD, CKSAAP and QSOrder, achieved better classification accuracies, which improved Acc from 83.5 to 88.7. These results indicate that these five types of features could indeed greatly help to distinguish the ACPs from non-ACPs.
Visualization of feature representations
To illustrate the feature representation ability of ACPredStackL, we used the t-SNE plot [66] to visualize representation map with two dimensions that were automatically learned by ACPredStackL. The results are shown in Figure 3. The original input representation map illustrated in Figure 3A shows that there are large overlaps between the ACPs and non-ACPs. Based on the representation map (Figure 3B) learned after the first level of ACPredStackL, we found that many points were classified as the wrong class. However, the representation map (Figure 3C) learned after the second level of ACPredStackL shows that the separation between the ACPs and non-ACPs was almost distinct and could be well distinguished. These results suggest that ACPredStackL is able to effectively learn good feature representations for classifying ACPs.
Figure 3 .
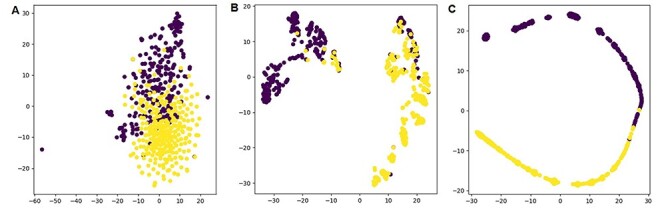
t-SNE plots of the input representation (panel A), feature representation after the first-level layer (panel B), and feature representation after the second-level layer (panel C).
10-fold cross validation test on the benchmark training datasets
To evaluate the performance of ACPredStackL, we first performed 10-fold cross-validation tests on the benchmark training datasets. The performance results are provided in Figure 4 and Supplementary Table S2. The results demonstrate that ACPredStackL outperformed all base classifiers in terms of MCC, Acc and Sn. ACPredStackL and SVMb achieved the highest Sp (0.925). Although ACPredStackL achieved a slightly lower AUC than SVMa and SVMb (0.966 versus 0.963), it performed better than the other five base classifiers in terms of AUC. To further evaluate the performance of ACPredStackL, we compared it with mACPpred [23], which was a state-of-the-art predictor developed based on the same benchmark datasets. From the results provided in Figure 4 and Supplementary Table S2, we conclude that ACPredStackL achieved a slightly better performance compared with mACPpred in terms of MCC (0.839 versus 0.836), Acc (0.918 versus 0.917), and Sn (0.913 versus 0.891), but a slightly lower performance in terms of AUC (0.963 versus 0.968) and Sp (0.925 versus 0.944). These results illustrate that ACPredStackL achieved a competitive performance in ACPs prediction by integrating the multi-features and assembling various base classifiers through the stacking ensemble strategy.
Figure 4 .
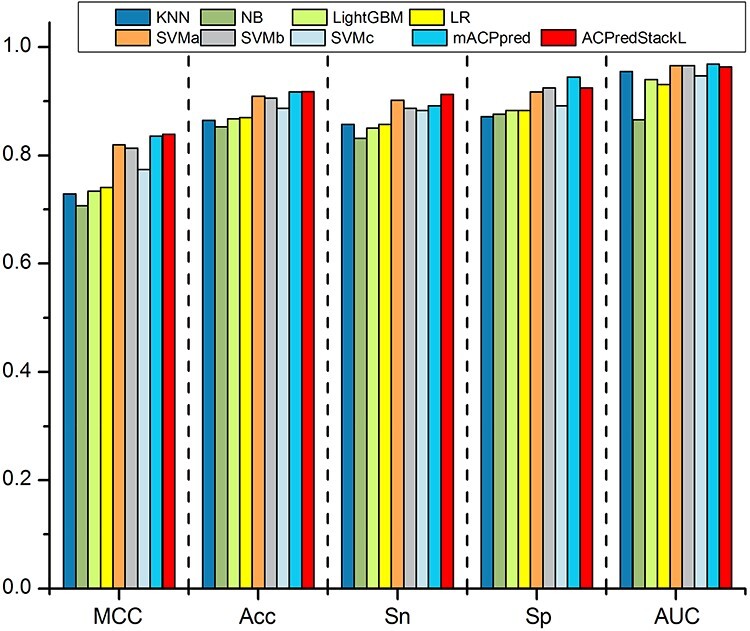
10-fold cross validation test on the benchmark dataset. Note that the values on the y axis represent performance scores of evaluation metrics.
Performance comparison on the independent test datasets
The prediction model with an excellent performance of cross-validation test may have poor generalization ability when assessed on the independent test datasets [23]. Therefore, we further conducted the independent test to validate and evaluate the robustness of ACPredStackL and compared its performance with a number of existing predictors, including iACP [17], SVMACP [12], RFACP [12], mACPpred [23], ACPred-Fuse [11] and ACPred-FL [20]. Among the compared predictors, SVMACP and RFACP are two machine learning-based methods proposed in the same work, which are constructed based on SVM and RF, respectively. The performance comparison results are shown in Table 3. The prediction results of ACPred-Fuse and ACPred-FL were obtained through their webservers, and the results of the other four predictors were retrieved from the literature [23]. The performance results show that ACPredStackL outperformed the other predictors on four out of five evaluation metrics, with the only exception of Sp, for which RFACP achieved best. These results indicate that ACPredStackL provides a better generalization ability and can achieve a more accurate performance than the existing methods. The detailed performance results of all base classifiers on the independent test dataset are provided in Supplementary Table S3. The results clearly show that ACPredStackL achieved the best performance than all other base classifiers in terms of four (out of five) evaluation metrics, with the only exception of Sp, which LightGBM achieved best. Altogether, these results demonstrate this ensemble learning method can improve the performance by combining multiple base learners.
Table 3.
Performance comparison of ACPredStackL, mACPpred, SVMACP, RFACP, iACP, ACPred-Fuse and ACPred-FL on the independent test dataset
| Methods | AUC | MCC | ACC | Sn | Sp |
|---|---|---|---|---|---|
| mACPpred | 0.967 | 0.829 | 0.914 | 0.885 | 0.943 |
| SVMACP | 0.896 | 0.592 | 0.768 | 0.554 | 0.981 |
| RFACP | 0.891 | 0.511 | 0.707 | 0.414 | 1.000 |
| iACP | 0.747 | 0.338 | 0.667 | 0.580 | 0.753 |
| ACPred-Fuse | - | 0.581 | 0.761 | 0.541 | 0.981 |
| ACPred-FL | - | −0.161 | 0.427 | 0.667 | 0.192 |
| ACPredStackL | 0.974 | 0.847 | 0.924 | 0.936 | 0.910 |
Note: Bold-font values denote that corresponding methods achieved the best performances on the corresponding evaluation metrics.
Performance evaluation of ACPredStackL on other benchmark datasets
In this section, we comprehensively evaluate and assess the performance of ACPredStackL on the other benchmark datasets and compare the results with several existing methods.
Performance evaluation on the Yi2019 dataset
As far as we know, ACP-DL [14] is the first deep learning-based predictor. This work has conducted 5-fold cross-validation experiments on two benchmark datasets named ACP740 and ACP240. Figure 5A and B show performance comparison results on the 5-fold cross-validation test using ACP740 and ACP240, respectively. The results of ACP-DL in Figure 5A and B were obtained from the corresponding literature [14]. As can be seen, compared with ACP-DL, ACPredStackL achieved a better performance in terms of Acc (87.44 versus 81.48), MCC (75.06 versus 63.05), Precision (88.73 versus 82.41), Sn (86.44 versus 82.61) and Sp (88.85 versus 80.59) on the ACP740 dataset; as well as higher Acc (86.30 versus 85.42), MCC (73.01 versus 71.44), Precision (89.73 versus 80.28) on the ACP240 dataset.
Figure 5 .
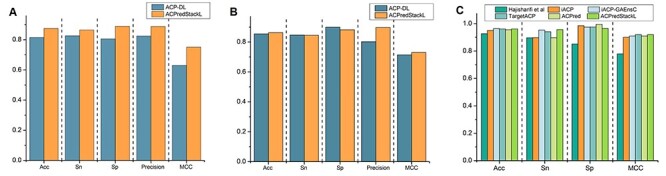
Performance evaluation on the Yi2019 and Hajisharifi2014 datasets. Panel A shows the performance results of ACPredStackL and ACP-DL on the 5-fold cross validation using ACP740. Panel B illustrates the performance results of ACPredStackL and ACP-DL on the 5-fold cross validation using ACP240. Panel C illustrates the performance results of the Hajisharifi’s method, iACP, iACP-GAEnsC, TargetACP, ACPred and ACPredStackL using the Hajisharifi2014 dataset. Note that the values on the y axis represents performance scores of evaluation metrics.
Performance evaluation on the Hajisharifi2014 dataset
Hajisharifi et al. [2] constructed a benchmark dataset containing 136 ACPs and 206 non-ACPs. Several state-of-the-art ACP predictors, including iACP [17], iACP-GAEnsC [13], TargetACP [21] and ACPred [22], are developed and evaluated based on this benchmark dataset through jackknife test. Therefore, we conducted the jackknife test to compare ACPredStackL with the aforementioned predictors. Figure 5C displays the performance comparison results on this benchmark dataset. The results of all the compared methods were obtained from the literature [13, 22]. As can be seen, ACPredStackL and TargetACP achieved the highest MCC value of 0.92, while ACPredStackL obtained the best Sn value of 95.65%. On the other hand, although ACPredStackL had a slightly lower value on Acc than iACP-GAEnsC (96.21% versus 96.45%) and TargetACP (96.21% versus 96.22%), it performed better than other ACPred, iACP and Hajisharifi et al’s method.
Performance evaluation on the Tyagi2013 dataset
Tyagi et al. constructed two benchmark datasets to evaluate their predictor named AntiCP [15]. The training dataset contained 225 ACPs and 225 non-ACPs, and the independent test dataset contained 50 ACPs and 50 non-ACPs, respectively. We conducted 10-fold cross-validation on the training dataset, and independent test on the test dataset. The performance comparison results between ACPredStackL and other methods are illustrated in Figure 6A and B. The results of all other compared methods were obtained from the literature [20], which conducted the same experiments in their original work to evaluate the performance of ACPred-FL. It should be noted that AntiCP contained two predictors: AntiCP_AAC and AntiCP_DC, which were built using the features of dipeptide composition (DC) and amino acid composition (AAC), respectively. Ten-fold cross-validation results (Figure 6A) show that ACPredStackL achieved a better performance than the other compared predictors in terms of all evaluation metrics. Moreover, the independent test results (Figure 6B) show that ACPredStackL achieved the highest accuracy of 0.926, the maximum MCC of 0.851 and Sn of 1.00, respectively.
Figure 6 .
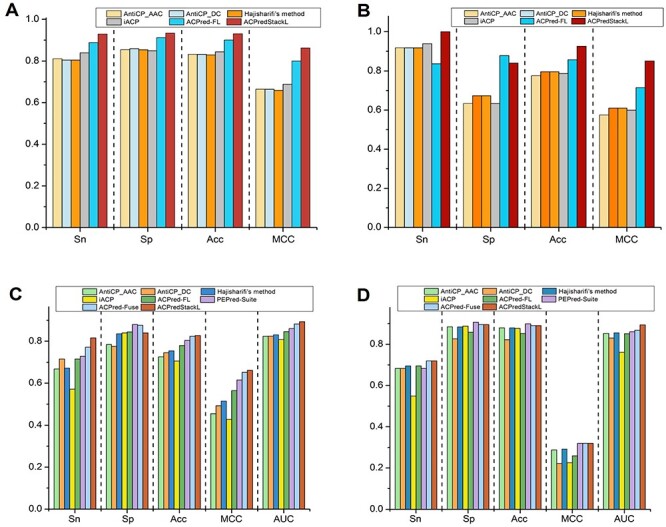
Panel A and B display the performance comparison of AntiCP_AAC, AntiCP_DC, Hajisharifi’s method, iACP, ACPred-FL and ACPredStackL on the Tyagi2013 dataset. Panel C and D illustrate performance comparison of AntiCP_AAC, AntiCP_DC, Hajisharifi’s method, iACP, ACPred_FL, PEPred_Suite and ACPRed_Fuse on the Rao2019 dataset. Note that the values on the y axis represents performance scores of evaluation metrics.
Performance evaluation on the Rao2019 dataset
Rao et al. [11] constructed two benchmark datasets to evaluate their predictor named ACPred-Fuse. The training dataset contained 250 ACPs and 250 non-ACPs, while the independent test dataset included 82 ACPs and 2628 non-ACPs, respectively. We compared ACPredStackL with other seven predictors on these two benchmark datasets. The performance results are illustrated in Figure 6C and D. The results of all the compared methods were obtained from the literature [11]. As can be seen Figure 6C, 10-fold cross-validation results show that ACPredStackL achieved the highest accuracy of 82.8%, as well as the maximum MCC of 0.661, AUC of 0.893 and Sn of 81.6, respectively. The independent test results (Figure 6D) shows that ACPredStackL achieved the same performance as ACPred-Fuse in terms of four evaluation metrics including Sn (72.0%), Sp (89.5%), Acc (89.0%) and MCC (0.320), while ACPredStackL achieved the maximum AUC of 0.894.
Performance evaluation on the Basith2020 dataset
The Basith2020 [36] dataset was an imbalanced one consisting of 246 ACPs and 1733 non-ACPs, respectively. It was constructed as an independent ACP dataset to evaluate the performance of different ACP predictors. In this study, we also used this dataset to further evaluate the performance of our method. The performance comparison results with other currently available ACP predictors are shown in Supplementary Table S4. AUC and MCC are often used as major metrics to evaluate the performance [67]. As can be seen from Supplementary Table S4, ACPredStackL achieved the highest AUC of 0.943. As a comparison, the other ten predictors achieved lower AUC scores ranging from 0.061 to 0.932. In terms of MCC, mACPpred achieved the best performance with an MCC of 0.642, while ACPredStackL achieved the second best MCC of 0.615. The other nine predictors achieved lower MCC ranging from −0.135 to 0.587. To evaluate the balanced performance of Sn and Sp, the balanced accuracy (BACC) was is another primary metric to assess the performance of different predictors [36]. In this regard, ACPredStackL achieved the best performance with a BACC of 0.878. In summary, among the 11 available predictors, ACPredStackL achieved the best performance in terms of BACC and AUC, and the second best MCC. These results demonstrate that ACPredStackL provides a competitive predictive performance of ACPs on the independent Basith2020 dataset.
Performance evaluation of ACPs identification from other peptides
Basith et al. [36] constructed independent data sets of six different types of peptides, including ACP, AHTPs (antihypertensive peptides), ATbPs (antitubercular peptides), AIPs (anti-inflammatory Peptides), QSPs (quorum sensing peptides) and CPPs (cell-penetrating peptides). For each dataset, all the positive samples were experimentally verified peptides. We thus used the experimentally verified peptides from the above six datasets to form a multi-peptide dataset, which contained 246 ACPs sequences and 885 other peptide sequences. We employed the CD-HIT program [40] with the sequence similarity threshold of 0.8 to remove identical peptides, and then used the remaining 189 ACPs sequences as the positive samples and 431 other five types of peptides as the negative samples to construct the new test dataset. Subsequently, we used this new dataset to evaluate the performance of different ACP predictors. The results are shown in Supp Table S5. As can be seen, ACPredStackL achieved the best performance with an MCC of 0.565, Acc of 77.9%, Sn of 87.3% and AUC of 0.884, respectively. The corresponding metrics of the other methods ranged from −0.281 to 0.509, 30.6% to 77.3%, 38.1% to 84.1% and 0.334 to 0.828, respectively. These results again demonstrate that the competitiveness of ACPredStackL in identifying ACPs from other types of peptides than the currently available ACP predictors.
Model interpretation
Interpreting and understanding the model’s output is vitally important in many bioinformatics applications. ACPredStackL combines six base classifiers, including KNN, NB, LightGBM, SVMa, SVMb and SVMc. Among these base classifiers, KNN, NB, SVMa, SVMb and SVMc are ‘Black-box’ classifiers. LightGBM can evaluate the importance of the features according to their weights for predictions; however, the relationship between each feature and the prediction result is unknown. Although ACPredStackL employed the F-score to rank the original features and select the optimal features for model training, we did not know how the selected optimal features would impact on the model predictions, and how exactly ACPredStackL made the prediction is also unknown to us. Recently, a unified framework termed SHapley Additive exPlanations (SHAP) [68] is proposed to interpret the output of the machine learning model. It assigns each feature an SHAP value that represents the effect on the prediction of the trained model with that feature. Let F denote the set of all features, for a feature 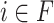 , its SHAP value
, its SHAP value 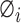 is defined as follows:
is defined as follows:
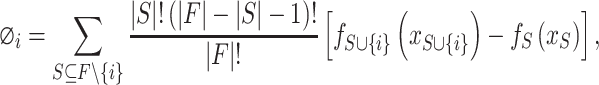 |
(6) |
where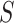 represents the feature subset of
represents the feature subset of 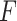 that excludes the feature
that excludes the feature 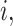 and
and 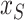 represents the values of the input features in
represents the values of the input features in 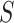 . For all feature subset
. For all feature subset 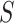 , we retrain two models
, we retrain two models 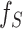 and
and 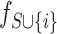 , and compare the predictions from the two models on the current input
, and compare the predictions from the two models on the current input 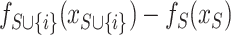 . According to Equation 6, the SHAP value of each feature can be computed and used to represent the effect on the model prediction.
. According to Equation 6, the SHAP value of each feature can be computed and used to represent the effect on the model prediction.
In this study, we applied the SHAP framework to interpret the predictions of our model. The distribution of the impact of the top 20 selected features on the output of ACPredStackL is illustrated in Figure 7. As can be seen, a number of features with high values achieved positive SHAP values, and other features had lower and negative SHAP values. Positive SHAP values indicate the prediction of ACPs, while negative SHAP values indicate the prediction of non-ACPs. Taking CTDD (163) in Figure 7 as an example, we found that its values were relatively high for most ACPs samples and low for most non-ACPs samples. Thus, for a new sample whose class (ACP or non-ACPs) is unknown, if CTDD (163)‘s value in the sample is very high then the predictive result will favour ACP, otherwise non-ACP. Certainly, the final predictive result will be made by all feature attributions in the sample.
Figure 7 .
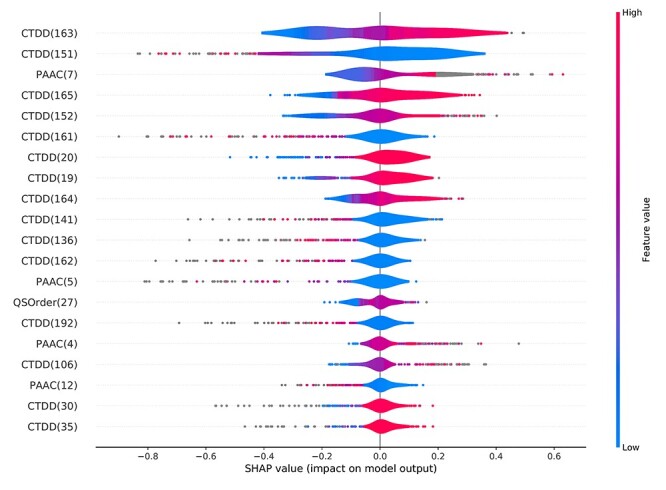
The top 20 features based on SHAP values generated for prediction of ACPs using ACPredStackL. Feature values are indicated by colors (low: blue; high: red). SHAP values greater than zero indicate prediction of ACPs, while SHAP values less than zero indicate prediction of non-ACPs. Note: each feature has a high dimension, the number within bracket behind each feature indicates which dimension of the feature.
To further illustrate the impact of these features on the model output, we plotted the distributions of the top six features for both ACPs and non-ACPs in Figure 8. The results indicate that the mean values of the six features were clearly distinct between the positive and negative samples. We further performed a student’s t-test to examine the statistical difference. The results indicate that the P-values were less than or equal to 0.01 for all of the top six features. This means that the distributions of each feature were significantly different between the positive and negative samples. Similar results for the distributions of the other 14 features in the positive and negative samples are illustrated in Supplementary Figure S1. Taken together, these results highlight that the features employed in ACPredStackL have a strong representation and discriminative ability for the prediction of ACPs.
Figure 8 .
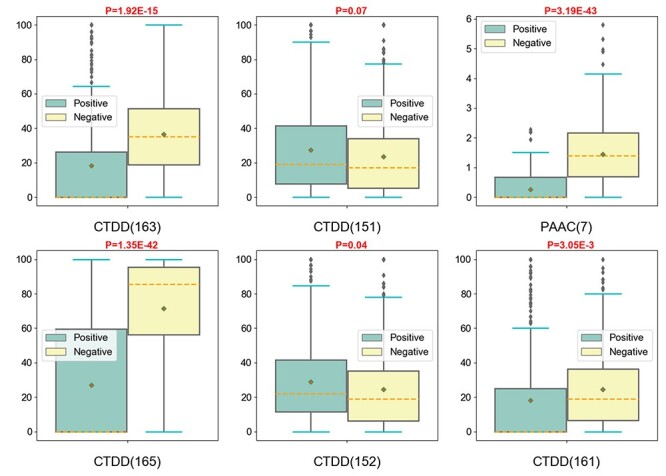
Distribution of the values of the six best-performing features between the positive and negative samples. Note that the values on the y axis represent feature values in samples.
Case studies
The explanation model based on the additive feature attribution methods [68] is defined as follows:
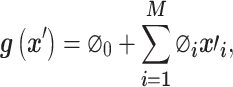 |
(7) |
where 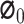 represents the base value of the model output, and
represents the base value of the model output, and 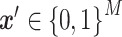 represents a sample vector with M features,
represents a sample vector with M features, 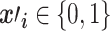 represents the
represents the 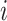 th feature of
th feature of 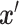 and
and 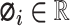 denotes the attribution values (in Equation 6) of the
denotes the attribution values (in Equation 6) of the 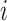 th feature. Accordingly, a model’s output is explained as a sum of SHAP values of each input feature.
th feature. Accordingly, a model’s output is explained as a sum of SHAP values of each input feature.
We performed the case studies by selecting four synthesized peptide sequences (Table 4) from [69] to further evaluate the predictive capability of ACPredStackL. Among these four peptides, Gradient2 is validated to be active against breast adenocarcinoma (MCF7) cells, and the other three are active against both of the MCF7 and lung adenocarcinoma (A549) cancer cell lines. To reduce redundancy and avoid over-estimation, we employed the CD-HIT program [40] with a sequence similarity threshold of 0.8 to check four peptides against the benchmark training datasets. The prediction results for these four case study peptides are provided in the Table 4. As can be seen, all the four peptides were correctly predicted as ACPs by ACPredStackL. The visualization of the model outputs is illustrated in Supplementary Figure S2. As can be seen from Table 4 and Supplementary Figure S2, the base SHAP value of the model output was 0.39 which represented the average model output over the training dataset. The output SHAP values for these four peptides were 0.69, 0.67, 0.70 and 0.68, respectively. The real numbers on the horizontal axis in each subfigure of Supplementary Figure S2 represents SHAP values, red bars denote the positive feature attributions that push the prediction higher, and blue bars denote the negative feature attributions that push the prediction lower. In Supplementary Figure S2, both the normalization feature value and the feature attribution range of each feature were below the horizontal axis, the sum of all feature attributions plus the base SHAP value was represented as the prediction output of the model. Taking the peptide Helical1 (Supplementary Figure S2A) as an example, six features including AAC (4), QSOrder (27), CTDD (20), PAAC (7), PAAC (4), QSOrder (44) made positive contributions to the prediction of ACPs. Four out of six features were included in the top 20 selected features (see Figure 7), including QSOrder (27), CTDD (20), PAAC (7) and PAAC (4). Thus, these results can explain how the ACPredStackL model made the final decision of the prediction. Thereby improving the model interpretability.
Table 4.
Four active peptides against cancer cells used in the case study
| Peptide ID | Peptide sequence | Prediction | Base SHAP value | Output SHAP values |
|---|---|---|---|---|
| Helical1 | FLWIKLGKLAGAVLKLILGLKKVV | ACP | 0.39 | 0.69 |
| AmphiArc1 | KWVKKVHNWLRRWIKVFEALFG | 0.67 | ||
| AmphiArc2 | KIFKKFKTIIKKVWRIFGRF | 0.70 | ||
| Gradient2 | AWLKRIKKFLKALFWVWVW | 0.68 |
Webserver implementation
To facilitate community-wide effort for the prediction of ACPs in a cost-effective and high-throughput manner, we have implemented an online webserver freely accessible (http://bigdata.biocie.cn/ACPredStackL/). The view page was implemented using HTML, bootstrap, jquery and ajax. The server side used the springboot framework to receive the requests from users, and then called the constructed ACPredStackL model to make ACPs predictions. The webserver is managed using Tomcat 7 and configured in a Linux environment on a 4-core cloud server machine with 16 GB of memory and a 500 GB hard disk.
Step-by-step instructions for submitting a job to the webserver is described as follows: First, users need to paste their peptide sequences in the FASTA format in the text area. In addition, users can alternatively upload a file containing peptide sequences in the FASTA format. Second, users need to click the ‘Submit’ button to submit the job. In addition, users also have the option to provide their email addresses so that they can receive a notification email once the submitted job is completed. Finally, after the submitted job is completed, user can view the prediction results from the job list page. In this case, a notification email will be automatically sent to users; users can view the job details by clicking a hyperlink contained in the email.
Discussions and Limitations
The existing predictors of ACPs identification were trained and tested on various benchmark datasets. All these predictors do not provide the stand-alone software; as such, we were not able to train them on selected benchmark datasets to compare with ACPredStackL. However, if a predictor provides the webserver that can predict ACPs from the manual input protein sequences, we compared ACPredStackL to the predictor via the webserver using benchmark independent test datasets (see Section ‘10-fold cross validation test on the benchmark training datasets’). ACPredStackL has achieved a competitive performance on the benchmark training datasets when compared with mACPpred. Further, ACPredStackL outperformed mACPpred, SVMACP, RFACP, iACP, ACPred-Fuse, ACPred-FL on four out of five evaluation metrics using independent test datasets. These results indicate that ACPredStackL has great capacity and utility.
Moreover, we compared the performance of ACPredStackL and other predictors on various test datasets (see Section ‘Performance evaluation of ACPredStackL on other benchmark datasets’), which almost covered all the benchmark datasets used in existing predictors. Compared with ACP-DL, the only deep learning-based method of ACPs identification, ACPredStackL achieved better performances in terms of all major evaluation metrics on the ACP740 dataset, and in terms of three out of five evaluation metrics on the ACP240 dataset, respectively. Generally speaking, deep learning-based predictors require sufficiently large amounts of training data in order to achieve better performance. However, there are only a small number of known ACPs currently available. Thus, the performance of deep learning methods such as ACP-DL is expected to be further improved in accordance with the increasingly available ACPs data in the future. In terms of the two primary performance metrics Acc and MCC, ACPredStackL outperformed a number of conventional machine learning based-predictors, including Hajisharifi et al, iACP, ACPred, AntiCP-AAC, AntiCP-DC and ACP-FL. Visualization representation maps learned by ACPredStackL clearly indicate that ACPredStackL is a reliable feature representation method. Altogether, these results demonstrate that ACPredStackL is a powerful predictor of ACPs.
All the existing predictors including ACPredStackL are ML-based approaches, which have the following limitations: First, ML-based predictors typically need sufficiently large training datasets. However, the number of currently available ACPs has limited the performance of existing predictors. For a newly discovered ACP, if its physicochemical or sequence features are significantly different from those in the current ACP databases, it would not be surprising that this APCs would be incorrectly predicted by the existing methods. Second, there is a need to develop new and informative feature representation schemes for training machine learning models from protein or peptide sequence information. In particular, ACP-specific feature representation schemes can better describe and represent the biological properties of ACPs and thus might be useful for further improving the performance of ACPs predictors.
Conclusion and future work
In this study, we have comprehensively reviewed all existing ACP identification methods and proposed ACPredStackL, a new stacking ensemble learning-based predictor for ACPs. We performed a comprehensive benchmarking evaluation of the currently available tools for the ACP prediction on multiple datasets, which almost covered all benchmark datasets used by current predictors. The results demonstrated that ACPredStackL improves the performance of ACP prediction by leveraging the merits of all assembled individual models. ACPredStackL outperforms the majority of the existing predictors on two comprehensive metrics (Acc and MCC). A user-friendly webserver of ACPredStackL is developed to facilitate high-throughput prediction and is freely available at http://bigdata.biocie.cn/ACPredStackL/. Overall, ACPredStackL is anticipated to be a powerful tool for the accurate and high-throughput prediction of ACPs from sequences information. We hope that this comprehensive review and the developed ACPredStackL tool will provide useful insights for ACPs prediction, facilitate efforts for identification and functional characterization of novel ACPs and inspire follow-up research in the future.
There exist more than 7000 naturally occurring peptides that are often associated with human physiology [70]. A number of peptide therapeutics are currently being evaluated in clinical trials. In this context, high-throughput computational methods are urgently needed to identify promising peptides with therapeutic potentials from sequences. However, most computational methods are tailored for specific type of peptides and can only be used to identify special peptides belong to a specific type. In future work, we plan to construct a general and more robust model that can be used to simultaneously predict multiple different types of peptides.
Key Points
We provide a comprehensive survey and summary of the existing machine learning-based methods for ACPs identification.
We propose a novel stacking ensemble learning-based method, termed ACPredStackL, to improve the predictive performance of ACPs.
Extensive benchmarking tests demonstrate that ACPredStackL achieves a better performance and outperforms the existing methods on multiple benchmark datasets.
A publicly available webserver (http://bigdata.biocie.cn/ACPredStackL/) is developed as an implementation of ACPredStackL to facilitate community-wide efforts for ACPs identification in a high-throughput manner.
Supplementary Material
Xiao Liang is currently a master student in the College of Information Engineering, Northwest A&F University, China. Her research interests are bioinformatics, data mining and machine learning.
Fuyi Li received his PhD in Bioinformatics from Monash University, Australia. He is currently a researcher fellow in the Peter Doherty Institute for Infection and Immunity, The University of Melbourne, Australia. His research interests are bioinformatics, computational biology, machine learning and data mining.
Jinxiang Chen is currently a master student in College of Information Engineering, Northwest A&F University, China. His research interests are bioinformatics, data mining and machine learning.
Junlong Li is a Bachelor student in the College of Information Engineering, Northwest A&F University, China. His research interests are bioinformatics, data mining and software engineering.
Hao Wu is an associate professor in the College of Information Engineering, Northwest A&F University, China. Her major research interests include bioinformatics and data mining.
Shuqin Li is a professor in the College of Information Engineering, Northwest A&F University, China. Her major research interests include bioinformatics, data mining and big data.
Jiangning Song is currently an associate professor and group leader in the Biomedicine Discovery Institute and Department of Biochemistry and Molecular Biology, Monash University, Melbourne, Australia. He is also affiliated with the Monash Centre for Data Science, Faculty of Information Technology, Monash University. His research interests include bioinformatics, computational biology, machine learning, data mining and pattern recognition.
Quanzhong Liu is an associate professor in the College Information Engineering, Northwest A&F University, Yangling, China. He received his PhD in Agricultural Electrisation and Automatization from Northwest A&F University, China. His research interests include data mining, machine learning, big data and bioinformatics.
Contributor Information
Xiao Liang, College of Information Engineering, Northwest A&F University, Yangling, 712100, China; Shaanxi Key Laboratory of Agricultural Information Perception and Intelligent Service, Yangling, Shaanxi 712100, China.
Fuyi Li, Monash Biomedicine Discovery Institute and Department of Biochemistry and Molecular Biology, Monash University, Melbourne, VIC 3800, Australia; Monash Centre for Data Science, Monash University, Melbourne, VIC 3800, Australia; Department of Microbiology and Immunology, Peter Doherty Institute for Infection and Immunity, University of Melbourne, Melbourne, Victoria, Australia.
Jinxiang Chen, College of Information Engineering, Northwest A&F University, Yangling, 712100, China.
Junlong Li, College of Information Engineering, Northwest A&F University, Yangling, 712100, China.
Hao Wu, College of Information Engineering, Northwest A&F University, Yangling, 712100, China.
Shuqin Li, College of Information Engineering, Northwest A&F University, Yangling, 712100, China; Shaanxi Key Laboratory of Agricultural Information Perception and Intelligent Service, Yangling, Shaanxi 712100, China.
Jiangning Song, Monash Biomedicine Discovery Institute and Department of Biochemistry and Molecular Biology, Monash University, Melbourne, VIC 3800, Australia; Monash Centre for Data Science, Monash University, Melbourne, VIC 3800, Australia; ARC Centre of Excellence in Advanced Molecular Imaging, Monash University, Melbourne, VIC 3800, Australia.
Quanzhong Liu, College of Information Engineering, Northwest A&F University, Yangling, 712100, China; Shaanxi Key Laboratory of Agricultural Information Perception and Intelligent Service, Yangling, Shaanxi 712100, China.
Funding
The National Natural Science Foundation of China (61972322); the National Health and Medical Research Council of Australia (NHMRC, grant, 1092262); the Australian Research Council (ARC, LP110200333 and DP120104460); the National Institute of Allergy and Infectious Diseases of the National Institutes of Health (R01 AI111965); Monash University (awarded a Major Inter-Disciplinary Research (IDR) project); and the Collaborative Research Program of Institute for Chemical Research, Kyoto University (2018-28).
References
- 1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. Ca-a Cancer J Clin 2019;69:7–34. [DOI] [PubMed] [Google Scholar]
- 2. Hajisharifi Z, Piryaiee M, Beigi MM, et al. Predicting anticancer peptides with Chou's pseudo amino acid composition and investigating their mutagenicity via Ames test. J Theor Biol 2014;341:34–40. [DOI] [PubMed] [Google Scholar]
- 3. Holohan C, Van Schaeybroeck S, Longley DB, et al. Cancer drug resistance: an evolving paradigm. Nat Rev Cancer 2013;13:714–26. [DOI] [PubMed] [Google Scholar]
- 4. Qin Y, Qin ZD, Chen J, et al. From antimicrobial to anticancer peptides: the transformation of peptides. Recent Pat Anticancer Drug Discov 2019;14:70–84. [DOI] [PubMed] [Google Scholar]
- 5. Tyagi A, Tuknait A, Anand P, et al. CancerPPD: a database of anticancer peptides and proteins. Nucleic Acids Res 2015;43:D837–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Thundimadathil J. Cancer treatment using peptides: current therapies and future prospects. Journal of amino acids 2012 2012;967347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Novkovic M, Simunic J, Bojovic V, et al. DADP: the database of anuran defense peptides. Bioinformatics 2012;28:1406–7. [DOI] [PubMed] [Google Scholar]
- 8. Thomas S, Karnik S, Barai RS, et al. CAMP: a useful resource for research on antimicrobial peptides. Nucleic Acids Res 2010;38:D774–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wang G, Li X, Wang Z. APD2: the updated antimicrobial peptide database and its application in peptide design. Nucleic Acids Res 2009;37:D933–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wei L, Zhou C, Su R, et al. PEPred-suite: improved and robust prediction of therapeutic peptides using adaptive feature representation learning. Bioinformatics 2019;35:4272–80. [DOI] [PubMed] [Google Scholar]
- 11. Rao B, Zhou C, Zhang G, et al. ACPred-fuse: fusing multi-view information improves the prediction of anticancer peptides. Brief Bioinform 2019. doi: 10.1093/bib/bbz088. [DOI] [PubMed] [Google Scholar]
- 12. Manavalan B, Basith S, Shin TH, et al. MLACP: machine-learning-based prediction of anticancer peptides. Oncotarget 2017;8:77121–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Akbar S, Hayat M, Iqbal M, et al. iACP-GAEnsC: evolutionary genetic algorithm based ensemble classification of anticancer peptides by utilizing hybrid feature space. Artif Intell Med 2017;79:62–70. [DOI] [PubMed] [Google Scholar]
- 14. Yi H-C, You Z-H, Zhou X, et al. ACP-DL: a deep learning long short-term memory model to predict anticancer peptides using high-efficiency feature representation. Mol Ther-Nucleic Acids 2019;17:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tyagi A, Kapoor P, Kumar R, et al. In Silico models for designing and discovering novel anticancer peptides. Sci Rep 2013;3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Vijayakumar S, Lakshmi PTV. ACPP: a web server for prediction and Design of Anti-cancer Peptides. Int J Peptide Res Therap 2015;21:99–106. [Google Scholar]
- 17. Chen W, Ding H, Feng P, et al. IACP: a sequence-based tool for identifying anticancer peptides. Oncotarget 2016;7:16895–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Li F-M, Wang X-Q. Identifying anticancer peptides by using improved hybrid compositions. Sci Rep 2016;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Xu L, Liang G, Wang L, et al. A novel hybrid sequence-based model for identifying anticancer peptides. Genes 2018;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wei L, Zhou C, Chen H, et al. ACPred-FL: a sequence-based predictor using effective feature representation to improve the prediction of anti-cancer peptides. Bioinformatics 2018;34:4007–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kabir M, Arif M, Ahmad S, et al. Intelligent computational method for discrimination of anticancer peptides by incorporating sequential and evolutionary profiles information. Chemom Intel Lab Syst 2018;182:158–65. [Google Scholar]
- 22. Schaduangrat N, Nantasenamat C, Prachayasittikul V, et al. ACPred: a computational tool for the prediction and analysis of anticancer peptides. Molecules 2019;24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Boopathi V, Subramaniyam S, Malik A, et al. mACPpred: a support vector machine-based meta-predictor for identification of anticancer peptides. Int J Mol Sci 2019;20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Agrawal P, Bhagat D, Mahalwal M, et al. AntiCP 2.0: an updated model for predicting anticancer peptides. Brief Bioinform 2020. doi: 10.1093/bib/bbaa153. [DOI] [PubMed] [Google Scholar]
- 25. Chou KC, Zhang CT. Prediction of protein structural classes. Crit Rev Biochem Mol Biol 1995;30:275–349. [DOI] [PubMed] [Google Scholar]
- 26. Dao F-Y, Lv H, Wang F, et al. Identify origin of replication in Saccharomyces cerevisiae using two-step feature selection technique. Bioinformatics 2019;35:2075–83. [DOI] [PubMed] [Google Scholar]
- 27. Chou K-C. Some remarks on protein attribute prediction and pseudo amino acid composition. J Theor Biol 2011;273:236–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chen X, Lin X. Big data deep learning: challenges and perspectives. IEEE Access 2:514–25. [Google Scholar]
- 29. Zhou Z. Ensemble Methods Foundations and Algorithms. CRC Press, 2012. [Google Scholar]
- 30. Polikar R. Ensemble based systems in decision making. IEEE Circ Sys Magazine 2006;6:21–45. [Google Scholar]
- 31. Liu B, Wang S, Dong Q, et al. Identification of DNA-binding proteins by combining auto-cross covariance transformation and ensemble learning. IEEE Trans Nanobioscience 2016;15:328–34. [DOI] [PubMed] [Google Scholar]
- 32. Mishra A, Pokhrel P, Hoque MT. StackDPPred: a stacking based prediction of DNA-binding protein from sequence. Bioinformatics 2019;35:433–41. [DOI] [PubMed] [Google Scholar]
- 33. Zhang Y, Yu S, Xie R, et al. PeNGaRoo, a combined gradient boosting and ensemble learning framework for predicting non-classical secreted proteins. Bioinformatics 2020;36:704–12. [DOI] [PubMed] [Google Scholar]
- 34. Li F, Chen J, Ge Z, et al. Computational prediction and interpretation of both general and specific types of promoters in Escherichia coli by exploiting a stacked ensemble-learning framework. Brief Bioinform 2020. doi: 10.1093/bib/bbaa049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yi HC, You ZH, Wang MN, et al. RPI-SE: a stacking ensemble learning framework for ncRNA-protein interactions prediction using sequence information. Bmc Bioinformatics 2020;21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Basith S, Manavalan B, Shin TH, et al. Machine intelligence in peptide therapeutics: a next-generation tool for rapid disease screening. Med Res Rev 2020;40:1276–314. [DOI] [PubMed] [Google Scholar]
- 37. Pirtskhalava M, Gabrielian A, Cruz P, et al. DBAASP v.2: an enhanced database of structure and antimicrobial/cytotoxic activity of natural and synthetic peptides. Nucleic Acids Res 2016;44:D1104–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Fan LL, Sun J, Zhou MF, et al. DRAMP: a comprehensive data repository of antimicrobial peptides. Sci Rep 2016;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zhao X, Wu H, Lu H, et al. LAMP: a database linking antimicrobial peptides. PLOS One 2013;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Huang Y, Niu B, Gao Y, et al. CD-HIT suite: a web server for clustering and comparing biological sequences. Bioinformatics 2010;26:680–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Chen Z, Zhao P, Li F, et al. iFeature: a python package and web server for features extraction and selection from protein and peptide sequences. Bioinformatics 2018;34:2499–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chou KC. Prediction of protein cellular attributes using pseudo-amino acid composition, Proteins-Structure Function and. Genetics 2001;43:246–55. [DOI] [PubMed] [Google Scholar]
- 43. Shen H-B, Chou K-C. PseAAC: a flexible web server for generating various kinds of protein pseudo amino acid composition. Anal Biochem 2008;373:386–8. [DOI] [PubMed] [Google Scholar]
- 44. Li ZR, Lin HH, Han LY, et al. PROFEAT: a web server for computing structural and physicochemical features of proteins and peptides from amino acid sequence. Nucleic Acids Res 2006;34:W32–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Schneider G, Wrede P. The rational design of amino-acid-sequences by artificial neural networks and simulated MOLECULAR EVOLUTION - DE-NOVO DESIGN of an idealized leader peptidase cleavage site. Biophys J 1994;66:335–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Grantham R. Amino acid difference formula to help explain protein evolution. Science (New York, NY) 1974;185:862–4. [DOI] [PubMed] [Google Scholar]
- 47. Wang M, Zhao X-M, Tan H, et al. Cascleave 2.0, a new approach for predicting caspase and granzyme cleavage targets. Bioinformatics 2014;30:71–80. [DOI] [PubMed] [Google Scholar]
- 48. Wei L, Luan S, Nagai LAE, et al. Exploring sequence-based features for the improved prediction of DNA N4-methylcytosine sites in multiple species. Bioinformatics (Oxford, England) 2019;35:1326–33. [DOI] [PubMed] [Google Scholar]
- 49. Li F, Li C, Wang M, et al. GlycoMine: a machine learning-based approach for predicting N-, C- and O-linked glycosylation in the human proteome. Bioinformatics 2015;31:1411–9. [DOI] [PubMed] [Google Scholar]
- 50. Li F, Li C, Revote J, et al. GlycoMine(struct): a new bioinformatics tool for highly accurate mapping of the human N-linked and O-linked glycoproteomes by incorporating structural features. Sci Rep 2016;6:34595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Chen Z, Zhao P, Li F, et al. iLearn: an integrated platform and meta-learner for feature engineering, machine-learning analysis and modeling of DNA, RNA and protein sequence data. Brief Bioinformatics 2020;21:1047–57. [DOI] [PubMed] [Google Scholar]
- 52. Whitney AWA. Direct method of nonparametric measurement selection. IEEE Trans Comput 1971;20:1100–3. [Google Scholar]
- 53. Aggarwal CC. Data classification: algorithms and applications. CRC Press, 2015. [Google Scholar]
- 54. Raschka S. MLxtend: providing machine learning and data science utilities and extensions to Python’s scientific computing stack. J Open Source Software 2018;3(24):638. [Google Scholar]
- 55. Jia C, Bi Y, Chen J, et al. PASSION: an ensemble neural network approach for identifying the binding sites of RBPs on circRNAs. Bioinformatics (Oxford, England) 2020. doi: 10.1093/bioinformatics/btaa522. [DOI] [PubMed] [Google Scholar]
- 56. Song J, Li F, Leier A, et al. PROSPERous: high-throughput prediction of substrate cleavage sites for 90 proteases with improved accuracy. Bioinformatics 2018;34:684–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Cheng L, Zhao H, Wang P, et al. Computational methods for identifying similar diseases. Mol Ther Nucleic Acids 2019;18:590–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Wei L, Hu J, Li F, et al. Comparative analysis and prediction of quorum-sensing peptides using feature representation learning and machine learning algorithms. Brief Bioinform 2018;21:106–19. [DOI] [PubMed] [Google Scholar]
- 59. Su R, Wu H, Liu X, et al. Predicting drug-induced hepatotoxicity based on biological feature maps and diverse classification strategies. Brief Bioinform 2019. [DOI] [PubMed] [Google Scholar]
- 60. Wei L, Su R, Luan S, et al. Iterative feature representations improve N4-methylcytosine site prediction. Bioinformatics 2019;35:4930–7. [DOI] [PubMed] [Google Scholar]
- 61. Li F, Leier A, Liu Q, et al. Procleave: predicting protease-specific substrate cleavage sites by combining sequence and structural information. Genomics Proteomics Bioinformatics 2020. doi: 10.1016/j.gpb.2019.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Jia C, Bi Y, Chen J, et al. PASSION: an ensemble neural network approach for identifying the binding sites of RBPs on circRNAs. Bioinformatics 2020. doi: 10.1093/bioinformatics/btaa522. [DOI] [PubMed] [Google Scholar]
- 63. Li F, Chen J, Leier A, et al. DeepCleave: a deep learning predictor for caspase and matrix metalloprotease substrates and cleavage sites. Bioinformatics 2020;36:1057–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Li F, Wang Y, Li C, et al. Twenty years of bioinformatics research for protease-specific substrate and cleavage site prediction: a comprehensive revisit and benchmarking of existing methods. Brief Bioinform 2019;20:2150–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Li F, Zhang Y, Purcell AW, et al. Positive-unlabelled learning of glycosylation sites in the human proteome. BMC Bioinformatics 2019;20:112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Maaten Lvd HG. Visualizing data using t-SNE. J Mach Learn Res 2008;9:2579–605. [Google Scholar]
- 67. Li F, Li C, Marquez-Lago TT, et al. Quokka: a comprehensive tool for rapid and accurate prediction of kinase family-specific phosphorylation sites in the human proteome. Bioinformatics 2018;34:4223–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Lundberg S, Lee S-I. A unified approach to interpreting model predictions, 31st Conference on Neural Information Processing Systems (NIPS), Long Beach, California. USA: ACM 2017;23:4765–74. [Google Scholar]
- 69. Gabernet G, Gautschi D, Mueller AT, et al. In silico design and optimization of selective membranolytic anticancer peptides. Sci Rep 2019;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Fosgerau K, Hoffmann T. Peptide therapeutics: current status and future directions. Drug Discov Today 2015;20:122–8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.