

Abstract
SARS-CoV-2 has caused millions of infections and hundreds of thousands of deaths globally. Presently, no cure for SARS-CoV-2 infection is available; thus, all hands are on deck for new drug discovery. Although, several studies have reported the potentials of some already approved drugs for the treatment of COVID-19. This study attempted to compare the potency and safety of some these trial drugs via in silico methods. The binding affinity and interactions of the trial drugs with proteins involved in viral polyprotein processing (Papain like protease (PLpro) and Chymotrypsin like-protease (3-CLpro), viral replication (RNA dependent RNA polymerase (RdRp)) and host protease were studied in this work. The pharmacokinetic properties and toxicity potentials of the trial drugs were also predicted using vNN Web Server for ADMET Predictions. From the results, Merimepodib and Dexamethaxone demonstrated the most significant inhibitory potential against the PLpro. The binding affinity (∆G°) for merimepodib was − 7.2 kcal/mol while the inhibition constant was 6.3 µM. The binding affinity of the inhibitors for CLpro ranged from − 5.6 to − 9.5 kcal/mol. whereas Lopinavir (− 7.7 kcal/mol) exhibited the strongest affinity for RdRp. Overall, our results showed that all the ligands have a higher affinity for the 3-Chymotrypsin like protease than the other proteins (PLpro, RdRp, and Host protease). Among these compounds lopinavir, merimepodib and dexamethasone could be inhibitors with potentials for the treatment of SARS-CoV-2. However, the only dexamethasone has attractive pharmacokinetic and toxicity properties probable for drug development; therefore, our study provides a basis for developing effective drugs targeting a specific protein in the SARS-CoV-2 life cycle.
Supplementary Information
The online version contains supplementary material available at 10.1007/s40203-021-00105-x.
Keywords: SARS-COV-2, Trial drugs, CLpro, RdRp, Safety
Introduction
The novel SAR-COV-2 outbreak has resulted in almost half a million death across the globe (Qamar et al. 2020). Between December 2019 and June 2020, more than 9473,215 persons have been infected and 484,249 number of death has been reported globally (WHO 2020). In sub-Sahara Africa, Nigeria in particular, thirty-six states including the Federal Capital Territory (FCT) have recorded cases of infected patients, with more than 22,614 already infected and 549 death recorded (WHO 2020). The report has it that three categories of people i.e. children, aged, and those with underlying medical history/low immunity (Shyamala et al. 2020) are most vulnerable to the disease. Consequently, many advanced nations of the world are now epicenters for the disease with no current approved cure (Shyamala et al. 2020). The current management strategy only involves the provision of symptomatic relief to patients (Shyamala et al. 2020).
The COVID-19 virus which causes severe acute respiratory syndrome (SARS) in humans is a positive-strand RNA virus that shares 80% homology with SAR-CoV and about 96% identity with bat coronavirus Bat CoV Rat G13 (Zhou et al. 2020). The genomic structure of the virus is a 5'-leader-UTR replicase-S(Spike)-E(Envelop)-M(Membrane)-N(Nucleocapsid)-3'ÚTR poly (A) tail, with the accessory genes responsible for viral pathogenesis interspersed within the structural genes at the 3' end of the genome (Fehr and Perlman 2015; Zhao et al. 2012). The spike protein is the point of attachment to the Angiotensin Converting Enzyme (ACE) receptor and it mobilizes the subsequent entry into host cells (Wu et al. 2020). The M and E proteins on the other hand function in the viral assembly while the N protein is responsible for viral RNA synthesis (Fehr and Perlman 2015; Song et al. 2019). With the knowledge of the viral genomic structure and the behavior in the human host, studies on drug development could focus on the design of therapies that could either be targeted to act on the host cells or directly on the virus itself (Wu et al. 2020). For instance, therapies could be targeted to block the pathways of human cells required for viral entry/or replication (Agrawal et al. 2015; Han et al. 2006; Li et al. 2003) or prevent viral replication through inhibition of critical viral enzymes (3-chymotrypsin like protease or papain-like proteases). Furthermore, inhibition of viral RNA synthesis (Inhibition of RNA dependent RNA polymerase and Helicase) and/or block the binding to host cells and self-assembly process (Wu et al. 2020). Strategy in the current fight against coronavirus involves testing existing broad-spectrum of already approved and trial drugs. Among the drugs already under investigation is Remdesivir (an adenosine analog), Ribavirin, and Favipiravir that abrupt viral RNA replication as a mechanism of antiviral activity (Hentig 2020). Other drugs include Lopinavir and ritonavir (HIV protease inhibitor) and Hydroxychloroquine (an anti-malarial) (Dayer et al. 2017; Hentig 2020; Shah et al. 2020). Figure 1 showed 2-D structures of Dexamethasone and other trial drugs of interest. Dexamethasone, a corticosteroid was recently welcomed by the WHO for the treatment of COVID-19 after the result of a trial from the United Kingdom showed the drug improved survival rate of patients critically ill with COVID-19 (Khan and Htar 2020; WHO 2020b). In this regard, our study focuses on computational investigations on the potency of these trial drugs against different target protein in the SARS-CoV-2 virus by obstructing the replication event of the virus life cycle and/or by prevention of viral RNA synthesis RNA-dependent RNA polymerase (RdRp)), and to evaluate their safety via ADMET prediction. Although a recent report identified the strong affinity of some of the trial drugs for the ACE, CLpro and spike glycoprotein (Adelusi et al. 2021), in this study, we focused on the inhibition of some accessory proteins vital for viral replication and survival in the host cell. We also examined the most suitable targets of each ligand studied in the viral protein.
Fig. 1.

2-D structures of the trial drugs
Methods
Software and database
Autodock tools 1.5.6, Autodock vina and discovery studio visualizer 2019, RCSB protein data bank database, Pubchem database, cactus online smiles translator and vNN Web Server for ADMET Predictions.
Preparation of protein and ligands
The candidate proteins (SARS-CoV-2 Papain like protease (PLpro) (PDB: 6wx4), Chymotrypsin-like main protease (3CLpro) (PDB:6YB7), SARS-CoV nsp 12 (PDB: 6nus), Host cell protease (TMPRSS1) (PDB:5ce1) were downloaded from the RCSB protein data bank database. While The SDS format of nine potential inhibitors namely: Remdesivir, Hydroxychloroquine, Dexamethasone, EIDD-2801, Favipiravir, Lopinavir, Merimepodib, and Ritonavir was obtained from the PubChem database and taken to cactus online smiles translator for PDB ligand download. Before docking analysis, interacting ligands, and water molecules were removed from the protein, thereafter saved in PDB format for docking analysis.
Docking studies
The docking analysis was performed using Autodock tools 1.5.6. On the Autodock tool, polar-H-atoms were first added to the proteins followed by Gasteiger charges calculation. The protein file was saved as pdbqt file and the grid dimensions were set. Discovery studio visualizer 2019 was used to visualize the interactions between the ligands and the protein. The binding affinity in the form of ∆G° was calculated using Autodock vina and ligands, which exhibits more negative free energy of binding and low Ki was considered more potent.
ADMET profiling
The canonical smiles of the potential inhibitors were obtained from the PubChem database. The ADMET property was predicted using vNN Web Server for ADMET Predictions. The major parameters assessed included; absorption, distribution, metabolism, excretion, and toxicity (ADMET) profile. The detailed result obtained is presented in Table 5, while the schematic workflow of this study is showed in Fig. 2.
Table 5.
ADMET Profile of the screened compounds
| Query | DILI | Cyto- toxicity | HLM | Cyp1A2 Inhibitor | Cyp3A4 Inhibitor | Cyp2D6 Inhibitor | Cyp2C9 Inhibitor | Cyp2C19 Inhibitor | BBB | P-gp Inhibitor | P-gp Substrate | hERG Blocker | MMP | AMES | MRTD (mg/day) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Remdesivir | Yes | Yes | Yes | No | No | No | No | No | No | Yes | Yes | No | No | No | 229 |
| Hydroxychloroquine | No | No | Yes | No | No | No | No | No | Yes | No | Yes | No | No | Yes | 478 |
| Dexamethasone | No | No | Yes | No | No | No | No | No | No | No | Yes | No | No | No | 8.4 |
| EIDD-2801 | Yes | No | Yes | No | No | No | No | No | No | No | No | No | No | No | 170 |
| Favipiravir | Yes | No | Yes | No | No | No | No | No | No | Yes | Yes | No | Yes | No | 327 |
| Lopinavir | Yes | No | Yes | No | Yes | No | No | No | No | Yes | Yes | Yes | No | Yes | 224 |
| Merimepodib | Yes | Yes | Yes | No | Yes | No | No | No | No | Yes | Yes | No | No | No | 367 |
| Ritonavir | Yes | Yes | Yes | No | Yes | No | No | No | No | Yes | Yes | No | No | No | 367 |
Fig. 2.
Schematic workflow of the study
Results and discussion
Approach targeting the viral proteases
In order to validate the docking protocol, two methods were used, viz, ligands were re-docked into the downloaded protein containing the full-length structure. The same protocol including the grid parameters was unchanged in the process. The docking pose was the same, and the method was accepted as correct. Also, the root mean square deviation of the ligands before and after docking determined using discovery studio was close to 1 which shows that the docking protocol was correct.
An important approach in the current research for the COVID-19 remedy involves inhibiting the production of nonstructural viral components to hamper the replication event of the causative virus life cycle (Kumar et al. 2020). Inhibition of the proteinase enzymes that process the viral polyprotein and virus maturation has been evaluated as an approach towards SARS treatment (Rabaan et al. 2017).
Chymotrypsin-like protease (3CLpro) or main protease (Mpro)-nsP5 and papain-like protease (PLpro)-nsP3, are the two main proteases involved in this process (Kumar et al. 2020; de Wit et al. 2016; Lee et al. 2005). In this study, we analyzed the potency of the trial drugs against the two proteases. Tables 1 and 2 showed the binding affinity (∆G°) along with the residue interaction of the trail drugs with SARS-CoV-2 PLpro and CLpro respectively. From the results, Merimepodib and Dexamethaxone demonstrated the most significant inhibitory potential against the PLpro. The binding affinity (∆G°) for merimepodib was − 7.2 kcal/mol while the inhibition constant was 6.3 µM. Merimepodib formed a hydrogen bond with Asn 146, Ala 131, and Asn 13, other hydrophobic interactions were formed with Ala 145, Ala 135, Lys 94, and Asp 37. The result of this study agrees with that of Bagherzadeh et al. (2020), who reported a strong inhibitory potential of merimepodib against SARS-CoV-2-PLpro. Dexamethasone with a binding affinity of − 7.1 kcal/mol against PLpro interacted via hydrogen bond with Tyr 95 and with Asn 146, Ala 145, Ala 144, Glu 143, Trp 93, Gly 142, Lys 94 via hydrophobic interaction (Figure S1). Dexamethasone, which was recently reported with outstanding potential after a trial result, has proven successful in the inhibition of viral proteases such as in the cases of the human immunodeficiency virus (HIV) and hepatitis C virus (HCV). Therefore, it could be a potential drug for the treatment of SARS-CoV-2 in line with earlier suggestions by Ghosh et al., (2016). Ritonavir (− 6.4 kcal/mol) and EIDD-2801 (− 6.1 kcal/mol) exhibited inhibition potential against PLpro somewhat greater than Remdesivir (− 5.9 kcal/mol), Lopinavir (− 5.6 kcal/mol), Favipiravir (− 5.5 kcal/mol) and Hydroxychloroquine (− 4.7 kcal/mol). However, the calculated binding affinity for all the inhibitors was negative, an indication that the molecules are potent and could serve as a starting point for developing effective drugs targeting SARS-CoV-2-PLpro.
Table 1.
Docking score and the interactions of the inhibitors with PLpro
| Ligands | Binding Affinity ΔG (Kcal/mol) | Inhibition Constant Ki (µM) 10–6 | Interacting Amino acids | Bond type |
|---|---|---|---|---|
| Merimepodib | − 7.2 | 6.3 | ASP 37, ALA 145, ALA 144, GLU 143, GLY 142, TRP 93, TYR 95, LYS 94, LYS 91, LEU 87, ASN 146, ARG 138, ALA 135, ASP 134, LEU 150, ALA 131, TYR 71, TYR 83, TYR 56, ASN 13 | Van der waals, Conventional Hydrogen Bond, Carbon Hydrogen Bond, Pi-Anion, Alkyl, Pi-Alkyl |
| Dexamethasone | − 7.1 | 7.5 | ASN 146, LEU 87, ALA 145, ALA 144, GLU 143, TRP 93, PRO 96, TYR 95, GLY 142, LYS 94, LYS 91 | Van der waals, Conventional Hydrogen Bond, Unfavorable Donor-Donor, Halogen (Fluorine), Pi-Alkyl, Alkyl |
| Ritonavir | − 6.4 | 23.9 | LYS 306, TYR 305, THR 257, TYR 251, GLY 256, PHE 258, THR 259, TYR 213, GLU 214, LYS 217, THR 313, TYR 310, LYS 218, GLU 307, ASN 308, SER 212 | Van der waals, Conventional Hydrogen Bond, Carbon Hydrogen Bond, Pi-Anion, Amide-Pi Stacked, Alkyl, Pi-Alkyl |
| EIDD-2801 | − 6.1 | 39.3 | LYS 94, TYR 95, THR 90, TRP 93, LYS 91, GLU 143, ALA 145, ALA 144, ASN 146, ARG 138, ASN 146, GLY 142, PRO 96 | Van der waals, Conventional Hydrogen Bond, Unfavorable Acceptor-Acceptor, Pi-Anion, Pi-Alkyl |
| Remdesvir | − 5.9 | 54.8 | GLN 174, CYS 155, HIS 175, TYR 154, ALA 153, PHE 79, ARG 82, ASN 156, TYR 171, THR 74, THR 75, ASP 76, PRO 77 | Van der waals, Attractive Charge, Conventional Hydrogen bond, Carbon hydrogen Bond, Pi-Pi T-shaped |
| Lopinavir | − 5.6 | 90.3 | VA 188, TYR 233, THR 231, PRO 316, GLU 318, LEU 317, LYS 315, THR 313, ILE 314, LYS 218, GLY 219, GLN 215 | Van der waals, Conventional Hydrogen Bond, Carbon Hydrogen Bond, Pi-Sigma, Alkyl, Pi-Alkyl |
| Favipiravir | − 5.5 | 107 | LYS 254, TYR 251, GLU 252 | Conventional Hydrogen Bond, Pi-Pi T-shaped, Pi-Alkyl |
| Hydroxychloroquine | − 4.7 | 403 | SER 170, GLU 203, LEU 199. TYR 207, LYS 232, MET 208, MET 206, ARG 166, VAL 202 | Van der waals, Conventional Hydrogen Bond, Pi-Sulfur, Pi-Alkyl, Alkyl |
Table 2.
Docking score and the interactions of the Inhibitors with 3CLpro
| Ligands | Binding Affinity ΔG (Kcal/mol) | Inhibition Constant Ki (µM) 10–6 | Interacting Amino acids | Bond Type |
|---|---|---|---|---|
| Lopinavir | − 9.5 | 0.14 | THR 196, ASP 197, THR 198, ASN 238, LYS 236, TYR 237, LEU 272, LEU 271, GLY 275, LEU 287, MET 276, ARG 131, TYR 239, VAL 204, ASP 289, THR 199, LEU 286, GLU 290, GLU 288, LYS 137 | Van der waals, Conventional Hydrogen Bond, Carbon Hydrogen Bond, Pi-Sigma, Pi-Sulfur, Pi-Alkyl |
| Dexamethasone | − 9.4 | 0.16 | TYR 237, THR 199, GLU 290, ASP 289, ARG 131, ASP 197, LYS 137, GLU 288, LEU 286, LEU 287, TYR 239, LEU 272 | Van der waals, Conventional hydrogen bond, Alkyl |
| Merimepodib | − 8.8 | 0.44 | ASN 238, LYS 137, ASP 289, ARG 131, GLU 288, GLY 275, LEU 272, TYR 237, LEU 286, LEU 287, TYR 239, VAL 204, THR 199, ASP 197, THR 198 | Van der waals, Conventional Hydrogen Bond, Carbon Hydrogen Bond, Pi-Anion, Pi-Cation, Pi-Alkyl |
| Remdesvir | − 8.2 | 1.2 | LEU 272, MET 276, LEU 287, THR 196, TYR 239, LEU 286, GLU 288, GLU 290, ASP 289, ARG 131, THR 199, TYR 237, THR 198, ASP 197, ASN 238 | Van der waals, Conventional Hydrogen Bond, Pi-Sigma, Pi-Sulfur, Pi-Alkyl |
| Ritonavir | − 8.1 | 1.4 | LEU 286, LEU 287, MET 276, LYS 137, ARG 131, THR 199 | Van der waals, Conventional Hydrogen Bond, Carbon Hydrogen Bond, Pi-Sigma, Alkyl, Pi-Alkyl |
| EIDD-2801 | − 7.8 | 2.3 | LEU 272, THR 199, LEU 287, THR 198, ARG 131, ARG 131, GLU 288, LYS 5, GLU 290, ASP 289, LYS 137, LEU 286, TYR 239 | Van der waals, Conventional Hydrogen Bond, Pi-Anion |
| Hydroxychloroquine | − 6.9 | 10.4 | MET 276, GLY 275, LEU 271, LEU 272, LEU 286, ARG 131, ASP 289, LYS 137, GLU 290, LYS 5, GLU 288, THR 199, LEU 287, TYR 239 | Van der waals, Conventional Hydrogen Bond, Pi-Cation, Pi-Anion |
| Favipiravir | − 5.6 | 90.3 | LEU 271, ASN 277, ASN 274, GLY 275, ARG 279, LEU 220, ASN 221, TRP 218, PHE 219, GLU 270, SER 267 | Van der waals, Conventional Hydrogen Bond, Halogen (Fluorine), Pi-Pi T-shaped, Pi-Alkyl |
The inhibitory potentials of the compounds against the SARS-CoV-2-CLpro as revealed in Table 2 showed the order of potency of all the trial inhibitors. The binding affinity for the inhibitors ranged from − 5.6 to − 9.5 kcal/mol. Lopinavir (− 9.5 kcal/mol) had greater affinity for CLpro, followed by Dexamethaxone (− 9.4 kcal/mol) > Merimepodib (− 8.8 kcal/mol) > Remdesivir (− 8.2 kcal/mol) > Ritonavir (− 8.1 kcal/mol) > EIDD-2801 (− 7.8 kcal/mol) > Hydroxychloroquine (− 6.9 kcal/mol) > Favipiravir (− 5.6 kcal/mol). The amino acid residues within the catalytic traid of Mpro include His 41, Cys 145, Glu 166, Ala 285, and Leu 286. These ligands/compounds exhibited one or more hydrogen and hydrophobic interactions with residues within the binding pocket that were similarly reported for Nelfinavir (Chandel et al. 2020). According to Kumar et al. (2020), Lopinavir-Ritonavir (− 10.6 kcal/mol), exhibited higher binding affinity for CLpro than the other drugs viz; Tipranavir (− 8.7 kcal/mol), Raltegravir (− 8.3 kcal/mol), α-ketoamide13b (− 8.3 kcal/mol), Nelfinavir (− 8.2 kcal/mol), Dolutegravir (− 8.1 kcal/mol), Tenofovir-disoproxil (− 8.1 kcal/mol), Baloxavir-marboxil (− 8.1 kcal/mol), Letermovir (− 8.0 kcal/mol), and Maraviroc (− 8.0 kcal/mol) (See Figure S2 for CLpro interactions with the drugs).
Both Lopinavir and Ritonavir are HIV-AIDS protease inhibitors and thus help decrease the amount of HIV in the body by improving the body's natural immune system (Doyon et al. 2005). A previous study conducted by Mothay and Ramesh (2020) showed that Remdesivir (− 5.51 Kal/mol) exhibited lower binding affinity than Lopinavir (− 6.08 kcal/mol) and Ritonavir (− 5.96 kcal/mol). Similarly, dexamethasone exhibited a stronger affinity for CLpro than Remdesivir and was said to have strong inhibition potential (Khan and Htar 2020). EIDD-2801, a similar drug to remdesivir, works by mimicking ribonucleosides, the primary components of RNA molecules that cause debilitating errors when the drugs are incorporated into viral RNA during replication thereby preventing the spread of the virus. The drug has proven effective for the treatment of COVID-19 patients and other serious coronavirus infection (Sheahan et al. 2020). In this study, EIDD-2801 exhibited a stronger affinity for CLpro than Hydroxychloroquine and Favipiravir. Among the compounds, dexamethasone, Merimepodib, EIDD-2801, and ritonavir have a considerable affinity for SARS-CoV-2-PLpro and CLpro and therefore can serve as a potent inhibitor of both proteases.
Approach targeting viral RNA synthesis
In our attempt to study the potentials of the inhibitors to block viral RNA synthesis, we docked the ligands against the RNA-dependent RNA polymerase (RdRp) which is a vital enzyme for coronavirus replication/transcription. It is, therefore, a significant approach for the development of potent inhibitor. In this study, all the inhibitors interacted with one or more residues within the polymerase domain. The polymerase domain is comprised of a fingers domain (a.a. 398–581, 628–687), a palm domain (a.a. 582–627, 688–815), and a thumb domain (a.a. 816–919) (Kirchdoerfer and Ward 2019). Again, Lopinavir (− 7.7 kcal/mol) exhibited the strongest affinity for RdRp. Dexamethasone (− 7.3 kcal/mol), Remdesivir (− 7.2 kcal/mol), Merimepodib (− 7.2 kcal/mol), Ritonavir (7.0 kcal/mol), EIDD-2801 (− 6.2 kcal/mol), Favipiravir (− 5.8 kcal/mol) and Hydroxychloroquine (− 4.5 kcal/mol) also exhibited negative binding affinity (Table 3). Only a few studies in the literature have reported the interaction of these compounds with the RdRp. Although remdesivir as an ATP analog is a potent inhibitor of RNA-dependent RNA polymerases (RdRps). It can terminate RNA synthesis by replacing ATP during polymerization and thus known as chain terminator drug (Wu et al. 2020). Lopinavir had a similar binding position as remdesivir, it formed a hydrogen bond with Val 330 but alkyl and, Pi-alkyl interaction with Val 398, TYR 273, ALA 379, ALA 383, ALA 382, PHE 396 (see Figure S3). Wu et al. (2020) as well reported the inhibitory potential of remdesivir, according to the study; it binds to the bottom RNA template channel, a position that is for the acceptor template nucleotide.
Table 3.
Docking score and the interactions of the Inhibitors with RdRp
| Ligands | Binding Affinity ΔG (Kcal/mol) | Inhibition Constant Ki (µM) 10–6 | Interacting Amino acids | Bond Type |
|---|---|---|---|---|
| Lopinavir | − 7.7 | 2.8 | SER 397, PHE 396, PRO 378, LEU 270, MET 666, THR 324, PHE 326, ALA 382, ALA 379, VAL 398, PRO 328, LEU 271, LEU 329, VAL 330, ALA 383, TYR 273, GLY 327, VAL 341, LEU 387 | Van der waals, Conventional Hydrogen Bond, Carbon Hydrogen Bond, Unfavorable Acceptor-Acceptor, Alkyl, Pi-Alkyl |
| Dexamethasone | − 7.3 | 5.4 | THR 710, GLY 712, SER 709, TYR 129, LEU 240, ALA 125 | Conventional Hydrogen Bond, Unfavorable Donor-Donor, Alkyl, Pi-Alkyl |
| Remdesvir | − 7.2 | 6.3 | TYR 273, LEU 329, LEU 271, ALA 399, SER 325, LEU 387, ALA 383, PHE 396, ALA 379, ALA 382, VAL 398, PRO 378, PHE 326, VAL 330, THR 324, PRO 328 | Van der waals, Conventional Hydrogen Bond, Pi-Pi Stacked, Alkyl, Pi-Alkyl |
| Merimepodib | − 7.2 | 6.3 | ALA 125, TYR 129, GLN 724, HIS 725, TYR 728, GLY 712, THR 710, LEU 708, ASN 705, SER 709, TYR 732, LEU 240, HIS 133, VAL 128, ARG 132 | Van der waals, Conventional Hydrogen Bond, Carbon Hydrogen Bond, Pi-Pi Stacked, Pi-Pi T-shaped, Pi -Alkyl |
| Ritonavir | − 7.0 | 8.8 | PRO 461, PRO 677, ASN 459, LEU 460, PHE 326, ARG 349, PHE 321, THR 319, SER 318, SER 255, ARG 249, ALA 252, HIS 256, LEU 261, LEU 265, VAL 320, PRO 322, TRP 268, THR 394, CYS 395, PHE 396, PRO 323 | Van der waals, Conventional Hydrogen Bond, Carbon Hydrogen Bond, Pi-Cation, Alkyl, Pi-Alkyl, Pi-Sulfur, Pi-Pi T-shaped |
| EIDD-2801 | − 6.2 | 33.3 | ARG 836, ALA 797, ASP 618, LYS 798, ASP 760, SER 759, SER 814, ASP 761, VYS 813, PHE 812, TRP 617, GLU 811, HIS 810 | Van der waals, Conventional Hydrogen Bond, Carbon Hydrogen Bond, Pi-Anion |
| Favipiravir | − 5.8 | 64.8 | LYS 798, ASP 618, ALA 762, PHE 812, ASP 761, GLY 616, TRP 617, TRP 800, GLU 811, HIS 810 | Van der waals, Conventional Hydrogen Bond, Carbon Hydrogen Bond, Halogen (Floorine), Unfavorable Donor-Donor |
| Hydroxychloroquine | − 4.5 | 562.0 | LEU 514, TRP 509, LEU 371, PHE 368, ALA 375, PHE 506, LEU 372, TYR 515, LEU 514 | Van der waals, Pi-Pi Stacked, Pi-alkyl |
It is interesting to note that all the ligands showed a preference in their interaction with the targeted proteins. The ligands demonstrated a more negative binding affinity for CLpo than RdRp and a stronger affinity for RdRp than PLpro (Fig. 3). Hydroxychloroquine (− 4.7, − 6.9, − 4.5 kcal/mol), Remdesvir (− 5.9, − 8.2, − 7.2 kcal/mol), Dexamethasone (− 7.1, − 9.4, − 7.3 kcal/mol), EIDD-2801 (− 6.1, − 7.8, − 6.2 kcal/mol), Favipiravir (− 5.5, − 5.6, − 5.8 kcal/mol), Lopinavir (− 5.6, − 9.5, − 7.7 kcal/mol), Merimepodib (− 7.2, − 8.8, − 7.2 kcal/mol), Ritonavir (− 6.4, − 8.1, − 7.0 kcal/mol) showed affinity for PLpro, CLpro and RdRp respectively.
Fig. 3.
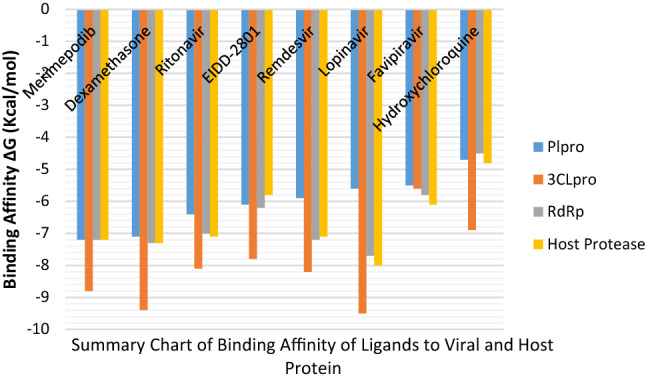
Clustered bar graph of the Binding Affinities of Ligands against the Viral and Host Protein
Approach targeting Host Cell Protease
The viral spike is the main protein that interacts with the host by binding to the Angiotensin Converting Enzyme (ACE) receptor, which upon cleavage by the host cell protease mediates the virus invasion, and determines viral tissue or host reaction (Millet and Whittaker 2015; Wu et al. 2020). Available studies have shown that inhibiting the host serine protease can prevent coronaviruses entry into the cell (Glowacka et al. 2011). Therefore, rendering important therapeutic target against coronavirus. Our study on the interactions that existed between the ligands and the host serine protease gives insight into the potency of each compound as a potential target inhibitor of host cell-mediated viral entry/or replication. Lopinavir again exhibited the most significant affinity for the host protease. The order of affinity is Lopinavir (− 8.0 kcal.mol) > Dexamethasone (− 7.3 kcal/mol) > Merimepodib (− 7.2 kcal/mol) > Remdesivir (− 7.1 kcal/mol) = Ritonavir (− 7.1 kcal/mol) > Favipiravir (− 6.1 kcal/mol) > EIDD-2801 (− 5.8 kcal/mol) > Hydroxychloroquine (− 4.8 kcal/mol) (Table 4).
Table 4.
Docking score and the interactions of the inhibitors with Host cell protease
| Ligands | Binding Affinity ΔG (Kcal/mol) | Inhibition Constant Ki (µM) 10–6 |
Interacting Amino acids | Bond Type |
|---|---|---|---|---|
| Lopinavir | − 8.0 | 1.7 | ALA 281, GLY 282, ARG 130, ALA 284, GLN 283, ILE 363, TYR 52, PRO 50, TRP 73, PRO 53, LEU 51, PHE 66, VAL 65, ILE 135, LYS 68, LEU 132 | Van der waals, Conventional Hydrogen Bond, Carbon Hydrogen Bond, Pi-Sigma, P-i-Pi Stacked, Alkyl, Pi-Alkyl |
| Dexamethasone | − 7.3 | 5.4 | GLY 378, HIS 203, SER 353, SER 376 | Conventional Hydrogen Bond, Pi-Alkyl |
| Merimepodib | − 7.2 | 6.3 | TYR 52, PHE 66, LEU 51, PRO 53, PRO 50, ILE 363 | Carbon Hydrogen Bond, Pi-Sigma, Pi-Pi Stacked, Alkyl, Pi-Alkyl |
| Remdesivir | − 7.1 | 7.5 | SER 353, SER 376, VAL 375, GLY 380, GLY 378, CYS 381, ALA 348, CYS 349, TRP 377, GLN 350, TYR 301, ASN 254, SER 251, ARG 208, PRO 245, ASN 250, PRO 206, TYR 243, HIS 203, GLY 351 | Van der waals, Conventional Hydrogen Bond, Unfavorable Positive-Positive Amide-Pi Stacked, Pi-Alkyl |
| Ritonavir | − 7.1 | 7.5 | HIS 186, GLN 350, HIS 203, ARG 208, PRO 206, PHE 246, ASN 250, PRO 245, TYR 243, TYR 301, ASN 254, SER 353, TRP 377, SER 376, GLY 378, GLY 380, VAL 375, CYS 381, CYS 349, ALA 348, ASN 298, GLY 351, LEU 187, GLN 305 | Van der waals, Conventional Hydrogen Bond, Carbon Hydrogen Bond, Unfavorable Donor-Donor, Pi-Cation, Pi-Pi T-shaped, Alkyl, Pi-Alkyl |
| Favipiravir | − 6.1 | 39.3 | CYS 349, TRP 377, ALA 348, CYS 381, ALA 348, GLY 380, GLY 378, GLN 350, SER 376, HIS 203, GLY 351, SER 353, VAL 375, GLY 388, VAL 389, ASP 347 | Van der waals, Conventional Hydrogen Bond, Carbon Hydrogen Bond, Unfavorable Donor-Donor, Amide-Pi Stacked, Pi-Alkyl |
| EIDD-2801 | − 5.8 | 64.8 | HIS 203, GLY 351, CYS 188, LEU 187, SER 353, GLN 350, VAL 375, GLY 388, VAL 389, ALA 348, CYS 349, GLY 380, CYS 381, GLY 378, TRP 377, SER 376 | Van der waals, Conventional Hydrogen Bond, Carbon Hydrogen Bond |
| Hydroxychloroquine | − 4.8 | 342.0 | GLN 305, ASN 298, GLN 350, TYR 301, TYR 302, ASN 209, TYR 182, ALA 185, LEU 185, HIS 186 | Van der waals, Convnetional Hydrogen Bond, Carbon Hydrogen Bond, Unfavorable Acceptor-Acceptor, Pi-Sigma, Pi-Alkyl |
One or more non-bond interaction (hydrogen and hydrophobic interactions) stabilized the interactions of Dexamethasone, Remdesivir, Favipiravir, Ritonavir, and EIDD-2801 with the residues within the catalytic triad (His 203 and Ser 353) as shown in Figure S4. It is obvious from our result that Merimepodib, lopinavir, and dexamethasone are outstanding in their potentials as an inhibitor of viral replication and invasion of the host cell.
Pharmacokinetics and toxicity profile of inhibitors
In Table 5, the ADMET prediction results of the inhibitors included the toxicity and pharmacokinetic properties such as absorption, distribution, and metabolism. All the inhibitors were stable to the human liver microsomal (HLM); for a drug to achieve its therapeutic function, it must not be metabolized quickly and so must be stable to the HLM. Only Merimepodib, lopinavir, and Ritonavir were inhibitors to Cyp 3A4, therefore may cause an elevation in the concentration of the corresponding drug substrate viz-a-viz leading to drug overdose (Murray 2006). Hydroxychloroquine and Dexamethasone were non-inhibitor to Pg while EIDD-2801 was neither a substrate nor an inhibitor to P-glycoprotein. P-glycoprotein (Pgp) function to extract foreign substances from the cell (Ambudkar et al. 2003), an event that increases the efflux of chemotherapeutic agents from the cell thus reducing the effective intracellular concentrations of such agents (Borst and Elferink 2002). Toxicity as a fundamental parameter for the continuation of research in drug design and development (Hage-Melim et al. 2020; Muster et al. 2008), was further assessed in this study. All the drugs except dexamethasone have one or more toxic potential. Only lopinavir showed cardiotoxic potential (hERG), while lopinavir and hydroxychloroquine have Ames toxicity, and remdesivir, merimepodib, and ritonavir are cytotoxic potentials. Despite the good pharmacokinetic property prediction for EIDD-2801, it has the potential to induce liver injury. The maximum recommended therapeutic dose (MRTD) for each compound was predicted and recorded as mg/day for an average adult with a bodyweight of 60 kg (Schyman et al. 2017). Although both lopinavir and dexamethasone were outstanding in their potentials to inhibit the key proteins involved in the various stages of the viral life cycle, it was only dexamethasone that was predicted to be safe.
Conclusion
In silico methods were used to investigate the potency of eight popular trial drugs against different target protein in the SARS-COV-2 by obstructing the replication event of the virus life cycle and/or by prevention of viral RNA synthesis RNA-dependent RNA polymerase (RdRp)), and to evaluate their safety via ADMET prediction.
The results showed that Merimepodib and Dexamethaxone are potential SARS-CoV-2 PLpro inhibitors while five drugs namely Dexamethaxone, Merimepodib, Remdesivir, Ritonavir, and EIDD-2801 could be effective inhibitors for SARS-CoV-2-CLpro with Dexamethaxone and Merimepodib showing outstanding properties. In addition, Lopinavir, Dexamethasone, Remdesivir, Merimepodib, and Ritonavir could be potential inhibitors to block viral RNA synthesis.
This study also revealed that all the inhibitors have more affinity for the viral chymotrypsin-like protease than the Papain-like protease. Furthermore, lopinavir, merimepodib, and dexamethasone could be inhibitors with potentials for the treatment of COVID-19 infection. The ADMET result revealed that only dexamethasone has attractive pharmacokinetic and toxicity properties probable for drug development.
Supplementary Information
Below is the link to the electronic supplementary material.
Author contributions
API and SB conceptualize the study, SB, API, FOS, AAE did the docking study, AAE, FOS did the ADMET study, API, AAE, and SB wrote the manuscript. All authors approved the manuscript.
Funding
The authors received no funding whatsoever from any source for the conduct of this research.
Declarations
Conflict of interests
The authors declare no exiting conflict of interest.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- Adelusi TI, Abdul-Hammed M, Ojo EM, Oyedele QK, Boyenle ID, Adedotun IO, Olaoba OT, Folorunsho AA, Kolawole OE. Molecular docking assessment of clinically approved antiviral drugs against Mpro, spike glycoprotein and angiotensin converting enzyme-2 revealed probable anti-SARS-CoV-2 potential. Trop J Nat Prod Res. 2021;5(4):778–791. doi: 10.26538/tjnpr/v5i4.30. [DOI] [Google Scholar]
- Agrawal AS, Garron T, Tao X, Peng BH, Wakamiya M, Chan TS, Couch RB, Tseng CK. Generation of a transgenic mouse model of Middle East respiratory syndrome coronavirus infection and disease. J Virol. 2015;89:3659–3670. doi: 10.1128/JVI.03427-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambudkar SV, Kimchi-Sarfaty C, Sauna ZE, Gottesman MM. P-glycoprotein: from genomics to mechanism. Oncogene. 2003;22:7468–7485. doi: 10.1038/sj.onc.1206948. [DOI] [PubMed] [Google Scholar]
- Bagherzadeh K, Daneshvarnejad K, Abbasinazari M, Azizian H (2020) In silico repositioning for Dual Inhibitor Discovery of SARS-CoV-2 (COVID-19) 3C-like Protease and Papain-like Peptidase. Med Chem 1–20. Preprint. 10.20944/preprints202004.0084.v1
- Borst P, Elferink RO. Mammalian ABC transporters in health and disease. Annu Rev Biochem. 2002;71:537–592. doi: 10.1146/annurev.biochem.71.102301.093055. [DOI] [PubMed] [Google Scholar]
- Chandel C, Raj S, Rathi B, Kumar D. In silico identification of potent FDA approved drugs against Coronavirus COVID-19 main protease: a drug repurposing approach. Chem Biol Lett. 2020;7(3):166–175. [Google Scholar]
- Dayer MZ, Taleb-Gassabi S, Dayer MS. Lopinavir; a potent drug against coronavirus infection: insight from molecular docking study. Arch Clin Infect Dis. 2017;12(4):e13823. doi: 10.5812/archcid.13823. [DOI] [Google Scholar]
- de Wit E, van Doremalen N, Falzarano D, Munster VJ. SARS and MERS: recent insights into emerging coronaviruses. Nat Rev Microbiol. 2016;14(8):523. doi: 10.1038/nrmicro.2016.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyon L, Tremblay S, Bourgon L, Wardrop E, Cordingley MG. Selection and characterization of HIV-1 showing reduced susceptibility to the non-peptidic protease inhibitor tipranavir. Antiviral Res. 2005;68:27–35. doi: 10.1016/j.antiviral.2005.07.003. [DOI] [PubMed] [Google Scholar]
- Fehr AR, Perlman S. Coronaviruses: an overview of their replication and pathogenesis. Methods Mol Biol. 2015;1282(2015):1–23. doi: 10.1007/978-1-4939-2438-7_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh AK, Osswald HL, Prato G. Recent progress in the development of HIV-1 protease inhibitors for the treatment of HIV/AIDS. J Med Chem. 2016;59(11):5172–5208. doi: 10.1021/acs.jmedchem.5b01697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glowacka I, Bertram S, Muller MA, Allen P, Soilleux E, Pfefferle S, Steffen I, Tsagaye TS, He Y, Gnirss K, Neimeyer D, Schneider H, Drosten C, Pohlman S. Evidence that TMPRSS2 activates the severe acute respiratory syndrome coronavirus spike protein for membrane fusion and reduces viral control by the humoral immune response. J Virol. 2011;85:4122–4134. doi: 10.1128/JVI.02232-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hage-Melim LS, Federico LB, de Oliveira NKS, Francisco VCC, Correia LC, de Lima HB, Gomes SQ, Barcelos MP, Francischini IAG, da Silva CHT. Virtual screening, ADME/Tox predictions and the drug-repurposing concept for future use of old drugs against the COVID-19. Life Scis. 2020;256(117963):1–13. doi: 10.1016/j.lfs.2020.117963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han DP, Penn-Nicholson A, Cho MW. Identification of critical determinants on ACE2 for SARS-CoV entry and development of a potent entry inhibitor. Virology. 2006;350:15–25. doi: 10.1016/j.virol.2006.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hentig NV. Appraisal of promising repurposed drug candidates for the treatment of SARS-COV-2 infection and COVID-19. J Antivir Antiretrovir. 2020;12:195. doi: 10.35248/1948-5964.20.12.195. [DOI] [Google Scholar]
- Khan SU, Htar T. Deciphering the binding mechanism of dexamethasone against SARS-CoV-2 main protease: computational molecular modelling approach. ChemRxiv. 2020 doi: 10.26434/chemrxiv.12517535.v1. [DOI] [Google Scholar]
- Kirchdoerfer RN, Ward AB. Structure of the SARS-CoV nsp12 polymerase bound to nsp7 and nsp8 co-factors. Nat Commun. 2019;10(2342):1–9. doi: 10.1038/s41467-019-10280-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar Y, Singh H, Patel CN. In silico prediction of potential inhibitors for the Main protease of SARS-CoV-2 using molecular docking and dynamics simulation based drug-repurposing. J Infect Public Health. 2020;2020:1–14. doi: 10.1016/j.jiph.2020.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee TW, Cherney MM, Huitema C, Liu J, James KE, Powers JC, Eltis LD, James MNG. Crystal structures of the main peptidase from the SARS coronavirus inhibited by a substrate-like aza-peptide epoxide. J Mol Biol. 2005;353(5):1137–1151. doi: 10.1016/j.jmb.2005.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Moore MJ, Vasilieva N, Sui J, Wong SK, Berne MA, Somasundaran M, Sullivan JL, Luzuriaga K, Greenough TC, Choe H, Farzan M. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature. 2003;426:450–454. doi: 10.1038/nature02145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millet JK, Whittaker GR. Host cell proteases: critical determinants of coronavirus tropism and pathogenesis. Virus Res. 2015;202:120–134. doi: 10.1016/j.virusres.2014.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mothay D, Ramesh KV. Binding site analysis of potential protease inhibitors of COVID-19 using AutoDock. Virus Dis. 2020 doi: 10.1007/s13337-020-00585-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray M. Role of CYP pharmacogenetics and drug-drug interactions in the efficacy and safety of atypical and other antipsychotic agents. J Pharm Pharmacol. 2006;58:871–885. doi: 10.1211/jpp.58.7.0001. [DOI] [PubMed] [Google Scholar]
- Muster W, Breidenbach A, Fischer H, Kirchner S, Müller L, Pähler C. Computational toxicology in drug development. Drug Discov Today. 2008;13:303–310. doi: 10.1016/j.drudis.2007.12.007. [DOI] [PubMed] [Google Scholar]
- Qamar MT, Alqahtani SM, Alamri MA, Chen L (2020) Structural basis of SARS-CoV-2 3CLpro and anti-COVID-19 drug discovery from medicinal plants. J Pharmaceutical Anal 10(4):1–16 [DOI] [PMC free article] [PubMed]
- Rabaan AA, Alahmed SH, Bazzi AM, Alhani HM. A review of candidate therapies for Middle East respiratory syndrome from a molecular perspective. J Med Microbiol. 2017;66:1261–1274. doi: 10.1099/jmm.0.000565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schyman P, Liu R, Desai V and Wallqvist A (2017) vNN Web Server for ADMET Predictions. Front. Pharmacol. 8:889. 10.3389/fphar.2017.00889 [DOI] [PMC free article] [PubMed]
- Shah B, Modi P, Sagar SR (2020) In silico studies on therapeutic agents for COVID-19: drug repurposing approach. Life Sci 252(117652):1–12 [DOI] [PMC free article] [PubMed]
- Sheahan TP, Sims AC, Zhou S, Graham RL, Pruijssers AJ, Agostini ML, Leist SR, Schäfer A, Dinnon KH, Stevens LJ, Chappell JD, Lu X, Hughes TM, George AS, Hill CS, Montgomery SA, Brown AJ, Bluemling GR, Natchus MG, Saindane M, Kolykhalov AA, Painter G, Harcourt F, Tamin A, Thornburg NJ, Swanstrom R, Denison MR, Baric RS (2020) An orally bioavailable broad-spectrum antiviral inhibits SARS-CoV-2 in human airway epithelial cell cultures and multiple coronaviruses in mice. Sci Transl Med 12(541):1–20 [DOI] [PMC free article] [PubMed]
- Shyamala DK, Sivakumari K, Rajesh S. Identification of potential drug candidates from herbal medicinal plants to treat COVID-19 by molecular docking interaction of drug compounds against main protease (Mpro) of Corona virus. J Xidian Univ. 2020;14(5):4945–4951. [Google Scholar]
- Song Z, Xu Y, Bao L, Zhang L, Yu P, Qu Y, Zhu H, Zhao W, Han Y, Qin C. From SARS to MERS, thrusting coronaviruses into the spotlight. Viruses. 2019;11(1):59. doi: 10.3390/v11010059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHO (2020a) Coronavirus disease (COVID-19) situation report – 158. https://www.who.int/docs/default-source/coronaviruse/situation-reports/20200626-covid-19-sitrep-158.pdf?sfvrsn=1d1aae8a_2. Accessed 26 June 2020
- WHO (2020b) WHO welcomes preliminary results about dexamethasone use in treating critically ill COVID-19 patients. https://www.who.int/news/item/16-06-2020-who-welcomes-preliminary-results-about-dexamethasone-use-in-treating-critically-ill-covid-19-patients. Accessed 16 June 2020
- Wu C, Liu Y, Yang Y, Zhang P, Zhong W, Wang Y, Wang Q, Xu Y, Li M, Li X, Zheng M, Chen L, Li H. Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods. Acta Pharmaceutica Sinica B. 2020 doi: 10.1016/j.apsb.2020.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao L, Jha BK, Wu A, Elliott R, Zeibuhr J, Gorbalenya AE, Silverman RH, Weiss SR. Antagonism of the interferon-induced OAS-RNase L pathway by murine coronavirus ns2 protein is required for virus replication and liver pathology. Cell Host Microbe. 2012;11(6):607–616. doi: 10.1016/j.chom.2012.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou P, Yang XL, Wang XG, Hu B, Zhang L, Zhang W, Si HR, Zhu Y, Li B, Huang CL, Chen HD, Chen J, Luo Y, Guo H, Jiang RD, Liu MQ, Chen Y, Shen XR, Wang X, Zheng XS, Zhao K, Chen QJ, Deng F, Liu LL, Yan B, Zhan FX, Wang YY, Xiao GF, Shi ZL. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;2020:1–5. doi: 10.1038/s41586-020-2012-7Medline. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.