

A tumor microenvironment chip reveals how platelets may fuel cancer metastasis and chemoresistance and unravels a new treatment.
Abstract
Platelets extravasate from the circulation into tumor microenvironment, enable metastasis, and confer resistance to chemotherapy in several cancers. Therefore, arresting tumor-platelet cross-talk with effective and atoxic antiplatelet agents in combination with anticancer drugs may serve as an effective cancer treatment strategy. To test this concept, we create an ovarian tumor microenvironment chip (OTME-Chip) that consists of a platelet-perfused tumor microenvironment and which recapitulates platelet extravasation and its consequences. By including gene-edited tumors and RNA sequencing, this organ-on-chip revealed that platelets and tumors interact through glycoprotein VI (GPVI) and tumor galectin-3 under shear. Last, as proof of principle of a clinical trial, we showed that a GPVI inhibitor, Revacept, impairs metastatic potential and improves chemotherapy. Since GPVI is an antithrombotic target that does not impair hemostasis, it represents a safe cancer therapeutic. We propose that OTME-Chip could be deployed to study other vascular and hematological targets in cancer.
INTRODUCTION
Platelets have long been recognized as anuclear blood constituents that regulate hemostasis and thrombosis. Platelets are also the first responders in the pathobiology of cancer: They play both structural and functional roles as reporters and transporters within the tumor-vascular organ (1, 2). To date, in vivo systems have been widely used in our understanding of cancer metastasis and its control through platelet function. For example, both mouse models (3–5) and human clinical studies of ovarian cancer (6) have shown that platelets extravasate into the solid tumor microenvironment and facilitate proliferation. A few animal studies have proposed the link between platelet extravasation and metastasis in ovarian cancer (5, 7). Some reports also indicate that platelet surface receptor GPVI interacts with circulating tumor cell surface glycoprotein galectin-3 and potentiates its growth (7–13). However, a more detailed understanding of pathophysiological outcomes of transendothelial platelet extravasation in cancer remains limited. Whether extravasated platelets can affect primary solid tumors through their GPVI remains questionable. In addition, because vascular shear regulates platelet GPVI activation (14), it remains to be tested whether shear can also regulate platelet interaction with cancer cells (15). Extravasated platelets are also known to release tumor-promoting growth factors and cytokines/interleukins (ILs) (5, 7) and induce chemoresistance in in vivo models (9, 16). Therefore, antiplatelet therapeutics in cancer offered along with chemotherapy can be helpful. However, while the in vivo models have provided foundational knowledge of platelet interactions with cancer cells, new preclinical models are still needed that can provide a reductionist approach to systematically investigating tumor-platelet-drug cross-talk.
Microphysiological models, also termed organ-on-chip platforms, are particularly well suited for mimicking longitudinal cancer events and preclinical drug discovery as they offer a bottoms-up (simple to incrementally complex) and dissectible approach in biological system design, define tumor microenvironment and three-dimensional (3D) architecture, and enable imaging and molecular analyses with high spatiotemporal resolution. Now, there is an unmet need for in vitro microphysiological models that can integrate the analysis of vascular and blood compartments to that of the tumor microenvironment. Studies with these models can be conducted using exclusively human-derived primary tumor and blood cells or their CRISPR gene-edited counterparts. In prior studies, we have demonstrated the utility of organ-on-chip for faithfully modeling vascular dysfunction and platelet hyperactivity (17–19). More recently, we demonstrated that ovarian cancer-on-chip (termed OvCa-Chip) reproduces platelet extravasation from the blood vessel into tumors (20). However, our prior design could not analyze the after platelet extravasation events that potentially can increase cancer cell proliferation, induce chemoresistance, and enhance metastasis. This was partly because OvCa-Chip lacked the technological capability of including a tumor microenvironment that can visualize and analyze invasion dynamics.
Addressing this critical deficiency here, we engineered an ovarian tumor microenvironment organ-on-chip platform (OTME-Chip), which, in addition to the tumors interfacing platelet-perfused vascular endothelial tissue, also incorporates an adjacent well-defined collagen hydrogel–based extracellular matrix (ECM) microenvironment. This novel integration of a highly organized hydrogel architecture adjacent to the tumor cell chamber enables precise visualization of cancer cell invasion dynamics in response to the biophysical and biological effects of platelets extravasated through the endothelium into the tumor microenvironment. Using CRISPR-Cas9 knockout (KO) cancer cells, our platform revealed that platelets might promote ovarian cancer metastasis and chemoresistance through a shear-dependent interaction of their GPVI with galectin-3 expressed on the cancer cells. Last, we explored a novel treatment opportunity to arrest ovarian cancer metastasis and increase the effects of chemotherapy via an antiplatelet drug that is relatively safe and currently undergoing clinical trials for other disease conditions. These results were further validated by performing next-generation sequencing and subsequent differential gene expression analysis of platelet-interacted cancer cells extracted from the OTME-Chip. Together, our work suggests that OTME-Chip, integrated with gene editing and next-generation RNA sequencing (RNA-seq) tools, is a platform to advance the discovery of novel antiplatelet therapeutics against tumor metastasis and chemoresistance.
RESULTS
Formation of OTME-Chip
We were inspired to design an organ-chip microfluidic platform that served as a 3D organomimetic in vitro model of the tumor microenvironment, interfaced with a blood vessel and flow of platelets (Fig. 1A). Here, our primary goal was to enable the investigation of cancer cell–platelet dynamics after platelets extravasate through the endothelium into the ovarian tumors. We kept a few elements of the existing platform of our recently published OvCa-Chip—a two-chamber organ-on-chip technology consisting of cancer cells cocultured with a 3D endothelial vessel, separated by a matrix-coated polydimethylsiloxane (PDMS) membrane (see Materials and Methods for details)—but to enable longitudinal studies of cancer progression and to analyze the effect of extravasated platelets on cancer cell proliferation and invasiveness, we completely reengineered the top tumor chamber of the device by adding two adjacent ECM chambers separated by an array of PDMS micropillars on the sides (Fig. 1B). This biosystem design strategy, now termed OTME-Chip, was inspired by a different organ-on-a-chip technology that modeled metastatic invasiveness of colorectal and breast cancer cells through artificially created ECM-mimicking materials (21, 22). This design does not permit blood perfusion or an intimate analysis of the interactions between the cancer cells, vascular cells, and blood constituents. However, in the OTME-Chip, the fusion of OvCa-Chip to this pillar structure design provides us the first opportunity to investigate the critical platelet–cancer cell interactions under flow, while still keeping the technology relatively simple and easy to adapt. First, we established the ECM compartments by injecting a pregel solution made of collagen type I and incubated the chip to form a semisolid hydrogel limited by micropillars. Next, we seeded the lower chamber of the device with human ovarian microvascular endothelial cells (HOMECs) to form an intact 3D vascular lumen. This was followed by the seeding of A2780 epithelial ovarian cancer cells in the central region of the top tumor chamber that formed the interface of a blood vessel and ovarian tumor tissue with the ECM. Further, we sustained this coculture with perfusion of freshly derived platelets from human blood inside the vascular lumen under microvascular shear for 3 days. Within 3 days, platelets increasingly extravasate into the ovarian cancer cells through the endothelium within the device (20).
Fig. 1. Microengineering of the OTME-Chip.
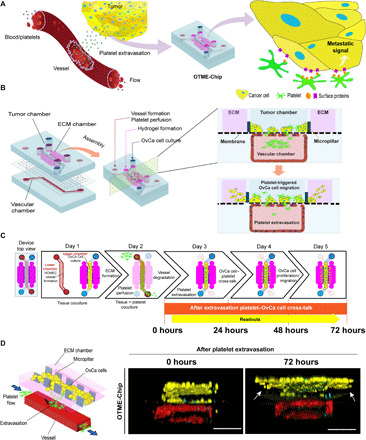
(A) A conceptual infographic of the human tumor microenvironment showing that the cancer cells interact with the neighboring blood vessels, making them permeable, and recruiting platelets into their vicinity. These extravasated platelets actively bind their ligands to the tumor surface receptors and result in tumor proliferation and chemoresistance, which can potentially be arrested by antiplatelet drugs. (B) Engineering drawing of the microdevice containing two PDMS compartments separated by a thin porous membrane that reproduces the microarchitecture of the tumor-vascular interface (left). On the right, cross-sectional side view of the OTME-Chip describes tissue organization inside the chip. (C) Illustration showing the timeline and steps of OTME-Chip formation, platelet extravasation into the tumors, and following consequences. Cancer cell dynamics and molecular readouts are analyzed every 24 hours after platelet extravasation. (D) Schematic diagram of OTME-Chip that shows tumor invasion dynamics can be systematically visualized and characterized after platelet extravasation from the bottom vascular chamber into the top tumor chamber. On the right, cross-sectional side view of 3D confocal scan of OTME-Chip showing cancer cells (yellow), endothelial cells (red), and platelets (cyan) at 0 hours (left) and 72 hours (right) after platelet extravasation. Scale bars, 100 μm.
After the platelets have extravasated into the tumor chamber, we now focused on new analyses on platelet interactions with cancer cells and consequences. We further progressed the coculture of the ovarian cancer cells with extravasated platelets together inside OTME-Chip for another 3 days with medium perfusion that is typical of ovarian interstitial flow (~0.5 to 1 dyne cm−2) (23). We then performed cellular and molecular analytics every 24 hours (Fig. 1C). After platelet extravasation from the vessel into the tumor compartment, we observed invasion of cancer cells through the pillars into the laterally created hydrogel ECM in the OTME-Chip (Fig. 1D). In contrast, no such invasion by cancer cells within ECM was detected when they were kept devoid of platelets inside the tumor chamber (Control-Chip). Therefore, these data support the ability of our platform to model platelet-tumor effects within the tumor microenvironment.
Platelets interact with cancer cells through binding of GPVI to galectin-3 under shear
In several cancers, including ovarian cancer, the expression of galectins is up-regulated and induces platelet adhesion and hyperactivity (12, 24, 25). Adhered and activated platelets, in turn, prime cancer cells for metastasis. Tumor galectin-3 specifically contains a collagen-like domain. On the other hand, the collagen receptor on platelets, GPVI, regulates platelet activation under shear (26). Therefore, we examined whether OTME-Chip may reveal the contribution of GPVI–galectin-3 binding to the platelet–cancer cell interaction and its metastatic consequences under shear (Fig. 2A). We first examined the expression and activity of galectin-3 over the course of our 72-hour timeline of exposure of cancer cells to extravasated platelets and found that galectin-3 expression was conserved and did not change over time (Fig. 2B).
Fig. 2. Platelet GPVI expression is shear dependent and it binds to tumor galectin-3 in OTME-Chip.
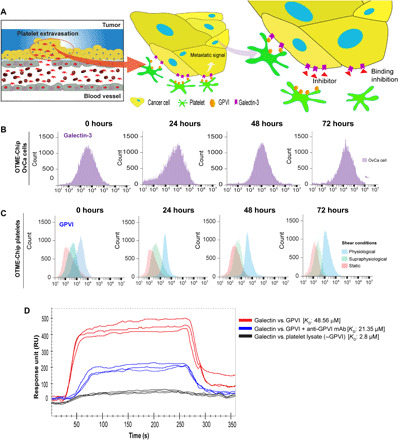
(A) Infographic describing platelet extravasation into the tumors inside the chip and platelet GPVI interaction with ovarian tumors through binding to galectin-3, which results in a prometastatic and chemoresistive tumor microenvironment. Therefore, GPVI inhibitors may be a therapeutic target to arrest ovarian cancer metastasis. Representative flow cytometry tracings at variable time points from cells recovered from the OTME-Chip, corresponding to (B) galectin-3 expression on the surface of ovarian cancer cells and (C) GPVI expression on the surface of extravasated platelets. (D) Surface plasmon resonance tracings (n = 3 independent experiments) show strong binding affinity between GPVI and galectin-3 proteins isolated from the platelets and cancer cells (red) against platelets exposed to anti-GPVI monoclonal antibody (mAb) (blue). GPVI-free platelet lysate shows no binding with galectin-3 (black). Binding affinity (KD) values are indicated in brackets.
Next, we analyzed the expression of platelet GPVI within OTME-Chip. Since platelet GPVI is known to be shear sensitive (27), we decided to compare platelet GPVI expression under variable shear. We found that when we perfused the platelets at a typical physiological shear (~1 dyne cm−2), GPVI expression was high, but, in contrast, both static condition and perfusion at a supraphysiological shear (~5 dyne cm−2) resulted in a diminished GPVI expression (Fig. 2C). Our data suggest that at low physiological shear, GPVI–galectin-3 interactions may be potent because of the high availability of GPVI.
Thereafter, to evaluate platelet GPVI binding with tumor galectin-3, immediately after exposing the platelets to shear within the OTME-Chip and extravasation, we isolated and purified the GPVI protein from the platelets and galectin-3 from the ovarian cancer cells. A rapid increase in the binding signal was detected when platelet-derived GPVI protein was perfused on a surface plasmon resonance chip coated with galectin-3 protein isolated from the tumors (Fig. 2D). In contrast, the binding was absent when a GPVI-free platelet lysate (immune pulled down) was used and substantially diminished when an inhibitory anti-GPVI monoclonal antibody was perfused along with GPVI. In summary, our analyses suggest that platelet GPVI binds to tumor galectin-3.
Consequences of GPVI and galectin-3 interaction in ovarian cancer assessed with OTME-Chip
We then investigated the possibility that the GPVI–galectin-3 interaction may serve as a mediator for platelet-promoted tumor metastasis in ovarian cancer. To test this hypothesis, we established an ovarian cancer cell line with galectin-3 knocked out (KO-g3) using the CRISPR-Cas9 editing method (Fig. 3A). We compared the parameters that govern cancer cell metastasis (ECM invasion, proliferation, cytokine profile, and transcriptional readouts) in OTME-Chips that were either composed of original ovarian cancer cells (OTME-Chip) or galectin-3 knocked down cancer cells (KO–OTME-Chip). We found rapid hydrogel ECM invasion by the wild-type ovarian cancer cells when cocultured with extravasated platelets (OTME-Chip) relative to controls devoid of platelets (Control-Chip). However, we observed diminished ECM invasion in KO–OTME-Chips in the presence of extravasated platelets (Fig. 3, B and C). Cancer cell proliferation increased in OTME-Chips relative to controls during 40 to 72 hours of extravasated platelets and tumor interaction, and proliferation was found diminished in galectin-3 KO–OTME-Chips (Fig. 3D). We observed this OvCa cell proliferation to be proportional to their ECM hydrogel invasion within our observation timeline.
Fig. 3. Platelet GPVI promotes ovarian metastasis through galectin-3 binding to cancer cells in OTME-Chip.
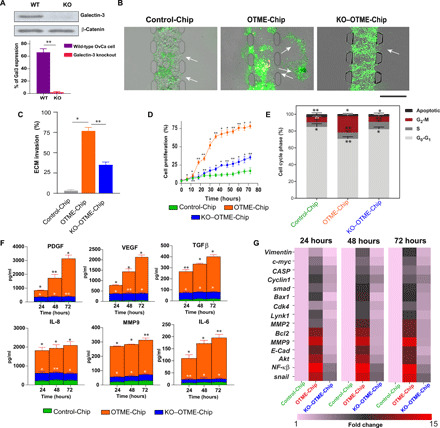
(A) Western blot and corresponding densitometry analyses shows highly reduced galectin-3 (Gal3) protein in the KO cancer cells against wild-type (WT) controls. (B) Representative fluorescence microscopy images showing cancer cell (green) invasion (marked by arrows) into hydrogel ECM due to extravasated platelets (yellow). Scale bar, 200 μm. (C) Bar graph showing the quantification of ECM invasion in Control-Chip, OTME-Chip, and KO–OTME-Chip at 48 hours. Analysis of (D) cancer cell proliferation, (E) flow cytometry–based cell cycle phases, (F) excreted growth factors (top) and cytokines (bottom) over time, and (G) real-time polymerase chain reaction (RT-PCR) heatmaps showing expressional alteration of cell proliferation and metastasis regulatory genes in Control-Chip, OTME-Chip, and KO–OTME-Chip. *P < 0.05 and **P < 0.01; n = 3 individual experiments; error bars are means ± SEM. One-way analysis of variance (ANOVA) is done followed by Dunnett’s multiple comparisons test.
G2-M extension and reduced apoptosis are a hallmark of metastasis of several carcinomas (28, 29). Cell cycle analysis of these OTME-Chips showed rapid alterations in cell cycle phases. A prominent G2-M phase extension and reduced apoptosis were observed in wild-type ovarian cancer cells due to platelets (OTME-Chip) as compared to their platelet-less counterpart (Control-Chip), and this extended G2-M was found to be reduced in KO-g3 cancer cells even in the presence of platelets (KO–OTME-Chip; Fig. 3E). We examined the effluents obtained from the tumor chambers of OTME-Chips and found a time-dependent increase in the concentration of growth factors platelet-derived growth factor (PDGF), vascular endothelial growth factor (VEGF), and transforming growth factor–β (TGFβ) and cytokines IL-8, matrix metalloproteinase 9 (MMP9), and IL-6 in effluents due to extravasated platelets that were significantly reduced in KO–TME-Chips (Fig. 3F).
We also hypothesized that GPVI–galectin-3 interaction eventually leads to the activation of the downstream nuclear factor κβ (NF-κβ) pathway in cancer cells. This NF-κβ signaling acts in combination with TGFβ/SMAD pathway to trigger tumor metastasis and rapid proliferation. Gene expression analysis of TME-Chips revealed a rapid up-regulation in the expression of proliferation and metastasis genes in the wild-type ovarian cancer cells due to extravasated platelets compared to KO-g3 cancer cells and platelet-less controls (Fig. 3G). A time-dependent rapid increase in the expression of BCL2, MMP9, E-CADHERIN, AKT, NF-kβ, and SNAIL and a moderate increase in VIMENTIN, C-MYC, CASP, CYCLIN1, SMAD, BAX, CDK4, LYNK1, and MMP2 similar to hallmark cell proliferation and metastasis regulator genes are observed in cancer cells over the coculture time due to platelets. These data strengthen the characterization of GPVI–galectin-3 interaction as an initiating factor behind the transcriptional signaling that is necessary for the proliferation and metastasis of ovarian cancer.
OTME-Chip predicts that pharmaceutical inhibition of GPVI arrests metastasis and supports chemotherapy
We followed up with investigating the potential consequences of pharmaceutical targeting of GPVI on platelets in cancer. There are currently no US Food and Drug Administration (FDA)–approved anti-GPVI drugs in the market. However, Revacept is an inhibitor that has been successfully evaluated against collagen-mediated platelet adhesion in phase 1 clinical trials of atherosclerosis and stroke (30). Revacept inhibited the platelet GPVI and tumor galectin-3 interaction in a colon carcinoma cell line (12). This drug is a fusion protein consisting of the Fc part of human immunoglobulin G (IgG), a short hinge region, a specific linker sequence, and the extracellular part of human platelet GPVI with a molecular mass of 150 kDa. We hypothesized that the specific inhibition of platelet GPVI with antiplatelet drug Revacept would arrest the tumor’s metastatic potential within our in vitro OTME-Chip. Subsequent to platelet perfusion in OTME-Chip and extravasation of platelets into the ovarian cancer cells, we exposed the tumors to Revacept at its clinically relevant concentration (40 μg/ml) (12). Thereafter, these chips were analyzed for 3 days, as described in our Materials and Methods (fig. S1). We found that Revacept-treated OTME-Chips (Rx–OTME-Chips) exhibited significantly reduced GPVI–galectin-3 binding kinetics (Fig. 4A). Revacept significantly reduced ECM invasion and proliferation rate of the cancer cells, suggesting that GPVI inhibition was also preventing the consequence of GPVI–galectin-3 interaction (Fig. 4, B to D). To confirm that our results are not dependent on A2780 cancer cells alone, we also tested a moderately invasive ovarian cancer cell line OVCAR3 (31, 32) and again observed an increased cancer cell invasion due to platelets but inhibited by Revacept (fig. S2).
Fig. 4. Revacept (GPVI inhibitor) arrests ovarian metastasis in OTME-Chip.
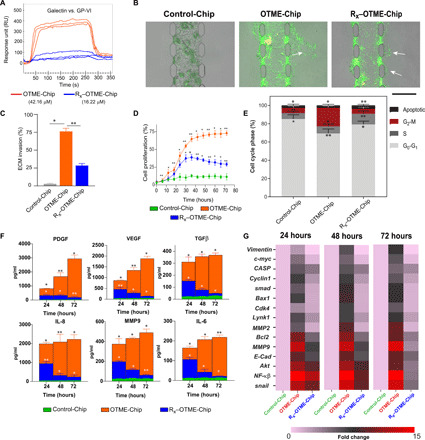
(A) Surface plasmon resonance tracings (n = 3 independent experiments) show strong binding affinity between GPVI and galectin-3 proteins isolated from the platelets and cancer cells (red) against platelets exposed to Revacept (blue). Binding affinity (KD) values indicated in brackets. (B) Representative fluorescence microscopy images showing cancer cell (green) invasion into hydrogel ECM due to extravasated platelets (yellow). Scale bar, 200 μm. (C) Bar graph showing the quantification of ECM invasion in Control-Chip, OTME-Chip, and Rx–OTME-Chip. Analysis of (D) cancer cell proliferation, (E) flow cytometry–based cell cycle phases, (F) excreted growth factors (top) and cytokines (bottom) over time, and (G) RT-PCR heatmaps showing expressional alteration of cell proliferation and metastasis regulatory genes in Control-Chip, OTME-Chip, and Rx–OTME-Chip. *P < 0.05 and **P < 0.01; n = 3 individual experiments; error bars are means ± SEM. One-way ANOVA is done followed by Dunnett’s multiple comparisons test.
Cell cycle analysis revealed a reduced G2-M phase in cancer cells from Rx–OTME-Chips compared to untreated controls (Fig. 4E). Further, Revacept reduced the growth factors (PDGF, VEGF, and TGFβ) and cytokines (IL-8, MMP9, and IL-6) (Fig. 4F). The transcriptional analysis of cancer cells obtained from the chips showed that the platelet-induced up-regulation of metastasis and cell proliferation genes declined rapidly over time in Rx–OTME-Chips (Fig. 4G).
Furthermore, since platelets have been shown to promote tumor chemoresistance (33, 34), we examined the effect of GPVI–galectin-3 interaction and blocking this interaction by Revacept on chemoresistance in ovarian cancer cells. We treated our OTME-Chips with either no drug (OTME-Chip) or anticancer drug cisplatin alone near its clinically relevant concentration (6 μg/ml) (Cx–OTME-Chip) (35) or cisplatin compounded with Revacept (CxRx–OTME-Chip). We found that cisplatin alone had a modest effect, whereas the cisplatin-Revacept combination had a significant effect in reducing tumor invasiveness (Fig. 5A), cell proliferation (Fig. 5B), G2-M phase (Fig. 5C), and concentration of growth factors and cytokines (Fig. 5D). Last, transcriptional analysis of cancer cells obtained from the chips showed that the platelet triggered up-regulation of metastasis and cell proliferation genes altered overtime modestly when cisplatin alone is included within OTME-Chips (Cx–OTME-Chip); however, Revacept significantly attenuated this overexpression (CxRx–OTME-Chip; Fig. 5E). Together, our OTME-Chip’s capability to assess anticancer drugs systematically provides us these preclinical datasets, which suggest that GPVI inhibition could be a potential strategy against ovarian cancer metastasis and chemoresistance.
Fig. 5. Revacept (GPVI inhibitor) decreases chemoresistance in OTME-Chip.
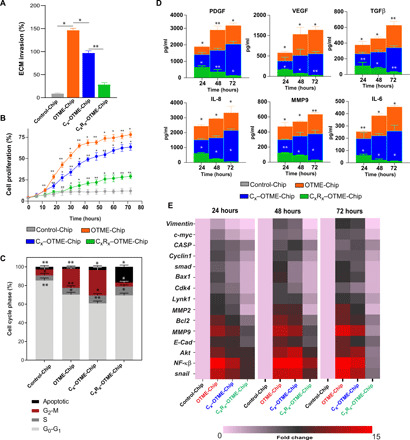
Analysis of (A) ECM invasion, (B) cancer cell proliferation, (C) flow cytometry–based cell cycle phases, (D) excreted growth factors (top) and cytokines (bottom) over time, and (E) RT-PCR heatmaps showing expressional alteration of cell proliferation and metastasis regulatory genes in Control-Chip, OTME-Chip, Cx–OTME-Chip, and CxRx–OTME-Chip. *P < 0.05 and **P < 0.01; n = 3 individual experiments; error bars are means ± SEM. One-way ANOVA is done followed by Dunnett’s multiple comparisons test.
Validation of cancer cell–platelet-drug cross-talk within OTME-Chip with next-generation RNA-seq
We have recently shown that a collaboration of organ-chip technology with RNA-seq analysis is a powerful tool to validate preclinical studies and derive unbiased clinical predictions (36). Therefore, to solidify our insights into the platelet-tumor signaling nexus and the therapeutic effect of Revacept, we performed transcriptomic profiling of different chip-derived tumors through RNA-seq (Fig. 6A). Through RNA-seq and differential gene analysis, we detected expression of 12,452 genes in the OTME-Chip relative to Control-Chip. Similarly, we detected expression of 5566, 6909, and 3821 genes in the Cx–OTME-Chip, CxRx–OTME-Chip, and KO–OTME-Chip, respectively, relative to Control-Chip (Fig. 6B). The reduction in differentially expressed genes under the different drug-treated and KO conditions suggests that the drug treatments were attenuating the transcriptomic expression and transformed the physiological behavior of the chips close to untreated controls. Among the different chip conditions, there were 15 groups of genes that were either unique to each condition or were common between chip conditions for the different permutations (Fig. 6B). Comparing genes common to two chip conditions at a time, we observed that the OTME-Chip and CxRx–OTME-Chip conditions had the highest number of common genes (~5500; Fig. 6C). In addition to the NF-κB and TGFβ/SMAD pathways that have been elucidated before (5, 7, 8) and in this work (Fig. 3), the RNA-seq analysis allowed us to investigate other signaling pathways that were up-regulated under the different chip conditions via Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway clustering. When compared to the platelet-free tumors (Control-Chip), we observed that in addition to the already known NF-κB and TGFβ/SMAD pathways, epithelial-mesenchymal transition (EMT) and metastasis regulatory signaling pathways such as Hippo (KEGG:04390), MAPK (mitogen-activated protein kinase) (KEGG:04010), mTOR (mechanistic target of rapamycin) (KEGG:04150), Notch (KEGG:04330), phosphatidylinositol 3 kinase-protein kinase B (PI3-Akt) (KEGG:04151), and wingless-related integrated site (Wnt) (KEGG:04310) pathways were up-regulated (Fig. 6D). This result suggests that targeting these other pathways could further open new avenues toward the development of anticancer therapeutics. To further visualize the efficacy of different treatments, we generated volcano plots and highlighted genes belonging to the aforementioned signaling pathways (Fig. 6E). As expected, both the chemotherapy drug cisplatin and the antiplatelet Revacept reduced the expression of these genes. This down-regulation in expression was also confirmed by generating heatmaps, showing holistic differences in expression profiles of the different chip conditions (Fig. 6F). OTME-Chip had a complementary expression profile compared to the Control-Chip (Fig. 6F). Consequently, we observed that cisplatin therapy down-regulated the expression of these pathways and that a combination therapy of cisplatin and Revacept further reduced their expression. This down-regulation in the CxRx–OTME-Chip was close to the KO-g3 cancer cells available from KO–OTME-Chip, where the GPVI–galectin-3 interaction was disturbed via KO. This result unbiasedly validates that GPVI–galectin-3 interaction is pivotal in platelet-mediated tumor promotion and establishes the therapeutic efficacy of antiplatelet drug Revacept against the progression of cancer with this preclinical platform.
Fig. 6. RNA-seq and differential gene expression analysis reveals the efficacy of antiplatelet therapy to attenuate tumor-promoting pathways.
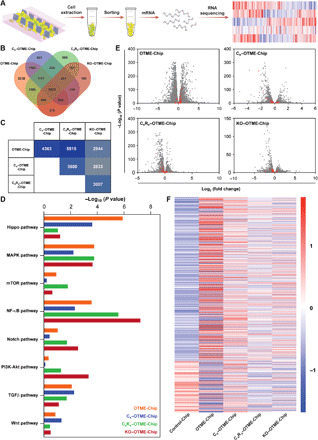
(A) Schematic of cancer cell isolation, cell sorting, and mRNA isolation process used in the study. Samples were then assessed for quality and processed for RNA sequencing. (B) Figure shows Venn diagram of differentially expressed genes among all groups. RNA sequencing and differential gene expression analysis revealed enrichment of 12,452, 5566, 6909, and 3821 genes in the OTME-Chip, Cx–OTME-Chip, CxRx–OTME-Chip, and KO–OTME-Chip, respectively, with respect to Control-Chip. (C) Group-wise comparison of common genes for all combinations. OTME-Chip and CxRx–OTME-Chip had the highest number common genes (~5500 genes). (D) KEGG pathway clustering revealed differential presence of EMT regulatory pathways among groups relative to Control-Chip (P < 0.05). (E) Volcano plots showing differentially expressed genes for all groups relative to Control-Chip (black dots). Red dots signify genes regulating EMT and metastasis. (F) Heatmap showing row-scaled z scores of ~900 genes belonging to the pathways as depicted in (E). For each group, n = 3 number of samples were sequenced.
DISCUSSION
Although several other groups have pioneered the creation of bioengineered models of tumors alone, tumor-vessel constructs, and tumor-ECM constructs, the primary focus of these studies did not include examining platelet-tumor interactions and consequences (22, 37–41). Notably, platelets are one of the first blood cells to interact with cancer cells, and it is increasingly getting appreciated clinically that thrombocytosis is associated with a poor prognosis in several cancers (42, 43). Specifically, in ovarian cancer, animal models convincingly support that platelets extravasate into the OTME and increase proliferation and EMT in ovarian cancer cells (4, 5, 33). In the current study, we built upon our prior expertise in leveraging organ-chip technology to integrate blood cell function into modeling processes in cancer. Here, we created a novel OTME-Chip that demonstrates a biomimicry of the consequences of the platelet–cancer cell interaction in ovarian cancer that is hard to achieve via conventional in vivo or in vitro models. The addition of hydrogel compartments adjacent to the cancer cell–vessel interface within the chip permitted time-lapse visualization of perfused platelets within the vessel, their extravasation through the endothelium, and consequential cancer cell invasiveness that was not previously obtained through ectopic in vivo experiments. Furthermore, isolating cancer cells from the chip during different time points of platelet perfusion have allowed us to identify the platelet-mediated increase in tumor proliferation and specific alterations in cancer cell cycle phases. Although this study is limited to ovarian cancer, this platform can be expanded to other cancer models and include more complex formulations of matrices and interstitial microenvironment.
We demonstrated that GPVI is a critical mediator in the adhesion of extravasated platelets to cancer cells. The counter receptor for GPVI is galectin-3, which is expressed on cancer cells. We showed that platelet GPVI expression is shear dependent, and, therefore, its interaction with the tumors could be dependent on the hemodynamic state. Replacing wild-type cancer cells with KO-g3 cells in OTME-Chip identified the key regulatory role of GPVI–galectin-3 interaction in metastasis. Our in vitro results are supported by several clinical observations and animal studies. For example, overexpression of galectin-3 increased tumor burden in A2780 ovarian cancer xenografted mice. Increased expression of galectin-3 has also been detected in advanced stages in patients with ovarian cancer (44). Increased levels of soluble GPVI have also been reported in plasma samples (11). GPVI blockade inhibited lung, colon, and breast cancer metastasis in mouse models (11, 12, 45). Studies in colorectal and breast cancer have described an increase in growth factors and cytokines in the TME after platelet extravasation into the tumors, and this has been predicted due to GPVI–galectin-3 interaction (7, 10, 12). In ovarian cancer, a similar elevation of growth factors and cytokines in TME is observed due to extravasated platelets (5). In addition, the extravasated platelets have been identified to cause proliferation of tumors via TGFβ/SMAD pathway, but the regulation of GPVI–galectin-3 interaction on activation of NF-κβ signaling has not been explored. Analysis of effluents from tumor microenvironment collected from the chips during the hours of platelet-tumor interaction by transcriptomics and multiplexing methods detected alterations in gene expression and protein profile. This identified the up-regulation of a series of metastasis triggering growth factors and cytokines due to GPVI–galectin-3 interaction.
Antiplatelet therapy has been tested in several clinical trials of cancer in the past (46). For example, acetylsalicylic acid or aspirin has been suggested to reduce platelet aggregation on tumors and inhibit metastasis. In an in vitro study, aspirin and P2Y12 inhibition attenuated platelet-induced ovarian cancer cell invasion (47). However, these drugs have a major drawback that they can increase bleeding risk in patients. Therefore, antiplatelet therapeutics that may be antimetastatic but do not alter hemostasis is an important unmet need. Platelet GPVI is an excellent target since it is known to be antithrombotic but does not cause bleeding (48). With our OTME-Chip, we have provided preclinical evidence that Revacept inhibited platelet-cancer cell interactions and its consequences and, therefore, is a potential drug to be advanced into clinical trials of ovarian cancer. Last, we showed that the GPVI–galectin-3 interaction induces chemoresistance in cancer cells, and Revacept can inhibit this effect. The inhibitory effect of Revacept treatment on cancer cell invasion independently and in combination with cisplatin was also verified in detail through the RNA-seq analysis. Therefore, our study has a translational value in the context that it motivates Revacept to be tested in a clinical trial against ovarian and other cancers.
Although, in this study, we focused on cancer metastasis triggered by the extravasated platelets, it is possible that blood immune cells such as tumor-associated macrophages, myeloid-derived suppressor cells, and regulatory T cells can also support tumor metastasis by forming a cumulative signaling along with platelets that remodel the tumor microenvironment (49–51). These immune cells could be included in this OTME-Chip study to identify their influence on tumors and metastasis. We did not include pericytes while forming the vascular tissue inside the chip, and pericytes can potentially exert influence on platelet extravasation (52). We also did not perfuse whole blood in the vascular chamber because it is very difficult to maintain blood fluidity in vitro for a prolonged period. Last, unlike some other tumor models where cancer cells are fully embedded within the matrix, cancer cells in our model were seeded over the matrix to prevent nonspecific interactions between platelets and cancer cells and to enable robust visual and molecular analytics. This relative simplicity and a provision to do analytics is a strength of our OTME-Chip, which is also not radically different from its prior versions (20) and consistent with prior models (39). Overall, this preliminary device design is a suitable proof of concept that highlights out OTME-Chip as a powerful tool in the investigation of blood cell influence on cancer and metastasis. An interesting future opportunity this platform offers is to also include the interaction of platelets with circulating tumor cells that may possibly disseminate from the solid tumor into the vascular chamber of this chip.
In conclusion, we demonstrated that complex interaction between the tumors and blood platelets and the resulting metastatic process could be modeled in a bioengineered tumor microenvironment with vascular components on a chip. We uncovered that this platform could be used in the identification of complex tumor–blood cell interactions, metastatic signaling, and the evaluation of drugs with an integrated genomic and proteomic approach. In addition, the scope of application of desired anticancer or antiplatelet drugs individually or in combination also makes our device a potential platform to perform drug evaluation studies against platelet-triggered metastasis and chemoresistance of cancer. Opportunity to include ECM in the chip also added additional capability to investigate the tissue invasiveness and the invasion behavior of tumors due to platelets under different conditions. We expect that this OTME-Chip technology could further be advanced toward personalized cancer medicine by integrating induced pluripotent stem cells derived from malignant tumors and other patient-derived blood or immune cells, where we can dissect the interaction signaling underlying metastasis. In summary, our human OTME-Chip model may offer a potential platform for studying the interaction between blood cells and cancer cells, acquiring functional readouts, and preclinical data of different antimetastasis drugs before initiating large animal studies and human clinical trials.
MATERIALS AND METHODS
Cell lines
HOMECs (ScienCell Research Laboratories) were cultured in endothelial cell medium (ECM; catalog no. 1001), and ovarian carcinoma A2780 cells (Sigma-Aldrich, catalog no. 93112519) were cultured in RPMI 1640 medium. Each was supplemented with penicillin (100 U ml−1) and streptomycin (100 μg ml−1). Tumor KO-g3 A2780 cells were developed using CRISPR-Cas9 methods and selected on puromycin (1 mM). Single guide RNAs (gRNAs) targeting the exon 3 of galectin-3 were designed for the KO study. The designed guides were screened in silico for off-target activity using the CRISPR search with mismatches, insetions and/or deletions (COSMID) webtool (53), and the gRNA (CATGATGCGTTATCTGGGTC) with the least number of off targets was selected for the KO experiments. The guide was delivered as a ribonucleoprotein complex with Cas9 to the A2780 cell line using an optimized nucleofection protocol.
Human platelets
Platelets were isolated from fresh human donor blood samples following previously mentioned protocol (20) using acid citrate dextrose buffer and Hepes-Tyrode’s buffer to preserve their cytoskeletal activity. Platelets were tagged using phycoerythrin-conjugated CD41 monoclonal antibody (1:200 dilution; Thermo Fisher Scientific, CAS-VIPL3) before perfusion into the device (~200 × 103 platelets μl−1). Blood samples were obtained via phlebotomy according to the policies of the U.S. Office of Human Research Protections and approved by the Texas A&M University Institutional Review Board (ID: IRB2016-0762D).
Drugs
Cisplatin (Thermo Fisher Scientific, USA) and Revacept (advanceCOR GmbH, Germany) were dissolved in dimethyl sulfoxide and phosphate-buffered saline (PBS)/4% mannitol/1% sucrose, respectively, to prepare master stocks (1 mg/ml). These were further diluted in RPMI 1640 medium to obtain respective working concentrations of cisplatin (6 μg ml−1) and Revacept (40 μg ml−1). They were perfused into the chip tumor chamber individually or in combination in the presence or absence of extravasated platelets.
Design and fabrication of the microfluidic OTME-Chip
OTME-Chips were adapted from previously reported OvCa-Chips (20), where the top portion of the device was changed from a single microchannel (1 mm in length by 1 mm in wide by 100 μm in height) to three parallel microchannels. The three microchannels were separated by hexagonal micropillars instead of a continuous PDMS wall, allowing for the formation of hydrogel fluid barriers that supported cell invasion. Each micropillar had dimensions of 250 μm by 100 μm by 100 μm and was equally spaced, leaving 100 μm between each micropillar. Drawings of the device were made using SOLIDWORKS 2019, from which photomasks were printed. Master molds were made using traditional soft lithography procedures (54). PDMS slabs and porous membrane were casted and cured, assembled, and processed according to previously developed protocols (20).
Pregelled hydrogel solutions were prepared on ice by neutralizing rat tail collagen I (3.57 mg ml−1) (Corning) with 1 N of NaOH before diluting to 2.5 mg ml−1 with cell culture medium. Pregelled (5 μl) hydrogel solution were injected into each ECM channel, and the device was stored in an incubator at 37°C for 1 hour for gel solidification. After gelation, hydrogel channels were hydrated by injecting a mixture of rat tail collagen type I (100 μg ml−1; BD Biosciences) and fibronectin (30 μg ml−1; BD Biosciences) inside the tumor and vessel chambers of chip and incubated at 37°C for at least 2 hours. This step also prepared cellular compartments for cell seeding. Before loading cells into their respective chambers, a wash with one-time PBS was performed to remove unbound residual collagen. HOMECs and A2780 OvCa cells were harvested and centrifuged, their supernatant removed, and pellets resuspended in respective fresh culture medium before loading into devices. HOMEC cell suspension (5 × 105 cells ml−1) was injected inside the vessel chamber and incubated at 37°C by following previous protocols (20) to obtain a confluent microvessel of HOMECs. After the microvessel was formed, the collagen-coated upper tumor chamber of the chip was seeded with cancer cell suspension (5 × 105 cells ml−1) and cultured for 24 hours to obtain a confluent monolayer. Platelets were perfused inside the vascular chamber using a syringe pump. The shear stress reported is at the wall that was calculated from the Navier-Stokes’ equation applicable for simple rectangular channels
where τw is wall shear stress, η is dynamic viscosity of fluid, Q is the flow rate, and h and w are the microfluidic channel’s height and width, respectively. After platelet extravasation, the cancer cells inside the tumor chamber were cultured under a continuous flow of RPMI 1640 medium for another 3 days (~1 dyne cm−2). Tumor chamber medium effluent was collected from the devices at 24, 48, and 72 hours. Cancer cells and platelets were isolated from the tumor chamber using Accutase treatment, and the CD41-tagged platelets were subsequently pulled down of from the cancer cell–platelet mixture using MojoSort human anti-CD41 magnetic nanobeads (BioLegend, USA) on a magnetic separator provided by the manufacturer. The magnetically separated platelet and tumor fractions were washed and kept for further downstream assays.
Immunostaining and fluorescence-activated cell sorting
Platelets and cancer cells isolated from the device were washed by centrifugation [300 relative centrifugal force (RCF) for 5 min] and resuspended in one-time PBS, followed by blocking with 2% bovine serum albumin (BSA) for 10 min and fixing with 2% paraformaldehyde for 15 min at room temperature. Fixed cells were permeabilized using 0.1% Triton X-100 for 5 min before immunostaining. Platelets were immunostained for GPVI surface marker using antihuman GPVI (Sigma-Aldrich, catalog no. ABS446) primary antibody, followed by incubation with appropriate fluorescent secondary antibody as per experimental need. Fixed and permeabilized cancer cells were stained for galectin-3 protein using antihuman galectin-3 primary antibody (Thermo Fisher Scientific, catalog no. A3A12) and compatible fluorescent-tagged secondary antibody before flow cytometry experiments. All flow cytometric analyses were performed using a BD Accuri C6 flow cytometer (BD Biosciences, USA), and data were processed using CellQuest Pro software.
Cell invasion
Cancer cells were live stained with PKH67 cytotracker-green (Sigma-Aldrich, USA) before seeding into the device. After extravasation, the ovarian cancer cells were incubated at 37°C with continuous perfusion of medium in tumor chamber for 3 days. Invasion dynamics of live-stained cancer cells (green) into the chip ECM chamber hydrogel was visualized using fluorescence microscopy (ZEISS Axio Observer; LD Plan Neofluar 10×, numerical aperture 0.4). Snapshots were taken at every 12 hours of interval with an exposure time of 200 ms. Images were analyzed and processed using software ZEN 2.3 lite (ZEISS). Measurement of cancer cell invasion through the hydrogel was performed using cell counter plugins in ImageJ. The ECM invasion of cancer cells was calculated as the ratio of area occupied by invaded cancer cells in ECM chamber versus the total area of ECM chamber (55).
Cancer cell proliferation assay
Isolated cancer cells were immunostained with monoclonal anti-Ki67 antibody Alexa Fluor 488 conjugate (Abcam). The fluorescence intensity was then measured with a plate reader using excitation/emission wavelengths of 488/519 nm. Platelet-free cancer cells obtained from other chips during similar time points were kept as control. Proliferation rate was measured as a percentage ratio of fluorescent intensities of experimental and control cells.
Cell cycle analysis
Cancer cells isolated via Accutase treatment from the chip were fixed in 70% ethanol at 20°C for 6 hours and permeabilized. Cells were then incubated with propidium iodide (100 μg ml−1) (Sigma-Aldrich) and ribonuclease A (50 μg ml−1) in the dark for 30 min at 37°C. Flow cytometry was performed on a BD Accuri C6 flow cytometer (BD Biosciences, USA), and cell cycle analysis was performed using CellQuest Pro software.
Growth factors and cytokines
Growth factors and cytokines present in OTME-Chip tumor chamber medium effluents were measured using multiplex (xMAP) magnetic bead–based technology provided by Millipore. Medium effluents collected from tumor chamber during different time points were analyzed for cytokines and growth factors using a MILLIPLEX MAP human cytokine/chemokine magnetic bead panel kit (Millipore, CAS-HCYTOMAG-60K) by following previously established protocols. Assays were run in a precalibrated Luminex MAGPIX reader. The mean fluorescent intensity of the magnetic beads correlating with the concentration (picograms per milliliter) of respective cytokines and growth factors was calculated using xPONENT 4.2 software, and data analysis was done with MILLIPLEX Analyst 5.1 software (Millipore). The concentration of cytokines obtained in each sample was normalized with respect to controls, and the assays were run in triplicate. Cytokine data for each group were normalized to corresponding cell number, respectively.
Quantitative real-time polymerase chain reaction
Cancer cell total RNA was isolated using the Arcturus PicoPure RNA Isolation Kit (Thermo Fisher Scientific, CAS-KIT0214) specifically designed to recover high-quality RNA consistently from fewer cells. Cells isolated from the chip were washed in prescribed buffer (0.9 ml of one-time PBS/10% BSA and 0.1 ml of 0.5 M EDTA), and total cellular RNA was isolated using the manufacturer’s protocol. Isolated RNA was treated with deoxyribonuclease, and its quality was checked to be standard 260/280 of >1.5 with spectrophotometry before 0.5 μg of RNA was processed further for the preparation of cDNA using the ProtoScript First Strand cDNA Synthesis kit (New England Biolabs). The cDNA prepared was further used for the subsequent quantitative real-time polymerase chain reactions (qRT-PCRs) using SYBR Green master mix (Applied Biosystems SYBR Green Select PCR Master Mix) and gene-specific primers. PCRs for respective genes were performed in qRT-PCR system (Thermo Fisher Applied BioSystems Quantstudio 3) keeping glyceraldehyde-3-phosphate dehydrogenase as an internal control. Gene expression was analyzed by quantifying the relative fold changes in mRNA levels of individual genes using comparative cycle threshold (CT)/ΔCT method.
Western blot
Galectin-3 knockdown in cancer cells was verified by Western blot assay. Briefly, cell lysates were prepared from equal amount of wild-type and KO A2780 cells, and protein extraction was performed using spin columns. Equal amount of protein (20 μg) from harvested cells was loaded onto 10 to 15% (w/v) polyacrylamide gels and separated by SDS–polyacrylamide gel electrophoresis, followed by transfer to polyvinylidene difluoride membrane. After transfer, the membrane was incubated with anti–galectin-3 antibody (1:10,000; Thermo Fisher Scientific, CAS-A3A12) and anti–β-actin antibody (1:10,000 dilution; Thermo Fisher Scientific, CAS-PA1-183), followed by incubation with horseradish peroxidase–conjugated anti-rabbit IgG (1:5000 dilution; Abcam, CAS-ab6721) secondary antibody. The protein bands were then visualized with chemiluminescence (Bio-Rad, USA), and densitometry quantification was performed using ImageJ analysis software.
Surface plasmon resonance
Physical interaction between platelet surface GPVI and tumor galectin-3 was determined via level-free analysis using surface plasmon resonance method (Nicoya Life, USA). Galectin-3 protein was pulled down from cancer cell and platelet lysates using biotinylated anti–galectin-3 monoclonal antibody and anti-GPVI monoclonal antibody, respectively. The proteins were diluted in HBS-EP-EDTA running buffer [0.01 M Hepes, 0.15 M NaCl, and 3 mM EDTA (pH 7.4)]. Streptavidin-coated chips purchased from Nicoya Life were used to immobilize biotinylated galectin-3 by injecting the protein into the device for 5 min using a flow rate of 20 μl min−1, thus immobilizing biotinylated galectin-3 on the streptavidin-coated chip. Successful immobilization was confirmed by observation of an increase of ~50 response units (RU) in the sensorgram signal. Purified GPVI (100 μl) was injected at a flow rate of 20 μl min−1 over the galectin-3–immobilized chip, and the resonance unit change in the sensorgram signal was recorded. For inhibition studies, Revacept (40 μg ml−1) was added with purified GPVI and injected into the device. The data obtained were analyzed by curve fitting of the Langmuir binding isotherm with the software.
RNA-seq and analysis
RNA from the chips were isolated according to the method described earlier. Isolated mRNA samples were then analyzed using the NextSeq 500 platform (Illumina), and sample preparation was performed using TruSeq RNA sample preparation with a paired-end read length of 2 × 75 bases (Molecular Genomics Workspace, Texas A&M University, College Station, TX). Then, we used hierarchical indexing for spliced alignment of transcripts 2 (HISAT2) to splice align the reads to latest Ensembl release 102 human genome/transcriptome (GRCh38.p13). To generate raw counts from alignment files (SAM), we used the Bioconductor package Subread. Differentially expressed genes were then evaluated for all groups using DESeq2 package in R. The cutoff to determine significant genes in all groups were false discovery rate–adjusted P value (q value) of <0.05. Last, KEGG pathway analysis was performed using the online database Database for Annotation Visualization and Integrated Discovery (v6.8) and visualized using Cytoscape and ClueGO (56).
Statistical analysis
All results are presented as means ± SEM, unless otherwise noted. Data were analyzed using one-way analysis of variance (ANOVA), followed by Dunnett’s multiple comparisons test using GraphPad Prism (GraphPad Software, San Diego, CA, USA). P < 0.05 was considered as statistically significant.
Acknowledgments
We thank M. Ungerer at advanceCOR GmbH, Germany for providing the Revacept drug as a gift. We thank J. A. Culpepper at Texas A&M University for managing phlebotomy of healthy individuals, S. Vitha at the Texas A&M Microscopy and Imaging Center for assisting with the confocal imaging, and G. Wright at Texas A&M Flow Cytometry Facility for assisting in fluorescence-activated cell sorting data analysis. Funding: Research reported in this publication was supported by the NIBIB of NIH under award no. R21EB025945, NSF CAREER award no. 1944322, and Texas A&M University President’s Excellence Award (T3 and X-Grant) to A.J. Portions of this work were also supported, in part, by NIH (P30 CA016672, P50 CA217685, CA177909, and R35 CA209904), MD Anderson Cancer Center Ovarian Cancer Moon Shot Program, the Blanton-Davis Ovarian Cancer Research Program, American Cancer Society Research Professor Award, and the Frank McGraw Memorial Chair in Cancer Research to A.S. and the Cancer Prevention and Research Institute of Texas (RR140081) to G.B. J.J.T. wishes to acknowledge support from the National Science Foundation Graduate Research Fellowship (grant no. 1650114). Author contributions: B.S.: Methodology, data curation, investigation, and writing—original draft preparation. T.M.: RNA-seq, bioinformatics, differential gene expression analysis, and writing—original draft preparation. J.J.T. and A.S.: Device design. M.C.: Gene editing. G.K.L.: PCR data. A.K.G., V.A.-K., A.K.S., and G.B.: Supervision. A.J.: Conceptualization, writing—reviewing and editing, and supervision. Competing interests: A.K.S: Consulting (Merck and Kiyatec), shareholder (BioPath), and research funding (M-Trap). The authors declare that they have no other competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/30/eabg5283/DC1
REFERENCES AND NOTES
- 1.Nemkov T., Reisz J. A., Xia Y., Zimring J. C., D’Alessandro A., Red blood cells as an organ? How deep omics characterization of the most abundant cell in the human body highlights other systemic metabolic functions beyond oxygen transport. Expert Rev Proteomics 15, 855–864 (2018). [DOI] [PubMed] [Google Scholar]
- 2.Becker R. C., Sexton T., Smyth S. S., Translational implications of platelets as vascular first responders. Circ. Res. 122, 506–522 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Haemmerle M., Bottsford-Miller J., Pradeep S., Taylor M. L., Choi H. J., Hansen J. M., Dalton H. J., Stone R. L., Cho M. S., Nick A. M., Nagaraja A. S., Gutschner T., Gharpure K. M., Mangala L. S., Rupaimoole R., Han H. D., Zand B., Armaiz-Pena G. N., Wu S. Y., Pecot C. V., Burns A. R., Lopez-Berestein G., Afshar-Kharghan V., Sood A. K., FAK regulates platelet extravasation and tumor growth after antiangiogenic therapy withdrawal. J. Clin. Invest. 126, 1885–1896 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cho M. S., Bottsford-Miller J., Vasquez H. G., Stone R., Zand B., Kroll M. H., Sood A. K., Afshar-Kharghan V., Platelets increase the proliferation of ovarian cancer cells. Blood 120, 4869–4872 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hu Q., Hisamatsu T., Haemmerle M., Cho M. S., Pradeep S., Rupaimoole R., Rodriguez-Aguayo C., Lopez-Berestein G., Wong S. T. C., Sood A. K., Afshar-Kharghan V., Role of platelet-derived Tgfβ1 in the progression of ovarian cancer. Clin. Cancer Res. 23, 5611–5621 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stone R. L., Nick A. M., McNeish I. A., Balkwill F., Han H. D., Bottsford-Miller J., Rupaimoole R., Armaiz-Pena G. N., Pecot C. V., Coward J., Deavers M. T., Vasquez H. G., Urbauer D., Landen C. N., Hu W., Gershenson H., Matsuo K., Shahzad M. M. K., King E. R., Tekedereli I., Ozpolat B., Ahn E. H., Bond V. K., Wang R., Drew A. F., Gushiken F., Lamkin D., Collins K., DeGeest K., Lutgendorf S. K., Chiu W., Lopez-Berestein G., Afshar-Kharghan V., Sood A. K., Paraneoplastic thrombocytosis in ovarian cancer. N. Engl. J. Med. 366, 610–618 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Labelle M., Begum S., Hynes R. O., Direct signaling between platelets and cancer cells induces an epithelial-mesenchymal-like transition and promotes metastasis. Cancer Cell 20, 576–590 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Orellana R., Kato S., Erices R., Bravo M. L., Gonzalez P., Oliva B., Cubillos S., Valdivia A., Ibañez C., Brañes J., Barriga M. I., Bravo E., Alonso C., Bustamente E., Castellon E., Hidalgo P., Trigo C., Panes O., Pereira J., Mezzano D., Cuello M. A., Owen G. I., Platelets enhance tissue factor protein and metastasis initiating cell markers, and act as chemoattractants increasing the migration of ovarian cancer cells. BMC Cancer 15, 290 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huong P. T., Nguyen L. T., Nguyen X. B., Lee S. K., Bach D. H., The role of platelets in the tumor-microenvironment and the drug resistance of cancer cells. Cancers 11, 240 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Leblanc R., Peyruchaud O., Metastasis: New functional implications of platelets and megakaryocytes. Blood 128, 24–31 (2016). [DOI] [PubMed] [Google Scholar]
- 11.Mammadova-Bach E., Gil-Pulido J., Sarukhanyan E., Burkard P., Shityakov S., Schonhart C., Stegner D., Remer K., Nurden P., Nurden A. T., Dandekar T., Nehez L., Dank M., Braun A., Mezzano D., Abrams S. I., Nieswandt B., Platelet glycoprotein VI promotes metastasis through interaction with cancer cell-derived galectin-3. Blood 135, 1146–1160 (2020). [DOI] [PubMed] [Google Scholar]
- 12.Dovizio M., Maier T. J., Alberti S., di Francesco L., Marcantoni E., Münch G., John C. M., Suess B., Sgambato A., Steinhilber D., Patrignani P., Pharmacological inhibition of platelet-tumor cell cross-talk prevents platelet-induced overexpression of cyclooxygenase-2 in HT29 human colon carcinoma cells. Mol. Pharmacol. 84, 25–40 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dumic J., Lauc G., Flogel M., Expression of galectin-3 in cells exposed to stress-roles of jun and NF-kappaB. Cell. Physiol. Biochem. 10, 149–158 (2000). [DOI] [PubMed] [Google Scholar]
- 14.Al-Tamimi M., Tan C. W., Qiao J., Pennings G. J., Javadzadegan A., Yong A. S. C., Arthur J. F., Davis A. K., Jing J., Mu F.-T., Hamilton J. R., Jackson S. P., Ludwig A., Berndt M. C., Ward C. M., Kritharides L., Andrews R. K., Gardiner E. E., Pathologic shear triggers shedding of vascular receptors: A novel mechanism for down-regulation of platelet glycoprotein VI in stenosed coronary vessels. Blood 119, 4311–4320 (2012). [DOI] [PubMed] [Google Scholar]
- 15.Trikha M., Zhou Z., Timar J., Raso E., Kennel M., Emmell E., Nakada M. T., Multiple roles for platelet GPIIb/IIIa and αvβ3 integrins in tumor growth, angiogenesis, and metastasis. Cancer Res. 62, 2824–2833 (2002). [PubMed] [Google Scholar]
- 16.Miyashita T., Tajima H., Makino I., Nakagawara H., Kitagawa H., Fushida S., Harmon J. W., Ohta T., Metastasis-promoting role of extravasated platelet activation in tumor. J. Surg. Res. 193, 289–294 (2015). [DOI] [PubMed] [Google Scholar]
- 17.Jain A., Barrile R., van der Meer A., Mammoto A., Mammoto T., Ceunynck K., Aisiku O., Otieno M. A., Louden C. S., Hamilton G. A., Flaumenhaft R., Ingber D. E., Primary human lung alveolus-on-a-chip model of intravascular thrombosis for assessment of therapeutics. Clin. Pharmacol. Ther. 103, 332–340 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jain A., Graveline A., Waterhouse A., Vernet A., Flaumenhaft R., Ingber D. E., A shear gradient-activated microfluidic device for automated monitoring of whole blood haemostasis and platelet function. Nat. Commun. 7, 10176 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jain A., van der Meer A. D., Papa A. L., Barrile R., Lai A., Schlechter B. L., Otieno M. A., Louden C. S., Hamilton G. A., Michelson A. D., Frelinger A. L. III, Ingber D. E., Assessment of whole blood thrombosis in a microfluidic device lined by fixed human endothelium. Biomed. Microdevices 18, 73 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Saha B., Mathur T., Handley K. F., Hu W., Afshar-Kharghan V., Sood A. K., Jain A., OvCa-Chip microsystem recreates vascular endothelium–mediated platelet extravasation in ovarian cancer. Blood Adv. 4, 3329–3342 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Carvalho M. R., Barata D., Teixeira L. M., Giselbrecht S., Reis R. L., Oliveira J. M., Truckenmüller R., Habibovic P., Colorectal tumor-on-a-chip system: A 3D tool for precision onco-nanomedicine. Sci. Adv. 5, eaaw1317 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Truong D., Fiorelli R., Barrientos E. S., Melendez E. L., Sanai N., Mehta S., Nikkhah M., A three-dimensional (3D) organotypic microfluidic model for glioma stem cells - Vascular interactions. Biomaterials 198, 63–77 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Avraham-Chakim L., Elad D., Zaretsky U., Kloog Y., Jaffa A., Grisaru D., Fluid-flow induced wall shear stress and epithelial ovarian cancer peritoneal spreading. PLOS ONE 8, e60965 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen J. H., Ni R. Z., Xiao M. B., Guo J. G., Zhou J. W., Comparative proteomic analysis of differentially expressed proteins in human pancreatic cancer tissue. Hepatobiliary Pancreat. Dis. Int. 8, 193–200 (2009). [PubMed] [Google Scholar]
- 25.Labrie M., De Araujo L. O. F., Communal L., Mes-Masson A. M., St-Pierre Y., Tissue and plasma levels of galectins in patients with high grade serous ovarian carcinoma as new predictive biomarkers. Sci. Rep. 7, 13244 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen Z., Mondal N. K., Ding J., Gao J., Griffith B. P., Wu Z. J., Shear-induced platelet receptor shedding by non-physiological high shear stress with short exposure time: Glycoprotein Ibα and glycoprotein VI. Thromb. Res. 135, 692–698 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Best D., Senis Y. A., Jarvis G. E., Eagleton H. J., Roberts D. J., Saito T., Jung S. M., Moroi M., Harrison P., Green F. R., Watson S. P., GPVI levels in platelets: Relationship to platelet function at high shear. Blood 102, 2811–2818 (2003). [DOI] [PubMed] [Google Scholar]
- 28.Bernal-Mizrachi L., Lovly C. M., Ratner L., The role of NF-κB-1 and NF-κB-2-mediated resistance to apoptosis in lymphomas. Proc. Natl. Acad. Sci. U.S.A. 103, 9220–9225 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mistry P., Deacon K., Mistry S., Blank J., Patel R., NF-κB promotes survival during mitotic cell cycle arrest. J. Biol. Chem. 279, 1482–1490 (2004). [DOI] [PubMed] [Google Scholar]
- 30.Ungerer M., Rosport K., Bültmann A., Piechatzek R., Uhland K., Schlieper P., Gawaz M., Münch G., Novel antiplatelet drug revacept (Dimeric Glycoprotein VI-Fc) specifically and efficiently inhibited collagen-induced platelet aggregation without affecting general hemostasis in humans. Circulation 123, 1891–1899 (2011). [DOI] [PubMed] [Google Scholar]
- 31.Guo Y., Cui W., Pei Y., Xu D., Platelets promote invasion and induce epithelial to mesenchymal transition in ovarian cancer cells by TGF-β signaling pathway. Gynecol. Oncol. 153, 639–650 (2019). [DOI] [PubMed] [Google Scholar]
- 32.Jiao Z., Huang L., Sun J., Xie J., Wang T., Yin X., Zhang H., Chen J., Six-transmembrane epithelial antigen of the prostate 1 expression promotes ovarian cancer metastasis by aiding progression of epithelial-to-mesenchymal transition. Histochem. Cell Biol. 154, 215–230 (2020). [DOI] [PubMed] [Google Scholar]
- 33.Ishikawa S., Miyashita T., Inokuchi M., Hayashi H., Oyama K., Tajima H., Takamura H., Ninomiya I., Ahmed A. K., Harman J. W., Fushida S., Ohta T., Platelets surrounding primary tumor cells are related to chemoresistance. Oncol. Rep. 36, 787–794 (2016). [DOI] [PubMed] [Google Scholar]
- 34.Radziwon-Balicka A., Medina C., O’Driscoll L., Treumann A., Bazou D., Inkielewicz-Stepniak I., Radomski A., Jow H., Radomski M. W., Platelets increase survival of adenocarcinoma cells challenged with anticancer drugs: Mechanisms and implications for chemoresistance. Br. J. Pharmacol. 167, 787–804 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bao L., Jaramillo M. C., Zhang Z., Zheng Y., Yao M., Zhang D. D., Yi X., Induction of autophagy contributes to cisplatin resistance in human ovarian cancer cells. Mol. Med. Rep. 11, 91–98 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mathur T., Flanagan J. M., Jain A., Tripartite collaboration of blood-derived endothelial cells, next generation RNA sequencing and bioengineered vessel-chip may distinguish vasculopathy and thrombosis amongst sickle cell disease patients. Bioeng. Transl. Med. e10211 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Agarwal P., Wang H., Sun M., Xu J., Zhao S., Liu Z., Gooch K. J., Zhao Y., Lu X., He X., Microfluidics enabled bottom-up engineering of 3D vascularized tumor for drug discovery. ACS Nano 11, 6691–6702 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen M. B., Whisler J. A., Fröse J., Yu C., Shin Y., Kamm R. D., On-chip human microvasculature assay for visualization and quantification of tumor cell extravasation dynamics. Nat. Protoc. 12, 865–880 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zervantonakis I. K., Hughes-Alford S. K., Charest J. L., Condeelis J. S., Gertler F. B., Kamm R. D., Three-dimensional microfluidic model for tumor cell intravasation and endothelial barrier function. Proc. Natl. Acad. Sci. U.S.A. 109, 13515–13520 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Truong D. D., Kratz A., Park J. G., Barrientos E. S., Saini H., Nguyen T., Pockaj B., Mouneimne G., LaBaer J., Nikkhah M., A human organotypic microfluidic tumor model permits investigation of the interplay between patient-derived fibroblasts and breast cancer cells. Cancer Res. 79, 3139–3151 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Veldhuizen J., Cutts J., Brafman D. A., Migrino R. Q., Nikkhah M., Engineering anisotropic human stem cell-derived three-dimensional cardiac tissue on-a-chip. Biomaterials 256, 120195 (2020). [DOI] [PubMed] [Google Scholar]
- 42.Menter D. G., Tucker S. C., Kopetz S., Sood A. K., Crissman J. D., Honn K. V., Platelets and cancer: A casual or causal relationship: revisited. Cancer Metastasis Rev. 33, 231–269 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schlesinger M., Role of platelets and platelet receptors in cancer metastasis. J. Hematol. Oncol. 11, 125 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kang H. G., Kim D. H., Kim S. J., Cho Y., Jung J., Jang W., Chun K. H., Galectin-3 supports stemness in ovarian cancer stem cells by activation of the Notch1 intracellular domain. Oncotarget 7, 68229–68241 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Haemmerle M., Stone R. L., Menter D. G., Afshar-Kharghan V., Sood A. K., The platelet lifeline to cancer: Challenges and opportunities. Cancer Cell 33, 965–983 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wojtukiewicz M. Z., Hempel D., Sierko E., Tucker S. C., Honn K. V., Antiplatelet agents for cancer treatment: A real perspective or just an echo from the past? Cancer Metastasis Rev. 36, 305–329 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cooke N. M., Spillane C. D., Sheils O., O’Leary J., Kenny D., Aspirin and P2Y12 inhibition attenuate platelet-induced ovarian cancer cell invasion. BMC Cancer 15, 627 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dutting S., Bender M., Nieswandt B., Platelet GPVI: A target for antithrombotic therapy?! Trends Pharmacol. Sci. 33, 583–590 (2012). [DOI] [PubMed] [Google Scholar]
- 49.Chen M. B., Hajal C., Benjamin D. C., Yu C., Azizgolshani H., Hynes R. O., Kamm R. D., Inflamed neutrophils sequestered at entrapped tumor cells via chemotactic confinement promote tumor cell extravasation. Proc. Natl. Acad. Sci. U.S.A. 115, 7022–7027 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Coffelt S. B., Wellenstein M. D., de Visser K. E., Neutrophils in cancer: Neutral no more. Nat. Rev. Cancer 16, 431–446 (2016). [DOI] [PubMed] [Google Scholar]
- 51.Wculek S. K., Malanchi I., Neutrophils support lung colonization of metastasis-initiating breast cancer cells. Nature 528, 413–417 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.McDowell S. A. C., Quail D. F., Immunological regulation of vascular inflammation during cancer metastasis. Front. Immunol. 10, 1984 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cradick T. J., Qiu P., Lee C. M., Fine E. J., Bao G., COSMID: A web-based tool for identifying and validating CRISPR/Cas off-target sites. Mol. Ther. Nucleic Acids 3, e214 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Huh D., Kim H. J., Fraser J. P., Shea D. E., Khan M., Bahinski A., Hamilton G. A., Ingber D. E., Microfabrication of human organs-on-chips. Nat. Protoc. 8, 2135–2157 (2013). [DOI] [PubMed] [Google Scholar]
- 55.Anguiano M., Castilla C., Maška M., Ederra C., Peláez R., Morales X., Muñoz-Arrieta G., Mujika M., Kozubek M., Muñoz-Barrutia A., Rouzaut A., Arana S., Garcia-Aznar J. M., Ortiz-de-Solorzano C., Characterization of three-dimensional cancer cell migration in mixed collagen-Matrigel scaffolds using microfluidics and image analysis. PLOS ONE 12, e0171417 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Carrow J. K., Cross L. M., Reese R. W., Jaiswal M. K., Gregory C. A., Kaunas R., Singh I., Gaharwar A. K., Widespread changes in transcriptome profile of human mesenchymal stem cells induced by two-dimensional nanosilicates. Proc. Natl. Acad. Sci. U.S.A. 115, E3905–E3913 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/30/eabg5283/DC1