

Abstract
For about half a century, the pharmacology of electroneutral cation-chloride cotransporters has been dominated by a few drugs that are widely used in clinical medicine. Because these diuretic drugs are so good at what they do, there has been little incentive in expanding their pharmacology. The increasing realization that cation-chloride cotransporters are involved in many other key physiological processes and the knowledge that different tissues express homologous proteins with matching transport functions have rekindled interest in drug discovery. This review summarizes the methods available to assess the function of these transporters and describe the multiple efforts that have made to identify new compounds. We describe multiple screens targeting KCC2 function and one screen designed to find compounds that discriminate between NKCC1 and NKCC2. Two of the KCC2 screens identified new inhibitors that are 3–4 orders of magnitude more potent than furosemide. Additional screens identified compounds that purportedly increase cell surface expression of the cotransporter, as well as several FDA-approved drugs that increase KCC2 transcription and expression. The technical details of each screen biased them toward specific processes in the life cycle of the transporter, making these efforts independent and complementary. In addition, each drug discovery effort contributes to our understanding of the biology of the cotransporters.
Keywords: drug screening, ion indicators, K-Cl cotransporter, Na-K-2Cl cotransporter, thallium
INTRODUCTION: THE TARGETS
The era of molecular biology not only provided clarity about the identity of several known transport mechanisms, but also led to the discovery of many novel ion channels and transporters. The cloning era clarified the composition of the electroneutral cation-chloride cotransporter family, with some members already known as functional units and new members discovered by homology cloning (1–6). The importance of cation-chloride cotransporters in physiology is supported by their link to human diseases and the development of genetically modified mouse models (Table 1). One key parameter that defines the functional properties of these transporters is their sensitivity to inhibitors and/or activators. The multiplicity of genes with very similar transport function, but somewhat specialized roles due to gene-specific tissue expression, highlights the need to develop new research tools and therapeutic agents. This review article was written to highlight the physiology and pathology of these transporters, describe the methodologies available to study their function, and describe recent drug discovery efforts.
Table 1.
SLC12 transporters, human diseases, mouse phenotypes, and historical inhibitors
| Gene | Transporter | Disease | Mouse Phenotype | Inhibitor | References |
|---|---|---|---|---|---|
| A1 | NKCC2 ↓ | Bartter | Lethality, polyurea | Furosemide | (7, 8) |
| A2 | NKCC1 ↓ | Deafness, lung and intestinal obstruction, saliva production | Deafness, intestinal obstruction, saliva production, male sterility, etc. | FurosemideBumetanide | (9–14) |
| A3 | NCC ↓NCC ↑ | GitelmanGordon | NaCl wasting, hypokalemia | Thiazides | (15, 16) |
| A4 | KCC1 | None | None | Furosemide | |
| A5 | KCC2 | SeizuresPossibly autism | Lethality, brain hyperexcitability, clonic/tonic seizures | Furosemide | (17–21) |
| A6 | KCC3 ↓KCC3 ↑ | HSMN/ACC | Peripheral neuropathyPeripheral neuropathy | Furosemide | (22–24) |
| A7 | KCC4 | None | Deafness, renal tubular acidosis | Furosemide | (25) |
HSMN/ACC, hereditary sensorimotor neuropathy/agenesis corpus callosum; KCC1–4, K-Cl cotransporter-1–4; NCC, Na-Cl cotransporter; NKCC1, Na-K-2Cl cotransporter-1; NKCC2, Na-K-2Cl cotransporter-2.
The first cation-chloride cotransporter gene is solute carrier family 12 A1 (SLC12A1), which encodes for the Na-K-2Cl cotransporter 2, or NKCC2. This protein is almost exclusively expressed in the thick ascending limb of Henle (hTAL), where it participates in 20%–25% of Na+-reabsorption. Recessive loss-of-function mutations in SLC12A1 result in Bartter syndrome, a salt wasting disorder associated with hypokalemic alkalosis, low blood pressure, and hypercalciuria (7). The NKCC2 knockout mouse closely recapitulates the human phenotype with severe polyuria leading to perinatal lethality. The early death can be minimized with treatment with indomethacin (8), a nonsteroidal anti-inflammatory drug that also helps patients with Bartter syndrome (26). Cotransporter function is reduced or eliminated with furosemide, bumetanide, or analogs, which are collectively referred to as “loop” diuretics.
SLC12A2 encodes a second Na-K-2Cl cotransporter: NKCC1. Compared with NKCC2, which has a highly restricted pattern of expression, NKCC1 is expressed widely throughout the body. It is found in many blood cells, in muscle, heart, brain, intestine, lung, etc. (Fig. 1). It is therefore not surprising that the NKCC1 knockout mouse displays a plethora of phenotypes including deafness and imbalance (inner ear deficit), intestinal obstruction, lack of saliva secretion, pain perception deficit, and male sterility (9, 10, 13, 27–29). As summarized in a recent review, deleterious mutations in NKCC1 have now been found in humans, leading to disorders reminiscent of the phenotypes observed in the genetically modified mouse models (30). These disorders include inner ear deficit, muscle weakness, lung and intestinal obstruction, and abnormal mucus secretion (11, 12, 14, 31). In addition, SLC12A2 is a haplo-insufficient gene, also linked to neurodevelopmental disorders such as intellectual disability and autism (32). In fact bumetanide, which also inhibits NKCC1, is currently tested in clinical trial for autism (33–35). Because inhibition of NKCC2 in the thick ascending limb of Henle results in enhanced K+ secretion in the distal tubule, close attention was being paid to the children’s serum potassium levels and when needed, K+ supplementation was given to prevent severe hypokalemia. The development of an inhibitor discriminating NKCC1 and NKCC2 would alleviate this unwarranted complication.
Figure 1.

Message RNA abundance of the seven cation-chloride cotransporters in tissues. Representative panels showing the abundance of Na-K-2Cl cotransporter-1 (NKCC1), Na-K-2Cl cotransporter-2 (NKCC2), Na-Cl cotransporter (NCC), and K-Cl cotransporter-1–4 (KCC1-4) in 19 selected tissues. As highlighted by the arrows, NKCC2 and NCC are almost exclusively expressed in kidney, whereas KCC2 expression is abundant in the central nervous system. Values are given as Normalized eXpression (NX). Note that NX values for NKCC2 and NCC in kidney far exceed the scale, which is represented by the broken bars. The NX values are given for these two bars. Data were collected and redrawn from the Human protein atlas (https://www.proteinatlas.org/).
SLC12A3 encodes the distal convoluted tubule Na-Cl cotransporter, NCC. Absence of a key tyrosine residue coordinating K+ in NKCC and KCC (36, 37) makes the transport of Na+ and Cl− through NCC independent of K+. Recessive loss-of-function mutations in SLC12A3 result in Gitelman syndrome, another salt wasting disorder associated with hypokalemic alkalosis (16). Several mouse models recapitulate the Gitelman phenotype: the Slc12a3 knockout mouse (15); the knockout of sterile 20-related proline-alanine-rich kinase (SPAK), a kinase that binds, phosphorylates, and activates NCC (38, 39); and the knockout of with-no-lysine kinase 4 (WNK4), a kinase that acts upstream of SPAK (40). Interestingly, gain of NCC function is observed in pseudohypoaldosteronism type II (PHAII) (41), a disorder caused by mutations in several proteins regulating NCC function. These proteins are the WNK4 and WNK1 kinases (41, 42) and culin-3 and Kelsh-like protein 3, proteins part of E3 ligase complex (43, 44). Patients with PHAII have increased blood pressure accompanied by hyperkalemia and metabolic acidosis. This disorder is also mimicked in mouse models leading to increased WNK4 expression (45) or increased SPAK activity (46). NCC is inhibited by thiazide diuretics such as hydrochlorothiazide and metolazone.
SLC12A4 encodes the first Na+-independent K-Cl cotransporter, KCC1 (4). KCC1 shares only 33%–36% amino acid identity with the three Na+-dependent cation chloride cotransporters, which have 75% amino acid identity among themselves. This clearly places this cotransporter in a subfamily apart from NCC, NKCC1, and NKCC2. This gene is expressed in many tissues with highest expression levels in smooth muscle cells and breast tissue (Fig. 1). Function of KCC1 is absent or negligible under isosmotic conditions and is activated under hypotonic, cell swelling, conditions or following N-ethylmaleimide (NEM) treatment (4). The mouse knockout exhibits no overt phenotype, unless another K-Cl cotransporter is also eliminated. Red blood cells lacking KCC1 and KCC3 (see SLC12A6) exhibit increased mean corpuscular volume leading to their increased susceptibility to osmotic lysis (47). Functional redundancy indicates the importance of KCC function in the volume control of red blood cells.
The SLC12A5 gene encodes KCC2, a K-Cl cotransporter mostly restricted to central neurons (Fig. 1). Compared with the other three K-Cl cotransporters, KCC2 exhibits basal transport activity under isosmotic conditions. The transporter serves as the “Cl− pump” driving intracellular Cl− well below its electrochemical potential equilibrium, thereby facilitating γ-aminobutyric acid (GABA)-mediated hyperpolarization and inhibition (48, 49). KCC2 transports Cl− out of the cell using the energy of the large K+ gradient maintained by the Na+/K+ pump. KCC2 readily prevents Cl− loading during sustained GABA release due to rapid and repetitive neuronal activity (50). Consequently, decreased KCC2 function leads to a significant increase in neuronal activity/firing (51). Although the full knockout of KCC2 in mice shows postnatal lethality due to respiratory failure (52), the knockout of the major isoform (KCC2b) survives for 2 wk after birth and demonstrates touch-evoked tonic-clonic seizure-like behavior consistent with brain hyper-excitability (21). In addition, seizures can be observed in older heterozygous animals, compared with their wild-type counterparts. Mutations in human KCC2 have been found in patients with epilepsy (18, 19). The development of the GABA inhibitory system is also critical for the proper development of brain, especially during the perinatal period. Thus, it is not surprising that mutations in KCC2 have also been reported in patients with neurodevelopmental disorders, like autism spectrum disorder (53). KCC2 expression levels are significantly reduced in patients with Rett syndrome (54, 55) and in the methyl CpG binding protein 2 (Mecp2) animal model of Rett syndrome (56). In addition, restoring KCC2 function rescues behavioral deficits in that animal model (57).
SLC12A6 expresses the K-Cl cotransporter-3 or KCC3. Once cloned and mapped to human chromosome 15 (5, 58, 59), it took only two years to link the transporter to a crippling peripheral neuropathy disorder predominant in Quebec (Canada) and to confirm the phenotype through the development of KCC3 knockout mice (22, 23). The disorder, originally known as Andermann syndrome or agenesis of corpus callosum with peripheral neuropathy, is now called hereditary sensorimotor neuropathy with agenesis of corpus callosum or hereditary sensorimotor neuropathy/agenesis corpus callosum (HSMN/ACC). Although the exact mechanism by which dysfunction in KCC3 results in HSMN/ACC, it is clear that it involves neurons (60, 61) and the maintenance of their volume (62, 63). Additional patients have been identified (64), including single copy SLC12A6 variants in patients with peripheral nerve disease (65). In addition, a patient and its mouse model with gain-of-function mutations in KCC3 also suffer from sensorimotor peripheral neuropathy (24). The mutation involves a key phosphorylation residue Thr991, which when substituted to alanine renders the transporter constitutively active.
SLC12A7 encodes for the fourth isoform of K-Cl cotransport, KCC4. The transporter is expressed in many tissues, although at relatively low levels (Fig. 1). The knockout mouse was generated in 2002 and showed early-onset deafness and renal tubular acidosis (25). Immunolocalization of the cotransporter in the inner ear revealed expression at the base of outer hair cells and in supporting Deiter’s cells. The transport of K+ at the base of hair cells is critical for K+ recycling and the maintenance of the endochoclear potential. In the kidney, the cotransporter is localized to the basolateral membrane of intercalated cells of type A, or acid producing cells. Absence of KCC4 leads to accumulation of intracellular Cl− resulting in inhibition of acid secretion and an alkaline urine (25). To date, there are no human disease-associated mutations in SLC12A7.
TECHNIQUES TO MEASURE CATION-CHLORIDE COTRANSPORTER FUNCTION
Direct Measurement of Ion Transport
Radioisotopes.
Isotopes have been extensively used in biology and physiology since the 1950s. Relevant isotopes for cation-chloride cotransporters are those tracing Na+, K+, and Cl− transport. Note that a limited number of ionic species can substitute for the natural substrates of the cotransporters. On the cationic side, the Na+ binding site is rather selective, compared with the more promiscuous K+ binding site. Indeed, Na+ can hardly be replaced by Li+. This is demonstrated in Fig. 2A, where K+ influx through NKCC1 occurs in Li+ at a rate that is 26% of the rate in Na+. The consensus in the field is that K+ can readily be substituted by Rb+ with little to no effect on transport activity. As seen in Fig. 2, B and C, Tl+ is also transported through NKCC1 and KCC2, although at about half the rate of K+. At this point, it is unclear whether Tl+ can also substitute for Na+, as the cation utilizes the K+ binding site. Interestingly for neuroscientists using Cs+ in their perforated patch pipette solution, the heavier cation moves easily through KCC2 as seen in Fig. 2D. This finding is a bit surprising, as the ionic radius of Cs+ is substantially greater than that of K+ and the coordination site in the cryo-EM structure seem to be rather tight (Fig. 3). On the anionic side, it is known that Br− is slightly better transported than Cl− by the sheep red blood cell K-Cl cotransporter (66). In contrast, as indicated in Fig. 2E, K+ flux by NKCC1 in the presence of Br− is reduced, compared with Cl−. Relevant for those thinking about using I− as strong interactors of halide sensitive dyes, this anion is poorly transported by the Na-K-2Cl cotransporter (Fig. 2E).
Figure 2.

Substitution of cations and anions as substrates for Na-K-2Cl cotransporter-1 (NKCC1) and K-Cl cotransporter-2 (KCC2). A: bumetanide-sensitive K+ influx in Xenopus laevis oocytes injected with mouse NKCC1 cRNA and incubated in either 96 mM NaCl, 96 mM LiCl, or 96 mM N-methyl glucamine (NMDG)Cl. All solutions had 4 mM KCl and 5 µCi/ml 86Rb. B: Xenopus oocytes injected with mouse NKCC1 cRNA were fluxed either in 96 mM NaCl with either 2 mM K2(SO4) or 2 mM Tl2(SO4) and 5 µCi/mL 86Rb. C: HEK293 cells overexpressing rat KCC2 were fluxed in 96 mM NMDGCl (Na+-free solution) with either 2 mM K2(SO4) or 2 mM Tl2(SO4) and 1 µCi/mL 86Rb. D: Xenopus oocytes injected with rat KCC2 cRNA were fluxed either in 96 mM NaCl with 1 and 8 mM KCl or CsCl and 5 µCi/mL 86Rb. E: X. laevis oocytes injected with mouse NKCC1 cRNA were fluxed in 96 mM NaCl, 96 mM NaBr, or 96 mM NaI. All solutions had 4 mM KNO3 and 5 µCi/mL 86Rb. For oocytes, bars represent means ± SEM (n = 20-25 oocytes). For HEK293 cells, bars represent means ± SEM (n = 3 dishes).
Figure 3.
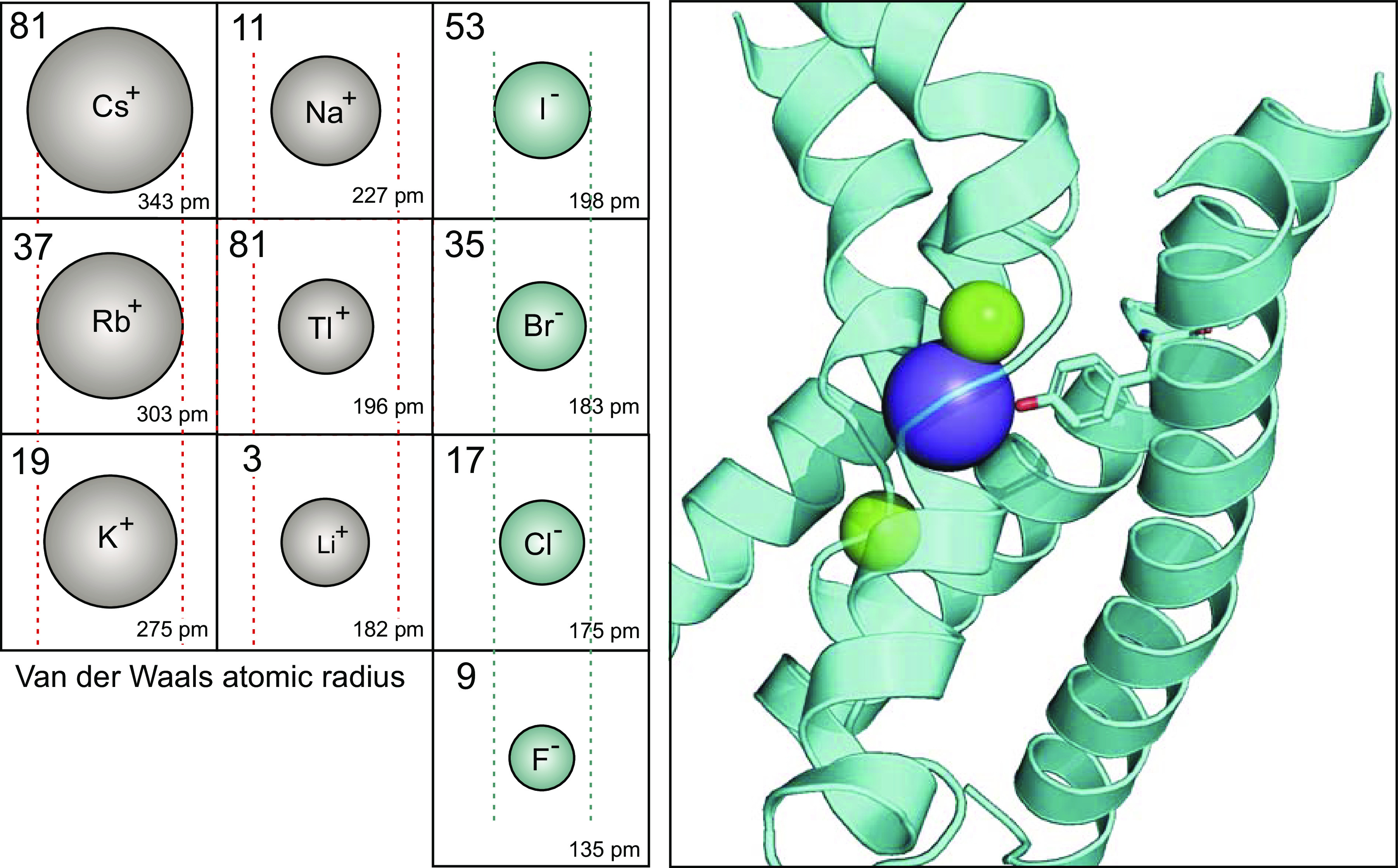
Atomic sizes of monovalent cations and anions. Left: for each ion, the atomic number is located in the top left corner, while the atomic radius (Van der Waals) in picometer (pm, or 10−12 m) is placed in the bottom right corner. The size of the drawn ion is proportional to its atomic radius. Lines (red) are placed around the size of the Rb+ ion, which is transported by Na-K-2Cl and K-Cl cotransporters. Cs+ is much larger, whereas K+ has an equivalent size. Na+, Tl+, and Li+ are much smaller. Note that cations are alkali metals (column 1 in periodic table), except for Thallium, which is a post-transition metal (column 13 in periodic table). The anions are generally smaller with I− of a size similar to Na+. Br− and Cl− are slightly smaller and Fl− is much smaller than I−. The atomic radius values were obtained from the Los Alamos National Laboratories (https://periodic.lanl.gov/list.shtml). Right: ribbon structure of portions of TM1, TM3, and TM6 showing the location of ions in the cryo-EM structure of hKCC1, PDB ID: 6KKT (37). K+ is in blue, whereas Cl− ions are in green. The tyrosine residue located in TM3 which coordinate K+ binding is drawn as stick.
Sodium.
A short-lived sodium isotope (24Na) was first used to follow Na+ in animal and human tissues (67–69), including urinary Na+ excretion (70). The isotope was also used to trace Na+ movement through the Na+/K+ pump (71, 72), and the Na-K-2Cl cotransporter (73). Its use was rapidly replaced by 22Na which has a much longer half-life (Table 2). This specific isotope has been used to trace Na+ movement through the Na+/H+ exchanger (74), the Na-K-2Cl cotransporter (75), the Na-Cl cotransporter (76), and the Na-HCO3 cotransporter (77).
Table 2.
Radioactive isotopes of Na+, K+ (Rb+), and Cl
| Isotope | Atomic No. | Half-Life | Decay Mode | Gamma Intensity |
|---|---|---|---|---|
| 24Na | 11 | 15 h | β− (1.39 MeV) | Yes |
| 22Na | 11 | 2.58 yr | β+ (89%, 0.54 MeV), ε (11%) | Yes |
| 42K | 19 | 12.4 h | β− (82%, 3.53 MeV) (18%, 2.03 MeV) | Low |
| 43K | 19 | 22.4 h | β− (0.83 MeV) | Yes |
| 83Rb | 37 | 83 days | ε (0.9 MeV) | Yes |
| 86Rb | 37 | 18.77 days | β− (1.77 MeV) | Low |
| 36Cl | 17 | 3 × 105 yr | β− (98%, 0.71 MeV), ε (2%) | No |
Atomic number or number of protons in the nucleus of the atom. Half-life is the rate of radioactive decay or time it takes for activity to be reduced by ½. Decay mode: β−, negative beta emission or energetic electron; β+ positron emission, ε, orbital electron capture or absorption of an inner atomic electron. Energy of decay is given in parenthesis. Where more than one decay exists, separate energy values and distribution are given. The last column indicates whether gamma ray energy is released.
Potassium.
Isotopes of potassium (42K and 43K) were originally used to label erythrocytes (78), trace the movement of the cation from blood to cerebrospinal fluid (79), measure the K+ permeability in cortical segments of the nephron (80), study the distribution of body K+ (81), or examine the stoichiometry of the sodium pump (71). Because of their short half-lives (12.4 h for 42K and 22.4 h for 43K), the use of these isotopes was cumbersome, as radioactive decay needed to be included in all flux calculations. Replacement of radioactive potassium isotopes with 86Rb, which has a half-life of 18.6 days (Table 2), greatly facilitated these experiments. Although it was shown that 86Rb had its own characteristic behavior and was not suitable for tracing potassium throughout the body (82), it is wildly used to trace the transmembrane movement of K+ through pumps, channels, and transporters (83–90). Unfortunately, Perkin Elmer discontinued the production and sale of 86Rb in 2019. Since then, our laboratory has been using 83Rb, an isotope with a half-life of 83 days (see Table 2). The isotope is produced by the Brookhaven National Laboratory and available through Oak Ridge National Laboratory.
Chloride.
In 1964, Goodford (91) used 36Cl to confirm that smooth muscle cells of the guinea pig colon had a relatively high Cl− content. The isotope was also used to trace Cl− movement during intestinal fluid secretion (92), mammary epithelium milk secretion (93), and chloride transport in erythrocytes (94). As for Na+ and K+ isotopes, 36Cl has been extensively used as a tracer to measure the unidirectional movement of the halide through transport proteins. For transporters that carry multiple ions, e.g., Na-K-2Cl, Na-Cl, and K-Cl cotransporters, 36Cl is useful to measure stoichiometry of transport. However, because the specific activity of 36Cl is so much lower than that of 86Rb (due to the high Cl− concentration vs. low K+ concentrations in external solutions), the use of the 86Rb is preferred. All radioactive isotopes relevant to cation-chloride cotransporters are listed in Fig. 4.
Figure 4.

Methods to assay the transport of Na+, K+, Cl− through Na-Cl cotransporter (NCC), Na-K-2Cl cotransporter (NKCC), and K-Cl cotransporter (KCC). Isotopes of Na+, Cl−, K+, and Rb+ are used to trace the transport of ions through the cotransporters. Isotopes of Rb+ are used to trace K+ movement. Nonradioactive Rb+ (85Rb+) can also be used as a tracer as the cold isotope is not naturally present in biological fluids. It can be detected using flame or atomic absorption spectrometry. Note that tissue/cell Na+ and K+ contents can also be determined by flame or atomic absorption spectrometry. Fluorescent indicators are also widely used. Indicators sensitive to Tl+ (another non-biological cation mimicking K+), Na+, K+, and Cl− are listed.
Atomic absorption spectrometry.
Adaptation of atomic absorption spectrometry to biological samples was originally made by Walsh in 1955 (95). The concentration of specific cations in a solution can be determined by first vaporizing (atomizing) the sample via a flame or an electrothermal graphite tube, then emitting light through the gas sample to measure its absorption by specific elements/atoms. Atomic absorption measures the amount of energy absorbed by a sample and this amount at discrete wavelengths (ion signature) is proportional to the concentration. The method was first used to measure amounts of calcium and magnesium in biological samples (96, 97). The movement of ions such as Na+ and K+ through biological membranes was also followed using atomic absorption spectrometry (98, 99). Nonradioactive Rb+ is also used as a tracer for K+ movement across membranes via quantitation using atomic absorption spectrometry (100, 101). The cold rubidium tracing method is preferred by those who cannot use radioactive materials. Its use will likely grow, as radioisotopes of Rb+ are increasingly difficult to obtain.
Fluorescent Indicators/Dyes
Sodium dyes.
The intracellular Na+ content of cells or tissues has been first determined using a variety of methods, including X-ray microanalysis, isotope equilibration methods, and flame absorption spectrometry. Although X-ray microanalysis was useful in comparing Na+ content from tissue to tissue and, for example, determine that tumor cells had a significantly higher Na+ content than normal cells (102), the reported values are expressed as a function of dry weight and not concentrations. Other methods like flame/atomic absorption spectrometry also require the knowledge of the water content of a cell/tissue to calculate its ion concentration. It also requires the knowledge of the extracellular fluid contribution, as the cation is abundant in the extracellular space. Determination of extracellular space contribution is typically made using membrane impermeant markers such as radiolabeled sucrose or inulin (103). Na+ concentrations in cells ranges from 5 mM to 15 mM (103), but local Na+ concentrations can transiently reach 40 mM in neuronal dendrites (104).
Multiple compounds that change fluorescence intensity with changes in intracellular Na+ have been developed (Fig. 4). They are SBFI (a benzofuranyl fluorophore linked to a crown ether chelator) (104, 105), CoroNa green (106), sodium green (107), and Ansante Natrium Green-2 or ANG-2 (108, 109). Although Na+ dyes are not completely insensitive to K+ and are often sensitive to changes in intracellular pH, ionic strength, and viscosity, they are useful in following changes in intracellular Na+. Note that unlike unidirectional ion flux measurements, a change in intracellular Na+ concentration due to Na+ transport is an indirect measurement that reflects net effect of transport in and out of the cell. Although the development of these new dyes is significant, they have not yet found their way into drug development.
Potassium dyes.
K+ dyes were created by modifying the chemistry of existing Na+ dyes. PBFI and Ansante Potassium Green (APG-2, APG4) were developed for greater sensitivity to K+ versus Na+. Although the APG4 dye has improved K+ selectivity over previous dyes, caution is required since the fluorescence is affected by intracellular proteins (molecular crowding) and cell volume (110).
Chloride dyes.
Both chemical dyes and genetically encoded [protein] dyes have been developed to survey Cl− in cells and tissues (Fig. 4). The first chemical dyes developed for measuring changes in intracellular Cl− were heterocyclic organic compounds containing a quaternary nitrogen group (111). The compounds are SPQ [6-methoxy-N-(3-sulfopropyl)quinolinium] (112), MQAE [N-(ethoxycarbonylmethyl)-6-methoxyquinolinium bromide] (113), and diH-MEQ [6-methoxy-N-ethylquinolinium iodide] (114). Dyes based on different scaffolds and with brighter fluorescence were also developed (114). One significant disadvantage of Cl− indicators is the fact that they are quenching indicators, meaning that their fluorescence decreases with increased concentration of the halide. These dyes have been used extensively before the appearance of yellow fluorescent protein (YFP)-based Cl− indicators (115). Note that generally, iodine produces stronger quenching and is often used as a substitute for Cl− in ion transport studies involving Cl− indicators. This is the case for studies of CFTR, which seem to readily transport iodine (116, 117). As indicated in the Radioisotopes section, however, the alternate halide is not as useful with cation-chloride cotransporters as iodine is a poor substrate for these transporters (Fig. 2E).
Genetically encoded Cl− dyes are based on yellow fluorescent protein and the cooperative binding of halide ions near the chromophore (118). Fluorescence decreases significantly as Cl− is raised from 0 mM to 150 mM. Clomeleon was engineered as the first ratiometric indicator for Cl− as it is a fusion protein of YFP-cyan fluorescent protein (CFP), with YFP being sensitive and CFP insensitive to the anion (119). Its KD for Cl− is 160 mM. Using cell-free protein engineering methods, mutations were made in the protein to significantly enhance the KD for Cl− without sacrificing the Förster Resonance Energy Transfer (FRET) properties. This effort led to the development of a new Superclomeleon protein with a brighter CFP, a shorter linker (enhancing FRET), and YFP mutations (Q69T/V153A) that reduce the KD for Cl− to ∼20 mM, which is far more compatible with physiological concentrations (120). A different fusion protein made use of an enhanced T203Y mutant YFP-fused to DsRed, allowing the simultaneous measurement of chloride and pH. The KD for Cl− was determined to be 13 mM, also within the physiological range (121). The sensor was used to assess intracellular Cl− in Drosophila renal epithelial cells (122) and SLC26A3 expressing Chinese hamster ovary cells (CHO) cells (123).
Thallium dyes.
(Tl+) is an interesting element and important in the context of high throughput screening (HTS). It is a metalloid element located with boron and aluminum in the IIIa (13th) column in the periodic table. Its atomic weight is 81 (Fig. 2). Its most common oxidation state is +1 making it “similar” to alkali metals. As indicated in Fig. 2, the atomic radius of Tl+ is 196 pm, which is a bit smaller than Na+ (227 pm), K+ (275 pm), and Rb+ (303 pm). Thus, Tl+ can replace both sodium and potassium ions in the glutamate transporter excitatory amino acid-1 (124); potassium in K+ channels (125) and Na-K-2Cl and K-Cl cotransporters (126, 127). Note that a smaller size cation does not necessarily translate into more effective transport. For NKCC1 and KCC2, the transport of Tl+ was reduced by ∼50% compared with K+, indicating that the natural cation is a much better substrate.
As it is the case for the transport of isotopes (see Radioisotopes section), the movement and detection of Tl+ directly report the transport of the cation across the plasma membrane. Thus, the use of Tl+ fluorescence indicators is quite different from the use of dyes that monitor the intracellular concentration of an ion. The first Tl+ indicator described was the coumarin benzothiazole-based Ca2+ indicator, BTC (125). The dye is conjugated to an acetoxymethyl (AM) group to facilitate its loading into cells. Because of its relatively low affinity for divalent cation, calcium-dependent changes in fluorescence are minimal when compared with the magnitude of the Tl+-dependent effects. In addition, Ca2+ binding decreases BTC fluorescence, whereas Tl+ binding increases it. The dye generates a strong positive signal upon interaction following Tl+ uptake into the cells. Other thallium-sensitive dyes are the zinc indicators fluozin-2 and 3 (127, 128), and Thallos, a fluorescein moiety coupled to an amino dicarboxylic acid metal binding unit (129, 130).
PHARMACOLOGY OF CATION-CHLORIDE COTRANSPORTER INHIBITION: A HISTORICAL VIEW
The pharmacology of cation-chloride cotransporters started with the clinical need for diuretics, and this was long before their molecular targets were identified. In conditions of decreased cardiac function such as in congestive heart failure, blood volume accumulates causing peripheral edema or abnormal accumulation of fluid in tissue spaces. This problem is typically treated first with beta-blockers (e.g., atenolol), angiotensin-converting enzyme (ACE) inhibitors (e.g., Lisinopril), or angiotensin-II receptor blockers (e.g., losartan), and second diuretics to eliminate the extra volume. The first agent used to induce diuresis was mercury (131), an element which use traces back to ancient times. In 1553, Paracelsus (Swiss physician, alchemist, and philosopher) recorded that mercuric chloride was a cure for dropsy (edema). This was followed by less toxic forms of mercury such as mersalyl (132). Possible sites of action of mercury are the distal tubule Na-K-2Cl and Na-Cl cotransporters (76), which have two highly conserved neighboring cysteine residues in the extracellular loop connecting TM7 and TM8. In the zebrafish cryo-EM structure of NKCC1, Cys496 and Cys507 form a disulfydral bridge, which likely stabilizes the cotransporter structure. Mercurial compounds could interfere with this bridge and inhibit cotransporter function.
Thiazides were the first non-mercurial compounds used in the clinic to induce diuresis (133). They were shortly followed by frusemide (134), now called furosemide, which has some chemical similarity with hydrochlorothiazide (Fig. 5). Interestingly, frusemide at its maximal dose produced more than twice the sodium excretion than any of the benzothiadiazine compounds, which at the time of discovery indicated a different mechanism of action. Bumetanide (3-(butylamino)-4-phenoxy-5-sulfamoylbenzoic acid) then came to clinical medicine in 1972 as a diuretic with higher potency than furosemide. A study published in the British Medical Journal reported its use in 27 edematous patients (135). The dosage of bumetanide required for similar effect was 1/40 the dosage of frusemide. The efficacy of furosemide as cornerstone loop diuretic is attributable in large measure to its active secretion by the proximal tubule organic anion transporters OAT1 and OAT3 (136). The resulting concentration of furosemide in the tubular fluid and the absence of albumin binding (90+% in plasma) in the tubular fluid greatly favors inhibition of TALH NKCC2 over systemic NKCC1.
Figure 5.

Two classes of diuretics (thiazide and loop) share structure similarities. Hydrochlorothiazide and furosemide have conserved phenol rings with a Cl− ion and a sulfonamide group. Furosemide and bumetanide share the sulfamoylbenzoic acid but the Cl− ion in furosemide is replaced with a phenoxy group. Conserved groups in structures are highlighted by the gray background.
Today, we now know that thiazide diuretics inhibits the Na-Cl cotransporter expressed in the distal convoluted tubule of the kidney, whereas furosemide and bumetanide target the Na-K-2Cl cotransporter expressed in the thick ascending limb of Henle (137, 138). As seen in Fig. 4, there are significant chemical similarities between chlorothiazide, furosemide, and bumetanide. Piretanide and torasemide are additional compounds with inhibitory effect on NKCC function. Ethacrynic acid, which was synthesized to mimic the sulfydryl reactivity of mercurial compounds, is also an inhibitor of Na-K-2Cl cotransport and is also used to treat fluid retention (139).
Furosemide was also key in the discovery of the K-Cl cotransport in sheep and human red blood cells (99, 140). Indeed, the cotransporter was originally defined as a NEM or cell swelling activatable furosemide-sensitive and Cl−-dependent K+ flux. The loop diuretic has, however, a much weaker effect on K-Cl cotransport, as it works 2–3 order of magnitude less efficiently than for inhibition of Na-K-2Cl cotransport. Interestingly, the potency of furosemides depends upon the level of extracellular K+. In the absence of external K+ (or Rb+), the EC50 for furosemide inhibition of K+ efflux in LK sheep red blood cells was greater than 1 mM, but in the presence of 23 mM external Rb+, the EC50 dropped to 50 µM (141). In addition, there was no furosemide effect in the absence of external Cl−, indicating that anion binding is also required for furosemide inhibition. This observation is consistent with an ordered model of binding where Cl− binds first followed by K+ (142). When Cl− is bound, furosemide can bind and affect both the Vmax and the apparent affinity for K+/Rb+ (141).
In toadfish, duck, and sheep red blood cells, 2,2′diisothiocyanatostilbene-2,2′ disulfonic acid (DIDS) or its derivative H2-DIDS strongly inhibits K-Cl cotransport (143). As with furosemide, DIDS inhibition required external Cl− and was enhanced by the presence of external K+. Non-competitive inhibition of K+ influx was observed for DIDS versus external K+ or Cl− concentrations. The effect of DIDS on red blood cell K-Cl cotransport might be species-specific, as human red blood cell K-Cl cotransporter activity was unaffected by the stilbene derivative (144).
WHAT ARE THE COMPOUNDS TRULY TARGETING?
There are no “true” potentiators or positive allosteric modulators of cation-chloride cotransporters. For K-Cl cotransport, the closest molecule to function as a potentiator is N-ethylmaleimide (NEM), which activates K-Cl cotransport-mediated K+ influx in LK sheep red blood cells by fourfold (101). Because Na-K-2Cl cotransport is otherwise inhibited by NEM (145, 146), it is currently believed that the thiol alkylating agent acts on a kinase inhibiting K-Cl cotransport and activating Na-K-2Cl cotransport. Work in the past 20 years has identified the WNK/SPAK kinase pathways as direct regulators of NKCC and KCC function (147–151). In 2006, we showed that NEM readily inhibits SPAK kinase activity using in vitro phosphorylation assays (152).
The NEM effect establishes that compounds can indirectly affect the function of a transport protein. The overall transport at the cell level depends upon the amount of protein expressed at the cell surface. For ion channel proteins, the current flowing through the membrane is typically defined by N × Po, where N is the total number of functional channels expressed at the plasma membrane, while Po is their open probability. For carriers, an equivalent parameter is Vmax or the maximum rate of transport. Expression of a protein at the cell surface involves many steps, including transcription/translation, forward trafficking, membrane residence time and internalization, degradation, and/or recycling. In addition to “N,” transport activity is also affected by post-translational modification and the channels or transporters at the membrane can be activated/deactivated by cellular processes. For the purpose of drug discovery, it is important to remember that all these steps are possible targets for manipulation, and the design of an experiment can favor one step over the other.
HIGH THROUGHPUT DRUG DISCOVERY SCREENING ACTIVITIES
First, recognition should be made to the original efforts of Hoechst AG, the German chemical and life-sciences company, for creating and testing in the 1950s and 1960s countless compounds that led to the discovery of chlorothiazide, furosemide, and bumetanide, drugs that are among the most utilized drugs in clinical medicine. It then took 30–40 years for Academia to be attracted to the development of new compounds. Drug discovery efforts were facilitated and justified by the cloning and molecular identification of the transporters, the study of their more diverse biological functions, and the need to discriminate pharmacologically between these transporters.
From West Point, Pennsylvania to Paris, France
In the early 1980s, the Merck Sharp & Dohme Research Laboratories in West Point, Pennsylvania teamed with a group of neurosurgeons at Albany Medical College to develop drugs that prevent the swelling associated with brain injury. They performed chemical modification around ethacrynic acid and developed a series of (aryloxy) alkanoic acid compounds, some of which had significant beneficial effect following brain injury. Among them, (indanyloxy) alkaloid acids were devoid of diuretic activity (153, 154). In collaboration with a group in Paris, they tested a subset of these compounds on N-ethylmaleimide stimulated K+ efflux in red blood cells and found four [(dihydroindenyl)oxy]alkanoic acids with potent inhibitory activity. One compound, called DIOA for simplicity, demonstrated an EC50 of 10 µM, without effect on NKCC1 (155). The compound was also active in inhibiting swelling-activated K+ efflux. It was noted that high concentrations of DIOA induced an increase in nonspecific membrane leak of K+, indicating some toxicity.
Drug Discovery at Vanderbilt University (Nashville, TN)
In 2009, we published the first HTS effort in developing modulators of KCC2 function (127). At the time, inhibitors were furosemide with an EC50 of 0.6 mM, and DIOA with an EC50 of 1 µM, and as activator was N-ethylmaleimide, which was effective at a dose of 1–2 mM. For our assay development, we used HEK293 cells transfected with the rat KCC2 cDNA (6) inserted into pIRES_puro2. Following puromycin selection, multiple individual clones were picked, expanded, and tested functionally using Tl+-induced fluozin-2 fluorescence increase. Expression of KCC2 was also confirmed by Western blot analysis. One of the clones was selected and used for the entire screen. The assay used an isosmotic solution containing 0.2 mM ouabain to prevent Tl+ uptake through the Na+/K+ pump. Compounds were preincubated for a period of 8 min before the addition of the Tl+-containing solution. The relatively short preincubation time did preclude any long-term effect of the drug. The fluorescence signal acquisition time that followed the addition of Tl+ was 5–6 min. A library of 234,560 compounds was screened yielding 4,933 primary hits (2%). Interestingly, a larger number of compounds increased (2,954) rather than decreased (1,880) the fluorescence signal. A secondary screen was done comparing KCC2-expressing HEK293 cells versus wild-type HEK293 cell and 465 inhibitory compounds showed specificity to KCC2-expressing cells. They were then re-tested in triplicate with 11-point concentration response curves and a total of 26 inhibitory compounds were selected. VU0240551 (D4) was identified as the most potent inhibitor with an IC50 of 560 nM. Unfortunately, as far as the activating compounds are concerned, the follow-up screens were unsuccessful to identify specific “agonist” of KCC2 (127).
Chemical modifications of the lead inhibitory compound were instructive as they pointed out to key chemical features of VU0240551. First, the position of the methyl group on the thiazol was critical as the compound was active with the methyl at position 4 (4-methyl thiazole), whereas completely inactive at position 5 (5-methyl thiazole). Second, the secondary amide could be replaced by a tertiary N-Me amide (VU0255011, ML077) with similar potency, suggesting additional possible modifications at this site. In fact, a follow-up chemical optimization study showed that replacing the methyl group of the tertiary amide by a cyclopropyl group, increased potency by 10-fold (156). This improved compound is VU0463271. The study also examined the pyridazine group and demonstrated intolerance to its replacement by pyridines, pyrazines, pyrimidines, and thiadiazoles (156). The KCC-specific inhibitors VU0240551 and VU0463271 are now widely used to study KCC2 function. Just in the past few months, we identified five studies making use of these inhibitors (157–161).
Drug Discovery at Union Chimique Belge, Braine L’Alleud, Belgium
Human liver adenocarcinoma SK-Hep cells were transfected with rat KCC2 and a clone was selected using 86Rb uptake (162). A screen was performed by pretreating these cells with NEM to activate the cotransporter and then exposing them to 5 mM RbCl for 10 min. Uptake of Rb+ was assayed using atomic absorption spectrometry. Because of the maximal activation of the cotransporter, this screen was conceptually biased to find inhibitors. Benzyl 1-acetyl-2-benzylprolinate (+)-2 was identified as an inhibitor with an EC50 of 350 nM. Structure activity relationship (SAR) efforts led to the identification of more potent inhibitors, but with increased lipophilicity. Conversely, SAR also identified a compound with slightly (two- to threefold) reduced potency over the original compound, but with decreased lipophilicity and much better pharmacokinetic properties. All compounds tested had some minimal inhibitory effect on NKCC1 function (162). Thus, this family of compounds represents a separate class of KCC inhibitors.
Search for KCC2 Agonists (Montreal, QC, Canada)
In 2013, Yves Dekoninck published the first screen that used measurements of intracellular Cl− levels as a readout for KCC2 activity (163). The cells utilized in this case were NG108-15 cells, a Mus musculus neuroblastoma/Rattus norvegicus glioma cell hybrid that natively expresses low levels of KCC2 expression. This low KCC2 level mimics the conditions of neuropathic pain and thus the screen was intended to identify compounds that increase KCC2 function. To assess KCC2 function, they transfected the chloride indicator: Clomeleon. They screened a library of 92,500 compounds and identified 78 positive hits. Secondary screens were done with cells that express NKCC1, KCC1, KCC2, and KCC4. They then focused on CL-058, (5Z)-5-[(2-hydroxyphenyl)methylidene]-2-piperidin-1-yl-1,3-thiazol-4-one, a three-ring compound with a middle thiazol group. After synthesis of 300 analogs, they identified a lead compound (CLP257) which had an added Fl− ion to the hydroxyphenyl group and that replaced the pyridine group with a pyridazine. The substitution increased the potency of the compound from an EC50 of 31.5 mM to 50 nN. Importantly, the effect seemed to be specific to KCC2 as Rb+ flux assays performed in Xenopus laevis oocytes expressing NKCC1, KCC1, KCC3, and KCC4 failed to demonstrate increased transport. Experiments were done at appropriate osmolarities, as KCC1, 3, and 4 require hypotonic swelling for activation. Effect of the compound on the GABA receptor was also tested using heterologous expression of receptor subunits in CHO cells.
CLP257 was shown to significantly increase the rate of Cl− accumulation upon elevation of extracellular K+, in two models of pain hypersensitivity. In these experiments, because freshly isolated spinal neurons were used to assay Cl−, the permeable MQAE Cl− indicator was used. Cl− load was imposed onto the neurons by delivery through the recording pipette. The neurons were from spinal cords treated with brain-derived neurotrophic factor (BDNF) or isolated from rats exposed to peripheral nerve injury. In both models, CLP257 application resulted in hyperpolarized EGABA, indicative of a higher rate of Cl− extrusion (163). Cell surface expression of KCC2 was determined to be higher after application of the compound, indicating that CLP257 affects protein turnover. Additional modifications were made to CLP257 to improve its pharmacokinetic profile and its usefulness in in vivo studies (163).
Note that Cardarelli et al. (164) challenged the idea of a direct effect of CLP257 on KCC2. They argued that CLP257 potentiates the GABAA receptor, providing an alternative explanation for the beneficial effect of the compound on pain perception following peripheral nerve injury. Additional evidence against CLP257 affecting KCC2 was the demonstration that the compound failed to interact with the protein by backscattering interferometry, while a direct interaction could be demonstrated between the inhibitor VU0463271 and the cotransporter (164). Also, several attempts to capture a CLP257-bound KCC2 structure failed (Guo, personal communication), whereas a VU0463271-bound hKCC1 cryo-EM structure was recently reported (165). It seems therefore that CLP257 is not a positive modulator of KCC2 function and its identification originated for some unknown reasons as a false-positive hit in the screen.
Search for Transcriptional Enhancers (Boston, MA)
The promoter region responsible for KCC2b expression, the forebrain and predominant isoform of KCC2, contains a typical 5′-cytosine-phosphate-guanine (CpG) island making it responsive to epigenetic modifications (Fig. 6A). In a 2012 study, Yeo et al. (166) demonstrated that bisphenol A, an environmental chemical released from polycarbonate plastics and epoxy resins, retarded the developmental upregulation in KCC2 mRNA expression in perinatal rat cortical neurons. This effect was rescued by using trichostatin A, a pan-histone deacetylase inhibitor or by reducing expression of HDAC1. Treatment of neurons with bisphenol A led to increased binding of MECP2 and decreased binding of H3K9ac to the KCC2b promoter, providing a mechanism for the decrease in KCC2b expression by the plastic toxin. In vivo experiments further confirmed the toxicity of bisphenol A on neuronal development (166).
Figure 6.

Targets and drugs associated with K-Cl cotransporter (KCC2) function. A: activity of the KCC2 promoter is affected by compounds that target histone acetylation and methylation of 5′-cytosine-phosphate-guanine (CpG) sites. The “plastic” toxin Bisphenol A inhibits the histone acetyltransferase which tend to increase KCC2 activity. Thus, the toxin reduces KCC2 expression. This phenomenon is reversed by Trichostatin A (Tri. A), which inhibits HDAC1. The screen of Jaenisch group (20) identified an inhibitor of GSK3β, a kinase that stimulates HDAC. Inhibition of GSK3β results in increased KCC2 expression. Methyl CpG binding protein 2 (Mecp2) binds to methylated CpG sites likely to stimulates CpG demethylases leading to stimulation of KCC2 expression. B: historical and new inhibitors to KCC2 function. The compounds are believed to interfere with the translocation of ions. Note that the phosphorylation events leading to inactivation of KCC2 or internalization of KCC2 are also targets for drug discovery. Cryo-EM model of KCC2 was drawn using Pymol and PDB ID: 7D8Z.
Jaenisch’s group (20) recently developed a sophisticated screen designed to identify compounds enhancing the transcription of KCC2. They introduced, via CRISPR/cas9, a luciferase reporter gene upstream of the stop codon in the SLC12A5 gene in human embryonic stem cells. They then differentiated the cells into neurons expressing KCC2. As the luciferase reporter protein was not fused to the KCC2 protein but expressed separately and in an equal amount to KCC2, the assay reports KCC2 with fidelity, but without affecting translation and function of the cotransporter. From a combination of three targeted chemical libraries, they screened 929 compounds and found 14 hits. Note that the compounds were added to the neurons for 1 wk, which seems to be an awfully long time to capture transcriptional effects and unfortunately likely to invite unspecific effects. The hit compounds included: KW-2449, an inhibitor of FLT3 (receptor-type tyrosine-protein kinase); BIO (6-bromoindirubin-3′-oxime), an inhibitor of the glycogen synthase kinase 3 β (GSK3b) pathway; and resveratrol. These compounds were shown to enhance KCC2 expression by twofold (20).
Consistent with an increase in KCC2 expression, these compounds led to a hyperpolarizing shift in EGABA in immature neurons, increased KCC2 expression in MECP2-null RTT (Rett syndrome) human neurons, and the rescue of functional and morphological deficits in RTT neurons (20). In vivo efficacy of KW-2449 and piperine was tested in a Mecp2 mutant mouse model of RTT (Mecp2 −/y) and the compounds improved the disease-related behavioral pathologies in this mouse model (20).
Recent Chemistry on Bumetanide (Genoa, Italy)
As indicated in a comprehensive review published in 2000, bumetanide inhibits both NKCC1 and NKCC2 with EC50s in the range of 10−7–10−6M (167). In addition, bumetanide is 40-fold more efficient in inducing diuresis in humans than its analog, furosemide (135). To target NKCC1 and avoid renal effects due to NKCC2 inhibition, Savardi et al. (168) synthetized new bumetanide analogs and tested them using a membrane-bound yellow fluorescent protein as Cl− indicator in HEK293 cells transfected with NKCC1 or NKCC2. They tested 135,000 compounds from several chemical libraries and selected 90 compounds for secondary screens. They selected two compounds out of these hits: ARN22392 and ARN22393, with moderate inhibitory effect on NKCC1. Upon chemical modifications, they identified a compound, ARN23746, with improved solubility and potency. Although the compound still had moderate effect on NKCC1 (38% at 10 µM and 95% at 100 µM) but was substantially weaker on NKCC2 (6.9% inhibition at 10 µM). The compound reduced GABA-mediated Ca2+-influx in neurons, consistent with the role that NKCC1 plays in accumulating Cl− and promoting GABA depolarization. ARN23746 had no significant off target activity and an improved pharmacokinetic profile, when compared with bumetanide. Finally, the compound was tested in vivo and shown to have no effect on diuresis, whereas it rescued the social deficits and repetitive behaviors in the valproic acid mouse model of autism, which is attributed to abnormal NKCC1 function (169). Thus, ARN23746 seems to be better (potentially better therapeutic) than bumetanide for in vivo studies involving NKCC1.
FUTURE PERSPECTIVES: DRUG DISCOVERY THROUGH COMPUTATIONAL DOCKING?
Over the past year, several cryo-EM structures of cation-chloride cotransporters have been published. The structure of the zebrafish NKCC1 came first (36), closely followed by the structures of the human KCC1 (37), mouse KCC4 (170), human KCC2, KCC2, and KCC4 (171). These major advances in the field now open the possibility of structure-based drug design. Structure-based or computer-aided drug design predicts the binding of small molecules to a protein, i.e., determine their position within the three-dimensional representation of the protein structure and estimate binding affinities. Computational approaches have the advantage of being relatively cheap compared with chemical screens. The cost largely consists of processor time on super computers.
Because the accuracy of a model is based on the number of simulations, it is useful to have some indication about the general vicinity of binding. This information can be gained through an iterative process with a first set of simulations identifying likely sites of binding, followed-up by additional simulations to refine the site of interaction. Note that a major limitation is the unique three-dimentional representation of a protein given by a crystal or cryo-EM structure. That representation only represents one state or configuration of a protein and that configuration might not be appropriate to model the binding of a drug. For instance, structures of membrane proteins typically favor inside versus outside configurations. This is the case for the several cation-chloride cotransporter structures just mentioned as well as for the crystal structures of related bacterial transporters (172). Based on the available structure of hKCC1, we simulated the binding of VU0463271 (and other compounds) using Autodock Vina, an open-source molecular docking software designed by the Scripps Institute (173). We started with the assumption that the drug must bind close to the pathway utilized by the ions during transport and were encouraged to find a putative binding site with very-high binding energy. Importantly, binding was coordinated by a tyrosine residue which is part of the K+ binding site, a key observation since the binding of VU0240551 is competitive with respect to K+ (127). One significant limitation of the docking experiment is the fact that the structure available was in inside-facing configuration with a closed extracellular pore. Would the binding be similar if the protein was in its outside-facing configuration with an open external pore? One way to answer this question would be to solve the structure of the transporter conjugated with the inhibitor. A paper recently uploaded to BioRxiv presents such a structure with the compound located as predicted within the ion permeation pathway, but closer to the external pore (165). In fact, the presence of the inhibitor likely stabilized the structure of the cotransporter in its outside-facing conformation. In the absence of published PDB coordinates, we cannot run molecular docking simulations and compare the positions and binding energies of the small inhibitory molecule in the two separate configurations.
Once the position of a small molecule within the structure of a protein is known, it becomes easier to test the effect of chemical modifications. This process can be automated, and the binding of thousands of chemical structures can be simulated to identify molecules with higher binding affinities. The process can also be done with multiple isoforms and small molecules can be identified based on strong binding with one isoform and much weaker binding with a separate but related isoform. These computer-based drug-screening efforts are great to generate hypotheses that can be tested experimentally.
CONCLUSIONS
The life cycle of a membrane transporter is complex and involves a multitude of steps that precede and follow the period of transport activity. A drug can affect the level of transport by tampering with any of these steps. Specificity of a drug toward the transport activity will depend upon the precise target and thus, the highest confidence for specificity is reached when the drug interferes directly with the transporter and its function. This is certainly the case for the thiazide and loop diuretics that inhibit NKCC1/NKCC2, and for the new compounds that inhibit KCC2 (Fig. 6B). That said, it is obvious that direct binding to the target alone does not guaranty specificity. Furosemide, for instance, is a potent inhibitor of NKCC1/NKCC2—but is also known to affect the activity of some GABAA receptors (174). Also, because of gene redundancy, paralogous genes typically encode proteins with high degree of conservation/homology, and therefore these proteins are likely to be affected by the same drugs. Not only furosemide and bumetanide affect the NKCC1 and NKCC2 transporters, but with lower affinity, the compounds also inhibit the related K-Cl cotransporters. High throughput screening efforts can identify compounds that discriminate between transporter isoforms, but success can also be obtained by screening large numbers of chemical variants. Also discussed in our last section of this review, we believe that computational docking simulations have the potential to help identifying small molecules that would bind to one isoform but be excluded from another.
The proteins involved in gene regulation, transcription, translation, trafficking, post-translational modification, internalization, and recycling are specific to individual transporters. What makes the process specific to each transporter protein is the unique combinatory sequence of all the regulatory players involved. Because each of these regulatory proteins participates in the regulation of many other proteins, their targeting makes it unspecific. Thus, targeting transcription factors or protein kinases might lead to the desired effect with a transporter under study, but will likely lead to unintended consequences. For instance, targeting GSK3β will not only lead to the desired upregulation of KCC2, but will also affect all the other processes regulated by the kinase. Let us remember, however, that most drugs currently used in medicine have known (and possibly unknown) side effects, and they are used because the benefit of using them to treat disorders far outweigh their unwarranted off target activities.
The pharmacology of Slc12 transporters is still limited to a few inhibitors. To date, all these compounds seem to act as full inhibitors rather than partial inhibitors. The development of partial inhibitors might be of use for diseases with abnormally high transporter activity. This would be for instance the case for patients with gain-of-function mutations in K-Cl cotransport (24, 175). Conversely, whether small molecules can be found to bind to the transporters and stimulate their function is still unknown. Any compound that would bind and stabilize the dimer, accelerate the translocation of ions, or increase the time of residency at the plasma membrane would increase the function of the transporter. Any compound that would indirectly affect these processes would also increase the function of the transporter. Functional screens are typically unbiased toward direct versus indirect mechanisms, and once a small molecule is identified as a modulator, additional work is needed to resolve the mechanism of action. As the physiology and pathophysiology of Slc12 transporters becomes increasingly clearer, the need to find additional modulators should only get bigger.
GRANTS
This work was supported in part by National Institute of Diabetes and Digestive and Kidney Diseases Grant DK093501 and by Leducq foundation for Cardiovascular Research Grant 17CVD05.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author.
AUTHOR CONTRIBUTIONS
E.D. prepared figures; drafted manuscript; edited and revised manuscript; and approved final version of manuscript.
REFERENCES
- 1.Delpire E, Rauchman MI, Beier DR, Hebert SC, Gullans SR. Molecular cloning and chromosome localization of a putative basolateral Na-K-2Cl cotransporter from mouse inner medullary collecting duct (mIMCD-3) cells. J Biol Chem 269: 25677–25683, 1994. doi: 10.1016/S0021-9258(18)47302-4. [DOI] [PubMed] [Google Scholar]
- 2.Gamba G, Miyanoshita A, Lombardi M, Lytton J, Lee W-S, Hediger M, Hebert SC. Molecular cloning, primary structure, and characterization of two members of the mammalian electroneutral sodium-(potassium)-chloride cotransporter family expressed in kidney. J Biol Chem 269: 17713–17722, 1994. doi: 10.1016/S0021-9258(17)32499-7. [DOI] [PubMed] [Google Scholar]
- 3.Gamba G, Saltzberg SN, Lombardi M, Miyanoshita A, Lytton J, Hediger MA, Brenner BM, Hebert SC. Primary structure and functional expression of a cDNA encoding the thiazide-sensitive, electroneutral sodium-chloride cotransporter. Proc Natl Acad Sci USA 90: 2749–2753, 1993. doi: 10.1073/pnas.90.7.2749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gillen CM, Brill S, Payne JA, Forbush BI. Molecular cloning and functional expression of the K-Cl cotransporter from rabbit, rat, and human. A new member of the cation-chloride cotransporter family. J Biol Chem 271: 16237–16244, 1996. doi: 10.1074/jbc.271.27.16237. [DOI] [PubMed] [Google Scholar]
- 5.Mount DB, Mercado A, Song L, Xu J, George JAL, Delpire E, Gamba G. Cloning and Characterization of KCC3 and KCC4, new members of the cation-chloride cotransporter gene family. J Biol Chem 274: 16355–16362, 1999. doi: 10.1074/jbc.274.23.16355. [DOI] [PubMed] [Google Scholar]
- 6.Payne JA, Stevenson TJ, Donaldson LF. Molecular characterization of a putative K-Cl cotransporter in rat brain. A neuronal-specific isoform. J Biol Chem 271: 16245–16252, 1996. doi: 10.1074/jbc.271.27.16245. [DOI] [PubMed] [Google Scholar]
- 7.Simon DB, Karet FE, Hamdan JM, Di Pietro A, Sanjad SA, Lifton RP. Bartter's syndrome, hypokalaemic alkalosis with hypercalciuria, is caused by mutations in the Na-K-2Cl cotransporter NKCC2. Nat Genet 13: 183–188, 1996. doi: 10.1038/ng0696-183. [DOI] [PubMed] [Google Scholar]
- 8.Takahashi N, Chernavvsky DR, Gomez RA, Igarashi P, Gitelman HJ, Smithies O. Uncompensated polyuria in a mouse model of Bartter's syndrome. Proc Natl Acad Sci USA 97: 5434–5439, 2000. doi: 10.1073/pnas.090091297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Delpire E, Lu J, England R, Dull C, Thorne T. Deafness and imbalance associated with inactivation of the secretory Na-K-2Cl co-transporter. Nat Genet 22: 192–195, 1999. doi: 10.1038/9713. [DOI] [PubMed] [Google Scholar]
- 10.Flagella M, Clarke LL, Miller ML, Erway LC, Giannella RA, Andringa A, Gawenis LR, Kramer J, Duffy JJ, Doetschman T, Lorenz JN, Yamoah EN, Cardell EL, Shull GE. Mice lacking the basolateral Na-K-2Cl cotransporter have impaired epithelial chloride secretion and are profoundly deaf. J Biol Chem 274: 26946–26955, 1999. doi: 10.1074/jbc.274.38.26946. [DOI] [PubMed] [Google Scholar]
- 11.Koumangoye R, Omer S, Kabeer MH, Delpire E. Novel human NKCC1 mutations cause defects in goblet cells mucus secretion and chronic inflammation. Cell Mol Gastroenterol Hepatol 9: 239–255, 2020. doi: 10.1016/j.jcmgh.2019.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Macnamara EF, Koehler AE, D'Souza P, Estwick T, Lee P, Vezina GN, MotUD Fauni H, Braddock SR, Torti E, Holt JM, Sharma P, Malicdan MCV, Tifft CJ; Undiagnosed Diseases Network. Kilquist syndrome: a novel syndromic hearing loss disorder caused by homozygous deletion of SLC12A2. Hum Mutat 40: 532–538, 2019. doi: 10.1002/humu.23722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pace AJ, Lee E, Athirakul K, Coffman TM, O'Brien DA, Koller BH. Failure of spermatogenesis in mouse lines deficient in the Na+-K+-2Cl− cotransporter. J Clin Invest 105: 441–450, 2000. doi: 10.1172/JCI8553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stodberg T, Magnusson M, Lesko N, Wredenberg A, Martin Munoz D, Stranneheim H, Wedell A. SLC12A2 mutations cause NKCC1 deficiency with encephalopathy and impaired secretory epithelia. Neurol Genet 6: e478, 2020. doi: 10.1212/NXG.0000000000000478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nicolet-Barousse L, Blanchard A, Roux C, Pietri L, Bloch-Faure M, Kolta S, Chappard C, Geoffroy V, Morieux C, Jeunemaitre X, Shull GE, Meneton P, Paillard M, Houillier P, De Vernejoul MC. Inactivation of the Na-Cl co-transporter (NCC) gene is associated with high BMD through both renal and bone mechanisms: analysis of patients with Gitelman syndrome and Ncc null mice. J Bone Miner Res 20: 799–808, 2005. doi: 10.1359/JBMR.041238. [DOI] [PubMed] [Google Scholar]
- 16.Simon DB, Nelson-Williams CJ, Bia M, Ellison D, Karet FEM, Molina A, Vaara I, Iwata F, Cushner HM, Koolen M, Gainza FJ, Gitelman HJ, Lifton RP. Gitelman’s variant of Bartter’s syndrome, inherited hypokalaemic alkalosis, is caused by mutations in the thiazide-sensitive Na-Cl cotransporter. Nat Genet 12: 24–30, 1996. doi: 10.1038/ng0196-24. [DOI] [PubMed] [Google Scholar]
- 17.Anacker AMJ, Moran JT, Santarelli S, Forsberg CG, Rogers TD, Stanwood GD, Hall BJ, Delpire E, Veenstra-VanderWeele J, Saxe MD. Enhanced social dominance and altered neuronal excitability in the prefrontal cortex of male KCC2b mutant mice. Autism Res 12: 732–743, 2019. doi: 10.1002/aur.2098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kahle KT, Merner ND, Friedel P, Silayeva L, Liang B, Khanna A, Shang Y, Lachance-Touchette P, Bourassa C, Levert A, Dion PA, Walcott B, Spiegelman D, Dionne-Laporte A, Hodgkinson A, Awadalla P, Nikbakht H, Majewski J, Cossette P, Deeb TZ, Moss SJ, Medina I, Rouleau GA. Genetically encoded impairment of neuronal KCC2 cotransporter function in human idiopathic generalized epilepsy. EMBO Rep 15: 766–774, 2014. doi: 10.15252/embr.201438840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Puskarjov M, Seja P, Heron SE, Williams TC, Ahmad F, Iona X, Oliver KL, Grinton BE, Vutskits L, Scheffer IE, Petrou S, Blaesse P, Dibbens LM, Berkovic SF, Kaila K. A variant of KCC2 from patients with febrile seizures impairs neuronal Cl− extrusion and dendritic spine formation. EMBO rep 15: 723–729, 2014. doi: 10.1002/embr.201438749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tang X, Drotar J, Li K, Clairmont CD, Brumm AS, Sullins AJ, Wu H, Liu XS, Wang J, Gray NS, Sur M, Jaenisch R. Pharmacological enhancement of KCC2 gene expression exerts therapeutic effects on human Rett syndrome neurons and Mecp2 mutant mice. Sci Transl Med 11: eaau0164, 2019. doi: 10.1126/scitranslmed.aau0164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Woo N-S, Lu J, England R, McClellan R, Dufour S, Mount DB, Deutch AY, Lovinger DM, Delpire E. Hyper-excitability and epilepsy associated with disruption of the mouse neuronal-specific K-Cl cotransporter gene. Hippocampus 12: 258–268, 2002. doi: 10.1002/hipo.10014. [DOI] [PubMed] [Google Scholar]
- 22.Boettger T, Rust MB, Maier H, Seidenbecher T, Schweizer M, Keating DJ, Faulhaber J, Ehmke H, Pfeffer C, Scheel O, Lemcke B, Horst J, Leuwer R, Pape HC, Volkl H, Hubner CA, Jentsch TJ. Loss of K-Cl co-transporter KCC3 causes deafness, neurodegeneration and reduced seizure threshold. EMBO J 22: 5422–5434, 2003. doi: 10.1093/emboj/cdg519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Howard HC, Mount DB, Rochefort D, Byun N, Dupré N, Lu J, Fan X, Song L, Rivière J-B, Prévost C, Horst J, Simonati A, Lemcke B, Welch R, England R, Zhan FQ, Mercado A, Siesser WB, George AL, McDonald MP, Bouchard J-P, Mathieu J, Delpire E, Rouleau GA. Mutations in the K-Cl cotransporter KCC3 cause a severe peripheral neuropathy associated with agenesis of the corpus callosum. Nat Genet 32: 384–392, 2002[Erratum inNat Genet32: 681, 2002]. doi: 10.1038/ng1002. [DOI] [PubMed] [Google Scholar]
- 24.Kahle KT, Flores B, Bharucha-Goebel D, Zhang J, Donkervoort S, Hegde M, Hussain G, Duran D, Liang B, Sun D, Bönnemann CG, Delpire E. Peripheral motor neuropathy is associated with defective kinase regulation of the KCC3 cotransporter. Sci Signal 9: ra77, 2016. doi: 10.1126/scisignal.aae0546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Boettger T, Hubner CA, Maier H, Rust MB, Beck FX, Jentsch TJ. Deafness and renal tubular acidosis in mice lacking the K-Cl co-transporter Kcc4. Nature 416: 874–878, 2002. doi: 10.1038/416874a. [DOI] [PubMed] [Google Scholar]
- 26.Garin EH, Fennell RS, Iravani A, Richard GA. Treatment of Bartter’s syndrome with indomethacin. Am J Dis Child 134: 258–261, 1980. doi: 10.1001/archpedi.1980.02130150016005. [DOI] [PubMed] [Google Scholar]
- 27.Evans RL, Park K, Turner RJ, Watson GE, Nguyen H-V, Dennett MR, Hand AR, Flagella M, Shull GE, Melvin JE. Severe impairment of salivation in Na+/K+/2Cl− cotransporter (NKCC1)-deficient mice. J Biol Chem 275: 26720–26726, 2000. doi: 10.1016/S0021-9258(19)61435-3. [DOI] [PubMed] [Google Scholar]
- 28.Grubb BR, Lee E, Pace AJ, Koller BH, Boucher RC. Intestinal ion transport in NKCC1-deficient mice. Am J Physiol Gastrointest Liver Physiol 279: G707–G718, 2000. doi: 10.1152/ajpgi.2000.279.4.G707. [DOI] [PubMed] [Google Scholar]
- 29.Sung K-W, Kirby M, McDonald MP, Lovinger DM, Delpire E. Abnormal GABAA-receptor mediated currents in dorsal root ganglion neurons isolated from Na-K-2Cl cotransporter null mice. J Neurosci 20: 7531–7538, 2000. doi: 10.1523/JNEUROSCI.20-20-07531.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Koumangoye R, Bastarache L, Delpire E. NKCC1: newly found as a human disease-causing ion transporter. Function (Oxf) 2: zqaa028, 2021. doi: 10.1093/function/zqaa028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Delpire E, Wolfe L, Flores B, Koumangoye R, Schornak CC, Omer S, Pusey B, Lau C, Markello T, Adams DR. A patient with multisystem dysfunction carries a truncation mutation in human SLC12A2, the gene encoding the Na-K-2Cl cotransporter, NKCC1. Cold Spring Harb Mol Case Stud 2: a001289, 2016. doi: 10.1101/mcs.a001289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McNeill A, Iovino E, Mansard L, Vache C, Baux D, Bedoukian E, Cox H, Dean J, Goudie D, Kumar A, Newbury-Ecob R, Fallerini C, Renieri A, Lopergolo D, Mari F, Blanchet C, Willems M, Roux A-F, Pippucci T, Delpire E. SLC12A2 variants cause a neurodevelopmental disorder or cochleovestibular defect. Brain 143: 2380–2387, 2020. doi: 10.1093/brain/awaa176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lemonnier E, Ben-Ari Y. The diuretic bumetanide decreases autistic behaviour in five infants treated during 3 months with no side effects. Acta Paediatr 99: 1885–1888, 2010. doi: 10.1111/j.1651-2227.2010.01933.x. [DOI] [PubMed] [Google Scholar]
- 34.Lemonnier E, Degrez C, Phelep M, Tyzio R, Josse F, Grandgeorge M, Hadjikhani N, Ben-Ari YA. Randomised controlled trial of bumetanide in the treatment of autism in children. Transl Psychiatry 2: e202, 2012. doi: 10.1038/tp.2012.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lemonnier E, Villeneuve N, Sonie S, Serret S, Rosier A, Roue M, Brosset P, Viellard M, Bernoux D, Rondeau S, Thummler S, Ravel D, Ben-Ari Y. Effects of bumetanide on neurobehavioral function in children and adolescents with autism spectrum disorders. Transl Psychiatry 7: e1056, 2017. doi: 10.1038/tp.2017.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chew TA, Orlando BJ, Zhang J, Latorraca NR, Wang A, Hollingsworth SA, Chen DH, Dror RO, Liao M, Feng L. Structure and mechanism of the cation-chloride cotransporter NKCC1. Nature 572: 488–492, 2019. doi: 10.1038/s41586-019-1438-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu S, Chang S, Han BL, Xu L, Zhang M, Zhao C, Yang W, Wang F, Li J, Delpire E, Ye S, Bai X-C, Guo J. Cryo-EM structures of the human cation-chloride cotransporter KCC1. Science 366: 505–508, 2019. doi: 10.1126/science.aay3129. [DOI] [PubMed] [Google Scholar]
- 38.Grimm PR, Taneja TK, Liu J, Coleman R, Chen YY, Delpire E, Wade JB, Welling PA. SPAK isoforms and OSR1 regulate sodium-chloride co-transporters in a nephron-specific manner. J Biol Chem 287: 37673–37690, 2012. doi: 10.1074/jbc.M112.402800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McCormick JA, Mutig K, Nelson JH, Saritas T, Hoorn EJ, Yang C-L, Rogers S, Curry J, Delpire E, Bachmann S, Ellison DH. A SPAK isoform switch modulates renal salt transport and blood pressure. Cell Metab 14: 352–364, 2011. doi: 10.1016/j.cmet.2011.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Castañeda-Bueno M, Cervantes-Pérez LG, Vázquez N, Uribe N, Kantesaria S, Morla L, Bobadilla NA, Doucet A, Alessi DR, Gamba G. Activation of the renal Na+:Cl− cotransporter by angiotensin II is a WNK4-dependent process. Proc Natl Acad Sci USA 109: 7929–7934, 2012. doi: 10.1073/pnas.1200947109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wilson FH, Disse-Nicodeme S, Choate KA, Ishikawa K, Nelson-Williams C, Desitter I, Gunel M, Milford DV, Lipkin GW, Achard JM, Feely MP, Dussol B, Berland Y, Unwin RJ, Mayan H, Simon DB, Farfel Z, Jeunemaitre X, Lifton RP. Human hypertension caused by mutations in WNK kinases. Science 293: 1107–1112, 2001. doi: 10.1126/science.1062844. [DOI] [PubMed] [Google Scholar]
- 42.Choate KA, Kahle KT, Wilson FH, Nelson-Williams C, Lifton RP. WNK1, a kinase mutated in inherited hypertension with hyperkalemia, localizes to diverse Cl−-transporting epithelia. Proc Natl Acad Sci USA 100: 663–668, 2003. doi: 10.1073/pnas.242728499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Boyden LM, Choi M, Choate KA, Nelson-Williams CJ, Farhi A, TokaHR, et al. Mutations in kelch-like 3 and cullin 3 cause hypertension and electrolyte abnormalities. Nature 482: 98–102, 2012. doi: 10.1038/nature10814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ohta A, Schumacher FR, Mehellou Y, Johnson C, Knebel A, Macartney TJ, Wood NT, Alessi DR, Kurz T. The CUL3-KLHL3 E3 ligase complex mutated in Gordon's hypertension syndrome interacts with and ubiquitylates WNK isoforms: disease-causing mutations in KLHL3 and WNK4 disrupt interaction. Biochem J 451: 111–122, 2013. doi: 10.1042/BJ20121903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yang SS, Morimoto T, Rai T, Chiga M, Sohara E, Ohno M, Uchida K, Lin SH, Moriguchi T, Shibuya H, Kondo Y, Sasaki S, Uchida S. Molecular pathogenesis of pseudohypoaldosteronism type II: generation and analysis of a Wnk4(D561A/+) knockin mouse model. Cell Metab 5: 331–344, 2007. doi: 10.1016/j.cmet.2007.03.009. [DOI] [PubMed] [Google Scholar]
- 46.Grimm PR, Coleman R, Delpire E, Welling PA. Constitutively active SPAK causes hyperkalemia by activating NCC and remodeling distal tubules. J Am Soc Nephrol 28: 2597–2606, 2017. doi: 10.1681/ASN.2016090948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rust MB, Alper SL, Rudhard Y, Shmukler BE, Vicente R, Brugnara C, Trudel M, Jentsch TJ, Hübner CA. Disruption of erythroid K-Cl cotransporters alters erythrocyte volume and partially rescues erythrocyte dehydration in SAD mice. J Clin Invest 117: 1708–1717, 2007. doi: 10.1172/JCI30630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rivera C, Voipio J, Payne JA, Ruusuvuori E, Lahtinen H, Lamsa K, Pirvola U, Saarma M, Kaila K. The K+/Cl− co-transporter KCC2 renders GABA hyperpolarizing during neuronal maturation. Nature 397: 251–255, 1999. doi: 10.1038/16697. [DOI] [PubMed] [Google Scholar]
- 49.Thompson SM, Deisz RA, Prince DA. Outward chloride/cation co-transport in mammalian cortical neurons. Neurosci Lett 89: 49–54, 1988. doi: 10.1016/0304-3940(88)90479-X. [DOI] [PubMed] [Google Scholar]
- 50.Zhu L, Lovinger D, Delpire E. Cortical neurons lacking KCC2 expression show impaired regulation of intracellular chloride. J Neurophysiol 93: 1557–1568, 2005. doi: 10.1152/jn.00616.2004. [DOI] [PubMed] [Google Scholar]
- 51.Zhu L, Polley N, Mathews GC, Delpire E. NKCC1 and KCC2 prevent hyperexcitability in the mouse hippocampus. Epilepsy Res 79: 201–212, 2008. doi: 10.1016/j.eplepsyres.2008.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hubner CA, Stein V, Hermans-Borgmeyer I, Meyer T, Ballanyi K, Jentsch TJ. Disruption of KCC2 reveals an essential role of K-Cl cotransport already in early synaptic inhibition. Neuron 30: 515–524, 2001. doi: 10.1016/S0896-6273(01)00297-5. [DOI] [PubMed] [Google Scholar]
- 53.Merner ND, Chandler MR, Bourassa C, Liang B, Khanna AR, Dion P, Rouleau GA, Kahle KT. Regulatory domain or CpG site variation in SLC12A5, encoding the chloride transporter KCC2, in human autism and schizophrenia. Front Cell Neurosci 9: 386, 2015. doi: 10.3389/fncel.2015.00386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Duarte ST, Armstrong J, Roche A, Ortez C, Pérez A, O'Callaghan Mdel M, Pereira A, Sanmartí F, Ormazábal A, Artuch R, Pineda M, García-Cazorla A. Abnormal expression of cerebrospinal fluid cation chloride cotransporters in patients with Rett syndrome. PLoS One 8: e68851, 2013. doi: 10.1371/journal.pone.0068851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hinz L, Torrella Barrufet J, Heine VM. KCC2 expression levels are reduced in post mortem brain tissue of Rett syndrome patients. Acta Neuropathol Commun 7: 196, 2019. doi: 10.1186/s40478-019-0852-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lozovaya N, Nardou R, Tyzio R, Chiesa M, Pons-Bennaceur A, Eftekhari S, Bui TT, Billon-Grand M, Rasero J, Bonifazi P, Guimond D, Gaiarsa JL, Ferrari DC, Ben-Ari Y. . Early alterations in a mouse model of Rett syndrome: the GABA developmental shift is abolished at birth. Sci Rep 9: 9276, 2019. doi: 10.1038/s41598-019-45635-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tang X, Kim J, Zhou L, Wengert E, Zhang L, Wu Z, Carromeu C, Muotri AR, Marchetto MC, Gage FH, Chen G. KCC2 rescues functional deficits in human neurons derived from patients with Rett syndrome. Proc Natl Acad Sci USA 113: 751–756, 2016. doi: 10.1073/pnas.1524013113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hiki K, D'Andrea RJ, Furze J, Crawford J, Woollatt E, Sutherland GR, Vadas MA, Gamble JR. Cloning, characterization, and chromosomal location of a novel human K+-Cl− cotransporter. J Biol Chem 274: 10661–10667, 1999. doi: 10.1074/jbc.274.15.10661. [DOI] [PubMed] [Google Scholar]
- 59.Race JE, Makhlouf FN, Logue PJ, Wilson FH, Dunham PB, Holtzman EJ. Molecular cloning and functional characterization of KCC3, a new K-Cl cotransporter. Am J Physiol Cell Physiol 277: C1210–C1219, 1999. doi: 10.1152/ajpcell.1999.277.6.C1210. [DOI] [PubMed] [Google Scholar]
- 60.Ding J, Delpire E. Deletion of KCC3 in parvalbumin neurons leads to locomotor deficit in a conditional mouse model of peripheral neuropathy associated with agenesis of the corpus callosum. Behav Brain Res 274: 128–136, 2014. doi: 10.1016/j.bbr.2014.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Shekarabi M, Moldrich RX, Rasheed S, Salin-Cantegrel A, Laganière J, Rochefort D, Hince P, Huot K, Gaudet R, Kurniawan N, Sotocinal SG, Ritchie J, Dion PA, Mogil JS, Richards LJ, Rouleau GA. Loss of neuronal potassium/chloride cotransporter 3 (KCC3) is responsible for the degenerative phenotype in a conditional mouse model of hereditary motor and sensory neuropathy associated with agenesis of the corpus callosum. J Neurosci 32: 3865–3876, 2012. doi: 10.1523/JNEUROSCI.3679-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Byun N, Delpire E. Axonal and periaxonal swelling precede peripheral neurodegeneration in KCC3 knockout mice. Neurobiol Dis 28: 39–51, 2007. doi: 10.1016/j.nbd.2007.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Flores B, Delpire E. Osmotic response of dorsal root ganglion neurons expressing wild-type and mutant KCC3 transporters. Cell Physiol Biochem 54: 577–590, 2020. doi: 10.33594/000000241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Uyanik G, Elcioglu N, Penzien J, Gross C, Yilmaz Y, Olmez A, Demir E, Wahl D, Scheglmann K, Winner B, Bogdahn U, Topaloglu H, Hehr U, Winkler J. Novel truncating and missense mutations of the KCC3 gene associated with Andermann syndrome. Neurology 66: 1044–1048, 2006[Erratum inNeurology67: 1528, 2006]. doi: 10.1212/01.wnl.0000204181.31175.8b. [DOI] [PubMed] [Google Scholar]
- 65.Park J, Flores BR, Scherer K, Kuepper H, Rossi M, Rupprich K, Rautenberg M, Deininger N, Weichselbaum A, Grimm A, Sturm M, Grasshoff U, Delpire E, Haack TB. De novo variants in SLC12A6 cause sporadic early-onset progressive sensorimotor neuropathy. J Med Genet 57: 283–288, 2020. doi: 10.1136/jmedgenet-2019-106273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lauf PK. Volume and anion dependency of ouabain-resistant K-Rb fluxes in sheep red blood cells. Am J Physiol Cell Physiol 255: C331–C339, 1988. doi: 10.1152/ajpcell.1988.255.3.C331. [DOI] [PubMed] [Google Scholar]
- 67.Davson H, Pollay M. The turnover of 24Na in the cerebrospinal fluid and its bearing on the blood-brain barrier. J Physiol 167: 247–255, 1963. doi: 10.1113/jphysiol.1963.sp007145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ennor AH, Rosenberg H. The separation and determination of 24Na and 32P in animal tissues. Biochem J 52: 591–593, 1952. doi: 10.1042/bj0520591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Miller H, Wilson GM. The measurement of exchangeable sodium in man using the isotope 24Na. Clin Sci 12: 97–111, 1953. [PubMed] [Google Scholar]
- 70.Iwai J, Ohanian EV, Dahl LK. Influence of thiazide on salt hypertension. Circ Res 40: I131–134, 1977. [PubMed] [Google Scholar]
- 71.Garrahan PJ, Glynn IM. The stoicheiometry of the sodium pump. J Physiol 192: 217–235, 1967. doi: 10.1113/jphysiol.1967.sp008297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tosteson DC, Hoffman JF. Regulation of cell volume by active cation transport in high and low potassium sheep red cells. J Gen Physiol 44: 169–194, 1960. doi: 10.1085/jgp.44.1.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kim HD, Tsai Y-S, Franklin CC, Turner JT. Characterization of Na+/K+/Cl− cotransport in cultured HT29 human colonic adenocarcinoma cells. Biochim Biophys Acta 946: 397–404, 1988[Erratum inBiochim Biophys Acta978: 349, 1989]. doi: 10.1016/0005-2736(88)90415-4. [DOI] [PubMed] [Google Scholar]
- 74.Weinman SA, Reuss L. Na+-H+ exchange and Na+ entry across the apical membrane of Necturus gallbladder. J Gen Physiol 83: 57–74, 1984. doi: 10.1085/jgp.83.1.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Paris S, Pouyssegur J. Growth factors activate the bumetanide sensitive Na+/K+/Cl− cotransport in hamster fibroblasts. J Biol Chem 261: 6177–6183, 1986. doi: 10.1016/S0021-9258(19)84545-3. [DOI] [PubMed] [Google Scholar]
- 76.Wilkinson DJ, Post MA, Venglarik C, Chang D, Dawson DC. Mercury blockade of thiazide-sensitive NaCl cotransport in flounder urinary bladder. Toxicol Appl Pharmacol 123: 170–176, 1993. doi: 10.1006/taap.1993.1234. [DOI] [PubMed] [Google Scholar]
- 77.Ruiz OS, Qiu YY, Cardoso LR, Arruda JA. Regulation of the renal Na-HCO3 cotransporter. VII. Mechanism of the cholinergic stimulation. Kidney Int 51: 1069–1077, 1997. doi: 10.1038/ki.1997.149. [DOI] [PubMed] [Google Scholar]
- 78.Hevesy G, Nylin G. Application of 42K labelled red corpuscles in blood volume measurements. Acta Physiol Scand 24: 285–292, 1952. doi: 10.1111/j.1748-1716.1952.tb00846.x. [DOI] [PubMed] [Google Scholar]
- 79.Domer FR. Transport of 42K from blood to cerebrospinal fluid in cats. J Physiol 158: 366–373, 1961. doi: 10.1113/jphysiol.1961.sp006773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.De Rouffignac C, Guinnebault M. Study, with the aid of K42 microinjections, of the potassium permeability of cortical segments of the nephron. Nephron 3: 175–197, 1966. doi: 10.1159/000179474. [DOI] [PubMed] [Google Scholar]
- 81.Johnson JE, Hartsuck JM, Zollinger RM, Moore FD. Radiopotassium equilibrium in total body potassium: studies using 43K and 42K. Metabolism 18: 663–668, 1969. doi: 10.1016/0026-0495(69)90079-1. [DOI] [PubMed] [Google Scholar]
- 82.Kilpatrick R, Miller H, Munro DS, Renschler H, Wislon GM. A comparison of the distribution of 42K and 86Rb in the rabbit. J Physiol 283: 71P–72P, 1955. [PubMed] [Google Scholar]
- 83.Bakkeren JA, Bonting SL. Studies on (Na+-K+)-activated ATPase. XXI. Changes in (Na+-K+)-activated ATPase activity and ouabain-sensitive 86Rb+ uptake rate in regenerating rat liver. Biochim Biophys Acta 150: 467–472, 1968. doi: 10.1016/0005-2736(68)90146-6. [DOI] [PubMed] [Google Scholar]
- 84.Henquin JC. The potassium permeability of pancreatic islet cells: mechanisms of control and influence on insulin release. Horm Metab Res Suppl 10: 66–73, 1980. [PubMed] [Google Scholar]
- 85.Kimelberg HK, Mayhew E. Increased ouabain-sensitive 86Rb+ uptake and sodium and potassium ion-activated adenosine triphosphatase activity in transformed cell lines. J Biol Chem 250: 100–104, 1975. doi: 10.1016/S0021-9258(19)41986-8. [DOI] [PubMed] [Google Scholar]
- 86.Malaisse WJ, Boschero AC, Kawazu S, Hutton JC. The stimulus secretion coupling of glucose-induced insulin release. XXVII. Effect of glucose on K+ fluxes in isolated islets. Pflugers Arch 373: 237–242, 1978. doi: 10.1007/BF00580830. [DOI] [PubMed] [Google Scholar]
- 87.McRoberts JA, Erlinger S, Rindler MJ, SaierMH, Jr.. Furosemide-sensitive salt transport in the Madin-Darby canine kidney cell line. Evidence for the cotransport of Na+, K+, and Cl. J Biol Chem 257: 2260–2266, 1982. doi: 10.1016/S0021-9258(18)34915-9. [DOI] [PubMed] [Google Scholar]
- 88.Parod RJ, PutneyJW, Jr.. An alpha-adrenergic receptor mechanism controlling potassium permeability in the rat lacrimal gland acinar cell. J Physiol 281: 359–369, 1978. doi: 10.1113/jphysiol.1978.sp012427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Rindler MJ, McRoberts JA, Saier MH. (Na+, K+)-cotransport in the Madin-Darby canine kidney cell line. Kinetic characterization of the interaction between Na+ and K+. J Biol Chem 257: 2254–2259, 1982. doi: 10.1016/S0021-9258(18)34914-7. [DOI] [PubMed] [Google Scholar]
- 90.Schackmann R, Schwartz A, Saccomani G, Sachs G. Cation transport by gastric H+:K+ ATPase. J Membrane Biol 32: 361–381, 1977. doi: 10.1007/BF01905228. [DOI] [PubMed] [Google Scholar]
- 91.Goodford PJ. Chloride content and 36Cl uptake in the smooth muscle of the guinea-pig taenia coli. J Physiol 170: 227–237, 1964. doi: 10.1113/jphysiol.1964.sp007326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Al-Awqati Q, Field M, Greenough WB III.. Reversal of cyclic AMP-mediated intestinal secretion by ethacrynic acid. J Clin Invest 53: 687–692, 1974. doi: 10.1172/JCI107606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Peaker M, Taylor JC. Milk secretion in the rabbit: changes during lactation and the mechanism of ion transport. J Physiol 253: 527–545, 1975. doi: 10.1113/jphysiol.1975.sp011205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Funder J, Wieth JO. Chloride transport in human erythrocytes and ghosts: a quantitative comparison. J Physiol 262: 679–698, 1976. doi: 10.1113/jphysiol.1976.sp011615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Walsh A. The application of atomic absorption spectra to chemical analysis. Spectrochim Acta 7: 108–117, 1955. doi: 10.1016/0371-1951(55)80013-6. [DOI] [Google Scholar]
- 96.Gow BS. Analysis of metals in saliva by atomic absorption spectroscopy. I. Calcium. J Dent Res 44: 885–889, 1965. doi: 10.1177/00220345650440052201. [DOI] [PubMed] [Google Scholar]
- 97.MacDonald MA, Watson L. The determination of magnesium in biological materials by atomic absorption spectrophotometry. Clin Chim Acta 14: 233–241, 1966. doi: 10.1016/0009-8981(66)90093-3. [DOI] [PubMed] [Google Scholar]
- 98.Armstrong WM, Bixenman WR, Frey KF, Garcia-Diaz JF, O'Regan MG, Owens JL. Energetics of coupled Na+ and Cl- entry into epithelial cells of bullfrog small intestine. Biochim Biophys Acta 551: 207–219, 1979. doi: 10.1016/0005-2736(79)90366-3. [DOI] [PubMed] [Google Scholar]
- 99.Lauf PK, Be T. A chloride dependent K+ flux induced by N-ethylmaleimide in genetically low K+ sheep and goat erythrocytes. Biochem Biophys Res Commun 70: 221–242, 1980. [DOI] [PubMed] [Google Scholar]
- 100.Anger JP, Vernhet L, Hichami A, Corlu A, Lepalabe E, Troussard A, Legrand A. Furosemide-sensitive K+ transport in transformed and nontransformed rat liver epithelial cells: regulation by protein kinase C and involvement in cell growth. Arch Int Pharmacodyn Ther 329: 307–318, 1995. [PubMed] [Google Scholar]
- 101.Lauf PK. Thiol-dependent passive K/Cl transport in sheep red cells. I. Dependence on chloride and external K+(Rb+) ions. J Membr Biol 73: 237–246, 1983. doi: 10.1007/BF01870538. [DOI] [PubMed] [Google Scholar]
- 102.Cameron IL, Smith NK, Pool TB, Sparks RL. Intracellular concentration of sodium and other elements as related to mitogenesis and oncogenesis in vivo. Cancer Res 40: 1493–1500, 1980. [PubMed] [Google Scholar]
- 103.Keenan MJ, Niedergerke R. Intracellular sodium concentration and resting sodium fluxes of the frog heart ventricle. J Physiol 188: 235–260, 1967. doi: 10.1113/jphysiol.1967.sp008136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Rose CR, Konnerth A. NMDA receptor-mediated Na+ signals in spines and dendrites. J Neurosci 21: 4207–4214, 2001. doi: 10.1523/JNEUROSCI.21-12-04207.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Baartscheer A, Schumacher CA, Fiolet JW. Small changes of cytosolic sodium in rat ventricular myocytes measured with SBFI in emission ratio mode. J Mol Cell Cardiol 29: 3375–3383, 1997. doi: 10.1006/jmcc.1997.0567. [DOI] [PubMed] [Google Scholar]
- 106.Meier SD, Kovalchuk Y, Rose CR. Properties of the new fluorescent Na+ indicator CoroNa Green: comparison with SBFI and confocal Na+ imaging. J Neurosci Methods 155: 251–259, 2006. doi: 10.1016/j.jneumeth.2006.01.009. [DOI] [PubMed] [Google Scholar]
- 107.Szmacinski H, Lakowicz JR. Sodium Green as a potential probe for intracellular sodium imaging based on fluorescence lifetime. Anal Biochem 250: 131–138, 1997. doi: 10.1006/abio.1997.2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Gregoriades JMC, Madaris AFJ, Alvarez FJ, Alvarez-Leefmans FJ. Genetic and pharmacologic inactivation of apical NKCC1 in choroid plexus epithelial cells reveals the physiological function of the cotransporter. Am J Physiol Cell Physiol 316: C525–C544, 2019. doi: 10.1152/ajpcell.00026.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Roder P, Hille C. ANG-2 for quantitative Na(+) determination in living cells by time-resolved fluorescence microscopy. Photochem Photobiol Sci 13: 1699–1710, 2014. doi: 10.1039/C4PP00061G. [DOI] [PubMed] [Google Scholar]
- 110.Rana PS, Gibbons BA, Vereninov AA, Yurinskaya VE, Clements RJ, Model TA, Model MA. Calibration and characterization of intracellular Asante potassium green probes, APG-2 and APG-4. Anal Biochem 567: 8–13, 2019. doi: 10.1016/j.ab.2018.11.024. [DOI] [PubMed] [Google Scholar]
- 111.Verkman AS. Development and biological applications of chloride-sensitive fluorescent indicators. Am J Physiol Cell Physiol 259: C375–C388, 1990. doi: 10.1152/ajpcell.1990.259.3.C375. [DOI] [PubMed] [Google Scholar]
- 112.Illsley NP, Verkman AS. Membrane chloride transport measured using a chloride-sensitive fluorescent probe. Biochemistry 26: 1215–1219, 1987. doi: 10.1021/bi00379a002. [DOI] [PubMed] [Google Scholar]
- 113.Verkman AS, Sellers MC, Chao AC, Leung T, Ketcham R. Synthesis and characterization of improved chloride-sensitive fluorescent indicators for biological applications. Anal Biochem 178: 355–361, 1989. doi: 10.1016/0003-2697(89)90652-0. [DOI] [PubMed] [Google Scholar]
- 114.Biwersi J, Verkman AS. Cell-permeable fluorescent indicator for cytosolic chloride. Biochemistry 30: 7879–7883, 1991. doi: 10.1021/bi00246a001. [DOI] [PubMed] [Google Scholar]
- 115.Verkman AS. Chemical and GFP-based fluorescent chloride indicators. In: Physiology and Pathology of Chloride Transporters and Channels in the Nervous System: From Molecules to Diseases, edited by Alvarez-Leefmans FJ, Delpire, E.London: Academic Press, 2009, p. 111–123. [Google Scholar]
- 116.Jayaraman S, Teitler L, Skalski B, Verkman AS. Long-wavelength iodide-sensitive fluorescent indicators for measurement of functional CFTR expression in cells. Am J Physiol Cell Physiol 277: C1008–C1018, 1999. doi: 10.1152/ajpcell.1999.277.5.C1008. [DOI] [PubMed] [Google Scholar]
- 117.Tabcharani JA, Chang XB, Riordan JR, Hanrahan JW. The cystic fibrosis transmembrane conductance regulator chloride channel. Iodide block and permeation. Biophys J 62: 1–4, 1992. doi: 10.1016/S0006-3495(92)81759-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Wachter RM, Remington SJ. Sensitivity of the yellow variant of green fluorescent protein to halides and nitrate. Curr Biol 9: R628–R629, 1999. doi: 10.1016/S0960-9822(99)80408-4. [DOI] [PubMed] [Google Scholar]
- 119.Kuner T, Augustine GJ. A genetically encoded ratiometric indicator for chloride: capturing chloride transients in cultured hippocampal neurons. Neuron 27: 447–459, 2000. doi: 10.1016/S0896-6273(00)00056-8. [DOI] [PubMed] [Google Scholar]
- 120.Grimley JS, Li L, Wang W, Wen L, Beese LS, Hellinga HW, Augustine GJ. Visualization of synaptic inhibition with an optogenetic sensor developed by cell-free protein engineering automation. J Neurosci 33: 16297–16309, 2013. doi: 10.1523/JNEUROSCI.4616-11.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Arosio D, Ricci F, Marchetti L, Gualdani R, Albertazzi L, Beltram F. Simultaneous intracellular chloride and pH measurements using a GFP-based sensor. Nat Methods 7: 516–518, 2010. doi: 10.1038/nmeth.1471. [DOI] [PubMed] [Google Scholar]
- 122.Sun Q, Wu Y, Jonusaite S, Pleinis JM, Humphreys JM, He H, Schellinger JN, Akella R, Stenesen D, Krämer H, Goldsmith EJ, Rodan AR. Intracellular chloride and scaffold protein Mo25 cooperatively regulate transepithelial ion transport through WNK signaling in the malpighian tubule. J Am Soc Nephrol 29: 1449–1461, 2018. doi: 10.1681/ASN.2017101091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Wasiluk T, Roueinfar M, Hiryak K, Torsiello M, Miner A, Lee J, Venditto M, Terzaghi W, Lucent D, VanWert AL. Simultaneous expression of ClopHensor and SLC26A3 reveals the nature of endogenous oxalate transport in CHO cells. Biol Open 8: bio041665, 2019. doi: 10.1242/bio.041665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Tao Z, Gameiro A, Grewer C. Thallium ions can replace both sodium and potassium ions in the glutamate transporter excitatory amino acid carrier 1. Biochemistry 47: 12923–12930, 2008. doi: 10.1021/bi8017174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Weaver CD, Harden D, Dworetzky SI, Robertson B, Knox RJ. A thallium-sensitive, fluorescence-based assay for detecting and characterizing potassium channel modulators in mammalian cells. J Biomol Screen 9: 671–677, 2004. doi: 10.1177/1087057104268749. [DOI] [PubMed] [Google Scholar]
- 126.Bakker-Grunwald T. Movement of thallous ion across the ascites cell membrane. J Membr Biol 47: 171–183, 1979. doi: 10.1007/BF01876115. [DOI] [PubMed] [Google Scholar]
- 127.Delpire E, Days E, Mi D, Lewis M, Kim K, Lindsley C, Weaver CD. Small molecule screen identifies inhibitors of the neuronal K-Cl cotransporter KCC2. Proc Natl Acad Sci USA 106: 5383–5388, 2009. doi: 10.1073/pnas.0812756106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Lewis LM, Bhave G, Chauder BA, Banerjee S, Lornsen KA, Redha R, Fallen K, Lindsley CW, Weaver CD, Denton JS. High-throughput screening reveals a small-molecule inhibitor of the renal outer medullary potassium channel and Kir7.1. Mol Pharmacol 76: 1094–1103, 2009. doi: 10.1124/mol.109.059840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Dutter BF, Ender A, Sulikowski GA, Weaver CD. Rhodol-based thallium sensors for cellular imaging of potassium channel activity. Org Biomol Chem 16: 5575–5579, 2018. doi: 10.1039/C8OB01098F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Kharade SV, Kurata H, Bender AM, Blobaum AL, Figueroa EE, Duran A, Kramer M, Days E, Vinson P, Flores D, Satlin LM, Meiler J, Weaver CD, Lindsley CW, Hopkins CR, Denton JS. Discovery, characterization, and effects on renal fluid and electrolyte excretion of the Kir4.1 potassium channel pore blocker, VU0134992. Mol Pharmacol 94: 926–937, 2018. doi: 10.1124/mol.118.112359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Stringham JS. On the diuretic effects of mercury in a case of syphilis. Med Phys J 18: 331–333, 1807. [PMC free article] [PubMed] [Google Scholar]
- 132.Barber HS. The mercurial diuretics. Postgrad Med J 14: 288–294, 1938. doi: 10.1136/pgmj.14.155.288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Novello FC, Sprague JM. Benzothiadiazine dioxides as novel diuretics. J Am Chem Soc 79: 2028–2029, 1957. doi: 10.1021/ja01565a079. [DOI] [Google Scholar]
- 134.Ingram TT. Today's drugs. Frusemide. Br Med J 2: 1640–1641, 1964. doi: 10.1136/bmj.2.5425.1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Asbury MJ, Gatenby PB, O’Sullivan S, Bourke E. potent new "loop" diuretic. Br Med J 1: 211–213, 1972. doi: 10.1136/bmj.1.5794.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Wang K, Kestenbaum B. Proximal tubular secretory clearance: a neglected partner of kidney function. Clin J Am Soc nephrol 13: 1291–1296, 2018. doi: 10.2215/CJN.12001017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Delpire E, Kaplan MR, Plotkin MD, Hebert SC. The Na-(K)-Cl cotransporter family in the mammalian kidney: molecular identification and function(s). Nephrol Dial Transplant 11: 1967–1973, 1996. doi: 10.1093/oxfordjournals.ndt.a027081. [DOI] [PubMed] [Google Scholar]
- 138.Hebert SC, Gullans SR. The electroneutral sodium-(potassium)-chloride co-transporter family: a journey from fish to the renal co-transporters. Curr Opin Nephrol Hypert 4: 389–391, 1995. doi: 10.1097/00041552-199509000-00002. [DOI] [PubMed] [Google Scholar]
- 139.Daley D, Evans B. Diuretic action of ethacrynic acid in congestive heart failure. Br Med J 2: 1169–1171, 1963. doi: 10.1136/bmj.2.5366.1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Dunham PB, Stewart GW, Ellory JC. Chloride-activated passive potassium transport in human erythrocytes. Proc Natl Acad Sci USA 77: 1711–1715, 1980. doi: 10.1073/pnas.77.3.1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Lauf PK. Thiol-dependent passive K/Cl transport in sheep red cells. IV. Furosemide inhibition as a function of external Rb+, Na+, and Cl. J Membr Biol 77: 57–62, 1984. doi: 10.1007/BF01871100. [DOI] [PubMed] [Google Scholar]
- 142.Delpire E, Lauf PK. Kinetics of Cl-dependent K fluxes in hyposmotically low K sheep erythrocytes. J Gen Physiol 97: 173–193, 1991. doi: 10.1085/jgp.97.2.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Delpire E, Lauf PK. Kinetics of DIDS inhibition of swelling-activated K-Cl cotransport in low K sheep erythrocytes. J Membrane Biol 126: 89–96, 1992. doi: 10.1007/BF00233463. [DOI] [PubMed] [Google Scholar]
- 144.Kaji D. Volume-sensitive K transport in human erythrocytes. J Gen Physiol 88: 719–738, 1986. doi: 10.1085/jgp.88.6.719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Muzyamba MC, Cossins AR, Gibson JS. Regulation of Na+-K+-2Cl− cotransport in turkey red cells: the role of oxygen tension and protein phosphorylation. J Physiol (Lond) 517: 421–429, 1999. doi: 10.1111/j.1469-7793.1999.0421t.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Palfrey HC, Leung S. Inhibition of Na-K-2Cl cotransport and bumetainde binding by ethacrinic acid, its analogues, and adducts. Am J Physiol Cell Physiol 264: C1270–C1277, 1993. doi: 10.1152/ajpcell.1993.264.5.C1270. [DOI] [PubMed] [Google Scholar]
- 147.Gagnon KB, England R, Delpire E. Volume sensitivity of cation-chloride cotransporters is modulated by the interaction of two kinases: SPAK and WNK4. Am J Physiol Cell Physiol 290: C134–C142, 2006. doi: 10.1152/ajpcell.00037.2005. [DOI] [PubMed] [Google Scholar]
- 148.Garzón-Muvdi T, Pacheco-Alvarez D, Gagnon KB, Vázquez N, Ponce-Coria J, Moreno E, Delpire E, Gamba G. WNK4 kinase is a negative regulator of K+-Cl- cotransporters. Am J Physiol Renal Physiol 292: F1197–F1207, 2007. doi: 10.1152/ajprenal.00335.2006. [DOI] [PubMed] [Google Scholar]
- 149.Piechotta K, Garbarini NJ, England R, Delpire E. Characterization of the interaction of the stress kinase SPAK with the Na+-K+-2Cl− cotransporter in the nervous system: evidence for a scaffolding role of the kinase. J Biol Chem 278: 52848–52856, 2003. doi: 10.1074/jbc.M309436200. [DOI] [PubMed] [Google Scholar]
- 150.Piechotta K, Lu J, Delpire E. Cation-chloride cotransporters interact with the stress-related kinases SPAK and OSR1. J Biol Chem 277: 50812–50819, 2002. doi: 10.1074/jbc.M208108200. [DOI] [PubMed] [Google Scholar]
- 151.Vitari AC, Deak M, Morrice NA, Alessi DR. The WNK1 and WNK4 protein kinases that are mutated in Gordon's hypertension syndrome, phosphorylate and active SPAK and OSR1 protein kinases. Biochem J 391: 17–24, 2005. doi: 10.1042/BJ20051180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Gagnon KB, England R, Delpire E. Characterization of SPAK and OSR1, regulatory kinases of the Na-K-2Cl cotransporter. Mol Cell Biol 26: 689–698, 2006. doi: 10.1128/MCB.26.2.689-698.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.CragoeEJ, Jr, Gould NP, WoltersdorfOW, Jr, Ziegler C, Bourke RS, Nelson LR, Kimelberg HK, Waldman JB, Popp AJ, Sedransk N. Agents for the treatment of brain injury. I. (Aryloxy)alkanoic acids. J Med Chem 25: 567–579, 1982. doi: 10.1021/jm00347a017. [DOI] [PubMed] [Google Scholar]
- 154.CragoeEJ, Jr, WoltersdorfOW, Jr, Gould NP, Pietruszkiewicz AM, Ziegler C, Sakurai Y, Stokker GE, Anderson PS, Bourke RS, Kimelberg HK, Nelson LR, Barron KD, Rose JR, Szarowski D, Popp AJ, Waldman JB. Agents for the treatment of brain edema. 2. [(2,3,9,9a-Tetrahydro-3-oxo-9a-substituted-1H-fluoren-7-yl)oxy]alkanoic acids and some of their analogs. J Med Chem 29: 825–841, 1986. doi: 10.1021/jm00155a038. [DOI] [PubMed] [Google Scholar]
- 155.Garay RP, Nazaret C, Hannaert PA, CragoeEJ, Jr.. Demonstration of a [K+Cl−]-cotransport system in human red cells by its sensitivity to [(dihydroindenyl)oxy]alkanoic acids: regulation of cell swelling and distinction from the bumetanide-sensitive [Na+, K+, Cl−]-cotransport system. Mol Pharmacol 33: 696–701, 1988. [PubMed] [Google Scholar]
- 156.Delpire E, Baranczak A, Waterson AG, Kim K, Kett N, Morrison RD, Daniels JS, Weaver CD, Lindsley CW. Further optimization of the neuronal K-Cl cotransporter KCC2 antagonist ML077: Development of a highly selective and more potent in vitro probe. Bioorg Med Chem Lett 22: 4532–4535, 2012. doi: 10.1016/j.bmcl.2012.05.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Delmotte Q, Hamze M, Medina I, Buhler E, Zhang J, Belgacem YH, Porcher C. Smoothened receptor signaling regulates the developmental shift of GABA polarity in rat somatosensory cortex. J Cell Sci 133: jcs247700, 2020. doi: 10.1242/jcs.247700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Ferrini F, Perez-Sanchez J, Ferland S, Lorenzo LE, Godin AG, Plasencia-Fernandez I, Cottet M, Castonguay A, Wang F, Salio C, Doyon N, Merighi A, De Koninck Y. Differential chloride homeostasis in the spinal dorsal horn locally shapes synaptic metaplasticity and modality-specific sensitization. Nat Commun 11: 3935, 2020. doi: 10.1038/s41467-020-17824-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Jin X, Kim WB, Kim MN, Jung WW, Kang HK, Hong EH, Kim YS, Shim WJ, Han HC, Colwell CS, Kim YB, Kim YI. Estrogen inhibits salt-dependent hypertension by suppressing GABAergic excitation in magnocellular AVP neurons. Cardiovasc Res. In press. doi: 10.1093/cvr/cvaa271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Raol YH, Joksimovic SM, Sampath D, Matter BA, Lam PM, Kompella UB, Todorovic SM, González MI. The role of KCC2 in hyperexcitability of the neonatal brain. Neurosci Lett 738: 135324, 2020. doi: 10.1016/j.neulet.2020.135324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Yin C, Ishii T, Kaneda M. Two types of Cl transporters contribute to the regulation of intracellular Cl concentrations in ON- and OFF-type bipolar cells in the mouse retina. Neuroscience 440: 267–276, 2020. doi: 10.1016/j.neuroscience.2020.06.004. [DOI] [PubMed] [Google Scholar]
- 162.Pégurier C, Bosman N, Collart P, Delporte ML, Leclercq K, Lengelé S, Kanduluru AK, Meunier S, Pacico N, Vadali LR, Wagner A, Wolff C, Provins L. Benzyl prolinate derivatives as novel selective KCC2 blockers. Bioorg Med Chem Lett 20: 2542–2545, 2010. doi: 10.1016/j.bmcl.2010.02.092. [DOI] [PubMed] [Google Scholar]
- 163.Gagnon M, Bergeron MJ, Lavertu G, Castonguay A, Tripathy S, Bonin RP, Perez-Sanchez J, Boudreau D, Wang B, Dumas L, Valade I, Bachand K, Jacob-Wagner M, Tardif C, Kianicka I, Isenring P, Attardo G, Coull JA, De Koninck Y. Chloride extrusion enhancers as novel therapeutics for neurological diseases. Nat Med 19: 1524–1528, 2013. doi: 10.1038/nm.3356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Cardarelli RA, Jones K, Pisella LI, Wobst HJ, McWilliams LJ, Sharpe PM, Burnham MP, Baker DJ, Chudotvorova I, Guyot J, Silayeva L, Morrow DH, Dekker N, Zicha S, Davies PA, Holenz J, Duggan ME, Dunlop J, Mather RJ, Wang Q, Medina I, Brandon NJ, Deeb TZ, Moss SJ. The small molecule CLP257 does not modify activity of the K(+)-Cl(−) co-transporter KCC2 but does potentiate GABA(A) receptor activity. Nat Med 23: 1394–1396, 2017. doi: 10.1038/nm.4442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Zhao Y, Shen J, Wang Q, Zhou M, Cao E. Inhibitory and transport mechanisms of the human cation-chloride cotransport KCC1. BioRxiv, 2020. doi: 10.1101/2020.07.26.221770. [DOI] [Google Scholar]
- 166.Yeo M, Berglund K, Hanna M, Guo JU, Kittur J, Torres MD, Abramowitz J, Busciglio J, Gao Y, Birnbaumer L, Liedtke WB. Bisphenol A delays the perinatal chloride shift in cortical neurons by epigenetic effects on the Kcc2/Slc12a5 promoter. Proc Natl Acad Sci USA 110: 4315–4320, 2013. doi: 10.1073/pnas.1300959110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Russell JM. Sodium-potassium-chloride cotransport. Physiol Rev 80: 211–276, 2000. doi: 10.1152/physrev.2000.80.1.211. [DOI] [PubMed] [Google Scholar]
- 168.Savardi A, Borgogno M, Narducci R, La Sala G, Ortega JA, Summa M, Armirotti A, Bertorelli R, Contestabile A, De Vivo M, Cancedda L. Discovery of a small molecule drug candidate for selective NKCC1 inhibition in brain disorders. Chem 6: 2073–2096, 2020. doi: 10.1016/j.chempr.2020.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Tyzio R, Nardou R, Ferrari DC, Tsintsadze T, Shahrokhi A, Eftekhari S, Khalilov I, Tsintsadze V, Brouchoud C, Chazal G, Lemonnier E, Lozovaya N, Burnashev N, Ben-Ari Y. Oxytocin-mediated GABA inhibition during delivery attenuates autism pathogenesis in rodent offspring. Science 343: 675–679, 2014. doi: 10.1126/science.1247190. [DOI] [PubMed] [Google Scholar]
- 170.Reid MS, Kern DM, Brohawn SG. Cryo-EM structure of the potassium-chloride cotransporter KCC4 in lipid nanodiscs. eLife 9: e52505, 2020. doi: 10.7554/eLife.52505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Xie Y, Chang S, Zhao C, Wang F, Liu S, Wang J, Delpire E, Ye S, Guo J. Structures and an activation mechanism of human potassium-chloride cotransporters. Sci Adv 6: eabc5883, 2020. doi: 10.1126/sciadv.abc5883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Yamashita A, Singh SK, Kawate T, Jin Y, Gouaux E. Crystal structure of a bacterial homologue of Na+/Cl−-dependent neurotransmitter transporters. Nature 437: 215–223, 2005. doi: 10.1038/nature03978. [DOI] [PubMed] [Google Scholar]
- 173.Delpire E, Guo J. The cryo-EM structures of DrNKCC1 and hKCC1: a new milestone in the physiology of cation-chloride cotransporters. Am J Physiol Cell Physiol 318: C225–C237, 2020. doi: 10.1152/ajpcell.00465.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Korpi ER, Luddens H. Furosemide interactions with brain GABAA receptors. Br J Pharmacol 120: 741–748, 1997. doi: 10.1038/sj.bjp.0700922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Flores B, Schornak CC, Delpire E. A role for KCC3 in maintaining cell volume of peripheral nerve fibers. Neurochem Int 123: 114–124, 2019. doi: 10.1016/j.neuint.2018.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]