

Abstract
Non-coding RNAs (ncRNAs) play crucial roles in multiple biological processes. However, only a few ncRNAs’ functions have been well studied. Given the significance of ncRNAs classification for understanding ncRNAs’ functions, more and more computational methods have been introduced to improve the classification automatically and accurately. In this paper, based on a convolutional neural network and a deep forest algorithm, multi-grained cascade forest (GcForest), we propose a novel deep fusion learning framework, GcForest fusion method (GCFM), to classify alignments of ncRNA sequences for accurate clustering of ncRNAs. GCFM integrates a multi-view structure feature representation including sequence-structure alignment encoding, structure image representation and shape alignment encoding of structural subunits, enabling us to capture the potential specificity between ncRNAs. For the classification of pairwise alignment of two ncRNA sequences, the F-value of GCFM improves 6% than an existing alignment-based method. Furthermore, the clustering of ncRNA families is carried out based on the classification matrix generated from GCFM. Results suggest better performance (with 20% accuracy improved) than existing ncRNA clustering methods (RNAclust, Ensembleclust and CNNclust). Additionally, we apply GCFM to construct a phylogenetic tree of ncRNA and predict the probability of interactions between RNAs. Most ncRNAs are located correctly in the phylogenetic tree, and the prediction accuracy of RNA interaction is 90.63%. A web server (http://bmbl.sdstate.edu/gcfm/) is developed to maximize its availability, and the source code and related data are available at the same URL.
Keywords: pairwise ncRNAs classification, ncRNAs clustering, multi-view structure feature representation, GcForest, deep fusion framework
1 Introduction
Non-coding RNAs (ncRNAs) play critical roles in a variety of cellular activities [1, 2]. They have significant associations with biological regulatory development and cell homeostasis and can be classified as multiple families with distinct functions [3], such as miRNAs for regulating gene expression [4], siRNAs for preventing the expression of disease-causing genes [5] and piRNAs for maintaining the integrity of germline DNA [6]. Recently, ncRNAs have been identified as innovative biomarkers of various diseases, including neurological, cardiovascular, developmental and cancer diseases, providing insights into the diagnosis and treatment of these diseases [7–9].
Given the significance, ncRNAs have attracted increasing attention in biological and biomedical research [10–12]. Conventional experimental methods were initially used to identify ncRNAs and infer their functions. However, due to tremendous labor and financial cost, the understanding of ncRNAs is still limited. Fortunately, one found RNAs with similar sequence or structure information tended to belong to the same family (a set of several similar genes, formed by duplication of a single original gene) and have similar biochemical functions [13–15]. This general understanding motivates various computational methods to infer the functions by assessing the similarity with well-studied ncRNAs [16].
Because the structure information of RNAs has higher conservation than sequences, current methods of comparison between different RNAs generally focus on structural characteristics [17, 18]. Such methods are classified into two categories, including the alignment-based and alignment-free methods. Specifically, the alignment-based methods rely on the string or tree representations of RNA secondary structure by dynamic programming. The most representative one is the Sankoff-based algorithm [19]. This algorithm simultaneously folds and aligns two or more RNA sequences based on free energy minimization, which has a reliable performance in prediction. However, the Sankoff-based algorithm has not been widely used due to the high computational complexity and time complexity. Given this, several other algorithms have been developed [20], such as FOLDALIGN [21], the revised Dynalign algorithm [22], and TOPAS [23]. These algorithms separate the folding and alignments processes, thereby reducing the computational complexity. Nevertheless, the alignment-based methods are still time-consuming due to unavoidable secondary structure alignments. By contrast, the alignment-free methods are based on the numerical representations of structure information of RNAs, improving the efficiency of RNA comparison. Multiple representations have been used, such as RNA-TVcurve [24, 25], GraphClust [26], DotcodeR [27] and DotAligner [28]. However, the alignment-free methods are with lower accuracies than alignment-based methods in the study of ncRNA families, because the feature representations of each ncRNA cannot reflect a consensus structure of each ncRNA family and obtain the optimal sequence alignment between different ncRNAs.
Additionally, several deep learning methods have been applied to predict noncoding-variant effects, DNA-protein binding, and similarity of RNAs, based on sequences [29–31]. The most widely used is the convolution neural network (CNN), which has good feature abstraction capabilities. However, the unavoidable experimentation for the model architecture construction and hyperparameter selection is a significant challenge. Out of a solution to this problem, a multi-grained cascade forest (GcForest), a kind of deep forest [32] was proposed. Driven by data, it is possible for the cascade module to automatically adjust the structure without the manual design of the structure. GcForest can achieve good performance for small-scale data. However, memory problems with its abstract feature extraction front-end multi-grain scanning prevent it from being applied to large-scale data.
In this paper, we firstly apply multi-view structure representations, including sequence-structure alignment encoding representation (SSR), RNA structure image representation (SIR), and RNA shape alignment representation (SAR), to capture multi-view features of ncRNAs. Specifically, SSR integrates secondary structure information into ncRNA sequences, avoiding the time-consuming of secondary structure alignment. The novel transformational gray image feature (i.e. SIR) transfers the probability of base pairing (i.e. the possibility of binding between two bases during the formation of a secondary structure) into a gray image, and the gray images of pairwise ncRNAs are aligned by subsampling or upsampling. Similar to SSR, SAR integrates the local shape structure information (i.e. stem, loop) into the alignments of ncRNA sequences. Then, we combine CNN module with GcForest module, and construct a deep fusion model, called GcForest fusion method (GCFM), for the classification of pairwise alignments of ncRNA sequences. This model can learn abstract features of ncRNAs at high levels and adjust part of model architecture automatically during training, improving the Accuracy of similarity assessment of pairwise ncRNAs.
2 Methods and materials
2.1 GCFM overview
The flowchart of GCFM is shown in Figure 1. It includes two steps: (i) construction of the multi-view structure feature representations. When we input two ncRNA sequences, three features, including SSR, SIR and SAR, can be extracted automatically. (ii) The overall integrating model. Based on the obtained multi-view structure feature representations, the features are extracted through the convolution module. The final classification results (the two sequences are within the same family or not) are obtained through the GcForest module with cascading.
Figure 1.
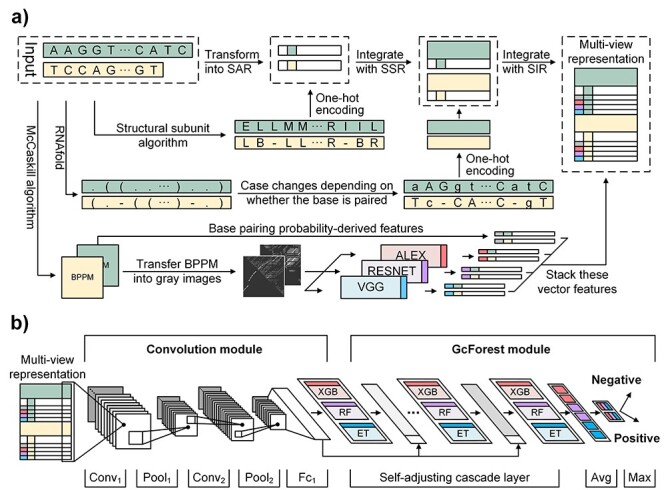
The framework of GCFM. (a) The whole flow chart of the multi-view structure feature representations. When two ncRNA sequences to be predicted are input, three feature representations (SSR, RNA SIR and SAR), will be extracted, respectively. BPPM represents the base-pairing probabilities matrix. (b) The overall architecture of the model. According to the obtained multi-view structure feature representations, the multi-view features are extracted through the convolution module. The final classification results are obtained through the GcForest module with cascading.
2.2 Datasets construction
We firstly downloaded ncRNAs sequences of humans from the Ensembl database (ftp://ftp.ensembl.org/pub/release-99/fasta/homo_sapiens/ncrna/) and the Genomic tRNA database (http://gtrnadb.ucsc.edu/genomes/eukaryota/Hsapi19/Hsapi19-seq.html) used by Aoki [31]. These sequences cover nine widely studied families, and the detailed information of each family is listed in Table 1.
Table 1.
The number of ncRNAs contained in each family and the number used in the different tasks
| Name | snRNA | snoRNA_C/D | snoRNA_H/ACA | YRNA | miRNA | tRNA | scaRNA | 5s_rRNA | Vault_RNA |
|---|---|---|---|---|---|---|---|---|---|
| 2069 | 324 | 161 | 835 | 1910 | 420 | 52 | 15 | 6 | |
| CLF | 100RD | 100RD | 100RD | 100RD | 100RD | 100RD | – | – | – |
| CLU | 10RD | 10RD | 10RD | 10RD | 10RD | 10RD | – | – | – |
CLU
|
10RD | 10RD | 10RD | 10RD | 10RD | 10RD | 10RD | 10RD | 6 |
Notes: CLF and CLU represent classification tasks and clustering tasks, respectively. RD means random selection. CLU means clustering with unknown ncRNA families.
means clustering with unknown ncRNA families.
Then, for classification of the relation of pairwise ncRNAs, we, respectively, chose 100 ncRNAs randomly from six families (snRNA, snoRNA_C/D, snoRNA_H/ACA, YRNA, miRNA and tRNA), as shown in Table 1. Based on the 600 ncRNAs, we got 179 700 pairwise ncRNAs (according to the combinatorial number formula, 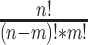 , where
, where 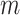 is equal to 2, and
is equal to 2, and 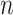 is equal to 600). For each pairwise ncRNA, we labeled it as a positive sample (i.e. ‘1’) if the two ncRNAs are in the same family (based on the prior family information of the Ensembl and Genomic tRNA databases). Otherwise, we labeled it as a negative sample (i.e. ‘0’). Ninety percent of pairwise ncRNAs (random selection) will be employed for training the model, and the left ten percent will be used for testing. This process will be repeated 10 times (10-fold cross-validation).
is equal to 600). For each pairwise ncRNA, we labeled it as a positive sample (i.e. ‘1’) if the two ncRNAs are in the same family (based on the prior family information of the Ensembl and Genomic tRNA databases). Otherwise, we labeled it as a negative sample (i.e. ‘0’). Ninety percent of pairwise ncRNAs (random selection) will be employed for training the model, and the left ten percent will be used for testing. This process will be repeated 10 times (10-fold cross-validation).
2.3 Multi-view structure feature representations
The nucleotides in the RNA chain follow the Watson–Crick pairing rules, forming complex structures associated with the functions of RNA. Here, we construct multi-view structure feature representations (Procedure 1), including SSR, SIR and SAR.
Procedure 1 Construction of multi-view structure feature representations
Input: two ncRNA sequences:
Output: Multi-view structure feature representations
calculate the secondary structure
by RNAfold
pairing probability matrix
for
by McCaskill algorithm
calculate the sequence representation of shape
according to Section 2.3.3
for each
do
for each
do
obtain
by changing the
according to
(Eq.1)
calculate extra pairing probability-derived features vector
including
and
according to Section 2.3.2
end for
obtain primary
by one-hot encoding
obtain
one-hot encoding representation
stack
and
into
for each
do
get adjusted image
according to
by Eq.2
end for
extract image feature
from
according to Section 2.3.2
stack
into
end for
stack the two
of each ncRNAs
return
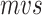
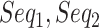
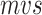
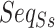
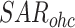
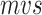
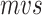
2.3.1 Sequence-structure alignment encoding representation (SSR)
We incorporate RNA secondary structure information into nucleotide sequence information for a novel sequence-structure representation. It fuses the secondary structure feature (represent as point and bracket) into a nucleotide sequence, called 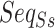 , by changing the upper or lower case of nucleotide letters: the paired nucleotide letters (if a nucleotide can combine with another one according to Watson–Crick pairing rules, they are considered as paired and represented by a bracket in secondary structure sequence) maintain the uppercase, and the unpaired are represented by lowercase (Equation 1). Additionally, the secondary structure sequence Ss is obtained by RNAfold [33] of the Vienna RNA package (Figure 1a). The new representation contains both secondary structure and alignments of ncRNA nucleotide sequences, called SSR.
, by changing the upper or lower case of nucleotide letters: the paired nucleotide letters (if a nucleotide can combine with another one according to Watson–Crick pairing rules, they are considered as paired and represented by a bracket in secondary structure sequence) maintain the uppercase, and the unpaired are represented by lowercase (Equation 1). Additionally, the secondary structure sequence Ss is obtained by RNAfold [33] of the Vienna RNA package (Figure 1a). The new representation contains both secondary structure and alignments of ncRNA nucleotide sequences, called SSR.
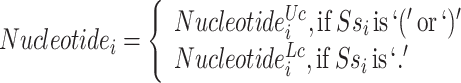 |
(1) |
where Uc is denoted as changing to uppercase, Lc means changing to lowercase.
For pairwise ncRNAs, the two 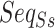 of them are different lengths mostly. Thus, aligning the two sequences with the same length is required to construct a uniform input shape of the feature. In our study, the
of them are different lengths mostly. Thus, aligning the two sequences with the same length is required to construct a uniform input shape of the feature. In our study, the 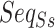 alignment is implemented by DAFS [34] with the fill of hyphen (-) for the alignment gap. Then, we used one-hot encoding (with nine chars, A, C, G, T, a, c, g, t, -) of each
alignment is implemented by DAFS [34] with the fill of hyphen (-) for the alignment gap. Then, we used one-hot encoding (with nine chars, A, C, G, T, a, c, g, t, -) of each 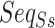 of the pairwise ncRNAs to obtain SSR. The SSR integrates into multi-view structure feature representation by stacking on the vertical direction with the other two part (i.e. SIR and SAR), as shown in the dotted box in Figure 1a.
of the pairwise ncRNAs to obtain SSR. The SSR integrates into multi-view structure feature representation by stacking on the vertical direction with the other two part (i.e. SIR and SAR), as shown in the dotted box in Figure 1a.
Additionally, we compare SSR with another one-hot encoding representation of nucleotide-only sequences to a previous study [31] under the same CNN architecture. The validity of the secondary structure information contained in the SSR can be seen in the Supplementary S1, which suggests the feasibility of our innovative secondary structure alignment approach by using the alignment information of nucleotide sequences.
2.3.2 RNA structure image representation (SIR)
The RNA secondary structure is generally measured by free energy, in which the most commonly used is minimum free energy (MFE) [35]. However, the predicted structure by MFE is not fully consistent with the real structure in nature [36]. The folding of RNA’s primary structures into secondary structures is a dynamic process, each base has the probability to pair with others in this process. SIR which ensembles the probability of all base pairing is widely needed to considering all possible secondary structures.
McCaskill’s partition function [37] is used to predict the thermal average probabilities of RNA base pairs rather than one single structure. The base pair probability matrix 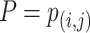 of RNA sequence can be predicted by McCaskill’s algorithm, which is implemented by the RNAfold program of the Vienna RNA package in this study. To consider all possible secondary structures of RNA, we transform
of RNA sequence can be predicted by McCaskill’s algorithm, which is implemented by the RNAfold program of the Vienna RNA package in this study. To consider all possible secondary structures of RNA, we transform 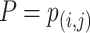 into an image by Equation 2.
into an image by Equation 2.
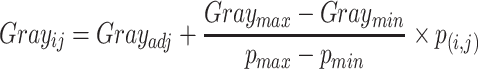 |
(2) |
where 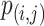 is the probability of base pairing of nucleotide
is the probability of base pairing of nucleotide 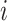 and
and 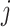 .
.
To highlight the low pairing probability, we introduce Gray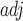 to adjust the display of gray image (Supplementary S2), enabling the original pair with a low probability to be discovered. This RNA SIR ensembles all possible structures, providing a novel insight into RNA structure analysis. Additionally, given that the lengths of pairwise RNA sequences are inconsistent, we use smooth filtering for subsampling and bicubic smooth for upsampling to normalize the images. After the transformation, the size of the images was consistent.
to adjust the display of gray image (Supplementary S2), enabling the original pair with a low probability to be discovered. This RNA SIR ensembles all possible structures, providing a novel insight into RNA structure analysis. Additionally, given that the lengths of pairwise RNA sequences are inconsistent, we use smooth filtering for subsampling and bicubic smooth for upsampling to normalize the images. After the transformation, the size of the images was consistent.
Based on the generated RNA structure images, we test the ability of eight common pretraining models to discriminate ncRNAs derived from image features. Results in Supplementary S2 showed that three models, VGG16, ResNet101 and AlexNet, can discriminate the ncRNA families. After that, we used the three models to extract the features of the RNA structure images. Each model calculates image features by forward-propagating and restricts the output feature shape by adding a fully connected layer. In this way, the image can be transferred into a vector with a fixed length, which can classify different ncRNA preliminary and reduce our model training complexity.
In addition, according to the base-pairing probability matrix 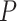 , the three-dimensional base-pairing probability-derived features can be calculated. For each position
, the three-dimensional base-pairing probability-derived features can be calculated. For each position 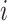 , we got three kinds sum of base pairing probability, nucleotide
, we got three kinds sum of base pairing probability, nucleotide 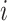 pairing at left side probability
pairing at left side probability 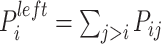 , pairing at right
, pairing at right 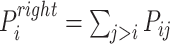 and unpaired probability
and unpaired probability 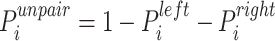 .
.
2.3.3 RNA shape alignment representation based on RNA structural subunit (SAR)
From a micro-perspective, local secondary structure can form different RNA structural subunits, such as hairpin, stem, loop, budge and multiloop. The global RNA secondary structure composed of these local shapes, implies a wealth of information, contributing to RNA function and evolution. Therefore, it is of great significance to integrate shape information into a feature representation.
The sequence of RNA shape is formed by a local secondary structure and can be calculated by a dynamic programming algorithm (Supplementary S3). For pairwise ncRNAs, the sequences of RNA shape can be aligned as same as the way of SSR, according to the hyphen for the alignment gap filled by DAFS. Then, as same as the process of SSR, the two sequences of RNA shape are represented by one-hot encoding, and stacking on the vertical direction with the other two part.
2.4 Architecture of GCFM
In this paper, we integrate the convolution module and the cascade forest, GcForest, to construct a deep ensembling learning architecture of GCFM (Figure 1b). The convolution module can not only extract high-level features but also reduce the dimension of features. The GcForest algorithm is a novel deep ensemble learning framework with no need to adjust a large number of parameters.
2.4.1 Convolution unit module
To combine the advantages of the convolution module and GcForest, we use the convolution unit module for preliminary feature learning and dimensionality reduction. Consequently, the extracted high-level abstract features are of sufficient and critical information with functions and with small dimensions, which is suitable for learning of GcForest.
The architecture of the convolution module employed in this paper consists of a three-layer fully connected network with one hidden layer following two convolution layers and two pooling layers. In our GCFM, the feature of the convolution unit module is computed by following formulae:
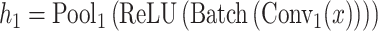 |
(3) |
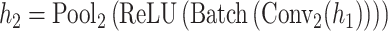 |
(4) |
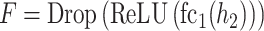 |
(5) |
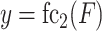 |
(6) |
where 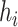 represents the i stacked part of the convolution module,
represents the i stacked part of the convolution module, 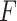 is the output vector,
is the output vector, 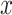 is the multi-view structure feature representations and
is the multi-view structure feature representations and 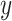 is the output label. In the calculation process of
is the output label. In the calculation process of 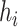 , the convolution layer extracts the pattern of the input features and outputs relevant features. The batch normalization layer regularizes the outputs of the convolution layer to a fixed mean and variance. ReLU is used for the regularized data from the batch normalization layer. The pooling layer is used to compress the input data of ReLU and preserve the main features. For expression of
, the convolution layer extracts the pattern of the input features and outputs relevant features. The batch normalization layer regularizes the outputs of the convolution layer to a fixed mean and variance. ReLU is used for the regularized data from the batch normalization layer. The pooling layer is used to compress the input data of ReLU and preserve the main features. For expression of 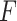 , the fully connected layer fc
, the fully connected layer fc is employed to learn all the weights to integrate better features and transform the output features into vectors, Dropout is only used in the model training. The last fully connected layer fc
is employed to learn all the weights to integrate better features and transform the output features into vectors, Dropout is only used in the model training. The last fully connected layer fc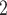 is used to evaluate the convolution ability in the initial process of model selection.
is used to evaluate the convolution ability in the initial process of model selection.
By adjusting the hyperparameters within ranges shown in Supplementary S4, we finally selected the parameter settings for the convolutional modules of the best test results. For 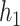 and
and 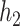 , the input channel of the convolution layer is 1 and 64, the output channel of the convolution layer is 64 and 128, the size of the convolution kernel is 15
, the input channel of the convolution layer is 1 and 64, the output channel of the convolution layer is 64 and 128, the size of the convolution kernel is 15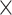 38 and 15
38 and 15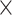 1, the size of pooling window is 10
1, the size of pooling window is 10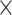 1 and 14
1 and 14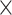 1, the stride of pooling applications are both eight and pooling methods are both max pooling. For
1, the stride of pooling applications are both eight and pooling methods are both max pooling. For 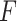 , the number of units in fully connected layer fc
, the number of units in fully connected layer fc is a 3/4 ratio to the number of units in the input layer. The weights of convolution layers are initialized by the value drawn independently from scaled Gaussian distribution whose mean is 0, and the standard deviation is
is a 3/4 ratio to the number of units in the input layer. The weights of convolution layers are initialized by the value drawn independently from scaled Gaussian distribution whose mean is 0, and the standard deviation is 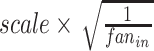 , where
, where 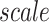 is a constant that determines the scale of the standard deviation and can be set as 1.0,
is a constant that determines the scale of the standard deviation and can be set as 1.0, 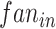 is the number of input units. Notably, the convolution module is mainly for preliminary feature learning, extraction and dimensionality reduction. Thus, the parameters can be changed optionally.
is the number of input units. Notably, the convolution module is mainly for preliminary feature learning, extraction and dimensionality reduction. Thus, the parameters can be changed optionally.
2.4.2 GcForest module
GcForest is a fire-new deep ensemble architecture based on a non-differentiable non-neural network-style module, which has fewer hyperparameters and better performance than other widely used deep neural networks. The model complexity of GcForest can be automatically determined in a data-dependent way.
Here, the GcForest module contains several cascade layers with the same components. Each layer consists of three algorithms, XGboosting, RandomForest and ExtraTrees (Figure 1b). These algorithms are implemented by the xgb class of xgboost and ensemble class of scikit-learn. After several adjustments within ranges shown in Supplementary S4, it is found that the following parameter settings have given better results and kept the complexity of the cascade forest small. For XGboosting, the maximum tree depth is 5, the number of boosted trees is 10, the boosting learning rate is 0.1 and the objective function is softmax. For RandomForest and ExtraTrees, the same parameter is set except default values: the maximum tree depth is 5, and the number of trees is 10. Five-fold cross-validation is used for the class vector generation of each classification. The number of cascade levels is automatically determined, but the cascade level will stop automatically growing when the accuracy does not increase in three rounds. And the maximum number of cascade layers allowed in our experiment is 7 to limit the complexity of this part of the self-adjusting model architecture. The default parameters settings of GcForest turned out to be good for classification, and it is no need to be adjusted excessively. Additionally, robustness analysis of the GcForest module under different CNN modules can also be found in Supplementary S4.
2.5 Evaluation criteria
We evaluate GCFM on two aspects: the performance on classification and the performance on clustering. In classification evaluation, we obtain the prediction labels of GCFM classification for each pair of ncRNAs in test data. According to prediction labels, for each pairwise ncRNAs, the true positive (TP) is accumulated if the real label and the prediction label are both positive. The true negative (TN) is defined if those labels are both negative, false positive (FP) is that the label of real is positive, but the prediction is not, false negative (FN) is opposed to the FP.
In clustering evaluation, we obtain the clusters generated by a clustering method for a set of ncRNAs. A pair of ncRNAs is counted as TP if they are clustered in the same cluster by a clustering algorithm, and meanwhile, they belong to the same ncRNA family in reality. Similarly, to the case in classification, TN, FP and FN are defined.
We calculate accuracy and F-value as follows:
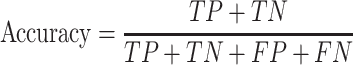 |
(7) |
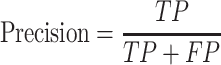 |
(8) |
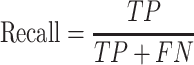 |
(9) |
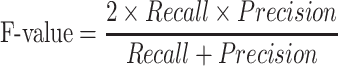 |
(10) |
3 Result
Here, we evaluated the performance of the GCFM. Firstly, multiple feature representations are used to compare with multi-view feature representations of GCFM. Then, the cascade module integrated with the CNN architecture (i.e. the architecture of GCFM) is used to explore whether there is a performance improvement over other existing methods. Next, we studied whether the multi-view feature representation of GCFM could improve the performance of ncRNA classification. Furthermore, we performed clustering based on the ncRNAs classification matrix generated from GCFM to evaluate the performance of classification.
3.1 GCFM suggests great performance on the classification of ncRNA
To our knowledge, only the study by Aoki et al. has focused on the classification using the alignments of ncRNA sequences. Therefore, our method was compared with this study in terms of both feature representations and model architecture.
The two input ncRNA sequences were aligned by DAFS, Clustal Omega [38] to eliminate factors of unequal length. The aligned sequences were encoded using one-hot encoding and word2vec as a matrix representation and then used as the input of CNN. The performance of several existing feature representations of ncRNA sequences is shown in the upper part of the Table 2 (under the same CNN architecture). Results showed the multi-view feature representations outperform other feature representations, both Accuracy and F-values were improved.
Table 2.
The performance on classification pairwise ncRNAs
| Methods | Accuracy | F-value |
|---|---|---|
| CNN with Clustal Omega (one-hot encoding) | 0.9580 | 0.8500 |
| CNN with DAFS (one-hot encoding) | 0.9710 | 0.9010 |
| CNN with DAFS (word2vec) | 0.9800 | 0.9310 |
| CNN with SSR, SIR and SAR | 0.9923 | 0.9954 |
| GCFM’s architecture with SSR | 0.9945 | 0.9834 |
| GCFM’s architecture with SIR | 0.9076 | 0.9459 |
| GCFM’s architecture with SAR | 0.9589 | 0.9697 |
| GCFM | 0.9991 | 0.9973 |
Notes: ’CNN with Clustal Omega (one-hot encoding)’ represents the CNN model with the input of Clustal-Omega alignments and one-hot encoding representation, ’CNN with DAFS (one-hot encoding)’ represents the CNN model with the input of DAFS alignments and one-hot encoding representation, ’CNN with DAFS (word2vec)’ denotes the CNN model with the input of DAFS alignments and word2vec distributed representation, ’CNN with SSR, SIR and SAR’ represents the CNN model with the input of our multi-view structure representation, ’GCFM’s architecture with SSR,’ ’GCFM’s architecture with SIR’ and ’GCFM’s architecture SAR,’ respectively, denotes the model architecture of GCFM with the input of SSR, with the input of SIR and with the input of SAR, ”GCFM” represents deep ensembling learning architecture with the input of multi-view structure representation (i.e. SSR, SIR and SAR). Bold font is used to indicate the best performance.
The impact of the GcForest module on model classification capabilities can be obtained by removing the GcForest module and keeping CNN module and feature representations constant. Results demonstrated the model capability of classification is the best while integrating the GcForest module.
Besides, we compared the three individual features within of changes into the multi-view feature representations. SSR yielded the best performance for ncRNA classification since it has accurate nucleotide sequence information than SIR and SAR. SAR performed better than SIR due to the abundant shape of secondary structure sequence information it contains. The last line in the bottom half of the Table 2 demonstrated the improvement of multi-view feature representation integrating SSR, SIR and SAR. The multi-view feature representation performed better due to the integrated information from multiple perspectives.
Moreover, ncRNA data from Rfam [39] were employed to verify the classification ability of GCFM. Rfam contains 3125 ncRNA families. We randomly selected 19 families from 30 families with more than ten thousand ncRNAs for model training and testing. The dataset construction (both positive and negative datasets) is the same as ’Datasets construction’ Section. Results showed that GCFM performs well with 97.33% accuracy and 98.62% F-value base on 10-fold cross-validation. When using the model without the GcForest module (only convolutional module), we found that the Accuracy and F-value are reduced by 3% and 4%, respectively, suggesting the great performance of GCFM.
Furthermore, we demonstrated the time-consumption of GCFM (Figure 2). Our architecture has two parts, the CNN and GcForest modules. The sum of CNN and GcForest time-consumption is our architecture time-consumption, while the CNN part represents the time-consumption of the method of Aoki et al. Results suggested that our method takes 15% more time than the method of Aoki et al. However, the f-value improved more than 6% in classification. We consider it is worthy to exchange a small amount of time for better performance.
Figure 2.
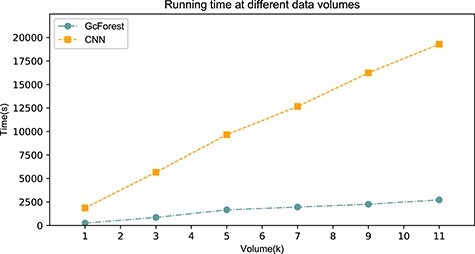
The time-consumption of CNN part and GcForest part under different data volumes.
3.2 GCFM contributes to accurate clustering of ncRNAs
The identification of ncRNA families is critical for the understanding of ncRNA functions. To further evaluate the performance of GCFM on capturing potential relationships of ncRNAs, we carried out clustering based on the relationship matrix from GCFM. A classification matrix with the size of 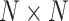 containing all pairwise ncRNAs relationships, where
containing all pairwise ncRNAs relationships, where 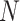 is the number of ncRNAs, obtained by classification method. Then, the clustering algorithm used to cluster the rows of the relational matrix as vectors with a length of
is the number of ncRNAs, obtained by classification method. Then, the clustering algorithm used to cluster the rows of the relational matrix as vectors with a length of 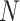 to obtain the final result of family affiliation. Different from most of the current popular unsupervised clustering methods of ncRNA families, the clustering can be supervised by introducing the result of classification.
to obtain the final result of family affiliation. Different from most of the current popular unsupervised clustering methods of ncRNA families, the clustering can be supervised by introducing the result of classification.
Specifically, we performed clustering for two datasets. The first dataset consists of 60 ncRNAs from six known families (randomly selected ten ncRNAs from each of the six families), as shown in Table 1. The second contains ncRNA with unknown families, and we randomly selected ten ncRNAs from each of nine families (including scaRNA, 5s_rRNA, and Vault_RNA) for the clustering tasks. Then, the classification matrix for these two datasets was generated by GCFM, respectively. Multiple widely used clustering algorithms, including K-means [40], spectral clustering [41, 42], Affinity propagation [43], birch [44], Mean-shift [45] and agglomerative clustering [46] were used here to ensure the stability of the results and to verify the validity of the relation matrix as clustering feature based on GCFM supervision. Our supervised clustering based on GCFM was compared with the previous supervised methods, CNNclust [31], and unsupervised methods, RNAClust [47] and Ensembleclust [48].
The results suggested that the clustering based on the relationship matrix from GCFM shows a great performance for both two datasets (Figure 3). In the clustering for ncRNA families, the F-value of clustering results based on the classification matrix from GCFM was over 10% higher than other models. Especially, in the clustering for the ncRNAs containing unknown families, the performance of clustering based on the classification matrix from GCFM dramatically outperforms other models with the improvement of an about 20% increase in accuracy and a 10% increase in F-value, showing strong robustness and generalization of GCFM classification of pairwise ncRNAs. This also indicates that GCFM can indeed capture potential relationships between ncRNAs and can enhance the performance of supervised clustering.
Figure 3.
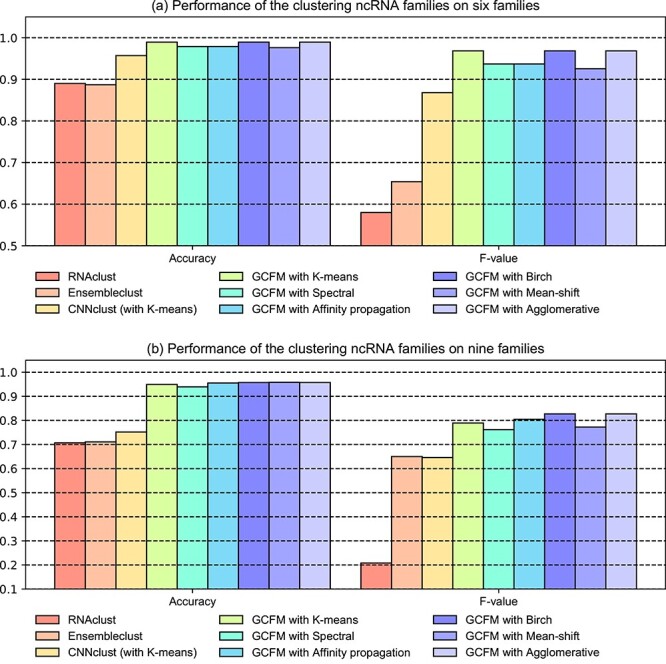
The performance of clustering ncRNA families. This bar plot shows the accuracy and F-value of nine different clustering methods on the two datasets.
3.3 GCFM helps infer ncRNA phylogenetic tree and RNA interaction accurately
To demonstrate the ability of GCFM and have a more systematic understanding of ncRNAs, two examples are shown here, including the construction of the phylogenetic tree and the prediction of ncRNA interaction. Phylogenetic tree construction which more visually shows the ability of relation prediction of GCFM between the pairwise ncRNAs and their family. We randomly selected three ncRNAs from each of the six families that were trained. Then, the phylogenetic tree was constructed based on the similarity calculated by GCFM using Neighbor-Joining algorithm [49]. The ncRNAs of the same family should be in the same branch or close to each other on a phylogenetic tree. By checking the family information of ncRNAs on the constructed tree, we found that 15 out of 18 ncRNAs show up in the right place (the right place means ncRNA belonging to the same family should be close to each other or within the same branch on the tree) in the phylogenetic tree (Figure 4). The correctness of tree construction suggests GCFM can measure the similarity between ncRNA accurately. To further demonstrate the universality of the similarity calculated by GCFM, we provided the results of other methods of phylogenetic tree construction in Supplementary S5. To show the feasibility of RNA interaction prediction, we predicted 1526 pairs of RNA interactions (from RNAInter [50]) by the trained model of our work. Based on the GCFM predictions, we screened them for interaction with a confidence level greater than 0.5 and found that 90.63% of them were correctly predicted to have interaction information.
Figure 4.
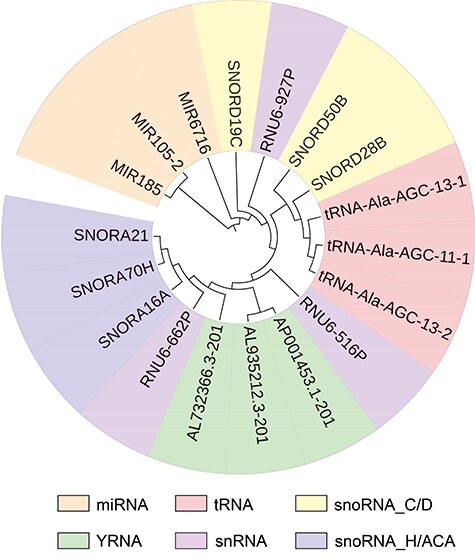
A phylogenetic tree for similarity calculation of pairwise ncRNAs based on GCFM.
4 GCFM web server
We developed a web server (http://bmbl.sdstate.edu/gcfm/) of GCFM to facilitate users, as shown in the screenshot of GCFM’s web server in Figure 5. The web server consists of three modules: (i) calculating whether the two ncRNAs are in the same family. When the sequences in the FASTA format of the two RNAs are input by a user, the server obtains similarities between two ncRNAs and evaluate whether they are in the same family. (ii) Clustering of multiple sequences based on a classification matrix and affinity propagation algorithm. The clustering function implements the derivation of bulk ncRNA family attribution and makes the classification matrix available for download, allowing users to further build phylogenetic trees based on the classification matrix, etc. (iii) Batch feature extraction. For the batch sequence input by users, features can be extracted automatically. For those who just want to use multi-view features, we provide unmatched feature extraction, where the extracted features can be applied to the study of multi-categorization of ncRNA and the study of interactions with ncRNA and protein. We provided usage examples of code and web server (Supplementary S6 and S7). Additionally, to facilitate the understanding of the multi-view structure feature representation construction, we provided three numerical examples of the construction of the multi-view structure feature representation in the resources available for download on the web server. More detail of multi-view structure feature representation can be found in Supplementary S8.
Figure 5.
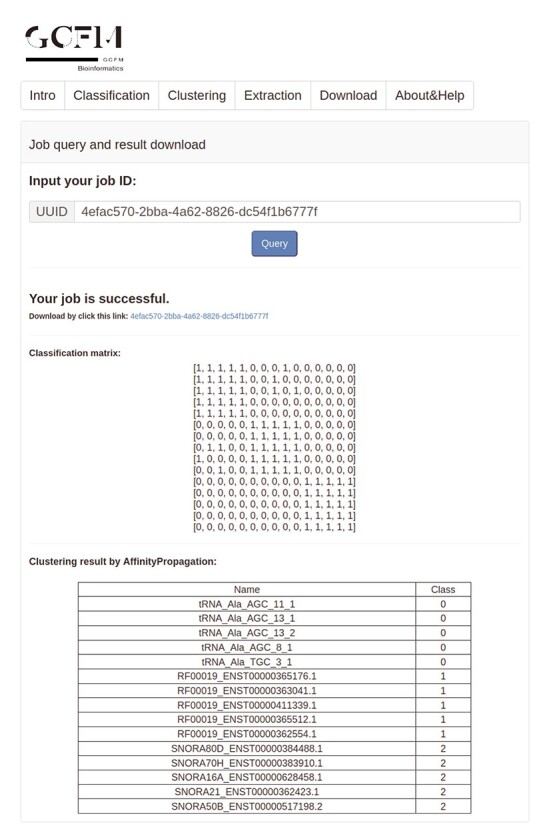
The screenshot of GCFM’s web server.
5 Discussion
Inferring the relationships of pairwise ncRNAs is a fundamental and critical step for understanding the function and evolution of ncRNAs. The mainstream methods for pairwise ncRNAs relationship inference are generally based on unsupervised learning. Inspired by the supervised learning method in Aoki’s work, we propose a supervised deep forest ensemble learning model for pairwise ncRNAs classification. Additionally, the integration of multi-view structure representations greatly improved classification performance. The web server is developed to facilitate users.
GCFM can capture the deep and abstract information automatically, providing insights into ncRNA clustering. The GCFM dramatically outperforms the state-of-the-art methods with strong robustness and generalization, as well as high accuracy of both classification and practical application of ncRNA clustering, not limited to species. One of our main contributions is the use of multi-view structure feature representations, including the SSR, SIR and SAR, based on RNA sequences, RNA secondary structure and RNA structural subunits. These features reflect the deep structure properties of RNA from micro- to macro-perspectives and can be highly beneficial for another RNA-related research. Another contribution is the integration of CNN and GcForest. In GCFM, the CNN unit reduces the data dimensionality and decreases the high memory cost of GcForest, and GcForest has strong learning power. The architecture of GCFM is validated of high effectiveness and efficiency. Especially, the GCFM only need fewer adjusted parameters compared with other traditional deep learning frameworks. Therefore, GCFM can be easily extended to the applications for classifier construction.
Additionally, GCFM has the ability to classify the relation of long sequences, such as the sequences of lncRNA. We found that several ncRNA sequences are longer than lncRNAs’, such as SNORA59B with 701 bp, and these sequences have been well-processed by GCFM.
In the future, we will explore the novel applications based on our proposed structure representation such as the evolution of analysis or deleterious mutation detection of ncRNAs. For the deleterious mutation detection, multi-view can highlight the changes in nucleotide bases. According to a simulated mutation on each position of the nucleotide sequence, the similarity of the simulated mutation sequence with the original sequences can be calculated by GCFM. If the result of them is dissimilar, the possibility of that position for harmful mutations may be large. Furthermore, we will exploit the architecture based on GcForest to multi-classification problems, such as the prediction of ncRNA family and ncRNA subcellular location.
Key Points
Based on three perspectives of a protein, including sequence, structure and shape, a multi-view structure feature representation is employed to provide a comprehensive feature of ncRNAs.
GcForest fusion method (GCFM) fuses convolution neural network and multi-grained cascade forest (GcForest) improving the Accuracy of ncRNA prediction. This method can contributes to the clustering of ncRNA sequences.
GCFM is suitable for exploring various relationships between RNAs even for lncRNAs with long lengths. GCFM shows a great performance for not only the prediction of ncRNA family affiliation but also the prediction of the interaction between ncRNAs.
Released as a web server, GCFM can be run on multiple OS platforms. GCFM is useful tool for feature construction based on sequence, machine learning model construction and performance evaluation.
Supplementary Material
Acknowledgments
The authors acknowledge the use of the high-performance computing facility, and associated support services at the University of JiLin, in the completion of this work. The authors thank all the participators and providers of online ncRNA resources. The authors also thank the editor and anonymous reviewers for handling this manuscript.
Ying Li (Ph.D.) is an associate professor at the College of Computer Science and Technology, Key Laboratory of Symbol Computation and Knowledge Engineering of Ministry of Education, Jilin University, Changchun, China. Her research topics include machine learning, bioinformatics and computational biology.
Qi Zhang is a graduate student at the College of Computer Science and Technology, Jilin University, Changchun, China. His research interests include computational biology and machine learning methods.
Zhaoqian Liu is a Ph.D. student in School of Mathematics, Shandong University, and now she is a visiting scholar at Ohio State University. Her research interest is computational methods in biology.
Cankun Wang is a biomedical informatics specialist at Ohio State University. His research interests include web development and computational methods in biology.
Siyu Han is a Ph.D. student in the Department of Computer Science, Faculty of Engineering, University of Bristol. His research interests include computational biology and machine learning methods.
Qin Ma (Ph.D.) is an associate professor in the Department of Biomedical Informatics, Ohio State University. Dr. Ma has over 10 years research experience in studying how functional machinery encoded in a genome.
Wei Du (Ph.D.) is an associate professor at the College of Computer Science and Technology, Jilin University, Changchun, China.
Contributor Information
Ying Li, College of Computer Science and Technology, Key Laboratory of Symbol Computation and Knowledge Engineering of Ministry of Education, Jilin University, Changchun, China.
Qi Zhang, College of Computer Science and Technology, Jilin University, Changchun, China.
Zhaoqian Liu, School of Mathematics, Shandong University, and now she is a visiting scholar at Ohio State University.
Cankun Wang, Ohio State University.
Siyu Han, Department of Computer Science, Faculty of Engineering, University of Bristol.
Qin Ma, Department of Biomedical Informatics, Ohio State University.
Wei Du, College of Computer Science and Technology, Jilin University, Changchun, China.
Funding
This work was supported by the National Natural Science Foundation of China (61872418, 61972175 and 71774154) and Natural Science Foundation of Jilin Province (20180101331JC and 20180101050JC).
References
- 1. Cech TR, Steitz JA. The noncoding RNA revolution—trashing old rules to forge new ones. Cell 2014; 157(1): 77–94. [DOI] [PubMed] [Google Scholar]
- 2. Meyers BC, Matzke M, Sundaresan V. The RNA world is alive and well. Trends Plant Sci 2008; 13(7): 311–3. [DOI] [PubMed] [Google Scholar]
- 3. Fu XD. Non-coding RNA: a new frontier in regulatory biology. Natl Sci Rev 2014; 1(2): 190–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Farazi TA, Spitzer JI, Morozov P, et al.. MiRNAs in human cancer. J Pathol 2011; 223(2): 102–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sioud M. Therapeutic siRNAs. Trends Pharmacol Sci 2004; 25(1): 22–8. [DOI] [PubMed] [Google Scholar]
- 6. Klattenhoff C, Theurkauf W. Biogenesis and germline functions of piRNAs. Development 2008; 135(1): 3–9. [DOI] [PubMed] [Google Scholar]
- 7. Esteller M. Non-coding RNAs in human disease. Nat Rev Genet 2011; 12(12): 861–74. [DOI] [PubMed] [Google Scholar]
- 8. Yoon JH, Abdelmohsen K, Gorospe M. Posttranscriptional gene regulation by long noncoding RNA. J Mol Biol 2013; 425(19): 3723–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mathieu È-L, Belhocine M, Dao LTM, et al.. Functions of lncRNA in development and diseases. Médecine/Sciences 2014; 30(8–9): 790–6. [DOI] [PubMed] [Google Scholar]
- 10. Hüttenhofer A, Vogel J. Experimental approaches to identify non-coding RNAs. Nucleic Acids Res 2006; 34(2): 635–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Emamjomeh A, Zahiri J, Asadian M, et al.. Identification, prediction and data analysis of noncoding RNAs: a review. Med Chem 2018; 15(3): 216–30. [DOI] [PubMed] [Google Scholar]
- 12. Wolfien M, Brauer DL, Bagnacani A, et al.. Workflow development for the functional characterization of ncRNAs. Methods Mol Biol 2019; 1912:111–32. [DOI] [PubMed] [Google Scholar]
- 13. Zhang Y, Huang H, Zhang D, et al.. A review on recent computational methods for predicting noncoding RNAs. Biomed Res Int 2017; 2017:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Altschul SF, Gish W, Miller W, et al.. Basic local alignment search tool. J Mol Biol 1990; 215(3): 403–10. [DOI] [PubMed] [Google Scholar]
- 15. Lindgreen S, Gardner PP, Krogh A. MASTR: multiple alignment and structure prediction of non-coding RNAs using simulated annealing. Bioinformatics 2007; 23(24): 3304–11. [DOI] [PubMed] [Google Scholar]
- 16. Rivas E, Eddy SR. Noncoding RNA gene detection using comparative sequence analysis. BMC Bioinformatics 2001; 2(1): 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mathews DH, Turner DH. Prediction of RNA secondary structure by free energy minimization. Curr Opin Struct Biol 2006; 16(3): 270–8. [DOI] [PubMed] [Google Scholar]
- 18. Childs L, Nikoloski Z, May P, et al.. Identification and classification of ncRNA molecules using graph properties. Nucleic Acids Res 2009; 37(9): e66–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Havgaard JH, Gorodkin J. RNA structural alignments, part I: Sankoff-based approaches for structural alignments. Methods Mol Biol 2014; 1097:275–90. [DOI] [PubMed] [Google Scholar]
- 20. Asai K, Hamada M. RNA structural alignments, part II: non-Sankoff approaches for structural alignments. Methods Mol Biol 2014; 1097:291–301. [DOI] [PubMed] [Google Scholar]
- 21. Havgaard JH, Torarinsson E, Gorodkin J. Fast pairwise structural RNA alignments by pruning of the dynamical programming matrix. PLoS Comput Biol 2005; 3(10): 1896–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Harmanci A, Sharma G, Mathews DH. Efficient pairwise RNA structure prediction using probabilistic alignment constraints in Dynalign. BMC Bioinformatics 2007; 8(1): 130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chen CC, Jeong H, Qian X, et al.. TOPAS: network-based structural alignment of RNA sequences. Bioinformatics 2019; 35(17): 2941–8. [DOI] [PubMed] [Google Scholar]
- 24. Li Y, Duan M, Liang Y. Multi-scale RNA comparison based on RNA triple vector curve representation. BMC Bioinformatics 2012; 13(1): 280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Li Y, Shi X, Liang Y, et al.. RNA-TVcurve: a web server for RNA secondary structure comparison based on a multi-scale similarity of its triple vector curve representation. BMC Bioinformatics 2017; 18(1): 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Heyne S, Costa F, Rose D, et al.. Graphclust: alignment-free structural clustering of local RNA secondary structures. Bioinformatics 2012; 28(12): 224–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kato Y, Gorodkin J, Havgaard JH. Alignment-free comparative genomic screen for structured RNAs using coarse-grained secondary structure dot plots. BMC Genomics 2017; 18(1): 935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Smith MA, Seemann SE, Quek XC, et al.. DotAligner: identification and clustering of RNA structure motifs. Genome Biol dec 2017; 18(1): 244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhou J, Troyanskaya OG. Predicting effects of noncoding variants with deep learning-based sequence model. Nat Methods 2015; 12(10): 931–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zeng H, Edwards MD, Liu G, et al.. Convolutional neural network architectures for predicting DNA-protein binding. Bioinformatics 2016; 32(12): 121–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Aoki G, Sakakibara Y. Convolutional neural networks for classification of alignments of non-coding RNA sequences. Bioinformatics 2018; 34(13): i237–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhou Z-H, Ji F. Deep Forest: Towards An Alternative to Deep Neural Networks. In: Proceedings of the Twenty-Sixth International Joint Conference on Artificial Intelligence, 2017, 3553–9.
- 33. Hofacker IL. Vienna RNA secondary structure server. Nucleic Acids Res 2003; 31(13): 3429–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sato K, Kato Y, Akutsu T, et al.. DAFS: simultaneous aligning and folding of RNA sequences via dual decomposition. Bioinformatics 2012; 28(24): 3218–24. [DOI] [PubMed] [Google Scholar]
- 35. Zuker M, Stiegler P. Optimal computer folding of large RNA sequences using thermodynamics and auxiliary information. Nucleic Acids Res 1981; 9(1): 133–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ye D, Lawrence CE. A statistical sampling algorithm for RNA secondary structure prediction. Nucleic Acids Res 2003; 31(24): 7280–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. McCaskill JS. The equilibrium partition function and base pair binding probabilities for RNA secondary structure. Biopolymers 1990; 29(6–7): 1105–19. [DOI] [PubMed] [Google Scholar]
- 38. Sievers F, Wilm A, Dineen D, et al.. Fast,scalable generation of high-quality protein multiple sequence alignments using Clustal omega. Mol Syst Biol 2011; 7(1): 539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kalvari I, Nawrocki EP, Argasinska J, et al.. Non-coding RNA analysis using the Rfam database. Curr Protoc Bioinformatics 2018; 62(1): e51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Arthur D, Vassilvitskii S. K-means++: The advantages of careful seeding. In: Proceedings of the Annual ACM-SIAM Symposium on Discrete Algorithms, 2007, 1027–35.
- 41. Ng AY, Jordan MI, Weiss Y. On spectral clustering: Analysis and an algorithm. In: Advances in Neural Information Processing Systems, 2002, 849–56.
- 42. Von Luxburg U. A tutorial on spectral clustering. Statistics and Computing 2007; 17(4): 395–416. [Google Scholar]
- 43. Frey BJ, Dueck D. Clustering by passing messages between data points. Science 2007; 315(5814): 972–6. [DOI] [PubMed] [Google Scholar]
- 44. Zhang T, Ramakrishnan R, Livny M. BIRCH: an efficient data clustering method for very large databases. SIGMOD Record (ACM Special Interest Group on Management of Data) 1996; 25(2): 103–14. [Google Scholar]
- 45. Comaniciu D, Meer P. Mean shift: a robust approach toward feature space analysis. IEEE Trans Pattern Anal Mach Intell 2002; 24(5): 603–19. [Google Scholar]
- 46. Day WHE, Edelsbrunner H. Efficient algorithms for agglomerative hierarchical clustering methods. J Classification 1984; 1(1): 7–24. [Google Scholar]
- 47. Jan E, Steffen H, Sebastian W , et al.. RNAclust: a tool for clustering of RNAs based on their secondary structures using LocARNA. RNAclust.pl Documentation. 1–9, 2010http://www.bioinf.uni-leipzig.de/∼kristin/Software/RNAclust/ (last accessed 21 May 2019). [Google Scholar]
- 48. Saito Y, Sato K, Sakakibara Y. Fast and accurate clustering of noncoding RNAs using ensembles of sequence alignments and secondary structures. BMC Bioinformatics 2011; 12(1): S48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Saitou N, Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 1987; 4(4): 406–25. [DOI] [PubMed] [Google Scholar]
- 50. Lin Y, Liu T, Cui T, et al.. RNAInter in 2020: RNA interactome repository with increased coverage and annotation. Nucleic Acids Res 2020; 48(D1): D189–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.