

Abstract
Innate lymphoid cells (ILCs) develop from common lymphoid progenitors (CLP), which further differentiate into the common ILC progenitor (CILP) that can give rise to both ILCs and NK cells. Murine ILC intermediates have recently been characterized, but the human counterparts and their developmental trajectories have not yet been identified, largely due to the lack of homologous surface receptors in both organisms. Here, we show that human CILPs (CD34+CD117+α4β7+Lin−) acquire CD48 and CD52, which define NK progenitors (NKPs) and innate lymphoid cell precursors (ILCPs). Two distinct NK cell subsets were generated in vitro from CD34+CD117+α4β7+Lin−CD48−CD52+ and CD34+CD117+α4β7+Lin−CD48+CD52+ NKPs, respectively. Independent of NKPs, ILCPs exist in the CD34+CD117+α4β7+Lin−CD48+CD52+ subset and give rise to ILC1s, ILC2s and NCR+ ILC3s, whereas CD34+CD117+α4β7+Lin−CD48+CD52− ILCPs give rise to a distinct subset of ILC3s that have lymphoid tissue inducer (LTi)-like properties. Additionally, CD48 expressing CD34+CD117+α4β7+Lin− precursors give rise to tissue-associated ILCs in vivo. We also observed that the interaction of 2B4 with CD48 induced differentiation of ILC2s, and together these findings show that expression of CD48 by human ILCPs modulates ILC differentiation.
Keywords: Innate lymphoid cells, development, NKPs, ILCPs, CD48, CD52, α4β7
One sentence summary:
Human common ILC progenitors acquire CD48 and CD52, defining innate lymphoid cell precursors and NK progenitors.
Introduction
Innate lymphoid cells (ILCs) have been classified into three major groups based on the expression of transcription factors that drive cell function (1). Group 1 ILCs (ILC1) express Tbet and produce IFN-γ upon activation (2, 3). They are functionally similar to the traditional NK cells but lack granule-based cytotoxicity (2, 3). Group 2 ILCs (ILC2) express GATA3 and produce IL-5 and IL-13 upon activation (4, 5). Group 3 ILCs (ILC3) express RORγt and following stimulation, express IL-22 (6, 7).
Each ILC group has multiple subpopulations, characterized by distinct surface receptor expression, gene profiles, and functionality. Expression of CD103, NKp44, and CD127 defines two ILC1 subsets that initiate IFN-γ responses against pathogens (2, 8). ILC2 subpopulations vary in their activation-induced cytokine production (IL-5/IL-13 or TGF-β/IL-10) (9–11), with the former being GATA3-dependent and having a role in maintaining homeostatic type 2 responses (4, 5), whereas the latter are ID3-dependent and regulate intestinal inflammation by attenuating other ILC populations (9). At least two human ILC3 subpopulations have been described, including the lymphoid tissue–inducer (LTi)-like cells and the natural cytotoxicity receptor (NCR)-expressing ILC3 cells (7, 12). Both ILC3 subsets are known to maintain intestinal tissue homeostasis, but evidence suggests that these two subsets also have distinct functions (12). These data highlight the previously underappreciated diversity within the ILC compartment and raise questions about if these differences are alternative states of activation and ILC plasticity, or conversely, whether they are different cell types that arise through distinct developmental trajectories (13–15).
In mice, ILCs develop from common lymphoid progenitors (CLP), which further differentiates into the common ILC progenitor (CILP). The CILPs, in turn, develop into either an NK progenitor (NKP) or the common ‘helper’ innate lymphoid cell progenitor (CHILP), the latter of which has the capacity to differentiate into all helper ILCs subtypes including ILC1, ILC2 and ILC3 (1, 16). Murine CLP (Lin−Thy-1−Sca1lowc-kitlowFlt3+IL-7Ra+), CILP (Lin−IL-7Ra+α4β7+CD25−CXCR6+) and CHILP (Id2+IL-7Ra+α4β7+CD25−PLZF+/−) are now well characterized (reviewed in (1, 17)). In contrast, the developmental intermediates of human ILCs are less clearly defined. This gap in knowledge is due in part from the study of murine ILC progenitors (ILCP) using transcription factor reporter mice and the use of receptors (Thy-1 and Sca1) not commonly used to characterize human hematopoietic stem cells (HSCs) and developmental intermediates (16). We demonstrated previously that functionally mature ILC3s could develop in vitro from CD34+ cells in the presence of instructive cytokines including IL-7, IL-15, SCF, and FLT3L (18). More recently, tonsillar-derived α4β7+CD34+ cells were shown to co-express c-kit (CD117) and RORγt and gave rise to ILC3 cells (19). Others have observed that CD117 expression on Lin− progenitors mark cells that potentially develop into all ILC subpopulations (18, 20). Using a similar approach, Renoux et al (21) identified a Lin−CD34+CD38+CD123−CD45RA+CD7+CD10+CD127− progenitor that specifically differentiates into the NK lineage and lacks ILC potential.
In addition to the commonly recognized surface antigens used to characterize human HSCs (CD34, CD38, CD117 and CD45), these cells and their downstream progenitors express signaling lymphocyte activation molecule (SLAM) family receptors including CD150, CD48, and 2B4 (CD244) (22). These receptors are located on chromosome 1 (23) and are present in a stage-specific manner, such that long-term repopulating HSCs are CD150+ but lack CD48 or CD244. In contrast, multipotent hematopoietic progenitors lack CD150 and CD48, but express CD244. Finally, more restricted progenitors express varying combinations of CD244 and CD48 but are CD150− (22). SLAM family member expression has been used to characterize HSCs and progenitors phenotypically, but their function in this context is largely unstudied. Herein, we use single cell RNA sequencing (scRNA-seq) and lineage differentiation assays to define the human CILP, NKP, and ILCP in vitro and in vivo. We provide evidence that NK and ILC progenitors can be better defined using CD48 and CD52 staining, and that 2B4 ligation by CD48 is required for the development of ILC2 lineage cells, at the cost of NK cell development.
Results
Human common ILC progenitors (CILPs) are contained in CD34+α4β7+CD117+ HSCs
We previously demonstrated that umbilical cord blood (UCB)-derived CD34+ cells cultured in the presence of IL-3, IL-7, IL-15, SCF, and FLT3L give rise to NK cells and ILC3s over 3–4 weeks (18). Based on this, we reasoned that CD34+ cells would transit through intermediates that could develop into all ILC subtypes. Using a panel of CD56, CD94, CD117, CD161, CD294, CD336 and a lineage cocktail (containing CD1a, CD3, CD4, CD5, CD11c, CD14, CD19, CD34, TCRαβ, TCRɣδ, FcεRI, CD123, CD303) we identified Lin− ILCs, including NK cells, ILC1, −2 and −3 (fig. S1A and S1B) in these cultures. Consistent with previous data (5, 11, 20, 24, 25), NK cells expressed CD56 and CD94, and ILC2s expressed CD117 and CD294 (fig. S1A and S1B). Based on prior studies (19, 20), we characterized the ILC3 compartment using CD56, CD117, CD161 and CD336 (fig. S1A and S1B). Finally, ILC1 cells were defined as lacking CD56, CD94, CD294 and CD336, but partially expressing CD117 and CD161 (fig. S1A and S1B). This surface expression pattern was validated using intracellular transcription factor staining, demonstrating Tbet in NK cells, GATA3 in ILC2s, and RORγt in ILC3 (fig. S1C). Further confirmation of cell identity was established by cytokine production, showing IFN-γ by NK cells and ILC1s, IL-13 by ILC2s, and IL-22 by ILC3s (fig. S1D).
We investigated whether CILPs were present in UCB CD34+ and integrin α4β7+ compartments (1, 26, 27). Nearly all UCB-derived CD34+ cells were CD117+, whereas only ~3% of these were α4β7+ (fig. S2A, S2B). However, after five days of culture in media containing FLT3L, LDL, SCF and TPO, which is a cytokine cocktail used to expand HSCs, a higher fraction of purified CD34+ cells acquired α4β7 (fig. S2B and S2C) (28). These 5 day-expanded CD34+ HSCs were FACS-sorted into CD34+α4β7+ and CD34+α4β7− subsets, followed by culture in cytokines that support ILC and NK development (fig. S2D). Compared with the CD34+α4β7− HSC subset, CD34+α4β7+ cells were more likely to differentiate into CD117+ ILC precursors (fig. S2D and S2E) (20, 21, 25). The CD34+α4β7+ progenitors were also more likely to develop into CD94+ NK cells as well as CD294+ ILC2s (fig. S2F and S2G). Similarly, cells differentiating from the CD34+α4β7+ population expressed RORγt, an ILC3-associated transcription factor (fig. S2H). Thus, the human CILP capable of developing into all ILC subtypes, is contained within the CD34+α4β7+ cell fraction.
Expression of signaling lymphocyte activation molecule (SLAM) family receptor CD48 on CILPs mark the ILCPs
We aimed to further characterize the ILCPs and identified a subpopulation of CD34+α4β7+Lin− cells that co-express CD48 (Figure 1A). When compared with CD34+α4β7+Lin−CD48− cells, the CD34+α4β7+Lin−CD48+ cells expressed higher mRNA transcripts associated with ILCs including GATA3, ID2, RORγt and Tbet (Figure 1B–1E). These two populations were FACS purified and differentiated with ILC- and NK cell-supporting cytokines. The CD34+α4β7+Lin−CD48+ cells gave rise to NK cells and ILCs, whereas CD34+α4β7+Lin−CD48− cells developed into NK cells, but not ILCs (Figure 1F–1H). Thus, this finding indicates that the CD34+α4β7+Lin−CD48+ progenitors contain ILCPs.
Fig. 1. Expression of CD48 by CD34+α4β7+ HSC progenitors.
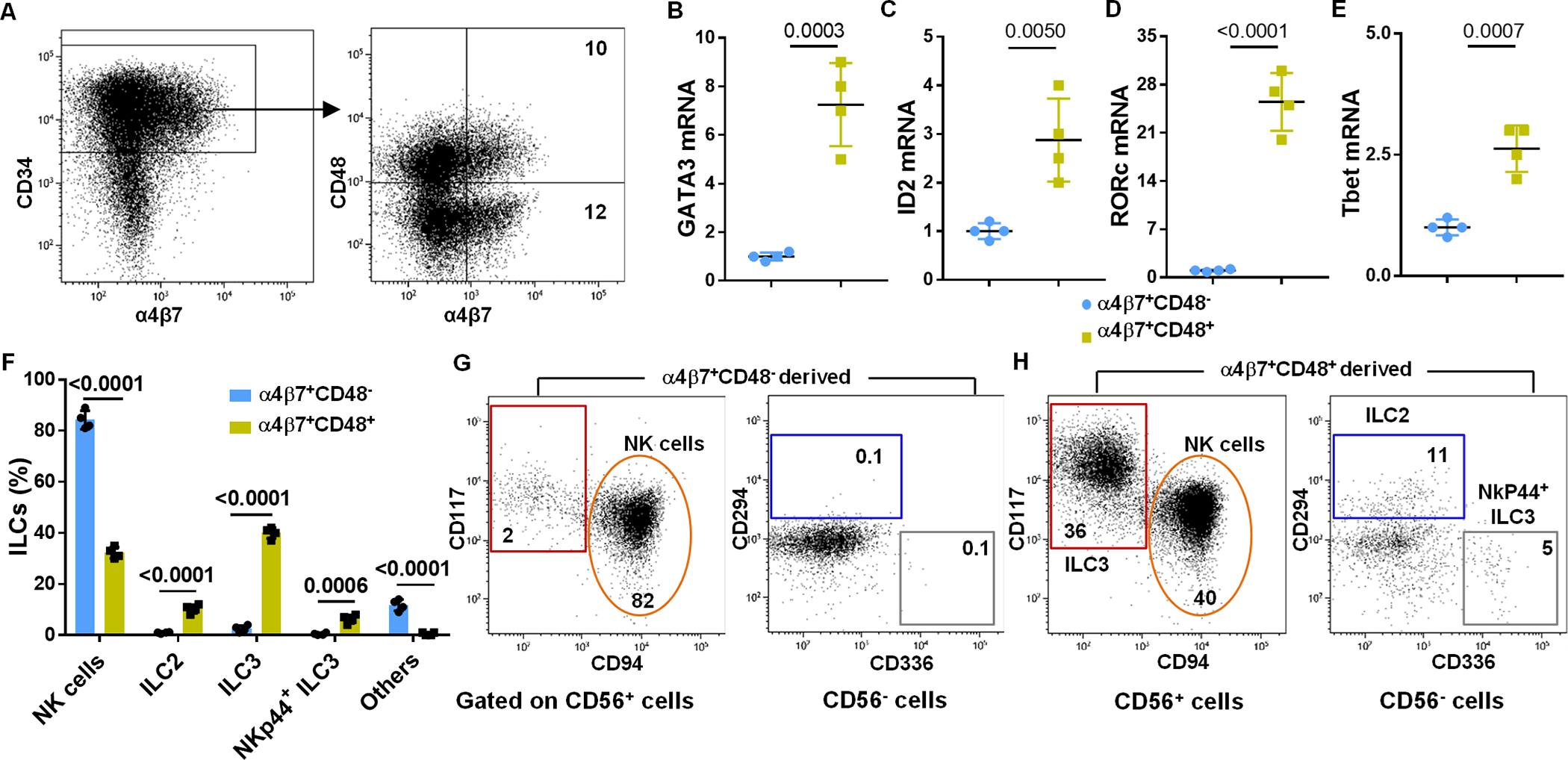
UCB-derived CD34+ cells were expanded for 5 days, FACS sorted into CD34+α4β7+CD48− and CD34+α4β7+CD48+ subsets and gene expression analysis were performed using qPCR. Cells were also cultured for twenty-one days under conditions that favor ILC differentiation. (A) Sorting gating strategy is shown (representative of n=4 experiments). (B-E) GATA3, ID2, RORγt and Tbet mRNA expression analysis by qPCR in the CD34+α4β7+CD48+ subset relative to the CD34+α4β7+CD48− subset (n=3 (B) or 4 (C, D and E) /group). (F) Differentiating cells were stained for ILC surface markers at day 21 of culture and the percentages of CD94+ NK cells, CD294+ ILC2, CD56−CD336+ ILC3 and CD56+CD117+ ILC3 derived from CD34+α4β7+CD48+ and CD34+α4β7+CD48− are shown (n=4/group). (G and H) Differentiating CD34+α4β7+CD48− and CD34+α4β7+CD48+ subsets were stained for ILC surface markers at day 21 of culture and CD94+ NK cells, CD294+ ILC2, CD56−CD336+ ILC3 and CD56+CD117+ ILC3 is shown, values represent the percentages of ILCs in the culture (representative of n=4 experiments). (B-F) Data are shown as mean ± SD, paired t-tests and p-values are depicted.
CD34+α4β7+Lin−CD48+ ILCPs give rise to tissue-associated ILCs in vivo
The development of CD34+α4β7+Lin−CD48− and CD34+α4β7+Lin−CD48+ cells was next tested in vivo using humanized NSG mice. Mice engrafted with CD34+α4β7+Lin−CD48− cells contained human CD45+ cells only in their bone marrow, whereas mice engrafted with CD34+α4β7+Lin−CD48+ cells showed human CD45+ cells in their bone marrow, spleen, liver and lung (Figure 2A). The reconstituting CD45+ cells from both progenitors lacked T cells, B cells and monocytes (fig. S2I). The bone marrow from both groups of mice contained NK cells, but lacked ILC2 and ILC3 cells (Figure 2B). The spleen, liver, and lung mainly showed human NK cells that were Tbet+ and IFN-γ+ (Figure 2B–2D). Mice that received CD34+α4β7+Lin−CD48+ cells also had Gata3- and IL-13-expressing ILC2s, as well as RORγt- and IL-22-expressing ILC3s within these tissues (Figure 2B–2D). Taken together, these results demonstrate that in vivo CD34+α4β7+Lin−CD48− cells give rise NK cells, but not ILCs, while CD34+α4β7+Lin−CD48+ cells give rise tissue-associated NK cells, ILC2s and ILC3s.
Fig. 2. CD34+α4β7+CD48+ precursors give rise to tissue-associated ILCs in vivo.
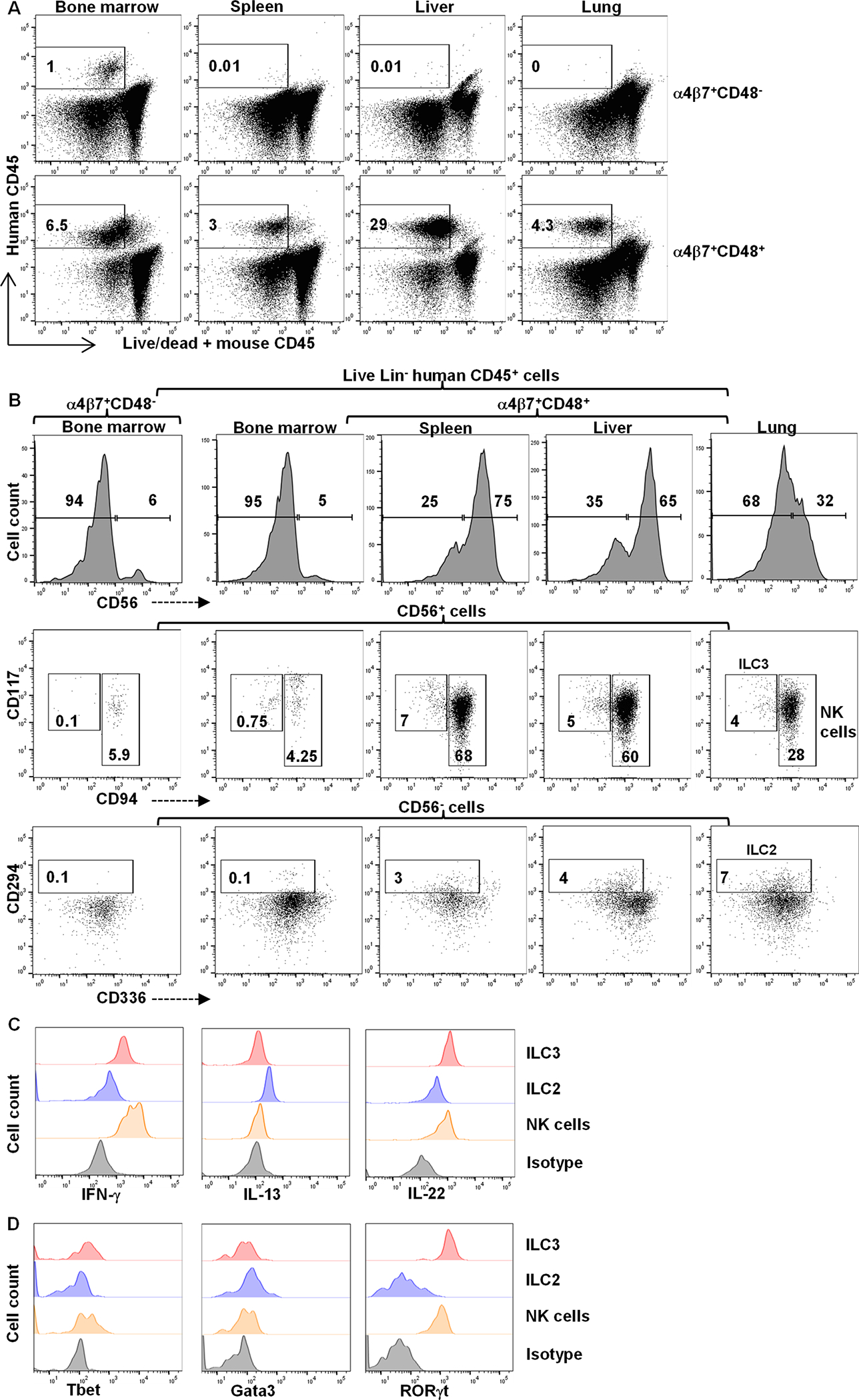
UCB-derived CD34+ HSCs were expanded for 5 days, FACS-sorted into CD34+α4β7+CD48+ and CD34+α4β7+CD48− cells and infused in to immunodeficient NSG mice. After 4 weeks, mice were sacrificed, lymphocytes were isolated and analyzed by flow cytometry. (A) Representative dot plot showing Lin− human CD45+ cells in murine tissues based on progenitor population (n=5/group). (B) Representative plots depicting human CD56 expression within the Lin− human CD45+ cells (top row), human CD117+CD94− ILC3s and CD94+ NK cells within the Lin− human CD45+CD56+ cells (middle row) and human CD294+ ILC2s within the Lin− human CD45+CD56− cells (bottom row) in murine tissues (n=5/group). (C and D) Human CD94+ NK cells, CD294+ ILC2s and CD117+CD94− ILC3s from murine lung were shown for INF-ɣ, IL-13 and IL-22 (C) or Tbet, Gata3 and RORɣt (D) (n=5/group). Values in A and B represent the percentage of positives.
Expression of CD52 in UCB-derived progenitors distinguishes ILCPs
To identify an additional antigen to further resolve the CD34+α4β7+Lin−CD48− and CD34+α4β7+Lin−CD48+ cell populations into various ILCPs, scRNA-seq was performed on differentiating CD34+ cells and from this, we identified CD52 as a candidate gene differentially expressed by the ILCP fraction. CD52, also known as CAMPATH-1 antigen, is a surface glycoprotein expressed on mature lymphocytes, macrophages and monocytes, but not HSCs (29, 30). Accordingly, freshly isolated UCB-derived CD34+ HSCs lacked CD52, however after 5 days of culture some CD34+α4β7+ cells expressed CD52, concurrent with CD48 acquisition (Figure 3A). Similar to freshly isolated UCB-derived HSCs, non-mobilized peripheral blood CD34+ HSCs lacked α4β7, CD48 and CD52 expression. In contrast, CD34+α4β7+ ILCPs in secondary lymphoid tissues (tonsil) expressed CD48 and CD52 (fig. S3).
Fig. 3. Expression of CD52 in CILPs identifies NKPs and ILCPs.
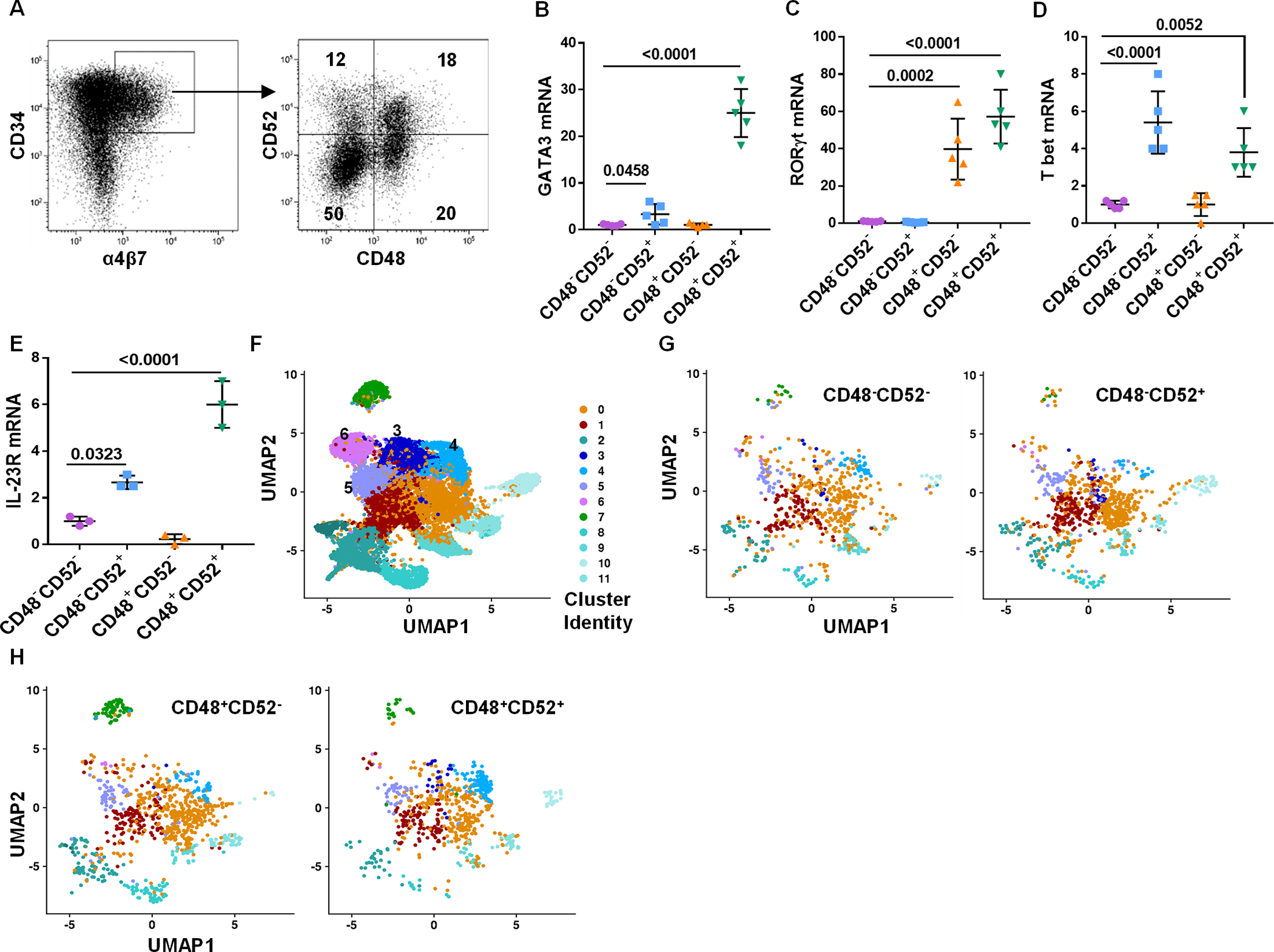
UCB-derived CD34+ HSCs were expanded for 5 days and CD34+α4β7+ progenitors were FACS-sorted into four (CD48−CD52−, CD48−CD52+, CD48+CD52− and CD48+CD52+) hematopoietic progenitor populations and were studied. Differentiating CD34+ cells were also used as a control data. (A) Gating strategy to sort CD34+α4β7+ into four subsets is shown and is representative of n=5 independent experiments. (B-E) qPCR and quantitative expression of GATA3 (B), RORγt (C), Tbet (D) and IL-23R (E) in hematopoietic progenitors is shown relative to the CD48−CD52− subset (n=5 (B, C and D) or 3 (E) /group). (F) UMAP of differentiating and terminally differentiated ILCs data, and values are cluster number (G) UMAP of CD48−CD52− (left) and CD48−CD52+ (right) samples after pre-processing and integration to differentiating ILCs data. (H) UMAP of CD48+CD52− (left) and CD48+CD52+ (right) samples after pre-processing and integration to differentiating ILCs data. (B-E) Data is shown as mean ± SD, One-way ANOVA and p-values are shown.
Gating on cultured UCB CD34+α4β7+Lin− cells showed four populations based on CD52 and CD48 expression (Figure 3A). scRNA-seq data from these FACS-sorted subpopulations corroborated the flow cytometry data, showing differential expression of CD52 and CD48 in these four UCB-derived CD34+ HSCs subsets (fig. S4A), and global transcriptome analysis demonstrated distinct expression patterns between these subpopulations (fig. S4B). Furthermore, relative to the CD34+α4β7+CD48−CD52− population, both the CD34+α4β7+CD48−CD52+ and CD34+α4β7+CD48+CD52+ subsets expressed Tbet and GATA3, while the CD34+α4β7+CD48+CD52− and CD34+α4β7+CD48+CD52+ cells also expressed RORγt (Figure 3B–3D). Given that RORγt was expressed by both the CD34+α4β7+CD48+CD52− and CD34+α4β7+CD48+CD52+ populations, we tested for IL-23R, as it functionally distinguishes NCR+ and LTi-like ILC3 cell development (12, 31). As shown in figure 3E, there was considerably higher expression of IL-23R in the CD34+α4β7+CD48+CD52+ subset relative to the CD34+α4β7+CD48+CD52− cells.
Using scRNAseq, we were unable to further cluster the gene signatures from the four CD34+α4β7+ cell populations (fig. S4C). Instead, we examined the terminally differentiated progeny of the bulk culture and identified 13 distinct cell clusters (Figure 3F), and this analysis was used to determine which clusters were present in the CD34+α4β7+ progenitors (Figure 3G and H). Assessment of the top 10 upregulated genes in each cluster from the differentiated sample (Figure 3F) showed that most expressed genes were not clearly related to NK or ILC cells (table S1). Clusters 3 and 4 showed higher expression of NK-associated transcripts (including PRF1, GZMK, XCL1, XCL2, CCL3 and CCL4), whereas clusters 5 and 6 expressed ILC-related genes (such as KLRB1, KRT81 and TNFRSF18) (fig. S5A), suggesting that clusters 3 and 4 are likely to be NK cells and clusters 5 and 6 are likely to be ILCs. Further analysis of clusters 3–6 in the CD34+α4β7+ progenitor subsets (Figure 4A) showed that cluster 3 is most abundant in the CD34+α4β7+CD48−CD52+ population. Conversely, cluster 4 was dominant in the CD34+α4β7+CD48+CD52+ population. Clusters 5 and 6 were equally distributed across all four subsets (Figure 4B). Comparison of differentially expressed genes within these four clusters also identified distinct genes (table S2). Despite assignment to the same cluster, differences were noted in gene expression across the CD34+α4β7+CD48−/+CD52−/+ subsets (Figure 4C–F). Taken together, these data suggest that clusters 3 and 4 of CD34+α4β7+ subsets correspond to the NKPs, and that clusters 5 and 6 represents ILCPs (9, 24).
Fig. 4. Differential gene expression by CD34+α4β7+ NKPs and ILCPs.
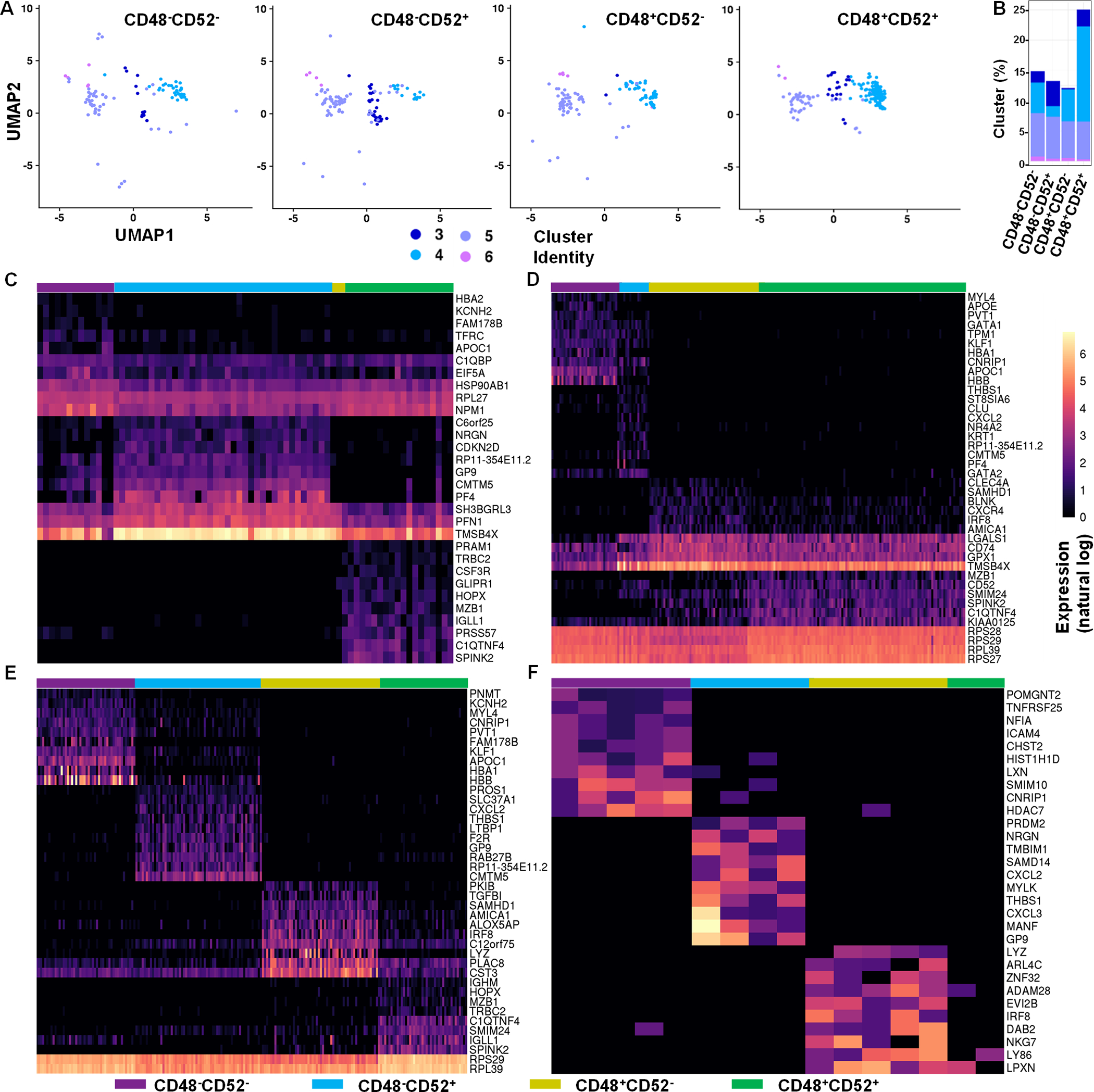
UCB-derived CD34+ HSCs were expanded for 5 days and CD34+α4β7+ hematopoietic progenitors were FACS-sorted into four (CD48−CD52−, CD48−CD52+, CD48+CD52− and CD48+CD52+) hematopoietic progenitor populations and were studied. (A) UMAP of CD48−CD52−, CD48−CD52+, CD48+CD52− and CD48+CD52+ samples showing clusters 3–6 after pre-processing and integration to terminally differentiated ILCs data. (B) Bar graph shows the percentage of cells in clusters 3–6 for each CD48−CD52−, CD48−CD52+, CD48+CD52− and CD48+CD52+ sample. (C-F) Heatmap shows the top 10 differentially expressed genes for CD48−CD52−, CD48−CD52+, CD48+CD52− and CD48+CD52+ samples within clusters 3 (C), 4 (D), 5 (E) and 6 (F). Color bar shows expression on a natural log scale. Genes are ordered by average normalized expression within each sample.
Purified ILCPs differentiate to give rise to distinct and functionally active ILCs
We purified the four CD34+α4β7+ populations based on CD48 and CD52 expression and cultured them under conditions that favored NK and ILC development to validate the gene expression findings. Gene expression data suggested the presence of clusters corresponding with NKPs (clusters 3–4) and ILCPs (clusters 5–6) in the CD34+α4β7+CD48−CD52− subset (Figure 4A), but these cells differentiated into non-ILC cells (Figure 5A and 5B). Likewise, the CD34+α4β7+CD48−CD52+ precursors contained clusters 5 and 6 cells but did not give rise ILCs and instead differentiated into NK cells and non-ILC cells (Figure 5A and 5B). In contrast, the presence of clusters 3 and 4 within the CD34+α4β7+CD48+CD52− population gave rise to ILC3 cells, but not NK cells (Figure 5A and 5B). Lastly, as above, the CD34+α4β7+CD48+CD52+ cells gave rise to NK cells and multiple ILC types, including CD56−CD117+/− ILC1, CD294+CD117+ ILC2 and CD56+CD94−CD117+ ILC3 (Figure 5A and 5B). Interestingly, single cell culture of CD34+α4β7+CD48−CD52− and CD34+α4β7+CD48+CD52− progenitors on irradiated OP9 stromal cells resulted in lack of cell proliferation. Cells proliferated in ~10% (39/384) and ~36% (140/384) of the wells seeded with a single CD34+α4β7+CD48−CD52+ and CD34+α4β7+CD48+CD52+ progenitors, respectively. The CD34+α4β7+CD48−CD52+ single cell culture predominantly (30/39) generated Lin+CD117+CD294+ cells, whereas the remaining wells (9/39) exclusively generated NK cells (fig. S5B). Previously, CD117+CD294+FcεRI+ mast cells were shown to develop from Lin−CD34hiα4β7+CD117+ progenitors (32, 33). The CD34+α4β7+CD48+CD52+ progenitors generated either NK cells alone (11.6%), ILC1s alone (13.9%), ILC2s alone (11.6%), ILC3s alone (44.2%) or a combination of these subsets (18.6%) (fig. S5C).
Fig. 5. CD34+α4β7+CD48+/− ILC precursors differentiate into functionally mature ILCs.
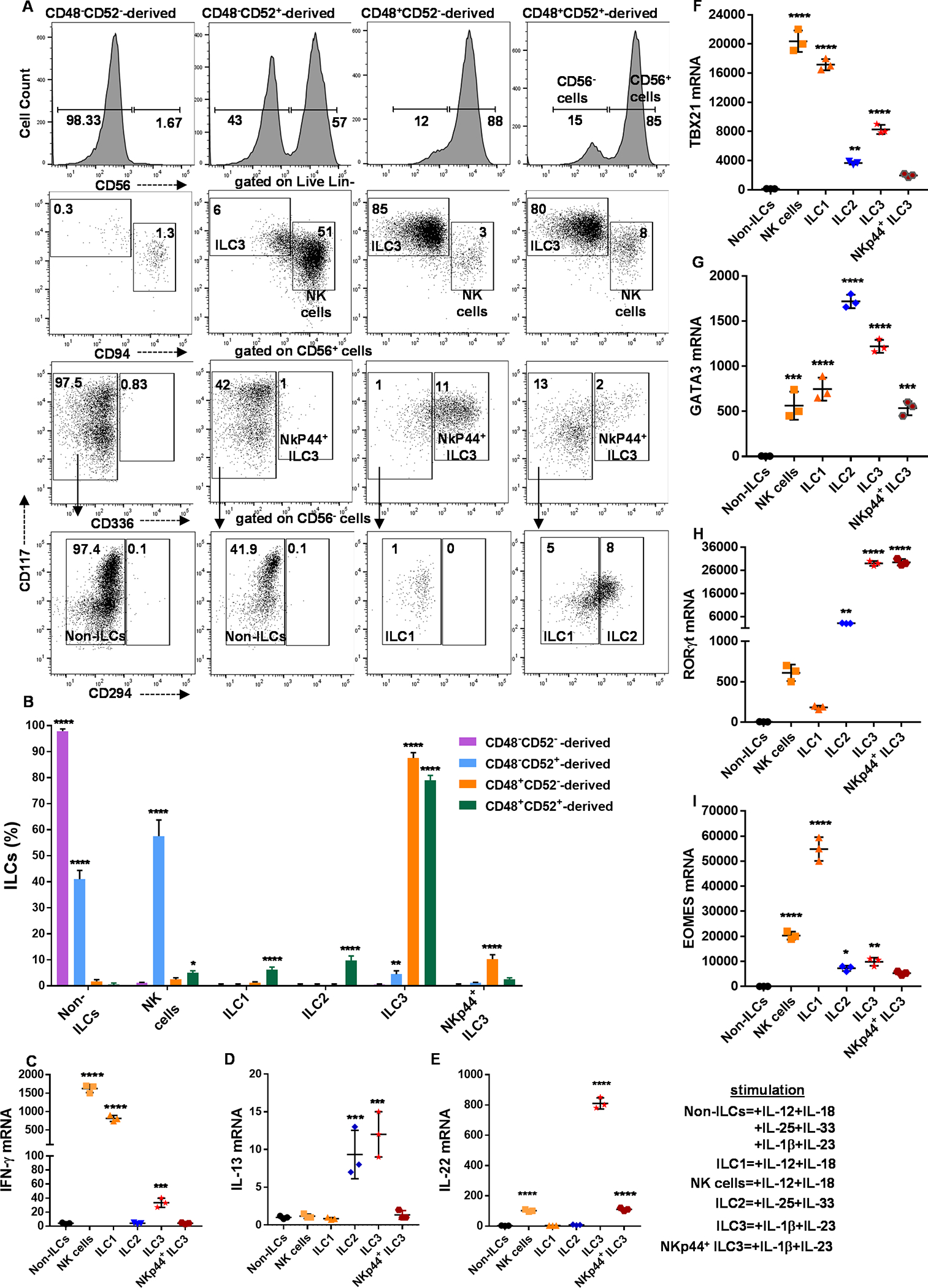
UCB-derived CD34+ HSCs were expanded for 5 days, and CD34+α4β7+ hematopoietic progenitors were sorted using FACS (as shown in figure 3A) following twenty-one days culture under conditions that favor ILC differentiation. (A and B) The output of the sorted populations (CD48−CD52−, CD48−CD52+, CD48+CD52− and CD48+CD52+) is shown. Cells were stained for NK cell or ILCs surface markers: NK cells, ILC1, ILC2, ILC3 and NKp44+ ILC3 are shown in dot plots (A) or bar graphs (B). Results are representative of n=4 experiments. (C-E) ILCs were sorted and stimulated for 16 hours to assess INF-ɣ (C), IL-13 (D) and IL-22 (E) production using qPCR. (F-I) ILCs were sorted to assess Tbet (F), GATA3 (G), RORɣt (H) and EOMES (I) expression using qPCR. (C-I) The expression for NK cells and ILCs were relative to that of non-ILCs, (n=3/group). (B-I) Data is shown as mean ± SD, and one-way ANOVA (*=p<0.05; **=p<0.01; ***=p<0.001; ****=p<0.0001).
We further tested the progeny of the above populations for functional differences. Activation of NK cells and ILC1 cells show that both upregulate IFN-γ mRNA (Figure 5C). Stimulation of ILC2s and ILC3s led to induction of mRNAs for IL-13 and IL-22, respectively (Figure 5D, 5E). FACS sorted NK cells and ILCs were assessed for transcription factors and showed preferential expression of Tbet, GATA3 and RORC by NK cells/ILC1s, ILC2s and ILC3s, respectively (Figure 5F–5H). Finally, we found that ILC1s upregulated Eomes (Figure 5I), corroborating previous reports (34, 35). In summary, the expression pattern of CD48 and CD52 by CD34+α4β7+Lin− cells define individual progenitor populations that give rise to distinct human NK cells, ILC1, 2 and 3 cells.
CD34+α4β7+CD48−CD52+-derived NK cells are distinct from their CD34+α4β7+CD48+CD52+-derived counterparts
Our findings indicated that both CD34+α4β7+CD48−CD52+ and the CD34+α4β7+CD48+CD52+ cells can give rise to NK cells, so both subsets were purified and differentiated in vitro for further investigation. There were similarities in CD56 and CD94 expression, but CD34+α4β7+CD48+CD52+-derived NK cells expressed higher levels of CD336 and lower levels of CD244, CD335 and CD337 compared with CD34+α4β7+CD48−CD52+-derived NK cells (Figure 6A). Moreover, CD34+α4β7+CD48−CD52+-derived NK cells showed more CD107a-mediated degranulation against K562 target cells (Figure 6B). Transcriptome analysis showed similar expression of the NK-associated genes including TBX21 (Tbet), NCAM1 (CD56), KLRD1 (CD94) and KLRB1 (CD161) in these subsets (fig. S6A). In spite of these similarities, scRNA-seq showed distinct patterns between the two NK subsets (Figure 6C), and CD34+α4β7+CD48−CD52+-derived NK cells expressed higher levels of CCL5, FCGR3A (CD16a), TNFSF14 and TYROBP (DAP12) (Figure 6C, 6D). Conversely, CD34+α4β7+CD48+CD52+-derived NK cells expressed higher CD7, GZMB, HLA-Class I and KLRC1 (NKG2A) (Figure 6C, 6D). Ingenuity pathway analysis showed distinct signaling pathway activation between the NK subsets (fig. S6B and S6C). Collectively, these results show that the CD34+α4β7+CD48−CD52+- and CD34+α4β7+CD48+CD52+-derived NK cells are distinct and may represent CD56dim and CD56bright NK cells, respectively (36–38).
Fig. 6. α4β7+CD48+- and α4β7+CD48−-Derived NK cells are distinct cell types.
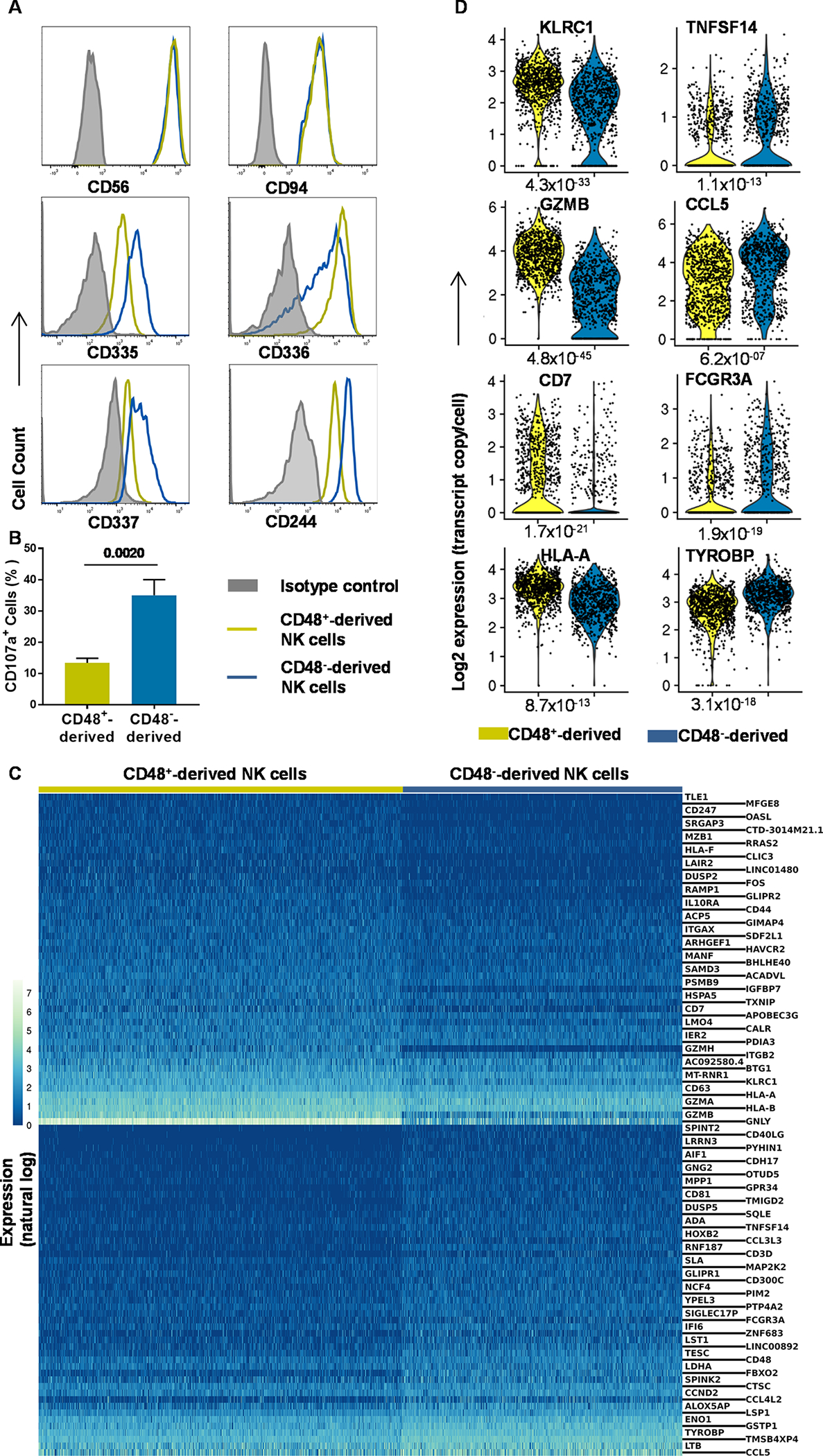
UCB-derived CD34+ HSCs were expanded for 5 days and FACS-sorted into CD34+α4β7+CD48+ and CD34+α4β7+CD48− cells and cultured for 21 days off-stroma. (A) Differentiating cells were stained for CD56, CD94, CD244, CD335, CD336 and CD337. (Representative of n=4 experiments). (B) NK cells were cultured with K562 in 5:1 ratio for CD107a assessment and the percentage of positive is shown, (n=3/group). (C) scRNA-seq and heatmap showing the top 50 differentially expressed genes between CD34+α4β7+CD48+- and CD34+α4β7+CD48−-derived NK cells. Color bar shows expression on a natural log scale. Genes are ordered by average normalized expression with respect to the higher expressing sample. (D) Violin plots derived from scRNA-seq data showing the distributions of CD7, CCL5, FCGR3A, GZMB, HLA-A, KLRC1, TNFSF14 and TYROBP in CD34+α4β7+CD48+- and CD34+α4β7+CD48−-derived NK cells. (B) Data are shown as mean ± SD, paired t-tests and p-values are depicted.
Separate ILC2s and ILC3s subsets differentiate from CD34+α4β7+CD48+ ILCPs
We also sought to identify phenotypically and functionally distinct ILC2 and ILC3 cell populations that can differentiate from CD34+α4β7+CD48+ ILCPs. We used CD11a, CD117 and CD294 (11) to identify two subsets (CD11alowCD117high and CD11ahighCD117low) of CD34+α4β7+CD48+CD52+-derived CD294+ ILC2s (Figure 7A). Stimulation of the CD11ahighCD117lowCD294+ ILC2 population was associated with IL-13 and IL-10 expression (Figure 7B, 7C). Compared with other ILCs or NK cells, both ILC2 subsets upregulated TGFB1 after stimulation (Figure 7D). Interestingly, TGF-β induced more IL-10 expression in the CD11alowCD117highCD294+ ILC2s (Figure 7E).
Fig. 7. Peculiar ILC2 and ILC3 subsets generated in vitro.
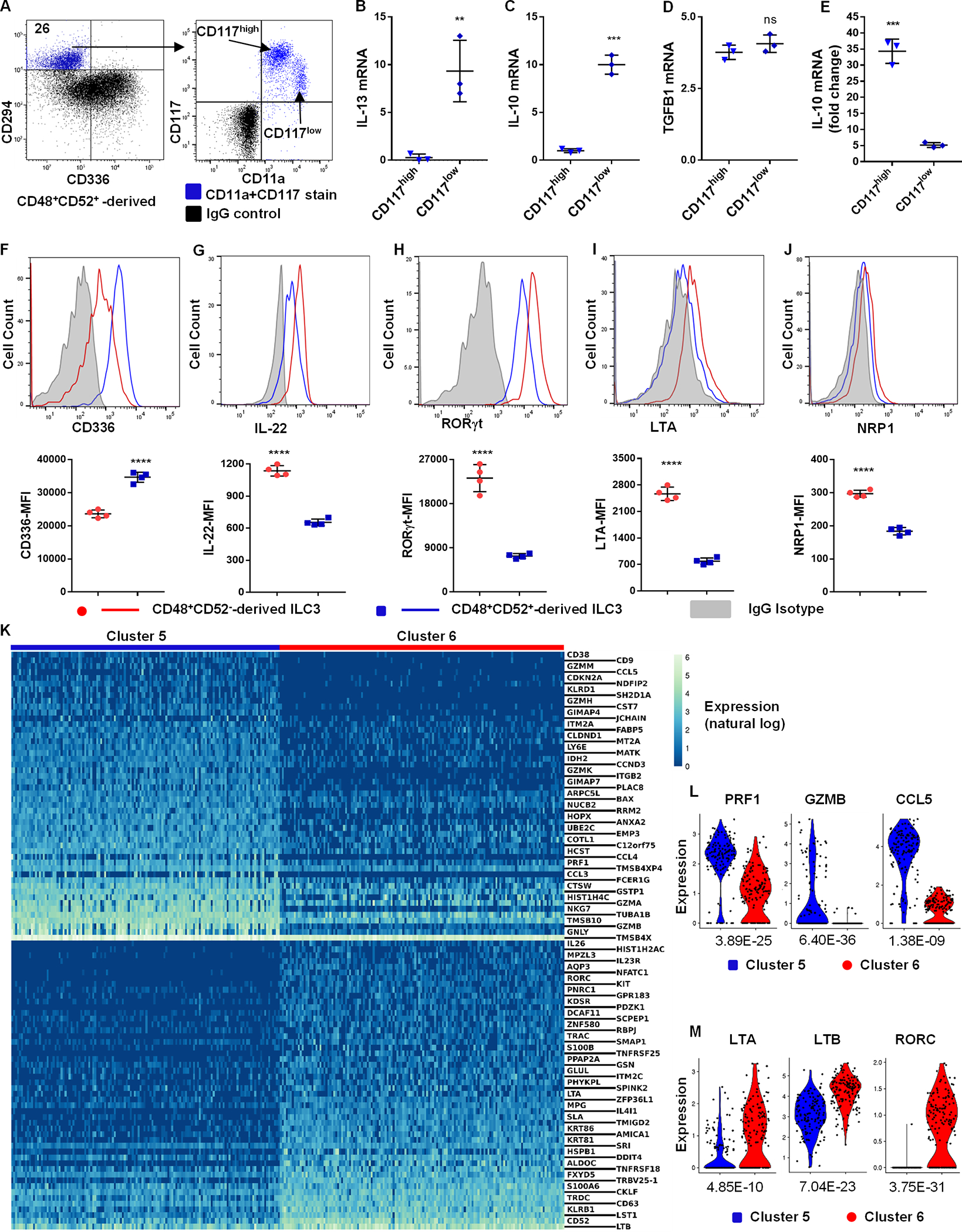
UCB-derived CD34+α4β7+ progenitors were FACS-sorted into four subsets (figure 3A), differentiated for 21 days off-stroma, and ILC2s or ILC3s were analyzed. (A) CD294+ ILC2s were displayed for CD11a and CD117. (Representative, n=4), values are the percentage of positive. (B-D) CD294+CD117high and CD294+CD117low ILC2s were stimulated with IL-25+IL-33 and IL-13 (B), IL-10 (C) and TGFB1 (D) were analyzed using qPCR (n=3/group). (E) CD294+CD117high and CD294+CD117low ILC2s were stimulated with TGF-β, and IL-10 mRNA was depicted as fold change compared to unstimulated cells (n=3/group). (F-J) Surface staining for CD336 (F) and NRP1 (J) as well as intracellular staining for IL-22 (G), RORγt (H) and LTA (I) in stimulated CD34+α4β7+CD48+CD52+- and CD34+α4β7+CD48+CD52−-derived ILC3. Representative histograms and MFI in bar graphs (n=4/group). (K) scRNA-seq and heatmap showing the top 50 differentially expressed genes in clusters 5 vs 6 (figure 3F) of ILC3s. Genes are ordered by average normalized expression with respect to the higher expressing sample. (L and M) Violin plots showing the distributions of PRF1, GZMB, CCL5, LTA, LTB and RORC in clusters 5 vs 6 (figure 3F) ILC3s. p-values are displayed. (B-J) Mean ± SD, paired t-tests (**=p<0.01; ***=p<0.001; ****=p<0.0001).
CD34+α4β7+CD48+CD52−-derived ILC3s showed less CD336 expression compared with CD34+α4β7+CD48+CD52+-derived ILC3s (Figure 7F). Previous reports indicate that LTi-like ILC3 cells express higher IL-22, RORγt, LTA and NRP1 compared with the NCR+ ILC3 (8, 12) and these were higher on CD34+α4β7+CD48+CD52−-derived ILC3s (Figure 7G–7J). As above, transcriptome analysis of the progeny of CD34+-differentiated cells (as in figure 3F) resulted in two distinct ILC3 subsets (cluster 5 and 6). Comparison of the top 50 upregulated genes within these two mature ILC3 subsets showed that cluster 5 expressed relatively more NK-associated genes including granzymes, PRF1, CCL3 and CCL5 (Figure 7K, 7L). Concomitantly, previous reports demonstrate that NCR+ ILC3 cells express NK cell-associated receptors in addition to ILC3-restricted genes (8, 12). In contrast, cluster 6 upregulated most of the ILC3-associated genes including KIT, KLRB1, LTA, LTB and RORC (Figure 7K, 5M). Moreover, IPA indicated activation of distinct signaling pathways between cluster 5 and 6 ILC3s (fig. S7A and S7B). Together these data provide evidence that the CD34+α4β7+CD48+CD52−-derived ILC3s are LTi-like ILC3, whereas the CD34+α4β7+CD48+CD52+-derived ILC3s represent NCR+ ILC3s.
2B4 receptor signaling enhances the development of ILC2 from ILCPs
The 2B4 (CD244) receptor modulates NK and T cell activation through CD48 binding (39, 40). Consistent with prior reports (41), nearly all freshly isolated UCB-derived CD34+ HSCs express CD244, whereas only a proportion show expression of CD48 (Figure 8A). CD244 surface density on freshly isolated CD34+CD48− HSCs is similar to CD34+CD48+ HSCs, but expression was upregulated on the latter progenitors after three days of in vitro culture (Figure 8B). We hypothesized that CD244 signaling may influence ILC differentiation from CD34+α4β7+CD48+ cells. Indeed, SLAM-associated protein (SAP), known as an adapter protein for CD244 signaling (23) was upregulated within CD34+α4β7+CD48+ cell population, suggesting activation of CD244 signaling (Figure 8C). During development, the addition of anti-CD244 or anti-CD48 blocking antibody abrogated ILC2 differentiation, whereas the proportion and absolute number of NK cells were increased (Figure 8D, 8E and fig. S8A, S8B). Conversely, activation of CD244 signaling using an agonist antibody increased ILC2 differentiation at the expense of NK cells (Figure 8F, 8G and fig. S8A, S8B). Moreover, culturing CD34+α4β7+CD48+ progenitors on a layer of irradiated CD48-expressing OP9 cells (vs. control OP9 stromal cells) showed an enhanced ILC2 generation (fig. S8C). We used CRISPR-Cas9 to knock out CD244 in CD34+α4β7+CD48+ progenitors followed by single cell culture on irradiated OP9 feeders to confirm this observation. CD244 expression was lost in 92% of the single cell cultures, whereas 94% of the single cell cultures containing control gRNA expressed CD244 (Figure 8H and fig. S8D). Strikingly, ILC2 development was completely lost in in the absence of CD244 (Figure 8I, 8J). In contrast, NK cells and ILC3s differentiated from CD244 knockout progenitors (fig. S8E and S8F). We identified two separate ILC2 populations once again based on CD11a and CD117 expression. Both ILC2 subsets increased proportionally with CD244 activation, suggesting that CD244 signaling acts upstream of ILC2 subtype specification. These data demonstrate that CD244 signaling influences ILC differentiation by modulating the development of CD34+α4β7+CD48+ progenitors into ILC2 cells.
Fig. 8. CD244 activation enhances ILC2 development.
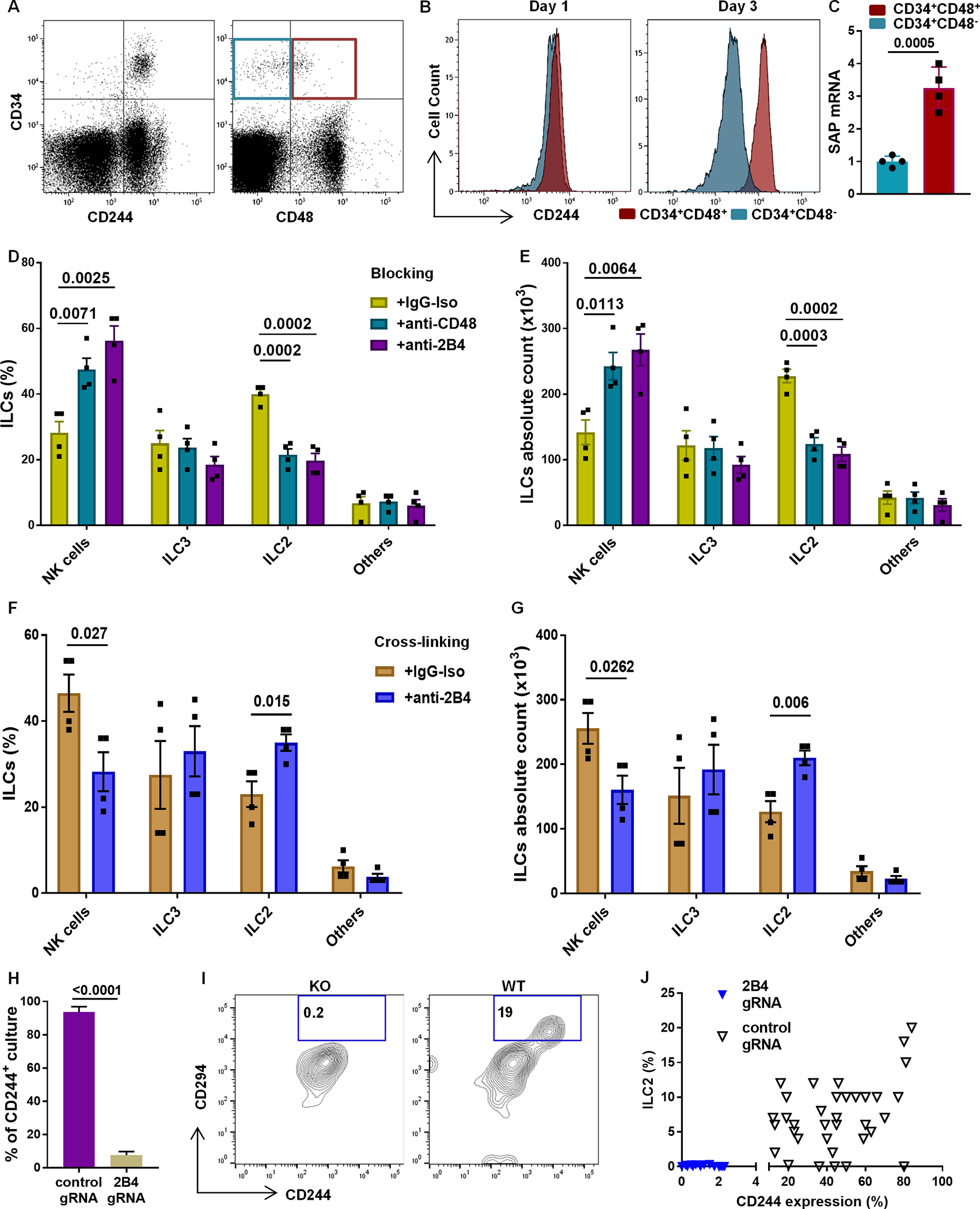
(A) Freshly isolated UCB-derived CD34+ HSCs were stained for CD244 and CD48 (representative, n=4). (B) CD244 in CD34+CD48− cells (aqua) and in CD34+CD48+ cells (red) for day 1 and day 3 UCB-derived CD34+ HSCs (representative, n=4). (C) SAP mRNA expression in day 5 CD34+CD48− and CD34+CD48+ cells (n=4/group). (D and E) Anti-CD244 and anti-CD48 blocking antibodies or isotype IgG controls? were added to differentiating CD34+α4β7+CD48+ cultures to block CD244 signaling. (F and G) CD34+α4β7+CD48+ cells were plated on anti-CD244 or IgG coated plates twice for 4 days prior to 21 days of differentiation to activate CD244 signaling via cross-linking. Cells were stained for ILCs, and the percentage (D and F) or absolute number (E and G) of CD94+ NK cells, CD294+ ILC2 and CD117+ ILC3 are shown (n=4/group). (H) CD244+ cultures are shown for cells transfected with control or CD244 gRNA (n=3/group). (I) Representative ILC2 (CD294) staining for cultures transfected with control or CD244 gRNA. (J) Dot plot showing generation of ILC2s from cultures that express (control gRNA) or lack (CD244 gRNA) CD244. (C-H) Mean ± SD, paired t-tests (C, F, G and H) or one-way ANOVA (D and E) and p-values are depicted.
Discussion
The exact developmental trajectory that human HSCs take to become ILCs is not fully defined, and human common ILCPs have not yet been characterized (17). To decipher ILC development in mice, transcription factor reporter and knockout animals have been used (16). Given that this approach is not possible in humans, we used scRNAseq, immunophenotyping, in vivo adoptive transfer and functional analysis to investigate the developmental trajectories of human HSCs. We describe developmental intermediates with progressively narrowing capacities to give rise to various NK and ILC subsets. All human ILCs are believed to arise from CILPs that are downstream of CLPs (20, 42). We found that human CILPs enriched within the Lin−α4β7+ compartment of CD34+ progenitors. Although these cells are rare in freshly isolated UCB, they expand with cell-specific cytokines. CD52 is expressed by mature lymphocytes and CD34+ HSCs lack this protein (29, 30, 43). However, after several days in culture, a portion of CD34+ progenitors express CD52, which is concurrent with α4β7 and CD48 acquisition. CD34+ cells that express α4β7, CD48 and CD52 were present in tonsillar tissue, but were not found in UCB or peripheral blood, suggesting the requirement of secondary lymphoid tissues for the differentiation of these progenitors (42). In murine models, CILP gave rise to CHILPs and NKPs, with the former developing exclusively into ILCs and not NKs, and the latter showing the opposite characteristics (1, 16). In contrast, studies using freshly isolated human tonsils provide evidence that these same intermediates are not necessarily occurring in humans (44, 45). Here, we show that human CILPs are further defined by CD48 and CD52 expression and can be delineated into four progenitor populations with unique gene expression and developmental potential. The data herein shows that CD34+α4β7+Lin−CD48−CD52+cells express Tbet and give rise to an NK population that has cytotoxic features and most closely resembles CD56dim NK cells. We also found that the cells derived from CD34+α4β7+Lin−CD48+CD52− cells are Tbet−RORγt+ and give rise to LTi-like ILC3 cells. In this manner, these populations appear to be restricted progenitors. However, the CD34+α4β7+Lin−CD48+CD52+ fraction expressing Tbet, GATA3 and RORγt develops into a few different cell types, including CD56bright NK cells, ILC1, ILC2 and ILC3 cells. We have made an exhaustive attempt to further resolve this fraction into progenitors destined to become either NK cells or ILCs. In spite of differential expression of multiple candidate molecules (AMICA1, CCR7, CD44, CD53, CD63, CD99, HLA-A, IL2RG, KRT1 and NOTCH1/2,) at a transcript level, we were unable to identify these and others as suitable antigens by using flow cytometry. Although our study does not quite formally define NKPs and ILCPs as “restricted” cell types, the finding that two NK cell subsets develop from distinct progenitors is consistent with the work of others (44, 45). Data obtained using scRNA-seq analysis indicated that there were distinct cell subpopulations with gene clusters that resembled mature NK cells and/or ILCs. Interestingly, these clusters could be found within each of the four progenitor populations (based on CD48 and CD52), but only those derived from the CD34+α4β7+Lin−CD48+CD52+ fraction gave rise to the ILC1, ILC2 and ILC3 cells. In this manner, these results leave open the possibility of a human CHILP that gives rise to only ILC1, −2 or −3 cells, as described in mice (1, 16). Similarly, the developmental potential of the CD34+α4β7+Lin−CD48−/+ cells was determined in vivo using humanized mice, showing that CD34+α4β7+Lin−CD48− cells engraft only in the bone marrow and give rise to NK cells, whereas CD34+α4β7+Lin−CD48+ cells give rise to tissue-associated NK cells, ILC1s, ILC2s and ILC3s in the spleen, liver, and lung. Both CD34+α4β7+Lin−CD48− and CD34+α4β7+Lin−CD48+ cells do not develop into T cells, B cells, or monocytes in vivo in the immune deficient mice.
Human NK cells are one of the best characterized subsets of innate lymphocytes and two subsets of NK cells (CD56bright and CD56dim cells) have been identified, but their developmental relationship are still poorly understood (36–38, 44, 45). Consistent with prior studies (44–47), we found two NK cell populations with similar expression of common NK cell surface markers such as CD56 and CD94, but variations in NKp30, NKp44, NKp46 and 2B4 expression. Both NK cell populations produced IFN-γ, but differed in their propensity for degranulation in response to K562 targets, reminiscent of CD56bright and CD56dim populations. The in vitro developmental system used allowed us to identify that these NK cell subpopulations are derived from two separate progenitors, with the resulting cells having differing transcriptomes, likely due to their distinct developmental origins (2, 3, 48). Likewise, these two NK subpopulation may also represent the already described circulating and tissue-associated NK cells (44, 49, 50), as shown by our in vivo data that CD34+α4β7+CD48+CD52+ progenitors, but not CD34+α4β7+CD48−CD52+ progenitors give rise NK cells in the tissues such as liver and lung. Comparison of ILC3s derived from the CD34+α4β7+Lin−CD48+CD52+ and CD34+α4β7+Lin−CD48+CD52− progenitors showed variations in overall transcriptomic expression and function. Such differences in human ILC3 subsets exist between the NCR+ ILC3 and the LTi-like ILC3 (12, 31), but the reasons for these are largely unknown. Unlike CD34+α4β7+Lin−CD48+CD52− ILC3 precursors, we find the CD34+α4β7+Lin−CD48+CD52+ ILC3 precursors to express both Tbet and GATA3, suggesting that these may modulate RORγt, as evidenced by less RORγt expression and IL-22 production in the CD34+α4β7+Lin−CD48+CD52+-derived ILC3 cells. Previous reports indicate that GATA3 antagonizes RORγt and enhances Tbet during NCR+ ILC3 development (51, 52). NCR+ ILC3 development also depends upon IL-23 signaling, leading to Tbet upregulation, which inhibits RORγt expression (31). The developmentally programed expression of IL-23R in CD34+α4β7+Lin−CD48+CD52+ ILC3 precursors, but not in CD34+α4β7+Lin−CD48+CD52− ILC3 precursors agrees with this report. Our findings suggest that CD34+α4β7+Lin−CD48+CD52+-derived and CD34+α4β7+Lin−CD48+CD52−-derived ILC3s are distinct subsets of ILC3s. Lastly, the expression of GATA3 and the production of IL-13 by ILC3s, both observed here, likely has implications for ILC3 plasticity (13, 14).
The SLAM family receptors distinguish HSCs and hematopoietic progenitors (22). We show the SLAM family receptor CD48 is not only a surface marker for human ILCPs, but also participates in CD244 signaling that in turn influences ILC development. All HSC progenitors expressed CD244, with relatively higher expression in the CD34+α4β7+Lin−CD48+ fraction. CD48 is the primary ligand for CD244 and is present on murine hematopoietic cells, with the exception of quiescent long-term HSCs (40, 53). The HSCs from CD48 knockout mice display low engraftment and differentiation potential (53). CD244 signaling modulates the proliferation and activation of mature lymphocytes, such as T cells and NK cells (39, 40). We reasoned that differential expression of CD48 by CD34+α4β7+Lin−CD48+ progenitors might indicate the necessity of CD244 signaling for their differentiation. Confirming this hypothesis, progenitors lacking CD244 showed a complete absence of ILC2s but the generation of NK cells was unchanged, which is consistent with a previous finding that NK cell development is intact in CD244 knockout mice (54). Conversely, augmented CD244 signaling enhanced ILC2 development, but CD244 blockade prevented ILC2 differentiation and increased the generation of NK cells. These findings suggest that CD244 signaling induces ILC2 development from ILCPs, and that ILC2 cells attenuate NK cell development or expansion, perhaps involving inhibitory factors such as TGFB1 and IL-10. In support of this concept, CD244 signaling led to the development of two ILC2 subsets, one of which expressed IL-10 upon stimulation with TGF-β. Recent murine studies report ‘regulatory’ ILCs that influence the function of other ILCs by IL-10 production (9, 10, 15).
Like all research, there are limitations to this work. We have largely used an in vitro differentiation system to obtain sufficient cells to perform experiments including scRNA-seq at various stages of development. Whether these conditions sufficiently mirror the events that occur in vivo could be questioned. However, we were able to confirm the presence of CD48 and CD52 on progenitors in tonsillar tissue, but not in blood or UCB, pointing the likely importance of the local tissue microenvironment in cell differentiation and the associated challenges in mimicking this. Moreover, we were not able to further resolve the CD34+α4β7+Lin−CD48+CD52+ that gives rise to multiple ILC types (ILC1, −2, −3 cells) and NK cells. However, the presented scRNA-seq and single cell culture data would support the existence of a common ILCPs independent of additional progenitor populations with restricted differentiation potential.
In summary, CD48 and CD52 expression by CD34+α4β7+ HSCs marks human ILCPs and NKPs. Using scRNA-seq we characterize the NK cell and ILC progenitors and demonstrate that various subpopulations of these cells have distinct developmental origins. CD244 triggering favors ILC2 development at the cost of NK cell differentiation. Together these findings define the developmental trajectories of specific human ILC subtypes and suggest that these subtypes are developmentally programmed and are not only a consequence of alternative activation states or ILC plasticity.
Materials and Methods
Study Design
This study was designed to investigate the developmental intermediates of human ILCs as they grow from HSCs. De-identified human peripheral blood, UCB and tonsil tissues all of which were deemed exempt by University of Colorado institutional review board (COMIRB) were used to perform this work. Animal experiments were approved by the ethical committee at University of Colorado Anschutz, IACUC protocol (00251).
Isolation and expansion of CD34+ HSCs
Mononuclear cells were isolated by density gradient centrifugation using Lymphoprep (Stemcell). UCB-derived CD34+ HSCs were positively enriched using MACS CD34+ enrichment kit (Milteny). The cells (purity, >95%) were suspended (5×104 cells/ml) in Stemspan II cell culture media (Stemcell) supplemented with 1% penicillin + streptomycin, stem cell factor (SCF, 100 ng/ml, R&D), FMS-like tyrosine kinase 3 (Flt3L, 100 ng/ml, Stemcell), thrombopoietin (TPO, 50 ng/ml, R&D) and low density lipoprotein (LDL, 10 μg/ml, Stemcell) and cultured in 24 well plates for 5 days of expansion. After 5 days of expansion the cells were expanded three-fold on average, while the proportion of CD34+ cells remained >95%.
Differentiation of CD34+ HSCs
After 5 days of culture, the expanded cells were considered for further differentiation experiments. Where specified, expanded CD34+ HSCs were FACS sorted into different subsets including CD34+α4β7+, CD34+α4β7−, CD34+α4β7+CD48+/− and CD34+α4β7+CD48+CD52+/−. For up to 28 days of differentiation, cells were cultured in B0 differentiation media (as previously described (25)), supplemented with SCF (20 ng/ml, R&D Systems), IL-3 (5 ng/ml, Stemcell), IL-7 (20 ng/ml, R&D), IL-15 (10 ng/ml, NIH), IL-23 (10 ng/ml, R&D) and Flt3L (10 ng/ml, Stemcell). In some experiments (specified in figure legends) cells were also cultured in the presence or absence of pre-plated and irradiated EL08.1D2 stromal cells. After a week of culture, IL-3 was excluded from the B0 differentiation media supplements. For plating progenitors on the stromal cells, 100 progenitor cells were plated per well of 96-well plates on the irradiated EL08.1D2 cells in 150 μl B0 differentiation media. Alternatively, cells were also plated without stroma using 96-well U-bottom plate and 1×103 cells were cultured per well. Culturing, maintenance and preparation of irradiated stromal layer of EL08.1D2 cells on 96-well plate culture was as described earlier (55).
In vivo generation of ILCs in NOD scid gamma mice (NSG) immunodeficient mice model
CD34+α4β7+CD48− and CD34+α4β7+CD48+ progenitors were FACS-sorted from day 5 expanded CD34+ HSCs as shown in figure 1A. 5×105 of CD34+α4β7+CD48− or CD34+α4β7+CD48+ progenitors were injected via tail vein into 10 (n=5/group) sublethally irradiated (3Gy) 4 weeks-old mice. Mice were intraperitoneal injected with IL-2, IL-7, IL-15, SCF and IL-23 (300 ng each) four times in weekly bases and sacrificed for analysis four weeks post-infusion. Isolation of lymphocytes from mice organs was as previously described (20)
CD244 cross-linking, blocking and knockout
To study potential stimulatory effects of CD244 activation in CD244-expressing CD48+ progenitors, anti-CD244 antibody clone C1.7 (ThermoFisher Scientific) was used to initiate cross-linking. For this purpose, anti-CD244 or isotype IgG antibody was coated as 2 μg/ml in PBS on flat bottom 96-well culture plates for two hours at room temperature. Following blocking by 5% FBS-containing culture media and three cycles of washing with PBS, cells were plated using B0 differentiation media on the coated plates to facilitate CD244 cross-linking. After 48 h of culture, cells were collected and transferred to a newly coated plate. Cells were finally collected after a total of 96 h of cross-linking and plated for further differentiation in 96-well U-bottom cell culture plate off-stroma. Alternatively, to activate CD244 by its natural ligand, human CD48 expressing OP9 stromal cells were generated using lentivirus transduction. The progenitor cells (100 cells) were plated per well of 96-well plates on the layer of irradiated CD48 expressing OP9 cells in 150 μl B0 differentiation media. To further investigate the stimulatory effects of CD244 signaling, both CD244, and its primary ligand, CD48, were blocked in differentiation cultures using 5 μg/ml neutralizing antibodies against CD244 (clone eBioPP35, eBiosciences) and CD48 (clone eBio156–4H9, eBiosciences). Moreover, CD244 was deleted from CD34+α4β7+CD48+ progenitors using the CRISPR-Cas9 system. For this purpose, 1×106 cells were electroporated in Amaxa 4D-Nucleofector system (Lonza) with a complex of Cas9 enzyme, tracrRNA and custom CD244 or control gRNA. The Alt-R Cas9 Nuclease V3, universal tracrRNA, CD244 gRNA and non-human control gRNA were purchased from Integrated DNA Technologies. The electroporated cells were plated as 1 cell per well of 96-well plates on the layer of irradiated OP9 cells in 150 μl B0 differentiation media for further differentiation.
scRNA-seq sample processing
UCB-derived CD34+ HSCs from 2 donors were expanded separately for 5 days as described above, and CD34+α4β7+ cells were sorted into four populations: CD48−CD52−, CD48−CD52+, CD48+CD52− and CD48+CD52+ (as in Figure3A) for transcriptome analysis. Day-1, −5, −9, −13, −18 and −23 differentiating CD34+ cells from 3 donors were also analyzed and used as a reference sample that contains the NKP, ILCP and their progeny. Terminally differentiated CD56+CD94+ NK cells from 2 donors were also analyzed to compare CD48−-derived and CD48+-derived NK cells. A total of 15,827 cells from 7 donors (table S3 for metrics) were captured using the Chromium™Single Cell 3’ Solution (10x Genomics). The resulting libraries were then sequenced as paired-end 151-bp reads on the Illumina NovaSeq 6000 platform at the University of Colorado’s Genomics Core Facility. Read mapping and quantification was performed using Cell Ranger (10x Genomics).
scRNA-seq analysis
Expression data for CD34+α4β7+ cells (n = 3,203) and differentiating CD34+ cells (n = 10,959) with more than 250 unique genes was imported into an R environment; the data was filtered and analyzed using the R package Seurat (56). 31,443 genes were retained after excluding genes expressed in less than one cell. 3,196 CD34+α4β7+ cells and 10,937 differentiating CD34+ cells remained for further analysis after excluding cells based on the following criteria: UMI counts greater than 60,000, mitochondrial percentages greater than 12%, or a UMI to unique gene count ratio less than 1 or greater than 8. This filtering criteria was chosen for the purpose of removing doublets or cells that lysed prematurely during library preparation. Gene counts were then normalized for each cell by dividing the total number of counts within each cell, multiplying by a scaling factor of 10,000, adding one, and then taking the natural log. Samples were then integrated, using the functions provided by Seurat, to improve downstream analysis using UMAP. Cell cycle state was determined for each cell, using Seurat’s CellCycleScoring function, and subsequently regressed out. Cells were then projected onto the top 30 principal components derived from principal component analysis (PCA). The dimensionality was then further reduced to two dimensions using uniform manifold approximation and projection (UMAP). 13 clusters were demarcated in the UMAP space by a shared nearest neighbor modularity optimization-based clustering algorithm. A similar protocol was used to analyze the CD48−- and CD48+-derived NK cells. Data for 937 CD48+- and 728 CD48−-derived NK cells with 20,465 genes were present after importing the data into R. 923 CD48+-derived NK cells and 712 CD48−-derived NK cells remained after excluding cells based on the following criteria: unique gene counts greater than 7,500, UMI counts greater than 60,000, mitochondrial percentages greater than 10%, or a UMI to unique gene count ratio less than 1 or greater than 9. Initial dimensionality reduction was performed using 32 principal components prior to UMAP.
Quantitative PCR (qPCR)
To perform qPCR, total RNAs were extracted from cells using RNeasy kit (Qiagen) and reverse transcribed into cDNAs using advanced iSuperscript cDNA synthesis kit (Bio-Rad). The qPCR reaction and analyses were performed as described before (27). The details of the primer assays used for this study is depicted in table S4.
Flow cytometry
Flow cytometry was used to analyze α4β7+ ILC progenitors, CD117+ and CD127+ ILC precursors as well as mature ILCs. The gating strategy for ILCs was as shown (fig. S1). To evaluate the intracellular IL-13, IL-22 and IFN-γ expressions in ILCs, cells were stimulated with 10 ng/ml PMA (Sigma) + 1 μg/ml ionomycin (Sigma) or 10 ng/ml of IL-12+IL-18 (for ILC1, R&D Systems), IL-25+IL-33 (for ILC2, Shenandoah) and IL-1β (Stemcell)+IL-23 (R&D Systems) (for ILC3) overnight and in the presence of 2 μg/mL brefeldin A (BD Biosciences) for the last 4 h. For intracellular staining, cells were first stained for surface markers and fixed, followed by permeabilization and staining for intracellular proteins. For CD107a degranulation assay, sorted NK cells were incubated with K562 cells at effector:target of 5:1, and the detail of the assay is previously described (57). All flow cytometry data were acquired in LSR II (BD Biosciences) and analyzed using FlowJo (BD Biosciences) or Kaluza (Beckman Coulter) analysis software. As negative controls, fluorochrome conjugated isotype-matched antibodies from the respective companies were utilized. Viability of cells was analyzed using flow cytometry with the fixable viability dye eFluor™ 780 (eBioscience). The detail of the antibodies used for surface or intracellular proteins staining is as mentioned in table S5.
Statistical Methods
One-way ANOVA and paired t-tests were used to compare differences between categorical values. For scRNA-seq analysis Wilcox test was applied on any gene that had an absolute natural-log fold-change of at least 0.25 and was expressed in more than 20% of cells in at least one of the groups being compared. A p-value of 0.05 was used as a cutoff prior to selecting differentially expressed genes shown in figures. IPA was performed using significantly differentially expressed genes. Pathways were considered significant at p <= 0.05.
Supplementary Material
Fig. S1. Representative ILC staining strategy.
Fig. S2. Generation of ILCs from CD34+α4β7+ HSC Progenitors.
Fig. S3. Surface expression of CD48, CD52, CD117 and α4β7 by peripheral blood- and tonsil-derived CD34+ HSCs.
Fig. S4. Gene expression profiling in four subsets of hematopoietic progenitors.
Fig. S5. RNA-seq and single cell culture demonstrated in vitro differentiation of NK cells and ILCs from CD34+ HSCs.
Fig. S6. Similar expression of NK cell-associated markers in CD34+α4β7+CD48+- and CD34+α4β7+CD48--derived NK cells.
Fig. S7. Differentially activated pathways in two distinct ILC3 subsets.
Fig. S8. The effect of CD244 signaling in the development of ILCs.
Table S1. Top 10 differentially expressed genes for each cluster within the terminally differentiated CD34+ HSCs-derived ILCs.
Table S2. Table showing differentially expressed genes for clusters 3-6 that are common to both terminally differentiated cells and CD34+α4β7+ hematopoietic progenitors.
Table S3. Description of samples used for scRNA-seq analysis.
Table S4. Lists and description of primer assays used in the study.
Table S5. Lists and description of antibodies used in the study.
Data file S1: Raw data
Acknowledgments
We thank the clinical immunology cell sorting facility, the genomics and microarray core, and the cancer center shared resource cores (P30CA046934) (University of Colorado) for technical assistance. The α4β7 antibody is a donation from National institute of health (NIH).
Funding
This work was supported by the grant from the NIH (NIH 1R01AI100879)
Footnotes
Competing interests
D.M.T. and M.R.V. are inventors on patent application (application no. 63/084,440) held/submitted by the regents of the University of Colorado, a body corporate that covers compositions and methods for producing and using ILCs to treat health conditions. All other authors declare that they have no competing interests.
Data and material availability
All data used to draw conclusions in this article are present in the main paper and/or the supplementary materials. The scRNA-seq datasets were deposited in the GSE accession: GSE160009.
References
- 1.Diefenbach A, Colonna M, Koyasu S, Development, Differentiation, and Diversity of Innate Lymphoid Cells. Immunity. 41, 354–365 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fuchs A, Vermi W, Lee JS, Lonardi S, Gilfillan S, Newberry RD, Cella M, Colonna M, Intraepithelial Type 1 Innate Lymphoid Cells Are a Unique Subset of IL-12- and IL-15-Responsive IFN-γ-Producing Cells. Immunity. 38, 769–781 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Björklund AK, Forkel M, Picelli S, Konya V, Theorell J, Friberg D, Sandberg R, Mjösberg J, The heterogeneity of human CD127+ innate lymphoid cells revealed by single-cell RNA sequencing. Nat. Immunol. 17, 451–460 (2016). [DOI] [PubMed] [Google Scholar]
- 4.Hoyler T, Klose CSN, Souabni A, Turqueti-Neves A, Pfeifer D, Rawlins EL, Voehringer D, Busslinger M, Diefenbach A, The Transcription Factor GATA-3 Controls Cell Fate and Maintenance of Type 2 Innate Lymphoid Cells. Immunity. 37, 634–648 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mjösberg JM, Trifari S, Crellin NK, Peters CP, van Drunen CM, Piet B, Fokkens WJ, Cupedo T, Spits H, Human IL-25- and IL-33-responsive type 2 innate lymphoid cells are defined by expression of CRTH2 and CD161. Nat. Immunol. 12, 1055–1062 (2011). [DOI] [PubMed] [Google Scholar]
- 6.Cella M, Fuchs A, Vermi W, Facchetti F, Otero K, Lennerz JKM, Doherty JM, Mills JC, Colonna M, A human natural killer cell subset provides an innate source of IL-22 for mucosal immunity. Nature. 457, 722–725 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Takatori H, Kanno Y, Watford WT, Tato CM, Weiss G, Ivanov II, Littman DR, O’Shea JJ, Lymphoid tissue inducer–like cells are an innate source of IL-17 and IL-22. J. Exp. Med. 206, 35–41 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bernink JH, Krabbendam L, Germar K, de Jong E, Gronke K, Kofoed-Nielsen M, Munneke JM, Hazenberg MD, Villaudy J, Buskens CJ, Bemelman WA, Diefenbach A, Blom B, Spits H, Interleukin-12 and −23 Control Plasticity of CD127+ Group 1 and Group 3 Innate Lymphoid Cells in the Intestinal Lamina Propria. Immunity. 43, 146–160 (2015). [DOI] [PubMed] [Google Scholar]
- 9.Wang S, Xia P, Chen Y, Qu Y, Xiong Z, Ye B, Du Y, Tian Y, Yin Z, Xu Z, Fan Z, Regulatory Innate Lymphoid Cells Control Innate Intestinal Inflammation. Cell. 171, 201–216.e18 (2017). [DOI] [PubMed] [Google Scholar]
- 10.Seehus CR, Kadavallore A, La Torre BD, Yeckes AR, Wang Y, Tang J, Kaye J, Alternative activation generates IL-10 producing type 2 innate lymphoid cells. Nat. Commun. 8, 1900 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hochdörfer T, Winkler C, Pardali K, Mjösberg J, Eur. J. Immunol., in press, doi: 10.1002/eji.201848006. [DOI] [PubMed] [Google Scholar]
- 12.Melo-Gonzalez F, Hepworth MR, Functional and phenotypic heterogeneity of group 3 innate lymphoid cells. Immunology. 150, 265–275 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lim AI, Verrier T, Vosshenrich CA, Di Santo JP, Developmental options and functional plasticity of innate lymphoid cells. Curr. Opin. Immunol. 44, 61–68 (2017). [DOI] [PubMed] [Google Scholar]
- 14.Gronke K, Kofoed-Nielsen M, Diefenbach A, Innate lymphoid cells, precursors and plasticity. Immunol. Lett. 179, 9–18 (2016). [DOI] [PubMed] [Google Scholar]
- 15.Cherrier DE, Serafini N, Di Santo JP, Innate Lymphoid Cell Development: A T Cell Perspective. Immunity. 48, 1091–1103 (2018). [DOI] [PubMed] [Google Scholar]
- 16.Constantinides MG, McDonald BD, Verhoef PA, Bendelac A, A committed precursor to innate lymphoid cells. Nature. 508, 397–401 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Juelke K, Romagnani C, Differentiation of human innate lymphoid cells (ILCs). Curr. Opin. Immunol. 38, 75–85 (2016). [DOI] [PubMed] [Google Scholar]
- 18.Tang Q, Ahn Y-O, Southern P, Blazar BR, Miller JS, Verneris MR, Development of IL-22-producing NK lineage cells from umbilical cord blood hematopoietic stem cells in the absence of secondary lymphoid tissue. Blood. 117, 4052–4055 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Montaldo E, Teixeira-Alves LG, Glatzer T, Durek P, Stervbo U, Hamann W, Babic M, Paclik D, Stölzel K, Gröne J, Lozza L, Juelke K, Matzmohr N, Loiacono F, Petronelli F, Huntington ND, Moretta L, Mingari MC, Romagnani C, Human RORγt+CD34+ Cells Are Lineage-Specified Progenitors of Group 3 RORγt+ Innate Lymphoid Cells. Immunity. 41, 988–1000 (2014). [DOI] [PubMed] [Google Scholar]
- 20.Lim AI, Li Y, Lopez-Lastra S, Stadhouders R, Paul F, Casrouge A, Serafini N, Puel A, Bustamante J, Surace L, Masse-Ranson G, David E, Strick-Marchand H, Le Bourhis L, Cocchi R, Topazio D, Graziano P, Muscarella LA, Rogge L, Norel X, Sallenave J-M, Allez M, Graf T, Hendriks RW, Casanova J-L, Amit I, Yssel H, Di Santo JP, Systemic Human ILC Precursors Provide a Substrate for Tissue ILC Differentiation. Cell. 168, 1086–1100.e10 (2017). [DOI] [PubMed] [Google Scholar]
- 21.Renoux VM, Zriwil A, Peitzsch C, Michaëlsson J, Friberg D, Soneji S, Sitnicka E, Identification of a Human Natural Killer Cell Lineage-Restricted Progenitor in Fetal and Adult Tissues. Immunity. 43, 394–407 (2015). [DOI] [PubMed] [Google Scholar]
- 22.Kiel MJ, Yilmaz ÖH, Iwashita T, Yilmaz OH, Terhorst C, Morrison SJ, SLAM Family Receptors Distinguish Hematopoietic Stem and Progenitor Cells and Reveal Endothelial Niches for Stem Cells. Cell. 121, 1109–1121 (2005). [DOI] [PubMed] [Google Scholar]
- 23.Engel P, Eck MJ, Terhorst C, The SAP and SLAM families in immune responses and X-linked lymphoproliferative disease. Nat. Rev. Immunol. 3, 813–821 (2003). [DOI] [PubMed] [Google Scholar]
- 24.Klose CSN, Flach M, Möhle L, Rogell L, Hoyler T, Ebert K, Fabiunke C, Pfeifer D, Sexl V, Fonseca-Pereira D, Domingues RG, Veiga-Fernandes H, Arnold SJ, Busslinger M, Dunay IR, Tanriver Y, Diefenbach A, Differentiation of Type 1 ILCs from a Common Progenitor to All Helper-like Innate Lymphoid Cell Lineages. Cell. 157, 340–356 (2014). [DOI] [PubMed] [Google Scholar]
- 25.Ahn Y, Blazar BR, Miller JS, Verneris MR, Lineage relationships of human interleukin-22-producing CD56+ ROR t+ innate lymphoid cells and conventional natural killer cells. Blood. 121, 2234–2243 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Possot C, Schmutz S, Chea S, Boucontet L, Louise A, Cumano A, Golub R, Notch signaling is necessary for adult, but not fetal, development of RORγt+ innate lymphoid cells. Nat. Immunol. 12, 949–958 (2011). [DOI] [PubMed] [Google Scholar]
- 27.Tufa DM, Yingst AM, Shank T, Shim S, Trahan GD, Lake J, Woods R, Jones KL, Verneris MR, Transient Expression of GATA3 in Hematopoietic Stem Cells Facilitates Helper Innate Lymphoid Cell Differentiation. Front. Immunol. 10 (2019), doi: 10.3389/fimmu.2019.00510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fares I, Chagraoui J, Gareau Y, Gingras S, Ruel R, Mayotte N, Csaszar E, Knapp DJHF, Miller P, Ngom M, Imren S, Roy D-C, Watts KL, Kiem H-P, Herrington R, Iscove NN, Humphries RK, Eaves CJ, Cohen S, Marinier A, Zandstra PW, Sauvageau G, Pyrimidoindole derivatives are agonists of human hematopoietic stem cell self-renewal. Science (80-. ). 345, 1509–1512 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rao SP, Sancho J, Campos-Rivera J, Boutin PM, Severy PB, Weeden T, Shankara S, Roberts BL, Kaplan JM, Human Peripheral Blood Mononuclear Cells Exhibit Heterogeneous CD52 Expression Levels and Show Differential Sensitivity to Alemtuzumab Mediated Cytolysis. PLoS One. 7, e39416 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Buggins AGS, Mufti GJ, Salisbury J, Codd J, Westwood N, Arno M, Fishlock K, Pagliuca A, Devereux S, Peripheral blood but not tissue dendritic cells express CD52 and are depleted by treatment with alemtuzumab. Blood. 100, 1715–20 (2002). [PubMed] [Google Scholar]
- 31.Klose CSN, Kiss EA, Schwierzeck V, Ebert K, Hoyler T, D’Hargues Y, Göppert N, Croxford AL, Waisman A, Tanriver Y, Diefenbach A, A T-bet gradient controls the fate and function of CCR6-RORγt+ innate lymphoid cells. Nature. 494, 261–5 (2013). [DOI] [PubMed] [Google Scholar]
- 32.Moon TC, Campos-Alberto E, Yoshimura T, Bredo G, Rieger AM, Puttagunta L, Barreda DR, Befus AD, Cameron L, Expression of DP2 (CRTh2), a Prostaglandin D2 Receptor, in Human Mast Cells. PLoS One. 9, e108595 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dahlin JS, Malinovschi A, Öhrvik H, Sandelin M, Janson C, Alving K, Hallgren J, Lin− CD34hi CD117int/hi FcεRI+ cells in human blood constitute a rare population of mast cell progenitors. Blood. 127, 383–391 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lugthart G, Melsen JE, Vervat C, van Ostaijen-ten Dam MM, Corver WE, Roelen DL, van Bergen J, van Tol MJD, Lankester AC, Schilham MW, Human Lymphoid Tissues Harbor a Distinct CD69 + CXCR6 + NK Cell Population. J. Immunol. 197, 78–84 (2016). [DOI] [PubMed] [Google Scholar]
- 35.Zhang J, Marotel M, Fauteux-Daniel S, Mathieu A-L, Viel S, Marçais A, Walzer T, T-bet and Eomes govern differentiation and function of mouse and human NK cells and ILC1. Eur. J. Immunol. 48, 738–750 (2018). [DOI] [PubMed] [Google Scholar]
- 36.Cooper MA, Fehniger TA, Caligiuri MA, The biology of human natural killer-cell subsets. Trends Immunol. 22, 633–40 (2001). [DOI] [PubMed] [Google Scholar]
- 37.Farag SS, Caligiuri MA, Human natural killer cell development and biology. Blood Rev. 20, 123–37 (2006). [DOI] [PubMed] [Google Scholar]
- 38.Jacobs R, Hintzen G, Kemper A, Beul K, Kempf S, Behrens G, Sykora KW, Schmidt RE, CD56bright cells differ in their KIR repertoire and cytotoxic features from CD56dim NK cells. Eur. J. Immunol. 31, 3121–7 (2001). [DOI] [PubMed] [Google Scholar]
- 39.Waggoner SN, Kumar V, Evolving role of 2B4/CD244 in T and NK cell responses during virus infection. Front. Immunol. 3 (2012), doi: 10.3389/fimmu.2012.00377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McArdel SL, Terhorst C, Sharpe AH, Roles of CD48 in regulating immunity and tolerance. Clin. Immunol. 164, 10–20 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sivori S, Falco M, Marcenaro E, Parolini S, Biassoni R, Bottino C, Moretta L, Moretta A, Early expression of triggering receptors and regulatory role of 2B4 in human natural killer cell precursors undergoing in vitro differentiation. Proc. Natl. Acad. Sci. 99, 4526–4531 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen L, Youssef Y, Robinson C, Ernst GF, Carson MY, Young KA, Scoville SD, Zhang X, Harris R, Sekhri P, Mansour AG, Chan WK, Nalin AP, Mao HC, Hughes T, Mace EM, Pan Y, Rustagi N, Chatterjee SS, Gunaratne PH, Behbehani GK, Mundy-Bosse BL, Caligiuri MA, Freud AG, CD56 Expression Marks Human Group 2 Innate Lymphoid Cell Divergence from a Shared NK Cell and Group 3 Innate Lymphoid Cell Developmental Pathway. Immunity. 49, 464–476.e4 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Oehler VG, Walter RB, Cummings C, Sala-Torra O, Stirewalt DL, Fang M, Radich JP, Wood B, Blood, in press (available at http://www.bloodjournal.org/content/116/21/2743.abstract).
- 44.Freud AG, Mundy-Bosse BL, Yu J, Caligiuri MA, The Broad Spectrum of Human Natural Killer Cell Diversity. Immunity. 47, 820–833 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Scoville SD, Freud AG, Caligiuri MA, Modeling human natural killer cell development in the era of innate lymphoid cells. Front. Immunol. (2017), doi: 10.3389/fimmu.2017.00360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Daussy C, Faure F, Mayol K, Viel S, Gasteiger G, Charrier E, Bienvenu J, Henry T, Debien E, Hasan UA, Marvel J, Yoh K, Takahashi S, Prinz I, de Bernard S, Buffat L, Walzer T, T-bet and Eomes instruct the development of two distinct natural killer cell lineages in the liver and in the bone marrow. J. Exp. Med. 211, 563–577 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Satoh-Takayama N, Lesjean-Pottier S, Vieira P, Sawa S, Eberl G, Vosshenrich CAJ, Di Santo JP, IL-7 and IL-15 independently program the differentiation of intestinal CD3 − NKp46 + cell subsets from Id2-dependent precursors. J. Exp. Med. 207, 273–280 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Spits H, Bernink JH, Lanier L, NK cells and type 1 innate lymphoid cells: partners in host defense. Nat. Immunol. 17, 758–764 (2016). [DOI] [PubMed] [Google Scholar]
- 49.Melsen JE, Lugthart G, Lankester AC, Schilham MW, Human Circulating and Tissue-Resident CD56bright Natural Killer Cell Populations. Front. Immunol. 7 (2016), doi: 10.3389/fimmu.2016.00262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Crinier A, Milpied P, Escalière B, Piperoglou C, Galluso J, Balsamo A, Spinelli L, Cervera-Marzal I, Ebbo M, Girard-Madoux M, Jaeger S, Bollon E, Hamed S, Hardwigsen J, Ugolini S, Vély F, Narni-Mancinelli E, Vivier E, High-Dimensional Single-Cell Analysis Identifies Organ-Specific Signatures and Conserved NK Cell Subsets in Humans and Mice. Immunity. 49, 971–986.e5 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.CSA, Ethiopa Agricultural Sample Survey 2010/2011 (2003 E.C.) (September - January 2010/11) Volume VII, Report on Crop and Livestock Product Utilization (Private Peasant Holdings, Meher Season) | HarvestChoice (2011), p. 190. [Google Scholar]
- 52.Corne JM, Green S, Sanderson G, Caul EO, Johnston SL, A multiplex RT-PCR for the detection of parainfluenza viruses 1–3 in clinical samples. J. Virol. Methods. 82, 9–18 (1999). [DOI] [PubMed] [Google Scholar]
- 53.Boles NC, Lin KK, Lukov GL, Bowman TV, Baldridge MT, Goodell MA, CD48 on hematopoietic progenitors regulates stem cells and suppresses tumor formation. Blood. 118, 80–87 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Vaidya SV, Stepp SE, McNerney ME, Lee J-K, Bennett M, Lee K-M, Stewart CL, Kumar V, Mathew PA, J. Immunol., in press, doi: 10.4049/jimmunol.174.2.800. [DOI] [Google Scholar]
- 55.Grzywacz B, Kataria N, Sikora M, Oostendorp RA, Dzierzak EA, Blazar BR, Miller JS, Verneris MR, Coordinated acquisition of inhibitory and activating receptors and functional properties by developing human natural killer cells. Blood (2006), doi: 10.1182/blood-2006-04-020198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Butler A, Hoffman P, Smibert P, Papalexi E, Satija R, Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 36, 411–420 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Angelos MG, Ruh PN, Webber BR, Blum RH, Ryan CD, Bendzick L, Shim S, Yingst AM, Tufa DM, Verneris MR, Kaufman DS, Aryl hydrocarbon receptor inhibition promotes hematolymphoid development from human pluripotent stem cells. Blood. 129, 3428–3439 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Representative ILC staining strategy.
Fig. S2. Generation of ILCs from CD34+α4β7+ HSC Progenitors.
Fig. S3. Surface expression of CD48, CD52, CD117 and α4β7 by peripheral blood- and tonsil-derived CD34+ HSCs.
Fig. S4. Gene expression profiling in four subsets of hematopoietic progenitors.
Fig. S5. RNA-seq and single cell culture demonstrated in vitro differentiation of NK cells and ILCs from CD34+ HSCs.
Fig. S6. Similar expression of NK cell-associated markers in CD34+α4β7+CD48+- and CD34+α4β7+CD48--derived NK cells.
Fig. S7. Differentially activated pathways in two distinct ILC3 subsets.
Fig. S8. The effect of CD244 signaling in the development of ILCs.
Table S1. Top 10 differentially expressed genes for each cluster within the terminally differentiated CD34+ HSCs-derived ILCs.
Table S2. Table showing differentially expressed genes for clusters 3-6 that are common to both terminally differentiated cells and CD34+α4β7+ hematopoietic progenitors.
Table S3. Description of samples used for scRNA-seq analysis.
Table S4. Lists and description of primer assays used in the study.
Table S5. Lists and description of antibodies used in the study.
Data file S1: Raw data
Data Availability Statement
All data used to draw conclusions in this article are present in the main paper and/or the supplementary materials. The scRNA-seq datasets were deposited in the GSE accession: GSE160009.