

Abstract
An additive genetic model is usually employed in case-control-based genome-wide association studies. The model usually encodes "AA", "Aa" and "aa" ("a" represents the minor allele) as three different numbers, implying the contribution of genotype "Aa" to the phenotype is different from "AA" and "aa". From the perspective of biological phenomena, the coding is reasonable since the phenotypes of lives are not "black and white". A case-control based study, however, has only two phenotypes, case and control, which means that the phenotypes are "black and white". It suggests that a recessive/dominant model may be an alternative to the additive model. In order to investigate whether the alternative is feasible, we conducted comparative experiments on several models used in those studies through chi-square test and logistic regression. Our simulation experiments demonstrate that a recessive model is better than the additive model. The area under the curve of the former has increased by 5% compared with the latter, the discrimination of identifying risk single nucleotide polymorphisms has been improved by 61%, and the precision has also reached 1.10 times that of the latter. Furthermore, the real data experiments show that the precision and area under the curve of the former are 16% and 20% higher than the latter respectively, and the area under the curve of dominant model of the former is 13% higher than the latter. The results indicate a recessive/dominant model may be an alternative to the additive model and suggest a new route for case-control-based studies.
Introduction
The single nucleotide polymorphisms (SNPs) may lead to changes in individual characteristics or phenotypes, resulting in changes in disease risk or physiological characteristics [1]. How to explore the association between genes and diseases from the changes in genomes is a meaningful work. Ozaki K. et al. published such study firstly in 2002, which explored the association between functional SNPs in lymphotoxin-α gene and the susceptibility to myocardial infarction [2]. After this study, a landmark genome-wide association study (GWAS) was born in 2005—Klein R.J. et al. investigated a group of patients with age-related macular degeneration (AMD) and found two SNPs with significantly altered allele frequency compared to the healthy controls [3]. GWAS tested hundreds of thousands to millions of genetic variants in the human genomes to identify genotype-phenotype associations and has revolutionized the field of genetics of complex diseases in the past decade [4,5]. Since the publication of AMD GWAS, GWASes have achieved great success [6–8], and more than 50,000 significant genome-wide associations between genetic variants and common diseases/traits have been reported [9]. The emergence of International HapMap Project [10] and Biobank [11] promoted the development of GWAS further. As of 2017, more than 3,000 human GWAS had tested more than 1,800 diseases and traits, and revealed thousands of SNP associations [12]. The most commonly used method for GWAS is the case-control setup, which compares two large groups of individuals, a case group affected by a disease and a healthy control group. Early statistical power calculations indicated that this method might be better than linkage studies at detecting weak genetic effects [13].
There are usually three genetic models of genes: additive model (AM), dominant model (DM), and recessive model (RM). From AMD research in 2005 to the most recent study of genetic variants in infant and early childhood growth [3,14–22], the most commonly used model in GWAS is additive [23,24]. The genotypes "AA", "Aa" and "aa" ("a" is the minor allele) are coded as three different numbers in a genome dataset with AM. The coding implies that the contribution of genotype "Aa" to phenotype is different from "AA" and "aa". It is reasonable since the phenotypes of lives are not "black and white". But, for a case-control-based GWAS, the phenotype of an individual is either case or control, which indicates that the genotypes in the study are "black and white". Thus, we believe that RM/DM may be an alternative to AM in case-control-based GWASes. Our comparison experiment of simulation and real data show that RM/DM can better represent the phenotypic manifestations of case-control-based GWAS datasets than AM. And it has higher area under the curve (AUC), precision, discrimination and accuracy.
Materials and methods
Simulation data
Simulation data are used for quantitative analysis to evaluate the performance of all models to detect associations between diseases and genes. This study used PLINK 1.07 [25] to generate simulation data. A total of two scenarios were simulated:
Scenario 1: A total of 500 independent datasets were simulated. Each dataset consists of 1,000 cases, 1,000 controls, and 10,000 SNPs (100 of which are disease SNPs). The generating parameters of the datasets were combined by 10 odds ratios (ORs) (= 1.1, 1.2,…, 2.0) and 10 minor allele frequencies (MAFs) (= 0.05, 0.10,…, 0.5), that is, 100 combinations in total. For each combination, the data generation was repeated five times.
Scenario 2: The numbers of samples and MAFs are same as in scenario 1. Instead of specifying ORs, the penetrances of "AA" and "aa" were set to 0.01 and 0.1 respectively, and that of "Aa" was changed from 0.01 to 0.1 with step of 0.01. Thus, it includes a total of 100 combinations of MAFs and penetrances. Similarly, the data simulation was repeated five times for each combination, and thus produced 500 datasets.
Real data
This study employed a real coronary artery disease (CAD) dataset to test the performance of RM, DM and AM models in identifying disease risk SNPs on real datasets. The dataset comes from one of the WTCCC1 research datasets of the Wellcome Trust Case Control Consortium (WTCCC) [26], which is a case-control dataset with 1,988 cases and 1,500 controls, containing 490,032 SNPs of the 22 autosomes and 10,536 SNPs of the sex chromosome X. We pre-processed the data to ensure its quality, including MAF test, Hardy-Weinberg equilibrium test, allele deletion test for each SNP, and SNP deletion test for each individual. The thresholds of the tests are 0.05, 0.01, 0.05 and 0.05, respectively. In addition, the SNPs in the exclusion list provided by WTCCC had also been removed. Only the 22 autosomes were used in our experiments. After pre-processing, the CAD dataset contains a total of 363,590 valid SNPs.
Approaches
Tools of data generation and pre-processing
The simulation data in scenario 1 were generated by PLINK 1.07, and the data in scenario 2 were generated by a modified PLINK 1.07. We modified the PLINK to add a new generation mode implemented by specifying the penetrances of genotypes, since the original PLINK does not support this function. The modified PLINK is available on https://github.com/spvm2000/mPLINK. The pre-processing including data formats transformation was implemented by coPLINK [27].
Analysis approaches
The essence of GWAS is to explore the associations in phenotype-SNP data, and identify the SNPs with a score greater than the threshold as risky. This study employed the classic and basic chi-square test and logistic regression to calculate the association scores between phenotypes and SNPs to evaluate the performance of the models in identifying disease risk SNPs in simulation and real data. The scores of the two approaches produce P values which are negatively correlated with SNP risk. In order to transfer positive correlations from negative, the score of chi-square and logistic were subjected to an operation −log10(•).
T test
For the purpose of indicating the AUC differences of the models are statistically significant, we performed a two-tailed t test on the comparison results of AUCs through the analysis tool set of Microsoft Excel.
Evaluation measurements
A discrimination odds ratio (dOR) and precision ratio (PR) were defined, and AUC was employed as the evaluation measurements for this study to compare the performance of all models in identifying disease risk SNPs.
Discrimination and discrimination odds ratio
If a genetic disease is caused by a gene mutation, it means that the base sequence of the gene has been partially or completely changed compared to a normal individual (that is, the gene has a risk SNP). In other words, there is a difference in genotype between the case and the control, or there is a difference in the degrees of association between the genotypes and phenotype of the risk SNPs and non-risk SNPs. Moreover, the association degrees of the risk SNPs are usually stronger than those of the non-risk SNPs. Here, we defined a measurement named as "discrimination" (represented by the letter "d") to describe this difference, which is defined as the ratio of the average score of the risk SNPs to that of the non-risk SNPs, namely,
| (1) |
where, represents the average score of the disease risk SNPs and is the average score of the non-risk SNPs. When the scores (or the absolute values of the scores) are positively correlated with the degrees of associations, the larger the d, the greater the discrimination, indicating that the greater the difference in the associations between the two types of SNPs and the disease, is more conducive to identifying risk SNPs. Therefore, the discrimination of a model can represent the model’s capability to identify disease risk SNPs. In order to compare the discriminations of two models (or methods), we also defined a measurement called "discrimination odds ratio" (dOR), which is defined as follows:
| (2) |
where, d1 and d2 are the discriminations of the two models/methods, respectively. Obviously, under the premise of that the discrimination is positively related to the association, dOR > 1 indicates that the model/method 1 is better than the model/method 2.
Precision ratio
Similarly, we define the precision ratio (PR) as:
| (3) |
where, Precision1 and Precision2 represent the identifying precisions of the two models/methods, respectively. In this way, we can infer whether the identifying precision of model/method 1 is better than model/method 2 according to whether the PR is greater than 1.
AUC
AUC is defined as the area under a receiver operating characteristic curve (ROC) and is often used as an important evaluation measurement for model/method comparison in machine learning [28]. Here, we employed AUC to compare the performance of models AM, RM and DM in identifying risk SNPs.
Experiments
Experiments of AUC
We calculated the true positive rates (TPRs) and the false positive rates (FPRs) of each simulation dataset firstly, and then calculated their means. Finally, we plotted the ROCs based on these means and calculated the AUCs from the ROCs.
Experiments of discrimination odds ratio over simulation datasets
The experiments calculated dOR means of all simulation datasets for scenario 1 and 2 on the non-additive models to AM of all approaches.
Experiments of precision ratio over simulation datasets
The P value of chi-square and logistic were corrected by BH (Benjamini & Hochberg), and a P-cutoff method with a cutoff of 0.05 was applied to infer the significance of the SNPs. Similar to the experiments of discrimination odds ratio, we calculated the PR means of the simulation datasets for the scenarios on the non-additive models to AM of the approaches.
Experiments of real data
In this experiment, RM, DM and AM models were used to identify risk SNPs, and BH correction was made to the P value of chi-square and logistic. In order to evaluate the disease risk SNPs identified by the approaches, we queried the SNP database [29] of the National Center for Biotechnology Information (NCBI) to obtain the genes where the SNPs are located. In addition, we consulted literatures to determine whether the genes are at risk of disease and infer the risk of the SNPs.
Results
Comparison over AUC
Fig 1A and 1B show the three models’ ROC curves derived from the two approaches and the AUC lists calculated from the ROCs based on the datasets generated from simulation scenario 1 and 2, respectively.
Fig 1. Receiver operating characteristic curves and areas under the curves on simulation datasets.

Four significant figures are given here in order to display the area differences among the approaches.
Fig 1 intuitively shows that the RM’s ROC curves of the two approaches are almost overlap, and the curves are obviously more skewed to the upper left corner than AM, which means that the accuracy of RM is higher than AM. The figure also shows DM is weaker than AM. The AUCs on Fig 1 indicate that the AUC of RM is significantly higher than AM (average 0.8124 vs. 0.7581 and 0.9953 vs. 0.9705, respectively), and DM is greatly worse than AM (average 0.6479 vs. 0.7581 and 0.9408 vs. 0.9705, respectively). To test whether the differences are statistically significant, we tested them by a two-tailed t test. The P values of RM vs. AM in scenario 1 and 2 are 3.84×10−32 and 2.12×10−10, respectively, and those of DM vs. AM in the scenarios are 2.00×10−81 and 1.72×10−17, respectively. From the figure, we can calculate that the grand average AUC of RM and AM in the two scenarios are 0.9039 and 0.8643, that is, the AUC of RM is 5% higher than that of AM.
Performance of identifying risk SNPs comparison over simulation datasets
Suppose the SNP scores (or the absolute values of the scores) are positively correlated to the degrees of association, then the discrimination odds ratio dOR > 1 indicates that the discrimination of model/method 1 is better than model/method 2. PR is similar. Table 1 shows the dORs and PRs of the non-additive models to AM of the approaches on the simulation datasets. From the table, we can calculate the grand average dOR of RM and DM to AM are 1.61 and 0.78 respectively, which indicates that the risk SNP discrimination of RM is 1.61 times that of AM, that is, RM is significantly better than AM in identifying risk SNPs.
Table 1. dORs and PRs on simulation datasets.
| Approach | Scenario 1 | Scenario 2 | ||||||
|---|---|---|---|---|---|---|---|---|
| RM vs. AM | DM vs. AM | RM vs. AM | DM vs. AM | |||||
| dOR | PR | dOR | PR | dOR | PR | dOR | PR | |
| Chi-square | 1.38 | 1.14 | 0.68 | 0.41 | 1.85 | 1.08 | 0.84 | 0.90 |
| Logistic | 1.45 | 1.16 | 0.70 | 0.45 | 1.74 | 1.01 | 0.89 | 0.92 |
| Average# | 1.42 | 1.15 | 0.69 | 0.43 | 1.80 | 1.05 | 0.87 | 0.91 |
#The grand average dOR of RM vs. AM and DM vs. AM in the two scenarios are 1.61 and 0.78, respectively. And, their grand average PRs are 1.10 and 0.67, respectively.
Furthermore, Table 1 shows the grand average PR of RM to AM is 1.10, which means that the average risk SNP identifying precision of RM is 10% higher than that of AM. Meanwhile, the table shows the grand average PR of DM to AM is 0.67, which indicates the identifying precision of DM is weaker than AM.
Comparison over real dataset
The real dataset CAD was analyzed based on AM, RM and DM. With the P-cutoff of 0.05, we obtained the CAD risk SNP counts after querying SNP database on NCBI and consulting literatures, as shown in Table 2. The detailed results are listed in S1-S4 Tables in S1 File. Table 2 shows that the average PR of RM to AM is 1.16, which is higher than 1.00 of DM to AM (P value with two-tailed t test is 2.13×10−2). It indicates that RM is better than AM and DM on the CAD dataset.
Table 2. Risk SNP counts and PRs on CAD.
| Approach | RM | DM | AM | PR | |
|---|---|---|---|---|---|
| RM vs. AM | DM vs. AM | ||||
| Chi-Square | 35 (72)# | 36 (86) | 58 (141) | 1.18 | 1.02 |
| Logistic | 31 (64) | 36 (86) | 63 (147) | 1.13 | 0.98 |
| Average | 33 (68) | 36 (86) | 60.5 (144) | 1.16 | 1.00 |
# The numbers in the parentheses are the risk SNP counts based on the P threshold of 0.05.
Based on the results of the real dataset, we obtained the three models’ ROC curves derived from the two approaches and the AUC list calculated from the ROCs (Fig 2).
Fig 2. Receiver operating characteristic curves and areas under the curves on real dataset.
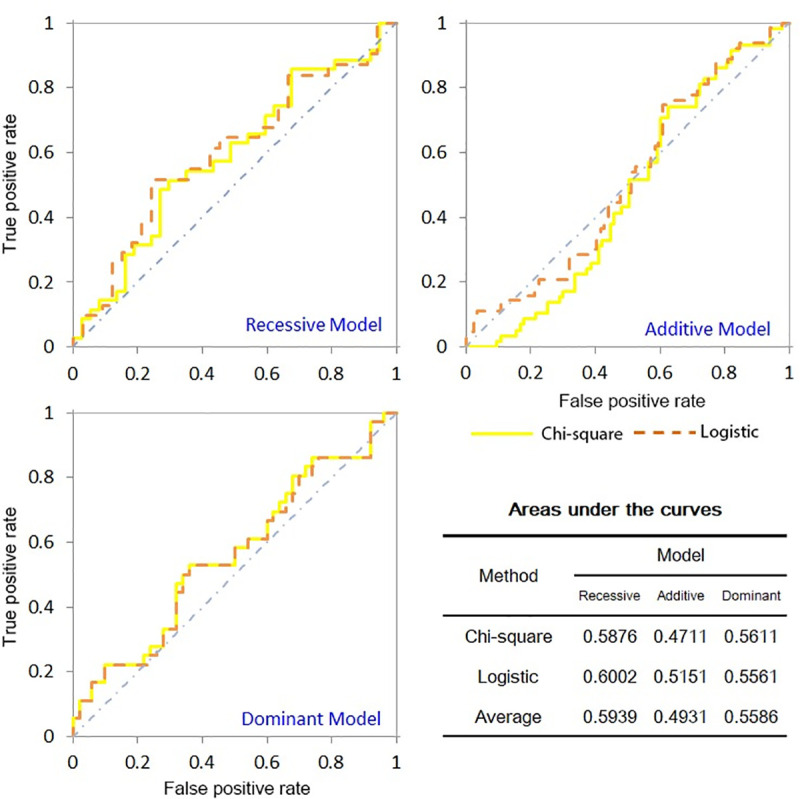
From Fig 2, we can learn that the AUC of RM and DM are markedly higher than AM (20% and 13% higher respectively).
Discussion
In the field of genetics, it is a high probability event that an improper genetic model assumption will lead to improper conclusions. Although AM is usually used in case-control-based GWAS, the dataset simplifies the phenotypes as two types, which means RM/DM may be an alternative to AM.
The comparative experiment of chi-square test and logistic regression based on simulation and real data verified this conjecture. The experiments show that RM is better than AM in terms of AUC, discrimination and precision. The real data experiments show that the AUC of DM is higher than that of RM, too. Existing works also approve this conjecture. In the study on risk of obesity and type 2 diabetes, Andrew R. Wood et al. identified two risk loci on obesity and type 2 diabetes, FTO and CDKAL1 [24] by employing a RM model. For the purpose of investigating whether novel nsCL/P risk loci could be identified by analyzing dominant/recessive genetic effects in SNP data from GWASes, the study of Bohmer A.C. et al. show that 18 loci are significant in DM and six loci are significant in RM among the 24 candidate loci, although they could not observe a new variant [30].
Our experiments show that RM is better than AM overall. Moreover, the real data experiments indicate the AUC of RM and DM are both higher than AM. The results suggest that AM is not necessarily choice in a case-control-based GWAS. How to evaluate AM, RM or DM is more suitable? The four-model strategy by Horita N. and Kaneko T. is a useful reference [31]. In addition, Bagos and Pantelis G. reviewed various methods and provided useful inspiration for us to genetic model selection in GWAS [32]. Moreover, it is a good idea that Kwak and Minjung used asymptotic property of the suptest to maintain the robustness of the Cochran–Armitage trend test when the genetic model is unknown [33].
Supporting information
(DOCX)
Data Availability
All relevant data are within the manuscript and its Supporting Information files.
Funding Statement
This study was supported by the National Natural Science Foundation of China (http://www.nsfc.gov.cn/) under grant number 31660321 which was awarded to H.M LIU. No author received salary or other funding from commercial companies. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Strachan T, Read A (2011) Human molecular genetics (4th ed.): Garland Science. [Google Scholar]
- 2.Ozaki K, Ohnishi Y, Iida A, Sekine A, Yamada R, et al. (2002) Functional SNPs in the lymphotoxin-α gene that are associated with susceptibility to myocardial infarction. Nature Genetics 32: 650–654. doi: 10.1038/ng1047 [DOI] [PubMed] [Google Scholar]
- 3.Klein RJ, Zeiss C, Chew EY, Tsai J-Y, Sackler RS, et al. (2005) Complement factor H polymorphism in age-related macular degeneration. Science 308: 385–389. doi: 10.1126/science.1109557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Visscher PM, Brown MA, McCarthy MI, Yang J (2012) Five years of GWAS discovery. The American Journal of Human Genetics 90: 7–24. doi: 10.1016/j.ajhg.2011.11.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Visscher PM, Wray NR, Zhang Q, Sklar P, McCarthy MI, et al. (2017) 10 years of GWAS discovery: biology, function, and translation. The American Journal of Human Genetics 101: 5–22. doi: 10.1016/j.ajhg.2017.06.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hirschhorn JN (2009) Genomewide association studies—illuminating biologic pathways. New England Journal of Medicine 360: 1699. doi: 10.1056/NEJMp0808934 [DOI] [PubMed] [Google Scholar]
- 7.Klein RJ, Xu X, Mukherjee S, Willis J, Hayes J (2010) Successes of genome-wide association studies. Cell 142: 350–351. doi: 10.1016/j.cell.2010.07.026 [DOI] [PubMed] [Google Scholar]
- 8.Speakman J, Loos R, O’Rahilly S, Hirschhorn J, Allison D (2018) GWAS for BMI: A treasure trove of fundamental insights into the genetic basis of obesity. International Journal of Obesity: 1. doi: 10.1038/s41366-018-0147-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.MacArthur J, Bowler E, Cerezo M, Gil L, Hall P, et al. (2016) The new NHGRI-EBI Catalog of published genome-wide association studies (GWAS Catalog). Nucleic acids research 45: D896–D901. doi: 10.1093/nar/gkw1133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Consortium IH (2003) The international HapMap project. Nature 426: 789. doi: 10.1038/nature02168 [DOI] [PubMed] [Google Scholar]
- 11.Greely HT (2007) The uneasy ethical and legal underpinnings of large-scale genomic biobanks. Annu Rev Genomics Hum Genet 8: 343–364. doi: 10.1146/annurev.genom.7.080505.115721 [DOI] [PubMed] [Google Scholar]
- 12.Yang Y, Chang T, Chen T, Lin W, Lin C, et al. (2018) Replication of results from a cervical cancer genome-wide association study in Taiwanese women. Scientific Reports 8: 1–5. doi: 10.1038/s41598-017-17765-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Risch N, Merikangas K (1996) The future of genetic studies of complex human diseases. Science 273: 1516–1517. doi: 10.1126/science.273.5281.1516 [DOI] [PubMed] [Google Scholar]
- 14.Wang WYS, Barratt BJ, Clayton DG, Todd JA (2005) Genome-wide association studies: theoretical and practical concerns. Nature Reviews Genetics 6: 109–118. doi: 10.1038/nrg1522 [DOI] [PubMed] [Google Scholar]
- 15.Zhang Y, Liu JS (2007) Bayesian inference of epistatic interactions in case-control studies. Nature genetics 39: 1167–1173. doi: 10.1038/ng2110 [DOI] [PubMed] [Google Scholar]
- 16.Miller DJ, Zhang Y, Yu G, Liu Y, Chen L, et al. (2009) An algorithm for learning maximum entropy probability models of disease risk that efficiently searches and sparingly encodes multilocus genomic interactions. Bioinformatics 25: 2478–2485. doi: 10.1093/bioinformatics/btp435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wan X, Yang C, Yang Q, Xue H, Fan X, et al. (2010) BOOST: A fast approach to detecting gene-gene interactions in genome-wide case-control studies. The American Journal of Human Genetics 87: 325–340. doi: 10.1016/j.ajhg.2010.07.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang J, Lee SH, Goddard ME, Visscher PM (2011) GCTA: A tool for genome-wide complex trait analysis. American Journal of Human Genetics 88: 76–82. doi: 10.1016/j.ajhg.2010.11.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mitha F, Herodotou H, Borisov N, Jiang C, Yoder J, et al. (2011) SNPpy—database management for SNP data from genome wide association studies. Plos One 6. doi: 10.1371/journal.pone.0024982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gyenesei A, Moody J, Laiho A, Semple CA, Haley C, et al. (2012) BiForce Toolbox: powerful high-throughput computational analysis of gene–gene interactions in genome-wide association studies. Nucleic Acids Research 40: 628–632. doi: 10.1093/nar/gks550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Putnam PP, Zhang G, Wilsey PA (2013) A comparison study of succinct data structures for use in GWAS. Bmc Bioinformatics 14: 369. doi: 10.1186/1471-2105-14-369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Helgeland Ø, Vaudel M, Juliusson PB, Holmen OL, Juodakis J, et al. (2019) Genome-wide association study reveals dynamic role of genetic variation in infant and early childhood growth. Nature communications 10: 1–10. doi: 10.1038/s41467-018-07882-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tam V, Patel N, Turcotte M, Bossé Y, Paré G, et al. (2019) Benefits and limitations of genome-wide association studies. Nature Reviews Genetics 20: 467–484. doi: 10.1038/s41576-019-0127-1 [DOI] [PubMed] [Google Scholar]
- 24.Wood AR, Tyrrell J, Beaumont R, Jones SE, Tuke MA, et al. (2016) Variants in the FTO and CDKAL1 loci have recessive effects on risk of obesity and type 2 diabetes, respectively. Diabetologia 59: 1214–1221. doi: 10.1007/s00125-016-3908-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, et al. (2007) PLINK: A tool set for whole-genome association and population-based linkage analyses. The American journal of human genetics 81: 559–575. doi: 10.1086/519795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Consortium WTCC (2007) Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature 447: 661. doi: 10.1038/nature05911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu H-M, Liu Z-F, Zheng J-P, Yang D, Hu S-Z, et al. (2020) coPLINK: A complementary tool to PLINK. PloS one 15: e0239144. doi: 10.1371/journal.pone.0239144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hanley JA, McNeil BJ (1983) A method of comparing the areas under receiver operating characteristic curves derived from the same cases. Radiology 148: 839–843. doi: 10.1148/radiology.148.3.6878708 [DOI] [PubMed] [Google Scholar]
- 29.Sherry ST, Ward M-H, Kholodov M, Baker J, Phan L, et al. (2001) dbSNP: the NCBI database of genetic variation. Nucleic acids research 29: 308–311. doi: 10.1093/nar/29.1.308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bohmer AC, Golz L, Kreusch T, Kramer F-J, Potzsch B, et al. (2017) Investigation of dominant and recessive inheritance models in GWAS data of nonsyndromic cleft lip with or without cleft palate. Birth Defects Research. [DOI] [PubMed] [Google Scholar]
- 31.Horita N, Kaneko T (2015) Genetic model selection for a case–control study and a meta-analysis. Meta Gene 5: 1–8. doi: 10.1016/j.mgene.2015.04.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bagos Pantelis G (2013) Genetic model selection in genome-wide association studies: robust methods and the use of meta-analysis. Statistical Applications in Genetics & Molecular Biology 12. doi: 10.1515/sagmb-2012-0016 [DOI] [PubMed] [Google Scholar]
- 33.Kwak Minjung (2014) Robust suptest for the genetic association study under genetic model uncertainty. Journal of the Korean Statistical Society 43: 463–474. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(DOCX)
Data Availability Statement
All relevant data are within the manuscript and its Supporting Information files.