

Abstract
Swelling-activated volume-regulated anion channels (VRACs) are heteromeric channels comprising LRRC8A and at least one other LRRC8 paralog. Cryoelectron microscopy (cryo-EM) structures of nonnative LRRC8A and LRRC8D homohexamers have been described. We demonstrate here that LRRC8A homohexamers poorly recapitulate VRAC functional properties. Unlike VRACs, LRRC8A channels heterologously expressed in Lrr8c−/− HCT116 cells are poorly activated by low intracellular ionic strength (µ) and insensitive to cell swelling with normal µ. Combining low µ with swelling modestly activates LRRC8A, allowing characterization of pore properties. VRACs are strongly inhibited by 10 µM 4-[(2-butyl-6,7-dichloro-2-cyclopentyl-2,3-dihydro-1-oxo-1H-inden-5-yl)oxy]butanoic acid (DCPIB) in a voltage-independent manner. In contrast, DCPIB block of LRRC8A is weak and voltage sensitive. Cryo-EM structures indicate that DCPIB block is dependent on arginine 103. Consistent with this, LRRC8A R103F mutants are insensitive to DCPIB. However, an LRRC8 chimeric channel in which R103 is replaced by a leucine at the homologous position is inhibited ∼90% by 10 µM DCPIB in a voltage-independent manner. Coexpression of LRRC8A and LRRC8C gives rise to channels with DCPIB sensitivity that is strongly µ dependent. At normal intracellular µ, LRRC8A + LRRC8C heteromers exhibit strong, voltage-independent DCPIB block that is insensitive to R103F. DCPIB inhibition is greatly reduced and exhibits voltage dependence with low intracellular µ. The R103F mutation has no effect on maximal DCPIB inhibition but eliminates voltage dependence under low µ conditions. Our findings demonstrate that the LRRC8A cryo-EM structure and the use of heterologously expressed LRRC8 heteromeric channels pose significant limitations for VRAC mutagenesis-based structure-function analysis. Native VRAC function is most closely mimicked by chimeric LRRC8 homomeric channels.
Keywords: anion channel, cell volume, LRRC8
INTRODUCTION
The volume-regulated anion channel (VRAC) is expressed widely in vertebrate cells. The channel is activated and inactivated by cell swelling and shrinkage, respectively, and plays a central role in mediating the efflux of Cl− and organic solutes required for regulatory volume decrease (1–4). VRAC volume sensitivity or “volume set-point” is modulated by intracellular ionic strength (µ) (5–7). When intracellular µ is decreased, less cell swelling is required to activate VRAC and the rate of current activation is increased (5). At very low intracellular µ, VRAC activates without swelling and will even activate in shrunken cells (5). The sensitivity of VRAC and other swelling-activated transport pathways such as the KCl cotransporter to intracellular µ allows cells to concomitantly regulate their volume and cytoplasmic inorganic ion concentrations (4, 5).
During the late 1990s and early 2000s, multiple laboratories claimed to have identified the genes underlying VRAC activity (8–12). Unfortunately, none of these gene candidates withstood the test of experimental verification by other laboratories (13–16). A major breakthrough occurred in 2014 when two laboratories independently demonstrated using genome-wide RNA interference screening that VRAC is encoded by members of the leucine-rich repeat (LRR) containing 8 (Lrrc8) gene family (17, 18). The Lrrc8 gene family comprises five members termed Lrrc8a-e (18, 19). LRRC8/VRACs comprise the essential subunit LRRC8A that must be co-assembled with at least one other family member (18). Co-assembly of LRRC8A with different LRRC8 family members gives rise to VRACs with distinct functional properties (18, 20–26). However, subunit stoichiometry and assembly order are currently unknown.
High-resolution cryoelectron microscopy (cryo-EM) structures of LRRC8A (27–30) and LRRC8D (31) homomeric channels were described recently. These structures have confirmed and significantly expanded bioinformatic predictions and biochemical studies of LRRC8 channel structural properties (17–19). LRRC8 subunits comprise intracellular (I), transmembrane (TM), extracellular (E), and leucine-rich repeat (LRR) regions with intracellular N- and C-termini. The membrane domain consists of four TM helices with two large extracellular loops, EL1 and EL2, connecting TM1 and TM2, and TM3 and TM4. An intracellular loop, IL1, connects TM2 and TM3. A second intracellular loop, IL2, connects TM4 to the LRR region that contains 15 leucine-rich repeat motifs.
LRRC8 proteins are closely related to pannexins (19), which form large-pore heptameric membrane channels (32–34). All cryo-EM structures demonstrate that LRRC8A and LRRC8D form homohexameric channels (27–31). A low resolution cryo-EM structure reconstructed using the homohexameric LRCC8A structure as a reference map and imposing C3 symmetry suggests that heterohexameric channels are formed by co-assembly of LRRC8A and LRRC8C (27).
While the cryo-EM structures of LRRC8A and LRRC8D homohexamers have provided valuable insights into VRACs, these channels do not appear to exist in nature. Furthermore, their functional properties have not been characterized in detail. We demonstrate here that both the regulation and inhibitor block of LRRC8A channels poorly recapitulate those of native VRACs, LRRC8 heteromers, and LRRC8 chimeric homomeric channels. LRRC8A homohexameric channels thus have significant limitations for detailed understanding of VRAC structure-function relationships. The use of heteromeric LRRC8 channels also constrains structure-function analysis due to the unknown and likely uncontrollable assembly and stoichiometry of channel subunits. We propose that LRRC8 chimeras recently developed in our laboratory (26) provide an experimental system better suited for defining VRAC structure-function relationships.
METHODS
Molecular Biology
Human LRRC8A and LRRC8C cDNAs cloned into pCMV6 were purchased from OriGene Technologies. Mutant and chimera cDNA constructs were generated by using either the QuikChange Lightning Multi Site-Directed mutagenesis kit (Agilent Technologies) or the Phusion High-Fidelity PCR kit (New England BioLabs) and the restriction-free cloning method (35). All cDNAs were tagged on their carboxy terminus with Myc-DDK epitopes. Mutant and wild-type constructs were confirmed by DNA sequencing.
Transfection and Whole Cell Patch-Clamp Recording
Human colon cancer HCT116 cells in which the five Lrrc8 genes were disrupted by genome editing (i.e., Lrrc8−/−) were a gift from T. Jentsch. Wild-type HCT116 cells were purchased from American Type Culture Collection. Lrrc8−/− HCT116 cells were transfected using Turbofectin 8.0 (OriGene Technologies) with 0.125 μg GFP cDNA and 0.25 μg of various LRRC8 cDNAs. All experimental protocols were performed on at least two independently transfected groups of cells. Expression of heteromeric channels comprising the LRRC8A and LRRC8C paralogs was accomplished by transfecting cells with equal amounts of each cDNA.
Transfected cells were identified by GFP fluorescence and patch clamped in the whole cell mode at room temperature using bath and patch pipette solutions presented in Table 1. Cesium was used as the major cation in these solutions to minimize cation currents. The ionic strength of the patch pipette solution was reduced by reducing CsCl concentration from 126 mM (defined as normal µ) to 26 mM (defined as low µ).
Table 1.
Composition of patch pipette and solutions
| Patch pipette solutions |
Bath solutions |
|||
|---|---|---|---|---|
| Normal µ | Low µ | 300 mOsm (isotonic) | 250 mOsm (hypotonic) | |
| CsCl, mM | 126 | 26 | 75 | 75 |
| MgSO4, mM | 2 | 2 | 5 | 5 |
| Ca-gluconate2, mM | – | – | 1 | 1 |
| ATP-Na2, mM | 2 | 2 | – | – |
| GTP-Na2, mM | 0.5 | 0.5 | – | – |
| Glutamine, mM | 2 | 2 | – | – |
| EGTA, mM | 1 | 1 | – | – |
| HEPES, mM | 20 | 20 | 12 | 12 |
| Tris, mM | – | – | 8 | 8 |
| CsOH, mM | 12 | 12 | – | – |
| HCl, mM | – | – | 2 | 2 |
| Glucose, mM | – | – | 5 | 5 |
| Sucrose, mM | 16 | 175 | 115 | 70 |
| pH | 7.2 | 7.2 | 7.4 | 7.4 |
| Osmolarity, mOsm | 275 | 275 | 300 | 250 |
| Ionic strength, M | 0.162 | 0.062 | 0.108 | 0.108 |
µ, ionic strength. The pH of patch pipette and bath solutions was adjusted with CsOH and HCl, respectively.
Cells were swollen by exposure to a 250 mOsm bath solution following a 45-s equilibration with the patch pipette solution. Osmolarity was reduced by removal of sucrose (Table 1). The total volume of the bath chamber used in these studies was ∼250 μL. Bath changes were performed by flushing the chamber for 1 min with ∼2 mL of solution.
4-[(2-Butyl-6,7-dichloro-2-cyclopentyl-2,3-dihydro-1-oxo-1H-inden-5-yl)oxy]butanoic acid (DCPIB; Bio-Techne Corporation) was dissolved in DSMO and stored as a 100 mM stock solution at −20°C. Working DCPIB solutions were made fresh daily by diluting the DMSO stock into the appropriate bath solution. Final DMSO concentrations were 0.0001%–0.05%.
Cells were patch clamped in the standard whole cell mode using patch electrodes pulled from 1.5-mm–outer-diameter silanized borosilicate microhematocrit tubes. Mean ± standard deviation (SD) pipette resistances were 1.8 ± 0.5 MΩ and 5.1 ± 1.0 MΩ with normal and low µ patch pipette solutions, respectively. Recordings were not performed on cells where access resistance was >2-fold that of the pipette resistance. Means ± SD whole cell access resistance was 4.2 ± 1.0 MΩ with a normal-µ pipette solution and 4.8 ± 1.4 MΩ with a low-µ pipette solution. Means ± SD cell capacitance was 13.2 ± 2.9 pF.
Currents were measured with an Axopatch 200 A (Axon Instruments) patch-clamp amplifier. Electrical connections to the patch-clamp amplifier were made using Ag/AgCl wires and 3 M KCl/agar bridges. Series resistance was compensated by >85% to minimize voltage errors. Data acquisition and analysis were performed using pClamp 10 software (Axon Instruments).
Patch-clamped cells were visualized throughout the recording period by video-enhanced differential interference contrast microscopy using a Nikon TE300 microscope and Nikon Plan Fluor 60×/0.7 numerical aperture extra-long working distance lens. Images were captured using a Dage-MTI CCD camera. The diameter of cells was measured at a single focal plane located at the point of maximum cell diameter. Cell morphology was assumed to approximate a sphere, and cell volume was calculated as 4/3 × π × r3, where r is the cell radius. Cells that exhibited volume changes in the absence of bath osmotic perturbations were not used for analysis of whole cell current properties.
Quantification of Current Properties
Changes in current amplitude were quantified using a voltage-ramping protocol. Membrane voltage was held at −30 mV throughout all experiments. Ramps were initiated by stepping membrane voltage to −100 mV and then ramping membrane voltage over 1 s to +100 mV. This was followed by a step back to −30 mV for 4 s before the ramp was repeated.
Step changes in membrane voltage were used to determine current-voltage relationships. Membrane voltage was stepped from −30 mV to −80 mV for 0.5 s followed by steps from −120 mV to +120 mV in 2-s, 20-mV increments. Current-voltage plots were generated using the steady-state current measured after initiating the voltage step.
Initial rates of current change were quantified by linear regression analysis of the first 10–15 current recordings after changes in current amplitude were first detected. Cells were exposed to DCPIB for 300 s, and peak inhibition was determined at the end of the exposure period. DCPIB half-maximal inhibitory concentration (IC50) and Hill coefficients were determined by fitting concentration-response relationships to the Hill equation using Prism-GraphPad software.
Statistical Analyses
Data are presented as means ± standard error of the mean (SE); n represents the number of patch-clamped cells from which currents were recorded. Statistical significance was determined using Student’s two-tailed t test for unpaired means.
RESULTS
LRRC8A homohexameric channels (8A) are largely insensitive to swelling when cells are patch clamped with a normal-µ pipette solution. Figure 1A shows the effect of swelling on whole cell current activation in LRRC8A-expressing cells under normal intracellular µ conditions. The inset to the figure shows relative cell volume changes during the recording period. Mean cell volume increased ∼3.4-fold over the course of the experiment. The initial rate of current change after cell swelling was induced was 0.16 pA/pF/min (Fig. 2), which was not significantly (P > 0.5) different from a hypothetical mean of 0. Means ± SE relative whole cell current 400 s after induction of cell swelling was 1.20 ± 0.16 (n = 6 cells), which was not significantly different (P > 0.3) from a hypothetical mean of 1.0 (Fig. 1A).
Figure 1.
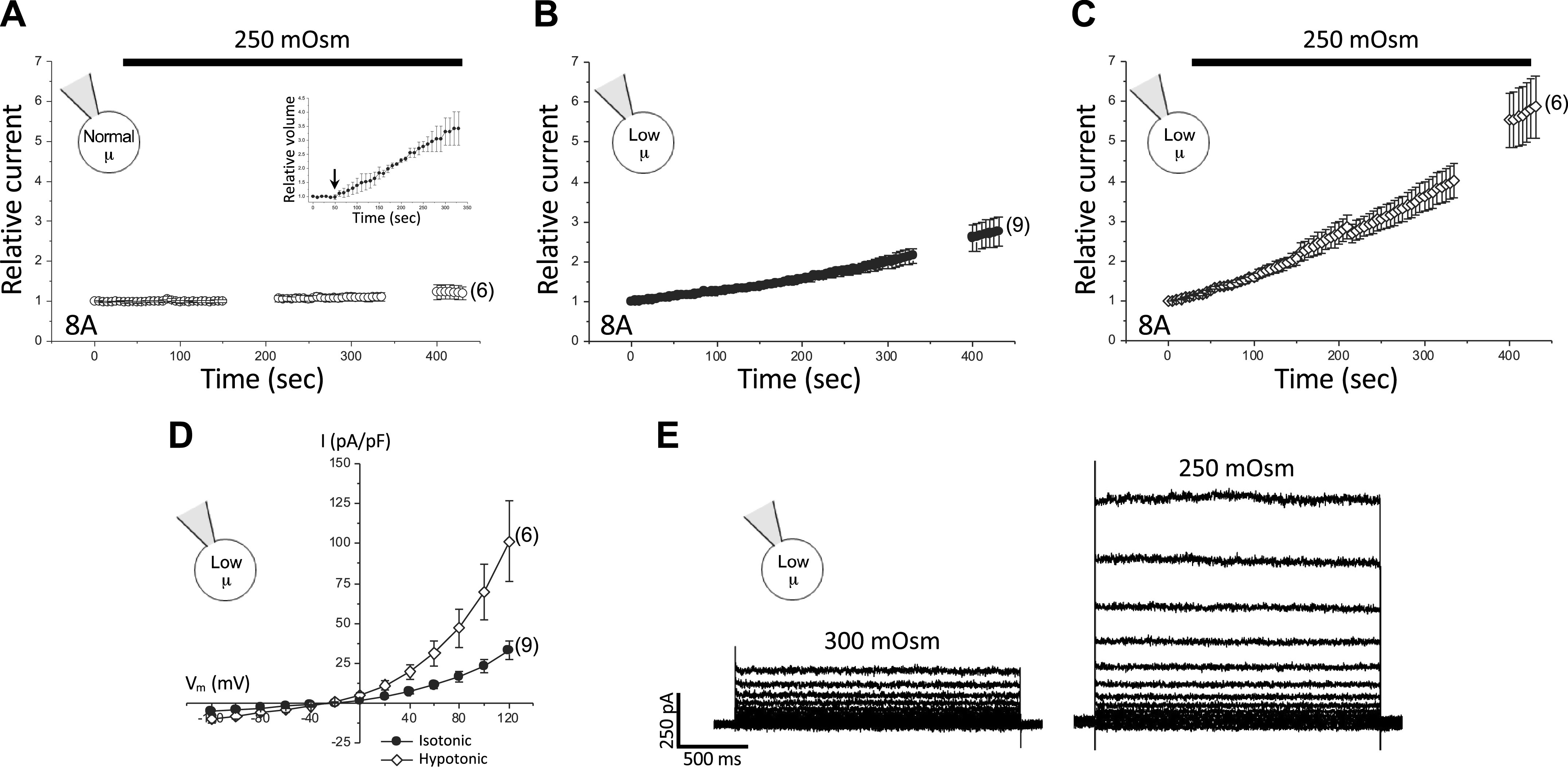
Effect of intracellular µ and cell swelling on LRRC8A homohexameric channel activity. A–C: time course of relative current changes measured at +100 mV in LRRC8A-expressing LRRC8−/− HCT116 cells equilibrated with a normal-µ patch pipette solution and exposed to a 250 mOsm hypotonic bath (A), cells equilibrated with a low-µ patch pipette solution and maintained in 300 mOsm isotonic bath medium (B), and cells equilibrated with a low-µ patch pipette solution and swollen by 250 mOsm bath medium (C). Inset in A shows relative cell volume changes measured over the course of the experiment. Arrow shows when bath Osmolarity was reduced to 250 mOsm. Current-to-voltage relationships (D) and representative traces of LRRC8A currents (E) from cells equilibrated with a low-µ patch pipette solution maintained in 300 mOsm bath medium or swollen by exposure to 250 mOsm bath medium. Values in A–D are means ± SE. n is shown in parentheses.
Figure 2.

Initial rates of current activation induced by cell swelling, low intracellular µ, and combined swelling and low intracellular µ in LRRC8A homohexameric channels, LRRC8A + LRRC8C heteromeric channels, LRRC8C-LRRC8A(IL125) homomeric chimeric channels, and native VRACs in wild-type HCT116 cells. Values are means ± SE. n is shown in parentheses. VRAC, volume-regulated anion channel.
Whole cell current activated very slowly (Fig. 1B) when LRRC8A-expressing cells were equilibrated with a low-µ patch pipette solution. The initial rate of current activation was 1.5 pA/pF/min (Fig. 2). Mean ± SE relative whole cell current increased 2.8 ± 0.4-fold (n = 9) 430 s after whole cell access was obtained (Fig. 1B).
In the presence of low intracellular µ, cell swelling increased both the rate and magnitude of current activation (Figs. 1C and 2). The initial rate of current activation after induction of cell swelling was increased significantly (P < 0.03) to 8.3 pA/pF/min compared with current activation induced by low intracellular µ alone (Figs. 1C and 2). The mean ± SE relative whole cell current measured 400 s after whole cell swelling began was 5.9 ± 0.8 (n = 6) (Fig. 1C), which was significantly (P < 0.002) greater than that observed with low intracellular µ alone.
Current-to-voltage relationships and representative traces of 8A currents activated by low intracellular µ and combined low intracellular µ and cell swelling are shown in Fig. 1, D and E. The mean ± SE reversal potentials for the currents were −22.5 ± 1.8 mV (low intracellular µ; n = 5) and −22.7 ± 1.7 mV (low intracellular µ plus cell swelling; n = 6). These reversal potentials are close to the Nernst potential for Cl− of −27.7 mV, indicating that the 8A channel has a high selectivity for Cl− over cations.
As a comparison, we quantified the effect of cell swelling and low intracellular µ on LRRC8A + LRRC8C (8A + 8C) heteromeric channels. When cells were equilibrated with a normal-µ pipette solution, 8A + 8C heteromeric channels activated at an initial rate of 7.6 pA/pF/min when exposed to a 250 mOsm bath solution (Fig. 2). The mean ± SE relative whole cell current measured 400 s after whole cell swelling began was 27.9 ± 10.9 (n = 7).
When cells were equilibrated with a low-µ pipette solution, 8A + 8C heteromeric channels activated rapidly at an initial rate of 110 pA/pF/min, which was almost two orders of magnitude faster (P < 0.02) than that observed with 8A homohexameric channels (Fig. 2). Cell swelling had no significant (P > 0.2) effect on the rate of current activation of 8A + 8C heteromeric channels exposed to a low-µ pipette solution (Fig. 2).
We recently described several homomeric chimeric LRRC8 constructs that generate channel activity more closely resembling native VRACs (26). All LRRC8 paralogs contain an intracellular loop (IL1) that connects transmembrane domains 2 and 3 (27–31). The minimal functional chimera we developed consists of a 25-amino acid sequence (D182–E206) unique to LRRC8A IL1 inserted into the corresponding region of LRRC8C. This chimera is termed 8C-8A(IL125). As shown in Fig. 2, the rate of swelling-induced activation of the 8C-8A(IL125) chimera in cells equilibrated with a normal-µ pipette solution was ∼4 times faster (P < 0.001) than that of 8A + 8C heteromeric channels. The rate of channel activation induced by low intracellular µ was not significantly (P > 0.4) different from that of 8A + 8C heteromers. Cell swelling did not significantly (P > 0.9) alter the rate of 8C-8A(IL125) channel activation in cells equilibrated with a low-µ pipette solution (Fig. 2).
Finally, we characterized the effect of cell swelling and low intracellular µ on activation of native VRAC currents expressed in wild-type HCT116 cells. The rate of swelling-induced current activation was similar (P > 0.7) to that of the 8C-8A(IL125) chimera (Fig. 2). Native VRAC currents were activated ∼3-fold faster (P < 0.003) by low intracellular µ compared with both the 8C-8A(IL125) chimeric and (8A + 8C) heteromeric channels, and this activation was not significantly (P > 0.8) altered by cell swelling (Fig. 2).
8A + 8C heteromeric channels, 8C-8A(IL125) chimeric homomeric channels, and native VRACs are dramatically activated by cell swelling and low intracellular µ (Fig. 2) (4). In contrast, 8A homohexameric channels are insensitive to cell swelling in the presence of normal intracellular µ. Low intracellular µ causes slow activation of 8A channels, and this activation is further enhanced by cell swelling (Figs. 1 and 2).
Pharmacological approaches have provided important insights into ion channel structure-function relationships (36–39). Micromolar concentrations of 4-[(2-Butyl-6,7-dichloro-2-cyclopentyl-2,3-dihydro-1-oxo-1H-inden-5-yl)oxy]butanoic acid (DCPIB) inhibit VRACs, albeit in a relatively nonselective fashion (40–42). A recent study by Kern et al. (30) described a cryoelectron microscopy (cryo-EM) structure of 8A homohexameric channels blocked by DCPIB. However, the effect of DCPIB on 8A homohexameric channel activity has not been described, limiting the utility of this inhibitor and the cryo-EM structure for structure-function analyses. We therefore quantified the effect of DCPIB on 8A homohexamers activated with a combination of low intracellular µ and cell swelling.
As shown in Fig. 3A, 8A homohexameric channels were poorly inhibited by 10 µM DCPIB. Furthermore, this inhibition exhibited a clear voltage dependence. At −100 mV and +100 mV, steady-state inhibition observed with 10 µM DCPIB was ∼40% and ∼13%, respectively (Fig. 3A; Table 2). The maximal inhibitory effect of DCPIB was observed at a concentration of 50 µM. Maximal inhibition was ∼61% at −100 mV and ∼45% at +100 mV (n = 3) (Fig. 3B). Half-maximal inhibitory concentrations (IC50) derived from the concentration-response curve (Fig. 3B) were 4.0 µM at −100 mV and 15.8 µM at +100 mV (Table 3).
Figure 3.

DCPIB inhibition of LRRC8A homohexameric channels. A: effect of 10 µM DCPIB on LRRC8A channels activated by cell swelling and low intracellular µ. B: dose-response relationships for inhibition of LRRC8A channels by DCPIB at +100 mV and −100 mV. Channels were activated by swelling and low intracellular µ. C: effect of 10 µM DCPIB on basal LRRC8A channel activity in cells equilibrated with a normal-µ pipette solution and swollen by 250 mOsm bath medium. D: effect of 0.01% DMSO on LRRC8A channels activated by cell swelling and low intracellular µ. Values are means ± SE. n is shown in parentheses. DCPIB, 4-[(2-butyl-6,7-dichloro-2-cyclopentyl-2,3-dihydro-1-oxo-1H-inden-5-yl)oxy]butanoic acid.
Table 2.
Effect of 10 µM DCPIB on LRRC8/VRAC activity
| Channel; intracellular µ | Peak inhibition 10 µM DCPIB/+100 mV | Peak inhibition 10 µM DCPIB/−100 mV |
|---|---|---|
| 8A; low µ | 13.4 ± 2.5% (5) | 40.3 ± 2.6% (5) |
| 8A; normal µ | 15.6 ± 5.0% (4) | 38.6 ± 3.7% (4) |
| 8A R103L; low µ | 2.8 ± 3.6% (5) | 23.1 ± 1.7% (5) |
| 8A R103F; low µ | 0.2 ± 1.6% (4) | 4.4 ± 1.1% (4) |
| Native VRAC; low µ | 50.3 ± 2.7% (5) | 53.7 ± 4.0% (5) |
| Native VRAC; normal µ | 88.4 ± 2.9% (3) | 88.2 ± 2.9% (3) |
| 8A + 8C; low µ | 3.5 ± 3.9% (6) | 23.0 ± 5.9% (6) |
| 8A + 8C; normal µ | 61.5 ± 8.6% (4) | 70.3 ± 6.4% (4) |
| 8A R103F + 8C; low µ | 26.1 ± 8.0% (5) | 26.3 ± 5.4% (5) |
| 8A R103F + 8C; normal µ | 72.4 ± 6.8% (4) | 77.1 ± 6.0% (4) |
| 8C-8A(IL125); low µ | 91.6 ± 6.2% (4) | 84.8 ± 10.6% (4) |
| 8C-8A(IL125); normal µ | 90.1 ± 6.0% (5) | 90.2 ± 7.1% (5) |
Values are means ± SE (n). Cells were equilibrated with normal- or low-µ patch pipette solutions. Swelling was induced by reducing bath Osmolarity to 250 mOsm. Native VRAC current was measured in wild-type HCT116 cells. All other recordings were obtained from LRRC8−/− HCT116 cells transfected with LRRC8 cDNAs. DCPIB, 4-[(2-butyl-6,7-dichloro-2-cyclopentyl-2,3-dihydro-1-oxo-1H-inden-5-yl)oxy]butanoic acid; VRAC, volume-regulated anion channel.
Table 3.
Hill parameters for DCPIB inhibition of 8A homohexameric channels, 8C-8A(IL125) homomeric chimeric channels, and native VRACs in wild-type HCT116 cells
| Channel; voltage | IC50 (µM) | Hill coefficient |
|---|---|---|
| 8A; −100 mV | 4.0 ± 0.4 | 1.1 ± 0.1 |
| 8A; +100 mV | 15.8 ± 4.3 | 1.2 ± 0.3 |
| 8C-8A(IL125); −100 mV | 1.7 ± 0.1 | 2.3 ± 0.2 |
| 8C-8A(IL125); +100 mV | 1.4 ± 0.3 | 2.0 ± 0.2 |
| Native VRAC; −100 mV | 2.5 ± 0.1 | 1.9 ± 0.1 |
| Native VRAC; +100 mV | 2.3 ± 0.0 | 1.7 ± 0.1 |
IC50 values and Hill coefficients were determined by fitting DCPIB concentration-response relationships to the Hill equation using Prism software. DCPIB, 4-[(2-butyl-6,7-dichloro-2-cyclopentyl-2,3-dihydro-1-oxo-1H-inden-5-yl)oxy]butanoic acid; VRACs, volume-regulated anion channels.
Native VRAC currents observed in calf bovine pulmonary artery endothelial cells, Xenopus oocytes, guinea pig atrial cardiomyocytes, rat pancreatic β-cells, and HEK-293 cells exhibit voltage-independent DCPIB inhibition, >85% inhibition by 10 µM DCPIB, and IC50 values of 2–5 µM (40–42). It is conceivable that the much weaker, voltage-dependent inhibition of 8A homohexameric channels by DCPIB is due to the low intracellular µ needed for activation. The small currents detected in 8A homohexameric channel-expressing cells equilibrated with a normal-µ pipette solution make characterization of DCPIB inhibition difficult. However, we occasionally observed transfected cells that had reasonably high levels of basal 8A homohexameric channel activity. Figure 3C shows the effect of 10 µM DCPIB on four 8A transfected cells equilibrated with a normal-µ pipette solution that were also swollen by exposure to a 250 mOsm bath. As reported earlier, swelling did not increase these currents further (data not shown). The effect of 10 µM DCPIB was similar to that observed with currents activated by combined low intracellular µ and swelling (Fig. 3A). At −100 and +100 mV, peak inhibition was ∼39% and ∼16%, respectively (Fig. 3C; Table 2) and was not significantly (P > 0.6) different from that observed in cells equilibrated with a low-µ pipette solution.
DCPIB is dissolved as a stock solution in DMSO. The final DMSO concentration in a bath solution containing 10 µM DCPIB is 0.01%. While unlikely, it is possible that this low concentration of DMSO has a direct effect on 8A activity that obscures the inhibitory action of DCPIB. To test this possibility, we quantified the effect of DMSO on 8A currents activated by cell swelling and low intracellular µ. As shown in Fig. 3D, 0.01% DMSO had no significant (P > 0.09) effect on whole cell current.
Finally, it is conceivable that DCPIB inhibition is cell type dependent. We therefore quantified the effect of 10 µM DCPIB on native VRAC current in wild-type HCT116 cells. As shown in Fig. 4A and Table 2, 10 µM DCPIB inhibited native VRAC current ∼50% in a voltage-independent manner when currents were activated by combined low intracellular µ and swelling. DCPIB inhibition was increased by increasing intracellular µ (Fig. 4B; Table 2). In cells equilibrated with a normal-µ pipette solution, maximal inhibition by 10 µM DCPIB was significantly (P < 0.001) increased to ∼90% at both −100 mV and +100 mV. The IC50 values derived from the concentration-response curve (Fig. 4C) were 2.5 µM at −100 mV and 2.3 µM at +100 mV (Table 3).
Figure 4.
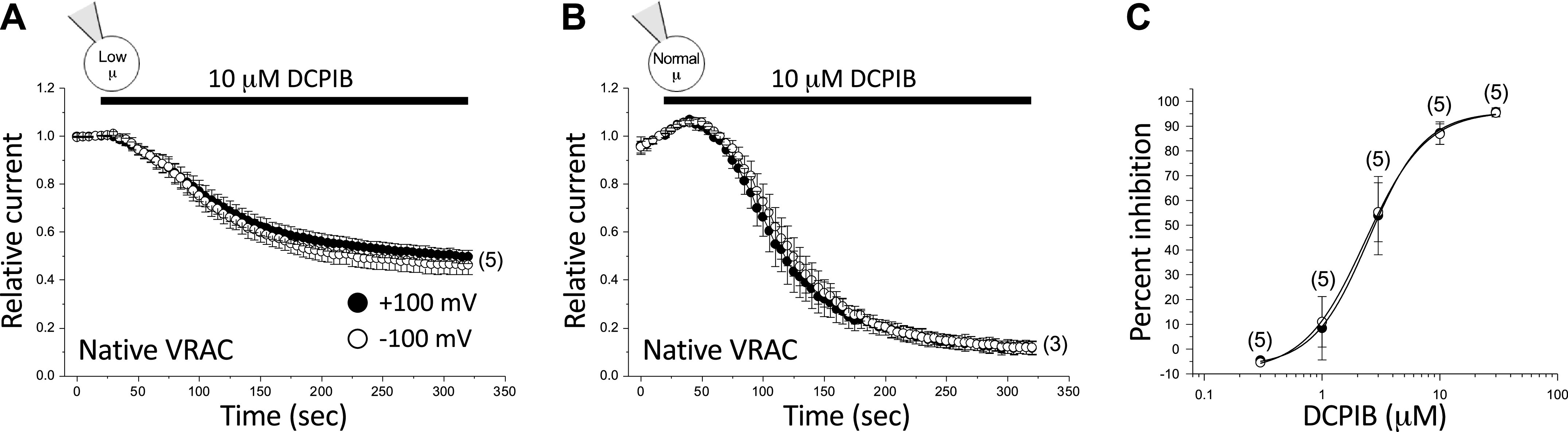
Effect of 10 µM DCPIB on native VRAC current in wild-type HCT116 cells. VRAC current was activated by swelling in cells equilibrated with a low-µ (A) or normal-µ (B) pipette solution. C: dose-response relationships for inhibition of native VRACs by DCPIB at +100 mV and −100 mV. Channels were activated by cell swelling in cells equilibrated with a normal-µ pipette solution. Values are means ± SE. n is shown in parentheses. DCPIB, 4-[(2-butyl-6,7-dichloro-2-cyclopentyl-2,3-dihydro-1-oxo-1H-inden-5-yl)oxy]butanoic acid; VRAC, volume-regulated anion channel.
As shown in Fig. 4, the rate of DCPIB inhibition of native VRAC currents is more rapid when cells were patch clamped with a normal intracellular µ pipette solution (Fig. 4). The mean ± SE initial rates of current inhibition were −60.1 ± 7.5%/min (n = 3) and −20.6 ± 3.4%/min (n = 5) in cells equilibrated with normal-µ and low-µ pipette solutions, respectively (P < 0.001).
The cryo-EM LRRC8A-DCPIB co-structure described by Kern et al. (30) shows that DCPIB blocks the channel pore by what the authors termed a “cork-in-bottle” mechanism. The butanoic acid group of DCPIB appears to interact with the ring of arginine residues at position 103 located on EL1 in the outer pore mouth. The remainder of the DCPIB molecule is extracellular to this region. Importantly, this arginine residue is only present in LRRC8A and LRRC8B. All other paralogs have a leucine or phenylalanine residue at the homologous position.
To assess the validity of this co-structure, we mutated R103 to either leucine or phenylalanine. The R103L and R103F mutations eliminated inhibition by 10 µM DCPIB at +100 mV (the degree of DCPIB inhibition observed was not significantly different from a hypothetical value of 0; P > 0.5) (Fig. 5; Table 2). At −100 mV, inhibition by 10 µM DCPIB was significantly (P < 0.0006) reduced to ∼23% and ∼4% for the R103L and R103F mutations, respectively (Fig. 5; Table 2).
Figure 5.

Effect of 10 µM DCPIB on 8A R103L (A) and 8A R103F (B) mutant homohexameric channels. Values are means ± SE. n is shown in parentheses. DCPIB, 4-[(2-butyl-6,7-dichloro-2-cyclopentyl-2,3-dihydro-1-oxo-1H-inden-5-yl)oxy]butanoic acid.
Data in Fig. 5 support the structural model of DCPIB inhibition proposed by Kern et al. (30). However, given the striking differences in the effect of DCPIB on 8A homohexameric channels (Fig. 3) versus native VRAC currents in HCT116 cells (Fig. 4) and multiple other cell types (40–42), it is unclear how relevant the 8A cryo-EM structure is for assessing VRAC structure-function relationships. To address this issue, we characterized the effect of the R103F mutation on 8A + 8C heteromeric channel activity. Surprisingly, DCPIB inhibition of the 8A + 8C heteromer was strongly affected by intracellular µ. When 8A + 8C heteromeric channels were activated by combined swelling and low intracellular µ, DCPIB inhibition was very weak and voltage dependent (Fig. 6A), resembling that of the 8A homohexamer (Fig. 3). Steady-state inhibition with 10 µM DCPIB was ∼23% at −100 mV. At +100 mV, no statistically significant (P > 0.4) inhibition was detected (Fig. 6A; Table 2). In striking contrast, DCPIB inhibition of swelling-activated 8A + 8C currents in cells equilibrated with a normal-µ pipette solution (Fig. 6B) was voltage independent and resembled that of the native VRAC current in wild-type HCT116 cells (Fig. 4B). Maximal inhibition observed was ∼62% at +100 mV and ∼70% at −100 mV (Fig. 6B; Table 2). These values were not significantly (P > 0.4) different.
Figure 6.

Effect of 10 µM DCPIB on wild-type 8A + 8C (A, B) and 8A R103F + 8C (C, D) mutant heteromeric channels. LRRC8−/− HCT116 cells were transfected with a 1:1 ratio of LRRC8A and LRRC8C cDNAs. Currents were activated by swelling in cells equilibrated with either a low-µ (A and C) or normal-µ (B and D) pipette solution. Values are means ± SE. n is shown in parentheses. DCPIB, 4-[(2-butyl-6,7-dichloro-2-cyclopentyl-2,3-dihydro-1-oxo-1H-inden-5-yl)oxy]butanoic acid.
Figure 6, C and D, shows the effect of the R103F mutation on 8A + 8C heteromeric channels under both normal and low intracellular µ conditions. When cells were equilibrated with a normal-µ pipette solution, the R103F mutation had no effect on DCPIB inhibition (Fig. 6D). At +100 mV and −100 mV, the steady-state inhibition with 10 µM DCPIB was ∼72% and ∼77%, respectively, in 8A R103F + 8C channels (Fig. 6D; Table 2), which was not significantly (P > 0.4) different from that observed with wild-type 8A + 8C heteromers (Fig. 6B; Table 2).
Surprisingly, in cells equilibrated with a low-µ pipette solution, the R103F mutation increased DCPIB inhibition at +100 mV and eliminated voltage dependence (Fig. 6C; Table 2). Maximal inhibition with 10 µM DCPIB was ∼26% and was not significantly (P > 0.9) different at +100 mV and −100 mV. At +100 mV, maximal DCPIB inhibition was significantly (P < 0.03) greater than that observed in wild-type 8A + 8C heteromeric channels (cf. Fig. 6, A and C).
As noted earlier, the R103 residue is located on EL1 connecting transmembrane domains 1 and 2 of LRRC8A (4, 17, 18). Homomeric 8C-8A(IL125) chimeric channels lack R103, which is replaced by a leucine residue located at the homologous position. If R103 is essential for DCPIB inhibition, as suggested by the cryo-EM structure generated by Kern et al. (30) and our 8A homohexamer patch-clamp data (Fig. 5), then 8C-8A(IL125) channels should be poorly blocked by this inhibitor. However, as shown in Fig. 7, 8C-8A(IL125) homomers were rapidly inhibited by DCPIB in a voltage-independent manner. Maximal inhibition observed with 10 µM DCPIB was ∼90% at both −100 mV and +100 mV (Fig. 7A; Table 2) when currents were activated by combined swelling and low intracellular µ. Increased intracellular µ had no obvious effect on DCPIB inhibition of 8C-8A(IL125) homomeric channels (Fig. 7B; Table 2). Estimated IC50 values for DCPIB inhibition were 1.4 µM and 1.7 µM at +100 mV and −100 mV, respectively (Fig. 7C; Table 3). These IC50 values and the degree of inhibition by 10 µM DCPIB are similar to what has been reported for native VRACs in multiple cell types (40–42), including VRAC in wild-type HCT116 cells (Fig. 4B; Table 3).
Figure 7.
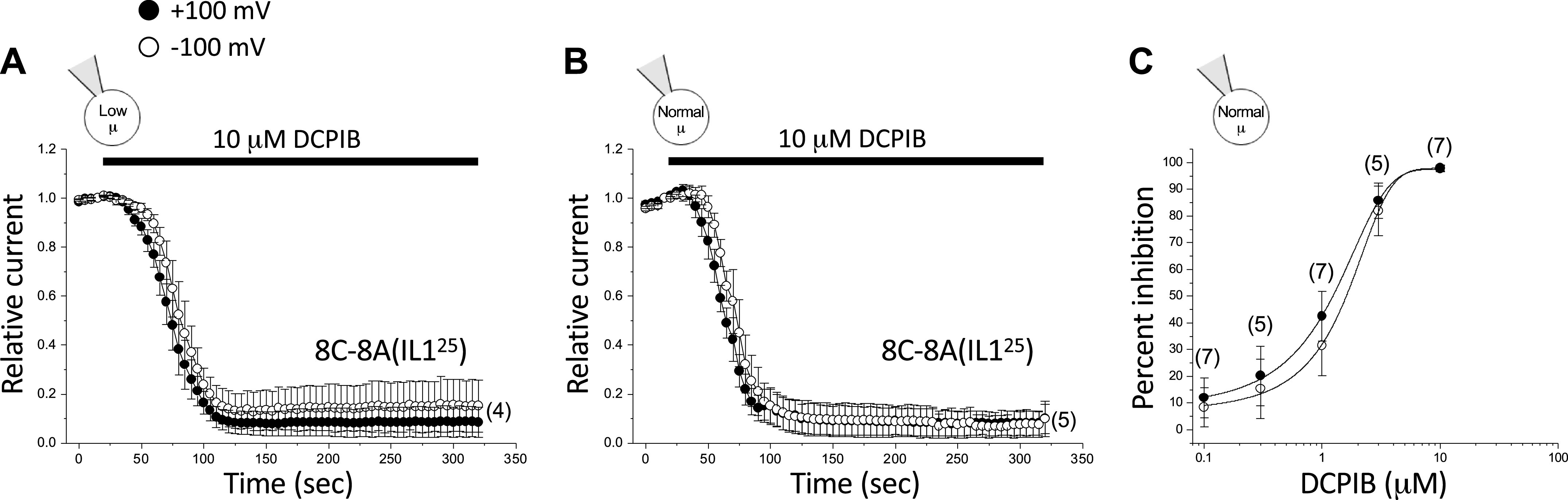
DCPIB inhibition of 8C-8A(IL125) homomeric channels. Effect of 10 µM DCPIB on 8C-8A(IL125) channels activated by cell swelling in the presence of a low-µ (A) and normal-µ (B) pipette solution. C: dose–response relationships for inhibition of 8C-8A(IL125) channels by DCPIB at +100 mV and −100 mV. Channels were activated by swelling in cells equilibrated with a normal-µ pipette solution. Values are means ± SE. n is shown in parentheses. DCPIB, 4-[(2-butyl-6,7-dichloro-2-cyclopentyl-2,3-dihydro-1-oxo-1H-inden-5-yl)oxy]butanoic acid.
DISCUSSION
The generation of cryo-EM structures of LRRC8A (27–30) and LRRC8D (31) homohexameric channels represents a major advancement in the VRAC field. However, using these structures to guide VRAC mutagenesis-based structure-function analysis warrants caution. Unlike native VRACs, LRRC8A homohexameric channels are insensitive to cell swelling in the presence of normal intracellular µ (Figs. 1A and 2) and are only slowly activated by swelling when intracellular µ is reduced (Figs. 1C and 2). Low intracellular µ rapidly and robustly activates native VRACs in the absence of cell swelling [Fig. 2; reviewed in (4)]. In contrast, low intracellular µ slowly and weakly activates LRRC8A channels (Figs. 1B and 2).
The lack of physiologically relevant regulation of LRRC8A homohexamers indicates that these channels adopt a nonnative VRAC configuration that disrupts conformational changes and/or interactions with regulatory partners required for cell volume and intracellular µ sensing. The functional properties of LRRC8D homohexamers are unknown and cannot be readily defined. LRRC8A is the only LRRC8 subunit trafficked to the plasma membrane when expressed alone. All other LRRC8 paralogs including LRRC8D are retained in the ER membrane unless they are co-expressed with LRRC8A (18, 26, 43).
Using a combination of low intracellular µ and cell swelling, we were able to reliably generate sufficient LRRC8A channel activity to test features of its cryo-EM structure using mutagenesis and electrophysiology. Because pharmacological tools are sensitive and widely used probes of ion channel pore structure (36–39), we focused our structure-function analysis on the model of LRRC8A block by DCPIB generated by Kern et al. (30). We found that DCPIB block of LRRC8A bears no resemblance to inhibition of LRRC8/VRACs under normal physiological conditions. DCPIB inhibition of LRRC8A is weak and strongly voltage dependent (Fig. 3). Half-maximal DCPIB concentrations determined by concentration-response data fitted with the Hill equation were 4.0 µM and 15.8 µM at −100 mV and +100 mV, respectively (Table 3). In contrast, DCPIB inhibits native VRACs (Fig. 4; Table 2) (39–41) and the 8C-8A(IL125) chimera (Fig. 7; Table 2) in a voltage-independent manner with IC50 values of 1.4–2.5 µM (Table 3). Heteromeric 8A + 8C LRRC8 channels are also inhibited by DCPIB in a voltage-independent manner when cells are patch clamped using a normal-µ pipette solution (Fig. 6B; Table 2).
Hill coefficients were close to 1 for LRRC8A homohexameric channels, suggesting a single DCPIB binding site (Table 3). This is consistent with the cryo-EM structure described by Kern et al. (30), which shows a single DCPIB molecule blocking the channel pore. In contrast, Hill coefficients for the 8C-8A(IL125) chimera and native VRAC are close to 2, suggesting that multiple DCPIB molecules are required to block these channels (Table 3). These results are similar to those of Friard et al. (42), which demonstrated that DCPIB inhibition of native VRACs in HEK-293 cells has a Hill coefficient of 2.9.
As predicted by the Kern et al. (30) cryo-EM structure, R103 is required for DCPIB inhibition of LRRC8A channels. Mutation of R103 to phenylalanine completely abolishes DCPIB inhibition in LRRC8A homohexamers (Fig. 5B). In contrast, the R103F mutation in 8A + 8C LRRC8 heteromers (Fig. 6, C and D) or its natural absence in the 8C-8A(IL125) chimeric channel (Fig. 7) does not reduce DCPIB inhibition. Taken together, the results shown in Figs. 3–7 and Tables 2 and 3 demonstrate that the LRRC8A homohexameric channel pore adopts a nonnative conformation that limits the utility of the LRRC8A cryo-EM structure for understanding the molecular bases of VRAC selectivity, gating, and regulation.
The concerns over using existing cryo-EM structures for structure-function analysis are further exacerbated by the heteromeric nature of VRACs. Heterologous expression of LRRC8/VRAC channels requires co-assembly of LRRC8A with at least one other LRRC8 protein. Subunit assembly order and stoichiometry are at present unknown and uncontrollable. Existing data suggest that stoichiometry is variable and dependent on subunit expression level (17, 18, 24, 26, 44, 45). It is thus possible, if not likely, that heterologous expression of LRRC8A and another LRRC8 paralog results in the formation of multiple channel types including channels with nonnative configurations. This possibility is supported by our data. Lowering intracellular µ in Lrrc8−/− HCT116 cells heterologously expressing 8A + 8C heteromeric channels induces a voltage dependence to DCPIB block and a striking reduction in DCPIB sensitivity (Fig. 6A; Table 2), which resembles that of LRRC8A homohexamers (Fig. 3; Table 2). In contrast, reduced µ causes only a modest reduction in DCPIB inhibition of native VRACs in wild-type HCT116 cells and has no effect on voltage dependence (Fig. 4A).
The precise molecular mechanisms underlying these effects of µ are unknown. However, the findings suggest that the structure of heterologously expressed 8A + 8C heteromeric channels is abnormally affected by intracellular ionic composition and possibly by other factors such as reactive oxygen species (20) and changes in regulatory signals and the activity of presumptive signaling pathways.
Homomeric LRRC8 chimeric channels exhibit functional properties that closely resemble those of native VRACs (Figs. 2 and 7; Tables 2 and 3) (26). It is noteworthy and interesting that simply replacing IL1 of LRRC8C, LRRC8D, or LRRC8E with the IL1 of LRRC8A gives rise to homomeric channels with physiologically relevant properties (26). Even more noteworthy is that a 25-amino acid sequence unique to the LRRC8A IL1 is sufficient for generating largely normal LRRC8/VRAC activity when it replaces the corresponding region in LRRC8C (Fig. 7; Tables 2 and 3) and LRRC8E (26). As we have discussed previously, in silico analysis of this 25-amino acid sequence indicates that it has characteristics of molecular recognition elements (MoREs) or molecular recognition features (MoRFs) (46–48). MoREs/MoRFs are small stretches of amino acids that mediate protein-protein interactions. It thus seems likely that the LRRC8A IL1 plays a central role in the proper assembly of LRRC8/VRAC heteromeric channels and possibly in their regulation.
To conclude, homohexameric LRRC8A channels exhibit regulation and pore properties that poorly recapitulate those of native VRACs. Structure-function analysis utilizing cryo-EM structures of these channels must therefore be interpreted with caution. Heterologously expressed heteromeric LRRC8 channels can also exhibit nonnative properties under certain conditions and at present have an unknown and likely variable subunit assembly order and stoichiometry. We propose that homomeric LRRC8 chimeric channels provide unique opportunities for generating more physiologically relevant VRAC cryo-EM structures and for conducting structure-function analysis and biochemical studies of channel assembly, pore conformation, gating, and regulation by cell volume changes and intracellular ionic strength.
GRANTS
This work was supported by National Institute of Diabetes, Digestive, and Kidney Diseases Grant R01 DK51610 to K.S.
DISCLOSURES
K.S. is cofounder and CEO of Novo Biosciences, Inc. None of the other authors have any conflicts of interest, financial or otherwise, to disclose.
AUTHOR CONTRIBUTIONS
T.Y., J.S.D., and K.S. conceived and designed research; T.Y. and E.E.F. performed experiments; T.Y., E.E.F., J.S.D., and K.S. analyzed data; T.Y., E.E.F., J.S.D., and K.S. interpreted results of experiments; T.Y., E.E.F., and K.S. prepared figures; K.S. drafted manuscript; T.Y., E.E.F., J.S.D., and K.S. edited and revised manuscript; T.Y., E.E.F., J.S.D., and K.S. approved final version of manuscript.
REFERENCES
- 1.Best L, Brown PD. Studies of the mechanism of activation of the volume-regulated anion channel in rat pancreatic b-cells. J Membr Biol 230: 83–91, 2009. doi: 10.1007/s00232-009-9189-x. [DOI] [PubMed] [Google Scholar]
- 2.Jentsch TJ. VRACs and other ion channels and transporters in the regulation of cell volume and beyond. Nat Rev Mol Cell Biol 17: 293–307, 2016. doi: 10.1038/nrm.2016.29. [DOI] [PubMed] [Google Scholar]
- 3.Nilius B, Prenen J, Voets T, Eggermont J, Droogmans G. Activation of volume-regulated chloride currents by reduction of intracellular ionic strength in bovine endothelial cells. J Physiol 506: 353–361, 1998. doi: 10.1111/j.1469-7793.1998.353bw.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Strange K, Yamada T, Denton JS. A 30-year journey from volume-regulated anion currents to molecular structure of the LRRC8 channel. J Gen Physiol 151: 100–117, 2019. doi: 10.1085/jgp.201812138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cannon CL, Basavappa S, Strange K. Intracellular ionic strength regulates the volume sensitivity of a swelling-activated anion channel. Am J Physiol Cell Physiol 275: C416–C422, 1998. doi: 10.1152/ajpcell.1998.275.2.C416. [DOI] [PubMed] [Google Scholar]
- 6.Emma F, McManus M, Strange K. Intracellular electrolytes regulate the volume set point of the organic osmolyte/anion channel VSOAC. Am J Physiol Cell Physiol 272: C1766–C1775, 1997. doi: 10.1152/ajpcell.1997.272.6.C1766. [DOI] [PubMed] [Google Scholar]
- 7.Jackson PS, Churchwell K, Ballatori N, Boyer JL, Strange K. Swelling-activated anion conductance in skate hepatocytes: regulation by cell Cl− and ATP. Am J Physiol Cell Physiol 270: C57–C66, 1996. doi: 10.1152/ajpcell.1996.270.1.C57. [DOI] [PubMed] [Google Scholar]
- 8.Duan D, Winter C, Cowley S, Hume JR, Horowitz B. Molecular identification of a volume-regulated chloride channel. Nature 390: 417–421, 1997. doi: 10.1038/37151. [DOI] [PubMed] [Google Scholar]
- 9.Gill DR, Hyde SC, Higgins CF, Valverde MA, Mintenig GM, Sepulveda FV. Separation of drug transport and chloride channel functions of the human multidrug resistance P-glycoprotein. Cell 71: 23–32, 1992. doi: 10.1016/0092-8674(92)90263-C. [DOI] [PubMed] [Google Scholar]
- 10.Krapivinsky GB, Ackerman MJ, Gordon EA, Krapivinsky LD, Clapham DE. Molecular characterization of a swelling-induced chloride conductance regulatory protein, pICln. Cell 76: 439–448, 1994. doi: 10.1016/0092-8674(94)90109-0. [DOI] [PubMed] [Google Scholar]
- 11.Paulmichl M, Li Y, Wickman K, Ackerman M, Peralta E, Clapham D. New mammalian chloride channel identified by expression cloning. Nature 356: 238–241, 1992. doi: 10.1038/356238a0. [DOI] [PubMed] [Google Scholar]
- 12.Valverde MA, Díaz M, Sepúlveda FV, Gill DR, Hyde SC, Higgins CF. Volume-regulated chloride channels associated with the human multidrug-resistance P-glycoprotein. Nature 355: 830–833, 1992. doi: 10.1038/355830a0. [DOI] [PubMed] [Google Scholar]
- 13.Li CH, Breton S, Cannon C, Emma F, Sanchez-Olea R, Morrison R, Bear C, Strange K. Recombinant pICln forms highly cation-selective channels when reconstituted into artificial and biological membranes. J Gen Physiol 112: 727–736, 1998. doi: 10.1085/jgp.112.6.727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nilius B, Droogmans G. Amazing chloride channels: an overview. Acta Physiol Scand 177: 119–147, 2003. doi: 10.1046/j.1365-201X.2003.01060.x. [DOI] [PubMed] [Google Scholar]
- 15.Strange K. Molecular identity of the outwardly rectifying, swelling-activated anion channel: time to re-evaluate pICln. J Gen Physiol 111: 617–622, 1998. doi: 10.1085/jgp.111.5.617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wine JJ, Luckie DB. Cell-volume regulation: P-glycoprotein - a cautionary tale. Curr Biol 6: 1410–1412, 1996. doi: 10.1016/S0960-9822(96)00744-0. [DOI] [PubMed] [Google Scholar]
- 17.Qiu Z, Dubin AE, Mathur J, Tu B, Reddy K, Miraglia LJ, Reinhardt J, Orth AP, Patapoutian A. SWELL1, a plasma membrane protein, is an essential component of volume-regulated anion channel. Cell 157: 447–458, 2014. doi: 10.1016/j.cell.2014.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Voss FK, Ullrich F, Munch J, Lazarow K, Lutter D, Mah N, Andrade-Navarro MA, von Kries JP, Stauber T, Jentsch TJ. Identification of LRRC8 heteromers as an essential component of the volume-regulated anion channel VRAC. Science 344: 634–638, 2014. doi: 10.1126/science.1252826. [DOI] [PubMed] [Google Scholar]
- 19.Abascal F, Zardoya R. LRRC8 proteins share a common ancestor with pannexins, and may form hexameric channels involved in cell-cell communication. Bioessays 34: 551–560, 2012. doi: 10.1002/bies.201100173. [DOI] [PubMed] [Google Scholar]
- 20.Gradogna A, Gavazzo P, Boccaccio A, Pusch M. Subunit-dependent oxidative stress sensitivity of LRRC8 volume-regulated anion channels. J Physiol 595: 6719–6733, 2017. doi: 10.1113/JP274795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lutter D, Ullrich F, Lueck JC, Kempa S, Jentsch TJ. Selective transport of neurotransmitters and modulators by distinct volume-regulated LRRC8 anion channels. J Cell Sci 130: 1122–1133, 2017. doi: 10.1242/jcs.196253. [DOI] [PubMed] [Google Scholar]
- 22.Planells-Cases R, Lutter D, Guyader C, Gerhards NM, Ullrich F, Elger DA, Kucukosmanoglu A, Xu G, Voss FK, Reincke SM, Stauber T, Blomen VA, Vis DJ, Wessels LF, Brummelkamp TR, Borst P, Rottenberg S, Jentsch TJ. Subunit composition of VRAC channels determines substrate specificity and cellular resistance to Pt-based anti-cancer drugs. EMBO J 34: 2993–3008, 2015. doi: 10.15252/embj.201592409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schober AL, Wilson CS, Mongin AA. Molecular composition and heterogeneity of the LRRC8-containing swelling-activated osmolyte channels in primary rat astrocytes. J Physiol 595: 6939–6951, 2017. doi: 10.1113/JP275053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Syeda R, Qiu Z, Dubin AE, Murthy SE, Florendo MN, Mason DE, Mathur J, Cahalan SM, Peters EC, Montal M, Patapoutian A. LRRC8 proteins form volume-regulated anion channels that sense ionic strength. Cell 164: 499–511, 2016. doi: 10.1016/j.cell.2015.12.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ullrich F, Reincke SM, Voss FK, Stauber T, Jentsch TJ. Inactivation and anion selectivity of volume-regulated anion channels (VRACs) depend on C-terminal residues of the first extracellular loop. J Biol Chem 291: 17040–17048, 2016. doi: 10.1074/jbc.M116.739342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yamada T, Strange K. Intracellular and extracellular loops of LRRC8 are essential for volume-regulated anion channel function. J Gen Physiol 150: 1003–1015, 2018. doi: 10.1085/jgp.201812016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Deneka D, Sawicka M, Lam AKM, Paulino C, Dutzler R. Structure of a volume-regulated anion channel of the LRRC8 family. Nature 558: 254–259, 2018. doi: 10.1038/s41586-018-0134-y. [DOI] [PubMed] [Google Scholar]
- 28.Kasuya G, Nakane T, Yokoyama T, Jia Y, Inoue M, Watanabe K, Nakamura R, Nishizawa T, Kusakizako T, Tsutsumi A, Yanagisawa H, Dohmae N, Hattori M, Ichijo H, Yan Z, Kikkawa M, Shirouzu M, Ishitani R, Nureki O. Cryo-EM structures of the human volume-regulated anion channel LRRC8. Nat Struct Mol Biol 25: 797–804, 2018. doi: 10.1038/s41594-018-0109-6. [DOI] [PubMed] [Google Scholar]
- 29.Kefauver JM, Saotome K, Dubin AE, Pallesen J, Cottrell CA, Cahalan SM, Qiu Z, Hong G, Crowley CS, Whitwam T, Lee WH, Ward AB, Patapoutian A. Structure of the human volume regulated anion channel. Elife 7: 38461, 2018. doi: 10.7554/eLife.38461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kern DM, Oh S, Hite RK, Brohawn SG. Cryo-EM structures of the DCPIB-inhibited volume-regulated anion channel LRRC8A in lipid nanodiscs. Elife 8: e42636, 2019. doi: 10.7554/eLife.42636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nakamura R, Numata T, Kasuya G, Yokoyama T, Nishizawa T, Kusakizako T, Kato T, Hagino T, Dohmae N, Inoue M, Watanabe K, Ichijo H, Kikkawa M, Shirouzu M, Jentsch TJ, Ishitani R, Okada Y, Nureki O. Cryo-EM structure of the volume-regulated anion channel LRRC8D isoform identifies features important for substrate permeation. Commun Biol 3: 240, 2020. doi: 10.1038/s42003-020-0951-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Deng Z, He Z, Maksaev G, Bitter RM, Rau M, Fitzpatrick JAJ, Yuan P. Cryo-EM structures of the ATP release channel pannexin 1. Nat Struct Mol Biol 27: 373–381, 2020. doi: 10.1038/s41594-020-0401-0. [DOI] [PubMed] [Google Scholar]
- 33.Michalski K, Syrjanen JL, Henze E, Kumpf J, Furukawa H, Kawate T. The Cryo-EM structure of pannexin 1 reveals unique motifs for ion selection and inhibition. Elife 9: e54670, 2020. doi: 10.7554/eLife.54670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Qu R, Dong L, Zhang J, Yu X, Wang L, Zhu S. Cryo-EM structure of human heptameric Pannexin 1 channel. Cell Res 30: 446–448, 2020. doi: 10.1038/s41422-020-0298-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bond SR, Naus CC. RF-Cloning.org: an online tool for the design of restriction-free cloning projects. Nucleic Acids Res 40: W209–W213, 2012. doi: 10.1093/nar/gks396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kharade SV, Sheehan JH, Figueroa EE, Meiler J, Denton JS. Pore polarity and charge determine differential block of Kir1.1 and Kir7.1 potassium channels by small-molecule Inhibitor VU590. Mol Pharmacol 92: 338–346, 2017. doi: 10.1124/mol.117.108472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Linsdell P. Cystic fibrosis transmembrane conductance regulator chloride channel blockers: pharmacological, biophysical and physiological relevance. World J Biol Chem 5: 26–39, 2014. doi: 10.4331/wjbc.v5.i1.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lolicato M, Arrigoni C, Mori T, Sekioka Y, Bryant C, Clark KA, MinorDL, Jr.. K2P2.1 (TREK-1)-activator complexes reveal a cryptic selectivity filter binding site. Nature 547: 364–368, 2017. doi: 10.1038/nature22988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhorov BS, Tikhonov DB. Computational structural pharmacology and toxicology of voltage-gated sodium channels. Curr Top Membr 78: 117–144, 2016. doi: 10.1016/bs.ctm.2015.12.001. [DOI] [PubMed] [Google Scholar]
- 40.Best L, Yates AP, Decher N, Steinmeyer K, Nilius B. Inhibition of glucose-induced electrical activity in rat pancreatic b-cells by DCPIB, a selective inhibitor of volume-sensitive anion currents. Eur J Pharmacol 489: 13–19, 2004. doi: 10.1016/j.ejphar.2004.02.030. [DOI] [PubMed] [Google Scholar]
- 41.Decher N, Lang HJ, Nilius B, Bruggemann A, Busch AE, Steinmeyer K. DCPIB is a novel selective blocker of I(Cl,swell) and prevents swelling-induced shortening of guinea-pig atrial action potential duration. Br J Pharmacol 134: 1467–1479, 2001. doi: 10.1038/sj.bjp.0704413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Friard J, Tauc M, Cougnon M, Compan V, Duranton C, Rubera I. Comparative effects of chloride channel inhibitors on LRRC8/VRAC-mediated chloride conductance. Front Pharmacol 8: 328, 2017. doi: 10.3389/fphar.2017.00328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lee CC, Freinkman E, Sabatini DM, Ploegh HL. The protein synthesis inhibitor blasticidin S enters mammalian cells via leucine-rich repeat-containing protein 8D. J Biol Chem 289: 17124–17131, 2014. doi: 10.1074/jbc.M114.571257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gaitan-Penas H, Gradogna A, Laparra-Cuervo L, Solsona C, Fernandez-Duenas V, Barrallo-Gimeno A, Ciruela F, Lakadamyali M, Pusch M, Estevez R. Investigation of LRRC8-mediated volume-regulated anion currents in Xenopus oocytes. Biophys J 111: 1429–1443, 2016. doi: 10.1016/j.bpj.2016.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yamada T, Wondergem R, Morrison R, Yin VP, Strange K. Leucine-rich repeat containing protein LRRC8A is essential for swelling-activated Cl- currents and embryonic development in zebrafish. Physiol Rep 4: e12940, 2016. doi: 10.14814/phy2.12940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mohan A, Oldfield CJ, Radivojac P, Vacic V, Cortese MS, Dunker AK, Uversky VN. Analysis of molecular recognition features (MoRFs). J Mol Biol 362: 1043–1059, 2006. doi: 10.1016/j.jmb.2006.07.087. [DOI] [PubMed] [Google Scholar]
- 47.Oldfield CJ, Cheng Y, Cortese MS, Romero P, Uversky VN, Dunker AK. Coupled folding and binding with alpha-helix-forming molecular recognition elements. Biochemistry 44: 12454–12470, 2005. doi: 10.1021/bi050736e. [DOI] [PubMed] [Google Scholar]
- 48.Xue B, Dunker AK, Uversky VN. Retro-MoRFs: identifying protein binding sites by normal and reverse alignment and intrinsic disorder prediction. Int J Mol Sci 11: 3725–3747, 2010. doi: 10.3390/ijms11103725. [DOI] [PMC free article] [PubMed] [Google Scholar]