

In-depth profiling of transmembrane proteins in the brain leads to the identification of GPCR regulators in a disease model.
Abstract
Transmembrane proteins play vital roles in mediating synaptic transmission, plasticity, and homeostasis in the brain. However, these proteins, especially the G protein–coupled receptors (GPCRs), are underrepresented in most large-scale proteomic surveys. Here, we present a new proteomic approach aided by deep learning models for comprehensive profiling of transmembrane protein families in multiple mouse brain regions. Our multiregional proteome profiling highlights the considerable discrepancy between messenger RNA and protein distribution, especially for region-enriched GPCRs, and predicts an endogenous GPCR interaction network in the brain. Furthermore, our new approach reveals the transmembrane proteome remodeling landscape in the brain of a mouse depression model, which led to the identification of two previously unknown GPCR regulators of depressive-like behaviors. Our study provides an enabling technology and rich data resource to expand the understanding of transmembrane proteome organization and dynamics in the brain and accelerate the discovery of potential therapeutic targets for depression treatment.
INTRODUCTION
As the most complex organ of the mammalian body, the brain has been intensively characterized at the molecular level in a system-wide fashion using a variety of transcriptomic or imaging approaches. The Allen Brain Atlas (https://portal.brain-map.org/) hosts a plethora of in situ hybridization (ISH) and microarray-based databases to describe the regional or cellular gene expression profiles of adult and developing mammalian brains (1–4). The Human Protein Atlas program (www.proteinatlas.org), together with a recent brain atlas project, has concertedly established high-quality transcriptome and protein imaging resources to map the spatial expression of transcripts and proteins across multiple mammalian brain regions (5, 6). Moreover, given that all the functions of the brain are ultimately mediated by proteins and that poor correlation between mRNA and protein abundances has been observed in various cell types and tissues (7–9), large-scale mass spectrometry (MS)–based proteomic surveys have been launched to map protein expression patterns in an unbiased manner across multiple regions of mouse and human brains (10, 11). However, despite our increasing ability to interrogate the molecular organization of the brain, in-depth and quantitative profiling of transmembrane protein expression is a notable exception.
G protein–coupled receptors (GPCRs), ion channels, and transporters constitute three prominent cell-surface transmembrane protein families that play essential roles in mediating neuronal signal processing and plasticity in the brain (12–14). A number of their family members, especially GPCRs, represent the most successful targets of molecular therapeutics for central nervous system (CNS) disorders (12, 15). However, these transmembrane proteins are especially challenging to measure using conventional proteomics techniques owing to their strong hydrophobicity, relatively low abundance, and fast turnover (8). Notably, GPCRs are notoriously underrepresented in current MS-based proteomic surveys. For example, a proteome atlas of 29 healthy human tissues profiled 103 GPCRs in total, 64 of which were from brain tissue of the 831 GPCRs encoded in the human genome (8). A similarly low coverage of GPCR identification (ID) was reported in another global proteomic analysis of human cells (56 GPCRs among 14,237 identified proteins) (16).
Here, we present a new proteomic approach for deep and accurate profiling of low-abundance transmembrane protein families in the region-resolved mouse brain. Our approach integrates three innovations compared to conventional proteomic workflows. First, we performed cell membrane fractionation to reduce the abundant cytosolic proteins and enrich transmembrane proteins. Second, we carried out single-shot data-independent acquisition (DIA) MS analysis rather than conventional data-dependent acquisition (DDA) analysis as DIA MS is an emerging technology with superior accuracy and reproducibility in proteomic quantification (17, 18). Last, we created a GPCR family–targeted hybrid library using deep learning tools for DIA MS data mining so as to achieve an unprecedented depth of transmembrane protein profiling. Using this approach, we were able to identify and quantify 143 GPCRs, 170 ion channels, and 176 transporter proteins across 10 mouse brain regions.
By matching our multiregional proteomics profiling data with the genome-wide transcriptomics and ISH data, we identified region-enriched GPCRs and other transmembrane proteins with considerably discordant mRNA and protein distribution over multiple brain regions. Through protein coexpression analysis, we predicted an endogenous GPCR interaction network in the mouse brain and validated the colocalization of a GPCR protein and its unknown interacting partner in neuronal cell culture and brain tissue. Furthermore, we used this new workflow to reveal the landscape of transmembrane proteome remodeling in 11 mouse brain regions of a chronic stress–induced depression model, which led to the rapid discovery of two novel GPCR regulators of depressive-like behaviors.
RESULTS
Profiling the transmembrane proteome in multiple mouse brain regions by single-shot DIA MS analysis
To profile transmembrane protein expression in the region-resolved mouse brain, we collected 10 anatomically dissected adult mouse brain regions. Nine and eight regions overlap with those documented in the Allen Mouse Brain Atlas and the recently published brain transcriptome atlas (6), respectively (Fig. 1A). To increase the proteome coverage of transmembrane proteins, we isolated cell membrane fractions, performed membrane protein extraction and digestion under optimal conditions, and analyzed protein digests from individual brain regions with single-shot DIA MS (Fig. 1B and fig. S1A). For each brain region, we also fractionated the pooled replicates and performed DDA MS analysis of prefractionated samples to build a project-specific spectral library for DIA MS data mining (Fig. 1B, left).
Fig. 1. Transmembrane proteome profiling of the region-resolved mouse brain with DIA MS analysis.
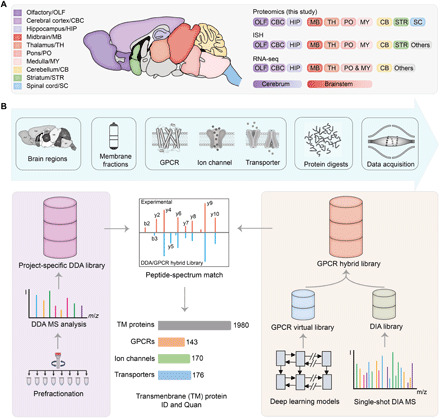
(A) A summary of the brain regions of the resting-state mice examined in this study (left) and the overlapping regions analyzed using ISH by Allen Brain Atlas or RNA-seq by Mulder and colleagues (6) (right). (B) Overall workflow of mouse brain DIA MS analysis and data mining. For each brain region, membrane proteins [represented by three transmembrane protein families this study focuses on] were isolated, extracted, and digested before MS analysis (top). A project-specific DDA library was built from DDA MS analysis of prefractionated multiregional brain tissues (left route). Meanwhile, a GPCR family–targeted hybrid library was built by merging an initial DIA library derived from the DIA MS data and a GPCR virtual library predicted from 524 mouse genome–encoded GPCR sequences using deep learning models (right route). The DIA MS data were searched with the two libraries to yield transmembrane protein ID and quantification results.
This DDA experiment–derived spectral library (DDA library for short) comprised a total of 134,560 peptide precursors mapped to 7995 protein groups, of which 2510 were transmembrane proteins (with at least one transmembrane domain). Mouse genome encodes 524 GPCRs, 316 ion channels, and 296 transporters (protein lists in table S1). Among them, 135 GPCRs, 207 ion channels, and 207 transporters were present in the DDA library (Fig. 2A and fig. S1B). Although the proteome coverages of three transmembrane protein families were lower than those detected at the transcript level by ISH or RNA sequencing (RNA-seq), our results significantly outnumber the most comprehensive mouse brain proteomic survey reported to date (fig. S1B) (10).
Fig. 2. Deep proteome coverage and reproducible quantification of GPCR, ion channel, and transporter family members in the mouse brain.
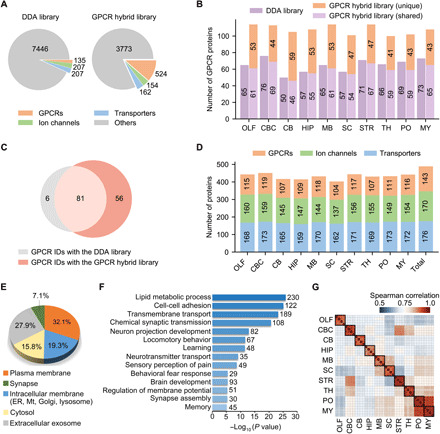
(A) Number of protein IDs in the DDA library and the GPCR hybrid library. The full complement of 524 genome-encoded GPCRs are included in the latter. (B) Number of GPCR IDs in each brain region yielded with the DDA library (purple) or the GPCR hybrid library after data filtering (light purple, shared IDs between the two libraries; orange, unique IDs only yielded with the hybrid library). OLF, olfactory bulb; CBC, cerebral cortex; CB, cerebellum; HIP, hippocampus; MB, midbrain; SC, spinal cord; STR, striatum; TH, thalamus; PO, pons; MY, medulla. (C) Comparison of GPCR IDs from 10 brain regions yielded with the two libraries. (D) Number of protein IDs for three families in each region and in total. GPCR IDs are concatenated from two libraries, and ion channel and transporter IDs were detected with the DDA library. (E) Subcellular localization of all protein IDs according to gene ontology cellular component classification. ER, endoplasmic reticulum; Mt, mitochondria. (F) Many enriched biological processes (P < 10−5) in all protein IDs are related to neuronal cell activity or brain functions. (G) Spearman correlation of protein quantification between replicates of each region.
Deepening the GPCR subproteome coverage with a targeted hybrid library strategy
Given that GPCRs are particularly challenging to map with conventional proteomics techniques (8), we developed a targeted hybrid library strategy to deepen the coverage of a selected transmembrane protein family (see Materials and Methods for details). Briefly, we created a GPCR family–targeted hybrid library (GPCR hybrid library for short) using deep learning models (Fig. 1B, right) (19, 20), which contains the full complement of 524 GPCRs encoded in the mouse genome (Fig. 2A). To control the false discovery rate (FDR) when using a targeted hybrid library, we implemented an additional data filtering criteria (Cscore, >1.0) to restrict the subgroup FDR of GPCR peptide ID as assessed using a decoy library approach (fig. S2, A to C). This strategy was applied to processing our 10–brain region DIA MS data.
An average of 108 GPCR proteins were identified per region using the GPCR hybrid library after data filtering, whereas only an average of 65 GPCR proteins were identified with the DDA library (Fig. 2B). Moreover, we observed similarly high quantification reproducibility for all proteins identified with the two libraries (fig. S2D). By combining GPCR IDs from two libraries, we substantially increased the GPCR subproteome coverage in the mouse brain through single-shot DIA MS analysis. Of the 143 concatenated GPCR IDs, 56 were exclusively detected using the targeted hybrid library strategy (Fig. 2C). Although we previously demonstrated this strategy in processing data from a few brain regions as a proof of a concept (21), our current study proved that this hybrid spectral library can deepen the GPCR subproteome coverage to surpass a large-scale project-specific DDA library.
In addition to GPCRs, our transmembrane proteome profiling identified and quantified 170 ion channels and 176 transporters across 10 brain regions (Fig. 2D). Of all protein groups profiled in our study, 58.5% are located in plasma membrane, intracellular membrane, or synapse (Fig. 2E). Most of them are enriched in biological processes closely related to neuronal cell activity or brain functions, such as neuron projection development, neurotransmitter transport, sensory perception of pain, and fear response that are known to be mediated by the three transmembrane protein families (Fig. 2F). In regard to the quantification performance of our new workflow, all proteins profiled between independent replicates of each brain region showed strong correlation and low quantification deviation [median coefficient of variation (CV), 8.3%], which indicates superior quantification consistency by our DIA MS analysis over the previous mouse brain proteomic analysis (median CV, 28.1%) (Fig. 2G and fig. S2E) (10).
Comparison of the transcriptome and proteome profiles for transmembrane proteins
Our transmembrane proteome profiling enables a global view of transmembrane protein expression across different mouse brain regions that can be compared with region-resolved gene expression at the transcriptome level (table S2). The principal components analysis (PCA) and the hierarchical clustering tree based on our quantification of all transmembrane proteins revealed four clusters of brain regions: pons/medulla, spinal cord/midbrain, cerebral cortex/striatum/thalamus, and olfactory bulb/hippocampus (HIP)/cerebellum (Fig. 3A). In contrast, the transcriptome profiles of transmembrane protein–coding genes showed a different pattern of regional connectivity, with cerebral cortex/HIP/amygdala being clustered more tightly and cerebellum standing out as an outlier (Fig. 3B).
Fig. 3. Multiregional expression profiles of transmembrane protein–coding genes at the transcriptome and proteome levels.
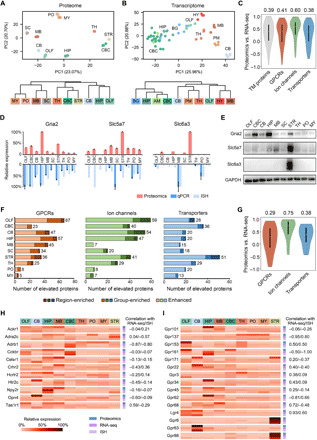
(A and B) PCA (top) and HCA (bottom) showed different patterns of regional expression of transmembrane protein–coding genes based on proteomics data (this study) (A) versus RNA-seq data (6) (B). (C) Spearman correlation of expression profiles across at least five brain regions between the proteomic and RNA-seq measurement for all transmembrane (TM) proteins and three transmembrane protein families. Median correlation coefficients are shown above the plots. (D) Examples of discordant expression profiles measured by proteomics, qPCR, and ISH. (E) Validation of regionally elevated expression of transmembrane proteins with inconsistent mRNA profiles by immunoblotting. (F) ID of regionally elevated GPCRs, ion channels, and transporters in the three categories (region-enriched, group-enriched, and enhanced). Total numbers of elevated proteins in each region are provided to the right. (G) Spearman correlation of expression profiles across at least five brain regions indicated the lowest protein-to-mRNA correlation for region/group-enriched GPCRs. Median correlation coefficients are shown above the plots. (H and I) Relative expression profiles of region/group-enriched GPCRs with negative mRNA-to-protein correlation (H) and region/group-enriched orphan receptors (I). The correlation coefficient of proteomic versus RNA-seq or proteomic versus ISH measurement is annotated for each GPCR. No correlation available for three receptors of which the expression profiles were overlapped between less than five regions.
An overall modest correlation between mRNA and protein abundances was observed for 1738 transmembrane protein–coding genes (median Spearman correlation coefficient, 0.39) shared between the RNA-seq and proteomics data (table S3). Among them, 392 genes (22%) showed negative correlation, indicating substantial difference in their mRNA and protein expression patterns (Fig. 3C). We also analyzed 1733 transmembrane protein–coding genes shared between the ISH and proteomics data and found an even weaker correlation (median correlation coefficient, 0.22; 32% genes showing negative correlation) (fig. S3A and table S3). Moreover, GPCR and transporter family members showed lower mRNA-to-protein correlation than ion channels (Fig. 3C and fig. S3A).
To validate the distinct interregional protein expression profiles revealed by our proteomics analysis, we selected one ion channel [Gria2 (glutamate receptor 2)] and two transporters [Slc6a3 (sodium-dependent dopamine transporter) and Slc5a7] with inconsistent RNA expression profiles to be examined by immunoblotting and immunostaining. Slc6a3 protein was predominantly expressed in the striatum, yet its mRNA was mainly detected in the midbrain and pons by quantitative polymerase chain reaction (qPCR) and ISH (Fig. 3D). Immunoblotting showed almost exclusive presence of Slc6a3 protein in the striatum (Fig. 3E). Immunostaining of the brain slice confirmed this result and further revealed the distribution of Slc6a3 in the axons of projection neurons (fig. S3, C to E). For Gria2 and Slc5a7, their protein expression patterns were validated to be appreciably different from their mRNA expression (Fig. 3, D and E). Therefore, our DIA MS-based proteome profiling provides a high-throughput and accurate measure of transmembrane protein distribution, which, in many cases, is largely discordant with the transcript distribution.
Brain region–enriched GPCRs and other transmembrane proteins
Using the region-averaged proteomic quantification data (table S4), we identified regionally elevated GPCR, ion channel, and transporter family members that were classified into three categories: region-enriched proteins (twofold higher abundance than all other brain regions), group-enriched proteins (two to four brain regions with twofold higher abundance than all other regions), and enhanced proteins (twofold higher abundance than the median of all other brain regions) (Fig. 3F and table S5). The GPCR family contains the largest number of regionally elevated proteins (three categories together) (Fig. 3F), in accordance with the largest variation of interregional protein abundances observed for GPCRs (fig. S3F). The striatum has the most region-enriched GPCRs, whereas most group-enriched GPCRs are shared among the olfactory bulb, midbrain, and HIP (table S5). Notably, the interregional mRNA-to-protein expression correlation was much weaker for region/group-enriched GPCRs than for region/group-enriched ion channels or transporters (Fig. 3G and fig. S3B).
Our study suggests that this subset of GPCRs enriched in certain brain regions experience unusually pronounced regulation of protein synthesis, degradation, or transport. This is exemplified by 11 region/group-enriched GPCRs with negative mRNA-to-protein correlation, including members from the chemokine receptor, adrenergic receptor, and neuropeptide Y receptor families (Fig. 3H). We also analyzed 15 region/group-enriched orphan GPCRs for which native ligands are unknown and physiological functions are largely unexplored (22, 23). The multiregional protein distribution for eight enriched orphan GPCRs were negatively correlated with their mRNA distribution (Fig. 3I). For instance, Gpr161 gene transcription mainly occurred in the HIP and olfactory bulb, but most of its protein product was likely to be transported to the cerebral cortex in addition to the olfactory bulb. The extensive efferent projections from the HIP and olfactory bulb to the prefrontal, cingulate, retrosplenial, and the olfactory cortex (24–26) might explain the enriched expression of Gpr161 protein in the cerebral cortex. For Gpr101, Gpr34, and Gpr62, their transcripts were distributed evenly across multiple regions, yet the protein product was highly enriched in one specific region. Together, the protein distribution patterns of these brain region–enriched GPCRs would shed new light on their posttranscriptional regulation and uncharacterized functions in the brain.
GPCR interaction prediction based on multiregional protein coexpression analysis
Protein coexpression or coregulation analysis based on the quantitative proteome profiling data can be exploited to infer the composition of protein complexes and their interaction networks (27, 28). Thus, we reasoned that it may be possible to find unknown protein-protein interactions (PPIs) from our multiregional proteomics resource. As a proof of concept, for each measured GPCR in our dataset, we extracted their potential interacting partners with correlated expression profiles over at least five brain regions. Using a commonly applied cutoff for positive correlation [Pearson correlation coefficient (PCC), >0.7] (27, 28), we initially identified 16,074 potential PPIs for 124 GPCRs. Examples are shown for cannabinoid receptor 1 (CB1) and metabotropic glutamate receptor 2 of which an array of known interactions with other GPCRs, ion channels, and classical signaling partners was identified (fig. S4A). Among the PPIs inferred from our coexpression analysis, we noticed a cluster of membrane-associated periodic skeleton (MPS) components (spectrin and ankyrin) and signaling molecules RTK (receptor tyrosine kinase) TrkB (tropomyosin-related tyrosine kinase B) and kinases Src (neuronal proto-oncogene tyrosine-protein kinase Src) and Fyn (tyrosine-protein kinase Fyn)] to pair with specific GPCRs, such as CB1 (fig. S4, A and B). This finding, in general, agreed with the recently reported colocalization of MPS components with CB1 and RTKs in neurons to form a cytoskeleton-dependent signaling platform (29).
To infer the unknown GPCR interaction network in the brain, we filtered our coexpression data to retain potential GPCR interactions with transmembrane proteins at a higher stringency (PCC, >0.9). This resulted in a high-quality predicted GPCR interaction network entailing 2828 unique PPIs between 120 GPCRs and 1159 transmembrane proteins (Fig. 4A and table S6). For CB1, we picked up an uncharacterized and most strongly correlated partner, leucine-rich repeat transmembrane protein FLRT3, for validation. Immunostaining of CB1 and FLRT3 in the hippocampal neuron revealed the colocalization of FLRT3 with a fraction of CB1-positive fibers (Fig. 4B). Partial colocalizaiton of CB1 and FLRT3 was also observed in the brain slice of HIP (Fig. 4C). To further verify the interaction between CB1 and FLRT3, we carried out a proximity ligation assay (PLA) (30) to confirm the close physical distribution between the two proteins in both CB1-expressing Chinese hamster ovary (CHO) cells and primary neurons (fig. S4C). In addition, immunoprecipitation (IP) was performed to verify the in vitro interaction between CB1 and FLRT3 (fig. S4D). These validation results clearly demonstrate the capacity of our proteomics-derived coexpression analysis to reveal new GPCR interactions that occur endogenously in the brain.
Fig. 4. A predicted GPCR interaction network from multiregional protein coexpression analysis.
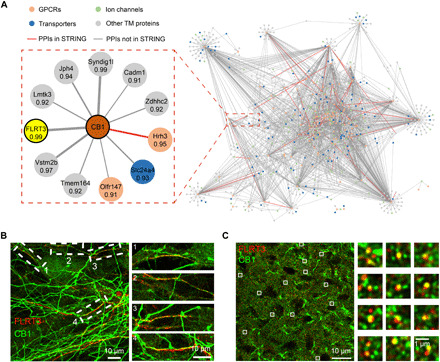
(A) A predicted interaction network between 120 GPCRs and 1159 transmembrane proteins in the mouse brain from high-stringency coexpression analysis (PCC >0.9). An enlarged PPI module for CB1 is shown, with the correlation coefficient for each putative PPI annotated. The most strongly correlated partner (FLRT3) was selected for experimental validation. (B) Colocalization of FLRT3 (red) with a fraction of CB1 (green) in the rat hippocampal neuron. Both endogenous proteins were stained with their antibodies. (C) Confocal image of FLRT3 (red) and CB1 (green) immunocytostaining in the hippocampal brain slice. Robust CB1-positive fibers were observed; a fraction of CB1-positive puncta colocalized with FLRT3 signals, which also showed a puncta staining pattern. Zoomed-in images of the boxed regions in (B) and (C) are shown on the right.
Profiling the brain region–resolved transmembrane proteome in a mouse depression model
To further demonstrate the power of our transmembrane proteome profiling technology for neurobiology, we applied our established workflow to a well-established mouse depression model to generate new insights into the pathophysiology of depression and accelerate the discovery of potential drug targets. Here, we adopted the chronic unpredictable mild stress (CUMS) model, in which CUMS mice were exposed chronically to a battery of unpredictable stressors and developed depressive-like symptoms after 21 days of consecutive exposure (Fig. 5A). Specifically, CUMS mice exhibited reduced sucrose preference and increased immobility in behavioral tests, which indicated the depressive-like anhedonia [as measured by the sucrose preference test (SPT)] and despair [as measured by the tail suspension test (TST) and forced swimming test (FST)] (fig. S5).
Fig. 5. Transmembrane proteome profiling of the mouse brain in the CUMS model of depression.
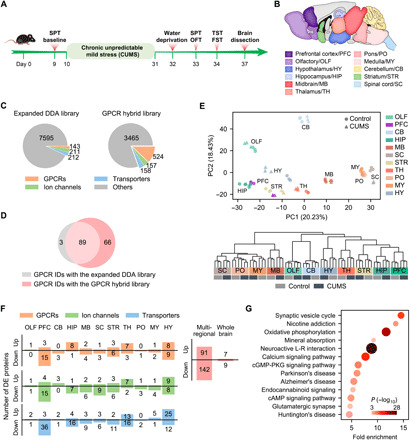
(A) Procedure for establishing the CUMS model. (B) Anatomically dissected brain regions of the control and CUMS mice for proteomic analysis. (C) Number of protein IDs in the expanded DDA library and the GPCR hybrid library specifically built for the CUMS model. (D) Comparison of GPCR IDs from 11 brain regions of the CUMS model with the two libraries. (E) PCA (top) and HCA (bottom) of regional expression of all detected transmembrane proteins revealed tight clustering of control and CUMS groups for most of brain regions analyzed. Notice the separation of control and CUMS groups for PFC and HY in the PCA plot. (F) Number of up- and down-regulated GPCRs, ion channels, and transporters identified in each region (left). Number of unique DE proteins from the GPCR/ion channel/transporter families identified in total by multiregional analysis and by whole brain analysis (right). (G) Significantly enriched pathways (P < 0.001) in the DE GPCRs, ion channels, and transporters identified by multiregional analysis. cGMP, guanosine 3′,5′-monophosphate; PKG, cGMP-dependent protein kinase.
For both the CUMS and control mice, we collected 11 anatomically dissected brain regions in triplicate, including the previously examined nine regions and two new regions [prefrontal cortex (PFC) and hypothalamus (HY)] (Fig. 5B). Cell membrane fractionation, protein extraction, and digestion were performed using the same protocol, and each protein digest was analyzed by single-shot DIA MS (Fig. 1B). For MS data mining, we constructed a largest DDA library specific for the CUMS model and a GPCR hybrid library derived from the DIA MS data, which contains the full complement of 524 mouse GPCRs (Fig. 5C) (see Materials and Methods for details).
After data filtering to control the subgroup FDR, an average of 74 GPCR proteins were identified and quantified per region with the GPCR hybrid library, representing an average gain of 25 GPCRs relative to the DDA library (fig. S6A). In total, 158 unique GPCRs were profiled in at least one brain region from control or CUMS mice, including 66 GPCRs exclusively detected with the targeted hybrid library strategy (Fig. 5D). In addition, our study enabled in-depth profiling of 180 ion channels and 179 transporters in the brain regions from control or CUMS mice (fig. S6B). Superior protein quantification consistency between experimental replicates of each brain region was achieved for both control and CUMS groups (median CV, 3.78 to 10.08%) (fig. S6C).
ID of differentially expressed transmembrane proteins in the depression model
Both the PCA and unsupervised hierarchical clustering analysis (HCA) based on the quantification of 1998 transmembrane proteins revealed tight clustering of six replicates of the control and CUMS groups for most of the brain regions analyzed (Fig. 5E). It implies that the molecular architecture of the transmembrane proteome was retained in most brain regions between the control and depressed states. We noticed the separation of control and CUMS groups for PFC and HY in the PCA plot, indicating larger perturbation of the transmembrane proteome here than in the other regions (Fig. 5E, top).
Our study identified discrete sets of differentially expressed (DE) transmembrane proteins from the GPCR, ion channel, and transporter families in each brain region of the CUMS model (Fig. 5F and table S7). In accordance with the PCA plot, the largest number of DE transmembrane proteins were found in PFC and HY, two brain regions critical for mood regulation and stress response (31–34). A total of 91 up-regulated and 142 down-regulated unique proteins from the three families were identified from our brain proteome profiling of the CUMS model, which uncovered the most comprehensive landscape of transmembrane proteome remodeling associated with depression pathogenesis. In sharp contrast, only 16 dysregulated proteins were identified when analyzing the whole mouse brain of the CUMS model (Fig. 5F and table S7). Therefore, the localized transmembrane proteome remodeling can be only captured by anatomical dissection combined with our high-sensitivity quantitative proteomics.
This group of 233 DE transmembrane proteins identified in our study is enriched in 13 pathways, including synaptic vesicle cycle, nicotine addiction, oxidative phosphorylation, calcium signaling, and adenosine 3′,5′-monophosphate (cAMP) signaling (Fig. 5G). In addition, they are enriched in molecules known to be involved in the development of Parkinson’s disease and Alzheimer’s disease, which may indicate shared molecular mechanisms between neurodegeneration and depression, as noted previously (35).
Discovery of two GPCR proteins as novel regulators of depression
Given our particular interest in mining the GPCR family in search of the molecular regulators, we examined 35 GPCRs showing significant differential expression in PFC, HIP, or HY, three regions engaged in mood disorder and development of depression (31–34). After an extensive literature search, we were excited to find 19 DE GPCRs identified from the three regions turned out to be disclosed regulators of depressive-like behaviors and serve as potential antidepression targets, all uncovered by pharmacological intervention and/or genetic manipulation in vivo (Fig. 6; individual target references in table S8) (34, 36–57). Adenosine receptor A1 (A1R) was significantly down-regulated in the PFC and HIP of CUMS mice (Fig. 6), in line with the findings that A1R overexpression in forebrain neurons led to pronounced resilience toward depressive-like behaviors (36), and coadministration of a regular antidepressant and an A1R agonist produced a synergistic antidepressant effect in mouse behavioral tests (37). CB1 was significantly down-regulated in HY (>1.5-fold) and modestly down-regulated in PFC and HIP of CUMS mice (>1.3-fold) (Fig. 6). Consistently, genetic deficiency (38) or antagonism (39) of CB1 caused an increase in depressive-like behavior, whereas cannabinoids with CB1 agonist activity alleviated depressive-like symptoms in animal models (40, 41). Nociceptin receptor (NOP) was significantly up-regulated in PFC of CUMS mice (Fig. 6), in line with the finding that targeting NOP with its antagonist has translated to antidepressant-like effects in rodent models and to an antidepressant efficacy in patients with major depressive disorder (42).
Fig. 6. DE GPCRs identified in three specific brain regions from the CUMS model and their link to depression.
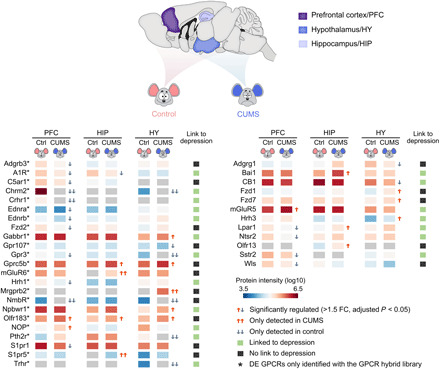
Protein intensity data were derived from DIA MS analysis with the GPCR virtual library (left) or the DDA library (right). GPCRs linked or never linked to depression in literature are annotated in green or black symbols. FC, fold change.
We then selected two DE GPCRs that have never been implicated in depression for functional evaluation. Neuromedin B (NMB) receptor (NmbR) was only detected in PFC and HY of control mice, suggesting suppressed expression in the two regions of CUMS mice (Fig. 6). The NmbR mRNA level was down-regulated by 1.6- and 1.9-fold in the two regions (P < 0.05 for both) as revealed by qPCR (fig. S7A). To examine whether modulating the NmbR activity would affect the acute depressive-like behaviors in mice, we stereotactically infused an NmbR agonist NMB into the medial prefrontal cortex (mPFC). Significantly reduced immobile time was observed in mice at 1 and 24 hours after infusion of a higher dose of NMB (1.25 μg each side) in both TST and FST, indicating a rapid and lasting anti-despair effect (fig. S7, B and C). Infusion of NMB at the higher dose into mPFC did not induce anxiety behavior or aberrant locomotion in the open-field test (OFT) (fig. S7D).
Next, we evaluated the effect of NMB in a validated mouse model of depression, chronic restraint stress (CRS) (58), based on multiple behavioral tests (Fig. 7A). Mice exposed to 14-day consecutive CRS received a local infusion of NMB into mPFC (1.25 μg each side), which was sufficient to reverse the depressive-like behaviors as measured in TST, FST, and SPT at 1 and 24 hours after infusion (Fig. 7, B and D). No change was observed in the general locomotion or anxiety behavior of NMB-treated CRS mice in the OFT (fig. S7E). Moreover, preinfusion of an NmbR antagonist, PD168368, completely blocked the antidepressant effect of NMB in the TST at 1 hour after infusion (Fig. 7E). Given that NMB also functions as an agonist for gastrin-releasing peptide (GRP) receptor (GrpR), which is closely related to NmbR (59), we evaluated the potent cognate agonist of GrpR, the GRP, in the CRS model. Local infusion of GRP into mPFC at doses comparable to or fivefold higher than NMB produced no antidepressant effect in the TST (Fig. 7E). These results suggested that NMB elicited a fast-acting antidepressant response in vivo mainly through the activation of NmbR.
Fig. 7. Discovery of novel GPCR regulators of depressive-like behaviors in the mouse model.
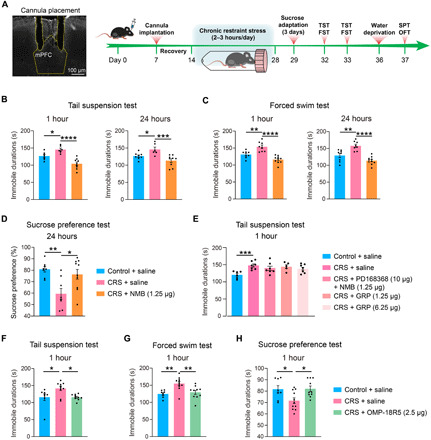
(A) Procedure for establishing the CRS model and performing stereotactical infusion of selected compounds into the mPFC before multiple behavioral tests. (B to D) Antidepressant effects of stereotactic infusion of NMB (1.25 μg each side) into the PFC at 1 and 24 hours after infusion as measured in the TST (B), FST (C), and SPT (D). (E) Abolished antidepressant effect of NMB (1.25 μg each side) by preinfusion of PD168368 (10 μg each side) into the PFC and no antidepressant effect of infusion of GRP (1.25 and 6.25 μg each side) into the PFC at 1 hour after infusion as measured in the TST. (F to H) Antidepressant effects of stereotactic infusion of OMP-18R5 (2.5 μg each side) into the PFC at 1 hour after infusion as measured in the TST (F), FST (G), and SPT (H). Data are means ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
Among several Frizzled (FZD) receptors found to be dysregulated in CUMS mice (Fig. 6), we selected Fzd7, which was up-regulated by 1.54-fold in HY and by 1.31-fold in PFC (P < 0.05 in both regions), for further examination. The Fzd7 mRNA level was not significantly altered in the two regions of the CUMS model (fig. S7A). For pharmacological evaluation, we produced an antibody (OMP-18R5) initially identified by binding to human FZD7 and later found to interact with four other FZD receptors (fig. S7f) (60). Stereotactic infusion of OMP-18R5 into the mPFC significantly reduced the immobile time in the TST at 1 hour after infusion for the medium dose tested (2.5 μg each side), while a lower (0.25 μg) or higher (12.5 μg) dose had no such effect (fig. S7J). Concordantly, infusion of OMP-18R5 (2.5 μg each side) into the mPFC significantly suppressed the depressive-like behaviors in the CRS model as measured in TST, FST, and SPT at 1 hour after infusion (Fig. 7, F to H), while the immunoglobulin G (IgG) isotype control had no such effects (fig. S7, G to I). Unlike NMB, OMP-18R5 did not retain its antidepressant effect at 24 hours after infusion either in the naïve mice or the CRS model (fig. S7, J and K). Thus, our results revealed a rapid antidepressant response in mice induced by the FZD antibody, which indicated the regulatory role of Fzd7 or other FZD receptors in depression.
DISCUSSION
Transmembrane proteins at the cell surface play central roles in mediating cell-cell communication, vital for synaptic transmission and plasticity in the CNS (13, 14). Profiling the distribution and dynamics of the transmembrane proteome in intact brain tissues, however, has remained a daunting task due to technical difficulties in extracting, processing, and identifying these highly hydrophobic and low-abundance proteins (8). As a result, most of the global proteomics of cells and tissues in CNS are biased toward the abundant soluble proteins in the cytosol or nucleus (10, 11, 61, 62). Our study provides a new workflow for sensitive and accurate profiling of the transmembrane proteome, especially the GPCR proteins, in brain tissues. With this approach, we have identified and quantified 143 and 158 GPCRs from the region-resolved mouse brain at a resting state and in the CUMS depression model, respectively, which more than doubles the largest number of GPCR IDs reported in mouse brain tissue (10). The unprecedented depth of mapping GPCRs and other transmembrane proteins in this study is attributed not only to cell membrane fractionation but also to our application of the DIA MS technology empowered by the targeted hybrid library strategy (Figs. 2, B and C, and 5D).
A limitation of our approach is the lack of cell type specificity when profiling the transmembrane proteome in the heterogeneous brain tissues. Although this may be beneficial for unbiased searches for potential drug targets, as showcased in our ID of key regulators of depression, we envision that this drawback could be overcome by combing our approach with an innovative APEX (ascorbic acid peroxidase) labeling technique for resolving cell type–specific cell surface proteomes in transgenic animals (14). In addition, note that we identified considerably more GPCRs and comparable numbers of ion channels and transporters using the targeted hybrid library versus the project-specific DDA library. Because the targeted hybrid library was built on single-shot DIA MS data and in silico prediction results, it is possible to avoid laborious prefractionation-based DDA MS analysis so as to work on exceedingly small amounts of cells or tissues.
Because transmembrane proteins, especially GPCRs, are major targets for drug development, we applied our approach to capturing regional changes of the brain transmembrane proteome in a mouse depression model. Just by looking into the DE GPCRs in three brain regions, we identified 19 regulators of depressive-like behaviors that have been disclosed over the past 15 years (Fig. 6). The extraordinary efficiency of our proteomic screen of potential drug targets is further illustrated by the discovery of two novel GPCR regulators of depression, NmbR and Fzd7. The role of NmbR in CNS is mainly associated with thermoregulation and smooth muscle contraction (59, 63), while the class FZD GPCR family is known to mediate Wnt/β-catenin signaling in embryonic development and tissue homeostasis (64, 65). Neither receptor has been linked to depression or other anxiety disorders. It merits further investigation whether the NmbR agonist and the Fzd antibody exert a rapid antidepressant effect through mediating specific neural circuit formation and whether they share cellular or molecular basis with the recently approved antidepression drug ketamine (58, 66, 67).
In summary, our study presents an enabling technology to characterize the regional distribution and dynamics of the transmembrane proteome in brain tissues. This technology, when applied to a disease model and combined with in vivo pharmacology tests, allows for exceptionally efficient discovery of molecular regulators that drive disease phenotypes. We anticipate more mechanistic studies to evaluate the potential of NmbR and Fzd7 as drug targets for treating clinical depression. We would also encourage neurobiologists to further examine the data resources provided in multiple aspects (brain region–enriched profiles, the PPI network, and protein dysregulation in the CUMS model), which could lead to more unexpected findings to enhance our understanding of brain physiology and disease pathogenesis mediated by transmembrane proteins.
MATERIALS AND METHODS
Animals and brain tissue preparation
The C57BL/6 mice (Shanghai Jiesijie, Laboratory Animal Technology Company, China) were housed under a 12-hour light-dark cycle with ad libitum free access to water and food. The room temperature and humidity for mice were 22° to 25°C and 40 to 50%. All experimental mice were male adults (8 to 16 weeks of age) and were habituated for 1 week at least before the experiments. All experimental procedures were approved by the Institutional Animal Care and Use Committee at ShanghaiTech University and performed in accordance with National Institutes of Health guidelines. The mice were euthanized with 2% chloral hydrate and rapidly dissected to obtain 10 main brain regions (olfactory bulb, cerebral cortex, cerebellum, HIP, midbrain, spinal cord, striatum, thalamus, pons, and medulla) at a resting state. For the CUMS model, the control and CUMS mice were euthanized and dissected to obtain 11 brain regions (olfactory bulb, PFC, cerebellum, HIP, midbrain, spinal cord, striatum, thalamus, pons, medulla, and HY). The fresh brain regions were immediately transferred into individual tubes, frozen on liquid nitrogen, and stored at −80°C until further processing.
Cell membrane fractionation and protein digestion
Most of brain regions were dissected from two to three mice and pooled as a replicate, except for the thalamus and HY dissected from five mice per replicate. Brain region sampling and the total number of mice sacrificed for specific proteomics experiments are summarized in table S9. Each region was prepared in quadruplicate for the resting-state mice and in triplicate for the CUMS model (both control and CUMS groups). Brain regions were homogenized in the isolation buffer of 30 mM tris-HCl (pH 7.4), 0.1 mM EDTA, 0.5% bovine serum albumin (BSA), 300 mM sucrose, supplemented with EDTA-free complete protease inhibitor cocktail tablets (Roche). The homogenate was centrifuged for 15 min at 3000g at 4°C. The cell pellet was resuspended in the isolation buffer, homogenized, and centrifuged for 20 min at 10,000g at 4°C. The supernatant was collected and subjected to ultracentrifugation for 1 hour at 160,000g at 4°C. The resulting membrane pellet was washed with 100 mM Na2CO3 and 100 mM tris-HCl (pH 7.4) separately and ultracentrifuged at 160,000g for 1 hour at 4°C. The membrane pellet was resuspended in the lysis buffer of 5% SDC (sodium deoxycholate) and 50 mM NH4HCO3 and heated at 95°C for 5 min. Protein concentration of the membrane extract was determined by BCA assay (TIANGEN, Beijing, China).
About 20 μg of protein extract from each brain region replicate was reduced with 15 mM dithiothreitol at 56°C for 30 min and alkylated with 40 mM iodoacetamide at room temperature for 30 min. Protein samples were diluted with 50 mM NH4HCO3 to a final SDC concentration of 0.5% before trypsin (Promega, Madison, USA) was added at an enzyme-to-protein ratio of 1:50 (w/w) for incubation at 37°C overnight. After quenching the digestion with 1% FA (formic acid), the supernatant was desalted with the C18 microspin column (The Nest Group, USA) and lyophilized under vacuum. The protein digest from each replicate was spiked in with an iRT (indexed retention time) reference kit (Biognosys, Zürich, Switzerland) and split into two, one half for the DIA MS analysis and the other half for the high-pH reversed-phase (RP) fractionation and DDA MS analysis.
High-pH RP fractionation of protein digests
Protein digests from the four replicates of each brain region from the resting-state mice were pooled and loaded onto an equilibrated, high-pH, RP fractionation spin column (Pierce). Peptides bound to the hydrophobic resin were desalted by washing with water. A step gradient of acetonitrile (5 to 50%) in a volatile high-pH elution solution was then applied to elute peptides into eight fractions sequentially. For the CUMS model, protein digests from five or six brain regions of CUMS mice were pooled and fractionated into eight fractions using the same procedure. All fractionated samples were dried under vacuum centrifuge and stored at −80°C before nano–liquid chromatography–tandem MS (nanoLC-MS/MS) analysis.
NanoLC-MS/MS analysis
DDA MS analysis
The fractionated peptide samples were dissolved in solvent A (0.1% formic acid) and separated on an analytical column (200 mm by 75 μm) and in-house packed with C18-AQ 3-μm C18 resin (Dr. Maisch GmbH, Germany) on a nanoflow EASY-nLC 1000 system (Thermo Fisher Scientific), using a gradient of 5 to 35% solvent B (0.1% formic acid in acetonitrile) over 120 min at a flow rate of 300 nl/min. DDA MS analysis was performed on an Orbitrap Fusion Tribrid mass spectrometer (Thermo Fisher Scientific). The acquisition method was set to the following parameters: survey scan resolution, 60,000 at 400 mass/charge ratio (m/z); AGC (automatic gain control) target, 4 × 105; maximum injection time, 50 ms; the range for survey scans, 300 to 1700 m/z; top 12 precursors selected for MS2 data acquisition; MS/MS scan resolution, 30,000; AGC target, 1 × 105; maximum injection time, 50 ms; isolation window, 1.6 m/z; normalized collision energy (NCE), 30 ± 5%; dynamic exclusion time, 60 s.
DIA MS analysis
Protein digest from each brain region replicate was analyzed using an EASY-nLC 1200 system (Thermo Fisher Scientific) coupled to a Q-Exactive HF-X mass spectrometer (Thermo Fisher Scientific). In the DIA mode, the precursor ions were fragmented in 35 variable windows covering a range of 300 to 1250 m/z. The resolution of Orbitrap analyzer was set to 60,000 for MS1 and 30,000 for MS2. The AGC was set to 3 × 106 in MS1 and 1 × 106 in MS2, with a maximum injection time of 20 ms in MS1 and 50 ms in MS2. The NCE was 30%. LC gradient was set as follows: 5 to 26% solvent B (0.1% formic acid and 80% acetonitrile) in 90 min; 26 to 45% solvent B in 30 min; 45 to 100% solvent B in 4 min; and 100% solvent B in 6 min.
Project-specific DDA library generation
The 80 DDA raw files acquired from fractionated protein digests from 10 brain regions were combined and searched using Proteome Discoverer 2.2 (Thermo Fisher Scientific) against mouse Swiss-Prot sequence database (July 2017, 16,905 entries) supplemented with a contaminant database and a protein entry containing the iRT peptide sequences. The search parameters included cabamidomethyl (C) as a fixed modification; acetylation (at protein N terminus), and oxidation (M) as variable modifications; trypsin/P as the specific enzyme; maximal two missed cleavages per peptide allowed; a precursor ion mass tolerance of 10 parts per million; and a fragment ion tolerance of 0.02 Da. FDRs on PSM (peptide spectrum match)/peptide/protein levels were all set to 1%. The resulting search result file was imported to Spectronaut 12.0 (Biognosys, Zürich, Switzerland) to generate a project-specific spectral library with default settings. In the CUMS model data analysis, the additional DDA raw files acquired from pooled brain region digests were searched in the same manner, and the research result was merged with the original DDA library to generate an expanded DDA library specific for the CUMS model.
Initial DIA library generation
The initial DIA library was generated by searching all DIA raw data acquired from 10 brain regions (40 runs in total) of the resting-state mice or from 11 regions (66 runs in total) of the CUMS model against mouse Swiss-Prot sequence database (July 2017, 16,905 entries) plus the iRT kit FASTA format sequences using Spectronaut 12.0. Standard settings were adopted as follows: Trypsin/P as the specific protease; maximal two missed cleavages per peptide allowed, carbamidomethyl (C) as fixed modification; and oxidation (M) and acetylation (at protein N terminus) as variable modifications; FDRs on PSM/peptide/protein levels were all set to 1%.
Targeted hybrid library generation
Deep learning model training and testing
Here, we used pDeep2 (19) and DeepRT (20) to predict fragment ion intensities and calibrated retention time (RT) values, respectively, from given peptide sequences. To train these two deep learning models with our project-specific data, we filtered the initial DIA library to retain a fraction of high-quality data if they met the following criteria: a peptide length of 7 to 26 residues, a charge state of +1 to +5, and at least six fragment ions assigned in the PSM. The filtered data were split at a ratio of 9:1, and 90% of peptide precursor entries were used for model retraining and 10% of entries for model testing. Retraining pDeep2 and DeepRT models were conducted according to a previously described procedure (21) for the 10-region mouse brain data and the CUMS model’s brain data separately. The performance of retrained pDeep2 was evaluated on the basis of the similarity between predicted and experimental MSMS spectra (PCC), while the performance of retrained DeepRT based on the deviation of predicted iRT from experimental iRT for 95% data points (ΔiRT95%) and the linear regression between predicted and experiment iRT (R2) as previously described (21). Retrained models based on the 10-region brain data or the CUMS model data demonstrated overall excellent performance in the model testing (fig. S1, C to F).
In silico digestion of the GPCR protein family members
The protein sequences of all 524 mouse genome–encoded GPCRs (listed in table S1) were subjected to in silico digestion under the following condition: trypsin specificity, no more than one missed cleavage, a peptide length from 7 to 33 residues, and a charge state at 2 or 3. A fixed carbamidomethyl modification on Cys were installed on all peptide sequences, and no variable modification was considered. This best-performing digestion condition was selected from 12 different sets of conditions in a previous study to yield the largest number of GPCR IDs with the highest percentage of bona fide IDs (21). For decoy library generation, we reversed the protein sequences of 524 mouse GPCRs and applied the same procedure to generate decoy GPCR peptide precursors. In silico digestion was also performed for the ion channel and transporter family members encoded in the genome.
Construction of GPCR hybrid libraries and subgroup FDR control
The validated pDeep2 and DeepRT models were used to predict the MS/MS fragmentation pattern and calibrated retention time for each GPCR peptide precursor yielded from in silico digestion. The outputs were combined to generate a GPCR virtual library, which was then merged with the initial DIA library to construct a GPCR family–targeted hybrid library. Targeted hybrid libraries for the ion channel and transporter families were also constructed in the same procedure. Notice that this targeted hybrid library strategy was the most effective in profiling GPCRs as very little increase in the subproteome coverages was observed for the ion channel and transporter families when searching data with their targeted hybrid libraries (fig. S2, F and G).
Meanwhile, a decoy GPCR hybrid library was generated by merging a decoy GPCR virtual library (built on the decoy GPCR peptide sequences) with the same initial DIA library. This decoy hybrid library structurally resembling the GPCR hybrid library was used to assess the subgroup FDR for GPCR peptide ID from DIA MS data search with the GPCR hybrid library. This subgroup FDR was defined as
where the Hitsdecoy is the number of decoy GPCR peptide IDs from data search with the decoy hybrid library, and the Hitshybrid is the number of GPCR peptide IDs with the GPCR hybrid library. We filtered GPCR IDs in data search results on the basis of the Cscore value so as to restrict the subgroup FDR less than 5% at the peptide level for any data search with a GPCR hybrid library (figs. S2, A and C, and S6D). Search results were not filtered for data search with a conventional DDA library.
DIA MS data processing
The DIA MS data were processed using Spectronaut 12.0 in the library-dependent mode. The aforementioned project-specific DDA library, the targeted hybrid library built for a selected protein family, and the corresponding decoy hybrid library were imported separately along with a specific dataset. For the resting-state 10-region analysis, DIA MS raw data from all regions were imported; for the CUMS model analysis, the raw data from paired control and CUMS groups for each brain region were imported and processed. DIA MS data search was performed with default settings in Spectronaut: a precursor and protein Q value cutoff of 0.01 (meaning the global FDR of <1% at both levels), quantification based on MS2 area, local normalization, and decoy generation method “mutated.” The reference iRT peptides were used to calibrate the retention time. Protein ID and quantification reports were exported for further analysis. In the CUMS model experiment, proteins with a fold change of >1.5 and a P value (adjusted for multiple testing) of <0.05 between the control and CUMS groups of any brain region were regarded DE. DE GPCRs were merged from those identified using the DDA library or the GPCR hybrid library.
Bioinformatic analysis
Comparison of multiregional proteomic and transcriptomic datasets
In our DDA and DIA MS data search results, the first ID from each protein group was retained as a unique protein ID for further analysis. The multiregional ISH data were downloaded from the Allen Brain Atlas, and the RNA-seq data were downloaded from The Human Protein Atlas. Spearman correlation analysis was performed on 1733 transmembrane protein–coding genes, which were measured in at least five brain regions by both proteomic and ISH analysis, and on 1738 transmembrane protein–coding genes measured in at least five brain regions by both proteomic and RNA-seq analysis.
PCA and HCA
PCA was implemented with the python package scikit-learn. The input matrix was z score–scaled, and the PCA parameters were set as default. HCA was implemented with the python package SciPy (68). The metric for hierarchical clustering was set to cosine, and the method was set to average.
Protein coexpression analysis
Pearson correlation analysis was performed between each GPCR with any of the other proteins codetected in at least five brain regions in our proteomics data. Putative PPI pairs were retained if their coexpression PCCs were above 0.7. Known GPCR interacting partners were retrieved from STRING database (69) with a score cutoff of 0.4 (https://string-db.org/api). Cytoscape (version 3.8.0) (70) was used to create a predicted interaction map for GPCRs and transmembrane proteins based on high-stringency coexpression profiles (PCC, >0.9).
Pathway enrichment
Pathway enrichment analysis of DE proteins identified in the CUMS model was implemented with DAVID Bioinformatics Resources 6.8 (https://david.ncifcrf.gov/). Pathways with P < 0.001 were regarded significantly enriched.
RT-qPCR analysis
Different brain regions from three to five mice were dissected and processed for RT-qPCR. The brain tissue was dissolved in 1 ml of TRIzol (Invitrogen, Life Technologies), and the total RNA was prepared according to the manufacturer‵s protocol. Total RNA was reverse-transcribed using the cDNA synthesis kit (Takara, China) into cDNAs, which were subjected to qPCR reactions on a CFX96 real-time qPCR system (Bio-Rad) using ChamQ SYBR Color qPCR Master Mix (Vazyme, China). Primers are as follows: Gria2, 5′-GCCGAGGCGAAACGAATGA-3′ (forward) and 5′-CACTCTCGATGCCATATACGTTG-3′ (reverse);
Slc5a7, 5′-ATGTCTTTCCACGTAGAAGGACT-3′ (forward) and 5′-TTGCCGCTGTTTTTGGTTTTC-3′ (reverse);
Slc6a3, 5′-TACGTGGGCTTCTTCTACAATGT-3′ (forward) and 5′-GTTGCTGCTATGTGCATCAGA-3′ (reverse); β-actin, 5′-GGCTGTATTCCCCTCCATCG-3′ (forward) and 5′-CCAGTTGGTAACAATGCCATGT-3′ (reverse). The relative quantification of gene expression was analyzed using the 2−ΔΔCt method. Data were normalized to β-actin.
Immunoblotting
Tissue lysates were extracted from each brain region in 5% SDS and 50 mM NH4HCO3 buffer supplemented with protease and phosphatase inhibitor cocktails (Roche). Total proteins of 20 μg were loaded and separated on 10% gels and transferred onto polyvinylidene difluoride membranes (Millipore, USA). Then, the membranes were blocked in 5% milk for 1 hour at room temperature and incubated overnight at 4°C with primary antibodies diluted in 5% milk. The primary antibodies were as follows: anti-Gria2 (ab133477, Abcam), anti-Slc6a3 (ab184451, Abcam), anti-Slc5a7 (sc-33713, Santa Cruz Biotechnology), anti–myelin basic protein (MAB386, Merck Millipore), anti–glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (60004-1, ProteinTech). The membranes were washed five times with TBST (Tris Buffered Saline with Tween 20) and then incubated with secondary antibodies for 1 hour at room temperature. After washing with TBST, enhanced chemiluminescence was added, and the membrane was scanned on the imager system (Bio-Rad, USA).
Immunostaining of rat hippocampal neurons and mouse brain slices
The hippocampal neuron culture was prepared from neonatal Sprague-Dawley rats as previously described (71). At 14 days of in vitro culture, hippocampal neurons were fixed with 4% (w/v) paraformaldehyde in phosphate-buffered saline (PBS) for 15 min. For brain slice preparation, C57BL/6 mice (8 weeks old) were first anesthetized with sodium pentobarbital (40 mg/kg, intraperitoneally). They were then perfused with normal saline (at 37°C), followed by ice-cold 4% (w/v) paraformaldehyde. The brain were postfixed, dehydrated in 30% sucrose solution, and cut into 20-μm-thick coronal sections with the freezing microtome (CM 1950, Leica).
Both cultural neurons and brain slices were rinsed in 0.01 M PBS (pH 7.4) and incubated with 10% BSA in PBS with 0.3% Triton X-100 at room temperature for 0.5 hours. They were then incubated overnight at 4°C with the primary antibodies diluted in 1% BSA with 0.1% Triton X-100 against Slc6a3 (rabbit, ab184451, Abcam;1:200), NeuN (mouse, MAB377, Merck; 1:1000), FLRT3 (goat, AF2795, R&D Systems; 1:200), and CB1 [Rb (rabbit), 93815, Cell Signaling Technology; 1:200]. After washing, the samples were incubated for 2 hours with the corresponding secondary antibodies (Invitrogen; 1:1000). Then, they were washed and mounted with Prolong gold (P36930, Life Technology). Z-stack images were collected on a laser scanning confocal microscope (Nikon A1R) with a voxel interval of 0.5 μm. Fiji software (https://imagej.net/Fiji) was used for imaging data processing.
In situ proximity ligation assay
The Duolink in situ PLA detection kit (catalog no. DUO92105-1KT, Sigma-Aldrich) was used, and the protocol was followed. Briefly, CHO cells or primary hippocampal cultured neurons were fixed, blocked, and incubated with the antibodies against CB1 and FLRT3 overnight, similar to the immunostaining procedure above. On the next day, all remaining incubation steps were performed at 37°C. Equal amounts of a plus PLA probe and a minus PLA probe, which bind to the CB1 and FLRT3 antibody respectively, were mixed and 1:10 diluted. After washing with PBS, cells were incubated with the probe pair for 1 hour then washed with 1 × washing buffer A. Ligase was diluted 1:40 with 1× ligation buffer and applied to cells for 0.5 hour. After washing, polymerase was then added to cells at 1:80 diluted in 1× amplification stock and incubated for 2.5 hours. Cells were washed twice with 1× washing buffer B, once with 0.01 × washing buffer B, and once with PBST (PBS with Tween 20). Then, cells were mounted with the mounting medium containing 4′,6-diamidino-2-phenylindole. Images were acquired as described above and further processed with Fiji software.
Immunoprecipitation
Following tetracycline induction, CB1-CHO cells were washed with ice-cold PBS and suspended in IP buffer containing 50 mM tris-HCl, 120 mM NaCl, 0.5% NP-40, 0.2% n-dodecyl-β-d-maltopyranoside, 5% glycerol, and 1× protease inhibitor cocktail (pH 7.5). The lysate was centrifuged, and the resulting supernatant was incubated with the rabbit anti-CB1 (93815, Cell Signaling Technology) antibody for 20 min at 4°C. Immunocomplex was incubated with Protein A Magnetic beads (Bio-Rad, USA) overnight on a rotating wheel at 4°C. The beads was then washed five times in wash buffer containing 20 mM tris-HCl, 100 mM NaCl, 1 mM EDTA, and 0.5% NP-40 (pH 8.0). The immunoprecipitates were mixed with the loading buffer and resolved by SDS–polyacrylamide gel electrophoresis (PAGE). Western blots were performed with relevant antibodies. Rabbit IgG was used as a negative control.
FZD7 antibody production
The human FZD7 antibody, OMP-18R5, was produced as previously described (60). Briefly, the genes encoding heavy and light chains of OMP-18R5 were cloned into the pTT5 vector and transfected into FreeStyle 293-F cells (Thermo Fisher Scientific). After 6-day culture, the supernatant from 2 × 108 cells was harvested by centrifugation at 6000g at 4°C for 20 min and clarified by centrifugation again at 4000g at 4°C for 30 min. The antibody was affinity-purified on an rProtein A chromatography column (GE Healthcare) and eluted with 100 mM Na citrate–HCl (pH 3.5). The eluted fraction was quickly neutralized by adding one-sixth volume of 1 M tris-HCl (pH 8.0), followed by buffer exchange to PBS (pH 7.4) in a 50-kDa concentrator (Merck Millipore). The protein purity was assessed by SDS-PAGE and analytical size exclusion chromatography as shown in fig. S7F.
Mouse depression model procedures
CUMS procedure
The CUMS procedure was performed as described previously (72, 73) with certain modifications. C57BL/6 mice were single-housed and received 3 weeks of unpredictable mild stressors. These stressors were performed in a random order with gradually increased intensity. Specific stressors included (i) food deprivation (24 hours), (ii) water deprivation (24 hours), (iii) soiled cage (200 ml of water in the sawdust bedding, 24 hours), (iv) 45° cage tilt (24 hours), (v) overnight illumination (12 hours), (vi) clipping the tail (1 hour), (vii) restraint in a 50-ml tube (6 to 8 hours), (viii) social attack (15 min), and (ix) foot shock (1.75 mA, 40 min). Control mice were group-housed in a neutral environment, and all mice lived under comparable environmental conditions. Two days after the CUMS procedure, behavioral tests were performed in the order of SPT, OFT, TST, and FST on individual mice, with at least 2-hour resting time between two different tests.
CRS procedure
C57BL/6 mice were individually restricted in 50-ml conical tubes for 2 to 3 hours per day in the light phase, and the procedure was performed for 2 weeks as previously described (58).
Compound administration to mouse models
Stereotactic surgery
Mice were anesthetized with isoflurane (1.0 to 1.5%) and oxygen (0.6 to 0.8 liters/min), fixed on a stereotaxic device (RWD, China) with the brain skull surface exposed. All surgical instruments were sterilized with alcohol. For cannula implantation, a 26-gauge double stainless steel cannula (RWD, China) with 3.0 mm in length was slowly implanted into the mPFC (anterior-posterior, +1.60 mm from the bregma; medial-lateral, ±0.35 mm from the sagittal structure; dorsal-ventral, −1.70 mm from the dura).
Compound infusion
Porcine NMB was purchased from Tocris (USA), and GRP and PD168368 were purchased from Cayman Chemical (USA). Fzd7 antibody OMP-18R5 was in-house–produced. All mice were recovered for at least 7 days after the surgery. Before compound infusion, a 26-gauge double internal injector with 3-mm projection was placed into the guide cannula. NMB (0.25/1.25 μg each side, 500 nl), GRP (1.25/6.25 μg each side, 500 nl), PD168368 (10 μg each side, 500 nl), OMP-18R5 (0.25 μg/2.5 μg/12.5 μg each side, 500 nl), or 0.9% saline (500 nl) was administered at a rate of 200 nl/min through the injector. Injector cannula was removed 5 min after infusion.
Animal behavioral tests
All mice were 10 to 16 weeks old for behavioral tests, which were performed between 14:00 p.m. and 21:00 p.m. In specific, SPT and OFT were performed during the dark phase, whereas FST and TST were performed during the light phase. For FST, mice were allowed to swim for 2 days in advance, 10 min per day. Behavioral data were analyzed by others blinded to the tests.
TST
Four mice were acoustically and visually isolated and were suspended in a computerized device for data recording. To ensure that mice could not contact or climb subjects, each mouse was suspended 50 cm above the floor by a tail tape that placed approximately 1 cm from the tip of the tail. The activities of mice were recorded for 6 min. The total duration of immobility recorded in the last 4 min was analyzed.
FST
Mice were put in a clear glass tank (40 cm in height and 20 cm in diameter) filled with water (26° to 27°C). The water depth was set to ensure that mice could not touch the bottom with their tails or hind limbs. Mice were allowed to swim for 6 min. Immobility of the mice was defined by floating in the water without struggling and only making movements necessary to keep their heads above the water. The last 4 min of the testing period was analyzed as the total immobility time.
SPT
For adaptation, mice were single-housed and exposed to a bottle of sucrose solution (2%) accompanied with a bottle of water for 3 days, followed by 24 hours of water deprivation. In the test, mice were exposed to two identical bottles, one filled with 1% sucrose solution and the other filled with water. The weight of sucrose solution or water consumption was measured after 2 hours of exposure. The positions of two bottles were switched every 24 hours in the adaptation period and after 1 hour in the test period. Sucrose preference was defined as the ratio of the weight of sucrose consumption versus the total weight of sucrose and water consumption during the 2-hour test. In the CUMS procedure, sucrose preference and body weight of mice were measured on days 1, 8, 15, and 22.
OFT
Mice were placed in a white open-field chamber (30 cm by 30 cm by 40 cm) and were free to move in a room with dim light. A video camera (Jiliang, China) was positioned above the chamber to track the movement of each animal for 5 min in the arena. The time spent in the central zone of the chamber as a measure of anxiety behavior and the total distance were automatically calculated.
Statistical tests
Data were analyzed by paired or unpaired t test and one-way or two-way analysis of variance (followed by a Bonferroni’s multiple comparison test). Significance was considered as *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
Acknowledgments
We thank X. Liu from Mammalian Core Facility of iHuman Institute, ShanghaiTech University for the support in protein expression. We are also thankful to W. Chen and W. Zhu from SIAIS at ShanghaiTech University for the assistance in MS data acquisition. Funding: This work was funded by ShanghaiTech University, the National Program on Key Basic Research Project of China (2018YFA0507004), National Natural Science Foundation of China (31971362, 81970878, 31771130, 1861128023, 31922029, 61890951, and 61890950), and Innovative Research Team of High-level Local Universities in Shanghai. Author contributions: W.S., J.H., and G.Z. conceived and supervised the project. S.L. performed the proteomic sample preparation, MS data acquisition, and immunoblotting experiment assisted by C.M. and X.D. Huoqing L. established the mouse models and performed behavioral tests. R.L. performed the deep learning model training, spectral library generation, and MS data processing assisted by S.L. and T.D. C.T., T.D., and Hui L. performed the immunostaining and PLA experiments with the help of Z.Z. and Yan L. C.P. and L.X. produced the FZD7 antibody, and L.X. also performed the qPCR analysis. C.F. helped with graphical preparation. F.X. supported the FZD7 antibody production. Yunxia L. and Y.Z. supported the MS data acquisition. W.S., S.L., R.L., Huoqing L., and C.T. wrote the manuscript with inputs from all authors. Competing Interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. The DDA/DIA MS raw data, spectral library files, and search result files generated during the current study are available in the ProteomeXchange Consortium via the iProX partner repository with the dataset identifier IPX0002210000. The pipeline TargetDIA and scripts for the targeted hybrid library generation and DIA MS data processing are available on GitHub at https://github.com/Shui-Group/TargetDIA. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/30/eabf0634/DC1
REFERENCES AND NOTES
- 1.Lein E. S., Hawrylycz M. J., Ao N., Ayres M., Bensinger A., Bernard A., Boe A. F., Boguski M. S., Brockway K. S., Byrnes E. J., Chen L., Chen L., Chen T. M., Chi Chin M., Chong J., Crook B. E., Czaplinska A., Dang C. N., Datta S., Dee N. R., Desaki A. L., Desta T., Diep E., Dolbeare T. A., Donelan M. J., Dong H. W., Dougherty J. G., Duncan B. J., Ebbert A. J., Eichele G., Estin L. K., Faber C., Facer B. A., Fields R., Fischer S. R., Fliss T. P., Frensley C., Gates S. N., Glattfelder K. J., Halverson K. R., Hart M. R., Hohmann J. G., Howell M. P., Jeung D. P., Johnson R. A., Karr P. T., Kawal R., Kidney J. M., Knapik R. H., Kuan C. L., Lake J. H., Laramee A. R., Larsen K. D., Lau C., Lemon T. A., Liang A. J., Liu Y., Luong L. T., Michaels J., Morgan J. J., Morgan R. J., Mortrud M. T., Mosqueda N. F., Ng L. L., Ng R., Orta G. J., Overly C. C., Pak T. H., Parry S. E., Pathak S. D., Pearson O. C., Puchalski R. B., Riley Z. L., Rockett H. R., Rowland S. A., Royall J. J., Ruiz M. J., Sarno N. R., Schaffnit K., Shapovalova N. V., Sivisay T., Slaughterbeck C. R., Smith S. C., Smith K. A., Smith B. I., Sodt A. J., Stewart N. N., Stumpf K. R., Sunkin S. M., Sutram M., Tam A., Teemer C. D., Thaller C., Thompson C. L., Varnam L. R., Visel A., Whitlock R. M., Wohnoutka P. E., Wolkey C. K., Wong V. Y., Wood M., Yaylaoglu M. B., Young R. C., Youngstrom B. L., Feng Yuan X., Zhang B., Zwingman T. A., Jones A. R., Genome-wide atlas of gene expression in the adult mouse brain. Nature 445, 168–176 (2007). [DOI] [PubMed] [Google Scholar]
- 2.Thompson C. L., Ng L., Menon V., Martinez S., Lee C. K., Glattfelder K., Sunkin S. M., Henry A., Lau C., Dang C., Garcia-Lopez R., Martinez-Ferre A., Pombero A., Rubenstein J. L. R., Wakeman W. B., Hohmann J., Dee N., Sodt A. J., Young R., Smith K., Nguyen T. N., Kidney J., Kuan L., Jeromin A., Kaykas A., Miller J., Page D., Orta G., Bernard A., Riley Z., Smith S., Wohnoutka P., Hawrylycz M. J., Puelles L., Jones A. R., A high-resolution spatiotemporal atlas of gene expression of the developing mouse brain. Neuron 83, 309–323 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hawrylycz M. J., Lein E. S., Guillozet-Bongaarts A. L., Shen E. H., Ng L., Miller J. A., van de Lagemaat L. N., Smith K. A., Ebbert A., Riley Z. L., Abajian C., Beckmann C. F., Bernard A., Bertagnolli D., Boe A. F., Cartagena P. M., Chakravarty M. M., Chapin M., Chong J., Dalley R. A., Daly B. D., Dang C., Datta S., Dee N., Dolbeare T. A., Faber V., Feng D., Fowler D. R., Goldy J., Gregor B. W., Haradon Z., Haynor D. R., Hohmann J. G., Horvath S., Howard R. E., Jeromin A., Jochim J. M., Kinnunen M., Lau C., Lazarz E. T., Lee C., Lemon T. A., Li L., Li Y., Morris J. A., Overly C. C., Parker P. D., Parry S. E., Reding M., Royall J. J., Schulkin J., Sequeira P. A., Slaughterbeck C. R., Smith S. C., Sodt A. J., Sunkin S. M., Swanson B. E., Vawter M. P., Williams D., Wohnoutka P., Zielke H. R., Geschwind D. H., Hof P. R., Smith S. M., Koch C., Grant S. G. N., Jones A. R., An anatomically comprehensive atlas of the adult human brain transcriptome. Nature 489, 391–399 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Miller J. A., Ding S. L., Sunkin S. M., Smith K. A., Ng L., Szafer A., Ebbert A., Riley Z. L., Royall J. J., Aiona K., Arnold J. M., Bennet C., Bertagnolli D., Brouner K., Butler S., Caldejon S., Carey A., Cuhaciyan C., Dalley R. A., Dee N., Dolbeare T. A., Facer B. A. C., Feng D., Fliss T. P., Gee G., Goldy J., Gourley L., Gregor B. W., Gu G., Howard R. E., Jochim J. M., Kuan C. L., Lau C., Lee C. K., Lee F., Lemon T. A., Lesnar P., McMurray B., Mastan N., Mosqueda N., Naluai-Cecchini T., Ngo N. K., Nyhus J., Oldre A., Olson E., Parente J., Parker P. D., Parry S. E., Stevens A., Pletikos M., Reding M., Roll K., Sandman D., Sarreal M., Shapouri S., Shapovalova N. V., Shen E. H., Sjoquist N., Slaughterbeck C. R., Smith M., Sodt A. J., Williams D., Zöllei L., Fischl B., Gerstein M. B., Geschwind D. H., Glass I. A., Hawrylycz M. J., Hevner R. F., Huang H., Jones A. R., Knowles J. A., Levitt P., Phillips J. W., Šestan N., Wohnoutka P., Dang C., Bernard A., Hohmann J. G., Lein E. S., Transcriptional landscape of the prenatal human brain. Nature 508, 199–206 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Uhlen M., Fagerberg L., Hallstrom B. M., Lindskog C., Oksvold P., Mardinoglu A., Sivertsson A., Kampf C., Sjostedt E., Asplund A., Olsson I., Edlund K., Lundberg E., Navani S., Szigyarto C. A. K., Odeberg J., Djureinovic D., Takanen J. O., Hober S., Alm T., Edqvist P. H., Berling H., Tegel H., Mulder J., Rockberg J., Nilsson P., Schwenk J. M., Hamsten M., von Feilitzen K., Forsberg M., Persson L., Johansson F., Zwahlen M., von Heijne G., Nielsen J., Ponten F., Proteomics. Tissue-based map of the human proteome. Science 347, 1260419 (2015). [DOI] [PubMed] [Google Scholar]
- 6.Sjostedt E., Zhong W., Fagerberg L., Karlsson M., Mitsios N., Adori C., Oksvold P., Edfors F., Limiszewska A., Hikmet F., Huang J., Du Y., Lin L., Dong Z., Yang L., Liu X., Jiang H., Xu X., Wang J., Yang H., Bolund L., Mardinoglu A., Zhang C., von Feilitzen K., Lindskog C., Pontén F., Luo Y., Hökfelt T., Uhlén M., Mulder J., An atlas of the protein-coding genes in the human, pig, and mouse brain. Science 367, (2020). [DOI] [PubMed] [Google Scholar]
- 7.Hosp F., Mann M., A primer on concepts and applications of proteomics in neuroscience. Neuron 96, 558–571 (2017). [DOI] [PubMed] [Google Scholar]
- 8.Wang D., Eraslan B., Wieland T., Hallström B., Hopf T., Zolg D. P., Zecha J., Asplund A., Li L.-H., Meng C., Frejno M., Schmidt T., Schnatbaum K., Wilhelm M., Ponten F., Uhlen M., Gagneur J., Hahne H., Kuster B., A deep proteome and transcriptome abundance atlas of 29 healthy human tissues. Mol. Syst. Biol. 15, e8503 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vogel C., Marcotte E. M., Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nat. Rev. Genet. 13, 227–232 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sharma K., Schmitt S., Bergner C. G., Tyanova S., Kannaiyan N., Manrique-Hoyos N., Kongi K., Cantuti L., Hanisch U. K., Philips M. A., Rossner M. J., Mann M., Simons M., Cell type- and brain region-resolved mouse brain proteome. Nat. Neurosci. 18, 1819–1831 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Carlyle B. C., Kitchen R. R., Kanyo J. E., Voss E. Z., Pletikos M., Sousa A. M. M., Lam T. K. T., Gerstein M. B., Sestan N., Nairn A. C., A multiregional proteomic survey of the postnatal human brain. Nat. Neurosci. 20, 1787–1795 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huang Y., Todd N., Thathiah A., The role of GPCRs in neurodegenerative diseases: Avenues for therapeutic intervention. Curr. Opin. Pharmacol. 32, 96–110 (2017). [DOI] [PubMed] [Google Scholar]
- 13.Burke K. J. Jr., Bender K. J., Modulation of ion channels in the axon: Mechanisms and function. Front. Cell. Neurosci. 13, 221 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li J., Han S., Li H., Udeshi N. D., Svinkina T., Mani D. R., Xu C., Guajardo R., Xie Q., Li T., Luginbuhl D. J., Wu B., McLaughlin C. N., Xie A., Kaewsapsak P., Quake S. R., Carr S. A., Ting A. Y., Luo L., Cell-surface proteomic profiling in the fly brain uncovers wiring regulators. Cell 180, 373–386.e15 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Santos R., Ursu O., Gaulton A., Bento A. P., Donadi R. S., Bologa C. G., Karlsson A., al-Lazikani B., Hersey A., Oprea T. I., Overington J. P., A comprehensive map of molecular drug targets. Nat. Rev. Drug Discov. 16, 19–34 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bekker-Jensen D. B., Kelstrup C. D., Batth T. S., Larsen S. C., Haldrup C., Bramsen J. B., Sørensen K. D., Høyer S., Ørntoft T. F., Andersen C. L., Nielsen M. L., Olsen J. V., An optimized shotgun strategy for the rapid generation of comprehensive human proteomes. Cell Syst. 4, 587–599.e4 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gillet L. C., Navarro P., Tate S., Röst H., Selevsek N., Reiter L., Bonner R., Aebersold R., Targeted data extraction of the MS/MS spectra generated by data-independent acquisition: A new concept for consistent and accurate proteome analysis. Mol. Cell. Proteomics 11, O111 016717 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ludwig C., Gillet L., Rosenberger G., Amon S., Collins B. C., Aebersold R., Data-independent acquisition-based SWATH-MS for quantitative proteomics: A tutorial. Mol. Syst. Biol. 14, e8126 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhou X. X., Zeng W. F., Chi H., Luo C., Liu C., Zhan J., He S. M., Zhang Z., pDeep: Predicting MS/MS spectra of peptides with deep learning. Anal. Chem. 89, 12690–12697 (2017). [DOI] [PubMed] [Google Scholar]
- 20.Ma C., Ren Y., Yang J., Ren Z., Yang H., Liu S., Improved peptide retention time prediction in liquid chromatography through deep learning. Anal. Chem. 90, 10881–10888 (2018). [DOI] [PubMed] [Google Scholar]
- 21.Lou R., Tang P., Ding K., Li S., Tian C., Li Y., Zhao S., Zhang Y., Shui W., Hybrid spectral library combining DIA-MS data and a targeted virtual library substantially deepens the proteome coverage. iScience 23, 100903 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Civelli O., Reinscheid R. K., Zhang Y., Wang Z., Fredriksson R., Schiöth H. B., G protein-coupled receptor deorphanizations. Annu. Rev. Pharmacol. Toxicol. 53, 127–146 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Alavi M. S., Shamsizadeh A., Azhdari-Zarmehri H., Roohbakhsh A., Orphan G protein-coupled receptors: The role in CNS disorders. Biomed. Pharmacother. 98, 222–232 (2018). [DOI] [PubMed] [Google Scholar]
- 24.Jay T. M., Witter M. P., Distribution of hippocampal CA1 and subicular efferents in the prefrontal cortex of the rat studied by means of anterograde transport of Phaseolus vulgaris-leucoagglutinin. J. Comp. Neurol. 313, 574–586 (1991). [DOI] [PubMed] [Google Scholar]
- 25.van Groen T., Wyss J. M., Extrinsic projections from area CA1 of the rat hippocampus: Olfactory, cortical, subcortical, and bilateral hippocampal formation projections. J. Comp. Neurol. 302, 515–528 (1990). [DOI] [PubMed] [Google Scholar]
- 26.Hintiryan H., Gou L., Zingg B., Yamashita S., Lyden H. M., Song M. Y., Grewal A. K., Zhang X., Toga A. W., Dong H.-W., Comprehensive connectivity of the mouse main olfactory bulb: Analysis and online digital atlas. Front. Neuroanat. 6, 30 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kustatscher G., Grabowski P., Schrader T. A., Passmore J. B., Schrader M., Rappsilber J., Co-regulation map of the human proteome enables identification of protein functions. Nat. Biotechnol. 37, 1361–1371 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Guo T., Luna A., Rajapakse V. N., Koh C. C., Wu Z., Liu W., Sun Y., Gao H., Menden M. P., Xu C., Calzone L., Martignetti L., Auwerx C., Buljan M., Banaei-Esfahani A., Ori A., Iskar M., Gillet L., Bi R., Zhang J., Zhang H., Yu C., Zhong Q., Varma S., Schmitt U., Qiu P., Zhang Q., Zhu Y., Wild P. J., Garnett M. J., Bork P., Beck M., Liu K., Saez-Rodriguez J., Elloumi F., Reinhold W. C., Sander C., Pommier Y., Aebersold R., Quantitative proteome landscape of the NCI-60 cancer cell lines. iScience 21, 664–680 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhou R., Han B., Xia C., Zhuang X., Membrane-associated periodic skeleton is a signaling platform for RTK transactivation in neurons. Science 365, 929–934 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schallmeiner E., Oksanen E., Ericsson O., Spångberg L., Eriksson S., Stenman U. H., Pettersson K., Landegren U., Sensitive protein detection via triple-binder proximity ligation assays. Nat. Methods 4, 135–137 (2007). [DOI] [PubMed] [Google Scholar]
- 31.Covington H. E. III, Lobo M. K., Maze I., Vialou V., Hyman J. M., Zaman S., Plant Q. L., Mouzon E., Ghose S., Tamminga C. A., Neve R. L., Deisseroth K., Nestler E. J., Antidepressant effect of optogenetic stimulation of the medial prefrontal cortex. J. Neurosci. 30, 16082–16090 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lin D., Boyle M. P., Dollar P., Lee H., Lein E. S., Perona P., Anderson D. J., Functional identification of an aggression locus in the mouse hypothalamus. Nature 470, 221–226 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Anthony T. E., Dee N., Bernard A., Lerchner W., Heintz N., Anderson D. J., Control of stress-induced persistent anxiety by an extra-amygdala septohypothalamic circuit. Cell 156, 522–536 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chung G., Kim S. J., Kim S. K., Metabotropic glutamate receptor 5 in the medial prefrontal cortex as a molecular determinant of pain and ensuing depression. Front. Mol. Neurosci. 11, 376 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Anisman H., Merali Z., Hayley S., Neurotransmitter, peptide, and cytokine processes in relation to depressive disorder: Comorbidity between depression and neurodegenerative disorders. Prog. Neurobiol. 85, 1–74 (2008). [DOI] [PubMed] [Google Scholar]
- 36.Serchov T., Clement H. W., Schwarz M. K., Iasevoli F., Tosh D. K., Idzko M., Jacobson K. A., de Bartolomeis A., Normann C., Biber K., van Calker D., Increased signaling via adenosine A1 receptors, sleep deprivation, imipramine, and ketamine inhibit depressive-like behavior via induction of homer1a. Neuron 87, 549–562 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cunha M. P., Pazini F. L., Rosa J. M., Ramos-Hryb A. B., Oliveira Á., Kaster M. P., Rodrigues A. L. S., Creatine, similarly to ketamine, affords antidepressant-like effects in the tail suspension test via adenosine A1 and A2A receptor activation. Purinergic Signal 11, 215–227 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Valverde O., Torrens M., CB1 receptor-deficient mice as a model for depression. Neuroscience 204, 193–206 (2012). [DOI] [PubMed] [Google Scholar]
- 39.Beyer C. E., Dwyer J. M., Piesla M. J., Platt B. J., Shen R., Rahman Z., Chan K., Manners M. T., Samad T. A., Kennedy J. D., Bingham B., Whiteside G. T., Depression-like phenotype following chronic CB1 receptor antagonism. Neurobiol. Dis. 39, 148–155 (2010). [DOI] [PubMed] [Google Scholar]
- 40.Bambico F. R., Katz N., Debonnel G., Gobbi G., Cannabinoids elicit antidepressant-like behavior and activate serotonergic neurons through the medial prefrontal cortex. J. Neurosci. 27, 11700–11711 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shen C. J., Zheng D., Li K. X., Yang J. M., Pan H. Q., Yu X. D., Fu J. Y., Zhu Y., Sun Q. X., Tang M. Y., Zhang Y., Sun P., Xie Y., Duan S., Hu H., Li X. M., Cannabinoid CB1 receptors in the amygdalar cholecystokinin glutamatergic afferents to nucleus accumbens modulate depressive-like behavior. Nat. Med. 25, 337–349 (2019). [DOI] [PubMed] [Google Scholar]
- 42.Post A., Smart T. S., Krikke-Workel J., Dawson G. R., Harmer C. J., Browning M., Jackson K., Kakar R., Mohs R., Statnick M., Wafford K., McCarthy A., Barth V., Witkin J. M., A selective nociceptin receptor antagonist to treat depression: Evidence from preclinical and clinical studies. Neuropsychopharmacology 41, 1803–1812 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Okajima D., Kudo G., Yokota H., Antidepressant-like behavior in brain-specific angiogenesis inhibitor 2-deficient mice. J. Physiol. Sci. 61, 47–54 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Witkin J. M., Overshiner C., Li X., Catlow J. T., Wishart G. N., Schober D. A., Heinz B. A., Nikolayev A., Tolstikov V. V., Anderson W. H., Higgs R. E., Kuo M. S., Felder C. C., M1and M2Muscarinic receptor subtypes regulate antidepressant-like effects of the rapidly acting antidepressant scopolamine. J. Pharmacol. Exp. Ther. 351, 448–456 (2014). [DOI] [PubMed] [Google Scholar]
- 45.Wang X. B., Zhang Y. Q., Xue R. R., Yang Z. Z., Zhang X. F., Corticotropin-releasing hormone 1 receptor antagonism attenuates chronic unpredictable mild stress-induced depressive-like behaviors in rats. Neuroreport 31, 1–8 (2020). [DOI] [PubMed] [Google Scholar]
- 46.Pinho-Ribeiro F. A., Borghi S. M., Staurengo-Ferrari L., Filgueiras G. B., Estanislau C., Verri W. A. Jr., Bosentan, a mixed endothelin receptor antagonist, induces antidepressant-like activity in mice. Neurosci. Lett. 560, 57–61 (2014). [DOI] [PubMed] [Google Scholar]
- 47.Ghose S., Winter M. K., McCarson K. E., Tamminga C. A., Enna S. J., The GABAβ receptor as a target for antidepressant drug action. Br. J. Pharmacol. 162, 1–17 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Valverde O., Célérier E., Baranyi M., Vanderhaeghen P., Maldonado R., Sperlagh B., Vassart G., Ledent C., GPR3 receptor, a novel actor in the emotional-like responses. PLOS ONE 4, e4704 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kano M., Fukudo S., Tashiro A., Utsumi A., Tamura D., Itoh M., Iwata R., Tashiro M., Mochizuki H., Funaki Y., Kato M., Hongo M., Yanai K., Decreased histamine H1 receptor binding in the brain of depressed patients. Eur. J. Neurosci. 20, 803–810 (2004). [DOI] [PubMed] [Google Scholar]
- 50.Iida T., Yoshikawa T., Kárpáti A., Matsuzawa T., Kitano H., Mogi A., Harada R., Naganuma F., Nakamura T., Yanai K., JNJ10181457, a histamine H3 receptor inverse agonist, regulates in vivo microglial functions and improves depression-like behaviours in mice. Biochem. Biophys. Res. Commun. 488, 534–540 (2017). [DOI] [PubMed] [Google Scholar]
- 51.Yamada M., Tsukagoshi M., Hashimoto T., Oka J. I., Saitoh A., Yamada M., Lysophosphatidic acid induces anxiety-like behavior via its receptors in mice. J. Neural Transm. (Vienna) 122, 487–494 (2015). [DOI] [PubMed] [Google Scholar]
- 52.Kajitani N., Miyano K., Okada-Tsuchioka M., Abe H., Itagaki K., Hisaoka-Nakashima K., Morioka N., Uezono Y., Takebayashi M., Identification of lysophosphatidic acid receptor 1 in astroglial cells as a target for glial cell line-derived neurotrophic factor expression induced by antidepressants. J. Biol. Chem. 291, 27364–27370 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nagata-Kuroiwa R., Furutani N., Hara J., Hondo M., Ishii M., Abe T., Mieda M., Tsujino N., Motoike T., Yanagawa Y., Kuwaki T., Yamamoto M., Yanagisawa M., Sakurai T., Critical role of neuropeptides B/W receptor 1 signaling in social behavior and fear memory. PLOS ONE 6, e16972 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hillhouse T. M., Shankland Z., Matazel K. S., Keiser A. A., Prus A. J., The quetiapine active metabolite N-desalkylquetiapine and the neurotensin NTS1 receptor agonist PD149163 exhibit antidepressant-like effects on operant responding in male rats. Exp. Clin. Psychopharmacol. 22, 548–556 (2014). [DOI] [PubMed] [Google Scholar]
- 55.Gellen B., Zelena D., Usdin T. B., Dobolyi A., The parathyroid hormone 2 receptor participates in physiological and behavioral alterations of mother mice. Physiol. Behav. 181, 51–58 (2017). [DOI] [PubMed] [Google Scholar]
- 56.Faron-Górecka A., Kuśmider M., Solich J., Kolasa M., Pabian P., Gruca P., Romańska I., Żurawek D., Szlachta M., Papp M., Antkiewicz-Michaluk L., Dziedzicka-Wasylewska M., Regulation of somatostatin receptor 2 in the context of antidepressant treatment response in chronic mild stress in rat. Psychopharmacology 235, 2137–2149 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zeng H., Schimpf B. A., Rohde A. D., Pavlova M. N., Gragerov A., Bergmann J. E., Thyrotropin-releasing hormone receptor 1-deficient mice display increased depression and anxiety-like behavior. Mol. Endocrinol. 21, 2795–2804 (2007). [DOI] [PubMed] [Google Scholar]
- 58.Yang Y., Cui Y., Sang K., Dong Y., Ni Z., Ma S., Hu H., Ketamine blocks bursting in the lateral habenula to rapidly relieve depression. Nature 554, 317–322 (2018). [DOI] [PubMed] [Google Scholar]
- 59.Ohki-Hamazaki H., Neuromedin B. Prog. Neurobiol. 62, 297–312 (2000). [DOI] [PubMed] [Google Scholar]
- 60.Gurney A., Axelrod F., Bond C. J., Cain J., Chartier C., Donigan L., Fischer M., Chaudhari A., Ji M., Kapoun A. M., Lam A., Lazetic S., Ma S., Mitra S., Park I. K., Pickell K., Sato A., Satyal S., Stroud M., Tran H., Yen W. C., Lewicki J., Hoey T., Wnt pathway inhibition via the targeting of Frizzled receptors results in decreased growth and tumorigenicity of human tumors. Proc. Natl. Acad. Sci. U.S.A. 109, 11717–11722 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bai B., Wang X., Li Y., Chen P.-C., Yu K., Dey K. K., Yarbro J. M., Han X., Lutz B. M., Rao S., Jiao Y., Sifford J. M., Han J., Wang M., Tan H., Shaw T. I., Cho J.-H., Zhou S., Wang H., Niu M., Mancieri A., Messler K. A., Sun X., Wu Z., Pagala V., High A. A., Bi W., Zhang H., Chi H., Haroutunian V., Zhang B., Beach T. G., Yu G., Peng J., Deep multilayer brain proteomics identifies molecular networks in Alzheimer’s disease progression. Neuron 105, 975–991.e7 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Johnson E. C. B., Dammer E. B., Duong D. M., Ping L., Zhou M., Yin L., Higginbotham L. A., Guajardo A., White B., Troncoso J. C., Thambisetty M., Montine T. J., Lee E. B., Trojanowski J. Q., Beach T. G., Reiman E. M., Haroutunian V., Wang M., Schadt E., Zhang B., Dickson D. W., Ertekin-Taner N., Golde T. E., Petyuk V. A., de Jager P. L., Bennett D. A., Wingo T. S., Rangaraju S., Hajjar I., Shulman J. M., Lah J. J., Levey A. I., Seyfried N. T., Large-scale proteomic analysis of Alzheimer’s disease brain and cerebrospinal fluid reveals early changes in energy metabolism associated with microglia and astrocyte activation. Nat. Med. 26, 769–780 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gajjar S., Patel B. M., Neuromedin: An insight into its types, receptors and therapeutic opportunities. Pharmacol. Rep. 69, 438–447 (2017). [DOI] [PubMed] [Google Scholar]
- 64.Nusse R., Clevers H., Wnt/β-catenin signaling, disease, and emerging therapeutic modalities. Cell 169, 985–999 (2017). [DOI] [PubMed] [Google Scholar]
- 65.Luo J., Sun P., Siwko S., Liu M., Xiao J., The role of GPCRs in bone diseases and dysfunctions. Bone Res 7, 19 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cui Y., Yang Y., Ni Z., Dong Y., Cai G., Foncelle A., Ma S., Sang K., Tang S., Li Y., Shen Y., Berry H., Wu S., Hu H., Astroglial Kir4.1 in the lateral habenula drives neuronal bursts in depression. Nature 554, 323–327 (2018). [DOI] [PubMed] [Google Scholar]
- 67.Yang Y., Wang H., Hu J., Hu H., Lateral habenula in the pathophysiology of depression. Curr. Opin. Neurobiol. 48, 90–96 (2018). [DOI] [PubMed] [Google Scholar]
- 68.Virtanen P., Gommers R., Oliphant T. E., Haberland M., Reddy T., Cournapeau D., Burovski E., Peterson P., Weckesser W., Bright J., van der Walt S. J., Brett M., Wilson J., Millman K. J., Mayorov N., Nelson A. R. J., Jones E., Kern R., Larson E., Carey C. J., Polat İ., Feng Y., Moore E. W., Plas J. V., Laxalde D., Perktold J., Cimrman R., Henriksen I., Quintero E. A., Harris C. R., Archibald A. M., Ribeiro A. H., Pedregosa F., van Mulbregt P.; SciPy 1.0 Contributors , SciPy 1.0: Fundamental algorithms for scientific computing in Python. Nat. Methods 17, 261–272 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Szklarczyk D., Gable A. L., Lyon D., Junge A., Wyder S., Huerta-Cepas J., Simonovic M., Doncheva N. T., Morris J. H., Bork P., Jensen L. J., von Mering C., STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 47, D607–D613 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Shannon P., Markiel A., Ozier O., Baliga N. S., Wang J. T., Ramage D., Amin N., Schwikowski B., Ideker T., Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhong G., He J., Zhou R., Lorenzo D., Babcock H. P., Bennett V., Zhuang X., Developmental mechanism of the periodic membrane skeleton in axons. eLife 3, e04581 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Schweizer M. C., Henniger M. S. H., Sillaber I., Chronic mild stress (CMS) in mice: Of anhedonia, ‘anomalous anxiolysis’ and activity. PLOS ONE 4, e4326 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Cerniauskas I., Winterer J., de Jong J. W., Lukacsovich D., Yang H., Khan F., Peck J. R., Obayashi S. K., Lilascharoen V., Lim B. K., Földy C., Lammel S., Chronic stress induces activity, synaptic, and transcriptional remodeling of the lateral habenula associated with deficits in motivated behaviors. Neuron 104, 899–915.e8 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/30/eabf0634/DC1