

Summary
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is an emerging new type of coronavirus that is responsible for the COVID-19 pandemic and the unprecedented global health emergency. Whole-genome sequencing (WGS) of SARS-CoV-2 plays a critical role in understanding the disease. Performance variation exists across SARS-CoV-2 viral WGS technologies, but there is currently no benchmarking study comparing different WGS sequencing protocols. We compared seven different SARS-CoV-2 WGS library protocols using RNA from patient nasopharyngeal swab samples under two storage conditions with low and high viral inputs. We found large differences in mappability and genome coverage, and variations in sensitivity, reproducibility, and precision of single-nucleotide variant calling across different protocols. For certain amplicon-based protocols, an appropriate primer trimming step is critical for accurate single-nucleotide variant calling. We ranked the performance of protocols based on six different metrics. Our findings offer guidance in choosing appropriate WGS protocols to characterize SARS-CoV-2 and its evolution.
Subject areas: Biological sciences, Molecular biology, Microbiology, Virology, omics
Graphical abstract
Highlights
-
•
Benchmarking comparison on seven different SARS-CoV-2 genome sequencing protocols
-
•
Ranked protocol performances based on genome coverage and SNV detection
-
•
Raised concern of primer-masking on SNV calling from Qiagen Amplicon protocol
-
•
Proposed bioinformatics remedy on making correct SNV call from masked genome regions
Biological sciences; Molecular biology; Microbiology; Virology; Omics
Introduction
The severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2), a novel coronavirus that caused the coronavirus disease of 2019 (COVID-19) (Guan et al., 2020; Huang et al., 2020), has led to a pandemic with >180 million confirmed cases and more than 3,923,238 deaths worldwide as of June 28th, 2021 (World Health Organization website https://www.who.int/emergencies/diseases/novel-coronavirus-2019). SARS-CoV-2 can be rapidly transmitted from person to person, even during the asymptomatic stage (Rothe et al., 2020), which is challenging healthcare systems and the public health response.
The whole-genome sequencing (WGS) of SARS-CoV-2 has been used as a powerful tool to study COVID-19 since the first sequence was released on January 10, 2020 (Wu et al., 2020). Analysis of the SARS-CoV-2 genome allows for understanding the clinical outcome (Zhang et al., 2020), developing diagnostics (Author Anonymous, 2020) and vaccines (Thanh Le et al., 2020) for COVID-19, and enables the tracking of the evolution (Andersen et al., 2020) and spread of the virus by phylogenetic analysis (Forster et al., 2020), which can reveal the dynamics of subtype evolution. To uncover the complete or near-complete sequence of SARS-CoV-2, leading laboratories have used several sequencing protocols, including shotgun metagenomic approaches (Bedford et al., 2020; Butler et al., 2020), target-capture sequencing using Twist custom target enrichment (Maurano et al., 2020), and target whole-genome amplification sequencing by a multiplex ARTIC primer set (Long et al., 2020; Lu et al., 2020a). However, large variations in performance, e.g., genome coverage and single-nucleotide variant (SNV) detection, occur across different protocols. Although there were studies that have investigated the characterizations of certain WGS protocols and sequencing platforms for SARS-CoV-2 (Hourdel et al., 2020), there are no comprehensive benchmark studies that have compared different protocols using the same patient samples to evaluate the effects of factors such as variation in viral input, sequencing platform and depth, sample quality, and storage condition on SARS-CoV-2 WGS. Notably, SARS-CoV-2 WGS requires a viral RNA isolation from the clinical samples for sequencing library construction, and there can be orders-of-magnitude differences in viral load across different subjects. A large proportion of clinical samples contain extremely low viral copy number, which may impact the quality of WGS and the confidence in calls of SNV or indel detection. More recently, two variants (lineage B.1.1.7 and B.1.617.2, also known as Alpha and Delta, first reported in the UK and India, respectively), showed increased transmissibility (Kupferschmidt, 2021; Kupferschmidt and Wadman, 2021; Rambaut et al., 2020) and later spread throughout the United States.
The mutations in the viral RNA may cause potential consequences, including increased dissemination, milder or more severe disease, and possibly decreased susceptibility to therapeutic treatment or vaccines. An accurate identification of viral genome variants by WGS using next-gen sequencing technologies is critical for the new emerging strain surveillance.
We report a benchmarking study on SARS-CoV-2 WGS using clinical nasopharyngeal (NP) swab samples. We compared seven different library construction protocols and specifically evaluated the cross-protocol performance in sequencing read mappability, viral genome coverage percentage and uniformity, effect of sequence depth, SNV calling concordance (reproducibility), precision (positive predictive value), and sensitivity (proportion of consensus variants identified at different sequencing depths and viral copy number inputs) across protocols. Our findings offer guidance with resource value not only for the research community, but also for diagnostics in choosing the most suitable SARS-CoV-2 WGS protocols and bioinformatics methods for SNV detection and calling to associate SARS-CoV-2 variation with its epidemiological and clinical characteristics.
Results
Study design, sample characteristics and SARS-CoV-2 WGS library construction and sequencing, sequencing QC and mapping
All SARS-CoV-2 viral samples were collected using NP swabs from COVID-19 patients at Loma Linda University (LLU) Medical Center, and the patients’ clinical characteristics and sample information are presented in Table S1. To examine the impact of sample storage condition on the performance of SARS-CoV-2 viral WGS, we used RNA from the NP swab samples stored under two different conditions: RNA isolated either immediately from freshly obtained samples or a frozen (−80°C) NP swab preserved in Qiagen AVL buffer. We quantified the SARS-CoV-2 virus copy number for all clinical samples using SYBR green qRT-PCR (Table S2).
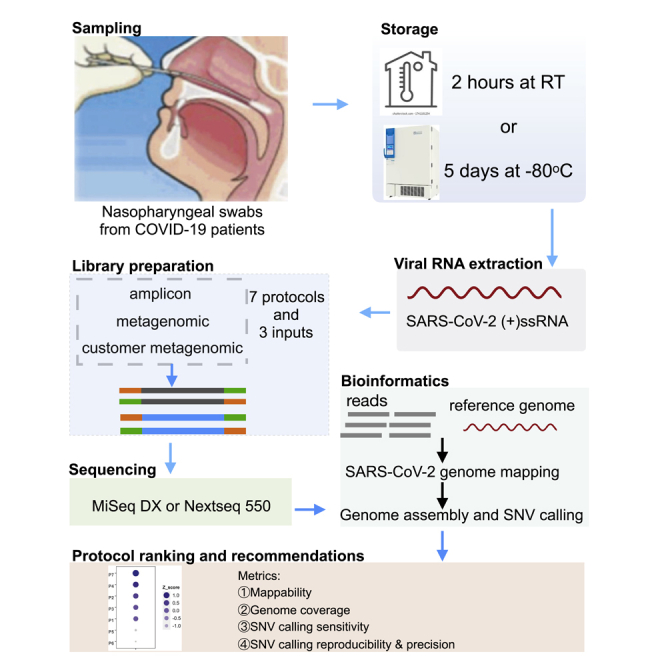
We compared seven SARS-CoV-2 WGS protocols, and constructed libraries using low (1,000 SARS-CoV-2 viral copy number, referred as 1K) and high viral copy inputs (250,000 and 1,000,000 SARS-CoV-2 viral copy number, referred as 250K and 1M, respectively) (Figure 1, Table S3). Protocol 1 (P1) is the QIAseq SARS-CoV-2 Primer Panel V1 (Qiagen) target whole-genome amplification of SARS-CoV-2 using the multiplex ARTIC V3 primer set. Protocol 2 (P2) is the QIAseq FX Single-cell RNA-seq library kit (Qiagen) coupled with human rRNA depletion. Protocol 3 (P3) is the QIAseq FX Single-cell RNA-seq library kit (Qiagen) coupled with both human and bacterial rRNA depletions. Protocol 4 (P4) is the Tecan Trio RNA-seq kit (NuGEN) coupled with human rRNA depletion and utilized single primer isothermal amplification (SPIA) technology for SARS-CoV-2 amplification. Protocols 5 and 6 (P5 and P6, respectively) refer to a cDNA synthesis recipe with a mix of random primers, oligo(dT), and four pairs of SARS-CoV-2 specific primers, followed by using either the Illumina DNA library preparation kit-DNA Nano (P5) or the Nextera XT (P6) kit. More recently, Qiagen released its modified SARS-CoV-2 Primer Panel kit (V2) which we also included as Protocol 7 (P7) and benchmarked it with the other six protocols.
Figure 1.

Schematic overview of the experimental design and workflow
Eight COVID-19 positive patient nasopharyngeal swab samples were used to construct the SARS-CoV-2 WGS libraries using seven protocols. Two different sample storage conditions were compared. For fresh samples, three different viral inputs, i.e., 1000 (1K, low) vs. either 250,000 or 1 million (250K or 1M, high) SARS-CoV-2 viral copies, were used from each same sample, whereas for frozen samples, two different viral inputs, i.e., 1000 (1K, low) vs. 1 million (1M, high) SARS-CoV-2 viral copies from each same sample, were used. P4 used different samples at low input vs. high input due to minimal total RNA amount required. The performances of protocols were benchmarked based on viral input, sequencing platform and depth, mappability, viral genome coverage and coverage uniformity, and sensitivity, reproducibility, as well as precision across seven protocols.
Overall, 56 libraries were generated across seven protocols (Tables S3 and S4). Of these, 36 libraries were sequenced on both MiSeqDx (300x2 bp and 150x2 bp, paired-end) and NextSeq 550 (150x2 bp, paired-end) (Table S4). The sequencing reads were mapped to SARS-CoV-2, human, and bacterial reference genomes. Overall, there was no substantive difference in the mapping rates between the MiSeqDx and NextSeq 550 platforms (Figure S1, Tables S5 and S6). In addition, as part of validation, we also constructed and sequenced the samples of additional 12 patients using two well-performing protocols (i.e., P2 and P7) based on our benchmarking comparison (i.e., 24 additional SARS-CoV-2 WGS libraries).
Sequence mapping to SARS-CoV-2 viral, human, and bacterial genomes
We compared the reads mapped to the SARS-CoV-2, human, and bacterial reference genomes between fresh and frozen samples across seven protocols (Figures 2A–2C). We observed statistically significant differences in mapping rates to the viral genome between fresh and frozen samples (p < 0.0001 —frozen samples generally had better viral mapping) after accounting for protocol, viral load, and input amount. However, we also observed that the lower mapping rate could be improved and compensated by deeper sequencing (i.e., ∼two times deeper for fresh samples vs. frozen) for certain protocols (Figure 2A). The significant differences in viral mapping were more easily seen in P1, P2, P3, and P4 generally corresponding to higher off-target bacterial sequence observed in fresh versus frozen samples (Figures 2A and 2C). One exception was the P1 for which fresh samples also resulted in higher (off-target) human mapping rates (Figure 2B). Overall, the above results suggested that frozen samples performed better than or equivalent to fresh samples in their mappability and on-target percentage to the SARS-CoV-2 genome for all protocols with P7 having very high mappability with either sample preservation storage methods (Figures 2A–2C).
Figure 2.

Comparison of reads mapped to SARS-CoV-2, human, bacterial genomes
The top panel box plots show the percentages of reads mapped to (A) SARS-CoV-2 genome, (B) human genome, and (C) bacterial genome, at two different sample storage conditions, i.e., fresh (FS) vs. frozen (FZ). The bottom boxplots show the percentages of reads mapped to (D) SARS-CoV-2 genome, (E) human genome, and (F) bacterial genome at different viral input, i.e., high (250K or 1M) vs. low (1K). Y axis shows the percentages of reads (log10 scaled, only for panels (A and D); X axis shows the protocol number. FS (fresh): RNA isolated from fresh samples; FZ (frozen): RNA isolated from frozen samples; Low: 1K viral input; High: 1M and 250K viral input.
Read mappability to the SARS-CoV-2 viral genome versus human and bacterial genomes were clearly different across protocols regardless of RNA prepared from fresh or frozen samples. The ARTIC amplicon-based target genome amplification technology P7 had the highest read mapping percentage (96.9% ± 7%) to the SARS-CoV-2 viral genome with the lowest read mapping percentage to human genome (0.17% ± 0.2%) among the seven protocols (Figure 2A). P1, an earlier version of the ARTIC amplicon-based protocol, had the second highest read mapping percentage (71% ± 30.2%) to the SARS-CoV-2 viral genome with a substantially higher mapping rate (17.4 ± 29.9%) to the human genome as compared to P7 (Figure 2A). All the other metagenomic approach-based protocols had starkly fewer reads mapped to the reference viral genome (<5.7%). For the QIAseq FX Single-cell RNA-seq library kit incorporated with human ribosomal RNA depletion (P2), the viral mapping rates were 1.6% ± 1.8%, and the mapping proportions to human genome were 80.9% ± 10.7%. When the same library kit was incorporated with both human and bacterial ribosomal RNA depletions (P3), the viral mapping rates dropped slightly to 1.2% ± 1.5%, and the mapping rates to the human genome also dropped to 69.4% ± 14.6%. Both P5 and P6 had the lowest percentage of reads mapped to the SARS-CoV-2 viral genome (0.03% ± 0.08%), and the highest mapping rates to human genome (96.1% ± 5% and 96.4% ± 2.5%, respectively) (Figure 2A). We also noticed that P2 had the highest rate of reads mapped to bacterial genome overall (5.9% ± 8.3%) among the seven protocols (Figure 2C).
To evaluate the variability of viral load on the SARS-CoV-2 WGS performance, we compared the SARS-CoV-2, human, and bacterial reference mapping rates between low (1K copies) and high viral inputs (250K and 1M copies) across seven protocols (Figures 2D–2F). In our study design, different levels of SARS-CoV-2 viral inputs were generated using either original undiluted patient NP sample-derived RNA or a dilution from the identical higher viral load samples across all protocols (Table S3). We found that high viral inputs (250K and 1M copies) had a higher mapping rate to the SARS-CoV-2 genome compared to low viral inputs across all protocols as expected, although the difference is smallest for P5, P6, and P7 [high vs. low, diff = 0.38, p = 0.01, linear mixed model (lmm, Table S7 and S8), Figure 2D)]. P1 and P7 performed well with P7 having better mapping rates than P1 at either input level (high viral input vs. low viral input mapping rates: P7, 99.7% ± 0.3% vs. 94.2 ± 9.7%) with significantly less sequencing reads mapped to either human or bacterial genomes (Figures 2D–2F), while P5 and P6 performed very poorly for both low and high viral inputs with extremely low SARS-CoV-2 viral mapping rates compared to other protocols (Figure 2D). For P4, low viral input resulted in orders of magnitude lower SARS-CoV-2 viral mapping rates compared to high viral inputs, e.g., 0.003% ± 0.01% vs. 2.1 ± 2.5%, suggesting P4 would require a higher viral input to obtain adequate SARS-CoV-2 viral genome coverage when sequencing depth is limited (Figure 2D). However, we recognize that the minimum RNA input restriction for the P4 protocol might be a factor in contributing to the poor mapping rates at low vial inputs. In general, the mapping rates to SARS-CoV-2 differed significantly by protocol (p < 2.20E-16, lmm), and by sample storage condition (p=2.45E-09, lmm, Table S7). According to the differences in their least square means, P7 had a statistically significant higher SARS-CoV-2 mapping rate compared to the other six protocols (P7 vs. P1, diff = 1.27, p = 1.52E-05, lmm, Table S8) followed by P1 (P1 vs. P2, diff = 3.25, p <2.20E-16, lmm, Table S8). The advantage in mapping rates of P7 and P1 is directly attributable to their viral amplicon-based design. Protocols P2 through P4 were less easily differentiated by mapping rate (Figure 2D).
Viral genome coverage and the effect of sequence read depth
To determine the impact of read depth to the SARS-CoV-2 genome coverage, we down-sampled all of the library-run datasets to 50,000 (50K), 100,000 (100K), 500,000 (500K), 1,000,000 (1M), 5,000,000 (5M), 10,000,000 (10M), 15,000,000 (15M), and ≥20,000,000 (20M) paired-end (PE) reads, and evaluated the SARS-CoV-2 viral genome coverage at different sequence depths across seven protocols (Figure 3). We noticed one sample (NP12) performed extremely differently compared to other samples across protocols likely due to a partial sample degradation. Therefore, we excluded this sample from the analysis for Figure 3. Both amplicon-based protocols P1 and P7 achieved significantly higher coverage for the SARS-CoV-2 at a threshold of >10X reads at each base (termed min10X), even with comparatively lower overall read depths compared to other protocols. Particularly, P7 consistently had higher percentages of SARS-CoV-2 genome coverage (min10X) regardless of viral input amount (Figures 3A and 3B). At low viral input and with only 1M sequencing reads, P7 had 94% ± 5.1% SARS-CoV-2 viral genome coverage (min10X). By comparison, P1, the other amplicon-based target genome amplification protocol, could only achieve 71% ± 23.7% viral genome coverage (min10X) at a sequencing depth even with 5M reads (Figure 3A). Interestingly, at low viral input, P2 and P3 achieved higher percentages of genome coverage (min10X) than P1 at 5M reads (Figure 3A). We found that P2 and P3 achieved almost complete coverage of SARS-CoV-2 genome (min10X, 95.6 ± 2.3% and 93.8 ± 5.2%, respectively) with 5M reads at low viral input (Figure 3A). P4 - P6 performed poorly with low viral input even at 15M read depth. The average performance of viral genome coverage (min10X) at the low input generally was lower than what was observed at the high input for each protocol, as would be expected. Notably, P2 achieved nearly 100% genome coverage (min10X) at the low input with 10M reads and reached nearly 100% genome coverage (min10X) at the high input with 1M reads (Figures 3A and 3B), making it much more similar to P7 at the read depth and input level. At low viral input, only P7 achieves reasonable coverage at lower read depth with P2 being a viable option at higher depths (>5M PE reads) with sample having low input.
Figure 3.

SARS-CoV-2 genome coverage across seven protocols at different sequence depths
The average genome coverage of samples at (A) low viral input (1K) and (B) high viral input (250K or 1M) with different down-sampled read depths. Bars represent the standard errors. The breath of coverage of SARS-CoV-2 genome was defined as the percentage of SARS-CoV-2 reference genome for which the genomic locations (bases) had minimal10X coverage. Seven down-sampled read depths (from 50K to 15M) plus the original total reads (20M∗) for each sample were provided. ∗For samples with sequencing reads more than 20M reads, the data only showed 20M down-sampled reads. The sequencing down-sampling were performed using seqtk (v1.0.r75) with ‘sample’ command. Sample NP12 was excluded in this figure.
Furthermore, when the samples contained high viral input, both P1 and P7 could achieve high viral genome coverage (min10X) at lower read depths as compared to all other protocols (Figure 3B). For example, when the sequencing depth was at only 50K PE, P1 had 93.9% ± 1.2% of genome coverage whereas P7 reached 98.3% ± 0.2% SARS-CoV-2 genome coverage (min10X, Figure 3B). In contrast, at high viral input, three RNA-seq based metagenomics protocols (P2, P3, and P4) had similar genome coverage levels across different sequencing depths (e.g., min10x at 1M read depth: P2, 99.3 ± 0.6%; P3, 97.5 ± 2.4%; and P4, 96.7 ± 1.6%, respectively, Figure 3B). Nevertheless, for P5 and P6, the observed genome coverage (whether >1X or >10X) was consistently low at even >20M reads, regardless of high or low viral input (Figures 3A and 3B), suggesting that P5 and P6 would not be suitable in practice to achieve necessary coverage for SNV detection.
SARS-CoV-2 viral genome coverage uniformity comparisons
To evaluate the coverage quality and regional bias relative to the viral reference genome between protocols, we computed the average coverage by genomic position across all samples across all protocols at a depth of 5M total reads (Figures 4A and 4B). As expected, due to their amplicon-based nature, P1 and P7 had the highest average coverage compared to all the other protocols (Figures 4A and 4B). A great majority of the regions across the SARS-CoV-2 genome had ∼1,000X coverage while some regions were near or exceeded 10,000X coverage for P1 and P7 at high viral input (Figure S2). The variations or “spikes” of the coverage in many regions across the viral genome were much more pronounced at the low viral input as compared to the high viral input derived from the same samples (Figure S3). For P2 and P3, the average coverage across the entire viral genome usually ranged from 300X to 600X (Figure 4A), whereas for P5 and P6, only certain regions were sequenced, and many regions showed no coverage in the samples with either low or high viral input (Figures 4A and 4B). Furthermore, we observed that protocols P2 and P3 had much higher coverage at the 3′ end of the viral genome, which was likely introduced by oligo (dT) primers during cDNA synthesis. Other than the 3′ end of the viral genome, P2 and P3 had comparably even coverage across the viral genome regardless of viral inputs (Figures 4A and 4B). However, the viral genome coverage was significantly different between the low and high viral inputs in P4 which had scarce or no coverage across whole genome at low viral input (Figures 4A and 4B). At high viral input, P4 had excellent coverage uniformity across the whole genome (Figure 4B). Finally, for completeness, in addition to greatly reduced viral genome coverage for P5 and P6, there was also a lack of coverage uniformity regardless of viral input (Figures 4A and 4B).
Figure 4.

SARS-CoV-2 Genome coverage uniformity across seven protocols
(A–D) (A) Average of SARS-CoV-2 genome coverage based on four samples at low viral input (1K); (B) Average of SARS-CoV-2 genome coverage based on four samples at high viral input (250K or 1M); for P1-P6, all the sequencing data were down-sampled to 5M reads. For P7, all the sequencing data were down-sampled to 1M reads. The boxplots in the panels (C) and (D) show the mean (M) with 2X standard deviation (SD) of coefficient of variation (CV) of the SARS-CoV-2 genome coverage uniformity at low viral input (C), or at high viral input (D). CV metric was computed using the SD and M of the coverage at each reference genome position. The inserted dash line square shows the boxplots of CV (M ± SD) for P1 vs. P7 at 1M read depth for low viral input (C) or high viral input (D). ∗P7 only had sequencing data available at 1M read depth, thus only 1M reads of sequencing data was used to calculate the CV.
We further compared the coverage uniformity using a quantitative metric, i.e., coefficient of variation (CV), across seven protocols (Figures 4C and 4D) computed using the average coverage depth by reference genome position. Amplicon-based P7 had the best uniformity of genome coverage at both low and high viral inputs and was the only protocol with a coverage CV at or less than one (i.e., 100%, Figures 4C and 4D), whereas protocols P1-P3 had ∼ three to four times higher CV compared to P7 at either viral inputs (Figures 4C and 4D). However, for P4 at high viral input, consistent with what was demonstrated in the coverage tracks (Figures 4A and 4B), the CV was very small (∼1, most similar to the P7, Figure 4D), indicating excellent uniformity in genome coverage. Conversely, at low viral input, P4 had much larger CV values (range of [9,18], Figure 4C). We also examined the relative impact of viral input (high vs. low, pairwise, derived from the same clinical sample) on coverage uniformity using our CV metric (Figure S4). We found that the uniformity and overall coverage of the SARS-CoV-2 genome as evaluated by CV improved remarkably for each of the six protocols (P4 was not evaluated due to distinct clinical samples used for low vs. high input) when higher viral inputs (e.g., 250K or 1M vs. 1K copies) were used (Figures 4A and 4B). Particularly, the coverage uniformity for P3 was the least affected by viral input changes. Coverage uniformity of P2 and P7 were also less impacted by low versus high viral input levels than other protocols (Figure S4B).
To gain a deeper understanding on the variations in read depth in certain regions and the differences of these variations between the two related amplicon-based protocols (i.e., P1 and P7), we compared the genome coverage profiles of three samples with high viral input (1M copies) for which the WGS libraries were constructed using P1 and sequenced at 5M read depth (Figure S5). We noticed that all three samples shared similar coverage patterns across the whole SARS-CoV-2 genome, suggesting that local high spikes in coverage were primer-set dependent. We examined the primer sets corresponding to those highly variable P1 coverage regions and found that the four mostly over-represented spiking regions in the three samples were associated with about 20 primer sets whose amplified genome regions were covered by multiple amplicons (Figure S5). Although the ARTIC V3 primer set was designed to cover each genome position with two amplicons (with the exception of the regions covered by amplicons 1 and 98), high coverage regions were associated with three or more amplicons. Consistent with this fact, the coverages for those regions were roughly equal to the sum of the coverages from three to four individual amplicons, supposing each amplicon had a similar amplification efficiency. Furthermore, at least one extra alternative primer pair was linked to each high coverage region, suggesting redundancy for some ARTIC V3 primers. In contrast, our study showed that P7 had a more uniform coverage across all regions compared to P1 (Figures 4A–4D, S2, and S3), even though there was no change made to the ARTIC V3 primers except for some modification of other related reagents in the P7 kit by Qiagen (proprietary, personal communication with Qiagen). In addition, for P1, our data suggested that the relatively low coverage in other regions, i.e., 1770–3303 and 23,609–24,856, might be due to the low efficiency of primers (Figure S5). Finally, it was unsurprising to observe low coverage at both the 5′- and 3′- regions for P7 and P1 since only one primer pair covered each of these regions.
Sensitivity of viral genome variant identification using consensus SNVs
Genome variants in clinical samples were called from Binary Alignment Map (BAM) files after removing duplications and primer sequences from amplicon reads. In order to evaluate the accuracy of variant calling, we called variants from clinical samples prepared from P1, P2, P3, P4, and P7 using VarScan 2 (v2.4.4) (Koboldt et al., 2012) and iVar (v1.2.2) (Grubaugh et al., 2019) against the reference SARS-CoV-2 genome NC_045512.2. The putative SNVs were defined as variants with 10X minimum coverage and >80% allele frequency, the default setting for VarScan. Our WGS data showed that five out of the eight clinical samples were able to produce SNV calls with the other three samples failing due to insufficient coverage. We analyzed the results from the independent preparation and library methods and found certain SARS-CoV-2 viral SNVs recurred across protocols and across patients. In particular, we identified 10 SNVs that were found by at least three distinct protocols in at least one patient, all at high allele frequency. Seven of these 10 were identified in more than one patient. We termed these 10 SNVs as the consensus SNVs for this study. These 10 consensus SNVs, for which our measurements of sensitivity were derived, are provided in Table S9. Notably, all consensus SNVs observed in patient samples had high (>90%) average allele frequency, which was to be expected for a haploid-type genome. Also, a separate set of 16 SNVs observed in the study that had observed allele frequency above 80% but were not classified as consensus SNVs were: 1) never replicated across protocols for the same sample; 2) never identified in more than one sample; and 3) all observed only when using low viral copy input (data not shown). Therefore, higher viral copy input was strongly associated with better cross-protocol SNV reproducibility as discussed in more detail in the next section.
Sensitivity results at high viral input for SNV calling are shown for three representative clinical samples (NP08, NP29, and NP30) in Figures 5A and 5B. All putative SNV data from high viral input at different read depths are provided in Figure 5C while Figure 5D illustrates the spatial organization of the consensus SNVs by sample. At high (1M) viral copy input level, 5M 150x2 paired-end reads were sufficient to detect nearly all consensus SNVs in the three representative clinical samples using any of P1–P4 except P7 for which only 1M reads were needed (Figure 5B). Although all five protocols were able to detect the majority of consensus SNVs from as low as 0.5M reads, P1 and P7 exhibited better performance than P2, P3, and P4 at low read depth due to a high percentage of on-target reads. We found that only 0.5M reads were sufficient for P7 to properly detect roughly 95% of the consensus SNVs (Figure 5B). However, P1 required 5M reads to detect more than 90% of the consensus SNVs, which was more than expected and contrasted greatly with P7 (Figure 5B). At 5M reads, P2 identified all consensus SNVs in all samples while P3 and P4 had sensitivity levels that exceeded P1 (Figures 5A and 5B).
Figure 5.

Influence of library protocol and read depth on SNV detection
(A) Comparison of SNV calling across P1, P2, P3, P4, and P7 protocols at various sequencing depth; all called consensus SNVs by sample are indicated using colored rectangles in the plot (some consensus SNVs are not associated with certain samples--see D).
(B) Sensitivity of SNV detection at different sequencing depth; only the data at high viral input (250K and 1M) were used and shown. Bars represent magnitudes of standard errors. Sensitivity was calculated using the consensus SNVs by sample as the set of positives. Each data point was the average of calling percentage by each protocol (n = 3). The calling percentage by protocol by read depth was calculated as the number of consensus SNVs called by sample divided by the total number of consensus SNVs previously established as present in each sample.
(C) SNVs called by P1, P2, P3, P4, and P7 at three different read depths (0.5M, 1M, 5M) in three samples at high viral inputs. The red dash line shows the threshold of SNV calling, i.e., frequency higher or equal to 80%.
(D) Consensus SNVs by sample depicted based on the NC_045512.2 genome; four sub-genome types were identified from all clinical samples in this study. Reference nucleotides are colored in black and the SNVs are colored in red.
However, at lower viral copy input, we observed that several consensus SNVs present in samples were not detected, i.e., either observed at low allele frequency or completely undetected across protocols, which indicated a reduced sensitivity in SNV detection with low viral input (Figures S6A–S6C). Particularly, at low viral copy input, we noticed that P4 did not make any SNV call at all due to its inadequate genome coverage. However, P2 achieved excellent sensitivity (almost 100%) at 5M PE reads while P7 was able to achieve ∼90% sensitivity on average at 0.5M PE reads even with low viral copy input (Figure S6D).
Reproducibility, precision of SNV detection, viral subtypes and phylogenetic analysis
To investigate the influence of viral input, sequencing read depth, and protocol on key quality parameters such as reproducibility and precision, we examined protocols P1, P2, P3, P4, and P7 using samples NP08, NP29, and NP30 which had WGS libraries constructed with both 1K and 1M viral inputs. We defined the reproducibility relative to an allele frequency threshold, i.e., a variant was reproducible between protocols A and B (or between input amounts for the same protocol) if the variant from protocol A’s library had an allele frequency equal to or greater than a threshold for which the variant was also identified by protocol B’s library at any allele frequency. We used the Jaccard index for reproducibility scoring. We observed that the protocol, read depth, viral input amount, and the sample itself impacted the reproducibility between protocols on called SNVs (Figure 6A). The sample itself and the viral input amount had the biggest impact on reproducibility, suggesting that there were characteristics about the sample, apart from viral input level, that influenced the SNV detections. The allele frequency threshold impact was muted once the threshold exceeded 50%. The average reproducibility across protocols using the Jaccard index was almost 90% when using 1M viral copies for input with reasonable allele frequency thresholds and when sequencing the sample to 5M PE reads (Figure 6B). Lowering either read depth to 1M or 0.5M PE reads or reducing the viral input to 1K copies noticeably reduced reproducibility by at least 20% and sometimes by 50% or more (Figure 6B).
Figure 6.

Influence of protocol, sample, read depth, input amount, and allele frequency calling threshold on SNV reproducibility
(A) By input amount (1K at the top vs. 1M at bottom) and read depth (0.5M, 1M, 5M), each graph indicates the reproducibility (%) of SNV calls between protocols (P1, P2, P3, P4, P7) for a given sample, where each point represents another protocol with which the indicated protocol (by color) was compared. Only at 1M viral copy input are results shown for 5M PE reads (bottom panel, right). X axis shows the samples (NP08, NP29, NP30), Y axis shows the SNV calling reproducibility.
(B) Jaccard score showing the average reproducibility between protocols across all combinations of protocols when varying read depth (0.5M, 1M, and 5M), viral input amount (1K, 1M), and allele frequency calling threshold; x-axis shows allele frequency (AF) threshold, y-axis shows the Jaccard score—average reproducibility (%); 0.5M/1K: 0.5M reads/1000 viral copies, 1M/1K: 1M reads/1000 viral copies; 0.5M/1M: 0.5M reads/1 million viral copies; 1M/1M: 1M reads/1 million viral copies; 5M/1M: 5M reads/1 million viral copies.
Regarding the precision of SNV calling, we noticed that at 1K viral copy input, a large number of low allele frequency SNVs (i.e., 5–80%, false) were putatively identified in each sample by P1, P2, P3, and P7 (P4 not evaluated at low viral copy input due to generally poor genome sequence coverage results), especially in sample NP08 (Figure S9). An increase in read depth did not improve the precision of SNV calling, nor did it reduce the number of low allele frequency SNVs (Figure S9). As SARS-CoV-2 is a haploid virus, we assumed that the vast majority of these low frequency putative variants were false SNVs. Consistent with this, none of these putative lower allele frequency SNVs were reproduced across at least three protocols (Figure S9). On the other hand, in certain clinical samples (NP08 and NP30), some putative false SNVs with high allele frequency (>80%) were called primarily by amplicon-based protocols (P1 or P7) at low viral copy input (Figures S6A–S6C, SNVs without an asterisk), nevertheless these SNVs were not reproduced even in the same samples with high viral copy input (Figures 5A and 5C).
As part of validation, we tested twelve additional clinical samples using P2 and P7 (one of the metagenomics RNA-seq or amplicon-based protocols, respectively) to compare the accuracy and sensitivity of their SNV detection. Consistent with what were observed above, at a higher viral input (>140,000 viral copies: corresponding to samples NP31, NP32, NP38, NP39, L2, L4, L7, and L8) (Table S10), both P2 and P7 were able to detect all of the same SNVs (SNV frequency >80% with >10X coverage). However, in samples NP31 and NP39, the metagenomics protocol P2 was able to detect an additional SNV at the position g.6543 which was masked by P7 (Figures S11A and S11B). At a lower viral input (<50,000 viral copies), P2 detected six of the seven SNVs in NP36, but none in NP34, NP35, and NP37. In addition, P7 detected fewer SNVs in NP34 and NP35 due to a partial genome coverage (Table S10, Figures S11A and S11B). In summary, these results further demonstrated a somehow compromised SNV detection by P7 due to primer masking and the subsequent incomplete primer trimming (see discussion), and the limitation of P2 in SNV detection when the viral copy number in the clinical sample was low.
Phylogenetic network analysis of complete SARS-CoV-2 genomes has been conducted to track the transmission of COVID-19 (Forster et al., 2020). Based on the SNVs identified in our tested samples, we performed phylogenetic analyses to explore the relatedness of the genotypes in our samples with the viral strains spread in the world. We identified 10 viral genome subtypes containing 6-13 SNVs within each genome subtype (Figure S7). From a global phylogeny generated via Nextstrain (Hadfield et al., 2018) (Figure S8A), We found that variants 3 and 7 from Loma Linda (CA, USA) were most related to the cohort from Australia, variant 6 was related to the cohort from MI (USA), variants 5 and 9 were related to the cases from CA, and variants 8 and 10 were mostly related to the cohort from Greece. Meanwhile, these variants pertain to clade 20A (variants 5 and 9), 20B (variants 8 and 10), and 20C (variants 3, 6, and 7), respectively (Figure S8B). As a note, limited by our sample collection time frame (all before June 2020), our variant analysis thus did not contain the newly emerged UK and India mutant strains which have been spread into the USA since December 2020.
In summary, we observed that nonreproducible candidate SNVs tended to have one or more of the following characteristics: 1) they tended to have allele frequency below 80% (94% of observed nonreproducible SNVs); 2) they tended to occur when using low viral input (85% of observed nonreproducible SNVs); and 3) they were observed at local pile-up depths of less than or equal to 50 bases (66% of observed nonreproducible SNVs). Our benchmarking study suggested that low viral copy input severely affected reproducibility of SNV calls as well as sensitivity and precision of SNV calls.
Discussion
The COVID-19 pandemic is causing a global health crisis. By June 2021, over 3.9 million deaths were attributable to COVID-19, and the number is continuously growing (World Health Organization website https://www.who.int/emergencies/diseases/novel-coronavirus-2019). There is an urgent need to better understand and track SARS-CoV-2 to improve the viral detection, tracing of the viral transmission, and the development of effective therapeutic approaches. Particularly, several newly emerged variants, such as lineage B.1.1.7 and lineage B.1.617.2, initially identified in the UK and India, respectively, are rapidly spreading in the world, and it was reported that these new strains had an increased transmissibility and could cause more reinfections (Kupferschmidt, 2021; Kupferschmidt and Wadman, 2021; Rambaut et al., 2020). It was speculated that these new strains might cause some challenges to the currently developed vaccines. Thus, generating full-length SARS-CoV-2 whole-genome sequence through next-generation sequencing (NGS) will allow better understanding of its evolution and enhance the treatment prevention strategies for COVID-19 (Andersen et al., 2020; Chan et al., 2020; Lu et al., 2020b; Paraskevis et al., 2020; Zhang and Holmes, 2020). Here, we compared seven WGS protocols for SARS-CoV-2 using clinical samples from infected patients, benchmarking the performances of these protocols in several aspects including the sequencing read mappability, genome coverage (percentage and uniformity, minimum sequences required); sample storage condition; effects of viral input, sequencing depth, length, and platform; sensitivity, reproducibility and precision of SNV calling and related assay factors (e.g., amount of viral input, sequencing depth, and bioinformatics pipeline).
The SARS-CoV-2 is a positive-sense single-stranded RNA virus, which has low stability once RNA enzymes are released after cellular destruction. The quality of virus RNA is critical for the detection and the overall genome sequencing. It has been reported that only 47–59% of the positive cases are identified by RT-PCR, possibly due to loss or degradation of virus RNA during the sampling process (Ai et al., 2020; Xie et al., 2020). Starting the RNA isolation immediately following NP swab sample collection may be ideal to minimize RNA degradation; however, immediate isolation is often impractical, especially when involving large cohorts of sampling at different time points. Therefore, we compared the samples isolated from two storage conditions, i.e., RNA isolated either immediately from the freshly prepared NP swabs or from the NP swabs in AVL buffer that were frozen at −80°C for 5-6 days. We found that although there were differences in the genome mappability between fresh and frozen samples across the protocols where the frozen samples performed slightly better than or equivalent to fresh samples in their on-target percentage to the SARS-CoV-2 genome (Figures 2A–2C), there was no practical difference in the on-target sequence mappability for P7. Furthermore, for other well-performing protocols such as P2, P3, and P4, one could overcome the differences in mappability by deeper sequencing (e.g., >2X deeper, Figures 3A and 3B). Thus, for the WGS of SARS-CoV-2 involving large numbers of samples we believe that using the RNA isolated from frozen samples (−80°C) can be a practical and better choice.
The ARTIC amplicon-based target whole-genome amplification of SARS-CoV-2 is considered as a highly sensitive and low-cost method which could provide high coverage for the viral genome with much less sequencing needed (St Hilaire et al., 2020). Several studies have used the ARTIC target whole-genome amplification technique for sequencing SARS-CoV-2 (Fauver et al., 2020; Gonzalez-Reiche et al., 2020; Long et al., 2020). The QIAseq SARS-CoV-2 Primer Panel protocols P1 and P7 were based on the ARTIC V3 primer set, but with a replacement of the 76_RIGHT primer by a substitute primer (i.e., 5′-TCTCTGCCAAATTGTTGGAAAGGCA-3′) (Itokawa et al., 2020). Consistent with the previous reports (Li et al., 2020), our study showed that P1 and P7 preferentially amplified SARS-CoV-2 genome up to 100-fold over human or bacterial genomes in human samples (Figures 2D–2F). Compared to the RNA-seq metagenomics-based technologies (i.e., P2, P3, and P4), P1 and P7 achieved more than 100-fold higher coverage for the SARS-CoV-2 genome depending on viral load and sequencing depth (Figures 2D–2F). At high viral input, as few as 50K reads were sufficient for P1 and P7 to achieve >90% viral genome coverage (min10X) (Figure 3B). We found that the P7 worked better than P1 for the samples with low viral copy number (Figures 3A and S3). Furthermore, we also noticed that P1 had noticeably more bias and large variations (spikes) in genomic coverage of several regions which were associated with the primer sets 19–21, 43–47, 75–77, and 88–90, respectively. However, these variations were significantly decreased with P7, which showed a much more uniform genome coverage at both low and high viral inputs (Figures 4C and S2, and S3).
Although the primer-panel based target amplicon sequencing has been shown as a cost-effective approach for sequencing the clinical COVID-19 samples to discover the individual genetic diversity (Xiao et al., 2020), we found there were some limitations for the ARTIC V3 amplicon-based target whole-genome amplification protocols. First, by design, the current ARTIC V3 amplicons only covered genome regions from positions 30 to 29,836, which would make it impossible for the ARTIC V3 amplicon-based protocols to detect a SNV outside of the PCR amplified regions. This scenario actually occurred in our benchmarking study and we found that a consensus variant, g.29868 G >A in sample NP29, was consistently detected by protocols P2, P3, and P4, but was missed by P1 and P7 (Figures 5A, 5C, and 5D). Second, a single-base mismatch between the primer and template may produce a PCR error such as chimeric PCR amplification (Potapov and Ong, 2017), which might lead to a false SNV call. For example, we found that P7, at low viral input, called a unique “false” SNV (g.28321 G > T) with almost 100% allele frequency and >1,000X coverage (Figure S9). However, this putative SNV was not detected in the same clinical sample prepared using either P7 at high viral input (1M) or P1, P2, P3, and P4 (Figures 5C and S9) at any input. Third, PCR amplified primer-originated “contaminated” sequences associated with the Qiagen protocols P1 and P7 may lead to an error in SNV calling. Coincidently, we had a consensus SNV (g.6543C >T) which was within the overlapping binding site to the right adjacent primers 21 and 21alt. Interestingly, this SNV was consistently called by P2, P3, P4, and P7 at the defined threshold (>80% frequency), but in P1 had a significantly lower variant allele frequency (Figures 5A and 5C) and was not called. Because the PCR primers could mask a SNV that was located in the primer-binding regions, proper primer trimming would be critical for accurately detecting SNVs within the primer binding regions. To understand how this inconsistency occurred, we analyzed the sequencing reads derived from the amplicons 21 and 22 (after adapter trimming) before and after primer trimming on the sequencing data generated from both P1 and P7. We found that for P1, after adapter trimming, only 7.18% of the reads contained g.6543C >T SNV; but after primer trimming using either iVar or CLC (Qiagen, https://www.qiagenbioinformatics.com/products/clc-genomics-workbench), the frequency of g.6543C >T became 43.09% or 6.46%, respectively, whereas many reads containing the primer sequences (g.6543 C) still remained and were not trimmed properly (Figure 7A). For P7, after adapter trimming, 63.03% of the reads contained the consensus SNV g.6543C >T; after primer trimming using either iVar or CLC, the frequency of g.6543C >T became 81.7% or 62.42%, respectively (Figure 7B). Per Qiagen protocols, during the library constructions for P1 and P7, the PCR amplified products were subject to an enzymatic random fragmentation which could generate primer-originated “contaminated” sequences, i.e., the reads containing partial primer sequences or reverse complementary complete/partial primer sequences that could not be removed by iVar or CLC. However, when applying Cutadapt (Martin, 2011), a trimming algorithm that removed all the partial or complete primer sequences by trimming only the end of the reads, i.e., “end-primer sequence trimming”, the frequency of the SNV calling for g.6543C >T dramatically increased to 91.14% (P1) and 95.79% (P7) (Figure 7).
Figure 7.

ARTIC V3 primer sequences masked an SNV call of g.6543C >T
A SNV in NP08 was found in the genome location at g.6543, which was within the primer n2019_21_R and n2019_21_R_alt sequences, and was also in the middle of amplicon 22. After sequencing, reads containing g.6543 could be covered by n2019_21_R and n2019_21R_alt sequences under 4 scenarios (right panel). Scenario 1: full primer sequences of n2019_21_R and n2019_21R_alt; Scenario 2: partial primer sequences of n2019_21_R and n2019_21R_alt; Scenario 3: full reverse-complementary sequences of n2019_21_R and n2019_21R_alt; Scenario 4: partial reverse-complementary sequences of n2019_21_R and n2019_21R_alt. Fastq files were first trimmed to remove adapters using Cutadapt with default settings, then subject to second round of trimming to remove SARS-CoV-2 ARTIC V3 amplicon primers, using iVar, Qiagen CLC package, and customized Cutadapt trimming. After trimming, the occurrence of primer sequences listed in each of the above scenarios were counted to provide evidence that g.6543C>T SNV call was compromised by primer-derived sequence “contamination”. Data was normalized to the number of counts per million reads. (A) Library prepared with P1 from NP08 at 1M viral input. (B) Library prepared with P7 from NP08 at 1M viral input. Y axis shows the CPM reads for P1 (A) or P7 (B); X axis shows reads containing g.6543 C allele in orange (false negative) and reads containing g.6543C >T allele in green (consensus SNV) in four different scenarios (illustrated in the right panel) using four different trimming methods (adapter trimming, iVar primer trimming, CLC primer trimming and non-internal primer trimming). Note: applying Cutadapt dramatically increased the allele frequency of g.6543C >T to 91.14% (P1) and 95.79% (P7).
In contrast to the ARTIC V3 amplicon-based target genome amplification, the RNA-seq based metagenomics sequencing protocols such as P2, P3 and P4 used an unbiased approach to cover the whole-genome. The metagenomics approach has been used for sequencing SARS-CoV-2 in several recent studies (Bedford et al., 2020; Butler et al., 2020; Lu et al., 2020a; Park et al., 2020). Obviously, a unique advantage of the metagenomic approach is its whole-genome coverage including all bases for the SARS-CoV-2 genome given an adequate sequencing depth. We found that when the samples contained a higher viral load (e.g., ∼250K or 1M copies), P2, P3, and P4 achieved almost complete SARS-CoV-2 genome coverage (min10X) with only ∼1M reads per sample (Figure 3B). When the samples NP08, NP29, and NP30 contained a lower viral input (<1K copies), we observed that only P2 achieved sufficient whole-genome coverage (min10X, Figure 3A) leading to 100% sensitivity for detecting the consensus SNVs at a depth of ∼x5M reads (Figure S6D). The protocol P4 is based on single primer isothermal amplification technology (SPIA, Tecan) coupled with the high-throughput sequencing, which can also generate a full-length SARS-CoV-2 genome. The SPIA has been shown to generate the full-length genomes for HIV, West Nile virus, bovine coronavirus, etc (Dafforn et al., 2004; Myrmel et al., 2017). However, for samples with low viral input (<1K copies), we observed that only 0.003% of the SPIA reads could map to the viral genome, suggesting that P4 might not be ideal if a low copy number of SARS-CoV-2 within sample is expected.
Detecting individual SARS-CoV-2 genome variation is critical in tracking the viral spread, evolution, and for understanding the potential drug resistance. Thus, we benchmarked and ranked the sensitivity, reproducibility, and precision of the SNV calling of SARS-CoV-2 across protocols (Figures 8 and S10). We found that the metagenomics protocol P2 was ranked consistently best in the sensitivity of SNV detection, followed by P7, P3, and P1 at either low or high viral input (Figures 8A and S10A). The rankings for reproducibility of SNV calling were very similar to rankings for sensitivity of SNV calling, although differences between top protocols were smaller and P4 at high viral input moved up in rank (Figures 8B and S10B). In contrast, there was a striking difference in the ranking order between the low viral input and high viral input regarding precision, i.e., P7 was ranked the best followed by P3 and P1 at low viral input; whereas at high viral input, P7 and P2 performed the best, followed by P3, P1, and P4 (Figures 8C and S10C). However, at low viral input, all protocols including P7 performed poorly for precision, thus the ranking may be somewhat random. At high input, the order is probably more meaningful. Therefore, one should not be surprised if the precision ranking for some protocols changes dramatically between low and high inputs. Overall, we observed that the viral input was a key factor impacting the SNV calling sensitivity, reproducibility, and precision for SARS-CoV-2, e.g., a low viral input adversely affected the SNV detection (Figures 5A, 5B, 6A, 6B, 8A–8C, S6, and S10A–S10C). As expected, limited copy number of the viral RNA requires extra PCR amplification, which inevitably introduces more noise and bias (Parekh et al., 2016) as well as potential errors (Potapov and Ong, 2017). Other studies have reported similar effects of viral input copy number on the sequencing and mutation detection quality in line with our observations (Grubaugh et al., 2019; Sah et al., 2013; Xu et al., 2014; Zhu et al., 2015).
Figure 8.

Z-score rankings of SARS-CoV-2 whole-genome sequencing protocols
Protocols were ranked individually using Z-score at each metric. Z-scores are plotted as circles with their size and color shade scaled to the Z-score value from large to small, and dark blue to light blue. Note that larger Z-score values imply better performance.
(A) Ranking of sensitivity of SNV detection (low and high inputs); sensitivity evaluates the ability of a protocol in detecting potential SNVs relative to the consensus SNVs defined (see results and methods)
(B) Ranking of reproducibility of SNV detection (low and high input); reproducibility metric measures the likelihood of SNVs detected in a given protocol that are also detected by another protocol in an independent experiment. Reproducibility and its calculation were defined in the methods.
(C) Ranking of precision of SNV detection (low and high inputs); precision metric measures the accuracy of SNVs detected by a protocol from all potential SNVs (based on a SNV call using allele frequency >5%).
(D) Ranking of SARS-CoV-2 genome mappability which measures the mapping efficiency of sequencing data to the viral genome.
(E) Ranking of SARS-CoV-2 genome coverage which measures the proportion of viral genome that can be covered at specific read depth.
(F) Ranking of uniformity of genome coverage which evaluates the evenness of coverage across viral genome. The reciprocal value of coefficient of variation (CV) was used for Z-score calculation in order to keep same ranking directionality (large value for better performance) as in other categories.
We also ranked the protocol performance based on mappability, minimal genome coverage and uniformity of genome coverage of the SARS-CoV-2 (Figures 8D–8F and S10D–S10F). Obviously, P7 and P1 performed much better than metagenomics protocols P2, P3, and P4 in the mappability at both low and high viral input (Figures 8D and S10D). In addition, P7 was ranked the best in uniformity of genome coverage at both low and high viral input, whereas P3, P4 and P2 also performed generally well (Figures 8F and S10F). However, for minimal genome coverage (% of genome with min10X), metagenomics protocols P2, P3 and P4 consistently outperformed the primer-panel based protocols P7 and P1 at both low and high viral inputs (Figures 8E and S10E)
In conclusion, our study showed that metagenomic approaches were more sensitive, reproducible, and accurate at moderate to higher read depths (e.g., 5M reads) for the SARS-CoV-2 SNV calling. Although amplicon approaches produced high coverage at lower read depths, they may yield less accurate detection (more false positives and false negatives), leading to reduced sensitivity compared to other methods for the reasons stated previously. Therefore, for protocols P2-P4, we recommend at least 1M viral copies for input and 5M PE reads so that reasonable levels of sensitivity, reproducibility, and precision are achieved. If lower read depths are preferred, then with P7, one can achieve satisfactory high levels of sensitivity, reproducibility, and precision in SNV calling with fewer reads (0.5M–1M PE reads), especially with a reasonable threshold for allele frequency and if the variants are within the amplicon design. In summary, we benchmarked SARS-CoV-2 WGS using seven NGS protocols and evaluated the differences in mappability, viral genome coverage, and variations in SNV calling sensitivity, reproducibility, and concordance across input amounts and between protocols. The results of our study will provide a thorough reference and resource on selecting appropriate WGS technologies for clinical SARS-CoV-2 samples, providing knowledge to mitigate the impact of COVID-19 on our society. It will be particularly valuable for the scientific community to better track any new emerging SARS-CoV-2 variants and strains.
Limitations of study
Due to the limited accessibility of clinical nasopharyngeal SARS-CoV-2 samples during the time when this study was carried on, one of the samples in the study could only reach to 250,000 viral copy number as the high viral input, which might add the variation within the group. Additional protocols, especially newly developed amplicon-based technology with shorter amplicons and not requiring an enzymatic random fragmentation step, should be considered in the future to extend the present findings.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Biological samples | ||
| Nasopharyngeal (NP) specimens collected from SARS-CoV-2 positive individuals | Loma Linda University Medical Center | IRB number 5200127 |
| Critical commercial assays | ||
| Trio RNA-Seq library preparation kit | Tecan | Cat No: 0357-08-FG |
| TruSeqNano DNA LT kit | Illumina | Cat No: 15041757 |
| Nextera XT DNA library prep kit | Illumina | Cat No:15032354 |
| QIAseq SARS-CoV-2 primer panel V2 | Qiagen | Cat No: 333896 |
| QIAamp viral RNA mini kit | Qiagen | Cat. No: 52,904 |
| TaqMan2019-nCoV assay kit | Applied Biosystems | Cat. No: A47532 |
| SuperScript III reverse transcriptase | Invitrogen | Cat. No: 18080093 |
| QIAseq SARS-CoV-2 primer panel V1 | Qiagen | Cat No: 333895 |
| QIAseq FX DNA library kit | Qiagen | Cat No: 180473 |
| QIAseq FX single cell RNA library kit | Qiagen | Cat No: 180735 |
| QIAseqFastSelectrRNA HMR kit | Qiagen | Cat No: 334387 |
| QIAseqFastSelect bacterial 5S/16S/23S kit | Qiagen | Cat No: 335927 |
| Fast SYBR green Master mix | Applied Biosystems | Cat No: 4385610 |
| Deposited data | ||
| All sequencing fastq files generated in this project | Short Reads Archive | SRA: PRJNA638938 |
| Code for genomic data processing | Github | https://github.com/oxwang/COVID19_MS1 |
| Oligonucleotides | ||
| SARS-CoV-2 specific primers R3: 5'-AGTCTACTTGACCATCAAC-3' | This paper | N/A |
| SARS-CoV-2 specific primers F4: 5'-CTTGAACTTCCTCTTGTCTG-3' | This paper | N/A |
| SARS-CoV-2 specific primers F1: 5'-ATTAAAGGTTTATACCTTCCC-3' | This paper | N/A |
| SARS-CoV-2 specific primers R1: 5'-TTTTTTTTTTTTGTCATTCTCC-3' | This paper | N/A |
| SARS-CoV-2 specific primers F25'-TTCTTATTTCACAGAGCA-3' | This paper | N/A |
| SARS-CoV-2 specific primers R2: 5'-AACATAACCATCCACTGAATATG-3' | This paper | N/A |
| SARS-CoV-2 specific primers F3: 5'-AAATGGGGTAAGGCTAGAC-3' | This paper | N/A |
| Software and algorithms | ||
| bwa (v0.7.12) | Li et al. 2009 | http://bio-bwa.sourceforge.net/bwa.shtml |
| samtools (v1.9) | Li et al. 2009 | https://github.com/samtools/samtools |
| Kraken (v1.1) | Wood et al. 2019 | https://github.com/DerrickWood/kraken2 |
| VarScan 2 (v2.4.4) | Koboldtet al. 2012 | https://sourceforge.net/projects/varscan/files/ |
| iVar (v1.2.2) | Grubaugh et al. 2019 | https://github.com/andersen-lab/ivar |
| lme4 (v 1.1.26) | Bates et al., 2015 | https://github.com/lme4/lme4 |
| lmerTest (v3.1.3) | Kuznetsovaet al. 2017 | https://github.com/runehaubo/lmerTestR |
| BCFTools (v1.9) | Li et al. 2009 | https://github.com/samtools/bcftools |
| Jalview (v2.11.1.0) | Waterhouse et al. 2009 | http://source.jalview.org/gitweb/?p=jalview.git;a=summary |
| Nextstrain | Hadfield et al. 2018 | https://github.com/nextstrain/ncov |
| Fastqc (v0.11.4) | Andrews, 2010 | http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ |
| CLC | Qiagen | https://www.qiagenbioinformatics.com/products/clc-genomics-workbench |
| Qualimap | Garcia-Alcaldeet al. 2012 | http://qualimap.conesalab.org |
| MultiQC | Ewels et al. 2016 | https://github.com/ewels/MultiQC |
| Cutadapt (v1.9.1) | Martin,2011 | https://github.com/marcelm/cutadapt |
| Auspice.us | Hadfield et al. 2018 | https://auspice.us/ |
| Other | ||
| MiSeqDx System | Illumina | https://www.illumina.com/systems/sequencing-platforms/miseqdx.html |
| QuantStudio 7 Flex Real-Time PCR System | Applied Biosystems | Cat No.44-856-98 |
| NextSeq 500 Sequencing System | Illumina | https://www.illumina.com/systems/sequencing-platforms/nextseq.html |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Charles Wang (chwang@llu.edu or oxwang@gmail.com).
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
The study was approved by the Institutional Review Board (IRB number 5200127) and the Institutional Biosafety Committee (IBC) of the Loma Linda University (LLU). The scope of the study information was presented to the subjects before sample collections. The IRB approved Waiver of Informed Consent (45 CFR 46.116) as this study imposes no more than minimal risk to the subjects. All the clinical specimens were collected at LLU Medical Center. Detailed information on COVID-19 patient clinical characteristics can be found in Table S1. Age and gender do not affect the results of this study.
Method details
Study design
Figure 1 illustrates our overall study design. Briefly, eight COVID-19 positive nasopharyngeal swab RNA samples, either freshly isolated or from frozen condition, were used to generate SARS-CoV-2 WGS libraries using seven protocols (Figure 1, Table S3). Two different SARS-CoV-2 inputs, low (1000 copies) vs. high (250,000 or 1 million copies) were used. Each pair-wise low vs. high input was obtained from the same clinical sample, except P4 for which the low viral input WGS libraries were obtained from different samples due to the limitation of minimal total RNA amount required (Table S3). For fresh samples, three different viral inputs, i.e., 1000 vs. either 250,000 or 1 million SARS-CoV-2 viral copies, were used from each same sample, whereas for frozen samples, two different viral inputs, i.e., 1000 vs. 1 million SARS-CoV-2 viral copies, each from the same sample were used. The seven protocols are described with more details in the result section. The SARS-CoV-2 WGS libraries were sequenced, pair-end, 300x2 or 150x2 bp, on two different Illumina platforms (MiSeqDx vs. NextSeq 550, Table S4). We benchmarked the performances of protocols on mappability, viral genome coverage (%) and uniformity, and sensitivity, reproducibility, and precision of SNV calling.
Clinical COVID-19 specimens and RNA isolation
The nasopharyngeal (NP) specimens were collected from SARS-CoV-2 positive individuals. A total of eight NP swab specimens were included in the evaluation. After collection, the NP swab was immediately immersed in 700 μl diluted AVL buffer (140 μL PBS + 560 μL AVL buffer) in an Eppendorf tube and incubated at room temperature for 10 min. For fresh samples, the RNA was extracted within 1-2 h of sample collection. For frozen samples, the tubes were placed in −80°C for 5-6 days before RNA isolation.
RNA was isolated from NP swabs with QIAamp Viral RNA Mini Kit (Qiagen, Germany) according to manufacturer’s instructions, and 5.6 μg carrier RNA in AVE buffer and 560 μL absolute ethanol were added to each sample and mixed well. The entire volume of lysate was passed through QIAamp Mini column and centrifuged at 6,000 g for 1 min, allowing RNA to bind to the column. Following the wash by AW1 and AW2 buffers, the RNA was eluted in 50 μL AVE buffer and stored at −80°C for down-stream procedures.
SARS-CoV-2 viral load determination by qRT-PCR
To confirm the presence of SARS-CoV-2 RNA and the viral copy number from clinical specimens, real-time RT-PCR was performed using SYBR green qRT-PCR method (Table S2). The 2019-nCoV primer and probe sets consisted of SARS-CoV-2-specific Orf1ab, spike (S) gene, nucleocapsid (N) gene, and human RNase P. Synthetic positive control (Applied Biosystems) containing the target sequences for each of the assays included in the 2019-nCoV Assay Kit, was included in each assay. Prior to quantitating SARS-CoV-2 viral load in clinical specimens, amplification efficiencies and limits of detection were assessed using six dilutions of 2019-nCoV positive control. All four RT-PCR reactions were performed for each sample in duplicate using the following cycling conditions on an Applied BiosystemsQuantStudio 7 Instrument (Applied Biosystems): Reverse transcription at 50°C for 5 min, initial activation at 95°C for 20 s, followed by 40 cycles of 95°C for 3 s and 60°C for 30 s, during which the quantitation of products (FAM) occurred. The efficiency of all four rRT-PCR reactions were higher than 99% (r2 ≥ 99.97%) and all three coronavirus reactions (Orf1ab, S, N) were required to be positive (Ct < 38), regardless of the quantity of RNase P detected. A result was deemed negative if all three reactions failed to detect the SARS-CoV-2 target genes (Ct ≥ 38) but RNase P was detected (Ct < 35). A result was deemed inconclusive if one or two of the three genes failed to amplify (Ct ≥ 38). A result was deemed invalid if all three coronavirus genes were negative and if there was evidence of insufficient input (e.g., RNase P Ct ≥ 35). The SARS-CoV-2 genome copy number in total RNA was quantitated by comparing the average coronavirus Ct across Orf1ab, S, and N to that of the positive control.
SARS-CoV-2 WGS library construction using QIAseq SARS-CoV-2 primer panel V1 (ARTICamplicon) library protocol (protocol 1)
The SARS-CoV-2 viral target genome amplicon libraries were constructed using QIAseq SARS-CoV-2 Primer Panel V1 (Qiagen, Germany) coupled with QIAseq FX DNA library kit (Qiagen) following the manufacturer’s protocols.
Briefly, 5 μL of total RNA of different viral input (1 million, 250,000 or 1,000 viral copies, respectively) was reverse transcribed to synthesize cDNA using random hexamers. 5 μL of cDNA was evenly split into two PCR pools (2.5 μL for each pool) and amplified into 400 bp amplicons using two sets of primers which cover 99% of the entire SARS-CoV-2 genome. The Qiagen primer panel was designed based on ARTIC V3 primers, with the exception that the right primer for amplicon 76 (nCoV-2019_76_RIGHT(−)) was replaced with a modified sequence, 5′-TCTCTGCCAAATTGTTGGAAAGGCA-3′ (Itokawa group). The PCR was performed per manufacturer’s instructions with 25-cycle amplification for 1 million and 250K viral copy samples, and 32-cycle amplification for 1,000 viral copy samples. After amplification, the contents of 2 PCR pools were combined into one single tube for each sample followed with an AMPure bead clean-up per manufacturer’s instructions. The purified amplicons were quantified using Qubit 3.0 (Life Technology) and normalized for DNA library construction.
Two inputs (40 ng and 1.8 ng) of purified amplicons were used for DNA library construction. Enzymatic fragmentation and end-repair were performed to generate 250 bp DNA fragments with an adenine on the 3′ end. Two different fragmentation times were applied depending on different DNA inputs, i.e., high input (1 million and 250K viral copy samples), 18 min; low input (1,000 viral copy samples), 12 min. The Illumina adaptors were ligated to the DNA fragments followed by AMPure bead clean up. The AMPure bead cleaned up DNA products were further amplified, i.e., 8 cycles for the 40 ng input of amplicons or 20 cycles for the 1.8 ng input of amplicons. The final libraries were quantified by Qubit 3.0 (Life Technology) and quality analyzed on a TapeStation 2200 (Agilent).
SARS-CoV-2 WGS library construction using QIAseqFX single-cell RNA-seq library protocols (protocols 2 and 3)
Two sets of libraries were constructed using the QIAseq FX Single-cell RNA library kit (Qiagen, Germany), with one set coupled with the QIAseq FastSelect rRNA HMR kit only (Protocol 2), and the other set coupled with both the QIAseq FastSelect rRNA HMR kit and the QIAseq FastSelect bacterial 5S/16S/23S kit (Protocol 3) following the manufacturer’s instructions. For fresh samples, three different viral inputs, i.e., 1000 vs. either 250,000 or 1 million SARS-CoV-2 viral copies, were used from each sample, whereas for frozen samples, two different viral inputs, i.e., 1000 vs. 1 million SARS-CoV-2 viral copies, were used.
Briefly, to deplete human ribosomal RNA, 1 μL of diluted (0.08x) FastSelect rRNA HMR (Qiagen, Germany) was added into 6 μL COVID-19 specimen RNA along with 3 μL NA denaturation buffer, followed by heating at 95°C for 3 min and then stepwise cooling to 25°C for 14 min. Afterward, reverse transcription was performed using both random primer and oligo(dT) primer, and the remaining library constructions were performed following the protocol of QIAseq FX single cell RNA library kit (Qiagen, Germany).
To deplete both human and bacterial ribosomal RNA, 1 μL of diluted (0.08x) FastSelect rRNA HMR (Qiagen, Germany) and 1 μL of diluted (0.08x) QIAseq FastSelect 5S/16S/23S (Qiagen) were added into 6 μL COVID-19 specimen RNA along with 3 μL NA denaturation buffer, followed by heated at 95°C for 3 min and then stepwise cooled to 25°C for 14 min. QIAseq Bead cleanup was carried out per the manufacturer’s instructions. Afterward, reverse transcription was conducted using both random primer and oligo (dT) primer, and the remaining library preparation steps were performed by following the protocol of QIAseq FX Single-cell RNA library kit (Qiagen).
After REPLI-g amplification, 200–1000 ng of input cDNAs were used for enzymatic fragmentation by incubating at 32°C for 15 min, followed by adaptor ligation and AMPureXP bead cleanup. Final libraries were eluted from the beads without amplification. All the libraries were quantified with Qubit 3.0 (Life Technologies) and quality analyzed on a TapeStation 2200 (Agilent).
SARS-CoV-2 WGS library construction using Tecan Trio RNA-Seq library protocol (protocol 4)
Eight RNA samples isolated from fresh and frozen specimens were used for Tecan Trio RNA-seq library construction (Tecan), following the Tecan Genomics protocol with integrated DNase treatment. For fresh sample, total RNA amounts containing 1 million or 250,000 SARS-CoV-2 viral copies for NP08 and NP17, and 1000 SARS-CoV-2 viral copies for NP12 and NP16 were used as input. For frozen samples, total RNA amounts containing 1 million or 250,000 SARS-CoV-2 viral copies for NP29 and NP30, and 1000 SARS-CoV-2 viral copies for NP26 and NP27 were used as initial input, respectively. All procedures were carried out using conditions specified in the Trio RNA-seq protocol.
The 10 μL of total RNA was treated with DNase, followed by cDNA synthesis using random hexamers. After purification by AMPure XP beads (Beckman Coulter), cDNAs were amplified on beads by single primer isothermal amplification (SPIA). Next, enzymatic fragmentation and end repair were performed to the cDNAs to generate blunt ends. The Illumina adaptors were ligated to cDNA fragments, followed by first round of library amplification. AnyDeplete probe mix was used to deplete the human ribosomal transcripts. The remained DNA libraries were amplified a second time for 6 cycles. Additionally, 9 cycles of amplification were carried out for the libraries with a yield lower than 10 ng.
After the second round of library amplification, double size selection by AMPure beads was performed to obtain library molecules with size ranging between 200 bp and 700 bp. Then, 22.5 μL of AMPure beads were added to the 50 μL library products. After incubation and magnetic separation, supernatant was collected and another 22.5 μL of AMPure beads were added. Following magnetic separation, the supernatant was removed, and the beads were washed with 70% alcohol. The final libraries were eluted in water. The libraries were quantified by Qubit 3.0 (Life Technology) and quality analyzed on a TapeStation 2200 (Agilent).
SARS-CoV-2 WGS library construction using metagenomics approach combining a customer cDNA synthesis recipe, Qiagen MDA and Illumina DNA library protocols (protocols 5 and 6)
The amount of RNA input was normalized based on the viral load determined by SYBR green qRT-PCR method. Specifically, total RNA amounts containing 1 million, 250,000 or 1000 SARS-CoV-2 viral copies were used to start the initial cDNA synthesis by SuperScript III reverse transcriptase (Invitrogen), using a mix of random primers, oligo(dT)18, and four pairs of SARS-CoV-2 specific primers that cover the 5′ AND-3′ ends. The sequences of the specific primers were: F1, 5′-ATTAAAGGTTTATACCTTCCC-3′; R1, 5′-TTTTTTTTTTTTGTCATTCTCC-3′; F2, 5′-TTCTTATTTCACAGAGCA-3′; R2, 5′-AACATAACCATCCACTGAATATG-3′; F3, 5′-AAATGGGGTAAGGCTAGAC-3′; R3, 5′-AGTCTACTTGACCATCAAC-3′; F4, 5′-AGCACACTTTCCTCGTGAAGG-3′; R4, 5′-CTTGAACTTCCTCTTGTCTG-3′. Reverse transcription (RT) primer annealing was conducted at 65°C for 5 min, then, incubated on ice for 1 min. RT was carried out at 25°C for 10 min, then at 55°C for 30 min in 10 μL volume. All RT products were used for cDNA amplification using QIAseq multiple displacement amplification (MDA) technology. After amplification, the cDNA was purified using equal volume of Agencout AMPure XP beads (Beckman Coulter). 100 ng or 1 ng purified cDNAs were used for library construction using either TruSeq DNA Nano library preparation kit (Illumina), i.e., P5, or Nextera XT DNA library preparation kit (Illumina), i.e., P6, respectively. All procedures were carried out following the protocols recommended by the manufacturers. The libraries were purified with Agencourt AMPure beads, quantitated by Qubit dsDNA HS assay (Life Technologies), and the quality was analyzed on a TapeStation 2200 with D1000 Screen tape (Agilent).
SARS-CoV-2 WGS library construction using QIAseq SARS-CoV-2 primer panel V2 (ARTICamplicon) library protocol (protocol 7)
The SARS-CoV-2 viral target genome amplicon libraries were constructed using the QIAseq SARS-CoV-2 Primer Panel V2 (Qiagen, Germany) coupled with QIAseq FX DNA library kit (Qiagen) following the manufacturer’s protocols. All the procedures including primers were identical as described in P1, except for some proprietary modifications on the buffers (Qiagen, personal communications).
SARS-CoV-2 WGS library sequencing
The libraries were multiplexed with different barcodes and pooled at 4 nM in equimolar amounts. The pooled libraries were quantified by Qubit prior to sequencing. The pooled libraries were clustered on Illumina NextSeq 550 high output flow cell at a final concentration of about 2.1 pM and MiSeqDx flow cell at 8.5 pM. The libraries were sequenced on an Illumina NextSeq 550, pair-end, 150x2 bp. The same libraries were also sequenced on an Illumina MiSeqDx, pair-end, 300x2 bp or 150x2 bp (Illumina, Inc., San Diego, CA).
SARS-CoV-2 WGS library construction and sequencing of additional clinical samples using P2 and P7
To further compare and validate the performance of the metagenomics vs. the amplicon-based protocols, two sets of libraries were constructed on 12 additional frozen NP patients’ samples with P2 and P7, respectively, using the same viral input for each sample (Table S10). Of the 12 additional samples, 8 were obtained and processed using the same method as described above. We also included 4 NP samples (L2, L4, L7, and L8) from the leftover solutions which were obtained from the pathology lab after clinical diagnosis testing (positive cases). Briefly, for the leftover samples obtained from the pathology lab, 140 μL of frozen solution from each clinical testing sample were mixed with 560 μL AVL buffer followed by subsequent RNA extraction. The libraries were constructed as described above. The P2 libraries were sequenced on a Nextseq 550, 150x2 bp, paired-end, and the P7 libraries were sequenced on a MiSeqDx, 150x2 bp, paired-end.
Sequence data processing, mapping, and mapping rate generation
Sample QC were reported by fastqc (Andrews, 2010), qualimap (Garcia-Alcalde et al., 2012), and MultiQC (Ewels et al., 2016). The raw reads were trimmed with cutadapt (v1.9.1). The trimmed reads were aligned to the Wuhan-Hu-1 reference using bwamem (v0.7.12) (Li and Durbin, 2009) with default settings. For the sequencing data generated using the ARTIC V3 based primer-panel protocols (P1 and P7), an extra primer trimming was performed using iVar (v1.2.2). The aligned reads were further de-duplicated by samtoolsrmdup (v1.9) (Li et al., 2009) to get the bam files. A Kraken2 (Wood et al., 2019) database was built based on the complete genomes in the NCBI RefSeq database for archaea, bacteria, protozoa, fungi, human, and viruses (SARS-CoV-2 genome included). To summarize the read mapping percentages to multiple taxa, the trimmed reads were classified into human, SARS-CoV-2, bacterial, and remaining reads (e.g., unclassified, archaeal, viral, fungi, protozoa) by using the Kraken2 database. The sequencing read mappability (mapping percentage) to the SARS-CoV-2 genome was computed for each of the 7 protocols at different sequencing depths.
SARS-CoV-2 SNV variant calling and generation of consensus SNVs
Variants were called on the bam files by VarScan 2 (v2.4.4) and BCFTools (v1.9). To accurately identify SNVs, we used samtoolsmpileup (parameters: -A -d 20000 -Q 0) and varscan2 (v2.4.4) (parameters: --p-value 0.99 –variants). Then, we filtered the low-confidence SNVs with snippy vcf_filter (parameters: --minqual 100 --mincov 10 --minfrac 0.8).
Samtools (v1.9) mpileup and BCFTools (v1.9) were used to generate the genome variants fastq file from the bam file, and the fastq file was converted into genome fasta file using Linux cat command. The variant fasta files generated from the same clinical sample but different protocols were piled up using Jalview (v2.11.1.0) (Waterhouse et al., 2009) alignment tool, from which one consensus fasta file was compiled for each clinical sample.
Phylogenetic analysis of SARS-CoV-2 variants
Phylogenetic analysis was performed using Nextstrain pipeline (https://github.com/nextstrain/ncov) (Hadfield et al., 2018) on a local Linux machine. Briefly, 10 SARS-CoV-2 consensus sequences from LLU NP08, NP17, NP29, and NP30, NP36, NP38, L2, L4, L7, L8 samples were combined with the public SARS-CoV-2 genome data (N = 417) from 15 countries downloaded from GISAID (http://gisaid.orgon) on August 15, 2020. The Nextstrain pipeline was run using the combined 417 public and 5 LLU SARS-CoV-2 WGS sequencing data. The output JASON file from Nextstrain pipeline was viewed using auspice (https://auspice.us) (Hadfield et al., 2018) (Figure S8).
Bioinformatics analysis for revealing a SNV masked by amplicon primers
In amplicon sequencing, a potential SNV allele could be located within an amplicon primer per se (masked SNV allele) and the amplified viral allele (potential SNV) from an adjacent second amplicon. We had a SNV, i.e., g.6543C >T (in NP08), which was covered by both n2019_21_R and n2019_21_R_alt primers. The full primer sequence could be removed from the end of a read by standard trimming or by iVar if it was located anywhere in a read. In both scenarios, the masked SNV allele reads would be removed and the potential SNV could be detected. However, P1 and P7 employed an enzymatic fragmentation step, which resulted in partial primer sequences at the end of some reads that could not be removed by either iVar or CLC Bio package. In addition, iVar may also remove the primer sequences located within the middle of a read, i.e., a “true” SNV derived from the reads amplified by a second adjacent amplicon. Under both circumstances, the potential SNV calling would be compromised. To reveal the masked SNV calling, we compared the g.6543 allele frequencies using different primer trimming methods on fastq file of NP08 at 1M viral input with 5M read depth. Briefly, the same fastq file was trimmed by quality trimming only, CLC Bio (default settings), iVar (default settings), and Cutadapt (ends trimming only), respectively. After trimming, the occurrences of full and partial sequences for primer n2019_21_R and n2019_21_R_alt, as well as their reverse complementary sequences that covered g.6543 were counted and the frequency of allele T was used to compare the efficiencies of trimming methods (Figure 7).
Methods used for protocol ranking
The ranking performances of SNV detection and viral genome mapping across seven protocols were evaluated individually for each of the six categories or metrics, using either Z score statistic based on harmonic mean (Figure 8) or displayed by individual sample/data point values (Figure S10), both derived from SNV calling and viral genome mapping data at low and high viral input. For the Z score based rankings, as each protocol contained multiple data points linked to different samples and read depths, a mean was taken as the initial ranking value for the given protocol and was used for Z score transformation. The Z score was calculated based on the average of all sample/data points per metric per protocol for either low or high input. To reduce the variation associated with viral input, only data points generated from 1K to 1M viral inputs were used for ranking. Sample NP08, NP29, and NP30 were used for SNV calling ranking evaluations on sensitivity, reproducibility, and precision. Other samples were also included in the mapping-based ranking evaluations on mappability, genome coverage, and uniformity of coverage. SNV detection sensitivity was measured by the percentage of consensus SNVs detected by a protocol at 1M and 5M PE reads. Reproducibility was measured between protocols at low and high input levels indicating whether the SNV calls using a VAF threshold of 80% were reproduced in a different protocol. There was one value per sample per protocol pair (P4, P5, and P6 were not used for low viral input results; P5 and P6 were not used for high viral input results). All data were based on sample sequencing output from 5M PE reads, except for P7 which did not have sequencing data at 5M depth and thus 1M PE read depth was used instead. Precision of SNV detection was measured with a VAF detection threshold of 20% or more using consensus SNVs as ground truth. Mappability was measured by the percentage of reads that aligned to the SARS-CoV-2 genome and was estimated overall, but not with respect to a particular read depth. The SARS-CoV-2 genome coverage was evaluated based on proportion of the viral genome which achieved at least 10X coverage from both 1M and 5M PE reads in each sample. For uniformity ranking evaluation, the reciprocal values of CV on genome coverage from all samples for each given protocol were included to compute the mean, which was used to calculate Z score (Figure 8). For the rankings displayed based on individual sample or data point for each of the six metrics, all sample/data point values were the same as used for the Z score based rankings, but no mean was calculated, which thus allowed to display the distribution of all samples and data point values for each of the six metrics at low and high viral inputs (see figure legends for detail in Figure S10).
Quantification and statistical analysis
Statistical analysis of factors impacting mappability
A linear mixed model (lmm) was created using lme4 package (Bates et al., 2015) in R to determine the significant factors explaining variations in mapping rates. The calculation details can be found in Tables S7 and S8. As mapping rates tended to be at extremes for different protocols (i.e., mapping rates were usually near 0 or 100%), we first transformed the mapping rate using the probit transform. The zero value in mapping rate was replaced with 1.00 × 10−9 for the probit transform. We then created a lmm using the transformed mapping rate as the dependent variable and employed fixed effects of protocol, viral copy input amount (low or high), and sample storage condition (fresh vs. frozen) and a random effect of the viral RNA input concentration. No interactions terms were significant and results from the simplest model with these factors were reported. p values were generated using the Satterthwaite approximation for degrees of freedom carried out by the lmerTest package in R. To identify which groups were statistically different from one another, pairwise comparisons were carried out for all the fixed effects using difflsmeans function of lmerTest package (Kuznetsova et al., 2017) in R. The degree of freedom was adjusted by Satterthwaite method.
To evaluate sequence mapping to SARS-CoV-2 viral, human, and bacterial genomes, data were presented as the mean ± one standard deviation. To evaluate sequence read depth on minimal viral genome coverage, data were presented as the mean ± one standard error.
Genome coverage and coverage uniformity calculation
The genome coverage was defined as the breath of coverage, which was measured as percentage of the SARS-CoV-2 reference genome for which the genomic positions (bases) were sequenced with minimal 10X coverage. Coverage uniformity on the SARS-CoV-2 genome was examined by comparing a quantitative metric, i.e., CV across seven protocols. CV was computed using the standard deviation and mean of the coverage at each reference genome position.
Calculations of sensitivity, reproducibility, and precision of SNV calling
Sensitivity– Variant calls from iVar were filtered for variant allele frequency or VAF (only putative variants with VAF >80% were considered as called variants). Sensitivity used the consensus SNV (discussed earlier) as the set of positives. Sensitivity was defined as TP/(TP + FN) (TP, true positive; FN, false negative). Protocols were ranked by average sensitivity using 5M reads for all protocols except P7, where 1M reads were used (P7 was not sequenced at 5M reads).
Reproducibility– Variant calls from iVar were filtered for variant allele frequency. For Figure 6, multiple VAF (0.1, 0.5, 0.8, 0.9) thresholds were considered for illustration (as well as different input levels and read depths) to establish variant calls by sample and by protocol. Given a set of variant calls, reproducibility between hypothetical protocol A and one of the other protocols was defined as the percent of variants identified by protocol A at VAF >0.8 that was also identified by the other protocol at any allele frequency for each sample. At low input, each protocol had three other protocols with which to compare (P4, P5, and P6 did not have calls at low input) with three samples, yielding nine data points for each protocol. At high input each protocol had four other protocols with which to compare (P5 and P6 did not have calls at low input) with three samples, yielding 12 data points for each protocol. Protocols were ranked by average reproducibility using 5M reads for all protocols except P7, where 1M reads were used (P7 was not sequenced at 5M reads).
We defined reproducibility relative to an allele frequency threshold: A variant was reproducible between protocols Ai and Aj if the variant from protocol Ai’s library had an allele frequency equal to or greater than a threshold and that variant was also identified by protocol Aj’s library at any allele frequency. For the summary measures of reproducibility between protocols, we averaged reproducibility values across Ai using the Jaccard index for reproducibility scoring.
Avg Reproducibility Protocol (Aj,α) = where n is the number of protocols and Ai and Aj are two sets of called SNVs.
Precision - Variant calls from iVar were filtered for variant allele frequency (putative variants with VAF >20% were considered as called variants, a lower threshold than that used for sensitivity, to better measure potential false calls). Precision used the consensus SNV (discussed earlier) as the set of positives. Calls of variants with VAF >20% that were not a consensus SNV were termed false positives (FP). Precision, also known as positive predictive value (PPV), was defined as TP/(TP + FP). Protocols were ranked by average precision using 5M reads for all protocols except P7, where 1M reads were used (P7 was not sequenced at 5M reads).
Acknowledgments
The authors are very grateful to Elder Michael Wan, Minister Tom Tui, Ms. Cherry Ngan of the House of Joy Christian Church; Pastor Ray Yang, Elder Bill Huang and Dr. George Liu of the Harvest Chinese Christian Church; for their generous donations of personal protective equipment for our study. The authors would like to thank the LLU Institutional Review Board Committee, particularly Mrs. Amy Casey and Dr. Travis Losey, for their expedited assistance in reviewing of the IRB application. The authors would also like to thank the following people and organizations for the assistances either in securing some critical reagents needed or providing technical consultation to our study during the COVID-19 pandemic, including Ms. Elizabeth Conzevoy, Dr. Sujash Chatterjee, Dr. Jonathan Shaffer, and Mr. Brian Dugan from Qiagen; Mr. Andrew Shaver, Mrs. Sinem Taylor, and Dr. Denise Stephens from Tecan Genomics. The genomic work carried out at the LLU Center for Genomics was funded in part by the United States National Institutes of Health (NIH) grant S10OD019960 (CW), the American Heart Association (AHA) grant 18IPA34170301 (CW), the Ardmore Institute of Health grant 2150141 (CW) and Dr. Charles A. Sims’ gift to the LLU Center for Genomics.
Author contributions
CW conceived and designed the study. TL, ZC, and WC performed experiments including RNA isolation, qRT-PCR, SARS-CoV-2 WGS library construction and sequencing. WC, TL, ZC, WJ, and CW drafted the manuscript. ZC, TL, WC, XC, WJ, MH, and ZY performed bioinformatics data analysis. WJ and TL conducted biostatistics tests and analysis. WC, ZC, TL, XC, and WJ prepared the methods for the manuscript. DT, CG, and DH helped the patient nasopharyngeal swab sample collection. DH helped coordinate the project meetings and IRB application. CW, WJ, JL, DH, DT, and CG helped revise the manuscript. All authors reviewed the manuscript. CW and WJ edited the manuscript. CW finalized and submitted the manuscript.
Declaration of interests
All the authors claim no conflicts of interests. Any mention of commercial products is for clarification and not intended as an endorsement.
Inclusion and diversity
We worked to ensure gender balance in the recruitment of human subjects. We worked to ensure ethnic or other types of diversity in the recruitment of human subjects. We worked to ensure that the study questionnaires were prepared in an inclusive way. One or more of the authors of this paper self-identifies as an under represented ethnic minority in science. The author list of this paper includes contributors from the location where the research was conducted who participated in the data collection, design, analysis, and/or interpretation of the work.
Published: August 20, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2021.102892.
Contributor Information
Wendell Jones, Email: wendell.jones@q2labsolutions.com.
Charles Wang, Email: oxwang@gmail.com, chwang@llu.edu.
Supplemental information
Data and code availability
-
•
The sequencing data have been uploaded to the NCBI SRA (Sequence Read Archive) under the BioProject accession “SRA: PRJNA638938”. The data is available to the public as of the date of publication.
-
•
We used many algorithms and code for the SARS-CoV-2 genome mapping, genome coverage, and SNV calling which have been published previously. All of our code are provided in the GitHub at the following link: https://github.com/oxwang/COVID19_MS1
-
•
Any additional information required for reanalyzing the reported data is available from the lead contact upon request.
References
- Author Anonymous First NGS-based COVID-19 diagnostic. Nat. Biotechnol. 2020;38:777. doi: 10.1038/s41587-020-0608-y. [DOI] [PubMed] [Google Scholar]
- Ai T., Yang Z., Hou H., Zhan C., Chen C., Lv W., Tao Q., Sun Z., Xia L. Correlation of chest CT and RT-PCR testing in coronavirus disease 2019 (COVID-19) in china: a report of 1014 cases. Radiology. 2020;296:E32–E40. doi: 10.1148/radiol.2020200642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen K.G., Rambaut A., Lipkin W.I., Holmes E.C., Garry R.F. The proximal origin of SARS-CoV-2. Nat. Med. 2020;26:450–452. doi: 10.1038/s41591-020-0820-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews S. FastQC: a quality control tool for high throughput sequence data. 2010. http://wwwbioinformaticsbabrahamacuk/projects/fastqc/
- Bates D., Mächler M., Bolker B., Walker S. Fitting linear mixed-effects models using lme4. J. Stat. Softw. 2015;67:1–48. [Google Scholar]
- Bedford T., Greninger A.L., Roychoudhury P., Starita L.M., Famulare M., Huang M.-L., Nalla A., Pepper G., Reinhardt A., Xie H. Cryptic transmission of SARS-CoV-2 in Washington State. medRxiv. 2020 doi: 10.1101/2020.04.02.20051417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler D.J., Mozsary C., Meydan C., Danko D.C., Foox J., Rosiene J., Shaiber A., Afshinnekoo E., MacKay M., Sedlazeck F.J. Host, viral, and environmental transcriptome profiles of the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) bioRxiv. 2020 doi: 10.1101/2020.04.20.048066. [DOI] [Google Scholar]
- Chan J.F., Yuan S., Kok K.H., To K.K., Chu H., Yang J., Xing F., Liu J., Yip C.C., Poon R.W. A familial cluster of pneumonia associated with the 2019 novel coronavirus indicating person-to-person transmission: a study of a family cluster. Lancet. 2020;395:514–523. doi: 10.1016/S0140-6736(20)30154-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dafforn A., Chen P., Deng G., Herrler M., Iglehart D., Koritala S., Lato S., Pillarisetty S., Purohit R., Wang M. Linear mRNA amplification from as little as 5 ng total RNA for global gene expression analysis. Biotechniques. 2004;37:854–857. doi: 10.2144/04375PF01. [DOI] [PubMed] [Google Scholar]
- Ewels P., Magnusson M., Lundin S., Kaller M. MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics. 2016;32:3047–3048. doi: 10.1093/bioinformatics/btw354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fauver J.R., Petrone M.E., Hodcroft E.B., Shioda K., Ehrlich H.Y., Watts A.G., Vogels C.B., Brito A.F., Alpert T., Muyombwe A. Coast-to-coast spread of SARS-CoV-2 in the Unitedstates revealed by genomic epidemiology. medRxiv. 2020 doi: 10.1101/2020.03.25.20043828. [DOI] [Google Scholar]
- Forster P., Forster L., Renfrew C., Forster M. Phylogenetic network analysis of SARS-CoV-2 genomes. Proc.Natl.Acad.Sci. U S A. 2020;117:9241–9243. doi: 10.1073/pnas.2004999117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Alcalde F., Okonechnikov K., Carbonell J., Cruz L.M., Gotz S., Tarazona S., Dopazo J., Meyer T.F., Conesa A. Qualimap: evaluating next-generation sequencing alignment data. Bioinformatics. 2012;28:2678–2679. doi: 10.1093/bioinformatics/bts503. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Reiche A.S., Hernandez M.M., Sullivan M., Ciferri B., Alshammary H., Obla A., Fabre S., Kleiner G., Polanco J., Khan Z. Introductions and early spread of SARS-CoV-2 in the New York city area. medRxiv. 2020 doi: 10.1101/2020.04.08.20056929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grubaugh N.D., Gangavarapu K., Quick J., Matteson N.L., De Jesus J.G., Main B.J., Tan A.L., Paul L.M., Brackney D.E., Grewal S. An amplicon-based sequencing framework for accurately measuring intrahost virus diversity using PrimalSeq and iVar. Genome Biol. 2019;20:8. doi: 10.1186/s13059-018-1618-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan W.J., Ni Z.Y., Hu Y., Liang W.H., Ou C.Q., He J.X., Liu L., Shan H., Lei C.L., Hui D.S.C. Clinical characteristics of coronavirus disease 2019 in China. N.Engl. J. Med. 2020;382:1708–1720. doi: 10.1056/NEJMoa2002032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadfield J., Megill C., Bell S.M., Huddleston J., Potter B., Callender C., Sagulenko P., Bedford T., Neher R.A. Nextstrain: real-time tracking of pathogen evolution. Bioinformatics. 2018;34:4121–4123. doi: 10.1093/bioinformatics/bty407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hourdel V., Kwasiborski A., Baliere C., Matheus S., Batejat C.F., Manuguerra J.C., Vanhomwegen J., Caro V. Rapid genomic characterization of SARS-CoV-2 by Directamplicon-based sequencing through comparison of MinION and Illumina iSeq100(TM) system. Front.Microbiol. 2020;11:571328. doi: 10.3389/fmicb.2020.571328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C., Wang Y., Li X., Ren L., Zhao J., Hu Y., Zhang L., Fan G., Xu J., Gu X. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020;395:497–506. doi: 10.1016/S0140-6736(20)30183-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itokawa K., Sekizuka T., Hashino M., Tanaka R., Kuroda M. A proposal of alternative primers for the ARTIC network’s multiplex PCR to improve coverage of SARS-CoV-2 genome sequencing. bioRxiv. 2020 doi: 10.1101/2020.03.10.985150. [DOI] [Google Scholar]
- Koboldt D.C., Zhang Q., Larson D.E., Shen D., McLellan M.D., Lin L., Miller C.A., Mardis E.R., Ding L., Wilson R.K. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012;22:568–576. doi: 10.1101/gr.129684.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kupferschmidt K. Fast-spreading UK virus variant raises alarms. Science. 2021;371:9–10. doi: 10.1126/science.371.6524.9. [DOI] [PubMed] [Google Scholar]
- Kupferschmidt K., Wadman M. Delta variant triggers new phase in the pandemic. Science. 2021;372:1375–1376. [Google Scholar]
- Kuznetsova A., Brockhoff B.P., Christensen R.H.B. lmerTest package: tests in linear mixed effects models. J. Stat. Softw. 2017;28:1–26. [Google Scholar]
- Li C., Debruyne D.N., Spencer J., Kapoor V., Liu L.Y., Zhou B., Pandey U., Bootwalla M., Ostrow D., Maglinte D.T. Highly sensitive and full-genome interrogation of SARS-CoV-2 using multiplexed PCR enrichment followed by next-generation sequencing. bioRxiv. 2020 doi: 10.1101/2020.03.12.988246. [DOI] [Google Scholar]
- Li H., Durbin R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., Genome Project Data Processing S. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long S.W., Christensen P.A., Bernard D.W., Davis J.R., Shukla M., Nguyen M., Saavedra M.O., Cantu C.C., Yerramilli P., Pruitt L. Molecular architecture of early dissemination and evolution of the SARS-CoV-2 virus in Metropolitan Houston, Texas. bioRxiv. 2020 doi: 10.1101/2020.05.01.072652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J., du Plessis L., Liu Z., Hill V., Kang M., Lin H., Sun J., Francois S., Kraemer M.U., Faria N.R. Genomic epidemiology of SARS-CoV-2 in Guangdong Province, China. Cell. 2020;181:997–1003.e9. doi: 10.1016/j.cell.2020.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu R., Zhao X., Li J., Niu P., Yang B., Wu H., Wang W., Song H., Huang B., Zhu N. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet. 2020;395:565–574. doi: 10.1016/S0140-6736(20)30251-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011;17:3. [Google Scholar]
- Maurano M.T., Ramaswami S., Westby G., Zappile P., Dimartino D., Shen G., Feng X., Ribeiro-dos-Santos A.M., Vulpescu N.A., Black M. sequencing identifies multiple, early introductions of SARS-CoV2 to New York City Region. medRxiv. 2020 doi: 10.1101/2020.04.15.20064931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myrmel M., Oma V., Khatri M., Hansen H.H., Stokstad M., Berg M., Blomström A.-L. Single primer isothermal amplification (SPIA) combined with next generation sequencing provides complete bovine coronavirus genome coverage and higher sequence depth compared to sequence-independent single primer amplification (SISPA) PloSOne. 2017;12:e0187780. doi: 10.1371/journal.pone.0187780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paraskevis D., Kostaki E.G., Magiorkinis G., Panayiotakopoulos G., Sourvinos G., Tsiodras S. Full-genome evolutionary analysis of the novel corona virus (2019-nCoV) rejects the hypothesis of emergence as a result of a recent recombination event. Infect.Genet.Evol. 2020;79:104212. doi: 10.1016/j.meegid.2020.104212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parekh S., Ziegenhain C., Vieth B., Enard W., Hellmann I. The impact of amplification on differential expression analyses by RNA-seq. Sci. Rep. 2016;6:25533. doi: 10.1038/srep25533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park W.B., Kwon N.J., Choi S.J., Kang C.K., Choe P.G., Kim J.Y., Yun J., Lee G.W., Seong M.W., Kim N.J. Virus isolation from the first patient with SARS-CoV-2 in korea. J. Korean Med.Sci. 2020;35:e84. doi: 10.3346/jkms.2020.35.e84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potapov V., Ong J.L. Examining sources of error in PCR by single-molecule sequencing. PLoS One. 2017;12:e0169774. doi: 10.1371/journal.pone.0169774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambaut A., Loman N., Pybus O., Barclay W., Barrett J., Carabelli A., Connor T., Peacock T., Robertson D., Volz E. Preliminary genomic characterisation of an emergent SARS-CoV-2 lineage in the UK defined by a novel set of spike mutations. GenomeEpidemiol. 2020 [Google Scholar]
- Rothe C., Schunk M., Sothmann P., Bretzel G., Froeschl G., Wallrauch C., Zimmer T., Thiel V., Janke C., Guggemos W. Transmission of 2019-nCoV infection from an asymptomatic contact in Germany. N.Engl. J. Med. 2020;382:970–971. doi: 10.1056/NEJMc2001468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sah S., Chen L., Houghton J., Kemppainen J., Marko A.C., Zeigler R., Latham G.J. Functional DNA quantification guides accurate next-generation sequencing mutation detection in formalin-fixed, paraffin-embedded tumor biopsies. Genome Med. 2013;5:77. doi: 10.1186/gm481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- St Hilaire B.G., Durand N.C., Mitra N., Pulido S.G., Mahajan R., Blackburn A., Colaric Z.L., Theisen J.W.M., Weisz D., Dudchenko O. A rapid, low cost, and highly sensitive SARS-CoV-2 diagnostic based on whole genome sequencing. bioRxiv. 2020 doi: 10.1101/2020.04.25.061499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thanh Le T., Andreadakis Z., Kumar A., Gomez Roman R., Tollefsen S., Saville M., Mayhew S. The COVID-19 vaccine development landscape. Nat. Rev. Drug Discov. 2020;19:305–306. doi: 10.1038/d41573-020-00073-5. [DOI] [PubMed] [Google Scholar]
- Waterhouse A.M., Procter J.B., Martin D.M., Clamp M., Barton G.J. Jalview Version 2—a multiple sequence alignment editor and analysis workbench. Bioinformatics. 2009;25:1189–1191. doi: 10.1093/bioinformatics/btp033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood D.E., Lu J., Langmead B. Improved metagenomic analysis with Kraken 2. Genome Biol. 2019;20:257. doi: 10.1186/s13059-019-1891-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu F., Zhao S., Yu B., Chen Y.M., Wang W., Song Z.G., Hu Y., Tao Z.W., Tian J.H., Pei Y.Y. A new coronavirus associated with human respiratory disease in China. Nature. 2020;579:265–269. doi: 10.1038/s41586-020-2008-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao M., Liu X., Ji J., Li M., Li J., Yang L., Sun W., Ren P., Yang G., Zhao J. Multiple approaches for massively parallel sequencing of SARS-CoV-2 genomes directly from clinical samples. Genome Med. 2020;12:57. doi: 10.1186/s13073-020-00751-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie C., Jiang L., Huang G., Pu H., Gong B., Lin H., Ma S., Chen X., Long B., Si G. Comparison of different samples for 2019 novel coronavirus detection by nucleic acid amplification tests. Int. J.Infect. Dis. 2020;93:264–267. doi: 10.1016/j.ijid.2020.02.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H., DiCarlo J., Satya R.V., Peng Q., Wang Y. Comparison of somatic mutation calling methods in amplicon and whole exome sequence data. BMC Genomics. 2014;15:244. doi: 10.1186/1471-2164-15-244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X., Tan Y., Ling Y., Lu G., Liu F., Yi Z., Jia X., Wu M., Shi B., Xu S. Viral and host factors related to the clinical outcome of COVID-19. Nature. 2020;583:437–440. doi: 10.1038/s41586-020-2355-0. [DOI] [PubMed] [Google Scholar]
- Zhang Y.Z., Holmes E.C. A genomic perspective on the origin and emergence of SARS-CoV-2. Cell. 2020;181:223–227. doi: 10.1016/j.cell.2020.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Q., Hu Q., Shepherd L., Wang J., Wei L., Morrison C.D., Conroy J.M., Glenn S.T., Davis W., Kwan M.L. The impact of DNA input amount and DNA source on the performance of whole-exome sequencing in cancer epidemiology. Cancer Epidemiol. Biomarkers Prev. 2015;24:1207–1213. doi: 10.1158/1055-9965.EPI-15-0205. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
The sequencing data have been uploaded to the NCBI SRA (Sequence Read Archive) under the BioProject accession “SRA: PRJNA638938”. The data is available to the public as of the date of publication.
-
•
We used many algorithms and code for the SARS-CoV-2 genome mapping, genome coverage, and SNV calling which have been published previously. All of our code are provided in the GitHub at the following link: https://github.com/oxwang/COVID19_MS1
-
•
Any additional information required for reanalyzing the reported data is available from the lead contact upon request.