

Abstract
Individuals that present with difficult-to-control asthma and sensitivity to one or more fungal species are categorized as a subset of severe asthma patients belonging to a group herein referred to as severe asthma with fungal sensitization (SAFS). We have previously reported the identification of numerous cytokines and chemokines that were elevated in human asthmatics that were sensitized to fungi vs. nonfungal sensitized asthmatics. Here, we show that the unique chemokine CX3CL1 (fractalkine) is elevated in both bronchoalveolar lavage fluid and sputum from human asthmatics sensitized to fungi, implicating an association with CX3CL1 in fungal asthma severity. In an experimental model of fungal-associated allergic airway inflammation, we demonstrate that the absence of CX3CR1 signaling unexpectedly resulted in a profound impairment in lung function. Histological assessment of lung tissue revealed an unrestricted inflammatory response that was subsequently characterized by enhanced levels of neutrophils, eosinophils, and inflammatory monocytes. Neutrophilic inflammation correlated with elevated IL-17A, proinflammatory cytokines (TNF-α, IL-1α, and IL-1β), neutrophil survival factors (granulocyte colony-stimulating factor), and neutrophil-targeting chemokines (CCL3 and CCL4). Eosinophilia correlated with elevated type 2 responses (IL-5 and IL-13) whereas inflammatory monocyte levels correlated with elevated type 1 responses (IFN-γ and CXCL9) and survival factors (macrophage colony-stimulating factor). Despite enhanced inflammatory responses, the immunoregulatory cytokine IL-10 and the natural inhibitor of IL-1 signaling, IL-1RA, were significantly elevated rather than impaired. Regulatory T-cell levels were unchanged, as were levels of the anti-inflammatory cytokines IL-35 and IL-38. Taken together, the CX3CL1/CX3CR1 axis preserves lung function during fungal-associated allergic airway inflammation through a nonclassical immunoregulatory mechanism.
Keywords: asthma, fungal, inflammation
INTRODUCTION
Asthma is an increasing health concern affecting more than 25 million individuals in the United States and more than 300 million individuals worldwide. In 2006, a new asthma phenotype termed “severe asthma with fungal sensitization” (SAFS) was described for individuals whose asthma was poorly controlled and who were sensitized to Alternaria, Aspergillus, Cladosporium, and/or Penicillium (1). Common characteristics in individuals having SAFS include younger onset of disease, higher IgE levels, higher steroid usage, and more frequent exacerbations and hospitalizations (2, 3). Studies have also shown that exposure to Penicillium, Aspergillus, and Cladosporium before the development of asthma symptoms is associated with increased risk of respiratory complications, whereas exposure to Alternaria was associated with an increased risk of exacerbation of current asthma (4). Likewise, a meta-analysis of studies conducted in the U.S. report a strong relationship between fungi in homes and respiratory health effects (5). Finally, data collected in Europe from 8 birth cohorts consisting of >31,000 children found that exposure to fungi during the first 2 yr of life was associated with increased risk of developing asthma (6). Individuals with SAFS often present with difficult-to-control asthma, due in part, to their robust association with neutrophilia (7). Unlike eosinophils, neutrophils and associated responses such as Th17/type 17 responses often do not respond to treatment with corticosteroids (8) and require a different strategy to manage. Thus a growing area of interest is the identification of factors that contribute to immunopathogenesis in allergic asthma, particularly SAFS, and determining whether such factors could be viable therapeutic targets.
We have previously reported an immune mediator profile of bronchoalveolar lavage fluid (BALF) from asthmatics that were sensitized to fungi compared with asthmatics that were atopic but not sensitized to fungi (9). From this analysis, we observed multiple mediators that were significantly higher in fungal (+) asthmatics compared with fungal (−) asthmatics, including various growth factors, chemokines, and cytokines. Among these, we identified IL-7 as a potential immunopathogenic factor in human asthma and experimental fungal-associated allergic airway inflammation (9). In addition, we have also recently identified the IL-1 signaling pathway as a potential contributor to immunopathogenesis in human fungal asthma and showed that treatment with the IL-1 receptor antagonist (IL-1RA) ameliorates asthmatic disease in an experimental animal model (10). An additional mediator we observed to be elevated in fungal-sensitized asthmatics was the unique chemokine (C-X3-C motif) ligand 1 (CX3CL1). CX3CL1 exists as both a soluble and membrane-bound molecule and is the only known ligand for chemokine (C-X3-C motif) receptor 1 (CX3CR1). The soluble form functions as a chemotactic factor for cells expressing CX3CR1 while the membrane-bound form serves to arrest circulating CX3CR1+ leukocytes and allow for diapedesis into sites of inflammation. In the current report, we sought to clarify the role of CX3CL1 in fungal-associated allergic airway inflammation.
MATERIALS AND METHODS
Subjects, Sputum Induction and Processing, and Luminex Analysis
Patients with mild to severe asthma were comprehensively characterized according to the National Heart, Lung, and Blood Institute (NHLBI) Severe Asthma Research Program (SARP) phenotype protocol at Wake Forest School of Medicine as previously described (11), and a description of this cohort was recently reported (12). The sputum induction method was adopted from the NHLBI Asthma Clinical Research Network and used in SARP (13). Bronchoalveolar fluid (BALF) and sputum samples were derived from both the SARP 1 and 2 cohorts. Per SARP protocol, BALF and sputum were not collected within a week of each other. Sputum was processed immediately after collection, and cell cytospins were stained for differential counts of at least 500 nonsquamous cells/subject slide. Total white blood cell count in sputum was determined via trypan blue staining and enumeration using a hemacytometer. Cell-free supernatants were aliquoted and stored at −80°C before use in Luminex analyses (see below). Biospecimens were randomly selected without a priori selection based on asthma severity, rather than based on whether they were skin test positive or negative for Alternaria, Aspergillus, and/or Cladosporium. Sputum supernatants were assayed for different inflammatory cytokine, chemokine, and growth factor protein concentrations using Milliplex Human Cytokine/Chemokine Panels I, II, III, and IV (cat nos. HCYTOMAG-60K-PX41, HCYP2MAG-62K-PX23, HCYP3MAG-63K, and HCY4MG-64K-PX21, respectively; MilliporeSigma). Standards for determination of linear curve plus two control samples representing high and low levels of each cytokine/chemokine were included in each assay. Human subjects were enrolled with informed consent under approved Wake Forest School of Medicine Institutional Review Board (IRB) BG01-425 (Wake Forest University IRB). Human samples were analyzed under University of Alabama (UAB) IRB X130827009 (UAB IRB for Human Use).
Mice
Wild-type (WT) CBL/6 and B6.129P2(Cg)-Cx3cr1tm1Litt/J mice (Cx3cr1-/- mice), 6–8 wk of age, were obtained from The Jackson Laboratory. All animals were housed in a specific pathogen-free, Association for Assessment and Accreditation of Laboratory Animal Care-certified facility and handled according to National Institutes of Health Public Health Service Office of Laboratory Animal Welfare Guide for Care and Use of Laboratory Animals (8th ed.) after review by the UAB and Tulane Institutional Animal Care and Use Committees (IACUC). All animal research was conducted under approved UAB IACUC Protocols 10199 and 20236 and approved Tulane IACUC Protocols 217 and 412.
Fungal-Associated Allergic Airway Inflammation Model
Aspergillus fumigatus isolate 13073 (ATCC) was maintained on potato dextrose agar for 5–7 days at 37°C. Conidia were harvested by washing the culture flask with 50 mL of sterile PBS supplemented with 0.1% Tween 20. The conidia were then passed through a sterile 40-μm nylon membrane to remove hyphal fragments and were enumerated on a hemacytometer. The repeated A. fumigatus exposure model was employed as previously described (10). Briefly, mice were lightly anesthetized with isoflurane and administered 1 × 107 live A. fumigatus conidia in a volume of 50 μL of PBS intratracheally. After resting for 7 days, mice were challenged intratracheally with 1 × 106 live A. fumigatus conidia in 50 μL of PBS daily for 5 consecutive days (days 7, 8, 9, 10, and 11), allowed to rest for 2 consecutive days (days 12 and 13), and then challenged intratracheally with 1 × 106 live A. fumigatus conidia in 50 μL of PBS daily for 3 consecutive days (days 14, 15, and 16). Twenty-four hours after the last A. fumigatus challenge (day 17), mice were euthanized for analysis via ketamine/xylazine (Vet One) overdose and aortic exsanguination.
Whole Lung Cytokine and Chemokine Analysis, Lung Cell Isolation and Culture, and Real-Time PCR
Following euthanization, the right lung was homogenized in PBS supplemented with Complete Mini protease inhibitor tablets (Roche Diagnostics), clarified by centrifugation (12,000 g for 10 min at 4°C), and stored at −80°C. From lung homogenate supernatants, IL-33, IL-1RA, CCL17, CCL22, IL-35, and IL-38 levels were quantified by ELISA (R&D Systems). For lung cell isolation, the lungs were collected and minced in IMDM media (MilliporeSigma) supplemented with 1× penicillin-streptomycin-glutamine (Mediatech), 10% heat-inactivated FBS (Invitrogen), and 0.4 mg/mL polymyxin B (Thermo Fisher Scientific), followed by incubation for 60 min with tissue culture grade type IV collagenase (1 mg/mL; MilliporeSigma) in a 37°C orbital shaker at 100 rpm. The cell suspension was filtered through sterile 70-μm and 40-μm nylon filters, and red blood cells were lysed with ACK buffer (Lonza) to create single-cell preparations. One million cells in a volume of 200 μL were cultured overnight with 1 million A. fumigatus conidia (1:1), followed by collection and clarification of supernatants. Supernatants from lung digest cells were analyzed for protein levels of 32 cytokines and chemokines using the Luminex-based Milliplex multiplex suspension cytokine array (MilliporeSigma), according to the manufacturer’s instructions. The data were analyzed using Bio-Plex Manager software (Bio-Rad). Lung digest cell supernatants were also used to quantify levels of IL-22 by ELISA (R&D Systems). Twenty-four hours after the last challenge, the left lungs were collected, homogenized in Trizol, and RNA isolated, and Muc5ac and Gob5 gene expression was quantified by real-time PCR and normalized to HPRT.
Lung Cell Flow Cytometry
Lung cells were isolated via BAL or by enzymatic digestion of whole lungs as previously described (9, 10). Cells were washed, and Fc receptors were blocked with Mouse BD Fc Block (BD Biosciences) at 4°C for 20 min. Thereafter, cells were stained with a single-color LIVE/DEAD Fixable Dead Cell Stain (Invitrogen), followed by labeling with specific immune cell surface markers. The following staining parameters were employed: eosinophils as CD45+CD11b+Siglec F+ Ly-6G– using anti-CD45 (BioLegend, cat no. 103115, clone 30-F11), anti-CD11b (BioLegend, cat no. 101237, clone M1/70), anti-Siglec F (BioLegend, cat no. 155507, clone S17007L), and anti-Ly-6G (BioLegend, cat no. 127621, clone 1A8); neutrophils as CD45+CD11b+Ly6G+ Siglec F– using anti-CD45, anti CD11b, anti-Ly-6G, and anti-Siglec F; inflammatory monocytes as CD45+CD11b+CD11c-CCR2+F4/80-Ly6C+ using anti-CD45, anti-CD11b, anti-CD11c (BioLegend, cat no. 117308, clone N418), anti-CCR2 (BioLegend, cat no. 150605, clone SA203G11), anti-F4/80 (BioLegend, cat no. 123146, clone BM8), and anti-Ly6C (BioLegend, cat no. 128012, clone HK1.4); T-regulatory cells as CD45+CD3+CD4+CD25+FoxP3+ using anti-CD45, anti-CD3 (BioLegend, cat no. 100216, clone 17A2), anti-CD4 (BioLegend, cat no. 100422, clone GK1.5), anti-CD25 (BioLegend, cat no. 101903, clone 3C7), and anti-FoxP3 (BioLegend, cat no. 126419, clone MF-14); IM1 as CD11c lo MHCII lo, IM2 as CD11c lo MHCII hi and IM3 as CD11c hi MHCII hi [anti-CD11c, BioLegend, cat no. 117308, clone N418; and anti-major histocompatibility complex (MHC) II, BioLegend cat no. 107630, clone M5/114.15.2]. Samples were acquired using a 4-laser, 20-parameter analytic BD LSRFortessa, and data were analyzed using FlowJo software (Tree Star, Inc.). Unstained lung leukocytes served as a control for background fluorescence and gating. Appropriately stained UltraComp eBeads (Thermo Fisher Scientific) served as single-color controls.
Pulmonary Function Assessment
Individual anesthetized A. fumigatus-exposed mice were intubated, and each animal was attached to a computer-controlled volume ventilator (flexiVent; SCIREQ). Regular breathing was set at 150 breaths per minute, with volume and pressure controlled by the flexiVent system based on individual animal weights. Positive end-expiratory pressure was set to 2 cmH2O and measured during each breath stroke. The single-frequency forced oscillation technique was employed to measure total/dynamic lung resistance (Rrs). The low-frequency/broadband forced oscillation technique was employed to measure Newtonian resistance (Rn; also known as airway hyperreactivity) All measurements were collected at baseline and after a linear dose response with methacholine challenge (10–40 mg/mL), as previously described (9, 10). Lung function was also assessed in naive WT and mutant mice, which confirmed no baseline anomalies and no differences between groups (data not shown).
Statistics
Data were analyzed using GraphPad Prism Version 7.0 statistical software (GraphPad Software). Comparisons between groups when data were normally or not normally distributed were made with the two-tailed unpaired Student’s t test, two-way-ANOVA or the Mann-Whitney U test, respectively. Significance was accepted at a value of P < 0.05.
RESULTS
Human Asthmatics Sensitized to Fungi Have Elevated CX3CL1 Levels in BALF and Sputum
Previous studies from our laboratory have reported that human asthmatics who are skin test positive for fungi demonstrate lung function measurements indicative of more severe disease [e.g., decreased forced expiratory volume in 1 s (FEV1) and concentration of methacholine producing 20% drop in FEV1 (PC20)] and increases in measurements of atopic disease (e.g., elevated serum IgE and blood eosinophil numbers) compared with subjects who were skin test negative (9). As part of this report, we undertook an analysis of sputum and BAL fluid from those patients in an effort to identify potential biomarkers of fungal asthma severity. We identified 12 mediators in BAL and have initiated mechanistic pursuits on several, such as IL-7 and various IL-1 family members (9, 10). One of the mediators identified in BAL fluid was the chemotactic protein CX3CL1 (9). Here, we extend this analysis and show that fungal-sensitized asthmatics exhibit increased levels of CX3CL1 in both BALF (Fig. 1A) and induced sputum (Fig. 1B). Thus CX3CL1 may function as a biomarker of disease pathology in fungal-associated allergic asthma.
Figure 1.

Human asthmatics sensitized to fungi have elevated CX3CL1 levels in bronchoalveolar lavage fluid (BALF) and sputum. BALF or sputum was collected from subjects with atopic asthma who were sensitized (BALF, n = 29; sputum, n = 55) or were not sensitized (BALF, n = 30; sputum, n = 83) to fungi. CX3CL1 was quantified in clarified BALF (A) and sputum (B) supernatants by MilliPlex. Data were normalized to the total protein content (BALF) or total white blood cell count (WBC; sputum) of each sample and are expressed as mean pg/mg protein or mean pg/total WBC (each symbol represents a single subject). P values were assessed using a two-tailed Student’s t test: *P < 0.05, **P < 0.01. Graphical data in A were previously reported in table format as mean pg/mg protein ± SE and the range provided in Ref. 9.
Signaling through CX3CR1 Preserves Lung Function during Experimental Fungal-Associated Allergic Airway Inflammation
Given the correlation between increased CX3CL1 levels in fungal-sensitized asthmatics and the increased severity of disease we have reported in these individuals (9, 10), we sought to determine the contribution of CX3CL1 in an experimental fungal-associated allergic airway inflammation animal model. We show that mice deficient in the receptor for CX3CL1 (Cx3cr1-/- mice) exhibited a profound increase in central airway resistance (airway hyperresponsiveness; Rn; Fig. 2A) as well as total lung resistance (Rrs; Fig. 2B) when compared with WT control mice. There was no difference in lung function between naïve WT and Cx3cr1-/- mice (Fig. 2, A and B). Histological analysis of lung sections from Cx3cr1-/- mice revealed an increase in inflammatory cells surrounding both blood vessels and the airways as well as infiltration into the alveolar spaces (Fig. 2C, 4th image; naïve Cx3cr1-/- mice, 2nd image) over that observed in WT mice (Fig. 2C, 3rd image; naïve WT mice, 1st image). Periodic acid-Schiff staining also revealed an increase in mucus within the airways, leading to complete occlusion in some instances, in Cx3cr1-/- mice (Fig. 2D, 4th image; naïve Cx3cr1-/- mice, 2nd image) compared with WT mice (Fig. 2D, 3rd image; naïve WT mice, 1st image). However, analysis of Muc5ac and Gob5 mRNA expression did not reveal an increase in expression in Cx3cr1-/- mice (Fig. 2E). Thus these data indicate that CX3CL1 plays an unexpected protective role in fungal-associated allergic airway inflammation.
Figure 2.
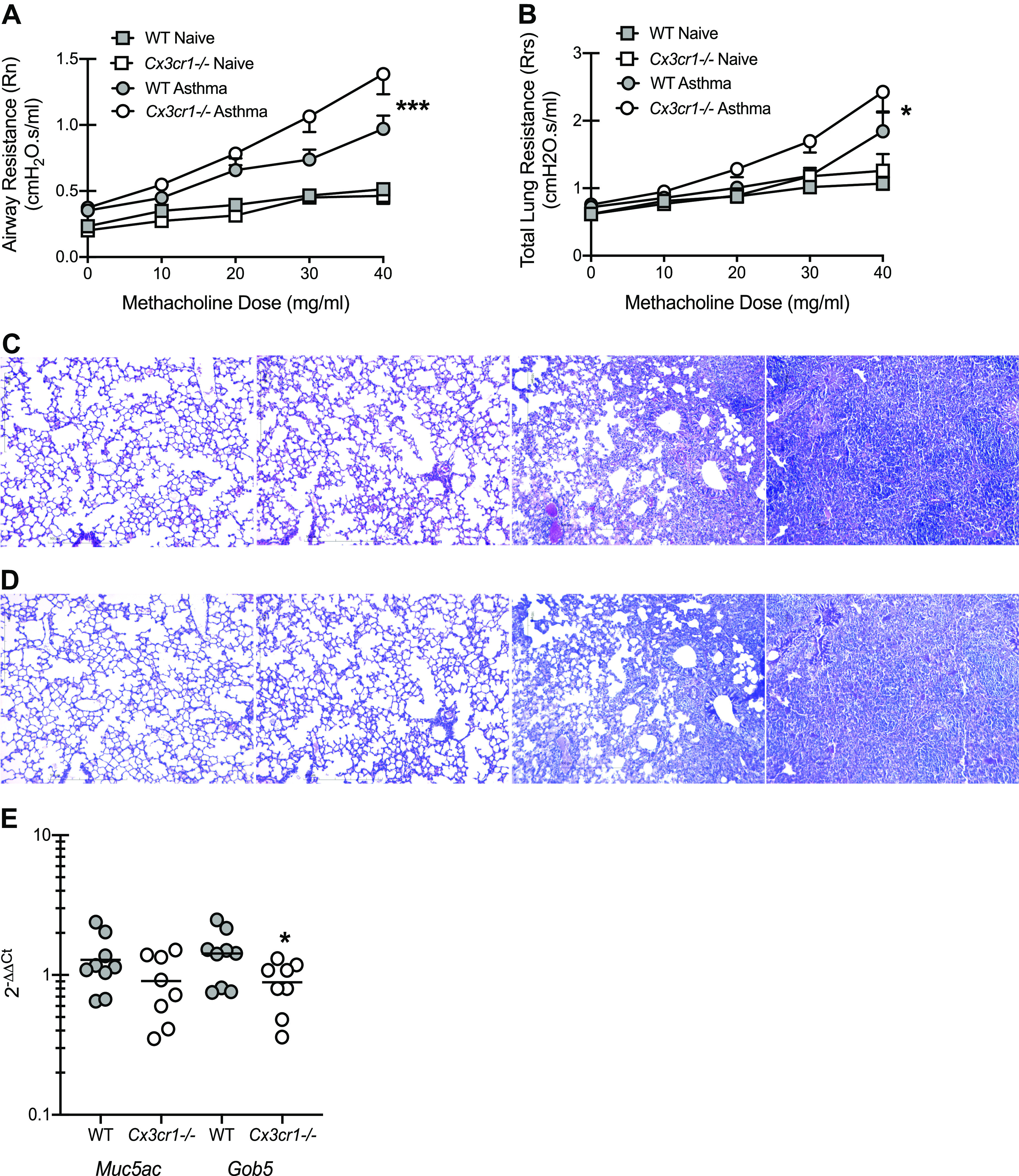
Signaling through CX3CR1 preserves lung function during experimental fungal-associated allergic airway inflammation. C57BL/6 [wild-type (WT)] and CX3CR1-deficient (Cx3cr1-/-) mice were chronically exposed to Aspergillus fumigatus as described in materials and methods. Twenty-four hours after the last organism challenge, airway (A; Newtonian) resistance and total lung resistance (B) was analyzed via mechanical ventilation using the flexiVent pulmonary function system. A and B illustrate cumulative data from 2 independent studies (n = 4–5 mice per group per study). Data expressed as means ± SE. *P < 0.05 and ***P < 0.001, respectively (two-way ANOVA). Representative hematoxylin and eosin (C)- and periodic acid-Schiff (D)-stained lung sections from both naïve (1st and 2nd images) and asthmatic (3rd and 4th images) WT mice (1st and 3rd images) and Cx3cr1-/- mice (2nd and 4th images). Original magnification: ×10. Bar = 100 μm. Twenty-four hours after the last challenge, the left lungs were collected and homogenized in Trizol, RNA was isolated, and Muc5ac and Gob5 gene expression was quantified by real-time PCR and normalized to HPRT (E). E illustrates cumulative data from 2 independent studies (n = 4–5 mice per group per study). Data expressed as means ± SE. *P < 0.05 (two-tailed Student’s t test).
The Absence of CX3CR1 Signaling Results in Increased Neutrophil Infiltration and Associated Responses during Fungal-Associated Allergic Airway Inflammation
The intriguing finding that signaling through CX3CR1 appeared to be beneficial for limiting fungal asthma severity prompted further interrogation of the immunologic factors underlying the increases in airway and lung resistance, cell infiltration, and mucus production. We have previously reported that reduction in neutrophils, a central effector against A. fumigatus and often associated with steroid resistant asthma, is associated with improvement in the severity of fungal-associated allergic asthma (10, 14). Results show that Cx3cr1-/- mice demonstrated a fourfold increase in neutrophils in the airway compared with WT mice (Fig. 3A). The enhanced levels of neutrophils correlated with numerous factors associated with neutrophil recruitment and proliferation, including IL-17A (Fig. 3B), granulocyte colony-stimulating factor (G-CSF) (Fig. 3C), the chemokines CCL3 and CCL4 (Fig. 3D), and the proinflammatory cytokine TNF-α (Fig. 3E). We have previously shown that signaling through the IL-1 receptor via IL-1α and IL-1β is required for optimal neutrophil recruitment to the lung during fungal asthma and that dysregulation of this signaling leads to an increase in neutrophilic infiltration (10). In turn, IL-1α and IL-1β were also elevated in Cx3cr1-/- mice (Fig. 3E). Levels of IL-7, which we have previously reported to drive IL-17A production during fungal asthma (9), were not different between WT and Cx3cr1-/- mice (1.2 ± 0.22 and 1.32 ± 0.19 pg/ml for WT and Cx3cr1-/- mice, respectively; P = 0.68). Thus signaling via CX3CR1 is critical for regulating the amount of neutrophils in the lungs via the regulation of proinflammatory cytokine and neutrophil-targeting chemokine levels.
Figure 3.

The absence of CX3CR1 signaling results in increased neutrophil infiltration and associated responses during fungal-associated allergic airway inflammation. A: C57BL/6 [wild-type (WT)] and CX3CR1-deficient (Cx3cr1-/-) mice were chronically exposed to Aspergillus fumigatus as described in materials and methods. Twenty-four hours after the last organism challenge, lung cells were isolated by bronchoalveolar lavage, enumerated, Fc-blocked, stained with a live/dead staining kit, and stained for neutrophils (CD45+, CD11b+, Ly-6G+, Siglec F−). A illustrates cumulative data from 2–3 independent studies (n = 3–4 mice per group per study). B–E: 24 h after last challenge, right lungs were collected and enzymatically digested and unfractionated lung cells were cocultured with A. fumigatus conidia for 24 h at a cell to organism ratio of 1:1. IL-17A (B), granulocyte colony-stimulating factor (G-CSF; C); CXCL1, CCL3, and CCL4 (D); and TNF-α, IL-1α, and IL-1β (E) levels were quantified in lung digest cell culture supernatants by MilliPlex. B–E illustrate cumulative data from 3 independent studies (n = 3–5 mice per group per study). *P < 0.05, **P < 0.01, and ***P < 0.001, respectively (two-tailed Student’s t test).
CX3CR1 Signaling Regulates the Magnitude of Type 2 Responses during Fungal-Associated Allergic Airway Inflammation
Allergic asthma is generally considered to be a type 2-dominant, eosinophilic disease process. In turn, we have shown that IL-1 signaling also drives eosinophil recruitment in our model (10); therefore, the elevated levels of IL-1α and IL-1β we observed in Cx3cr1-/- mice indicate the possibility of eosinophil involvement in disease exacerbation. Similar to neutrophils, we observed a threefold increase in eosinophils in the absence of CX3CL1 signaling (Fig. 4A). This correlated with a small, but significant, increase in the eosinophil survival factor (and type 2 cytokine) IL-5 (Fig. 4B). The type 2 cytokine IL-13, but not IL-4, was also elevated in asthmatic Cx3cr1-/- mice (Fig. 4C). However, there were no differences in the pro-eosinophil chemokine CCL11 (Fig. 4D). In contrast, CCL5, which may also function in eosinophil recruitment, was increased in Cx3cr1-/- mice (Fig. 4E). Interestingly, the pro-type-2 cytokine IL-33 and the pro-allergic chemokines CCL17 and CCL22, which we have previously reported as biomarkers of fungal asthma severity (9, 14), and are recognized biomarkers in human allergic bronchopulmonary aspergillosis (16, 17), were not different between WT and Cx3cr1-/- mice (Fig. 4F). Thus signaling via CX3CR1 may function in suppressing some aspects of type 2-mediated pathogenesis in fungal-associated allergic asthma.
Figure 4.

CX3CR1 signaling regulates the magnitude of type 2 responses during fungal-associated allergic airway inflammation. A: C57BL/6 [wild-type (WT)] and CX3CR1-deficient (Cx3cr1-/-) mice were chronically exposed to Aspergillus fumigatus as described in materials and methods. Twenty-four hours after the last organism challenge, lung cells were isolated by bronchoalveolar lavage, enumerated, Fc-blocked, stained with a live/dead staining kit, and stained for eosinophils (CD45+, CD11b+, Siglec F+, Ly-6G−). A illustrates cumulative data from 3–4 independent studies (n = 3–4 mice per group per study). B–E: 24 h after last challenge, right lungs were collected and enzymatically digested and unfractionated lung cells were cocultured with A. fumigatus conidia for 24 h at a cell to organism ratio of 1:1. IL-5 (B), IL-4 and IL-13 (C), CCL11 (D), and CCL5 (E) levels were quantified in lung digest cell culture supernatants by Milliplex. F: 24 h after last challenge, the left lungs were collected and homogenized and IL-33, CCL17, and CCL22 levels were quantified in clarified lung homogenates. B–E illustrate cumulative data from 2–3 independent studies (n = 3–5 mice per group per study). **P < 0.01 and ***P < 0.001 (two-tailed Student’s t test).
The Absence of CX3CR1 Signaling Results in Increased Type 1 Responses during Fungal-Associated Allergic Airway Inflammation
Recent studies have implicated a role for IFN-γ associated type 1 responses in severe asthma pathogenesis and corticosteroid insensitivity (18, 19). In turn, we have also shown that IL-1 signaling drives severity of experimental fungal asthma, which correlated with increased type 1 responses (10). Results show that type 1 responses, specifically the production of IFN-γ and the IFN-γ associated chemokine CXCL9, were elevated in Cx3cr1-/- mice (Fig. 5A). IFN-γ is one of two cytokines approved to treat invasive fungal infections and one mechanism of action is thought to be the activation of monocytes (20). To this end, we observed a threefold increase in inflammatory monocytes in the lungs of asthmatic Cx3cr1-/- mice (Fig. 5B). This increase was not associated with increases in the classic inflammatory monocyte chemokine CCL2 (Fig. 5C) (21). However, macrophage colony-stimulating factor (M-CSF), which functions as a survival factor for monocytes (22), was increased in the lungs of asthmatic Cx3cr1-/- mice (Fig. 5D). Thus signaling via CX3CR1 may function in suppressing some aspects of type 1-mediated pathogenesis in fungal-associated allergic asthma.
Figure 5.

The absence of CX3CR1 signaling results in increased type 1 responses during fungal-associated allergic airway inflammation. A: C57BL/6 [wild-type (WT)] and CX3CR1-deficient (Cx3cr1-/-) mice were chronically exposed to Aspergillus fumigatus as described in materials and methods. Twenty-four hours after last challenge, right lungs were collected and enzymatically digested and unfractionated lung cells were cocultured with A. fumigatus conidia for 24 h at a cell-to-organism ratio of 1:1. IFN-γ, CXCL9, and CXCL10 levels were quantified in lung digest cell culture supernatants by MilliPlex. A illustrates cumulative data from 3–4 independent studies (n = 3–5 mice per group per study). *P < 0.05 (two-tailed Student’s t test). B: 24 h after the last organism challenge, lung cells were isolated by bronchoalveolar lavage, enumerated, Fc-blocked, stained with a live/dead staining kit, and stained for inflammatory monocytes (CD45+, CD11b+, Ly-6C+, CCR2+). B illustrates cumulative data from 2 independent studies (n = 4–5 mice per group per study). C and D: CCL2 (C) and macrophage colony-stimulating factor (M-CSF; D) levels were quantified in lung digest cell culture supernatants by MilliPlex or ELISA. C and D illustrate cumulative data from 3–4 independent studies (n = 3–5 mice per group per study). **P < 0.01 (two-tailed Student’s t test).
Anti-Inflammatory Mechanisms Are Not Impaired in the Absence of CX3CR1 Signaling during Fungal-Associated Allergic Airway Inflammation
The observation that Cx3cr1-/- mice demonstrated worse lung function in the presence of profound increases in inflammatory cell recruitment and inflammatory mediator production indicated the possibility that induction of one or more immunoregulatory pathways required CX3CR1 signaling. Results in Fig. 6A show, however, that T-regulatory cells were not affected by the absence of CX3CR1 during experimental fungal asthma. In contrast, the immunoregulatory cytokine IL-10 was significantly elevated, rather than attenuated, in Cx3cr1-/- mice (Fig. 6B). We have recently reported that mice deficient in the natural inhibitor of IL-1 receptor signaling, IL-1 receptor antagonist (IL-1RA), had type 1 and type 17 responses that were associated with neutrophilic inflammation and increased airway hyperreactivity (10). However, likely owing to increased IL-1α and IL-1β levels, IL-1RA levels were similarly elevated in Cx3cr1-/- mice (Fig. 6C). The IL-12 family cytokine IL-35, another immunoregulatory cytokine often produced by T-regulatory cells (23), was not different between WT and Cx3cr1-/- mice (Fig. 6D). Finally, the IL-1 family cytokine IL-38, which functions in an antagonistic manner after receptor binding (24), was also not different between WT and Cx3cr1-/- mice (Fig. 6E). Thus major immunoregulatory pathways are not negatively affected by the absence of CX3CR1 signaling.
Figure 6.

Anti-inflammatory mechanisms are not impaired in the absence of CX3CR1 signaling during fungal-associated allergic airway inflammation. A: C57BL/6 [wild-type (WT)] and CX3CR1-deficient (Cx3cr1-/-) mice were chronically exposed to Aspergillus fumigatus as described in materials and methods. Twenty-four hours after the last organism challenge, lung cells were isolated by bronchoalveolar lavage, enumerated, Fc-blocked, stained with a live/dead staining kit, and stained for T-regulatory cells (CD4+, FoxP3+). A illustrates cumulative data from 2–3 independent studies (n = 2–3 mice per group per study). Twenty-four hours after last challenge, right lungs were collected and enzymatically digested and unfractionated lung cells were cocultured with A. fumigatus conidia for 24 h at a cell to organism ratio of 1:1. B and C: IL-10 (B) and IL-1RA (C) levels were quantified in lung digest cell culture supernatants by MilliPlex or ELISA. B and C illustrate cumulative data from 3–4 independent studies (n = 3–5 mice per group per study). **P < 0.01 and ***P < 0.001 (two-tailed Student’s t test). D and E: 24 h after last challenge, the left lungs were collected and homogenized and IL-35 (D) and IL-38 (E) levels were quantified in clarified lung homogenates by ELISA. D and E illustrate cumulative data from 3–4 independent studies (n = 3–5 mice per group per study). **P < 0.01 (two-tailed Student’s t test).
Modulation of Interstitial Macrophage Populations in the Absence of CX3CR1 Signaling during Fungal-Associated Allergic Airway Inflammation
Recent studies have identified additional resident tissue macrophage subsets in the lung (25–27). These populations, termed interstitial macrophages (IMs), have different transcription profiles and were shown to be located either in the interstitium of the bronchovascular bundles or along nerve fibers (25–27). In one report, three different IM populations were differentiated by expression of CD11c, CD206, and MHC II (26). A second report identified two IM populations separated by CX3CR1 and MHC II expression (25). Finally, a third report identified two IM populations based on CD206 expression (27). As these three reports appear to have overlapping populations, we characterized the three populations differentiated by CD11c, CD206 and MHC II (26). Results showed that the IM1 population (CD11c lo MHCII lo) was significantly increased in Cx3cr1-/- mice (Fig. 7A). In contrast, the IM2 population (CD11c lo MHCII hi) was significantly lower in Cx3cr1-/- mice (Fig. 7B). The IM3 population (CD11c hi MHCII hi) trended higher in Cx3cr1-/- mice (P = 0.12, Fig. 7C). Alveolar macrophages also trended higher in Cx3cr1-/- mice, but were not significantly different than WT mice (5.77 × 104 ± 2.71 × 104 vs. 1.77 × 105 ± 6.9 × 104 in WT vs. Cx3cr1-/- mice; P = 0.09). As the IM2 population has been shown to have immunoregulatory characteristics (25), the possibility exists that lower levels of this IM subset may result in the hyperinflammatory state observed in Cx3cr1-/- mice during fungal-associated allergic airway inflammation.
Figure 7.

Modulation of interstitial macrophage populations in the absence of CX3CR1 signaling during fungal-associated allergic airway inflammation. C57BL/6 [wild-type (WT)] and CX3CR1-deficient (Cx3cr1-/-) mice were chronically exposed to Aspergillus fumigatus as described in materials and methods. Twenty-four hours after the last organism challenge, lung cells were isolated by bronchoalveolar lavage, enumerated, Fc-blocked, stained with a live/dead staining kit, and stained for interstitial macrophage population 1 (IM1; CD11c lo MHCII lo; A), interstitial macrophage population 2 (IM2; CD11c lo MHCII hi; B), and interstitial macrophage population 3 (IM3; CD11c hi MHCII hi; C). A–C illustrate cumulative data from 3 independent studies (n = 4–5 mice per group per study). *P < 0.05 and **P < 0.01, respectively (two-tailed Student’s t test).
DISCUSSION
We have previously described positive correlations between several immunologic factors and the severity of disease in human asthmatic sensitive to fungi as well as their contributions to disease pathology in an experimental animal model of fungal-associated allergic airway inflammation (9, 10). In the current report, analysis of sputum and bronchoalveolar lavage fluid from fungal-sensitized asthmatics identified the chemokine (C-X3-C motif) ligand 1 as an additional potential immunological factor associated with allergic (fungal) asthma pathogenesis. CX3CL1 has not been widely studied in in vivo models of allergic asthma. In fact, a lone report using Leishmania homolog of activated C kinase (LACK) as an allergen has shown that Cx3cr1-/- mice or mice treated with anti-CX3CR1 antibody exhibited decreased airway resistance and suppression of T-helper type 2 responses (28). Similar findings were also observed in when more common allergens such as ragweed and house dust mite were employed (28). In vitro studies have shown that CX3CL1 production by airway smooth muscle cells can be induced by IFN-γ and TNF-α (29) leading to mast cell recruitment (30). Another study has shown that airway smooth muscle cell production of CX3CL1 is dependent on expression/function of the intermediate-conductance Ca2+-activated K+ (KCa) channel KCa3.1, which was insensitive to glucocorticoid treatment (31). Altogether, although the data are somewhat limited, CX3CL1 signaling appears to function in an immunopathogenic manner in asthma.
Much to our surprise, data presented here show a complete opposite role for CX3CL1 signaling in fungal asthma. The gold standard for experimental asthma models is the ability to measure changes in pulmonary function, specifically airway hyperresponsiveness (AHR). Our initial assessment of pulmonary function of Cx3cr1-/- mice following our model of A. fumigatus-induced allergic airway inflammation revealed increased airway hyperresponsiveness in the absence of CX3CR1, a finding in direct contradiction to the report on LACK antigen and other reports suggesting loss of CX3CR1 ameliorates disease pathology (28). A difference, however, between our study and others is that we measure total lung resistance (Rrs) and Newtonian resistance (Rn) via direct cannulation of the trachea and using a computer-controlled ventilator while other studies employed an indirect method of assessing AHR by measuring enhanced pause (Penh) via whole body plethysmography, a controversial method for assessing AHR (32, 33). To better visualize why CX3CR1 deficiency resulted in worse pulmonary function during fungal asthma, we analyzed lung sections for evidence of inflammation and mucus production. Histological analysis of Cx3cr1-/- mice confirmed pathological changes consistent with increased AHR including a dramatic inflammatory cell infiltration around the airways and in the alveolar spaces.
We next sought to better characterize the exacerbated immune response observed in Cx3cr1-/- mice during fungal asthma. Neutrophils are thought to be a significant contributor to fungal asthma severity (7, 34). Our previous work has shown that recognition of fungal beta-glucans via Dectin-1 contributed to neutrophil inflammation and worse severity of fungal-associated allergic airway inflammation/fungal asthma (14). We have subsequently reported that levels of the pro-neutrophil inflammatory cytokines IL-17A and IL-22 correlate with fungal asthma severity (35). More recently, we have shown that IL-1R1 signaling drives neutrophilic responses during fungal asthma (10). This has led us to collectively hypothesize that a Dectin-1 → IL-1α/β → IL-1R1 → IL-17A → G-CSF axis supports neutrophilic inflammation during severe fungal-associated allergic airway inflammation (10). Unfortunately, while it would be of interest to directly interrogate the role of neutrophils in airway hyperreactivity during our live organism fungal asthma model, this is not feasible, as the experimental manipulation of neutrophil levels (e.g., depletion) results in the development of invasive fungal infection (36). Nevertheless, data presented here demonstrate that in the absence of CX3CL1 signaling, we observed augmented neutrophil numbers in parallel with increased levels of IL-1α/β, IL-17A, and G-CSF. This suggests that CX3CL1 is critical for regulating the detrimental neutrophilic axis during fungal asthma. A study examining lung infection with Francisella tularensis has demonstrated increased neutrophils levels in the lungs of Cx3cr1-/- mice (37) suggesting that a regulatory effect of CX3CL1 signaling on neutrophil levels in the lungs is not limited to fungi. However, respiratory syncytial virus (RSV) infection in Cx3cr1-/- mice is characterized by impaired neutrophil recruitment (38); albeit this observation may be age dependent, as newborn Cx3cr1-/- mice demonstrate exacerbated neutrophil recruitment upon RSV challenge (39), further supporting a regulatory role for CX3CL1.
Our data suggest that neutrophils are not the only cell type in the allergic lung impacted by CX3CL1 signaling. Eosinophils are a hallmark of allergic asthma (40) and are a well-recognized biomarker of allergic bronchopulmonary aspergillus (ABPA) (41). Indeed, eosinophil deficiency has been shown to result in a marked improvement in fungal asthma severity (42). We have previously reported that in our model of fungal-associated allergic asthma, eosinophil levels may (9, 10) or may not (14, 35) correlate with severity. This is likely a result of the mixed inflammatory response generated in our model of fungal-associated allergic airway inflammation/fungal asthma (i.e., type 1, 2 and 17 responses, increased IgE levels, and increased neutrophils and eosinophils) (14). However, we have reported that eosinophils recognize, respond to, and kill A. fumigatus and are required for lung clearance (43); thus these cells are undoubtedly a factor in our model of fungal asthma. Similar to neutrophils, eosinophil levels were higher in the absence of CX3CL1 signaling. We found that the primary chemokine supporting eosinophil recruitment, CCL11, was not increased in Cx3cr1-/- mice. Likewise, other chemokines that often associate with allergic asthma and ABPA specifically, such as CCL17 and CCL22 (16, 17), were not different between WT and Cx3cr1-/- mice. However, other chemokines, such as CCL5/RANTES, may play a role in eosinophil recruitment (44) and CCL5 was elevated in Cx3cr1-/- mice. Instead, type 2 responses, and specifically the eosinophil survival factor IL-5, were elevated. However, the increase in IL-5, while significant, was moderate and may not fully explain the threefold increase in eosinophil numbers in Cx3cr1-/- mice. Nevertheless, IL-5 has become an attractive and efficacious target against eosinophil-driven allergic asthma (45). Inflammatory monocytes were an additional cell population that were increased in the allergic lung in the absence of CX3CL1 signaling, which correlated with increased IFN-γ and CXCL9 levels. Inflammatory monocytes are critical for CD4 T cell responses (21) and may also serve as a source of CXCL9 (46) during A. fumigatus exposure; thus, the increase in these cells may explain why these type 1 mediators are elevated. Monocyte development also requires M-CSF (47, 48), and the increase in this growth factor in Cx3cr1-/- mice could explain the increase in these cells during fungal asthma.
The observation that the absence of CX3CL1 signaling results in the enhancement of multiple immune responses (type 1, type 2, type 17, and proinflammatory) suggested that an immunoregulatory pathway was missing or impaired. We first examined CD4 T regulatory cell numbers in the airway as well as IL-10 production by lung digest cells. Despite observing no differences in T-regulatory cell numbers, IL-10 was surprisingly higher in Cx3cr1-/- mice. We speculate this is a result of the exacerbated inflammatory responses in the absence of CX3CL1 signaling. Alternatively, IL-10 may be produced by myeloid-derived suppressor cells (49), which resemble neutrophils in the lung, a cell population that was elevated in the lungs of asthmatic Cx3cr1-/- mice. We next examined whether there was a defect in IL-1RA production, as we have previously reported that the absence of IL-1RA results in worse fungal asthma severity due to enhanced type 1 and type 17 responses (10). However, IL-1RA was increased rather than decreased, and we suspect this is due to elevated levels of IL-1α and IL-1β levels, which are known to also induce IL-1RA as a mechanism of self-regulation (50). IL-35 is an additional regulatory cytokine primarily associated with Tregs that can regulate inflammation during allergic asthma (51). A recent study has shown that IL-35 levels were significantly lower in human subjects with eosinophilic asthma compared with neutrophil or paucigranulocytic asthma (52). However, we have previously reported that IL-35 mRNA levels were not modulated by the absence of Dectin-1 during fungal asthma (14) and, likewise, IL-35 protein levels were not different in the absence of CX3CR1 signaling in the current study. IL-38 is an IL-1 family member that has recently been shown to modulate eosinophil recruitment as well as Th17 responses during asthma (53). Although eosinophils numbers and IL-17A levels were both enhanced in the absence of CX3CL1 signaling, this was not due to lower IL-38 levels. Collectively, although signaling via the CX3CL1/CX3CR1 axis is beneficial for regulating multiple immune responses that contribute to airway hyperresponsiveness, the immunoregulatory pathway(s) induced by this axis appears unique and is a focus of ongoing studies.
Recent studies have begun to characterize and elucidate the roles of nonalveolar resident tissue macrophages in the lung. Gibbings et al. (26) identified three interstitial macrophage (IM) populations via expression of CD11c, CD206, and MHC II. By RNA-seq, all three of these populations expressed CX3CR1, yet have distinct gene signatures. Chakarov et al. (25) identified two IM populations via differential expression of CX3CR1 and MHC II. The CX3CR1 hi population was also MHC II hi but was CD206 lo/neg, which is suggestive of being the same as the Gibbings IM2 population. The Chakarov CX3CR1 hi MHC II hi CD206 low/neg population was thought to be more immunoregulatory based on IL-10 production (25). Finally, Schyns et al. (27) identified two IM populations separated by CD206 expression, of which the CD206 neg population was stated to overlap with the CX3CR1 hi MHC II hi population in Chakarov and the IM2 population in Gibbings. The Schyns CD206 high population is thought to overlap with Gibbings IM1. Although the putative IM2 immunoregulatory population was significantly lower in Cx3cr1-/- mice, IL-10 levels were actually higher in Cx3cr1-/- mice. This suggests that the IM2 population is not immunerogulatory via IL-10 production, nor are the other mechanisms we documented earlier, during fungal asthma. The Schyns CD206 high population (similar to the Gibbings IM1 population) was shown to produce CXCL1 and CXCL10 (27). We further found that the Gibbings IM1 population was significantly higher in Cx3cr1-/- mice, and although we demonstrated that CXCL1 and CXCL10 trended higher in Cx3cr1-/- mice, they were not significant. Ongoing studies will continue to investigate these relatively rare and unique cells during fungal asthma.
In summary, we have identified CX3CL1 as a putative biomarker of disease severity in human asthmatics that were sensitized to fungi. Interrogating the role of CX3CL1 signaling in experimental fungal-associated allergic airway inflammation revealed this pathway to regulate multiple immune responses. In the absence of CX3CL1 signaling, exacerbated type 1, type 2, and type 17 immune responses correlated with poorer lung function. Although appearing to function in an immunoregulatory capacity, CX3CL1 signaling did not promote various pathways known to modulate type 1, type 2, and/or type 17 immune responses. Understanding how the CX3CL1/CX3CR1 axis controls lung inflammation and lung function during fungal-associated allergic airway inflammation could reveal a novel therapeutic approach to limit disease severity in those asthmatics sensitize to fungal organisms.
GRANTS
This work was supported by National Heart, Lung, and Blood Institute Grants HL122426 and HL136211 (both to C.S.).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
M.S.G. and C.S. conceived and designed research; M.S.G., M.J., J.P.B., and Z.Y. performed experiments; M.S.G., S.M., A.T.H., D.A.M., and C.S. analyzed data; M.S.G., S.M., A.T.H., D.A.M., and C.S. interpreted results of experiments; M.S.G. and C.S. prepared figures; M.S.G. and C.S. drafted manuscript; M.S.G., S.M., A.T.H., D.A.M., and C.S. edited and revised manuscript; M.S.G., S.M., A.T.H., D.A.M., and C.S. approved final version of manuscript.
REFERENCES
- 1.Denning DW, O'Driscoll BR, Hogaboam CM, Bowyer P, Rm N. The link between fungi and severe asthma: a summary of the evidence. Eur Respir J 27: 615–626, 2006. doi: 10.1183/09031936.06.00074705. [DOI] [PubMed] [Google Scholar]
- 2.Goh KJ, Yii AC, Lapperre TS, Chan AK, Chew FT, Chotirmall SH, Koh MS. Sensitization to Aspergillus species is associated with frequent exacerbations in severe asthma. J Asthma Allergy 10: 131–140, 2017. doi: 10.2147/JAA.S130459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Medrek SK, Kao CC, Yang DH, Hanania NA, Parulekar AD. Fungal sensitization is associated with increased risk of life-threatening asthma. J Allergy Clin Immunol Pract 5: 1025–1031,2017. doi: 10.1016/j.jaip.2016.11.015. [DOI] [PubMed] [Google Scholar]
- 4.Sharpe RA, Bearman N, Thornton CR, Husk K, Osborne NJ. Indoor fungal diversity and asthma: a meta-analysis and systematic review of risk factors. J Allergy Clin Immunol 135: 110–122, 2015. doi: 10.1016/j.jaci.2014.07.002. [DOI] [PubMed] [Google Scholar]
- 5.Fisk WJ, Lei-Gomez Q, Mendell MJ. Meta-analyses of the associations of respiratory health effects with dampness and mold in homes. Indoor Air 17: 284–296, 2007. doi: 10.1111/j.1600-0668.2007.00475.x. [DOI] [PubMed] [Google Scholar]
- 6.Tischer CG, Hohmann C, Thiering E, Herbarth O, Muller A, Henderson J, Granell R, Fantini MP, Luciano L, Bergstrom A, Kull I, Link E, von BA, Kuehni CE, Strippoli MP, Gehring U, Wijga A, Eller E, Bindslev-Jensen C, Keil T, Heinrich J, as part of the ENRIECO consortium. Meta-analysis of mould and dampness exposure on asthma and allergy in eight European birth cohorts: an ENRIECO initiative. Allergy 66: 1570–1579, 2011. doi: 10.1111/j.1398-9995.2011.02712.x. [DOI] [PubMed] [Google Scholar]
- 7.Bush A. Kids, difficult asthma and fungus. J Fungi (Basel) 6: 55, 2020. doi: 10.3390/jof6020055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McKinley L, Alcorn JF, Peterson A, Dupont RB, Kapadia S, Logar A, Henry A, Irvin CG, Piganelli JD, Ray A, Kolls JK. TH17 cells mediate steroid-resistant airway inflammation and airway hyperresponsiveness in mice. J Immunol 181: 4089–4097, 2008. doi: 10.4049/jimmunol.181.6.4089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Reeder KM, Dunaway CW, Blackburn JP, Yu Z, Matalon S, Hastie AT, Ampleford EJ, Meyers DA, Steele C. The common gamma-chain cytokine IL-7 promotes immunopathogenesis during fungal asthma. Mucosal Immunol 11: 1352–1362, 2018. doi: 10.1038/s41385-018-0028-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Godwin MS, Reeder KM, Garth JM, Blackburn JP, Jones M, Yu Z, Matalon S, Hastie AT, Meyers DA, Steele C. IL-1RA regulates immunopathogenesis during fungal-associated allergic airway inflammation. JCI Insight 4: e129055, 2019. doi: 10.1172/jci.insight.129055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Moore WC, Meyers DA, Wenzel SE, Teague WG, Li H, Li X, D'Agostino R Jr, Castro M, Curran-Everett D, Fitzpatrick AM, Gaston B, Jarjour NN, Sorkness R, Calhoun WJ, Chung KF, Comhair SA, Dweik RA, Israel E, Peters SP, Busse WW, Erzurum SC, Bleecker ER, National Heart, Lung, and Blood Institute's Severe Asthma Research Program. Identification of asthma phenotypes using cluster analysis in the Severe Asthma Research Program. Am J Respir Crit Care Med 181: 315–323, 2010. doi: 10.1164/rccm.200906-0896OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hastie AT, Steele C, Dunaway CW, Moore WC, Rector BM, Ampleford E, Li H, Denlinger LC, Jarjour N, Meyers DA, Bleecker ER, NHLBI Severe Asthma Research Program (SARP). Complex association patterns for inflammatory mediators in induced sputum from subjects with asthma. Clin Exp Allergy 48: 787–797, 2018. doi: 10.1111/cea.13129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Moore WC, Hastie AT, Li X, Li H, Busse WW, Jarjour NN, Wenzel SE, Peters SP, Meyers DA, Bleecker, ER, National Heart, Lung, and Blood Institute's Severe Asthma Research Program. Sputum neutrophil counts are associated with more severe asthma phenotypes using cluster analysis. J Allergy Clin Immunol 133: 1557–1563,2014. doi: 10.1016/j.jaci.2013.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lilly LM, Gessner MA, Dunaway CW, Metz AE, Schwiebert L, Weaver CT, Brown GD, Steele C. The beta-glucan receptor dectin-1 promotes lung immunopathology during fungal allergy via IL-22. J Immunol 189: 3653–3660, 2012. doi: 10.4049/jimmunol.1201797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hartl D, Latzin P, Zissel G, Krane M, Krauss-Etschmann S, Griese M. Chemokines indicate allergic bronchopulmonary aspergillosis in patients with cystic fibrosis. Am J Respir Crit Care Med 173: 1370–1376, 2006. doi: 10.1164/rccm.200508-1271OC. [DOI] [PubMed] [Google Scholar]
- 17.Latzin P, Hartl D, Regamey N, Frey U, Schoeni MH, Casaulta C. Comparison of serum markers for allergic bronchopulmonary aspergillosis in cystic fibrosis. Eur Respir J 31: 36–42, 2008. doi: 10.1183/09031936.00078107. [DOI] [PubMed] [Google Scholar]
- 18.Gauthier M, Chakraborty K, Oriss TB, Raundhal M, Das S, Chen J, Huff R, Sinha A, Fajt M, Ray P, Wenzel SE, Ray A. Severe asthma in humans and mouse model suggests a CXCL10 signature underlies corticosteroid-resistant Th1 bias. JCI Insight 2: e94580, 2017. doi: 10.1172/jci.insight.94580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Raundhal M, Morse C, Khare A, Oriss TB, Milosevic J, Trudeau J, Huff R, Pilewski J, Holguin F, Kolls J, Wenzel S, Ray P, Ray A. High IFN-gamma and low SLPI mark severe asthma in mice and humans. J Clin Invest 125: 3037–3050, 2015. doi: 10.1172/JCI80911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Assendorp EL, Gresnigt MS, Sprenkeler EG, Meis JF, Dors N, van der Linden JW, Henriet, SS. Adjunctive interferon-gamma immunotherapy in a pediatric case of Aspergillus terreus infection. Eur J Clin Microbiol Infect Dis 37: 1915–1922, 2018. doi: 10.1007/s10096-018-3325-4. [DOI] [PubMed] [Google Scholar]
- 21.Hohl TM, Rivera A, Lipuma L, Gallegos A, Shi C, Mack M, Pamer EG. Inflammatory monocytes facilitate adaptive CD4 T cell responses during respiratory fungal infection. Cell Host Microbe 6: 470–481, 2009. doi: 10.1016/j.chom.2009.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Leonhardt J, Grosse S, Marx C, Siwczak F, Stengel S, Bruns T, Bauer R, Kiehntopf M, Williams DL, Wang ZQ, Mosig AS, Weis S, Bauer M, Heller R. Candida albicans beta-glucan differentiates human monocytes into a specific subset of macrophages. Front Immunol 9: 2818, 2018. doi: 10.3389/fimmu.2018.02818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang J, Zhang Y, Wang Q, Li C, Deng H, Si C, Xiong H. Interleukin-35 in immune-related diseases: protection or destruction. Immunology 157: 13–20, 2019. doi: 10.1111/imm.13044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Catalan-Dibene J, McIntyre LL, Zlotnik A. Interleukin 30 to interleukin 40. J Interferon Cytokine Res 38: 423–439, 2018. doi: 10.1089/jir.2018.0089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chakarov S, Lim HY, Tan L, Lim SY, See P, Lum J, , et al. Two distinct interstitial macrophage populations coexist across tissues in specific subtissular niches. Science 363: eaau0964, 2019. doi: 10.1126/science.aau0964. [DOI] [PubMed] [Google Scholar]
- 26.Gibbings SL, Thomas SM, Atif SM, McCubbrey AL, Desch AN, Danhorn T, Leach SM, Bratton DL, Henson PM, Janssen WJ, Jakubzick CV. Three unique interstitial macrophages in the murine lung at steady state. Am J Respir Cell Mol Biol 57: 66–76, 2017. doi: 10.1165/rcmb.2016-0361OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schyns J, Bai Q, Ruscitti C, Radermecker C, De Schepper S, Chakarov S, Farnir F, Pirottin D, Ginhoux F, Boeckxstaens G, Bureau F, Marichal T. Non-classical tissue monocytes and two functionally distinct populations of interstitial macrophages populate the mouse lung. Nat Commun 10: 3964, 2019. doi: 10.1038/s41467-019-11843-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mionnet C, Buatois V, Kanda A, Milcent V, Fleury S, Lair D, Langelot M, Lacoeuille Y, Hessel E, Coffman R, Magnan A, Dombrowicz D, Glaichenhaus N, Julia V. CX3CR1 is required for airway inflammation by promoting T helper cell survival and maintenance in inflamed lung. Nat Med 16: 1305–1312, 2010. doi: 10.1038/nm.2253. [DOI] [PubMed] [Google Scholar]
- 29.Sukkar MB, Issa R, Xie S, Oltmanns U, Newton R, Chung, KF. Fractalkine/CX3CL1 production by human airway smooth muscle cells: induction by IFN-γ and TNF-α and regulation by TGF-β and corticosteroids. Am J Physiol Lung Cell Mol Physiol 287: L1230–L1240, 2004. doi: 10.1152/ajplung.00014.2004. [DOI] [PubMed] [Google Scholar]
- 30.El-Shazly A, Berger P, Girodet PO, Ousova O, Fayon M, Vernejoux JM, Marthan R, Tunon-de-Lara JM. Fraktalkine produced by airway smooth muscle cells contributes to mast cell recruitment in asthma. J Immunol 176: 1860–1868, 2006. doi: 10.4049/jimmunol.176.3.1860. [DOI] [PubMed] [Google Scholar]
- 31.Chachi L, Shikotra A, Duffy SM, Tliba O, Brightling C, Bradding P, Amrani Y. Functional KCa3.1 channels regulate steroid insensitivity in bronchial smooth muscle cells. J Immunol 191: 2624–2636, 2013. doi: 10.4049/jimmunol.1300104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bates J, Irvin C, Brusasco V, Drazen J, Fredberg J, Loring S, Eidelman D, Ludwig M, Macklem P, Martin J, Milic-Emili J, Hantos Z, Hyatt R, Lai-Fook S, Leff A, Solway J, Lutchen K, Suki B, Mitzner W, Pare P, Pride N, Sly P. The use and misuse of Penh in animal models of lung disease. Am J Respir Cell Mol Biol 31: 373–374, 2004. doi: 10.1165/ajrcmb.31.3.1. [DOI] [PubMed] [Google Scholar]
- 33.Lundblad LK, Irvin CG, Hantos Z, Sly P, Mitzner W, Bates JH. Penh is not a measure of airway resistance. Eur Respir J 30: 805, 2007. doi: 10.1183/09031936.00091307. [DOI] [PubMed] [Google Scholar]
- 34.Sharma A, Laxman B, Naureckas ET, Hogarth DK, Sperling AI, Solway J, Ober C, Gilbert JA, White SR. Associations between fungal and bacterial microbiota of airways and asthma endotypes. J Allergy Clin Immunol 144: 1214–1227,2019. doi: 10.1016/j.jaci.2019.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Garth JM, Mackel JJ, Reeder KM, Blackburn JP, Dunaway CW, Yu Z, Matalon S, Fitz L, Steele C. Acidic mammalian Chitinase negatively affects immune responses during acute and chronic Aspergillus fumigatus exposure. Infect Immun 86, 2018. doi: 10.1128/IAI.00944-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stephens-Romero SD, Mednick AJ, Feldmesser M. The pathogenesis of fatal outcome in murine pulmonary aspergillosis depends on the neutrophil depletion strategy. Infect Immunol 73: 114–125, 2005. doi: 10.1128/IAI.73.1.114-125.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hall JD, Kurtz SL, Rigel NW, Gunn BM, Taft-Benz S, Morrison JP, Fong AM, Patel DD, Braunstein M, Kawula TH. The impact of chemokine receptor CX3CR1 deficiency during respiratory infections with Mycobacterium tuberculosis or Francisella tularensis. Clin Exp Immunol 156: 278–284, 2009. doi: 10.1111/j.1365-2249.2009.03882.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Johnson CH, Miao C, Blanchard EG, Caidi H, Radu GU, Harcourt JL, Haynes LM. Effect of chemokine receptor CX3CR1 deficiency in a murine model of respiratory syncytial virus infection. Comp Med 62: 14–20, 2012. [PMC free article] [PubMed] [Google Scholar]
- 39.Das S, Raundhal M, Chen J, Oriss TB, Huff R, Williams JV, Ray A, Ray P. Respiratory syncytial virus infection of newborn CX3CR1-deficient mice induces a pathogenic pulmonary innate immune response. JCI Insight 2, 2017. doi: 10.1172/jci.insight.94605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Matucci A, Vultaggio A, Maggi E, Kasujee I. Is IgE or eosinophils the key player in allergic asthma pathogenesis? Are we asking the right question? Respir Res 19: 113, 2018. doi: 10.1186/s12931-018-0813-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kozlova Y, Frolova E, Uchevatkina A, Filippova L, Aak O, Burygina E, Taraskina A, Vasilyeva N, Klimko N. Diagnostic markers of allergic bronchopulmonary aspergillosis in patients with severe asthma. Mycoses 63: 596–603, 2020. doi: 10.1111/myc.13083. [DOI] [PubMed] [Google Scholar]
- 42.Fulkerson PC, Fischetti CA, McBride ML, Hassman LM, Hogan SP, Rothenberg ME. A central regulatory role for eosinophils and the eotaxin/CCR3 axis in chronic experimental allergic airway inflammation. Proc Natl Acad Sci U S A 103: 16418–16423, 2006. doi: 10.1073/pnas.0607863103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lilly LM, Scopel M, Nelson MP, Burg AR, Dunaway CW, Steele C. Eosinophil deficiency compromises lung defense against Aspergillus fumigatus. Infect Immun 82: 1315–1325, 2014. doi: 10.1128/IAI.01172-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ravin KA, Loy M. The eosinophil in infection. Clinic Rev Allerg Immunol 50: 214–227, 2016. doi: 10.1007/s12016-015-8525-4. [DOI] [PubMed] [Google Scholar]
- 45.Walsh GM. Anti-IL-5 monoclonal antibodies for the treatment of asthma: an update. Expert Opin Biol Ther 20: 1237–1244, 2020. doi: 10.1080/14712598.2020.1782381. [DOI] [PubMed] [Google Scholar]
- 46.Guo Y, Kasahara S, Jhingran A, Tosini NL, Zhai B, Aufiero MA, Mills KAM, Gjonbalaj M, Espinosa V, Rivera A, Luster AD, Hohl TM. During Aspergillus infection, monocyte-derived DCs, neutrophils, and plasmacytoid DCs enhance innate immune defense through CXCR3-dependent crosstalk. Cell Host Microbe 28: 104–116,2020. doi: 10.1016/j.chom.2020.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dai XM, Ryan GR, Hapel AJ, Dominguez MG, Russell RG, Kapp S, Sylvestre V, Stanley ER. Targeted disruption of the mouse colony-stimulating factor 1 receptor gene results in osteopetrosis, mononuclear phagocyte deficiency, increased primitive progenitor cell frequencies, and reproductive defects. Blood 99: 111–120, 2002. doi: 10.1182/blood.V99.1.111. [DOI] [PubMed] [Google Scholar]
- 48.Terry RL, Miller SD. Molecular control of monocyte development. Cell Immunol 291: 16–21, 2014. doi: 10.1016/j.cellimm.2014.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Penaloza HF, Noguera LP, Ahn D, Vallejos OP, Castellanos RM, Vazquez Y, Salazar-Echegarai FJ, Gonzalez L, Suazo I, Pardo-Roa C, Salazar GA, Prince A, Bueno SM. Interleukin-10 produced by myeloid-derived suppressor cells provides protection to carbapenem-resistant Klebsiella pneumoniae sequence type 258 by enhancing its clearance in the airways. Infect Immun 87, 2019. doi: 10.1128/IAI.00665-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Huang HY, Wen Y, Kruessel JS, Raga F, Soong YK, Polan ML. Interleukin (IL)-1beta regulation of IL-1beta and IL-1 receptor antagonist expression in cultured human endometrial stromal cells. J Clin Endocrinol Metab 86: 1387–1393, 2001. doi: 10.1210/jc.86.3.1387. [DOI] [PubMed] [Google Scholar]
- 51.Branchett WJ, Cm L. Regulatory cytokine function in the respiratory tract. Mucosal Immunol 12: 589–600, 2019. doi: 10.1038/s41385-019-0158-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li W, Gao R, Xin T, Gao P. Different expression levels of interleukin-35 in asthma phenotypes. Respir Res 21: 89, 2020. doi: 10.1186/s12931-020-01356-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tsang MS, Sun X, Wong CK. The role of new IL-1 family members (IL-36 and IL-38) in atopic dermatitis, allergic asthma, and allergic rhinitis. Curr Allergy Asthma Rep 20: 40, 2020. doi: 10.1007/s11882-020-00937-1. [DOI] [PubMed] [Google Scholar]