

Abstract
The accurate quantification of tumor-infiltrating immune cells turns crucial to uncover their role in tumor immune escape, to determine patient prognosis and to predict response to immune checkpoint blockade. Current state-of-the-art methods that quantify immune cells from tumor biopsies using gene expression data apply computational deconvolution methods that present multicollinearity and estimation errors resulting in the overestimation or underestimation of the diversity of infiltrating immune cells and their quantity. To overcome such limitations, we developed MIXTURE, a new ν-support vector regression-based noise constrained recursive feature selection algorithm based on validated immune cell molecular signatures. MIXTURE provides increased robustness to cell type identification and proportion estimation, outperforms the current methods, and is available to the wider scientific community. We applied MIXTURE to transcriptomic data from tumor biopsies and found relevant novel associations between the components of the immune infiltrate and molecular subtypes, tumor driver biomarkers, tumor mutational burden, microsatellite instability, intratumor heterogeneity, cytolytic score, programmed cell death ligand 1 expression, patients’ survival and response to anti-cytotoxic T-lymphocyte-associated antigen 4 and anti-programmed cell death protein 1 immunotherapy.
Keywords: immune infiltrate, deconvolution, RNA sequencing, cancer, digital cytometry, immunotherapy
Introduction
The immune checkpoint blockade (ICB) immunotherapy has changed the paradigm of cancer treatment for several indications after decades of only partially effective therapies [1]. The malignant phenotype of cancer is defined not only by the intrinsic activities of cancer cells but also by the tumor microenvironment components, especially the tumor-infiltrating immune cells [2]. The exclusion of CD8 T cells from the tumor correlates with poor clinical outcome in colorectal cancer [3], while a high infiltrate of cytotoxic CD8 T cells has been mostly linked to prolonged survival [4], being also predictive of increased overall survival in patients with breast, head and neck and colorectal cancer with hepatic and lung metastases [5]. In contrast, the immunosuppressive regulatory T cells (Tregs) are associated with shorter survival in several cancer types [5]. The immunosuppressive macrophage M2-phenotype favors the growth and development of an invasive and pro-angiogenic phenotype [6] and has been linked with poor prognosis in cancer patients [5].
Despite the substantial advancements in clinical cancer care, most patients receiving anti-cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4) and anti-programmed cell death protein 1 (PD-1) ICB do not derive benefit. Therefore, there is an urgent need to identify and develop predictive biomarkers of ICB response, enabling a precision medicine approach in cancer immunotherapy for better understanding and overcoming the resistance mechanisms. Immune checkpoint inhibitor efficacy is affected by a combination of factors that involve the oncogenic drivers, the tumor mutational burden (TMB), microsatellite instability (MSI), the expression levels of the PD-ligand 1 (PD-L1) and the immune infiltrate [7]. The latter has been shown to influence the response to ICB [8]. Increasing evidence suggests that it is not only the density of immune infiltrating cells but also their phenotype that impact the response to ICB. Although the role of the immune microenvironment as a factor predicting response to ICB remains to be validated [7], it holds the promise to unveil the role of the immune system in cancer progression and response to therapy.
Flow cytometry, immunohistochemistry and single-cell RNA sequencing are the current gold standards studying the tumor immune infiltrate. Still, they have limitations related to sample availability, high throughput and high costs. Thus, several computational algorithms made possible to infer the immune tumor microenvironment (ITME) composition by using immunological-associated gene sets approaches [9–11], or by deconvolving the tumor immune content from bulk tumor gene expression [12–18] applying profiled molecular signatures based on gene expression signature data [13,14,17,19–22]. The first ones require a significant number of genes, whereas the second ones can be applied over a reduced set of molecular signature genes. Regardless of their potential, current methods present controversial results concerning their accuracy [14,23]. Specifically, current state-of-the-art molecular signature-based deconvolution methods [16,18–20] present limitations driven by (i) collinearity issues, due to highly correlated signature profiles, (ii) floating-point errors or lack of non-negative definition which violate the sum-to-one coefficients constraints and (iii) highly skewed gene expression input data. The collinearity issues may over or misestimate cell types. Moreover, floating-point error noise or the lack of non-negative coefficient constraints may deliver over cell type identification, resulting in biased-proportion estimates. Then, the skewed data distribution may provide biased proportion estimates when using ordinary least squares approaches. Consequently, current methods may estimate an inaccurate and biased proportion of the tumor immune cell type levels.
To overcome such limitations, we developed MIXTURE—a new deconvolution method applying a noise constrained Recursive Feature Selection algorithm for ν-support vector regression (v-SVR)—contrasted with the current state-of-the-art methods [17,19–21]. MIXTURE was evaluated on the LM22 signature—a 22 immune cells signature based on mature human hematopoietic populations and activation state [17], and on the TIL10 signature—a Tumor Infiltrate Lymphocyte signature of 10 immune cell types [20] and validated over simulated and flow cytometry derived cell type proportion data. Here, we show the MIXTURE’s superior performance analyzing both simulated and real benchmark datasets. We applied MIXTURE to evaluate the ITME association with known genomic features such as oncogenic drivers, TMB, MSI, intratumor heterogeneity (ITH) and PD-L1 expression in breast cancer (BRCA), lung cancer (LUAD), melanoma (SKCM), head and neck squamous cell carcinoma (HNSC) and colorectal cancer (COAD). Importantly, we correlated the ITME with both survival and response to immunotherapy in melanoma patients with known response to anti-CTLA-4 or anti-PD-1 immunotherapy. Our analysis shows that MIXTURE outperforms the competing methods in estimating, accurately and robustly, the ITME. Our study reveals new associations of the ITME with patients’ outcomes and validated biomarkers of response to ICB.
Materials and methods
Quantification of tumor-infiltrating immune cells
The linear deconvolution of the cell types present in a gene signature matrix (X), holding N genes for k cell types, associated with the components of a mixture of cell types present in a tumor gene expression profile (Y), involves solving the following regression model equation 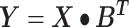 . The proportions for all cell types in the mixture sample is represented by the column vector
. The proportions for all cell types in the mixture sample is represented by the column vector 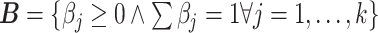 , i.e. the vector of regression coefficients satisfying both, the non-negativity and sum-to-one constraints [24].
, i.e. the vector of regression coefficients satisfying both, the non-negativity and sum-to-one constraints [24].
MIXTURE deconvolution algorithm
The MIXTURE algorithm re-estimates  by iteratively removing the columns of X associated with null coefficients from the regression step after all estimated coefficients result in
by iteratively removing the columns of X associated with null coefficients from the regression step after all estimated coefficients result in 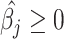 by using the v-SVR approach as a regression function similar to CIBERSORT. Floating-point errors affect the inequality comparison
by using the v-SVR approach as a regression function similar to CIBERSORT. Floating-point errors affect the inequality comparison  used to define the null coefficients, resulting in an overestimation of the number of positive coefficients, and negatively impacting the selection step. To avoid this, MIXTURE defines a noise constraint threshold
used to define the null coefficients, resulting in an overestimation of the number of positive coefficients, and negatively impacting the selection step. To avoid this, MIXTURE defines a noise constraint threshold 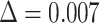 (chosen through a simulation process—see supplementary section ‘Noise Constrain Threshold Selection’) to iteratively set as zero or null
(chosen through a simulation process—see supplementary section ‘Noise Constrain Threshold Selection’) to iteratively set as zero or null 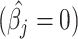 those estimated normalized coefficients satisfying
those estimated normalized coefficients satisfying 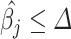 leading to a more accurate estimation of the cell types present in the sample. Specifically, the MIXTURE algorithm is based in the following procedure:
leading to a more accurate estimation of the cell types present in the sample. Specifically, the MIXTURE algorithm is based in the following procedure:
| Step | MIXTURE Function: Inputs (Y, X); Output: 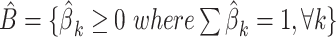
|
|---|---|
| 1 |
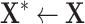
|
| 2 |
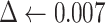
|
| 3 |
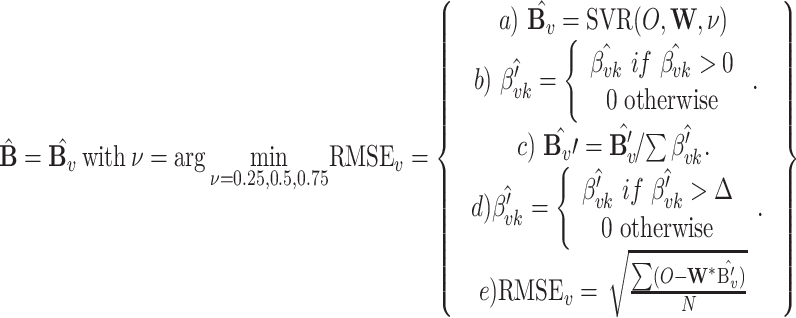
|
| 4 |
|
| 5 |
|
| 6 |
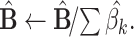
|
| 7 |
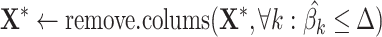
|
| 8 | GOTO 3 |
| 9 |
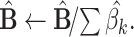 (this yields
(this yields 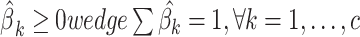 ) ) |
The algorithm receives as input the bulk tumor gene expression profile Y and the signature matrix X (Step1). Then (Step 2) set the Noise Threshold to a given value 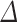 = 0.007. In Step 3, the optimization v-SVR process takes place by minimizing the root mean squared error between Y and their estimated profile
= 0.007. In Step 3, the optimization v-SVR process takes place by minimizing the root mean squared error between Y and their estimated profile 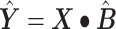 over three v values of 0.25, 0.5 and 0.7, and using
over three v values of 0.25, 0.5 and 0.7, and using  to define null coefficients. Then it returns the normalized vector
to define null coefficients. Then it returns the normalized vector 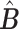 from the best ‘v’. If all
from the best ‘v’. If all 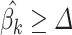 (Step 4) it returns the normalized
(Step 4) it returns the normalized 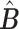 , removes all columns from X for whom
, removes all columns from X for whom 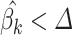 , and go to Step 3. The algorithm stops if all the elements of the normalized vector
, and go to Step 3. The algorithm stops if all the elements of the normalized vector 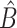 resulted >
resulted >  .
.
Benchmarking of deconvolution methods
We tested MIXTURE’s performance against four state-of-the-art algorithms, namely ABBAS, ABIS, CIBERSORT and quanTIseq, by using both LM22 and TIL10 signatures to estimate their corresponding cell types on the evaluated samples (Table 1). We assessed their ability to estimate the cell type content and proportion estimates from (i) the LM22 and TIL10 molecular signature profiles as pure cell samples (self-test), (ii) Simulated bulk samples built from a random gene expression from the molecular signature, LM22 or TIL10, and adding known proportions of randomly chosen cell type signature profiles, (iii) flow cytometry derived cell type proportion content of Follicular Lymphoma and Plasma Blood Mononuclear Cells samples and (iv) from full negative cell type content samples from pure cell lines (see supplementary material for details).
Table 1.
Summary of the compared software packages
| Software | Deconvolution method | Noise constraint | Recursive Feature Extraction | Signature | Platform | Refs |
|---|---|---|---|---|---|---|
| ABBAS | OLS | No | Yes | Any | R | [19] |
| ABIS | RLM | No | No | Any | R / WEB (http://timer.cistrome.org/) | [21] |
| quanTIseq | LSEI | No | No | Any | R | [20] |
| CIBERSORT | v-SVR | No | No | Any | R / WEB (https://cibersort.stanford.edu/) | [17] |
| MIXTURE | v-SVR | Yes | Yes | Any | R / Shiny / Python / Dash | - |
Abbreviations: OLS: ordinary least squares, RLM: robust linear model, LSEI: least squares with equality and inequality constraint, v-SVR: nu-support vector regression.
Validation and discovery data sets
To validate MIXTURE on real cancer samples, we evaluated BRCA, LUAD, SKCM, HNSC and COAD TCGA biopsies to look for associations with known genomic features like oncogenic drivers, TMB, ITH and MSI (see supplementary material for details).
Statistical analysis
We used the Wilcoxon test to compare the estimated coefficients between methods (paired or unpaired accordingly) with P < 0.05 as the significance threshold value. Since correlation is not a suitable method to compare predicted versus truth proportions (it does not allow estimation bias nor systematic error analysis), the Bland–Altman statistical method for assessing agreement between clinical measurements [25] was used to compare estimated proportions against the true simulated or flow cytometry derived ones [26]. The mean ± standard deviation was used to show the overall bias when appropriate, and loess smoothing to visualize bias. We applied the Pearson coefficient for correlation analysis and Cox Proportional Hazards models for survival analysis with the survival R library” To evaluate cell type content prediction accuracy, an analysis of sensitivity, positive predictability and F1-scores was conducted. For patient proportion analysis, Pearson’s chi-squared method was performed, and P-values were calculated by Monte Carlo simulation.
Code availability
In order to use MIXTURE, we have developed both R and Python algorithms available at https://github.com/elmerfer/MIXTURE.App.
Results
MIXTURE outperforms other deconvolution methods in a self-test analysis
We analyzed the performance of MIXTURE (Figure 1A) against the current state-of-the-art methods assuming that any method should be able to estimate the provided signature matrix proportions correctly (i.e. pure cell type-gene expression profile). That means that if we replace Y by each column of X (a pure cell type profile), the method should provide only one coefficient equal to 1 and the remaining coefficients equal to 0, as presented in Equation (1):
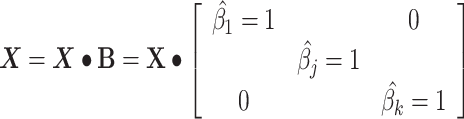 |
(1) |
Figure 1 .

Development and benchmarking of MIXTURE, a new deconvolution algorithm to estimate the immune cell infiltrate from transcriptomic data. (A) The MIXTURE workflow and data output overview with LM22 and TIL10 signatures. MIXTURE estimates the ITME of human samples from transcriptomic data. (B) Heatmaps showing auto correlation of ITME cell types in LM22 and TIL10 signature matrices. (C) ABBAS, ABIS, quanTIseq, CIBERSORT and MIXTURE self-check test. The estimated coefficient 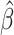 > 0 are presented in each rectangle for both LM22 and TIL10 signature matrices. The coefficients represent the estimated proportion for each cell type when its pure signature was used to feed the algorithm. (D) Distribution of the estimated NCTs for the LM22 signature. (E) Distribution of the estimated NCTs for the TIL10 signature. In both graphs, the X-axis represents the true number of coefficients fed into the algorithm and the Y-axis the estimated coefficients calculated by each method. LM22 signature matrix: BN = naïve B cells, BM = memory B cells, PC = Plasma cells, CD8 = CD8 T cells, CD4N = naïve CD4 T cells, CD4Mr = resting memory CD4 T cells, CD4Ma = activated memory CD4 T cells, FH = follicular helper T cells, Tr = regulatory T cells, TGD = gamma delta T cells (γδ T cells), NKr = resting Natural killer cells, NKa = activated Natural killer cells, M = Monocytes, M0 = M0 Macrophages, M1 = M1 Macrophages, M2 = M2 Macrophages, Dr = resting Dendritic cells, Da = activated Dendritic cells, Mr = resting Mast cells, Ma = activated Mast cells, E = Eosinophils and N = Neutrophils. TIL10 signature matrix: B = B cells, CD4 = CD4 T cells, CD8 = CD8 T cells, D = Dendritic cells, M1 = M1 Macrophages, M2 = M2 Macrophages, Mo = Monocytes, N = Neutrophils, NK = Natural killer cells, Tr = regulatory T cells.
> 0 are presented in each rectangle for both LM22 and TIL10 signature matrices. The coefficients represent the estimated proportion for each cell type when its pure signature was used to feed the algorithm. (D) Distribution of the estimated NCTs for the LM22 signature. (E) Distribution of the estimated NCTs for the TIL10 signature. In both graphs, the X-axis represents the true number of coefficients fed into the algorithm and the Y-axis the estimated coefficients calculated by each method. LM22 signature matrix: BN = naïve B cells, BM = memory B cells, PC = Plasma cells, CD8 = CD8 T cells, CD4N = naïve CD4 T cells, CD4Mr = resting memory CD4 T cells, CD4Ma = activated memory CD4 T cells, FH = follicular helper T cells, Tr = regulatory T cells, TGD = gamma delta T cells (γδ T cells), NKr = resting Natural killer cells, NKa = activated Natural killer cells, M = Monocytes, M0 = M0 Macrophages, M1 = M1 Macrophages, M2 = M2 Macrophages, Dr = resting Dendritic cells, Da = activated Dendritic cells, Mr = resting Mast cells, Ma = activated Mast cells, E = Eosinophils and N = Neutrophils. TIL10 signature matrix: B = B cells, CD4 = CD4 T cells, CD8 = CD8 T cells, D = Dendritic cells, M1 = M1 Macrophages, M2 = M2 Macrophages, Mo = Monocytes, N = Neutrophils, NK = Natural killer cells, Tr = regulatory T cells.
All methods were tested on LM22 and TIL10 molecular signatures to estimate the coefficient associated with each cell type present on each signature. To highlight the collinearity issues which produce tumor cell types misestimation, we performed the autocorrelation matrices 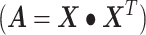 for each molecular signature. We found that both signature cell type profiles are highly correlated evidencing collinearity issues (Figure 1B). All methods, except MIXTURE, overestimate the number of cell types (NCTs) present in each profile for both signature matrices showing their lack of robustness to collinearity. Specifically, the ABBAS method provides between 6 to 9 and 4 to 6 cell types for LM22 and TIL10, respectively. The ABIS method provides between 10–13 and 4–8 cell types for each signature. The quanTiseq method is more robust to collinearity, thus providing better estimations and fewer false-positive detections on LM22. However, it misidentifies monocytes (M), CD4 T cells (CD4), M1 macrophages (M1) and Tregs. The CIBERSORT method provides the expected NCTs only for the 23% of the signature profiles and overestimates a maximum of 8 and 6 cell types for LM22 and TIL10 signatures, respectively (Figure 1C). The values outside the main diagonal (Figure 1C), which represent false-positive detections, should be zero. Most methods, except MIXTURE, provide values
for each molecular signature. We found that both signature cell type profiles are highly correlated evidencing collinearity issues (Figure 1B). All methods, except MIXTURE, overestimate the number of cell types (NCTs) present in each profile for both signature matrices showing their lack of robustness to collinearity. Specifically, the ABBAS method provides between 6 to 9 and 4 to 6 cell types for LM22 and TIL10, respectively. The ABIS method provides between 10–13 and 4–8 cell types for each signature. The quanTiseq method is more robust to collinearity, thus providing better estimations and fewer false-positive detections on LM22. However, it misidentifies monocytes (M), CD4 T cells (CD4), M1 macrophages (M1) and Tregs. The CIBERSORT method provides the expected NCTs only for the 23% of the signature profiles and overestimates a maximum of 8 and 6 cell types for LM22 and TIL10 signatures, respectively (Figure 1C). The values outside the main diagonal (Figure 1C), which represent false-positive detections, should be zero. Most methods, except MIXTURE, provide values 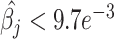 (3Q), evidencing the floating-point error in the definition of null coefficients, i.e. cell types that should reach
(3Q), evidencing the floating-point error in the definition of null coefficients, i.e. cell types that should reach 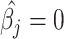 (Supplementary Table 1). These results show the unpredictable identification and proportion estimation of the immune cell types on tumor samples.
(Supplementary Table 1). These results show the unpredictable identification and proportion estimation of the immune cell types on tumor samples.
MIXTURE outperforms other methods on simulated data scenarios
One thousand simulated bulk mixture samples were built, for each signature matrix, by adding up between 2–8 (LM22) and 2–6 (TIL10) cell type expression profiles randomly chosen. They were weighted by normalized  drawn from a uniform distribution between 0.2 and 1, and then a random gene expression profile taken from the respective molecular signature was added. By doing this, bulk mixture samples with known cell type proportions and appropriate distributional characteristics were generated (see supplementary material for details). MIXTURE is the method that provides the most accurate number of present cell-types, reaching at most two more cell types than expected in 75% of the cases for both molecular signatures (Figure 1D and E). The ABBAS and ABIS methods provide more than the expected cell types in 100% of the cases, while CIBERSORT either over or underestimates the expected amount of cell types. The quanTIseq performs better than ABAS, ABIS and CIBERSORT, but worse than MIXTURE for the LM22 signature, and surprisingly provides no cell type identification for most of the cases when using simulated samples from its own signature TIL10 while MIXTURE performs with higher precision (Figure 1D and E).
drawn from a uniform distribution between 0.2 and 1, and then a random gene expression profile taken from the respective molecular signature was added. By doing this, bulk mixture samples with known cell type proportions and appropriate distributional characteristics were generated (see supplementary material for details). MIXTURE is the method that provides the most accurate number of present cell-types, reaching at most two more cell types than expected in 75% of the cases for both molecular signatures (Figure 1D and E). The ABBAS and ABIS methods provide more than the expected cell types in 100% of the cases, while CIBERSORT either over or underestimates the expected amount of cell types. The quanTIseq performs better than ABAS, ABIS and CIBERSORT, but worse than MIXTURE for the LM22 signature, and surprisingly provides no cell type identification for most of the cases when using simulated samples from its own signature TIL10 while MIXTURE performs with higher precision (Figure 1D and E).
We then compared the proportion estimates of the detected cell types against the simulated ones using Bland–Altman and correlation plots [25]. MIXTURE shows higher precision in predicting simulated proportion values of the mixture sample (Supplementary Table 2, Figure 2A and B). We found that ABBAS, ABIS and CIBERSORT tend to underestimate high-proportional values (prediction dependent bias) probably due to the overestimation of cell types, as seen in Figure 1D and E. When using the LM22 molecular signature, ABBAS and quanTIseq provided the highest false-positive predicted proportions, amount of 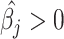 when the
when the 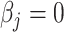 is expected by simulation and evidenced by vertical dots at
is expected by simulation and evidenced by vertical dots at 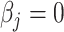 and the highest false-negative predicted proportions i.e.
and the highest false-negative predicted proportions i.e. 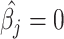 when the simulated was
when the simulated was 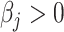 and evidenced as lying dots onto the X-axis in correlation plots. The quanTIseq method with the TIL10 signature shows bias towards both prediction extremes
and evidenced as lying dots onto the X-axis in correlation plots. The quanTIseq method with the TIL10 signature shows bias towards both prediction extremes 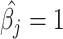 given a
given a 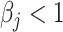 and
and 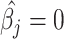 given a
given a 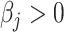 (i.e. false-negative) while CIBERSORT and MIXTURE tend to provide false-positives for both molecular signatures and false-negatives only for TIL10. However, MIXTURE estimations were much more robust than CIBERSORT estimations to noise inputs regardless of the used signature, with less bias and less sensitivity to prediction dependent bias and fewer false-positive and negative predictions. This shows that quanTIseq may miss-assign cell types, whereas ABIS and CIBERSORT may be affected by collinearity and floating-point noise (Figure 2A and B).
(i.e. false-negative) while CIBERSORT and MIXTURE tend to provide false-positives for both molecular signatures and false-negatives only for TIL10. However, MIXTURE estimations were much more robust than CIBERSORT estimations to noise inputs regardless of the used signature, with less bias and less sensitivity to prediction dependent bias and fewer false-positive and negative predictions. This shows that quanTIseq may miss-assign cell types, whereas ABIS and CIBERSORT may be affected by collinearity and floating-point noise (Figure 2A and B).
Figure 2 .
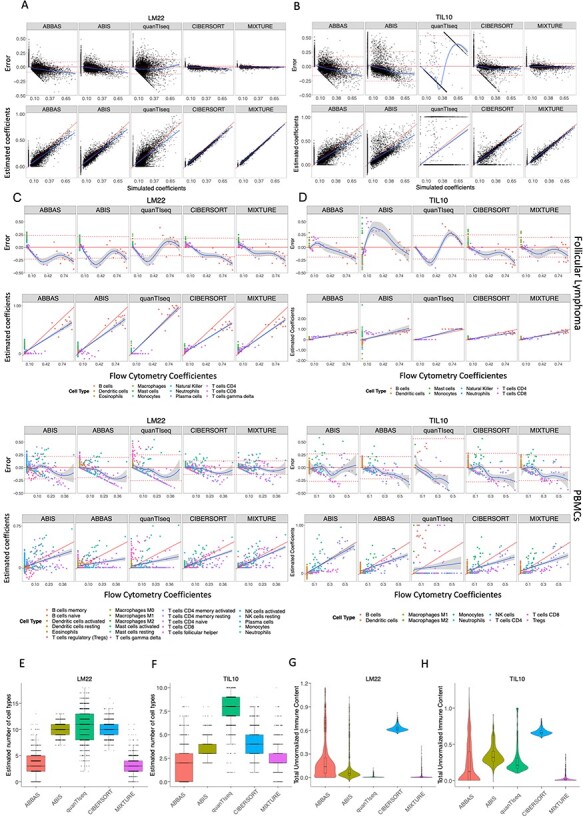
MIXTURE performance on cell type proportion estimation. (A-B) Bland–Altman and correlation plots for Scenario 2 using ABBAS, ABIS, quanTIseq, CIBERSORT and MIXTURE. The upper panels represent the differences between estimated against true simulated values for LM22 and TIL10 signatures, respectively. Horizontal lines indicate the mean ± standard deviation of errors and the loess smooth regression line represents prediction dependent bias. The lower panels show the correlation plots between estimated coefficients and true simulated proportion values. The red line represents the expected identity line, and the blue line is the linear regression line representing prediction depended bias. (C-D) Bland–Altman and correlation plots for Scenario 3 comparing estimated coefficients against true flow cytometry-derived immune content for FL (upper panels) and PBMCs (lower panels) datasets using LM22 and TIL10 signature, respectively by following the validation process from Newman et al. Bland–Altman plots show the difference between estimated coefficients and true coefficients (obtained by flow cytometry). The horizontal continuous and dashed lines represent the overall mean and standard deviation of errors, and the blue continuous line represents the loess smooth regression line to show prediction dependent bias. In correlation plots, the red straight line represents the identity and the blue line represents the regression line between true and estimated proportions. (E-H) For Scenario 4, deconvolution analysis of 1018 pure tumor cell lines from 55 different cancer types obtained from the Cancer Cell Line Encyclopedia (CCLE) was performed with ABBAS, ABIS, CIBERSORT, quanTIseq and MIXTURE methods. In figures E and F, the distribution of the number of total immune cell types estimated by each evaluated method is shown, while figures G and H show the distribution of total un-normalized immune content using LM22 or TIL10 signatures, respectively.
MIXTURE accurately estimates cell population proportions on real benchmark datasets
After proving that our method is robust to collinearity issues and floating-point errors on simulated data, we tested its performance on flow cytometry derived cell-type contents on real follicular lymphoma (FL) and peripheral blood mononuclear cells (PBMCs) benchmark data [17], following the same procedure as Newman et al. [17]. In the FL dataset, only 3 cell types (B cells, CD8 and CD4 T cells) among 12 cell types were present, whereas 9 cell types (naïve and memory B cells, CD8 T cells, naïve, memory resting and memory activated CD4 T, γδ-T cells, activated NK cells and Monocytes) out of 22 were present in the PBMCs dataset. These cell types were mapped to the LM22 and TIL10 cell types (Supplementary material Tables 3 and 4). Similar results, as obtained with simulated data, were obtained by using such datasets for LM22 (Figure 2C) and TIL10 (Figure 2D), where all methods provided comparable results between them on both signatures. The MIXTURE algorithm presented the lesser error standard deviation for FL data on both signatures, and equal or lesser than CIBERSORT on PBMCs data. In contrast, the ABBAS method presented the lesser standard deviation for PBMCs data but a significant bias-dependent error (Supplementary Table 5 and Figure 2C and D). In all cases, the error trend bias (i.e. smooth lines closer to the origin line in the Bland–Altman plots and regression line closer to the identity line in the correlation plots) was the smallest for MIXTURE. Considering the present cell types, those with available proportion values (i.e. coefficients 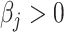 ) as the true positive ones and those cell types not present in the sample as the true negative ones, we evaluated cell type detection capabilities and prediction error distribution of each deconvolution method and molecular signature (Supplementary Table 6). In all the analyses, MIXTURE presented the smallest error rate and the highest F1 score, except for TIL10 and PBMCs data where the ABIS method provided the best performance scores, and MIXTURE resulted superior than CIBERSORT in error rate and similar for F1 score. However, MIXTURE presented better trend bias behavior than the ABIS method. On the other hand, when evaluating the distribution of the errors (Supplementary Figure 1) for null and true coefficients (i.e. zero and true cell type proportions), we found that for null coefficients, the third quartile (3Q) of the errors values of quanTIseq for LM22 (TIL10) was 0.0 (0.0) for both benchmark data, followed by MIXTURE with 0.01 (0.0) and 0.0 (0.08) for FL and PBMCs data, respectively. The error distribution associated with true cell type proportions present in the sample (
) as the true positive ones and those cell types not present in the sample as the true negative ones, we evaluated cell type detection capabilities and prediction error distribution of each deconvolution method and molecular signature (Supplementary Table 6). In all the analyses, MIXTURE presented the smallest error rate and the highest F1 score, except for TIL10 and PBMCs data where the ABIS method provided the best performance scores, and MIXTURE resulted superior than CIBERSORT in error rate and similar for F1 score. However, MIXTURE presented better trend bias behavior than the ABIS method. On the other hand, when evaluating the distribution of the errors (Supplementary Figure 1) for null and true coefficients (i.e. zero and true cell type proportions), we found that for null coefficients, the third quartile (3Q) of the errors values of quanTIseq for LM22 (TIL10) was 0.0 (0.0) for both benchmark data, followed by MIXTURE with 0.01 (0.0) and 0.0 (0.08) for FL and PBMCs data, respectively. The error distribution associated with true cell type proportions present in the sample (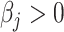 ) was much more symmetric for MIXTURE than for all the other methods, yielding the smallest interquartile range for both molecular signatures in FL data (Supplementary Table 7). Although both CIBERSORT and MIXTURE were very robust to the signature matrix as well as to the target dataset, MIXTURE provided accurate estimations with lesser error bias and a smaller standard deviation compared to CIBERSORT. In those cases where the error bias was lesser for ABBAS, the error trend bias was worse for this method compared to MIXTURE (Figure 2C and D). Moreover, MIXTURE presented better performance when using the LM22 molecular signature. These results confirm MIXTURE superior performance on real benchmark datasets.
) was much more symmetric for MIXTURE than for all the other methods, yielding the smallest interquartile range for both molecular signatures in FL data (Supplementary Table 7). Although both CIBERSORT and MIXTURE were very robust to the signature matrix as well as to the target dataset, MIXTURE provided accurate estimations with lesser error bias and a smaller standard deviation compared to CIBERSORT. In those cases where the error bias was lesser for ABBAS, the error trend bias was worse for this method compared to MIXTURE (Figure 2C and D). Moreover, MIXTURE presented better performance when using the LM22 molecular signature. These results confirm MIXTURE superior performance on real benchmark datasets.
MIXTURE provides consistent and higher robustness on false discovery for LM22 and TIL10 signatures
Since robust methods should not provide any immune cell type estimation when evaluating pure cell type samples CIBERSORT, quanTIseq and MIXTURE methods were challenged to estimate the number of immune cell types present in the 1018 pure tumor cell lines from the genomics of drug sensitivity in cancer database considered as negative controls as they are free from immune cell types i.e. all 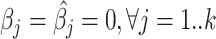 . (Scenario 4). In this test, MIXTURE provides the lesser amount of falsely detected immune cell types for both signatures (LM22 and TIL10), estimating a total NCTs ≤4 and NCTs ≤3 in 75% for LM22 and TIL10, respectively (Figure 2E and F). However, CIBERSORT provides NCTs ≥ 9 and NCTs ≥ 3 in 75% of the LM22 and TIL10 cell lines (Figure 2E and F). The quanTIseq method provides NCTs > 0 for both signatures, reaching median values of 11 and 8 cell lines for LM22 and TIL signatures, respectively. Despite this, the absolute estimated coefficient values (unnormalized
. (Scenario 4). In this test, MIXTURE provides the lesser amount of falsely detected immune cell types for both signatures (LM22 and TIL10), estimating a total NCTs ≤4 and NCTs ≤3 in 75% for LM22 and TIL10, respectively (Figure 2E and F). However, CIBERSORT provides NCTs ≥ 9 and NCTs ≥ 3 in 75% of the LM22 and TIL10 cell lines (Figure 2E and F). The quanTIseq method provides NCTs > 0 for both signatures, reaching median values of 11 and 8 cell lines for LM22 and TIL signatures, respectively. Despite this, the absolute estimated coefficient values (unnormalized 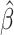 ) are lower for LM22 signature similar to MIXTURE, whereas for TIL10 the values are higher than those estimated by MIXTURE (Figure 2G and H). In contrast, MIXTURE provides the lowest NCTs as well as the lowest absolute estimated coefficient values (paired Wilcoxon test, P < 0.01) regardless of the molecular signature, whereas CIBERSORT is the one providing higher levels of absolute estimated coefficient for both evaluated molecular signatures (Figure 2G and H). Additionally, the distribution of the absolute values (non-normalized coefficients) for each cell type present on the molecular signatures predicted by each model, showed that ABIS provided negative and positive coefficients, whereas MIXTURE and quanTIseq provided the lowest values for LM22 and MIXTURE provided the lowest values for TIL10 (Supplementary Figure 2). These results confirm the superior performance of the MIXTURE-LM22 pair.
) are lower for LM22 signature similar to MIXTURE, whereas for TIL10 the values are higher than those estimated by MIXTURE (Figure 2G and H). In contrast, MIXTURE provides the lowest NCTs as well as the lowest absolute estimated coefficient values (paired Wilcoxon test, P < 0.01) regardless of the molecular signature, whereas CIBERSORT is the one providing higher levels of absolute estimated coefficient for both evaluated molecular signatures (Figure 2G and H). Additionally, the distribution of the absolute values (non-normalized coefficients) for each cell type present on the molecular signatures predicted by each model, showed that ABIS provided negative and positive coefficients, whereas MIXTURE and quanTIseq provided the lowest values for LM22 and MIXTURE provided the lowest values for TIL10 (Supplementary Figure 2). These results confirm the superior performance of the MIXTURE-LM22 pair.
Gene set based-unsupervised methods also show high false discovery rates on pure cell lines
Although the MIXTURE algorithm is not fully comparable against gene set based cell type estimation methods, we tested the later ones on pure cell lines to evaluate score-based methods like xCell [9], ImSig [11] and MCP counter [10] where, for instance, from the 34 distinct cell types (Lymphoid and Myeloid cell types) from xCell, using its web interface, it identified between 3 and 21 different cell types present in the cell lines samples. From the 7 immune cell types present in the ImSig method, all except the NK cells were detected in pure cell lines and similar results were achieved by the MCP counter with scores >0 for all the cell lines present in the model (Supplementary Figure 3). These results suggest that the gene set score-based immune content predictors presented high false-positive discovery rates, highlighting the relevance of the precision obtained by deconvolution methods to assess the immune infiltrate composition robustly.
MIXTURE analysis shows the predictive value of tumor-associated macrophages and CD8 T cells on TCGA breast cancer data
Tumor-associated macrophages (TAMs) are abundant immune cells in the tumor microenvironment capable of orchestrating inflammatory responses during breast cancer progression promoting tumor angiogenesis, matrix remodeling, invasion, immunosuppression, metastasis and chemoresistance [27,28]. Several clinical studies have shown the association between the high infiltrate of TAMs in the tumor microenvironment with poor prognosis in breast and other cancers [29] and TAMs modulating therapies [30] are currently evaluated in clinical studies which advocates for the efficient identification of the ITME composition.
We explored the ITME composition on confidently assigned PAM50 subtypes [31] in TCGA breast cancer primary tumors (Scenario 5). In this dataset, 1091 BRCA samples were classified as 18.5% Basal-like, 11% Her2-Enriched, 11.8% Luminal B, 24% Luminal A, 25.6% Not Assigned and 9% Normal-Like. Not assigned and Normal-like samples were left out, resulting in a total of 703 subjects for analysis. We used CIBERSORT, quanTIseq and MIXTURE hereafters because MIXTURE showed superior performance than ABBAS and ABIS, and because LM22 and TIL10 were built and validated for CIBERSORT and quanTIseq, respectively. The LM22 cell types were mapped to the 10 cell types of TIL10 for comparison (see Supplementary Table 3). Our analysis shows that the quanTIseq method did not provide comparable estimation profiles between LM22 and TIL10 while CIBERSORT and MIXTURE were able to identify the different immune cell types and an increased M2/M1 ratio in BRCA tumor biopsies for both signatures (Figure 3A). The correlation values for quanTIseq estimations show that only CD8 T cells and M1 macrophages resulted in higher than 0.5 between their proportion profiles on both molecular signatures. On the other hand, the B-cell proportion profiles correlated higher than 0.5 between signatures for CIBERSORT, whereas Natural Killer cells NK, CD8 T cells, M2 macrophages and neutrophils showed correlations higher than 0.5 by MIXTURE, suggesting their superior performance for both cell type signatures (Figure 3B). Since our results show the appropriateness of MIXTURE with the LM22 signature to explore the ITME, we proceeded to use the MIXTURE-LM22 pair hereafter to study the immune cell type proportion profiles and their association with survival according to the confident PAM50 intrinsic subtype assignation and by the Estrogen Receptor status (ER+/−). We found that high levels of M1 macrophages were positively associated with good outcome irrespective of the PAM50 subtype and ER status as well as CD4 activated memory T cells (Figure 3C). In contrast, high levels of M2 and M0 macrophages were associated with poor patient outcome in agreement with previously reported data [29,32] obtained through more expensive and labor-intensive technologies like mechanically dissociated gentleMACS system and flow cytometry. Also, high levels of γδ-T cells were found to be associated with a good outcome. However, controversial results were published for such cell types in different tumor types [33]. This is also the case for Tregs and CD8 T cells that were here associated with good outcomes for the Her2 subtype as reported in [34] by immunohistochemistry methods although its prognostic significance remains controversial for breast cancer [35]. Our results suggest that MIXTURE with the LM22 molecular signature provides an improved ITME estimation from bulk gene expression profiles according to what was reported by flow cytometry and IHC methods.
Figure 3 .
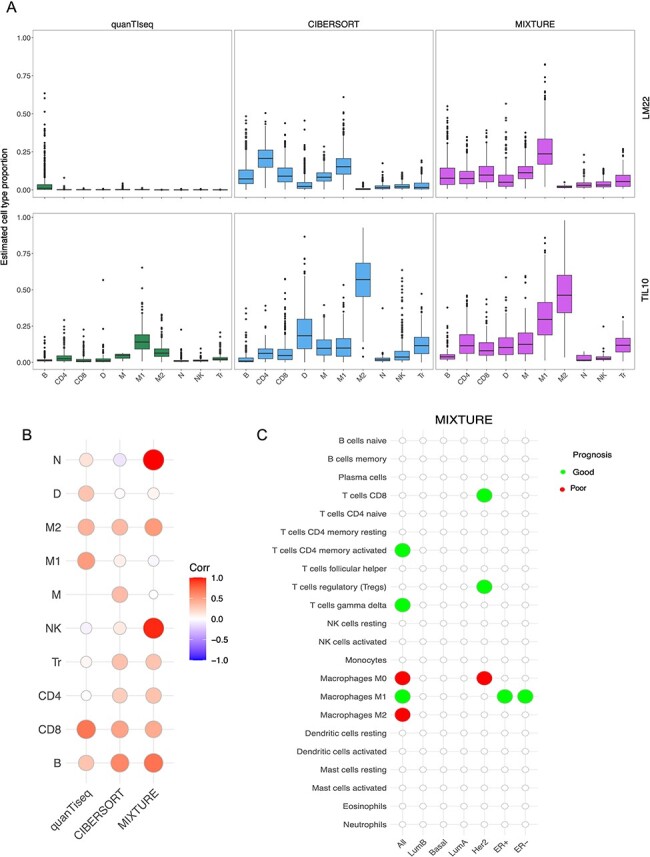
MIXTURE reveals the importance of the ITME in prognosis of breast cancer patients. (A) The TCGA-BRCA cohort classified by the PBCMC algorithm excluding the Not assigned and the Normal like subtype (n = 703) was analyzed. The estimated cell type proportions distribution (Y-axis) for each of the 10 cell types (X-axis) of the LM22 signature (upper panel) and TIL10 signatures (lower panel) are shown. The LM22 cell types where collapsed in order to match the TIL10 10 cell types (Supplementary Table 2). (B) Correlation matrix where red = 1.0 shows the highest correlation and blue = −1.0 shows the lowest correlation between the estimated cell type proportions by using LM22 and TIL10 signature matrices. (C) Cox regression analysis shows the association of each cell type proportion estimated by MIXTURE with patient prognosis for each molecular subtype using the LM22 signature. Green and red circles represent association with good and poor survival, respectively. TIL10 signature matrix: B = B cells, CD4 = CD4 T cells, CD8 = CD8 T cells, D = Dendritic cells, M1 = M1 Macrophages, M2 = M2 Macrophages, Mo = Monocytes, N = Neutrophils, NK = Natural killer cells and Tr = regulatory T cells.
MIXTURE analysis reveals the impact of known biomarkers on the immune infiltrate in lung cancer biopsies
Although antibodies targeting CTLA-4 and PD-1 co-inhibitory receptors have provided unprecedented opportunities to treat cancer patients [1], response to therapy is not predictable. Identifying predictive markers of therapeutic response is paramount to this treatment modality. Currently, genomic features like mutations in oncogenic drivers (EGFR and KRAS) and in tumor suppressor genes (TP53 and STK11), as well as TMB, are associated with different ITMEs in non-small cell lung cancer (NSCLC) patients evaluated by immunohistochemistry [36] or by whole genome/exome techniques [37]. Moreover, the TMB and PD-L1 expression are currently considered as prognostic biomarkers for ICB therapy for NSCLC [38]. Since strong adaptive immune response and a pronounced immune evasion phenotype is observed in lung cancer associated with these biomarkers, they were explored by applying MIXTURE to the TCGA-LUAD cohort. We found that tumor biopsies with similar immune infiltrate cluster together according to the presence of TP53 mutations, EGFR mutations, TMB and PD-L1 expression (Figure 4A). We also found that the presence of any mutation in EGFR drives a lower infiltrate of CD8 T cells and a higher infiltrate of monocytes and dendritic cells (DCs) compared to EGFR-wt biopsies (Supplementary Figure 4A). We found that Del19 and L858R-EGFR mutated tumors present lower infiltrate of CD8 T cells, M1 macrophages and activated memory CD4 T cells but higher infiltrates of monocytes, resting memory CD4 T cells compared to EGFR-wt biopsies (Figure 4B). Opposite to the effect of EGFR mutations in the composition of the ITME, the presence of any mutation in TP53 induces a higher infiltrate in CD8 T cells, M1 and M0 macrophages and a lower infiltrate of DCs compared to TP53-wt biopsies (Supplementary Figure 4B). Interestingly, MIXTURE analysis showed that the co-occurrence of TP53 mutations and EGFR mutations revert the inflamed immune infiltrate induced by TP53, confirming the immunosuppressive effect of EGFR mutations in lung cancer. Specifically, TP53-EGFR mutant tumors present a lower infiltrate of CD8 T cells and M1 macrophages compared to TP53 mutant tumors (Figure 4C and D). EGFR-mutant tumors also show an increased infiltrate in monocytes compared to wild-type or TP53 and STK11 mutant tumors (Figure 4E). The latter, a known tumor suppressor, does not seem to induce an inflamed immune infiltrate, as shown for TP53 by MIXTURE. Our analysis also indicates that CD8 T cells and M1 macrophages infiltrate correlate with PD-L1 mRNA levels on the TCGA-LUAD cohort (Figure 4F). In contrast, a lower infiltrate of activated DCs was found in high versus low PD-L1 expressing tumors. In addition, lower infiltrate of activated and resting DCs accompanied by a higher infiltrate of M0 macrophages was observed in high TMB (known biomarker for anti-PD-1 blockade therapy response) compared to low TMB tumors (Supplementary Figure 4C). We also found that tumors with high TMB display an immune infiltrate enriched in CD8 T cells and M1 macrophages with decreased infiltration of monocytes (Figure 4G).
Figure 4 .
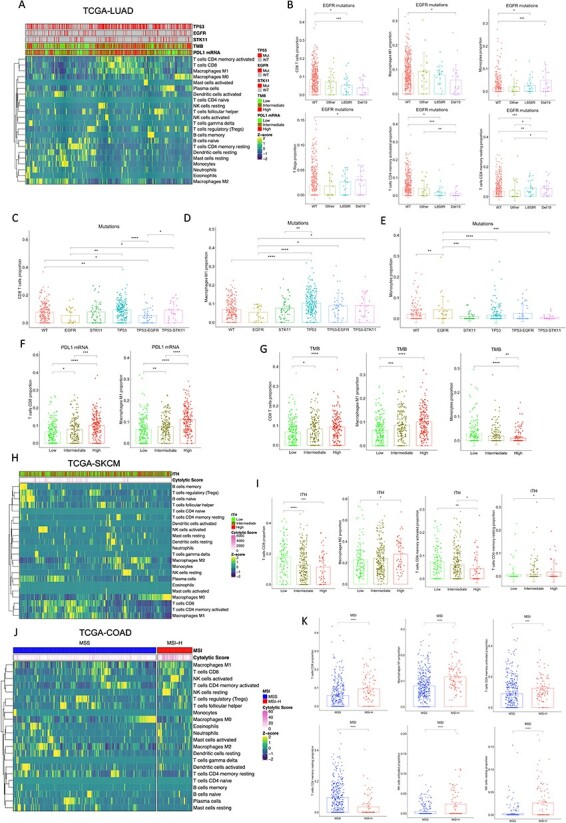
MIXTURE analysis reveals differential immune infiltrate driven by known biomarkers in cancer. (A) The heatmap represents the immune infiltrate composition analyzed with MIXTURE using the LM22 signature in the TCGA-LUAD cohort (n = 526). Mutations in EGFR, TP53, STK11, PD-L1 gene expression and TMB are represented. (B) The bar plots represent the relative cell abundance of CD8 T cells, M1 Macrophages, Monocytes, regulatory T cells, resting and activated memory CD4 T cells on tumor biopsies EGFR-wt (n = 459), EGFR L858R (n = 22), EGFR-Del19 (n = 22) or with other EGFR mutations (n = 23). (C-E) The bar plots represent the relative cell abundance of CD8 T cells (C), M1 Macrophages (D) and Monocytes (E) on tumor biopsies EGFRmut (n = 25), STK11mut (n = 49), TP53mut (n = 195), TP53-EGFRmut (n = 41), TP53-STK11mut (n = 22) or wild-type for all these genes (n = 193). (F) The bar plots represent the relative cell abundance of CD8 T cells and M1 Macrophages on tumor biopsies with low (n = 174), intermediate (n = 173) or high (n = 179) PD-L1 mRNA expression. (G) The bar plots represent the relative cell abundance of CD8 T cells, M1 Macrophages and monocytes on tumor biopsies with low (n = 171), intermediate (n = 169) or high (n = 177) TMB levels. (H) The heatmap represents the immune infiltrate compositionanalyzed with MIXTURE using the LM22 signature in the TCGA- SKCM cohort (n = 401) with transcriptomic data matched to cytolytic score and ITH data from [40].The cytolytic score and ITH levels are presented. (I) The bar plots represent the relative cell abundance of CD8 T cells, M2 Macrophages, resting and activated memory CD4 T cells on melanoma biopsies with low (n = 110), intermediate (n = 244) and high (n = 47) ITH score. (J) The heatmap represents the immune infiltrate composition analyzed with MIXTURE using the LM22 signature in the TCGA-COAD (n = 452) cohort with transcriptomic data matched to MSI status from [42]. The cytolytic score and MSI status, MSI-H (high) or MSS (stable), are presented. (K) The bar plots represent the relative cell abundance of CD8 T cells, M1 Macrophages, activated and resting memory CD4 T cells, activated and resting NK cells on MSI-H (n = 89) or MSS (n = 363) COAD biopsies. To compare between groups, a two-sided Mann Whitney–Wilcoxon test was used. ns = no statistical significance, * = P ≤ 0.05, ** = P ≤ 0.01, *** = P ≤ 0.001.
MIXTURE estimates an immunosuppressive tumor microenvironment in high intratumor heterogeneity melanoma
Cutaneous melanoma is the most highly mutated malignancy and has the highest objective response rates to checkpoint blockade [39]. The leading hypothesis in immunotherapy is that tumors with high TMB are more immunogenic because T cells respond efficiently to neoantigens, thus shaping the efficacy of anti-PD-1 therapy. In a recent study, melanoma patients from the TCGA-SKCM were grouped based on their TMB, copy number variation (CNV), and ITH estimated as the number of clones in the biopsy [40]. Neither TMB nor CNV load, as a single component, were significantly associated with patient survival. However, patients with low ITH had significantly better survival [40]. To elucidate the immune profiles of ITH-high, ITH-intermediate and ITH-low melanoma tumors, we stratified the SKCM-TCGA cohort (Scenario 5) through their ITH level and evaluated them with MIXTURE (Figure 4H). Our analysis showed a decreased infiltrate of CD8 and activated memory CD4 T cells and increased levels of M2-macrophages and memory CD4 T cells associated with ITH-high tumors (Figure 4I), confirming that ITH delineates an immunosuppressive microenvironment in the tumor, which predicts low response to ICB. These results were confirmed in HNSC biopsies from the TCGA (Supplementary Figure 5). The cytolytic score—the geometric mean expression of two key cytolytic effectors granzyme A and perforin-1—is an indirect measure of antitumor immunity upregulated upon CD8 T cell activation and following successful immunotherapy [41]. Critically, our analysis showed that there is a significantly higher cytolytic score in ITH-low melanoma biopsies compared to ITH-high tumors (Figure 4H and Supplementary Figure 6A) and resulted associated to several immune cell types such as CD8 T cells, macrophages (M0, M1 and M2) and memory CD4 T cells (Supplementary Figure 6B).
Colorectal tumors with high microsatellite instability present a highly reactive immune infiltrate
MSI—the spontaneous loss or gain of nucleotides from repetitive DNA tracts—is a tumor-agnostic predictive biomarker for pembrolizumab therapy guidance [42]. MSI has been detected in about 15% of all colorectal cancers [43], for which anti-PD-1 blocking antibodies, pembrolizumab and nivolumab, have shown clinical efficacy [44]. Understanding the immune infiltrate in high microsatellite instability (MSI-H) is crucial to elucidate the determinants of response to anti-PD-1 therapy. By stratifying the TCGA-COAD (Scenario 5) biopsies according to their MSI status [42], we found that MSI-H tumors presented a significantly higher cytolytic score compared to microsatellite-stable tumors (MSS) (Figure 4J and Supplementary Figure 7A (see supplementary materials for MSI stratification details). We then applied MIXTURE to elucidate the ITME of MSI-H and MSS biopsies (Figure 4J) and found a prominent infiltrate composed of CD8 T cells, M1 macrophages and activated memory CD4 T cells and low frequency of resting memory CD4 T cells as well as an increased number of resting and activated NK cells (Figure 4K) on tumors with MSI-H compared to MSS. There was also a higher proportion of neutrophils and a lower proportion of mast cells and plasma cells in MSI-H tumors compared to MSS tumors (Supplementary Figure 7B). This highly reactive immune infiltrate may explain the response to anti-PD-1 therapy in MSI-H tumors and highlights the importance of the ITME estimation as a predictive biomarker to select cancer patients for anti-PD-1 therapy.
MIXTURE reveals novel associations between the immune infiltrate and response to therapy in melanoma biopsies
Given the high financial costs and potential toxicities associated with ICB therapies, there is an urgent need to identify new and reliable biomarkers to distinguish better cancer patients likely to respond to immunotherapy where the understanding of the genetic landscapes of immunotherapy-sensitive and -resistant tumors results essential. Current monoclonal antibodies directed against CTLA-4 such as ipilimumab, yield considerable clinical benefit for metastatic melanoma patients. Additionally, overall mutational load, neoantigen load and expression of cytolytic markers in the immune microenvironment are significantly associated with clinical benefit [45]. By re-analyzing the transcriptomic data from the Van Allen tumor biopsies cohort with MIXTURE and dividing the biopsies into TMB-low and TMB-high, we found that TMB-high tumors present a higher number of CD8 T cells, M1 macrophages and follicular helper T cells compared to low TMB biopsies (Figure 5A). This inflammatory infiltrate has been correlated with improved response to anti-CTLA-4 immunotherapy [45–47].
Figure 5 .
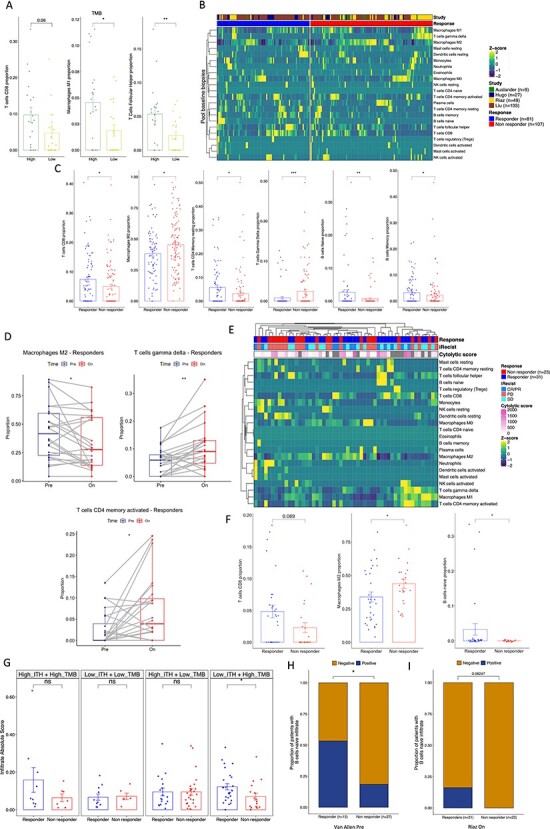
MIXTURE reveals novel associations of immune infiltrate in melanoma biopsies treated with immunotherapy. (A) The bar plots represent the proportion of CD8 T cells, M1 Macrophages and Follicular T Helper cells for patients from the Van Allen cohort (n = 40) split by the median into high versus low TMB. (B) The heatmap represents the immune infiltrate composition analyzed with MIXTURE using the LM22 signature in tumor biopsies from patients before anti-PD- 1 treatment (n = 188) from four different cohorts: Auslander (n = 9), Hugo (n = 27), Riaz (n = 49) and Liu (n = 107). Patients were grouped as responders (n = 81) and non-responders (n = 107) according to the iRECIST criteria. Partial Response (PR), Complete Response (CR), Mixed Response (MR) and Stable Disease (SD) were considered as a responder patient while Progressive Disease (PD) was considered as a non-responder patient. (C) The bar plots represent the proportion of CD8 T cells, M2 Macrophages, resting memory CD4 T cells, γδ T cells, naïve and memory B cells in responders and non-responder patients. (D) The paired box plots represent the proportions of M2 Macrophages, γδ T cells and activated memory CD4 T cells on paired analysis pre versus on anti-PD-1 treatment of responding patients calculated by MIXTURE (n = 24). (E) The heatmap represents the immune infiltrate composition analyzed with MIXTURE using the LM22 signature in tumor biopsies from patients on-treatment (n = 54), the cytolytic score and the iRECIST criteria CR/PR (n = 13), SD (n = 18) and PD (n = 23) are presented for each patient. CR/PR and SD were considered as a responder while PD was considered as a non-responder patient. (F) The bar plots represent the proportions of CD8 T cells, M2 Macrophages and naïve B cells in biopsies on treatment from patients grouped as responders (n = 31) and non-responders (n = 23) with the same criteria as in B. (G) The bar plots represent the immune infiltrate absolute score calculated by MIXTURE using the LM22 signature on tumor biopsies from the Liu cohort (n = 120). Patients were grouped as ‘high’ or ‘low’ TMB and ‘high’ or ‘low’ ITH. Both variables were used to define the groups (High_ITH + High_TMB, n = 16; R = 9 / NR = 7; Low_ITH + Low_TMB, n = 17; R = 11 / NR = 6; High_ITH + Low_TMB, n = 44; R = 18 / NR = 26; Low_ITH + High_TMB, n = 43; R = 26 / NR = 17). (H-I) The stacked bar plots show the proportion of patients with or without naïve B cells infiltrate calculated by MIXTURE on pre- treatment biopsies from the Van Allen cohort (H;n = 15 R, n = 27 NR) and in tumor biopsies on- treatment from the Riaz et al. cohort (I;n = 31 R, n = 23 NR). To compare between groups, two-sided Mann Whitney–Wilcoxon test was used. ns = no statistical significance, * = P ≤ 0.05, ** = P ≤ 0.01, *** = P ≤ 0.001.
Anti-PD-1 therapy is currently the most widely used immunotherapy in cancer. Approximately 70% of melanoma patients do not respond to anti-PD-1 treatment. Thus, a deeper understanding of the factors that correlate with response to therapy could help to prospectively identify patients likely to benefit from treatment and could also shed light on rational combinations of agents to increase response rates. Biopsies from responders (n = 81) and non-responders (n = 107) patients before treatment from four studies [48–51] that have annotated cohorts of baseline biopsies or paired baseline/on-treatment melanoma samples with anti-PD-1 therapy (Scenario 5). There was clear evidence of a different immune infiltrate that could affect treatment efficacy. Indeed, in biopsies from responding patients, there was a significantly higher infiltration of CD8 T cells, naïve and memory B cells and resting memory CD4 T cells. Likewise, there was a lower abundance of γδ-T cells and M2 macrophages (Figure 5B and C).
We wanted to study the change in the ITME before and during immunotherapy treatment in patients responding and non-responding patients [51]. MIXTURE revealed a significant decrease in M2 macrophages of responders patients whereas there was a significant increase in γδ-T cells and activated memory CD4 T cells (Figure 5D). We had previously analyzed the same biopsy cohort with CIBERSORT and did not find a selective infiltrate characterized by M2 macrophages and γδ-T cells in paired pretreatment/on-treatment samples [52]. This shows the improved performance of MIXTURE compared to CIBERSORT. In addition, when the tumor transcriptome of ‘on-treatment’ biopsies was analyzed (Figure 5E), the infiltrate of M2 macrophages resulted lower in responders patients, but a higher infiltrate of CD8 T cells and naïve B cells was found (Figure 5F). Also, the total immune infiltrate in tumors according to their TMB and ITH score and their response to immunotherapy was analyzed, as in the Liu et al. cohort. The total immune infiltrate was significantly higher in the group of responding patients with Low ITH + High TMB (Figure 5G). However, when we analyzed the ITH and TMB parameters separately, we only found a statistically significant difference in the total immune infiltrate when comparing responders versus non-responders subjects with High TMB (Supplementary Figure 8). These results confirm the importance of combining biomarkers of response to ICB to better select patients as it has recently been proposed [53].
MIXTURE analysis also revealed that the proportion of patients displaying a tumor infiltrate enriched in naïve B cells was higher in responders to anti-CTLA-4 at baseline (Figure 5H) and on-treatment with anti-PD-1 (Figure 5I) therapy. This is in accordance with recent studies that show that the presence of B cells in human tumors, in compartments called tertiary lymphoid structures (TLS), is associated with a favorable response to immunotherapy in melanoma [54].
MIXTURE reveals the modulation of the ITME after treatment with anti-CTLA-4 therapy
As discussed recently [50], the optimal role of anti-CTLA-4 in conjunction or sequentially with anti-PD-1 ICB is unclear. Thus, understanding the biology underlying the response to anti-PD-1 therapy in tumors with or without prior anti-CTLA-4 therapy may help to personalize the rational design of combination therapies for individual patients. To address this, we analyzed three RNAseq published datasets [48,50,51] from patients’ biopsies with clinically annotated data, naïve to Ipilimumab (Ipi-naïve; Figure 6A) or that progressed to Ipilimumab (Ipi-Progressive; Figure 6B) which were subsequently treated with anti-PD-1 therapy. We found that both the infiltrate of naïve B cells and memory B cells was not different between responding and non-responding patients in the Ipi-naïve group, while it was increased in responding patients in the Ipi-progressive group (Figure 6C). Likewise, the infiltrate of CD8 and resting memory CD4 T cells was significantly increased in responding patients in the Ipi-Progressive group, but not in the Ipi-naïve group. In addition, there was a higher infiltrate of γδ-T cells in non-responding patients of the Ipi-naïve group. Also, and confirming our previous observations, the M2 macrophage infiltrate was differentially lower in responding compared to non-responding patients in the Ipi-progressive but not in the Ipi-naïve group (Figure 6C). When we compared the ITME in Ipi-naïve versus Ipi-progressive patients in the anti-PD-1 responding group, we found that CD8 T cells, M1 macrophages and plasma cells were differentially higher in the Ipi-progressive group compared to the Ipi-naïve group while there was no statistical difference in the non-responding group (Figure 6D). These results agree with the previous findings [50,51], where there is evidence of immune response in the ITME at the time of progression following Ipilimumab therapy.
Figure 6 .
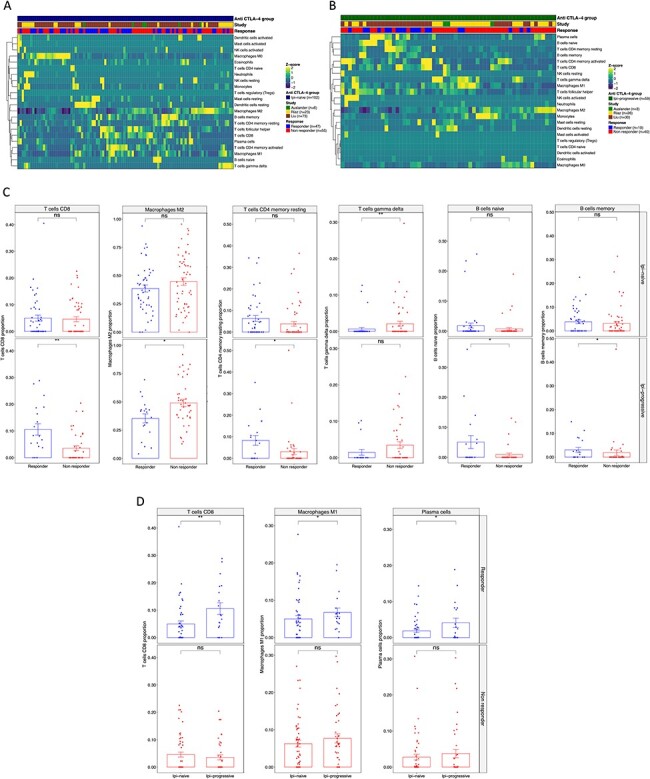
MIXTURE reveals the modulation of the ITME after first-line therapy with anti-CTLA-4 blockade. (A-B) The heatmap represents the immune infiltrate composition analyzed with MIXTURE using the LM22 signature in tumor biopsies from patients (A) before anti-PD1 treatment naïve to Ipilimumab (n = 102) or (B) progressive to Ipilimumab (n = 159) from Auslander (n = 9), Riaz (n = 49) and Liu (n = 107) cohorts. Patients are grouped as responders and non-responders according to the iRECIST criteria where PR, CR, MR and SD where considered as a responder patient while PD was considered as a non-responder patient. (C) The bar plots represent the proportions of CD8 T cells, M2 Macrophages, resting memory CD4 T cells, γδ T cells, naïve and memory B cells on biopsies from responders and non-responders patients Ipi-naïve (upper panel) or Ipi-Prog (lower panel). (D) The bar plot represents the CD8 T cells, M1 Macrophages and Plasma cells on tumor biopsies from Responding (upper panel) and non-Responding (lower panel) patients; Ipi-Naïve versus Ipi-Prog biopsies are compared. To compare between groups, two-sided Mann Whitney–Wilcoxon test was used. ns = no statistical significance, * = P ≤ 0.05, ** = P ≤ 0.01, *** = P ≤ 0.001.
Discussion
Monoclonal antibodies against PD-1 and CTLA-4 have shown robust antitumor activity but limited to a small number of patients. Understanding the interplay between the tumor and the immune surrounding cells is crucial to determine resistance mechanisms. Here we show that the tumor-infiltrating immune cells can be accurately quantified from RNA sequencing and/or microarray data applying MIXTURE, overcoming the limitations of the current state-of-the-art deconvolution methods by using a noise constraint threshold to prevent floating point errors jointly with the RFE process, improving the known robustness of the v-SVR to collinearity. Consequently, it reduces the number of false cell type detections and estimation bias, providing an accurate estimation of the cell types composition of the ITME. After proving MIXTURE’s superior performance against published methods using simulated and benchmark flow cytometry derived immune cell type content on bulk tissue data and on pure cancer cell lines-expression profiles, it was applied to publicly available datasets from the TCGA and from published cohorts of patients treated with immunotherapy and correlated the immune infiltrate with known biomarkers of prognosis and response to treatment.
First, we interrogated the TCGA-BRCA dataset to evaluate the impact of different immune cell types in the clinical outcome of BRCA patients on each molecular subtype, revealing that M2 and M0 macrophages proportions correlated with poor prognosis regardless of the BRCA molecular subtype contrasting with M1 macrophages, activated memory CD4 T cells and γδ-T cells. Interestingly, only the Her2 molecular subtype presented a better outcome associated with CD8 T cells and Tregs. Several M2-macrophage targeting strategies such as depletion, reprogramming and targeting functional molecules have been proposed [30]. The potent effector function, the broad range of activity, and safety profile of γδ-T cells make them an ideal cellular therapy to enhance current immunotherapy strategies and improve targeted therapy efficacy [55]. In addition, the association between activated memory CD4 T cells and good prognosis may be related to the antitumor properties of this population in breast cancer. Memory T cells have numerous functional properties such as the ability to respond to lower antigen concentrations, faster proliferation and the ability to recognize and reject tumor tissue [56].
Second, PD-L1 expression and genomic features such as mutations in oncogenic drivers, TMB, and MSI have been proposed as biomarkers of response to ICB [53]. Our study confirms that genomic alterations are associated with different immune profiles. By applying MIXTURE on stratified LUAD biopsies by PD-L1 mRNA expression or TMB levels, we found an increased infiltrate of CD8 T cells and M1 macrophages on high PD-L1 expression as well as high-TMB tumors. High-TMB tumors also have predicted lower infiltrate of monocytes, as recently shown [57].
Third, our MIXTURE analysis on SKCM and HNSC biopsies reveal that high ITH is immunosuppressive as tumors present high infiltrate of M2 or M0 macrophages and resting memory CD4 T cells and low infiltrate of CD8 T cells and activated memory CD4 T cells, among other cell types. These results highlight that ITH modulates the ITME and supports its role as a potential biomarker of response to immunotherapy. We also observed that a high-cytolytic score is associated with an inflamed ITME in SKCM patients, propounding that it could be considered together with the other biomarkers to better select patients for ICB treatment. Our study also revealed that both TMB and ITH modulate the total immune infiltrate of melanoma biopsies as well as the cell type composition. We found that, when using ITH or TMB as standalone biomarkers for stratifying patients, there were no differences in the total immune infiltrate of responder and non-responder patients to anti-PD-1 therapy. On the other hand, when patients were stratified combining both biomarkers, we found a higher immune infiltrate in patients that achieved clinical benefit only in the subgroup with high-TMB and low-ITH. These results confirm the importance of combining as many biomarkers of response to ICB as possible to better select patients [53].
Fourth, an additional example of improved subject stratification by incorporating the MIXTURE immune estimation is the use of MSI. Our ITME estimation on the TCGA colorectal cancer cohort confirms that MSI-H tumors exhibit the highest cytolytic score accompanied by higher proportions of CD8 T cells and M1 macrophages. This supports the notion of predictive potential of MSI biomarker for anti-PD-1 therapy in colorectal cancer due to its capacity to generate a more immunogenic tumor with an inflamed immune infiltrate more likely to respond to immunotherapy.
Fifth, melanoma biopsies from anti-PD-1 responding patients showed higher proportions of CD8 T cells, naïve B cells, memory B cells and memory resting CD4 T cells along with lower proportions of γδ-T cells and M2 macrophages, suggesting that patients whose tumors display a pro-inflammatory microenvironment have better chances to achieve clinical benefit. These results reveal the importance of the immune infiltrate composition in mechanisms underlying response and resistance to therapy as well as how the tumor microenvironment is modulated during treatment. For example, vast evidence indicates that M2-macrophages are key players of the immunosuppressive tumor microenvironment [58] and several M2-macrophage targeting strategies have been proposed to enhance the efficacy of the immune checkpoint inhibition [30]. We found that the number of patients showing infiltration of naïve B cells in the tumor microenvironment is higher in responder patients compared to non-responders both in anti-PD-1 and anti-CTLA-4 therapies. This could facilitate the generation of the recently described TLS associated with improved survival and response to immunotherapy in melanoma [54]. This finding supports the novel and relevant role of B cells in the tumor microenvironment as biomarkers of response to immunotherapies as well as potential therapeutic targets.
Last, our analysis on anti-PD-1 responding patients showed that the increase in immune infiltrate after Ipilimumab treatment is enriched in CD8 T cells, memory resting CD4 T cells, naïve and memory B cells and a decrease in M2 macrophages. These results may explain the clinical benefit achieved in Ipi-progressive patients and show that the better response rates depend not only on a higher immune infiltrate but on the nature and type of immune cells. These changes were not observed in the Ipi-naïve group. Moreover, when analyzing the differences between Ipi-naïve and Ipi-Progressive patients separately, the responders of the Ipi-progressive group showed an increase in CD8 T cells, M1 macrophages and plasma cells proportions compared to responders of the Ipi-naïve group while there were no differences between non-responder groups. These results highlight the impact of first-line treatments on subsequent response to second- and third-line therapies.
We conclude that MIXTURE performance and accuracy is highly relevant for the study of tumor biopsies to infer novel associations of the immune infiltrate with response to therapy and to propose novel predictive biomarkers and therapeutic targets. Our study shows that CD8 T cells and M2 macrophages, which are key cells with opposite roles in tumor biopsies, infiltrate differently in responders and non-responders to ICB. Accurate immune deconvolution is particularly important in the clinical setting because tumor genomic alterations used to select targeted therapies also delineate the ITME, and thus impact the response to ICB. Finally, immunotherapies provide improved survival for only a subset of cancer patients and the mechanisms of intrinsic resistance and acquired resistance remain to be elucidated, as well as the role of the immune infiltrate in these clinical scenarios.
Authors’ contributions
E.A.F. developed MIXTURE, designed validation experiments, interpreted the data, performed deconvolution analyses in simulated scenarios and method benchmarking, supervised and wrote the manuscript. Y.M. implemented quanTIseq software, performed deconvolution analyses in simulated scenarios and real datasets and contributed to manuscript writing. F.V. contributed to data interpretation and manuscript writing. J.M. implemented quanTIseq software and performed deconvolution analyses in simulated scenarios and real datasets and contributed to manuscript writing. D.R. analyzed the TCGA BRCA data with MIXTURE and contributed to manuscript writing. M.M. implemented the Python version of MIXTURE, performed the analysis over the cell lines and contributed to manuscript writing. M.B. supervised the statistical models and evaluation on results and contributed to manuscript writing. H.L. contributed to data interpretation, discussion, and manuscript writing. G.A.R. contributed to data interpretation, discussion and manuscript writing.
M.R.G. designed validation experiments, interpreted the data, supervised and wrote the manuscript.
All authors read and approved the final manuscript.
Key Points
We developed MIXTURE—a noise-constrained recursive feature selection algorithm for the accurate estimation of the immune cell types and proportions present in a tumor sample outperforming competing methods.
MIXTURE outperforms current state-of-the-art deconvolution methods and also provides increased robustness to cell type identification and proportion estimation, and is available to the wider scientific community.
We showed the efficacy of applying MIXTURE on real data to describe the association between known biomarkers and immune infiltrate differences.
By applying MIXTURE to patient biopsies, we identified novel associations between immune infiltrate and response to anti-PD-1 and anti-CTLA-4 immunotherapies.
Supplementary Material
Acknowledgments
This work was supported by the Argentinean National Council of Scientific Research (CONICET) to E.A.F., the Universidad Católica de Córdoba to E.A.F., The Harry J Lloyd Foundation to M.R.G., the National Cancer Institute (Argentina) to Y.D.M. and M.R.G., the Argentinean Agency for Promotion of Science and Technology to M.R.G. and the Grupo Español Multidisciplinar de Melanoma (GEM) to M.R.G., G.A.R. and M.R.G. thank Sales, Bunge & Born and Lounsbery Foundations for their kind donations.
Elmer A. Fernandez is a senior researcher at the National Council of Scientific Research (CONICET) of Argentina, working on the development and applications of statistical-computational-machine learning techniques for high-throughput biological data interpretation in translational medicine projects and biology. He currently leads the Bioscience Data Mining Group at CIDIE-CONICET-UCC in Córdoba, Argentina.
Yamil Mahmoud is a PhD student at the Translational Immuno Oncology Lab at the Institute of Biology and Experimental Medicine in Buenos Aires, Argentina. He works on the study of biomarkers of response and resistance to therapies in melanoma through an integrative approach focused on immuno-oncology, genomics and computational biology.
Florencia Veigas is a PhD student at the Translational Immuno Oncology Lab at the Institute of Biology and Experimental Medicine, who is studying the molecular mechanisms underlying resistance to immunotherapy in melanoma in order to identify predictive biomarkers of response and propose novel therapeutic targets.
Dario Rocha is a PhD student in Bioinformatics and M.Sc. student in applied statistics at the Universidad Nacional de Córdoba, Argentina, who is currently working in the analysis of cancer molecular data.
Matías Miranda is a degree student at the Universidad Catolica de Cordoba. He works on machine learning and their applications in bioinformatics and a junior data scientist at CoreBI.
Joaquin Merlo is a PhD student at Translational Immuno Oncology Lab at the Institute of Biology and Experimental Medicine. He works in the study of novel and validated biomarkers of melanoma progression and response to immunotherapies.
Mónica Balzarini is a principal researcher at CONICET. She leads the biostatistics group at the Agronomy Faculty at the National University of Córdoba.
Hugo D. Luján is a senior researcher at the Argentinian National Council for Scientific and Technical Research (CONICET) and full professor at the School of Medicine, Catholic University of Cordoba (UCC), Argentina. He works on deciphering the molecular networks controlling gene expression and cell differentiation of early branching eukaryotic microorganisms and host–pathogen interactions. He is the head of the Center for Research and Development in Immunology and Infectious Diseases (CIDIE CONICET/UCC).
Gabriel A. Rabinovich is a senior researcher at the National Council of Scientific Research (CONICET) of Argentina, working on the role of galectin/glycan interactions in cancer and autoimmune diseases. He currently leads the Immunopathology Lab at the Institute of Biology and Experimental Medicine in Buenos Aires, Argentina.
Maria Romina Girotti is a senior researcher at the National Council of Scientific Research (CONICET) of Argentina. Her research is focused on understanding the mechanisms of response and resistance to immunotherapy in cancer. She is the co-director of the Translational Immuno Oncology Lab at the Institute of Biology and Experimental Medicine in Buenos Aires, Argentina.
Contributor Information
Elmer A Fernández, National Council of Scientific Research.
Yamil D Mahmoud, Translational Immuno Oncology Lab at the Institute of Biology and Experimental Medicine in Buenos Aires, Argentina.
Florencia Veigas, Translational Immuno Oncology Lab at the Institute of Biology and Experimental Medicine.
Darío Rocha, Universidad Nacional de Córdoba, Argentina.
Matías Miranda, Universidad Catolica de Cordoba.
Joaquín Merlo, Translational Immuno Oncology Lab at the Institute of Biology and Experimental Medicine.
Mónica Balzarini, CONICET.
Hugo D Lujan, Argentinian National Council for Scientific and Technical Research.
Gabriel A Rabinovich, National Council of Scientific Research.
María Romina Girotti, Translational Immuno Oncology Lab at the Institute of Biology and Experimental Medicine in Buenos Aires, Argentina.
Availability
Mixture is available at https://github.com/elmerfer/MIXTURE.App.
Conflict of Interest
Authors declare no conflicts of interest.
References
- 1. Couzin-Frankel J. Breakthrough of the year 2013. Cancer immunotherapy. Science (80) 2013;342:1432–3. [DOI] [PubMed] [Google Scholar]
- 2. Gajewski TF, Schreiber H, Fu YX. Innate and adaptive immune cells in the tumor microenvironment. Nat Immunol 2013;14:1014–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Naito Y, Saito K, Shiiba K, et al. CD8+ T cells infiltrated within cancer cell nests as a prognostic factor in human colorectal cancer. Cancer Res 1998;58:3491–4. [PubMed] [Google Scholar]
- 4. Galon J, Costes A, Sanchez-Cabo F, et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science (80) 2006;313:1960–4. [DOI] [PubMed] [Google Scholar]
- 5. Fridman WH, Zitvogel L, Sautes-Fridman C, et al. The immune contexture in cancer prognosis and treatment. Nat Rev Clin Oncol 2017;14:717–34. [DOI] [PubMed] [Google Scholar]
- 6. Qian BZ, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell 2010;141:39–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Havel JJ, Chowell D, Chan TA. The evolving landscape of biomarkers for checkpoint inhibitor immunotherapy. Nat Rev Cancer 2019;19:133–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tumeh PC, Harview CL, Yearley JH, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014;515:568–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Aran D, Hu Z, xCell BAJ. Digitally portraying the tissue cellular heterogeneity landscape. Genome Biol 2017;18:220. 10.1186/s13059-017-1349-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Becht E, Giraldo NA, Lacroix L, et al. Estimating the population abundance of tissue-infiltrating immune and stromal cell populations using gene expression. Genome Biol 2016;17:218. 10.1186/s13059-016-1070-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Nirmal AJ, Regan T, Shih BB, et al. Immune cell gene signatures for profiling the microenvironment of solid tumors. Cancer Immunol Res 2018;6:1388 LP–1400. [DOI] [PubMed] [Google Scholar]
- 12. Cesano A. nCounter® PanCancer immune profiling panel (NanoString Technologies, Inc., Seattle, WA). J Immunother Cancer 2015;3:42. doi: 10.1186/s40425-015-0088-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Finotello F, Rieder D, Hackl H, et al. Next-generation computational tools for interrogating cancer immunity. Nat Rev Genet 2019;20:724–46. 10.1038/s41576-019-0166-7 [DOI] [PubMed] [Google Scholar]
- 14. Finotello F, Trajanoski Z. Quantifying tumor-infiltrating immune cells from transcriptomics data. Cancer Immunol Immunother 2018;67:1031–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gaujoux R, Seoighe C. CellMix: a comprehensive toolbox for gene expression deconvolution. Bioinformatics 2013;29:2211–2. [DOI] [PubMed] [Google Scholar]
- 16. Mariathasan S, Turley SJ, Nickles D, et al. TGFbeta attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 2018;554:544–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Newman AM, Liu CL, Green MR, et al. Robust enumeration of cell subsets from tissue expression profiles. Nat Methods 2015;12:453–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yoshihara K, Shahmoradgoli M, Martinez E, et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat Commun 2013;4:2612. 10.1038/ncomms3612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Abbas AR, Wolslegel K, Seshasayee D, et al. Deconvolution of blood microarray data identifies cellular activation patterns in systemic lupus erythematosus. PLoS One 2009;4:e6098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Finotello F, Mayer C, Plattner C, et al. Molecular and pharmacological modulators of the tumor immune contexture revealed by deconvolution of RNA-seq data. Genome Med 2019;11(34). 10.1186/s13073-019-0638-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Monaco G, Lee B, Xu W, et al. RNA-Seq signatures normalized by mRNA abundance allow absolute deconvolution of human immune cell types. Cell Rep 2019;26:1627–1640 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Newman AM, Steen CB, Liu CL, et al. Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat Biotechnol 2019;;37:773–782. 10.1038/s41587-019-0114-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Li B, Liu JS, Liu XS. Revisit linear regression-based deconvolution methods for tumor gene expression data. Genome Biol 2017;18:127. 10.1186/s13059-017-1256-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Avila Cobos F, Vandesompele J, Mestdagh P, et al. Computational deconvolution of transcriptomics data from mixed cell populations. Bioinformatics 2018;34:1969–79. [DOI] [PubMed] [Google Scholar]
- 25. Bland JM, Altman DG. Statistical methods for assessing agreement between two methods of clinical measurement. Lancet 1986;1:307–10. [PubMed] [Google Scholar]
- 26. Fernandez EA, Balzarini M, Valtuille R. Bedside linear regression equations to estimate equilibrated blood urea. Tech Probl Patients Hemodial 2011. Available from: http://www.intechopen.com/books/technicalproblems-in-patients-on-hemodialysis/bedside-linear-regression-equations-to-estimate-equilibrated-blood-urea [Google Scholar]
- 27. Kitamura T, Qian BZ, Pollard JW. Immune cell promotion of metastasis. Nat Rev Immunol 2015;15:73–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Velaei K, Samadi N, Barazvan B, et al. Tumor microenvironment-mediated chemoresistance in breast cancer. Breast 2016;30:92–100. [DOI] [PubMed] [Google Scholar]
- 29. Qiu SQ, Waaijer SJH, Zwager MC, et al. Tumor-associated macrophages in breast cancer: innocent bystander or important player? Cancer Treat Rev 2018;70:178–89. [DOI] [PubMed] [Google Scholar]
- 30. Mantovani A, Marchesi F, Malesci A, et al. Tumour-associated macrophages as treatment targets in oncology. Nat Rev Clin Oncol 2017;14:399–416. 10.1038/nrclinonc.2016.217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Parker JS, Mullins M, Cheang MC, et al. Supervised risk predictor of breast cancer based on intrinsic subtypes. J Clin Oncol 2009;27:1160–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hung CH, Chen FM, Lin YC, et al. Altered monocyte differentiation and macrophage polarization patterns in patients with breast cancer. BMC Cancer 2018;18:366. 10.1186/s12885-018-4284-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhao Y, Niu C, Cui J. Gamma-delta (gammadelta) T cells: friend or foe in cancer development? J Transl Med 2018;16(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Liu S, Foulkes WD, Leung S, et al. Prognostic significance of FOXP3+ tumor-infiltrating lymphocytes in breast cancer depends on estrogen receptor and human epidermal growth factor receptor-2 expression status and concurrent cytotoxic T-cell infiltration. Breast Cancer Res 2014;16:432. 10.1186/s13058-014-0432-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zhou Y, Shao N, Aierken N, et al. Prognostic value of tumor-infiltrating Foxp3+ regulatory T cells in patients with breast cancer: a meta-analysis. J Cancer 2017;8:4098–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Biton J, Mansuet-Lupo A, Pecuchet N, et al. TP53, STK11, and EGFR mutations predict tumor immune profile and the response to anti-PD-1 in lung adenocarcinoma. Clin Cancer Res 2018;24:5710–23. [DOI] [PubMed] [Google Scholar]
- 37. Skoulidis F, Byers LA, Diao L, et al. Co-occurring genomic alterations define major subsets of KRAS-mutant lung adenocarcinoma with distinct biology, immune profiles, and therapeutic vulnerabilities. Cancer Discov 2015;5:860–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hellmann MD, Ciuleanu TE, Pluzanski A, et al. Nivolumab plus Ipilimumab in lung cancer with a high tumor mutational burden. N Engl J Med 2018;378:2093–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Larkin J, Chiarion-Sileni V, Gonzalez R, et al. Five-year survival with combined Nivolumab and Ipilimumab in advanced melanoma. N Engl J Med 2019;381:1535–46. [DOI] [PubMed] [Google Scholar]
- 40. Wolf Y, Bartok O, Patkar S, et al. UVB-induced tumor heterogeneity diminishes immune response in melanoma. Cell 2019;179:219–235.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Rooney MS, Shukla SA, Wu CJ, et al. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell 2015;160:48–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hause RJ, Pritchard CC, Shendure J, et al. Classification and characterization of microsatellite instability across 18 cancer types. Nat Med 2016;22:1342–50. [DOI] [PubMed] [Google Scholar]
- 43. Boland CR, Goel A. Microsatellite instability in colorectal cancer. Gastroenterology 2010;138:2073–87e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ganesh K, Stadler ZK, Cercek A, et al. Immunotherapy in colorectal cancer: rationale, challenges and potential. Nat Rev Gastroenterol Hepatol 2019;16:361–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Van Allen EM, Miao D, Schilling B, et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science (80) 2015;350:207–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Rizvi NA, Hellmann MD, Snyder A, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science (80) 2015;348:124–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Snyder A, Makarov V, Merghoub T, et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med 2014;371:2189–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Auslander N, Zhang G, Lee JS, et al. Robust prediction of response to immune checkpoint blockade therapy in metastatic melanoma. Nat Med 2018. 10.1038/s41591-018-0157-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Hugo W, Zaretsky JM, Sun L, et al. Genomic and Transcriptomic features of response to anti-PD-1 therapy in metastatic melanoma. Cell 2016;165:35–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Liu D, Schilling B, Liu D, et al. Integrative molecular and clinical modeling of clinical outcomes to PD1 blockade in patients with metastatic melanoma. Nat Med 2019;25:1916–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Riaz N, Havel JJ, Makarov V, et al. Tumor and microenvironment evolution during immunotherapy with Nivolumab. Cell 2017;171:934–949.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Segovia M, Russo S, Jeldres M, et al. Targeting TMEM176B enhances antitumor immunity and augments the efficacy of immune checkpoint blockers by unleashing Inflammasome activation. Cancer Cell 2019;35:767–781.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Hegde PS, Chen DS. Top 10 challenges in cancer immunotherapy. Immunity 2020;52:17–35. [DOI] [PubMed] [Google Scholar]
- 54. Cabrita R, Lauss M, Sanna A, et al. Tertiary lymphoid structures improve immunotherapy and survival in melanoma. Nature 2020;577:561–5. [DOI] [PubMed] [Google Scholar]
- 55. Raverdeau M, Cunningham SP, Harmon C, et al. Gammadelta T cells in cancer: a small population of lymphocytes with big implications. Clin Transl Immunol 2019;8:e01080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Beckhove P, Feuerer M, Dolenc M, et al. Specifically activated memory T cell subsets from cancer patients recognize and reject xenotransplanted autologous tumors. J Clin Invest 2004;114:67–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Beaubier N, Bontrager M, Huether R, et al. Integrated genomic profiling expands clinical options for patients with cancer. Nat Biotechnol 2019;37:1351–60. 10.1038/s41587-019-0259-z [DOI] [PubMed] [Google Scholar]
- 58. DeNardo DG, Ruffell B. Macrophages as regulators of tumour immunity and immunotherapy. Nat Rev Immunol 2019;19(6):369–82. 10.1038/s41577-019-0127-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.