

Abstract
About 10–15% of all human cancer cells employ a telomerase-independent recombination-based telomere maintenance method, known as alternative lengthening of telomere (ALT), of which the full mechanism remains incompletely understood. While implicated in previous studies as the initiating signals for ALT telomere repair, the prevalence of non-canonical nucleic acid structures in ALT cancers remains unclear. Extending earlier reports, we observe higher levels of DNA/RNA hybrids (R-loops) in ALT-positive (ALT+) compared to telomerase-positive (TERT+) cells. Strikingly, we observe even more pronounced differences for an associated four-stranded nucleic acid structure, G-quadruplex (G4). G4 signals are found at the telomere and are broadly associated with telomere length and accompanied by DNA damage markers. We establish an interdependent relationship between ALT-associated G4s and R-loops and confirm that these two structures can be spatially linked into unique structures, G-loops, at the telomere. Additionally, stabilization of G4s and R-loops cooperatively enhances ALT-activity. However, co-stabilization at higher doses resulted in cytotoxicity in a synergistic manner. Nuclear G4 signals are significantly and reproducibly different between ALT+ and TERT+ low-grade glioma tumours. Together, we present G4 as a novel hallmark of ALT cancers with potential future applications as a convenient biomarker for identifying ALT+ tumours and as therapeutic targets.
Graphical Abstract
Graphical abstract.
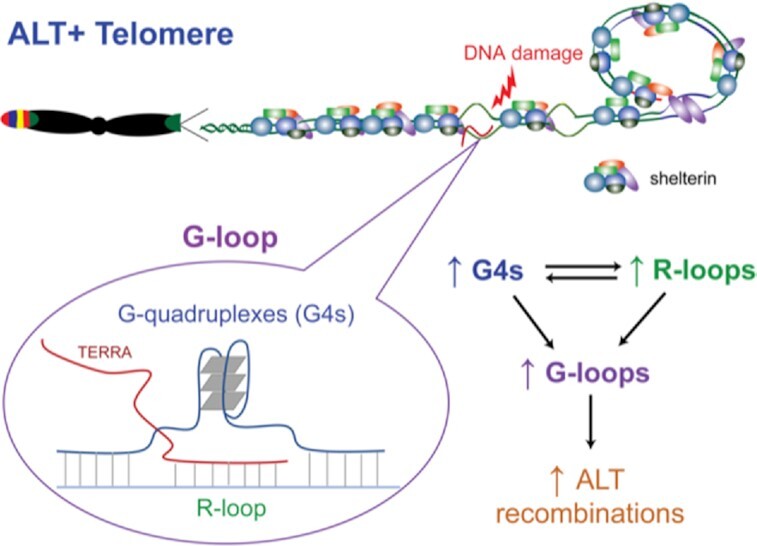
ALT+ cells promote a telomeric environment that leads to increased formation of G-quadruplexes and R-loops. Co-formation of G-quadruplexes and R-loops results in special structures, G-loops, which are postulated to facilitate the ALT recombination process.
INTRODUCTION
Telomeres are essential protective structures that cap the ends of chromosomes to maintain the termini's integrity and stability. All cancers require a functional telomere maintenance mechanism to sustain replicative immortality (1). While the majority (∼85–90%) of cancers utilize a telomerase-dependent (TERT+) method of telomere maintenance, a subset (∼10–15%) of cancers adopt telomerase-independent alternative-lengthening of telomeres (ALT or ALT+) mechanisms. Some notable features of ALT+ cells include long mean telomere lengths, a higher number of degenerate variant telomere repeats, elevated levels of ALT-associated Promyelocytic Leukemia (PML) bodies and extrachromosomal telomeric DNA (especially c-circles, single-stranded extrachromosomal C-rich telomeric circular DNA) (1,2). ALT is prevalent in certain cancers, including osteosarcomas, soft tissue sarcoma and central nervous system tumours (3,4). Clinically, the prevalence of ALT in several types of cancers, including pancreatic neuroendocrine tumour (5,6) and soft tissue sarcoma (7), has been associated with unfavourable prognosis and increased mortality. Despite being discovered more than two decades ago, the mechanisms behind ALT are still not fully understood, necessitating the need to further study the biology of ALT in hopes of spurring therapeutic development.
Specifically, the telomeric chromatin environments are significantly different between ALT+ and TERT+ cancer cells, and a permissive chromatin environment may be necessary for proper engagement of the ALT mechanism. Among the differences, a reduction in the compaction state of ALT telomeres is thought to facilitate the use of recombination, as well as enhanced transcriptional activity leading to the increased levels of telomeric repeat-containing RNA (TERRA) (8–11). While multiple factors may be involved in establishing the unique state of ALT telomeric chromatin, there are two well-characterized factors. First, over 80% of all ALT cancers are observed to harbour genetic dysfunction of the Alpha Thalassemia/Mental Retardation Syndrome X-linked Chromatin Remodeler (ATRX) or its partner Death Domain Associated Protein (DAXX), which function as a chromatin remodelling complex in histone H3.3 deposition and heterochromatin maintenance (12,13). Thus, ATRX has been suggested to suppress ALT, but the exact mechanisms are still unclear (14,15). An intriguing hypothesis proposed is that ATRX may be involved in suppressing DNA secondary structures (15). Second, ALT+ cancers carry higher numbers of degenerate variant telomeric repeats that cannot associate properly with shelterin, the six-membered telomeric DNA-binding protein complex. Instead, these variant repeats are recognized by orphan nuclear receptors that promote chromatin decompaction (16–18).
Telomere elongation in ALT undergoes a type of homology-directed repair (HDR) process related to that of the break-induced replication (BIR) pathway, specifically called break-induced telomere synthesis (BITS) (2). Two different BITS pathways have been identified, including the slower RAD51-dependent HOP2-MND1-mediated long-range homology search mechanism (19) and the faster RFC-PCNA DNA damage sensor-mediated mechanism (20). Additionally, replication defects at an ALT telomere can trigger the RAD52-dependent mitotic DNA synthesis (MiDAS) mechanism (21). While all these ALT pathways share a common prerequisite in that initiation of the recombination process requires some form of persistent DNA damage, the full mechanism of the induction of this damage signal remains unclear.
Given the ‘open’ and more permissive telomere environment in ALT, we and others speculate that there is a higher chance of forming non-canonical structures at these loci and that these structures may be the source of the persistent DNA damage for ALT initiation. Recently, multiple reports have implicated a role of telomeric R-loops (TelR-loops), DNA/RNA hybrid structures resulting from the association of TERRA transcribed from ALT telomeres (10,22,23), in the ALT mechanisms. Intriguingly, R-loop formation displaces the G-rich telomeric DNA strand, increasing the opportunity to form another non-canonical nucleic acid structure, the G-quadruplex (G4). G4s are formed by Hoogsteen bonding of adjacent guanine bases to form planar units, the G-quartets, which then stack on each other through π-stacking to form the overall columnar structure. While potential G4 forming sequences (pG4s) are widespread in the genome (24) and transcriptome (25), telomeric regions hold the largest pG4 reservoir due to their long consecutive G-rich repeats. We speculate that this is especially true in ALT+ cells due to their longer telomere tracks and more permissive telomeric chromatin environment. To expand our understanding of the biology of ALT, we set out to examine the relationship between ALT, R-loops and G4s.
We observed higher nuclear G4 and R-loop levels in ALT+ compared to TERT+ cells. Quantification of telomeric G4 and R-loop signals revealed their associations with telomere length and DNA damage signals. Additionally, our results revealed the increased formation of ALT-specific G4/R-loop linked structures, G-loops, which may play a role in stimulating ALT activity. We demonstrated that G4-specific antibodies and chemical probes can differentiate between low-grade glioma tumours that were typed as either ALT+ or TERT+ based on their ATRX status. Together, we present enrichment of nuclear G4s as hallmarks of ALT+ cancers and provide the proof-of-principle for exploiting these nucleic acid structures as future biomarkers and therapeutics.
MATERIALS AND METHODS
Cell line and culture
Cell lines (SKLU1, U2OS, SAOS-2, HELA, MCF7, HT1080) were obtained from American Type Culture Collection (ATCC). GM847 was obtained from the Coriell Institute cell repository. SUSM1 and HELA1.3 were obtained from Collins Lab (UC Berkeley, U.S.A.). Additional information of cell lines can be found in Supplementary Table S1. The cells were cultured in Dulbecco’s modified Eagle’s medium (Gibco) supplemented with 5% synthetic fetal bovine serum (FetalClone III; GE Life Sciences) and 1% penicillin-streptomycin mixture (Gibco). Incubation was done at 37°C in a humidified, 5% CO2 atmosphere-controlled incubator (HERAcell). All cell culturing was performed using standard procedures, including aspiration and washing with phosphate-buffered saline (PBS, Gibco), trypsinizing using Trypsin-EDTA (Gibco). Coulter Counter (Beckman Coulter) was used for cell counting. Transfection protocols can be found in the Extended Methods (Supplementary File).
Immunocytochemistry (ICC) Staining
Cell lines were fixed with 100% ice-cold methanol. The fixed cells on coverslips were then blocked in blocking solution (4% BSA/TBS) for 1 h. For any other primary antibodies used, 2 μg/ml of antibody diluted in 250 μl of block solution was used and incubated at 4°C overnight. For BG4 (G4-specific antibody) staining, samples were stained with 2 μg/ml BG4 for 3 h at room temperature before staining with 2 μg/ml anti-FLAG antibody overnight at 4°C. The coverslips were then washed three times with PBS-T (0.02% Tween) for 10 min each. Appropriate secondary antibodies were added at 1 μg/ml and incubated at room temperature for 2 h. The coverslips were washed three times with PBS-T for 10 min each, counterstained in DAPI and mounted using Fluoromount G (Southern Biotech). A detailed list of antibodies used in this study are listed (Supplementary Table S2). The slides were imaged on Zeiss LSM700 confocal. The raw images were then processed and quantified using CellProfiler (26) using a custom pipeline for nuclear foci. Large foci were detected by filtering signals within the 10–50 pixel range (One pixel is equivalent to 0.08 × 0.08 μm (0.0064 μm2)). Small foci were detected by excluding signals above 10 pixels and passing signals below 10 pixels. Nuclear intensity was detected by the total signal within the nuclear mask as defined by the DAPI signal. For the overlap analysis, EZColocalization (27), a plugin in ImageJ, was used to generate a metric matrix for threshold overlap score (TOS) using default settings.
Chromatin-immunoprecipitation (ChIP) assay
Cells were seeded at a density of 250 000–750 000 (depending on the cell line) cells per 10-cm dish. The cells were treated with ligands of interest the next day. Cells were washed once with PBS and crosslinked in 1% formaldehyde/fixation buffer (50 mM HEPES KOH, pH 7.5, 100 mM NaCl, 1 mM EDTA pH 8, 0.5 mM EGTA, pH 8) for 5 min and quenched with 0.125 M glycine for 5 min. Cells were then resuspended in shearing buffer (0.1% SDS, 1 mM EDTA, 10 mM Tris, pH 8.1) sonicated using Covaris m220 at 10% duty and chromatin shearing preset setting for 4 min. Sonicate was then spun down at 13 200 rpm for 10 min at 4°C, and the supernatant is moved to a new tube. Sonicate was then precleared and diluted 1:1 with 2× IP buffer (0.1% SDS, 1 mM EDTA, 10 mM Tris pH 8.1, 2% Triton-X, 300 mM NaCl), and 0.1% BSA was added to each IP. An aliquot was saved for the input control. About 4–5 μg of antibody was added to each ChIP tube and no antibody was added to the control tube. The IP was performed overnight at 4°C on the rotator. On the following day, 25 μl of 50% slurry of pre-blocked agarose beads (M2 anti-FLAG [Sigma, A2220] for G4-ChIP or Protein G Sepharose beads [GE Life Sciences, 17061802] for other ChIPs) and were incubated at 4°C on a rotator for 2 h. Beads were centrifuged at 5000 rpm and washed sequentially twice for each buffer for 5 min each: ChIP Wash Buffer A (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris-HCl, pH 8, 150 mM NaCl), Wash Buffer B (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris-HCl, pH 8, 500 mM NaCl), Wash Buffer C (0.25 M LiCl, 1% NP-40, 1% sodium deoxycholate, 1 mM EDTA, 10 mM Tris-HCl, pH 8) and TE (10 mM Tris-HCl, pH 8, 1 mM EDTA). The sample was incubated in 1% SDS/TE buffer at 65°C overnight to reverse the crosslink and and DNA-purified with a DNA purification kit (BioBasics) using the manufacturer ’s recommended protocol. The extended protocol for ChIP-ReChIP and ChIP in nuclease-treated samples can be found in the Extended Methods (Supplementary File).
Quantitative PCR for ChIP
Telomeric DNA was quantified using real-time quantitative PCR (q-PCR) on the BioRad Opticon 2. PCR reaction was set up using tel1b primer (5′-CGGTTTGTTTGGGTTTGGGTTTGGGTTTGGGTTTGGGTT-3′) with final concentration of 100 nM and tel2b primer (5′-GGCTTGCCTTACCCTTACCCTTACCCTTACCCTTACCCT-3′) with final concentration of 500 nM. Alu repeat was used as genome background control. Alu DNA was quantified using Alu forward primer (5′-GACCATCCCGGCTAAAACG-3′) and reverse primer (5′-CGGGTTCACGCCATTCTC-3′) at a final concentration of 100 nM. The master mix was made using SYBR mix (ABM), forward primer, reverse primer and PCR-grade H2O. Each ChIP sample was then quantified by running three technical repeats. Tel/Alu ChIP signal ratio was then computed, and the ratio of each cell line was then normalized to that of HELA. For comparisons between conditions, the ChIP signal was first normalized to the input then the Tel/Alu ratios were calculated from the normalized values. The input-normalized Tel/Alu ratios were then normalized against the appropriate controls (either GFP-empty vector control or non-treated control).
Proximity ligation assay (PLA)
Cells were seeded on coverslips in a 12-well plate at a density of 10 000 cells per well. One day post-seeding, cells were washed with PBS, fixed with ice-cold methanol for 10 min, and permeabilized with ice-cold acetone for 1 min. After blocking in 3% BSA, 0.1% Tween-20 in 4XSSC for 1 h at room temperature, cells were then incubated with or without 2 μg/ml BG4 in blocking solution for 3 h at room temperature on a rocker. Following 3× PBST wash for 5 min, cells were incubated with primary antibodies [1:250 rabbit anti-S9.6 antibody (Ab01137-23.0, Absolute Antibody) and 1:500 mouse anti-Flag-M2 antibody (F1804, Sigma)] at 4°C overnight. The next day, subsequent steps in proximity ligation assay were carried out with Duolink In Situ Kit (Sigma) following the manufacturer’s instructions. The samples were imaged on LeicaDM18 microscope at 100×. Negative controls were included in each experiment, treated identically but either without BG4 antibody incubation for 3 h or without anti-S9.6 antibody overnight.
C-circle assay
C-circle assay (CCA) coupled with qPCR quantification was performed using a modified version of a published protocol (28). Cells were then harvested by scraping and centrifugation. The cell pellets were then incubated at 56°C in 50 μl of Quick C-circle Preparation (QCP) Buffer (50 mM KCl, 10 mM Tris-HCl, 2 mM MgCl2, 0.5% IGEPAL CA-630 detergent, 0.5% Tween-20) supplemented with fresh 1 mg/ml Proteinase K (Sigma) and vortex intermittently for 1 h. DNA from the lysate was then purified using column purification (BioBasic). The purified DNA was then diluted 10× in elution buffer. About 10 μl of the diluted material is added to 9.25 μl of 2.16× CCA Buffer (final reaction concentration of 4 mM DTT, 4 μg/ml BSA, 0.1% Tween-20, 1 mM dATP, 1 mM dGTP, 1 mM dTTP, 1 mM dCTP, 1× phi29 buffer) and 0.75 μL phi29 polymerase (NEB). Two reaction tubes per sample were made: one with phi29 polymerase added (Phi+) and one without (Phi-). Samples were incubated at 30°C for 8 h then 70°C for 20 min. Real-time qPCR was performed using the Tel1b/Tel2b primer pair. The ratio of Phi+/Phi- signal was calculated for the raw CCA signal. Real-time qPCR was performed on a single-copy gene, ACTB, with the primer pair (forward: CTGGAACGGTGAAGGTGACA, reverse: AAGGGACTTCCTGTAACAACGCA) and used quantified for input differences. The raw CCA signals were normalized between samples using ACTB.
Cytotoxicity profiling
Cytotoxicity screening of ligands was performed as previously described (29). Cells were seeded at 3000–6000 cells per well in a 96-well plate and were grown for 24 h. Cells were then treated with varying doses of ligands. The plate was then inserted in the Incucyte ZOOM Live-cell Imaging System (Essen Bioscience) and imaged at 3 h intervals for 72 h. Phase confluency was measured for each well over the period and exported into numerical values. The confluency values were then normalized per experiment. Dose–response curves were then plotted using GraphPad Prism.
Immunohistochemistry (IHC) staining using BG4
FFPE slides were first deparaffinized by immersing in twice in Xylene (Sigma) for 10 min each. The slides were then immersed in 100% ethanol twice for 10 min each, followed by 95% ethanol for 5 min, 70% ethanol for 5 min and 50% for 5 min. The slides were rinsed with dH2O and rehydrated with PBS. A short antigen retrieval step was included by immersing the slides in preheated citrate buffer at 95°C for 2 min. The slides were then cooled and rinsed with dH2O, followed by PBS. For brief permeabilization, the slides were immersed in 0.2% Triton-X/PBS for 5 min. The staining areas on the tissue slides were circled using a hydrophobic marker. Blocking buffer (4% BSA/TBS) was dropped to the areas of interest and incubated at room temperature for 1 h. Then, the slides were drained, and BG4 antibody (final concentration: 5 μg/ml) in blocking buffer was added to each slide and incubated for 2.5 h. The slides were washed three times in PBS-T for 5 min each. Anti-FLAG antibody (Sigma) (final concentration: 3 μg/ml) was added to slides and incubated overnight at 4C in a humidified chamber. The slides were washed three times in PBS-T for 10 min each. The secondary antibody, AF-594 (Thermos) (final concentration: 2 μg/ml), was added to the slides and incubated for 1.5 h. The slides were washed three times in PBS-T for 10 min each. The slides were counterstained with DAPI for 5 min and washed twice in PBS for 5 min each. Flouromount G was used to mount a clean coverslip to the slide.
IHC using Naptho-Template assembled synthetic G-quartet (N-TASQ) probe
The deparaffinization steps were performed the same as described above. The slides were blocked with a blocking buffer for 30 min. The slides were drained, and N-TASQ in blocking buffer (final concentration: 40 μM) was added to each slide and incubated for 1 h. The slides were washed three times in PBS-T for 10 min each. The slides were counterstained with DAPI for 5 min and washed twice in PBS for 5 min each. Fluoromount G was used to mount a clean coverslip to the slide.
Statistical analysis
Statistical analysis of the comparison between ALT+ and TERT+ cell lines was performed using Student’s t-test on aggregated datasets from multiple cell lines. One-way or two-way ANOVA with multiple-test corrections was performed for comparison between conditions. All statistical analyses were performed using GraphPad Prism. All error bars represent SEM unless otherwise specified.
RESULTS
G4s are elevated in the nucleus of ALT+ cells compared to TERT+ cells
The ALT status of different cell lines was confirmed by measuring the absolute or relative telomere lengths with telomere restriction fragment (TRF) analysis or quantitative PCR, respectively, and examining PML levels by immunocytochemistry (ICC) (Supplementary Figure S1). These analyses classified four ALT+ (i.e. exhibiting longer telomeres and more PML bodies) and four TERT+ cell lines for further experiments. We also added HELA1.3, a HELA variant that is also a TERT+ cell line with telomere length comparable to those observed in ALT+ cell lines. While R-loop’s importance in ALT has previously been implicated through studies into the roles of R-loop-modifying factors such as RNaseH1 (22) and FANCM (23), quantitative immunofluorescence imaging detection and direct comparison in multiple cell lines are lacking. To study this, we performed ICC using the R-loop targeting antibody, S9.6 (30), in the panel of confirmed ALT+ and TERT+ cell lines. Antibody specificity was tested using RNaseH digestion (Supplementary Figure S2). We observed a higher number of large-sized nuclear S9.6 foci in ALT+ cells than TERT+ cells (Supplementary Figure S3); however, the foci intensity remained similar. We became interested in examining the levels of G4s, which are structurally compatible structures with R-loops, to determine whether their levels are similarly higher in ALT+ than TERT+ cell lines.
We next performed ICC staining of an expanded panel of ALT+ and TERT+ cell lines using the G4-specific antibody, BG4 (31). The BG4 antibody was validated through testing G4 ligand-induced increases in foci (Supplementary Figure S2). We observed qualitative and quantitative differences between the nuclei of ALT+ and TERT+ cells (Figure 1A). Notably, similar to that of R-loops, we determined that a higher number of large G4 foci and an increase in these foci’s intensity were associated with ALT+ cells compared to TERT+ cells (Figure 1B). Quantification of the signals outside the large foci (i.e. signals from small foci) and the broad nuclear signals (i.e. sum of all nuclear signals) were also revealed to be higher in ALT+ cells (Figure 1C and D). While the identities of these large foci (or nuclear bodies) were unclear, we hypothesized that telomeres could be a significant contributor to these signals due to the longer ALT+ telomere track being reservoirs of abundant G4-forming sequences.
Figure 1.

ALT+ cells harbour higher level of G4s than TERT+ cells. (A) Immunocytochemistry (ICC) straining experiments for G4 in ALT+ cells lines (within blue frame) versus TERT+ (within red frame) (B) Mean large G4 foci count (top) and foci intensity (bottom) of G4 foci per nucleus. (C) Mean small G4 foci count per nucleus as quantified. (D) Mean G4 total nuclear intensity per nucleus as quantified. Automated quantification of 100 cells per cell line was performed in parallel. Two-tailed t-tests were performed for comparing ALT+ versus TERT+ groups. Error bars represent the SEM.
Telomeric G4 and R-loop levels are correlated with telomere length in ALT+ cells and associated with DNA damage signals
Some G4 and R-loop foci from our analyses appeared to resemble the large telomere foci widely seen in ALT+ cells in multiple previous studies (3,4,32). While a limited number of reports on R-loop formation in ALT telomere exists, G4 formation in ALT telomere has been unexplored. We reasoned that the observed increases in G4 foci number and intensity could represent long clusters of telomeric G4s. To probe whether telomere accounts for some of the nuclear signals, we performed co-staining ICC experiments for G4 with an antibody against Telomeric Repeat Binding Factor 2 (TRF2), a member of the shelterin telomere-binding protein complex. It is also noteworthy that canonical repeats are interspersed within the ALT telomere and that shelterin proteins still associate with the degenerate repeats at ALT telomere but to a lesser extent. Thus, TRF2 clusters are expected within ALT cells. We found a high degree of nuclear colocalization, as evidenced by the increase in threshold overlap score (TOS) when shifting the threshold towards the top 10 percentile of pixels by intensity (Supplementary Figure S4).
For robust quantification of telomeric G4s and R-loops, we performed chromatin-immunoprecipitation coupled with real-time quantitative polymerase chain reaction (ChIP-qPCR) assay. We chose to focus on five cell lines within the panel with varying telomere lengths. We used Alu repeats as a genome background control between different cell lines and calculated Tel/Alu ratios to measure the level of telomeric localization. Both G4-ChIP and R-loop-ChIP showed higher Tel/Alu ChIP ratios in ALT+ compared to TERT+ cell lines. Additionally, we separately plotted the Tel/Alu ratios derived from G4-ChIP or R-loop-ChIP experiments against each cell line’s corresponding telomere length. Linear regression plot of G4-ChIP signals against telomere length showed a strong positive correlation between the two variables (R2 = 0.96) (Figure 2A and B). Likewise, R-loop-ChIP displayed a similar pattern (R2 = 0.97). We repeated the linear regression analyses with the addition of data from HELA1.3, a long-telomere TERT+ cell line. Notably, HELA1.3 was a clear outliner in the linear regression plots (Figure 2A and B, red line). Accordingly, after the addition of HELA1.3 to the analyses, the coefficients of determination (R2) were significantly decreased for both TelG4 (R2 = 0.42) and TelR-loop (R2 = 0.11). Thus, we concluded that while TelG4 and TelR-loop levels are highly correlative with telomere length, a longer telomere length does not fully explain the magnitude of TelG4 and TelR-loop signals in ALT+ cells.
Figure 2.

Levels of telomeric G4s and R-loops are higher in ALT+ cells and are broadly associated with telomere length and coupled with DNA damage signals. (A andB) Top; (A) G4-ChIP assay or (B) R-loop-ChIP assay in ALT+ cell lines (blue shades) versus TERT+ cell lines (red shades). Data shown are the means from three biological repeat experiments. T-test (two-tailed) was performed for the combined ALT+ versus TERT+ datasets. Bottom; linear regression plot of Tel/Alu ChIP signal ratio against telomere length (as measured by telomere restriction fragment (TRF) analysis). Line of best fit is plotted along with R2 values with HELA1.3 (red) or without HELA1.3 (black). (C) ChIP-Re-ChIP (γH2AX-G4) and quantified for telomeric DNA (Tel DNA) in ALT+ cells, GM847 and SKLU1. The ReChIP signal for each sample was then normalized to its respective beads only control. Two-way ANOVA with Sidak multiple correction was performed. Data shown are the means from three biological repeat experiments. Error bars represent the SEM. (D) ChIP-ReChIP (first ChIP for R-loop followed by second ChIP for G4) and quantified for Tel DNA in ALT+ cells, GM847 and SKLU1. ReChIP signal for each sample was then normalized to its respective beads only control. Two-way ANOVA with Sidak multiple correction was performed. Data shown are the means from three biological repeat experiments. Error bars represent the SEM.
ALT+ telomere harbours elevated levels of endogenous DNA damage signals (12), and these damage signals may present initiation signals for recombination-based repair. To probe whether TelG4s could be involved in the increased DNA damage signals, we first performed ChIP for the DNA damage response (DDR) marker, γH2AX, and confirmed higher telomere-enriched signals of DNA damage in ALT+ cells compared to TERT+ cells, as well as a strong positive correlation with telomere length (R2 = 0.91) (Supplementary Figure S5). To demonstrate that the increases in telomeric DNA damage signals are closely associated with G4 formation, we performed the ChIP-ReChIP assay, a sequential ChIP using γH2AX antibody in the first ChIP followed by Re-ChIP of the eluate with the BG4 antibody in the second ChIP reaction and subsequent quantification using qPCR (Figure 2C). The antibody order was chosen based on ReChIP compatibility screening of the antibodies in which BG4 was found to be more tolerant of the rigorous ReChIP procedure and thus applied for the second ChIP. ReChIP enrichment of telomeric DNA signals over beads-only control in both ALT+ cell lines, GM847 and SKLU1, indicated close proximity (within approximately 500 bp, the median ChIP sonicated chromatin length) between telomeric G4s and DNA damage signals. The similar telomere-length associated ChIP signal pattern for G4 and R-loop suggested that these structures may also be situated closely. To confirm this assertion, we performed ICC co-staining for G4 and R-loop, which showed strong colocalization between these structures in multiple ALT+ cell lines (Supplementary Figure S6). Importantly, ChIP-ReChIP experiments showed that G4 and R-loop structures are also co-enriched at the ALT telomere (Figure 2D).
Telomeric G4s and R-loops are interdependent in ALT+ cells
Given the elevated levels of both TelG4s and TelR-loops in ALT+ cells, as well as the mechanistic plausibility of their co-formation, we examined whether these two structures can affect each other’s formation or stability. To study the effect of R-loop changes on G4 abundance, we targeted biological pathways responsible for R-loop resolutions. Telomeric R-loops are resolved mainly in cells by RNaseH1 (22) and FANCM (23). We first overexpressed recombinant GFP-tagged RNaseH1 in GM847 ALT+ cells to reduce the cellular levels of R-loops. ICC staining for G4s in GM847 overexpressing RNaseH1 showed decreased G4 levels compared to those seen in control GFP-overexpressing cells (Figure 3A). Using ChIP, we observed parallel reductions of TelG4 and TelR-loops upon overexpression with RNaseH1 (Figure 3B).
Figure 3.

G4s and R-loops are interdependent. (A) ICC staining for GFP (green) and G4 (red) in GM847 transfected with GFP-vector control (within orange frame) or GFP-RNaseH1 (within green frame). Right, Quantification of GFP+ cells for mean BG4 foci count per nucleus. T-test (two-tailed) was performed. (B) G4-ChIP or R-loop-ChIP results from GM847 transfected with either GFP-vector or GFP-RNaseH1. Tel/Alu ChIP signals were then normalized against GFP-vector control. Data shown are the mean from three biological repeat experiments. Two-way ANOVA with Sidak multiple comparisons was performed. Error bars represent the SEM. (C) Left, R-loop ICC staining (green) in ALT+ cell lines (GM847, SKLU1, U2OS) treated with vehicle or G4 ligand, RHPS4 (1, 1.5 and 5 μM, respectively). Mean R-loop foci count (center) and intensity (right) per nucleus were quantified. About 100 cells per treatment were quantified. Two-way ANOVA with Sidak-corrected multiple comparisons was performed against the untreated controls. (D) G4 ICC staining (red) in GM847 with vehicle or R-loop inducer, PIP-199 0.75 μM. A total of 50 cells per treatment were quantified. T-test (two-tailed) was performed. (E) G4-ChIP or R-loop-ChIP of GM847 treated with vehicle, 0.75 μM PIP-199 or 1 μM RHPS4. ChIP signal was first normalized to input then the Tel/Alu ratio calculated for each condition was then normalized against that of the vehicle control. Data shown are the mean from three biological repeat experiments. Two-way ANOVA with Sidak-corrected multiple comparisons was performed against the vehicle control. Error bars represent the SEM.
We also tested whether changes in G4s can affect R-loop accumulation. Treatment with G4 ligands, small molecules that can bind and increase the stability of G4s, is a well-established method to study G4-related biological changes. We used sub-lethal doses of RHPS4, a pentacyclic acridine G4 ligand, which can target a multitude of G4s, including those within the telomeres (33). ICC staining experiments revealed significantly increased R-loops in ALT+ cells treated with RHPS4 (Figure 3C). In addition, we tested the effects of a newly identified small-molecule telomeric R-loop inducer, PIP-199, which indirectly increases R-loop accumulation through the inhibition of MM2-RMI1/2 interactions that disrupts the formation of the FANCM-BTR (BLM-TOP3-RMI1) complex (34). PIP-199 is not known to directly affect BLM helicase activity or G4 stability but instead breaks the cooperation of the FANCM and BTR complexes which have R-loop dissolution activity. ICC staining experiments revealed PIP-199-induced increases in G4 levels in ALT+ cells (Figure 3D). We performed parallel ChIP experiments to quantify the levels of TelG4 and TelR-loop in the vehicle-, PIP-199- and RHPS4-treated GM847 ALT+ cells. Treatments with both chemicals showed significant and parallel increases in G4 and R-loop levels, with PIP-199 (R-loop inducer) displaying a parallel 1.5–2 fold increases in TelR-loop and corresponding TelG4, and RHPS4 (G4 ligand) displaying a parallel 3–4 fold increases in TelG4 and corresponding TelR-loop (Figure 3E). Thus, we showed that both positive (using PIP-199) and negative changes (using RNaseH1) in TelR-loop levels induced corresponding changes in TelG4 stability in the same direction. Together, our ICC and ChIP experiments quantitatively showed that the two telomeric structures are interdependent.
Telomeric G4s and R-loops form G-loops to stimulate ALT activity cooperatively
Our observation of G4-R-loop interdependency suggested that they may form a linked structure, a G-loop, in which G4 and R-loop form on opposing strands (35). To confirm colocalization, we performed ICC co-staining experiments for G4 and R-loop and observed highly colocalized nuclear staining, as illustrated by the TOS increase when examining the top 10 percentile of pixels by intensity (Supplementary Figure S6). We employed the proximity ligation assay (PLA) with BG4 and S9.6 antibodies to probe for high-resolution colocalization of G4s and R-loops within the nanometer range. We observed a significantly higher number of G4:R-loop PLA signals, representing potential G-loop formation sites, in ALT+ cells compared with TERT+ cells (Figure 4A). As controls, these PLA signals were efficiently removed by in vitro RNaseH1 treatment. Additionally, we evaluated G4:R-loop PLA signals in GM847 and U2OS ALT+ cells overexpressing RNaseH1 and showed significant reductions in PLA foci compared to those of the vector control (Supplementary Figure S7). Reciprocally, we used the siRNA-mediated knockdown of FANCM to increase the cellular accumulation of R-loops. We demonstrated significant increases in G4:R-loop PLA foci in GM847 and U2OS cells treated with FANCM-siRNA compared to control-siRNA knockdown cells (Supplementary Figure S7).
Figure 4.

G4s and R-loops are spatially linked into G-loops at higher levels in ALT+ cells and cooperate to stimulate ALT activity. (A) Top, Proximity ligation assay for G4/R-loop colocalization in a panel of ALT+ and TERT+ cell lines. Left, Quantification of the mean PLA foci per nucleus. 200 cells per condition were quantified. In vitro RNAseH1 digestion was used as a control for each cell line. One-way ANOVA with Dunnett’s multiple test correction was performed comparing non-treated samples and RNaseH1-treated samples. Right, Pie charts depicting the distribution of PLA foci per nucleus. T-test (two-tailed) was performed for the combined set of ALT+ versus TERT+ cell lines. (B) C-circle assay (CCA) followed by qPCR in (Left) GM847 treated with either vehicle (black), RHPS4 1.25 μM (red), PIP-199 0.37 μM (blue) or cotreated with both RHPS4 and PIP-199 (purple) (Right) U2OS treated with either vehicle (black), RHPS4 5 μM (red), PIP-199 0.37 μM (blue) or cotreated with both RHPS4 and PIP-199 (purple). C-circle score from quantifying Tel DNA ratio between the presence and absence of phi polymerase was first normalized to single copy gene then normalized to the vehicle control. One-way ANOVA with Dunnett’s multiple test correction was performed. Data were collected from three biological repeat experiments. (C) Curve-shift analysis in ALT+ cells, GM847, SKLU1, U2OS and SAOS-2. RHPS4 cytotoxicity dose response curve was generated with treatment of either DMSO vehicle control or a nontoxic dose of PIP199 (0.75, 3, 0.75 and 1.5 μM, respectively) for each cell line. The LD50, dose kills half of the cell population, was calculated with 95% confidence intervals for each condition per cell line, from data collected in three biological repeat experiments. Error bars represent the SEM.
Since our combined results indicated higher endogenous G-loop levels in ALT+ cells, we surmised that these unique combined structures could play a role in the ALT telomere maintenance mechanism. To test this, we either single-treated or co-treated GM847 and U2OS cells with a previously determined sub-lethal dose of RHPS4 and PIP-199 (Supplementary Figure S8) to increase TelG4 and TelR-loop levels, respectively. After 24 h treatment, we measured changes in c-circle levels, the most widely used and sensitive ALT activity measure, using the rolling circle amplification and qPCR method (28). The sensitivity of the C-circle Assay (CCA)-qPCR method was demonstrated by the significant increase of Tel DNA signals with the addition of phi29 (φ) polymerase (+Phi) to that of a mock reaction control (-Phi) in ALT+ cells (∼10-fold in GM847 and ∼6.5-fold in U2OS) compared to TERT+ cells (∼1.4-fold in HELA 1.3) (Supplementary Figure S9). Single-agent treatments with G4 ligand or R-loop inducer induced a slight but reproducible increase in CCA score for GM847 ALT+ cells, while the co-treatment significantly increased the CCA score to ∼3.2-fold (Figure 4B). U2OS ALT+ cells showed a similar CCA score pattern, with a reproducible increase in telomeric c-circles following RHPS4 single-agent treatments, a higher increase after PIP-199 single-agent treatments, and the co-treatment with both agents resulted in the highest increase in CCA scores. These data suggest that an increase in TelG4 and TelR-loop levels positively correlates with ALT activity and that stabilization of the two linked structures induces ALT beyond a simple additive effect (GM847: +70% versus +220%; U2OS: +200% versus 350%). Conversely, HELA 1.3 did not display any significant changes to CCA scores after any combination of treatments, confirming that the ALT status rather than telomere length was driving the effects (Supplementary Figure S9).
While we observed a synergistic increase in ALT activity from increased Tel-G4 and Tel-R-loop levels, we anticipated that over-accumulations of TelG4 and TelR-loop can be cytotoxic and that targeting both structures at the same time could create synergism of cytotoxicity. We performed a curve-shift analysis (36) on the dose–response curve for cytotoxicity of RHPS4 in four ALT+ cell lines, with or without co-treatment with a previously determined sub-lethal dose (lethal dose [LD] of 5–15%) of PIP-199. Dose–response curves showed a leftward shift of the normalized RHPS4 cytotoxicity dose–response curve after the addition of a sub-lethal dose of PIP-199 in all four ALT+ cell lines: GM847 (LD50: 4.33 versus 6.39 μM), SKLU1 (LD50: 3.78 versus 6.20 μM), U2OS (LD50: 7.98 versus 17.44 μM) and SAOS-2 (LD50: 2.34 versus 4.73 μM) (Figure 4C). These results suggest that the drug-drug interaction is of synergistic nature (leftward shift) rather than additive (expected minimal or no shift) or antagonistic (rightward shift) nature. Together, the observations support the view that combination treatments of G4 and R-loop stabilizing chemicals may have therapeutic potential in the clinical management of ALT+ cancers.
Global nuclear G4 levels are higher in ALT+ (ATRX negative) low-grade gliomas
Our observation of the significant difference between nuclear G4 signals in ALT+ versus TERT+ cells suggested that G4 may be a feasible biomarker for differentiating between cancers with different telomere maintenance methods. Unlike R-loops that requires both DNA and RNA in a hybrid structure, we speculated that the nuclear G4 signals could be contributed by any combinations of G4-DNAs and G4-RNAs, given the higher level of both TERRA and telomeric DNA in ALT+ cells. To test this, we performed ChIP on nuclease-digested samples. As expected, R-loop-ChIP signals were RNaseH-sensitive, confirming that the majority of the signal represented DNA/RNA hybrids (Supplementary Figure S10). Interestingly, for G4-ChIP, while the signal appeared to be more DNAse-sensitive, RNase A treatment also resulted in decreases in the signal but to a lesser extent (Supplementary Figure S10). We reasoned that due to the amplified G4 signals contributed by both G4-DNAs and G4-RNAs, detection of G4s could offer a more convenient and sensitive approach to identifying ALT tumours in clinical settings. To explore the feasibility of this idea, we performed immunohistochemistry (IHC) staining with BG4 in low-grade glioma samples that are either ATRX-positive (ATRX+) or ATRX-negative (ATRX-) (Figure 5). Due to the high association between ATRX mutation status and ALT status (12), we designated ATRX+ as probable TERT+ and ATRX- as probable ALT+. As an alternative approach to BG4 staining, we used a G4-specific probe, Naptho-Template Assembled Synthetic G-quartet (N-TASQ) for a more robust analysis (37). The red-edge nature of N-TASQ also allows for a quicker staining protocol that uses one-step staining rather than the multi-step labelling protocol used for the BG4 antibody (37). We first confirmed that the ICC staining using N-TASQ in ALT+ cell lines showed similar patterns as those obtained with BG4 (Supplementary Figure S11). Primary low-grade glioma tumour samples (n = 39, 10 ATRX+ samples and 29 ATRX- samples) stored as formalin-fixed paraffin-embedded (FFPE) slides were first deparaffinized before staining. We performed IHC staining in parallel with either BG4 or N-TASQ (Figure 5 and Supplementary Figure S12). DAPI signal was used to generate masks for cellular nuclei. Due to the variable autofluorescence background between samples, we normalized the BG4 signal (in the red channel) against the background (in the green channel). On the other hand, such normalization would not be possible for N-TASQ due to its red-edge nature (38), where the N-TASQ signals could be detected in all channels. Thus, we used the channel with the lowest background (red) for the quantification of N-TASQ. In agreement with our hypothesis, we observed a significant difference in global G4 levels between the two groups of samples using both the BG4 antibody and the N-TASQ probe (Figure 5B and C). Linear regression analysis between BG4 and N-TASQ of matched samples revealed a moderate correlation (R = 0.66) between the two G4-specific tools, demonstrating the reproducibility with different labelling approaches (Figure 5D).
Figure 5.

Nuclear G4s mark highly probable ALT+ low-grade glioma tumours. (A) IHC staining using BG4 (top) or N-TASQ (bottom) in ALT+ or TERT+ low-grade glioma patient tumour samples as typed by ATRX status. (B) Quantification of staining scores (BG4 score or N-TASQ score) for each matched probable ALT+ or TERT+ samples. (C) Average staining scores for the ALT+ (n = 29) versus TERT+ (n = 10) groups. T-test (two-tailed) was performed. (D) Linear regression plot for BG4 score against N-TASQ score. Each point represents a tumour sample. The black line represents the line of the best fit (R = 0.66).
DISCUSSION
The requirement of persistent DNA damage for ALT telomere maintenance initiation implicates the presence of special structural conditions at the telomeric chromatin. We propose that the formation of non-canonical nucleic acid structures could establish an environment conducive to ALT recombination. While recent reports have demonstrated R-loops’ involvement, the roles of G4s in ALT are less certain. Our imaging data reveal significant differences in the nuclear G4 levels between ALT+ and TERT+ cell lines (Figure 1). We quantitatively show that the telomere accounts for a large portion of the elevated G4 signal in ALT+ cells, which are broadly associated with telomere length and DNA double-strand break (DSB) markers (Figure 2). We also demonstrate that G4s and R-loops positively influence each other's stability and suggest that they are likely to be linked as unique structures in the form of G-loops (Figures 3 and 4). Additionally, we show that G-loop levels are higher in ALT+ cells and that small molecule-mediated stabilization of both G4 and R-loop can cooperatively enhance ALT activity at sub-lethal doses and synergistically cause cytotoxicity at higher doses (Figure 4). Finally, due to the amplified G4 signals in ALT+ cells, we further provide the proof-of-principle of using G4s as sensitive markers for differentiating between ALT+ and TERT+ glioma tumour samples (Figure 5). We suggest that G4s are both actionable ALT biomarkers and exploitable therapeutic targets.
An often-unexplained phenomenon is the source of increased endogenous DNA damage levels present in ALT+ cancers. While DNA damage is detrimental to growth and proliferation in most cases, ALT+ cancer may present an exception in which an elevated baseline level of DNA damage is required to facilitate telomere maintenance by recombination. We contend that the formation of unique nucleic acid structures could be critical in sustaining this higher level of DNA damage in ALT+ cancers. Additionally, recent studies suggest that ATRX may be involved in regulating the stability of non-canonical nucleic acid structures, including R-loops (39) and G4s (40). Given that ATRX dysfunction is widely observed in ALT+ cancers, mutations disrupting ATRX may be the most common and efficient way in which ALT+ cells enhance the formation or stability of these non-canonical nucleic acid structures. Based on our observations, we have built a model supporting the existence of increased G4s and R-loops in ALT+ cancers. Proximal formation of G4s and R-loops can lead to the assembly of G-loops, which are more effective than either one of the two structures on their own to promote the contextual environment required for ALT maintenance by keeping a larger and more stable opened chromatin conformation (Figure 6). We reason that both structures’ coexistence may reduce the energy threshold required to keep a more favourable chromatin state for the formation of spontaneous DSB DNA damage (Figure 6). However, the resulting potential overabundance of DNA damage, which could cause negative consequences on cells, would require tight regulation through specific helicase activities. Recent studies have emphasized the FANCM-BTR complex (34) as a key regulator of ALT homeostasis, which may have evolved to process both G4s (by BLM in the BTR complex) (41) and R-loops (by FANCM) (23) within G-loops. In addition to promoting spontaneous DNA damage for ALT-BIR mechanisms, there is also an intriguing possibility that large G-loops can promote enhanced accessibility, facilitating telomere clustering in the intertelomeric recombination ALT pathway (19).
Figure 6.
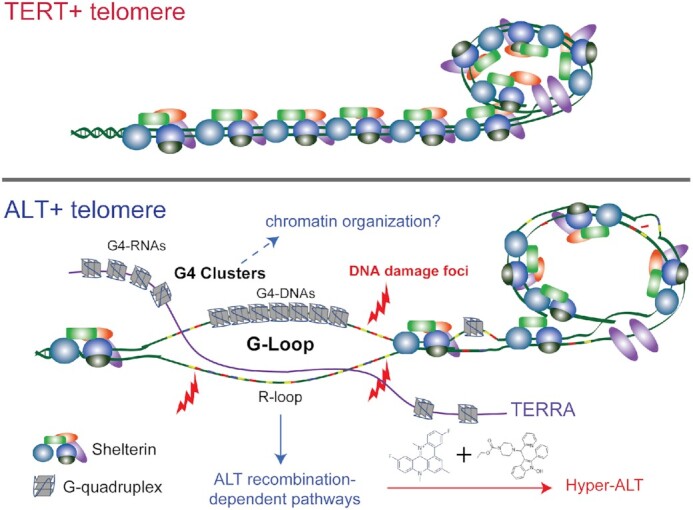
Proposed model of G-quadruplex formation in ALT. At the TERT+ telomere, there are proper shelterin binding and proper telomere structure. Telomere capping function is intact. At the ALT+ telomere, ALT-promoting events, including chromatin remodeler dysfunction (i.e. ATRX), long telomere tract with degenerate repeats, and reduced shelterin binding, contribute to a less compacted chromatin and dysfunctional telomere structure. This provides the opportunity for the formation of G4s and/or R-loops. TERRA (purple) associate with the C-rich telomere strand to form R-loop. The other displaced strand harbours long G-rich regions which associate with each other to form a stretch of G4s. The cooperative actions of G4 and R-loop leads to further opening with increased stability of the R-loops and G4s leading to the formation of the G-loop. The actions of endonuclease at the opening of G-loop leads to double-strand break which facilities the initiation of ALT-associated recombination-based repair mechanisms. Stabilization of G-loops through treatments with G4-ligands and R-loop inducers drive the cancer cell to a hyper-ALT state, manifested in excessive increases in ALT features and accumulation of toxic recombination intermediates. Other potential G4 structures at the ALT telomere include the G4 clusters, contributed by both G4-DNAs formed in telomeric DNAs and G4-RNAs formed in associated TERRAs. The roles of these G4 clusters are unclear but may participate in ALT-specific chromatin organization.
Based on our cytotoxicity screens using a combination of G4 ligand and R-loop inducer, we propose that TelG-loops may also be valuable therapeutic targets for ALT+ cancers (Figure 4C). The use of G4 ligands in the treatment of ALT+ cancers has been proposed earlier (15,42,43). Our results suggest that G4 ligand-induced cytotoxicity is, at least partially, due to the targeting of telomeric G4s. In addition to confirming the cytotoxic potential of targeting G4s, our data demonstrate the feasibility of inducing R-loop formation simultaneously with G4 stabilization for synergistic purposes in future clinical applications for ALT+ cancers. The enhanced ALT activity we have observed after co-stabilization of G4 and R-loop (Figure 4B) suggests that observed cytotoxicity is likely due in part to hyper-ALT phenotypes, manifested in excessive increases in ALT features and accumulation of toxic recombination intermediates (Figure 6). A consideration for future ALT-targeted therapies may include targeting multiple components within the G-loops. Our findings add credence to the newly proposed connections between G4s and R-loops (44–46). We have shown using proximity-ligation and genetic/pharmacological manipulations that G4s and R-loops are likely to be interacting with each other as G-loops. While our cytotoxicity screening and ALT activity output are complementary, they are not directly proportional. Thus, we expect that other targets may also be affected. We anticipate this G4/R-loop relationship can be extended beyond the telomere to G-rich regions in the rest of the genome and impact other important homeostatic processes in human biology. Biological processes, including transcription, replication and chromatin structure remodelling, are potentially conducive to transient G-loops formation. Thus, our findings provide incentives to study G-loops at other genomic loci to understand the coordinated stabilization of R-loops and G4 and their roles in other biological pathways.
In addition to the mechanistic connection between G4s, R-loop and ALT telomere, we also emphasize a G4-centric marker for ALT status. We suggest that the detection of G4s is a better method for imaging-based ALT status differentiation (ALT-calling) due to the abundance of G4 signals. As a result of ALT-promoting genetic/epigenetics modifying events, we suggest that there may be a global change in the steady-state levels of G4s. Developing ALT-calling methods using the abundant G4 signals may be more convenient and reliable than currently implemented methods. Current molecular diagnostics for testing for ALT status include telomere length measurement (TRF, Q-FISH, TelPCR, sequencing), absence of telomerase activity (TRAP), c-circle quantification, TERRA measurements (northern blot) and ATRX/DAXX expression (IHC, Western, sequencing) (47). All of these methods are labour-intensive and/or require large amounts of tumour materials. Likewise, the multi-step protocol for BG4-based G4 detection takes a longer time (1–2 days) to implement. In contrast, the one-step N-TASQ-based G4 detection drastically cuts the processing time to under 2 h. Despite the spectral limitations of N-TASQ, it has proven to be a viable tool for labelling clinical samples, with comparable results as BG4 and a strong correlation to ATRX mutation status (Figure 5). To our knowledge, our findings represent the only comparison to date between different imaging-based G4 detection methods (antibody versus chemical probes) in a set of clinical samples. The relatively high concordance between the two methods validates G4s as the likely targets in these imaging results. In anticipation of screening a more extensive clinical cohort, we expect that it is also possible to scale these G4-specific tools for tissue microarrays to increase throughput.
A closer examination of our imaging results reveals accumulations of both larger and smaller G4 foci in the ALT+ cell nuclei. Based on our ICC colocalization and ChIP results, we reason that a significant portion of these large foci may be clusters of telomeric G4s. While G4-DNAs form on the displaced telomeric DNA strand within the G-loop, the free single-stranded ends of TERRAs may also be folded into G4-RNAs. This occurrence would conceivably result in clusters of G4 signals, termed G4 clusters, contributed by a combination of both G4-DNAs and G4-RNAs. We reason that this may explain the amplified signals seen for G4s in ALT+ cells. While staining in other structures such as nucleoli or ALT-associated PML bodies cannot be ruled out as partially responsible for some of these signals, we emphasize that the telomere is likely a significant contributor, which our ChIP-qPCR data confirms. Only recently have studies began to shed light on the natures of some of these ALT-associated nuclear bodies. Notably, reports have revealed that these large foci may be nuclear condensates resulting from the telomere clustering, a phenomenon that is thought to promote the ALT recombination mechanisms (42,48). Thus, we have opted to focus our analyses on telomeric regions for our study.
While we suggest a role of G4s in stimulating ALT-based mechanisms, it remains unclear why we also observed global upregulation of G4 levels. As alluded to earlier, some additional signals may be attributed to G4-RNAs and the ALT-specific chromatin environment. Given the widespread distribution of potential G4-forming sequences in the human genome (49), it is conceivable that ALT-promoting mechanisms would lead to elevated steady-state levels of G4s at other genomic regions as well. Notably, recent studies have revealed that ATRX-mutated cancers harbour ribosomal DNA (rDNA) instability (50), suggesting a role for ATRX in suppressing genomic instability at the nucleoli. In addition to the factors mentioned above, another potential G4-promoting event may result from changes in the genome sequences. Strikingly, a recent study examining whole genome sequencing data from tumour samples reported a significantly higher number of insertions of telomeric repeats in extra-telomeric regions in ALT+ cells (51). It has also been proposed previously that aberrant recruitment of NR2C/F orphan nuclear transcription factors in ALT facilitates the telomere tethering with other genomic sites and the subsequent formation of telomere insertions (TTIs) (52). The existence of these TTIs has heavy implications on the formation of de novo G4s since every unit of a telomeric repeat contains a G-tract (triple G repeat). Depending on the surrounding sequences, insertions of one or more G-tracts could potentially allow the formation of a G4 that otherwise would not have been able to due to the sequence requirement. Thus, ALT+ cells, in addition to having the expansion of pG4s at the telomere, may also harbour pG4 expansions throughout the genome. This phenomenon raises an intriguing possibility of using G4s to track the evolution of ALT over time, where a long duration of ALT could induce more ALT-mediated ‘G4 genomic scars’. The existence of extra-telomeric G4s may also explain some ALT+ cancer cells’ vulnerability to G4 ligands treatments. Future studies in understanding the full spectrum of these targets would be informative in expanding potential ALT-targeted therapies.
While the variations in the IHC staining for gliomas may be attributed to tissue sample preparations and the staining procedures, another key consideration is the reliance on ATRX status as the ALT status reference. However, it is important to note that some ALT+ tumours (∼10–20%) do not harbour defective ATRX/DAXX (12,53). One such example presented in the multi-cell line comparison component of our study is the SKLU1 cell line, derived from lung epithelial carcinoma, which has functional ATRX/DAXX, yet displayed higher levels of G4 in the nuclei (Figure 1). Thus, in this aspect, G4s may present a more inclusive biomarker for ALT+ cancers. Our clinical results are also limited to low-grade gliomas tumour samples, representing one type of ALT-associated cancers. Future studies to comprehensively evaluate different ALT biomarkers in various ALT+ cancers should expand to include G4s as diagnostic and/or prognostic markers.
In summary, we emphasize the importance of nucleic acid structures in ALT+ cancers and propose that an elevated level of G4s is another hallmark of ALT+ cancers. Additionally, we have shown that G4s and R-loops are interdependent and can co-exist in G-loops, which plays a role in stimulating ALT recombination. Our study provides new insights into the ALT biology and G4/R-loop interplay and the proof-of-principle for exploiting G4s of novel biomarkers and therapeutics against ALT+ cancers.
DATA AVAILABILITY
All relevant data are available from the authors upon reasonable request.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Dr Shankar Balasubramanian (Cambridge University, UK) for the BG4 plasmid, Dr Robert Crouch (NIH, USA) for the RNaseH1 plasmid and Dr Kathleen Collins (UC Berkeley, USA) for the SUSM1 and HELA1.3 cell lines.
Author Contributions: S.Y.Y. and J.M.Y.W. devised the project and conceptualize the idea with input from PCS. S.Y.Y., J.L., H.K. and J.M.Y.W. performed optimization experiments. S.Y.Y. and J.M.Y.W. designed and performed ICC/IHC, ChIP, Re-ChIP, CCA, overexpression and cytotoxicity experiments. E.Y.C.C. and P.C.S. designed and performed PLA, overexpression and knockdown experiments. S.Y. collected and provided the tumour samples. D.M. produced and provided the N-TASQ probe. S.Y.Y. and J.M.Y.W. interpreted the results and co-wrote the manuscript with inputs from E.Y.C.C., P.C.S., S.Y. and D.M.
Contributor Information
Sunny Y Yang, Faculty of Pharmaceutical Sciences, University of British Columbia, Vancouver, V6T 1Z3, Canada.
Emily Y C Chang, Terry Fox Laboratory, BC Cancer, Vancouver, V5Z 1L3, Canada.
Joanne Lim, Faculty of Pharmaceutical Sciences, University of British Columbia, Vancouver, V6T 1Z3, Canada.
Harwood H Kwan, Faculty of Pharmaceutical Sciences, University of British Columbia, Vancouver, V6T 1Z3, Canada.
David Monchaud, Institut de Chimie Moléculaire, ICMUB CNRS UMR 6302, UBFC Dijon, 21078 Dijon, France.
Stephen Yip, Department of Pathology and Laboratory Medicine, University of British Columbia, Vancouver, V6T 2B5 Canada.
Peter C Stirling, Terry Fox Laboratory, BC Cancer, Vancouver, V5Z 1L3, Canada.
Judy M Y Wong, Faculty of Pharmaceutical Sciences, University of British Columbia, Vancouver, V6T 1Z3, Canada.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Cancer Online.
FUNDING
UBC Faculty of Pharmaceutical Sciences Research Reinvestment Funds.
Conflict of interest statement. None declared.
REFERENCES
- 1. Cesare A.J., Reddel R.R. Alternative lengthening of telomeres: models, mechanisms and implications. Nat. Rev. Genet. 2010; 11:319–330. [DOI] [PubMed] [Google Scholar]
- 2. Sommer A., Royle N.J. ALT: a multi-faceted phenomenon. Genes. 2020; 11:133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Heaphy C.M., Subhawong A.P., Hong S.-M., Goggins M.G., Montgomery E.A., Gabrielson E., Netto G.J., Epstein J.I., Lotan T.L., Westra W.H. et al. Prevalence of the alternative lengthening of telomeres telomere maintenance mechanism in human cancer subtypes. Am. J. Pathol. 2011; 179:1608–1615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Heaphy C.M., de Wilde R.F., Jiao Y., Klein A.P., Edil B.H., Shi C., Bettegowda C., Rodriguez F.J., Eberhart C.G., Hebbar S. et al. Altered telomeres in tumors with ATRX and DAXX mutations. Science. 2011; 333:425–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kim J.Y., Brosnan-Cashman J.A., An S., Kim S.J., Song K.-B., Kim M.-S., Kim M.-J., Hwang D.W., Meeker A.K., Yu E. et al. Alternative lengthening of telomeres in primary pancreatic neuroendocrine tumors is associated with aggressive clinical behavior and poor survival. Clin. Cancer Res. 2016; 23:1598–1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Singhi A.D., Liu T.-C., Roncaioli J.L., Cao D., Zeh H.J., Zureikat A.H., Tsung A., Marsh J.W., Lee K.K., Hogg M.E. et al. Alternative lengthening of telomeres and loss of DAXX/ATRX expression predicts metastatic disease and poor survival in patients with pancreatic neuroendocrine tumors. Clin. Cancer Res. 2016; 23:600–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lawlor R.T., Veronese N., Pea A., Nottegar A., Smith L., Pilati C., Demurtas J., Fassan M., Cheng L., Luchini C. Alternative lengthening of telomeres (ALT) influences survival in soft tissue sarcomas: a systematic review with meta-analysis. BMC Cancer. 2019; 19:232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Azzalin C.M., Reichenbach P., Khoriauli L., Giulotto E., Lingner J. Telomeric repeat containing RNA and RNA surveillance factors at mammalian chromosome ends. Science. 2007; 318:798–801. [DOI] [PubMed] [Google Scholar]
- 9. Schoeftner S., Blasco M.A. Developmentally regulated transcription of mammalian telomeres by DNA-dependent RNA polymerase II. Nat. Cell Biol. 2007; 10:228–236. [DOI] [PubMed] [Google Scholar]
- 10. Episkopou H., Draskovic I., Van Beneden A., Tilman G., Mattiussi M., Gobin M., Arnoult N., Londoño-Vallejo A., Decottignies A. Alternative Lengthening of Telomeres is characterized by reduced compaction of telomeric chromatin. Nucleic Acids Res. 2014; 42:4391–4405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jung A.R., Yoo J.E., Shim Y.-H., Choi Y.-N., Jeung H.-C., Chung H.C., Rha S.Y., Oh B.-K. Increased alternative lengthening of telomere phenotypes of telomerase-negative immortal cells upon trichostatin–a treatment. Anticancer Res. 2013; 33:821–829. [PubMed] [Google Scholar]
- 12. Lovejoy C.A., Li W., Reisenweber S., Thongthip S., Bruno J., de Lange T., De S., Petrini J.H.J., Sung P.A., Jasin M. et al. Loss of ATRX, genome instability, and an altered DNA damage response are hallmarks of the alternative lengthening of telomeres pathway. PLoS Genet. 2012; 8:e1002772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dyer M.A., Qadeer Z.A., Valle-Garcia D., Bernstein E. ATRX and DAXX: mechanisms and mutations. Cold Spring Harb. Perspect. Med. 2017; 7:a026567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Napier C.E., Huschtscha L.I., Harvey A., Bower K., Noble J.R., Hendrickson E.A., Reddel R.R. ATRX represses alternative lengthening of telomeres. Oncotarget. 2015; 6:16543–16558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Clynes D., Jelinska C., Xella B., Ayyub H., Scott C., Mitson M., Taylor S., Higgs D.R., Gibbons R.J. Suppression of the alternative lengthening of telomere pathway by the chromatin remodelling factor ATRX. Nat. Commun. 2015; 6:7538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lee M., Hills M., Conomos D., Stutz M.D., Dagg R.A., Lau L.M.S., Reddel R.R., Pickett H.A. Telomere extension by telomerase and ALT generates variant repeats by mechanistically distinct processes. Nucleic Acids Res. 2013; 42:1733–1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Conomos D., Stutz M.D., Hills M., Neumann A.A., Bryan T.M., Reddel R.R., Pickett H.A. Variant repeats are interspersed throughout the telomeres and recruit nuclear receptors in ALT cells. J. Cell Biol. 2012; 199:893–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bandaria Jigar N., Qin P., Berk V., Chu S., Yildiz A. Shelterin protects chromosome ends by compacting telomeric chromatin. Cell. 2016; 164:735–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cho N., Dilley Robert L., Lampson Michael A., Greenberg Roger A Interchromosomal homology searches drive directional ALT telomere movement and synapsis. Cell. 2014; 159:108–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Dilley R.L., Verma P., Cho N.W., Winters H.D., Wondisford A.R., Greenberg R.A. Break-induced telomere synthesis underlies alternative telomere maintenance. Nature. 2016; 539:54–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Min J., Wright W.E., Shay J.W. Alternative lengthening of telomeres mediated by mitotic DNA synthesis engages break-induced replication processes. Mol. Cell. Biol. 2017; 37:e00226-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Arora R., Lee Y., Wischnewski H., Brun C.M., Schwarz T., Azzalin C.M. RNaseH1 regulates TERRA-telomeric DNA hybrids and telomere maintenance in ALT tumour cells. Nat. Commun. 2014; 5:5220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Silva B., Pentz R., Figueira A.M., Arora R., Lee Y.W., Hodson C., Wischnewski H., Deans A.J., Azzalin C.M. FANCM limits ALT activity by restricting telomeric replication stress induced by deregulated BLM and R-loops. Nat. Commun. 2019; 10:2253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hänsel-Hertsch R., Spiegel J., Marsico G., Tannahill D., Balasubramanian S. Genome-wide mapping of endogenous G-quadruplex DNA structures by chromatin immunoprecipitation and high-throughput sequencing. Nat. Protoc. 2018; 13:551–564. [DOI] [PubMed] [Google Scholar]
- 25. Yang S.Y., Lejault P., Chevrier S., Boidot R., Robertson A.G., Wong J.M.Y., Monchaud D Transcriptome-wide identification of transient RNA G-quadruplexes in human cells. Nat. Commun. 2018; 9:4730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. McQuin C., Goodman A., Chernyshev V., Kamentsky L., Cimini B.A., Karhohs K.W., Doan M., Ding L., Rafelski S.M., Thirstrup D. et al. CellProfiler 3.0: Next-generation image processing for biology. PLoS Biol. 2018; 16:e2005970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Stauffer W., Sheng H., Lim H.N. EzColocalization: An ImageJ plugin for visualizing and measuring colocalization in cells and organisms. Sci. Rep. 2018; 8:15764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Henson J.D., Lau L.M., Koch S., Martin La Rotta N., Dagg R.A., Reddel R.R. The C-Circle assay for alternative-lengthening-of-telomeres activity. Methods. 2017; 114:74–84. [DOI] [PubMed] [Google Scholar]
- 29. Yang S.Y., Amor S., Laguerre A., Wong J.M.Y., Monchaud D Real-time and quantitative fluorescent live-cell imaging with quadruplex-specific red-edge probe (G4-REP). Biochim. Biophys. Acta. 2017; 1861:1312–1320. [DOI] [PubMed] [Google Scholar]
- 30. Ginno Paul A., Lott Paul L., Christensen Holly C., Korf I., Chédin F R-Loop formation is a distinctive characteristic of unmethylated human CpG Island promoters. Mol. Cell. 2012; 45:814–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Biffi G., Tannahill D., McCafferty J., Balasubramanian S. Quantitative visualization of DNA G-quadruplex structures in human cells. Nat. Chem. 2013; 5:182–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Schwartzentruber J., Korshunov A., Liu X.-Y., Jones D.T.W., Pfaff E., Jacob K., Sturm D., Fontebasso A.M., Quang D.-A.K., Tönjes M. et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature. 2012; 482:226–231. [DOI] [PubMed] [Google Scholar]
- 33. Salvati E., Leonetti C., Rizzo A., Scarsella M., Mottolese M., Galati R., Sperduti I., Stevens M.F.G., D’Incalci M., Blasco M. et al. Telomere damage induced by the G-quadruplex ligand RHPS4 has an antitumor effect. J. Clin. Invest. 2007; 117:3236–3247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lu R., O’Rourke J.J., Sobinoff A.P., Allen J.A.M., Nelson C.B., Tomlinson C.G., Lee M., Reddel R.R., Deans A.J., Pickett H.A. The FANCM-BLM-TOP3A-RMI complex suppresses alternative lengthening of telomeres (ALT). Nat. Commun. 2019; 10:2252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Duquette M.L. Intracellular transcription of G-rich DNAs induces formation of G-loops, novel structures containing G4 DNA. Genes Dev. 2004; 18:1618–1629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zhao L., Au J.L-S., Wientjes M.G. Comparison of methods for evaluating drug-drug interaction. Front. Biosci. 2010; 2:241–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Laguerre A., Hukezalie K., Winckler P., Katranji F., Chanteloup G., Pirrotta M., Perrier-Cornet J.-M., Wong J.M.Y., Monchaud D Visualization of RNA-Quadruplexes in Live Cells. J. Am. Chem. Soc. 2015; 137:8521–8525. [DOI] [PubMed] [Google Scholar]
- 38. Laguerre A., Wong J.M.Y., Monchaud D Direct visualization of both DNA and RNA quadruplexes in human cells via an uncommon spectroscopic method. Sci. Rep. 2016; 6:32141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Nguyen D.T., Voon H.P.J., Xella B., Scott C., Clynes D., Babbs C., Ayyub H., Kerry J., Sharpe J.A., Sloane-Stanley J.A. et al. The chromatin remodelling factor ATRX suppresses R-loops in transcribed telomeric repeats. EMBO Rep. 2017; 18:914–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wang Y., Yang J., Wild A.T., Wu W.H., Shah R., Danussi C., Riggins G.J., Kannan K., Sulman E.P., Chan T.A. et al. G-quadruplex DNA drives genomic instability and represents a targetable molecular abnormality in ATRX-deficient malignant glioma. Nat. Commun. 2019; 10:943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Budhathoki J.B., Ray S., Urban V., Janscak P., Yodh J.G., Balci H. RecQ-core of BLM unfolds telomeric G-quadruplex in the absence of ATP. Nucleic Acids Res. 2014; 42:11528–11545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Min J., Wright W.E., Shay J.W. Clustered telomeres in phase-separated nuclear condensates engage mitotic DNA synthesis through BLM and RAD52. Genes Dev. 2019; 33:814–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Amato R., Valenzuela M., Berardinelli F., Salvati E., Maresca C., Leone S., Antoccia A., Sgura A. G-quadruplex stabilization fuels the ALT pathway in ALT-positive osteosarcoma cells. Genes. 2020; 11:304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zhang J.-M., Zou L. Alternative lengthening of telomeres: from molecular mechanisms to therapeutic outlooks. Cell Biosci. 2020; 10:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Tan J., Lan L. The DNA secondary structures at telomeres and genome instability. Cell Biosci. 2020; 10:47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Bryan T.M. G-Quadruplexes at Telomeres: Friend or Foe. Molecules. 2020; 25:3686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Pompili L., Leonetti C., Biroccio A., Salvati E. Diagnosis and treatment of ALT tumors: is Trabectedin a new therapeutic option?. J. Exp. Clin. Cancer Res. 2017; 36:189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zhang H., Zhao R., Tones J., Liu M., Dilley R.L., Chenoweth D.M., Greenberg R.A., Lampson M.A. Nuclear body phase separation drives telomere clustering in ALT cancer cells. Mol. Biol. Cell. 2020; 31:2048–2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Hänsel-Hertsch R., Di Antonio M., Balasubramanian S. DNA G-quadruplexes in the human genome: detection, functions and therapeutic potential. Nat. Rev. Mol. Cell Biol. 2017; 18:279–284. [DOI] [PubMed] [Google Scholar]
- 50. Udugama M., Sanij E., Voon H.P.J., Son J., Hii L., Henson J.D., Chan F.L., Chang F.T.M., Liu Y., Pearson R.B. et al. Ribosomal DNA copy loss and repeat instability in ATRX-mutated cancers. Proc. Natl. Acad. Sci. 2018; 115:4737–4742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Sieverling L., Hong C., Koser S.D., Ginsbach P., Kleinheinz K., Hutter B., Braun D.M., Cortés-Ciriano I., Xi R., Kabbe R. et al. Genomic footprints of activated telomere maintenance mechanisms in cancer. Nat. Commun. 2020; 11:733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Marzec P., Armenise C., Pérot G., Roumelioti F.-M., Basyuk E., Gagos S., Chibon F., Déjardin J. Nuclear-receptor-mediated telomere insertion leads to genome instability in ALT cancers. Cell. 2015; 160:913–927. [DOI] [PubMed] [Google Scholar]
- 53. Barthel F.P., Wei W., Tang M., Martinez-Ledesma E., Hu X., Amin S.B., Akdemir K.C., Seth S., Song X., Wang Q. et al. Systematic analysis of telomere length and somatic alterations in 31 cancer types. Nat. Genet. 2017; 49:349–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All relevant data are available from the authors upon reasonable request.