

Abstract
Colorectal cancer (CRC) is multi-faceted with subtypes defined by genetic, histological, and immunologic features which are potentially influenced by inflammation, mutagens, and/or microbiota. CRCs with activating mutations in BRAF are associated with distinct clinical characteristics though the pathogenesis is not well understood. The Wnt-driven multiple intestinal neoplasia (MinApcΔ716/+) enterotoxigenic Bacteroides fragilis (ETBF) murine model is characterized by IL-17-dependent, distal colon adenomas. Herein, we report that addition of the BRAFV600E mutation to this model results in emergence of a distinct locus of mid-colon tumors. In ETBF-colonized BRAFV600ELgr5CreMin (BLM) mice, tumors have similarities to human BRAFV600E tumors, including histology, CpG island DNA hypermethylation, and immune signatures. In comparison to Min ETBF tumors, BLM ETBF tumors are infiltrated by CD8+ T cells, express interferon-gamma signatures, and are sensitive to anti-PDL1 treatment. These results provide direct evidence for critical roles of host genetic and microbiota interactions in CRC pathogenesis and sensitivity to immunotherapy.
INTRODUCTION
In the US, colorectal cancer (CRC) is the third leading cause of cancer death for both men and women (1). Classification of CRC tumors into subtypes using features such as location along the axis of the colon, differing genetic mutations, genomic instability [e.g., chromosomal instability (CIN) or microsatellite instability (MSI)], pathology and epigenetic biomarkers help define predictive factors for treatment and patient survival. Further, recent advances have implicated the microbiota as a key contributing factor of disease (2–4).
Characterization of both healthy and disease-state gut microbiomes has revealed complex, bidirectional relationships between microbes and host in which microbes play important roles in modifying the host immune response, metabolism, and protecting the host against pathogen invasion (5,6). Colonization with enterotoxigenic Bacteroides fragilis (ETBF) is associated with CRC patients and tumors (7–9). To study the role of microbes and the host immune response in CRC, we previously developed a murine model wherein ETBF colonization induces Wnt- and IL-17-dependent tumorigenesis in the distal colon of multiple intestinal neoplasia (MinApcΔ716/+) mice that are heterozygous for the adenomatous polyposis coli (Apc) gene (10). However, how or if mutations other than Apc+/− modify ETBF-driven tumorigenesis is unknown. To determine if other mutations commonly found in CRC have specific interactions with ETBF, we introduced into the Min ETBF model the BRAFV600E activating mutation of the MAPK pathway which comprises approximately 90% of all BRAF mutations in human CRC (11). The mouse obtained to carry out these experiments expresses human BRAFV600E at the mouse Braf locus.
Herein, we demonstrate that, compared to the Min ETBF model of distal tumorigenesis, ETBF colonization of BRAFF-V600ELgr5tm1(Cre/ERT2)CleMinApcΔ716/+ (BLM) mice results in a new more proximal locus of colon tumors, reminiscent of the predominant right-sided location of BRAF mutant tumors in patients. ETBF-induced tumorigenesis in mice with a different MAPK-activating mutation, KrasG12DLgr5CreMinApcΔ716/+, maintained only distal tumorigenesis, which is consistent with the preponderance of KRAS-mutant CRC being left-sided in humans. Additionally, BLM colon tumors display a serrated-like histopathology, changes in the mucus bilayer and increased mucus production. We also investigated changes in DNA methylation in the colon tissues of BLM mice compared to Min mice since in humans the BRAFV600E mutation is associated with the CpG methylator phenotype (CIMP) and found marked changes in DNA methylation in the BLM mice. Further, ETBF induced, in BLM mice, a colitis with an interferon-gamma (IFN-γ)-driven immune signature associated with a robust recruitment of Cd274 (coding PDL1)-expressing myeloid-derived suppressor cells (MDSC). Notably, strong infiltration of BLM tumors with CD8+ cells coincided with efficacy of PDL1 blockade in significantly reducing colon tumor numbers suggesting an adaptive immunosuppression mechanism induced by IFN-γ producing CD8+ tumor-infiltrating T cells. Together ETBF colonization of the BLM mouse model provides novel insight into oncogene:microbiota interactions potentially relevant to the pathogenesis of human BRAFV600E-mutated CRC.
RESULTS
BRAF V600E mutation impacts colon and mucosal architecture resulting in excess mucosal and systemic inflammation and DNA damage after ETBF colonization.
In wild-type (WT) C57Bl/6 mice, ETBF induces a rapid onset, predominantly distal colitis despite uniform colonization of ETBF along the axis of the colon (12). In initial experiments, we compared the colonic response of BRAFV600ELgr5Cre (BL mice, see Methods) and WT mice to ETBF colonization. Upon ETBF colonization, the mid-proximal colon regions (defined in Supplementary Figure 1A) of BL mice exhibit enhanced colitis and colon epithelial cell (CEC) shedding (Figure 1A). ETBF fecal colonization was similar between WT and BL mice (Supplementary Fig 1B). ETBF colonization of BL mice also led to a disruption of the ordered mucus bilayer in both the distal and mid-proximal regions of BL mice when compared to sham mice and compared to the unperturbed mid-proximal region of WT colons colonized by ETBF at the same time point (Supplementary Figure 1C). After long term ETBF colonization (15-43 weeks), we noted increases in spleen weight as well as splenic and liver inflammation in ETBF-colonized BL vs WT mice (Supplementary Figure 1D, 1E). Long term ETBF colonization (15-43 weeks) of BL mice did not result in tumor formation in the colon (WT N = 31; BL N = 31; 1-5 mice per experiment for > 10 experiments). Combined, these results suggest mice with an activating BRAFV600E mutation colonized by ETBF exhibit increased colon mucosal and systemic inflammation.
Figure 1. Inflammation and myeloid cells with a PMN-MDSC signature are increased in the mid-proximal colon of BRAFV600E relative to wild-type mice after ETBF colonization.
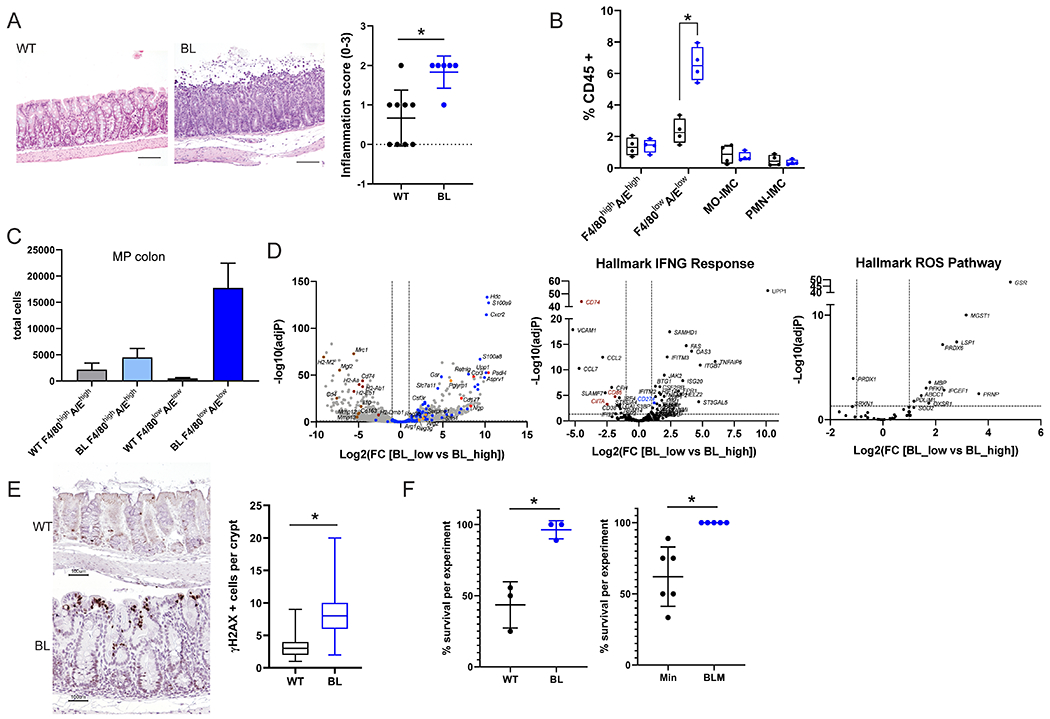
A) Representative H&E images of mid-proximal colon regions of WT and BL mice 7 days after ETBF colonization. Scale bar = 100 μM. Inflammation scores for the mid-proximal colon region in WT (N=9) and BL (N=6) for 2 experiments. B) FACS analysis of myeloid cells (F4/80highI-A/Ehigh cells; F4/80lowI-A/Elow cells; Mo, monocytes; PMN, polymorphonuclear cells; IMC, immature myeloid cells) from mid-proximal colon tissues 7 days post ETBF. C) Total number of F4/80highI-A/Ehigh and F4/80lowI-A/Elow cells sorted from the mid-proximal colon regions of WT and BL mice (N=3 each) 2 weeks post ETBF inoculation. D) Volcano plots of DESeq2 comparison of sorted F4/80lowI-A/Elow (low) and F4/80highI-A/Ehigh (high) cells isolated from BL mid-proximal in C. Dashed lines indicate cut-offs used for significantly differentially expressed genes (adjP < 0.01 and log2-fold change > 2). Left, genes from MDSC gene set (49); center, hallmark INFγ Response genes; and right, hallmark ROS pathway genes. Gene highlight colors: blue, MDSC; red, neutrophil-associated; orange, antibacterial enzymes and peptides; and brown, MHC-II associated. E) Representative γ H2AX IHC in the mid-proximal colons of mice 7 days after ETBF colonization. The number of γ H2AX positive cells per crypt that contain at least one positive cell were counted. N > 47 crypts for each genotype. Scale bar = 100 μM. F) Percent survival within 10 days of ETBF colonization for WT and BL (left, 3 experiments; total WT N=21, BL N=18 mice) and Min and BLM (right, 6 experiments; total Min N=39, BLM N=32 mice).* P < 0.05 by Fisher’s exact test. In A and F data is presented as mean +/− SD; for B and C box limits are set at the third and first quartile range with the central line at the median. *P <0.05 values for A-C were calculated by Mann-Whitney U test. Black, WT and blue, BL for all graphs. Additional information on distal colon in Supplementary Figure 1.
To further examine the impact of BRAFV600E on colon inflammation in ETBF-colonized mice, we performed flow cytometry analysis of mid-proximal and distal colon mucosal lamina propria leukocytes isolated from BL and WT mice 7 days after ETBF colonization, when acute colitis peaks in ETBF-induced inflammation in WT mice (12,13), and also at a later time point 2 weeks after ETBF colonization. Notably, ETBF-colonized BL mice displayed significantly higher levels of CD45+CD11b+Ly6G−Ly6C−F4/80lowI-A/Elow cells denoted F4/80lowI-A/Elow, in the mid-proximal region when compared to WT mice (Figure 1B). This increased influx of F4/80lowI-A/Elow cells was not observed in the distal colon of either BL or WT mice at 7 days after ETBF colonization nor in the sham controls regardless of region (Supplementary Figure 1F). There were no significant differences for other leukocyte populations examined for any of these colon regions (Supplementary Figure 1G). Cytokines previously identified as crucial for ETBF-driven tumorigenesis, II6, II17a, Cxcl2, and Cxcl5, were examined by qPCR and no differences were found between distal and mid-proximal regions in WT versus BL mice 7 days post colonization (Supplementary Figure 1H).
To better understand the lineage and functional natures of the F4/80lowI-A/Elow cell subset, we performed RNAseq analysis of cell-sorted CD45+CD11b+Ly6G−Ly6C−F4/80lowI-A/Elow (F4/80lowI-A/Elow) versus CD45+CD11b+Ly6G−Ly6C−F4/80highI-A/Ehigh (F4/80highI-A/Ehigh) cells from the mid-proximal colons of 2 week-ETBF-colonized BL and WT mice. Analysis of the absolute numbers of both cell subsets recovered by cell sorting confirmed the striking accumulation of F4/80lowI-A/Elow compared to F4/80highI-A/Ehigh in the mid-proximal colon of BL mice compared to WT (Figure 1C). In BL mice, the F4/80lowI-A/Elow cells have a predominant MDSC gene expression signature compared to F4/80highI-A/Ehigh including S100A8, S100A9, Arg1, Slc7a11 (coding xCT) (14–16) as well as the transcription factor Slfn4 (17) (Figure 1D, Supplementary Table 1). Metabolism of L-arginine by arginase 1 and transport of cysteine by xc− cystine/glutamate antiporter are fundamental hallmarks of the immunosuppressive functions of MDSC (18). T cells that do not express the xCT chain of the xc− cystine/glutamate antiporter are dependent on antigen-presenting cells producing cysteine from the reduction of cystine in the microenvironment. However, MDSCs that do not express the ASC transporter do not, in turn, export cysteine leading to the sequestration of this essential amino-acid for T cell activation (19). Deprivation of the tumor immune micro-environment (TiME) of L-arginine and cysteine by MDSC sequestration suppresses T cell functions. Furthermore, this MDSC signature is combined with expression of neutrophil-associated genes including Cxcr2, Ccr3, Cd177, Hdc, Ngp, Csf3r, Asprv1, Padi4 (20,21). Interestingly, this neutrophil-like cell subset remained Ly6G negative and F4/80low, resembling the macrophage-like neutrophils previously described in chronic inflammation (20). These gene expression profiles established that F4/80lowI-A/Elow cells represent polymorphonuclear MDSC (PMN-MDSC) and establishes that their early accumulation in the mid-proximal region of BL mice colon occurs in the same region that later develops mid-proximal tumors. Moreover, these PMN-MDSC were characterized by an IFN-γ gene signature (Figure 1D) suggesting that PMN-MDSC were activated and recruited by proinflammatory signals, such as IFN-γ (22). One of the IFN-γ target genes significantly upregulated in F4/80lowI-A/Elow compared to F4/80highI-A/Ehigh cells is Cd274 (coding PDL1) that is associated with MDSC immunosuppressive function by interacting with the T cell checkpoint PD1. These PMN-MDSC cells also express antibacterial enzymes and peptides such as Retnlg, Pglyrp1, Reg3b, Reg3g, Serpine1 highlighting that they may act in defense against ETBF and/or other microbiota members that induced their recruitment. F4/80lowI-A/Elow cells in BL mice were also enriched for MDSC and IFN-ɣ gene signatures relative to F4/80low I-A/Elow in WT mice where less IFN-γ production was observed (Supplementary Figure 1I). BL F4/80highI-A/Ehigh demonstrated a typical gene expression signature of pro-regenerative macrophages (Cd4, Mrc1, Mgl2, Il10, Cd163, Mmp12, Mmp13) expressing higher levels of MHC-II associated genes (H2-M2, H2-Eb1, H2-Aa, H2-Ab1, H2-Dmb1, CIITA, Cd74) and costimulatory molecules (Cd86) (Figure 1D).
BL F4/80low I-AElow cells were also enriched for the hallmark ROS pathway gene set (Figure 1D) and we indeed found that BL mice had more γH2AX foci, a marker of DNA damage, in the mid-proximal colon 7 days after ETBF colonization compared to WT mice (Figure 1E). Of interest, despite the altered mucosal morphology as well as increased inflammation and colon epithelial DNA damage, mice with the BRAFV600E mutation survived the acute inflammatory response to ETBF colonization significantly better than WT mice (Figure 1F).
BRAFV600E mutation in ETBF-colonized Min mice promotes mid-proximal colon tumorigenesis.
At age 916 weeks, baseline (sham) colon tumorigenesis in mice displaying ApcΔ716/+ only (Min) or both ApcΔ716/+ and BRAFV600ELgr5Cre mutations (BLM mice, see Methods) was generally rare and restricted to the distal 0-3 cm of the colon (N = 11 mice each genotype; median 0 for each genotype; tumor range 0-2 tumors Min mice vs 0-3 tumors BLM). Similarly, no increase in microadenoma numbers was observed in sham BLM colons compared to Min colons (Min mice N = 10, median = 0 tumors, range (0-2); BLM mice N = 8, median = 0 tumors, range (0-1)). Consistent with our previous work, tumor formation in Min mice colonized long-term with ETBF (6-13 weeks) occurred predominantly in the distal 3 centimeters of the colon (Figure 2A) (10). However, BLM mice with long-term ETBF colonization had significantly more tumors due to emergence of a new locus of tumor formation in the mid-proximal colon (Figure 2A, Supplementary Figure 2A). While BLM mice had significant increases in mid-proximal tumors relative to Min mice, there was no significant differences in distal colon tumor formation. However, total tumors for BLM mice were significantly elevated at the end of long-term ETBF experiments (Min median tumors = 10 (range 1-45); BLM median tumors = 21 (6-54); P < 0.0001, Mann-Whitney U test). Total colon microscopic adenomas or microadenomas were also significantly elevated in BLM mice when compared to Min mice in these long-term ETBF colonization experiments (Figure 2B). Of note, the altered mid-proximal colon tumorigenesis pattern was unique to BLM mice as ETBF colonization of mice with KrasG12D, a different MAPK-activating mutation common in human CRC (i.e., KrasG12DLgr5CreMinApcΔ716/+ mice), displayed tumorigenesis restricted to the most distal 0-3 cm of the colon similar to ETBF colonization of Min mice (Supplementary Figure 2B). No difference in fecal or mucosal ETBF colonization was detected between Min and BLM mice (Supplementary Figures 2C, 2D).
Figure 2. ETBF induces mid-proximal colon tumorigenesis with serrated-like morphology in BLM mice.
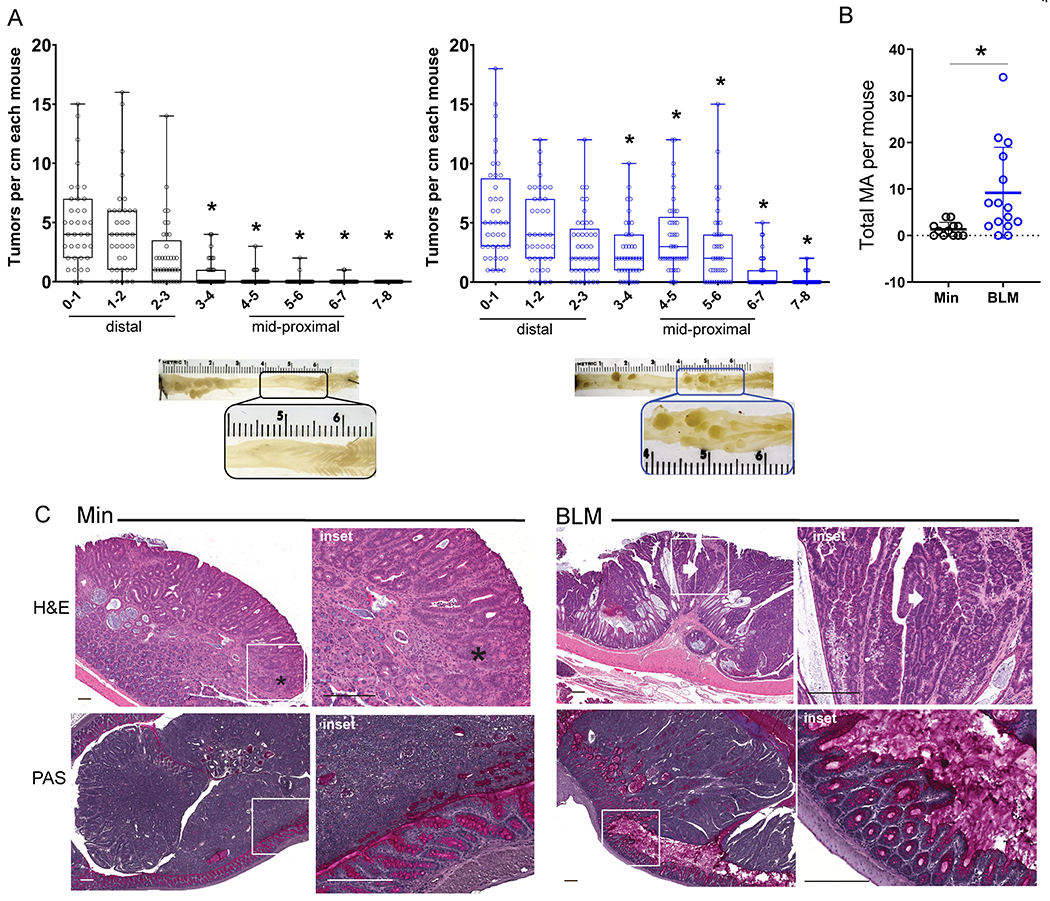
A) Tumor counts by centimeter from distal (0-1) to proximal (7–8) for each mouse in long-term ETBF-colonized Min and BLM colons at 9-13 weeks post inoculation. Min N=32; BLM N=41 for >10 experiments. *P<0.05 by pair-wise Mann-Whitney U test comparing corresponding colon centimeter sections. Box limits are set at the third and first quartile range with the central line at the median. Images below plots are examples of gross tumor appearance in the mid-proximal regions of ETBF-colonized Min (left) and BLM (right) colons. B) Number of microadenomas (MA) in ETBF-colonized Min and BLM colons 9-13 weeks post colonization. Data presented as mean +/− SD. *P<0.05 by Mann-Whitney U test. C) Top panels: Representative H&Es of formalin-fixed ETBF-induced Min and BLM tumor morphologies. Scale bars = 200 μm. Insets: Min ETBF tumor tubular adenoma (*), BLM ETBF tumor villiform adenoma (arrow); see also Supplementary Figure 2. Bottom panels: Representative PAS of methacarn-fixed, ETBF-induced Min and BLM tumor morphologies with preserved mucus layer. Scale bars 200 μm.
ETBF-colonized BLM mice also displayed a marked change in tumor histology. In contrast to the tubular adenoma morphology of colon tumors in ETBF-colonized Min mice, the tumors of ETBF-colonized BLM mice exhibited a mixture of villous and tubular adenoma histology and pink eosinophilic dysplasia, abundant cytoplasm and enlarged nuclei, similar to the histopathology of sessile serrated tumors in humans with BRAFV600E CRC (Figure 2C, Supplementary Figure 2E). As with BL mice, BLM mice display increased mucus production and a disordered bilayer after ETBF colonization when intact colons are fixed in methacarn solution and observed by PAS or MUC2 IHC (Figure 2C, Supplementary Figure 2F). Also similar to BL mice, ETBF-colonized BLM mice exhibited increased survival compared to ETBF-colonized Min mice within the first 10 days of colonization (Figure 1F; BLM 100% survival; N = 32; Min 60% survival; N = 35; P<0.0001, Fisher’s exact test). Altogether, these data indicate that the addition of BRAFV600E to the ETBF-colonized Min murine model results in a new locus of serrated-like, mid-proximal tumors.
ETBF colonization enhances CpG island DNA hypermethylation in tissues expressing BRAFV600E.
Changes in epithelial DNA methylation have been associated with both ETBF and the expression of BRAFV600E (23–25). Thus, we next used methylation-binding domain sequencing (MBD-seq) to examine the impact of ETBF on CpG island DNA hypermethylation in BLM and Min mice.
Analysis of all DNA hypermethylated regions in any ETBF sample (BLM or Min, epithelium or tumor) relative to sham epithelium indicated that the BLM and Min genotypes were associated with distinct DNA methylation patterns (Figure 3A). After ETBF colonization, BLM mid-proximal epithelium had a higher number of CpG islands displaying DNA hypermethylation relative to sham BLM mid-proximal epithelium than Min distal epithelium relative to sham Min distal epithelium (Figure 3B; Supplementary Table 2). ETBF-induced BLM tumors also had more CpG islands with DNA hypermethylation than Min tumors when compared to their respective controls (Figure 3C). Unsupervised hierarchical clustering of the distinct regions harboring significant DNA hypermethylation in either ETBF-colonized epithelium or ETBF-induced tumors compared to sham epithelium demonstrated that ETBF-associated DNA hypermethylation was similar in BLM epithelium and tumor samples (Figure 3A, Supplementary Figure 3A). In contrast, ETBF colonization had more of an effect on DNA hypermethylation in Min tumors than epithelium as demonstrated by ETBF-induced Min tumors clustering separately from the ETBF-colonized Min epithelium samples (Supplementary Figure 3B). CIMP-related genes, Cdkn2a and Gata5, as well as Polg, Fut4, and Ano1, all had relative DNA methylation levels that were consistent with the MBDseq data when validated by quantitative methylation-specific PCR (Supplementary Figure 3C).
Figure 3. The addition of BRAFV600E results in distinct ETBF colonization-associated CpG island DNA hypermethylation.
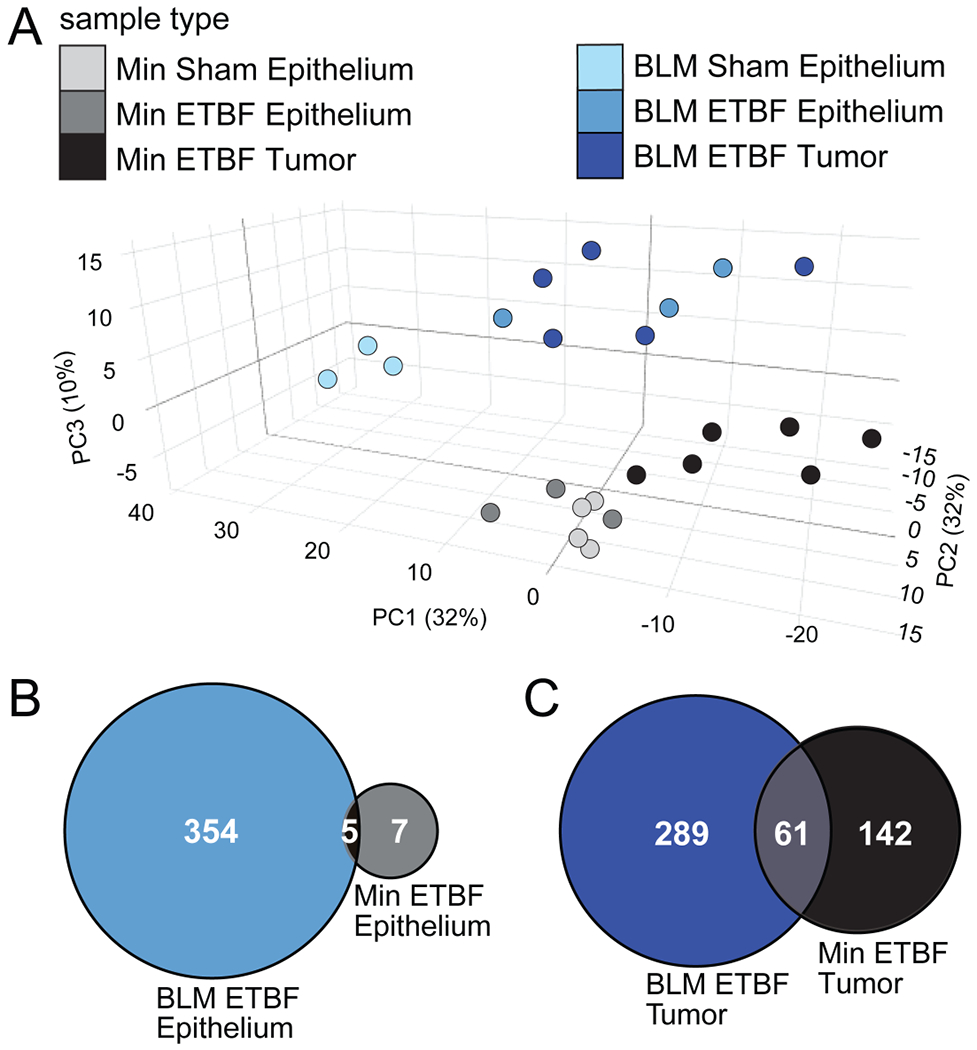
A) Principal component (PC) analysis of MBD-seq z-scores of the 747 regions in CpG islands with DNA hypermethylation in at least one of the tumor groups relative to sham epithelium. Epithelia (N=3-4) and tumor (N=5-6) samples were collected from distal and mid-proximal regions of Min and BLM colons, respectively, 9-13 weeks post inoculation. X, Y and Z axes show PC1, PC2, and PC3 that explain 32, 32 and 10% of the total variance, respectively. Proportional Venn diagrams of the number of overlapping regions with significant DNA hypermethylation in B) ETBF-colonized epithelia or C) ETBF-induced tumors relative to their respective sham epitheliums.
Gene expression analysis indicates an IFN-γ-associated gene expression signature in BLM epithelium and tumors compared to Min epithelium and tumors.
To further define the differences in ETBF-driven colon tumorigenesis between BLM and Min mice, we performed RNA sequencing on mRNA extracted from ETBF-induced BLM mid-proximal and Min distal colon tumors as well as BLM mid-proximal and Min distal normal epithelium from sham mice. Initial principal component analysis (PCA) using all expressed genes revealed clustering by genotype and ETBF status indicating that genotype as well as ETBF colonization and tumor phenotype lead to distinct gene expression signatures (Figure 4A, Supplementary Table 3).
Figure 4. BRAFV600E mutation promotes an IFN-associated gene - expression signature in the normal epithelium and tumors of BLM mice.
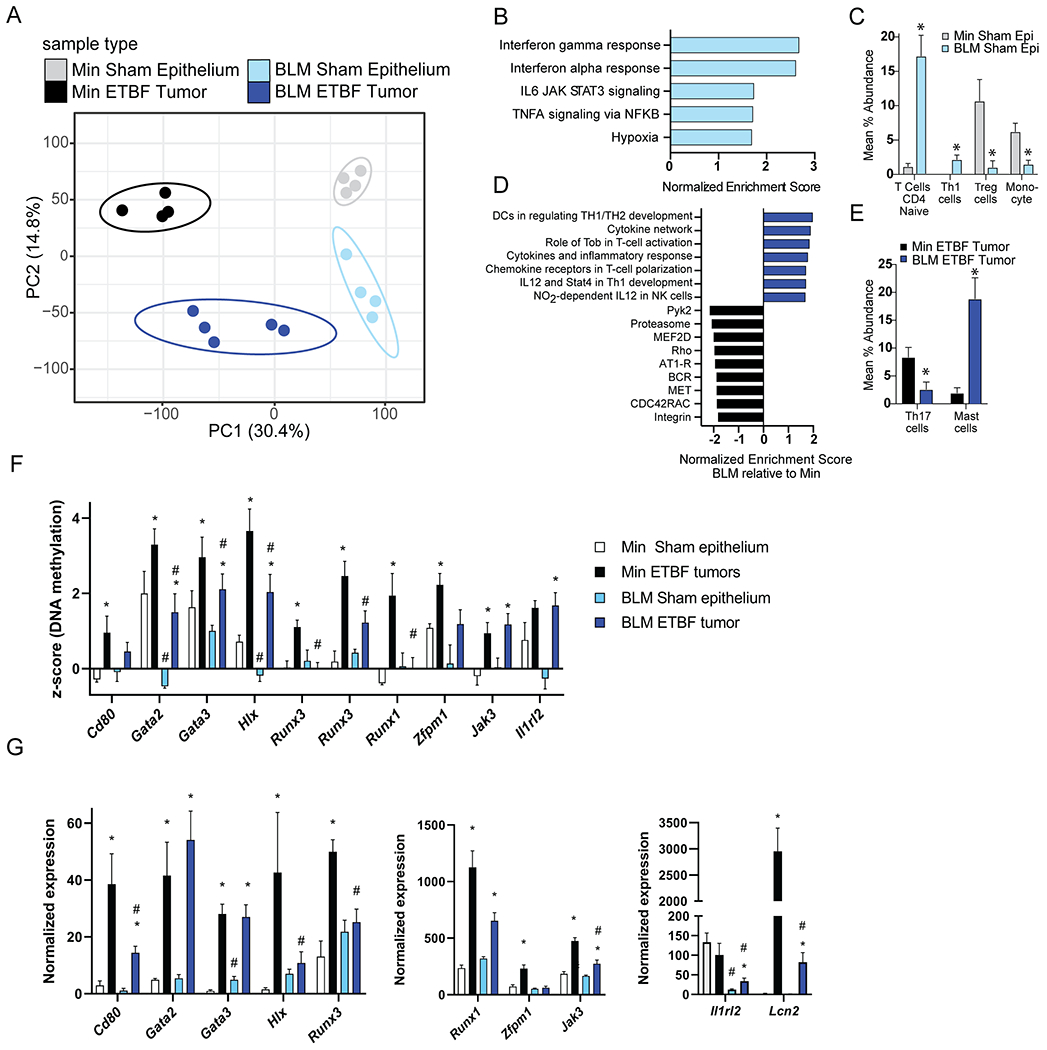
RNA-seq analysis of colon epithelia (N=4) and individual tumors (N=4-5) collected from distal and mid-proximal regions of Min and BLM colons, respectively, 9-13 weeks post-inoculation. Epithelia samples are from 2 independent experimental cohorts. Tumors are from 6 independent ETBF inoculation cohorts. A) PCA analysis of all expressed genes. Unit variance scaling is applied to rows; SVD with imputation is used to calculate principal components. X and Y axes show PC1 and PC2 that explain 30.4 and 14.8% of the total variance, respectively. Prediction ellipses are such that with probability 0.95, a new observation from the same group will fall inside the ellipse. B) Normalized enrichment score from GSEA of mid-proximal BLM versus distal Min sham epithelium using all genes from the RNA-seq data and Hallmark gene sets. All pathways with an FDR < 0.01 are listed (none were enriched in Min sham epithelium). C) Differential abundance results based on CIBERSORT to predict immune cell composition per RNA-seq sample. Bar graphs represent mean ± SEM of immune cell types with significant differences (*P < 0.05 by t-test) in BLM sham epithelium (Epi) compared to Min sham epithelium. D) Normalized enrichment score from GSEA of ETBF-induced mid-proximal BLM versus distal Min tumors using all genes from the RNA-seq data and gene sets derived from the BioCarta pathway database. All pathways with an FDR < 0.05 are listed. E) Differential abundance results based on CIBERSORT as in C for ETBF-induced BLM versus Min tumors. F) Mean ± SEM of DNA methylation levels (Z-scores) from MBD-seq data of indicated genes. N=3-4 epithelium. N=5-6 tumors. *P < 0.05 relative to respective sham epithelium. #P < 0.05 relative to respective Min sample. G) Mean ± SEM of normalized expression from RNA-seq data of indicated genes. N=4-5. *,# as in F. Color key is consistent for all panels: grey=Min Sham epithelium, black=Min ETBF tumor, light blue= BLM sham epithelium, dark blue=BLM ETBF tumor.
By performing gene set enrichment analysis (GSEA) using gene sets derived from hallmark pathways (26), we detected a significant change in the baseline mucosal immunity gene expression between BLM and Min sham epithelium. GSEA indicated that BLM sham epithelium is significantly enriched for the hallmark IFN-γ and IFN-α gene sets (Figure 4B, Supplementary Figure 4A). We also used CIBERSORT, a computational method for quantifying immune subsets from bulk tissue gene profiles, to survey potential infiltrating leukocytes and other immune components previously shown to correlate with both prognosis and therapeutic response (27). Consistent with the GSEA, CIBERSORT analysis demonstrated that the BLM sham epithelium is enriched in a Th1 immune signature while Min sham epithelium is enriched in regulatory T cell (Treg) and monocyte gene profiles (Figure 4C). These data suggest that the mucosa of BLM mice is poised for Th1 polarization prior to ETBF colonization.
Using gene sets derived from BioCarta pathways (28), GSEA analysis comparing ETBF-induced BLM mid-proximal and Min distal tumor profiles demonstrated significant enrichment in Th1-related expression gene sets in ETBF-induced BLM tumors compared to Min tumors, including genes associated with IL12 and Stat4-dependent signaling in Th1 development (Figure 4D). Relative to Min tumors, BLM tumors displayed decreased enrichment of pathways involved in cell motility (RHO, MET, CDC42RAC, Integrin) and activation of calcium signaling and protein kinase C (PYK2, MEF2D, AT1R, BCR) (Figure 4D). CIBERSORT analysis also found a predominance of Th17-related gene expression in ETBF-induced Min tumors compared to BLM tumors (Figure 4E). BLM tumors had increased expression of genes related to mast cells that can be activated by microbial pathogens (29), are pro-inflammatory, and previously shown to play a role in polyp formation in Min mice and CRC in humans (30). Quantitative RT-PCR confirmed that ETBF-induced Min tumors had higher expression of Th17-associated genes such as II6, II1b and Tgfb relative to ETBF-induced BLM tumors as well as IL-17-target genes Cxcl1, 2 and 5 (Supplementary Figure 4B). ETBF-induced Min tumors were also enriched for genes associated with MO-IMC function and recruitment including Arg1, Nos2 (which encodes iNOS), Ccl2, S100A8/9, and Mmp9 when compared to ETBF-induced BLM tumors (Supplementary Figure 4B). In contrast, relative to ETBF-induced Min tumors, ETBF-induced BLM tumors are enriched for the antimicrobial genes Reg3b, Reg3g (bactericidal peptides) and Ccl28 (homing of IgA-producing plasma cells at mucosal surface). Further, Muc2 and Cldn15 (claudin-15, barrier function) expression is enriched in both sham BLM epithelium and ETBF-induced BLM tumors relative to their respective Min controls. Enhanced expression of Muc2 and barrier function genes in the sham BLM mid-proximal epithelium is lost in ETBF-induced BLM mid-proximal tumors which are more similar to ETBF-induced Min distal tumors for these features (Supplementary Figure 4B).
Further, genes associated with DNA hypermethylation and altered gene expression in ETBF-induced Min distal tumors relative to Min distal sham epithelium were enriched for gene ontology pathways associated with regulation of alpha-beta T cell activation and leukocyte differentiation. Many of these genes had significantly lower DNA methylation levels in BLM epithelium and/or tumors than the respective Min samples (Gata2, Gata3, Hlx, Runx3, Runx1) and/or were not DNA hypermethylated in ETBF-induced BLM tumors relative to BLM epithelium (Cd80, Runx3, Runx1, Zfpm1) (Figure 4F). These DNA methylation differences were associated with gene expression differences between the Min and BLM sample types for some genes (Cd80, Gata3, Hlx, Runx3, Zfpm1) (Figure 4G). Though overall DNA hypermethylation in ETBF-induced BLM tumors was not strongly associated with genes involved in immune cell regulation, some immune genes with altered expression also had changes in DNA methylation. For example, Il1rl2 (encodes IL36R), a gene involved IL17-mediated fibrosis (31), was significantly DNA hypermethylated in ETBF-induced BLM tumors relative to sham BLM epithelium (Figure 4F). The DNA methylation change was associated with significantly altered Il1rl2 expression in ETBF-induced BLM tumors relative to sham BLM epithelium and Min distal tumors (Figure 4G). Expression of Lcn2 (Lipocalin2), a target gene of IL36R signaling, and mediator of epithelial protection to microbial inflammation (32) was also reduced in ETBF-induced BLM mid-proximal tumors relative to ETBF-induced Min distal tumors.
Lastly, similar to ETBF-induced Min tumors, ETBF-induced BLM tumors were found to be MSS and distinct from tumors from mismatch repair-deficient mice, which are MSI+ (Supplementary Table 4). Altogether the DNA methylation data demonstrates that when the BRAFV600E mutation is present, ETBF-colonization results in a greater number of CpG islands with DNA hypermethylation than in Min only tissue. Furthermore, there are differences in DNA hypermethylation of genes related to the regulation of T cells between ETBF-induced Min and BLM tumors. Altogether, our findings suggest that DNA methylation changes associated with BRAFV600E mutation contribute to the altered inflammatory response to ETBF in BLM mice.
ETBF-colonized BLM mice display increased tumor-infiltrating CD8+ T cells and F4/80+ myeloid cells.
Since BRAFV600E impacts the methylation and expression of some immune-related genes, we sought to characterize the nature of the tumor-infiltrating leukocytes in BLM and Min mice. We performed intracellular cytokine staining and flow cytometry analysis on enzymatically-digested tumors. ETBF-induced BLM tumors had a significant increase in CD3+ CD8+ T cells and IFN-γ-producing cells as compared to ETBF-induced Min tumors (Figure 5A). The monocytic immature myeloid cell (MO-IMC; CD11b+Ly6ChiLy6G−) signature characteristic of ETBF-induced Min tumors (33) was significantly decreased in BLM tumors (Supplementary Figure 5A). There was no difference in IL-17-producing cells or other immune cell types examined between the different genotypes. IHC staining of tumor sections confirmed the accumulation of the CD8+ cells (Figure 5B) and demonstrated the overall accumulation of F4/80+ myeloid cells (Supplementary Figure 5B) in ETBF-induced BLM tumors. Altogether these results indicate BLM tissues with ETBF treatment have a Type 1 immune microenvironment characterized by IFN-γ production and prevalent CD8+ cell infiltration.
Figure 5. IFN-γ-producing CD8+ cells infiltrate ETBF-induced tumors in BLM mice but not Min mice.
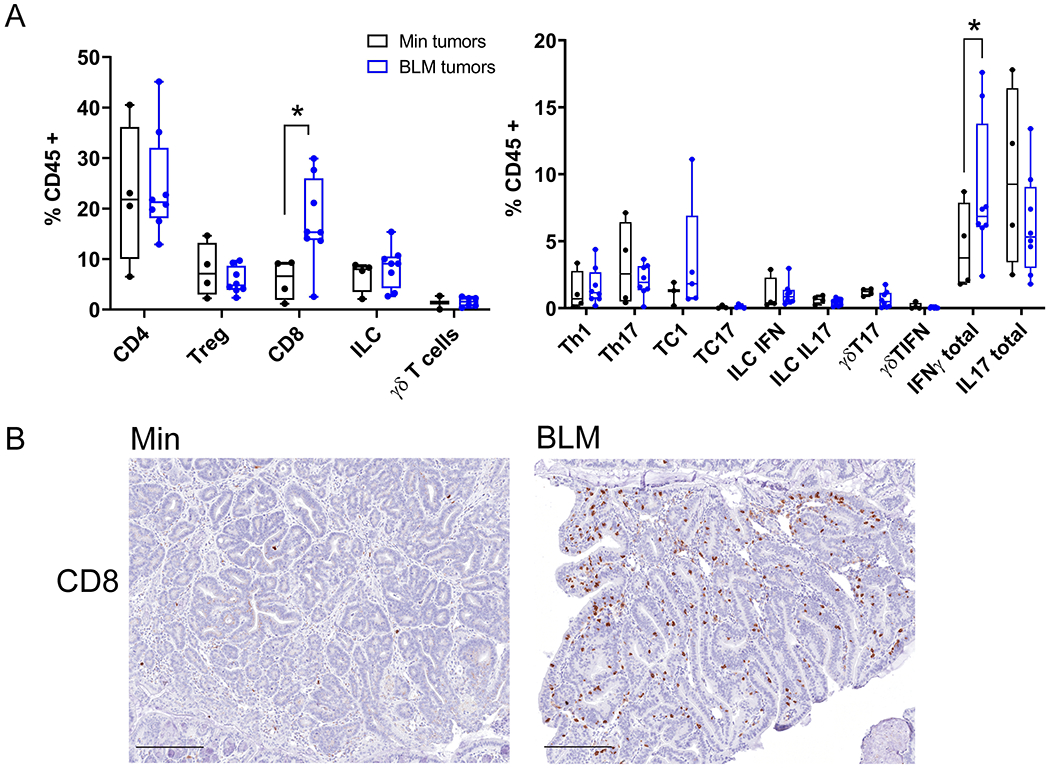
A) Left, Flow cytometry analysis of lamina propria leukocytes isolated from BLM (blue) and Min (black) ETBF-induced colon tumors collected 9-13 weeks post ETBF inoculation. Right, flow cytometry analysis of Intracellular cytokine staining of lamina propria leukocytes isolated from colon tumors in BLM (blue) and Min (black) mice. Each data point represents all tumors for one mouse pooled; Min (N=4) and BLM (N=7) mice from two different experiments. Box limits are set at the third and first quartile range with the central line at the median. *P<0.05 by pair-wise Mann-Whitney U test. B) Representative IHC of CD8+ infiltrating cells in BLM and Min ETBF tumor sections. N= 5-7 mice per group examined and included all tumors along axis of colon. Scale bar, 100μM.
Anti-PDL1 therapy reduces ETBF-driven tumors in BLM but not Min mouse colons.
ETBF-induced tumorigenesis in the mid-proximal region of BLM mice despite increased tissue infiltration with CD8+ T cells and an IFN-γ signature is reminiscent of adaptive immunosuppression described in human cancer whereby adaptive IFN-γ-induced upregulation of T cell checkpoint ligands in the tumor microenvironment suppresses the function of tumor-infiltrating effector T cells (34). Since our gene expression data confirmed higher expression of an IFN-γ-signaling pathway (Stat1, Jak2, Irf1) and IFN-γ-target T cell checkpoint genes (Ido1, Cd274/Pdl1) in ETBF-induced tumors in BLM mice relative Min mice, and Cd274 gene expression was upregulated in accumulating PMN-MDSCs compared to tissue residential macrophages in the early phase of ETBF colitis (Figure 1D), we next tested the hypothesis that checkpoint blockade using anti-PDL1 antibodies would inhibit tumor development in ETBF-colonized BLM mice. ETBF-colonized BLM and Min mice were treated with anti-PDL1 or isotype (control) antibodies (see Methods) and tumor numbers were evaluated 6 weeks after onset of ETBF colonization. We found that anti-PDL1 therapy significantly reduced the total number of ETBF-induced colon tumors in BLM mice whereas anti-PDL1 had no effect in Min mice (Figure 6A). This reduction was seen in both the distal (0-3 cm) and mid-proximal (4-7 cm) region of the colon in BLM ETBF mice treated with anti-PDL1 (Figure 6B).
Figure 6. Anti-PDL1 therapy specifically reduces colon tumorigenesis in ETBF-colonized BLM mice.
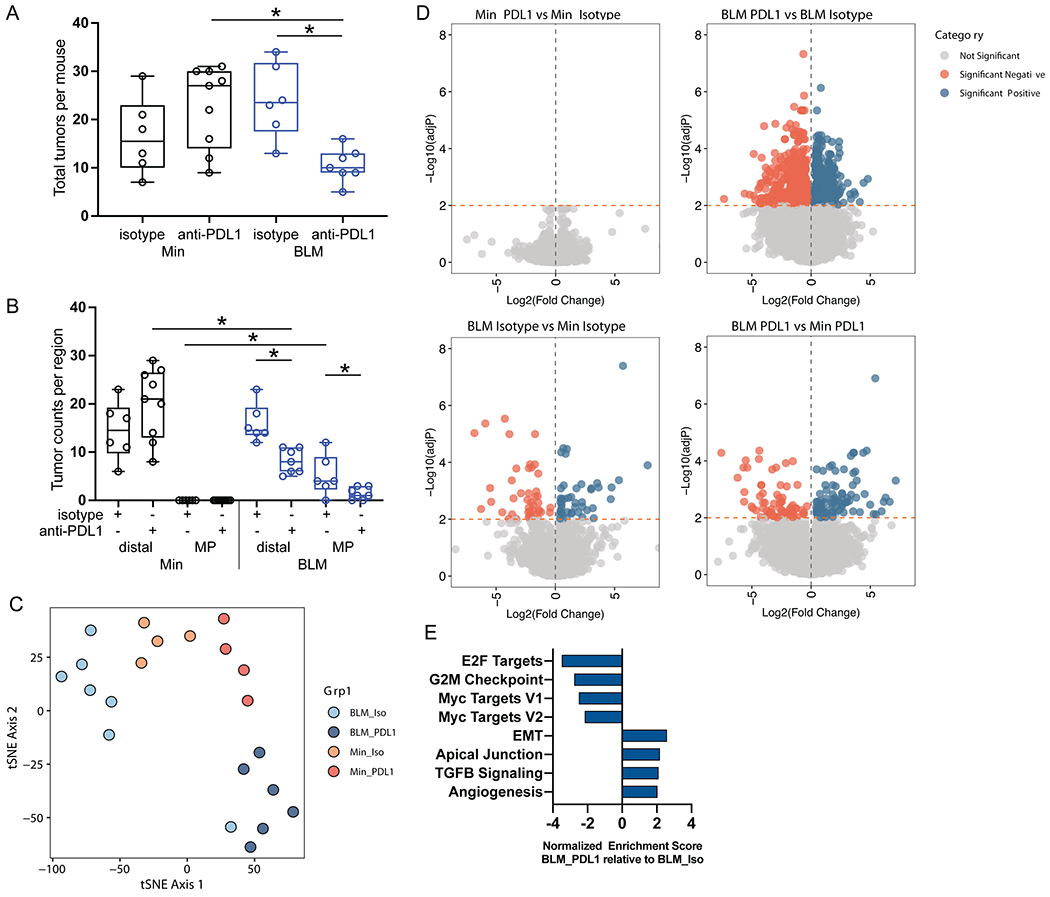
A) Total number of tumors in ETBF-colonized BLM and Min mice treated with anti-PDL1 (Min N=9; BLM N=7) and isotype control (Min N=6; BLM N=6). Mice are from two independent experiments. Box limits are set at the third and first quartile range with the central line at the median; each point is one mouse; black = Min, blue= BLM. *P<0.05 by Mann-Whitney U test. B) Tumors from A separated by distal (0-3 cm) and mid-proximal (4-7 cm) colon regions. Graphs configured and statistics as in A. C) tSNE plot generated using differentially expressed genes (adjP < 0.05 for any pairwise group comparison) in the RNAseq data from mRNA isolated from colon tumors collected from experiment in A. Total number of tumors from each group: Min isotype N=4, BLM isotype N= 7, Min PDL1 N=4, BLM PDL1 N=6. D) Volcano plots of DESseq2 data for comparisons indicated above each graph. Genes with significant differences in expression (adjP < 0.01) are highlighted in orange (downregulated) and blue (upregulated). E) Normalized enrichment score from GSEA analysis of anti-PDL1-treated BLM tumors compared to isotype treatment (Iso) using all genes from the RNA-seq data and hallmark gene sets. All pathways with an FDR < 0.001 are listed.
To further characterize the BLM mouse response to anti-PDL1 therapy, we performed RNA sequencing on mRNA extracted from ETBF-induced BLM mid-proximal and distal and Min distal colon tumors following isotype or anti-PDL1 therapy. Analysis of the RNA-seq data by t-Distributed Stochastic Neighbor Embedding (t-SNE) revealed that tumors from BLM mice treated with anti-PDL1 (BLM_PDL1) predominantly clustered separately from tumors isolated from isotype-treated BLM mice (BLM_Iso), whereas Min_Iso and Min_PDL1 tumors clustered more closely together (Figure 6C). Furthermore, BLM_PDL1 tumors had 334 and 510 genes significantly (adjP<0.01) up- and down-regulated, respectively, when compared to BLM_Iso tumors. In contrast, Min_PDL1 had no genes with significantly altered expression relative to Min_Iso tumors (Figure 6D, Supplementary Table 5). A heatmap of all differentially expressed genes (adjP < 0.05 for any pairwise group comparison) indicated the mid-proximal and distal tumors from BLM mice responded to anti-PDL1 therapy in a similar manner and the similarity between distal and mid-proximal tumors in the BLM_Iso group also suggests that the addition of the BRAFV600E mutation results in similar tumors regardless of region (Supplementary Figure 6A). GSEA with gene sets derived from hallmark pathways demonstrated that BLM_PDL1 tumors are significantly depleted for pathways related to cell proliferation (E2F targets, G2M checkpoint, Myc targets) relative to BLM_Iso tumors. BLM_PDL1 tumors were enriched for epithelial-mesenchymal transition (EMT), TGFβ signaling and angiogenesis pathways suggesting potential mechanisms connected to anti-PDL1 resistance (Figure 6E). The potential resistant state of these tumors is further confirmed by a lack of increase in CD8+ cells (Supplementary Figure 6B) and a reduction in expression of IFN-γ pathway genes (Supplementary Figure 6C) in BLM_PDL1 versus BLM_Iso tumors.
DISCUSSION
Understanding the interaction between genetics, microbes, and inflammation during cancer pathogenesis will aid in predicting and treating cancers. Mouse models of CRC often fail to recapitulate many physiological aspects of human disease. For example, the APC gene is mutated in 85% of sporadic human CRC, yet in so-called Min mice, Apc loss of heterozygosity leads predominantly to small bowel tumors. However, our group has found that upon infecting Min mice with ETBF, the combination of the B. fragilis toxin (BFT) and a Th17 inflammatory response induces the formation of numerous distal colon tumors. ETBF is detected in CRC patients significantly more often when compared to healthy individuals (7,8). ETBF may be an “oncogenic” bacterium candidate triggering in vivo tumorigenesis via inflammation, the production of BFT, and epigenetic modulation of epithelial gene expression (reviewed (2)). Thus, we hypothesize that the multifactorial etiology of human tumors may require similar such components in mouse models. To assist in testing specifically our hypothesis regarding a gene:microbe; inflammation interaction, genotypes in our experiments were co-housed to diminish, if not eliminate, the impact of the broader microbiome members on our results.
BRAFV600E mutation is found in an estimated 90% of BRAF-mutated tumors. BRAF mutation in MSS CRC patients has been associated with a serrated, poorly differentiated, mucinous tumor phenotype and a poor prognosis compared to other subtypes of CRC, including the more common BRAFmut MSI CRCs (35). Herein we tested the impact of the BRAFV600E mutation on ETBF colon tumorigenesis to better understand the pathogenesis of BRAF-mutated CRC. In other murine studies, introduction of the BRAFV600E mutation in all intestinal epithelial cells during gestation resulted in tumor formation predominantly in the small intestine (36). Other studies in which activation of mutant BRAF occurred in adult intestinal cells demonstrated that mutant BRAF inefficiently induced intestinal tumorigenesis without the expression of additional genes (37,38). Our group has previously demonstrated that in the presence of induced BRAFV600E mutation, xenografts derived from ageing transverse colon organoids (5 months in vitro) selected for CIMP-like promoter DNA hypermethylation and activation of the Wnt pathway demonstrate cell transformation with right-sided CRC characteristics (25). These events accelerated with advancing age of the initiating organoids. Thus, while the BRAF mutation likely alters the colon epithelium, key micro-environmental changes, like ETBF-induced inflammation and additional genetic abnormalities, like loss of APC function, are required for full tumorigenesis. Together this work suggests that, in vivo, environmental stress and/or aging is needed for changes in the methylation phenotype in cells bearing the BRAFV600E mutation. In this regard, we confirmed here that the BRAFV600E mutation alone does not result in increased colonic tumors in Min mice, yet upon ETBF colonization, introduction of BRAFV600E drove right-sided colon tumorigenesis with a serrated-like histology, similar to BRAF mutant tumors in humans. Interestingly, we also demonstrated that BRAFV600E mutation and ETBF colonization results in disruption of the mucus layer and significant changes in myeloid populations that persist through tumorigenesis. Overall, the immune and epithelial results did not differ between the mid-proximal and distal regions of BLM mice suggesting that the main drivers of these changes are BRAFV600E mutation combined with ETBF colonization. Disruption of the protective inner layer of mucus that is typically devoid of bacteria likely causes altered interactions between the microbiota and epithelial and immune cells, illustrated herein by the recruitment of PMN-MDSC and their activation by local proinflammatory cytokines. Interestingly, we have recently demonstrated that biofilms, dense communities of inner mucus-invasive bacteria encased in complex matrices, predominantly form on right-sided colon cancers in humans (39), which further underscores regional and genetic differences in the epithelial response to gut flora. These findings suggest that microbiota can play a critical role in the formation of BRAF mutant CRC.
BRAFV600E is associated with CIMP in human CRC. We observed that the BLM epithelium of uninfected mice had low baseline levels of DNA methylation demonstrating that the presence of the BRAFV600E mutation is not sufficient to cause DNA hypermethylation as has been suggested previously (25). However, exposure to ETBF induced significant CpG island DNA hypermethylation in both the BLM mid-proximal epithelium and tumors compared to the sham mid-proximal epithelium baseline. Importantly, this degree of DNA hypermethylation is not present in ETBF-colonized Min distal epithelium, suggesting that the presence of the BRAFV600E mutation and ETBF uniquely induces high levels of CpG island DNA hypermethylation outside of tumorigenesis. BRAFV600E activates constitutive MAPK signaling and can contribute to cell proliferation and survival. We noted that there was both an increase in survival of the acute ETBF infection of mice with BRAFV600E mutation 10 days after ETBF colonization (BL vs WT or BLM vs Min) as well as an increase in γH2AX foci detected by IHC. Previously, we have observed that oxidative damage to chromatin during inflammation leads to epigenetic silencing (23,40). We hypothesize that the increased survival of ETBF-colonized BRAFV600E mice compared to WT or Min controls may indicate that these mice have more intestinal epithelial cells that have undergone and survived oxidative DNA damages, which may, in turn, result in a greater impact of ETBF on epigenetic changes such as DNA methylation. Further, we speculate that enhanced survival fosters mucosal conditions favoring the emergence of a new locus of tumorigenesis in parallel with sensitivity to PDL1 blockade.
BRAF mutation in MSS CRC is associated with a particularly poor prognosis, however our findings suggest that this combination may delineate a population of CRC patients who could benefit from immune checkpoint blockade. The IFN-γ− and Th1-associated expression signatures found in the colon tissues of mice with the BRAFV600E mutation suggest that they are primed to have a predominantly Th1 immune environment. Sham mice bearing BRAFV600E do not show a significant increase of specific IFN-γ-producing cell subsets, including CD4+, CD8+, ILC or γδ-T cells, suggesting that the overall increase in IFN-γ-producing cells stimulated by ETBF results from a general type 1 polarization rather the induction of a specific cell type. Thus, the Th1-related gene expression patterns correlate with priming the BLM tissues to form tumors with high levels of CD8+ T cells and IFN-γ-activated, macrophage-like PMN-MDSCs (Ly6GnegF4/80lowI-A/Elow) (20) as well as a decrease in MO-IMC cells typically detected in Min ETBF tumors. Importantly, the IFN-γ activation of these PMN-MDSCs in BL mice was associated with the upregulation of the immunosuppressive ligand CD274 (aka PDL1). These findings are consistent with the association between BRAFV600E, CIMP and the presence of tumor-infiltrating lymphocytes (TILs) in human CRC (41). Importantly, these characteristics of a type 1 TiME correlated with BLM tumors being responsive to anti-PDL1 therapy. Similar observations were recently made in BRAF-mutated CRC and NSCLC patients that responded to anti-PD1 treatment even if they lack traditional biomarkers such as high tumor mutational burden or PDL1 expression that are normally associated with a positive response to checkpoint therapy (42). Thus, we posit that the BRAFV600E mutation conditions the epithelium to a distinct tumor pathogenesis and TiME during ETBF-driven tumorigenesis that gains responsiveness to checkpoint inhibition. Tumors that persisted in BLM mice after anti-PDL1 therapy are depleted in gene signatures related to cell proliferation, which is consistent with findings in melanomas from patients that responded to anti-PD1 checkpoint therapy (43). Responding melanomas were also found to have upregulation of IFN-γ gene signatures. However, persisting BLM_PDL1 tumors were depleted for IFN-γ and enriched for cancer-promoting pathways, including EMT and angiogenesis. Interestingly, EMT and angiogenesis pathways were enriched in melanomas and other tumor types, including colon cancer, with innate anti-PD1 resistance (44). The persistence of strong signatures associated with non-response in persisting BLM_PDL1 tumors raises important questions. For example, although we saw a response to anti-PDL1 therapy in BLM mice, the signatures we observed in the remaining tumors may herald a relapse, which was not tested in our experiments. Further, in our model the microbiome driver of tumorigenesis is still present and perhaps eliminating ETBF in parallel with anti-PDL1 therapy would change the outcome and/or durability of the immune checkpoint therapy response. Our in situ model of tumorigenesis provides the ideal system to test these hypotheses further. Lastly, despite some similarity between responses in our model and those reported in melanoma, immune checkpoint therapy responses may be tumor specific (42). Currently, studies on responses in cancer types other than melanoma are limited.
Our interest in how microbe-driven inflammation in a BRAF- and Apc-mutated background may change the landscape of the colonic epithelium, mucosal immunology and tumorigenesis potential shows parallels to human CRC development that may help us understand these pathways and better develop therapies and biomarkers for selecting therapies for patients. These observations support the hypothesis that specific microbe:oncogene interactions are crucial to CRC pathogenesis and to guide targeted therapies.
METHODS
Animal Model
C57BL/6J (originally from Jackson Laboratories) and MinApcΔ716/+ mice (Drs. David Huso and Bert Vogelstein (Johns Hopkins University, Baltimore, MD) were handled and inoculated with enterotoxigenic Bacteroides fragilis strain 86-5443-2-2 (ETBF) as in (10). Loxp-flanked BRAFF-V600E were obtained from Jackson labs (RRID:IMSR_JAX:017837) as were Leucine Rich Repeat Containing G Protein-Coupled Receptor 5 (Lgr5) CreERT2 knock-in mice (Lgr5tm1(Cre/ERT2)Cle; RRID:IMSR_JAX:008875) which were then crossed with the MinApcΔ716/+ mice to produce BrafF-V600E Lgr5tm1(Cre/ERT2)CleMinApcΔ716/+ (BLM) mice. Loxp-flanked KrasG12D mice (also known as B6.129S4-Krastm4Tyj/J; RRID:IMSR_JAX:008179) were also obtained from Jackson labs. Recombination in mice bearing Lgr5Cre was induced with tamoxifen as in (45) at 4 weeks of age. All mice were bred and maintained in a specific pathogen-free barrier facility in accordance with the Association for the Assessment and Accreditation of Laboratory Animal Care International. For all experiments, both male and female mice were used. Mice of different genotypes were cohoused upon weaning and for the majority of the experiments in order to maintain a uniform microbiome. The Johns Hopkins University Animal Care and Use Committee approved all experimental protocols.
Tissue harvested for molecular analysis or flow cytometry
Colon regions were defined as: distal (7 day experiments 0–2 cm measured from the rectum; longer time points 0-3 cm) and mid-proximal (the 2 cm region distal to the portion of the mouse colon with transverse folds commonly referred to as the “proximal” colon). Colons were flushed with PBS, measured, and either snap-frozen and stored at −80°C or processed immediately as appropriate for assay. Tumors were counted and mapped along the length of the colon using a dissecting scope and, for some assays, individual tumors were removed for snap-freezing or processing at the time of harvest. Samples specifically described as “epithelial” were collected by scraping debris-free mucosal surface of the dissected colon (tissues were verified to have no gross tumors under the dissecting scope), snap-frozen and stored at −80°C. Others have demonstrated that this method is effective for obtaining intestinal epithelial cells (46). Tissues for immune studies or profiling were snap-frozen whole or immediately processed as appropriate. Tissues were washed three times in PBS before using in the indicated protocol.
Tissue processing for Flow cytometry
Colon tissues were enzymatically processed using 400U/ml Liberase (Sigma Aldrich Cat# 5401127001) and 0.1 mg/ml DNAse1 (Sigma Aldrich Cat# 10104159001)). Splenocytes were isolated from Liberase-treated spleen samples using Lymphoprep density gradient (Accurate Chemical and Scientific Corporation). Leukocytes were isolated from single cell suspensions using 80/40/20 Percoll density gradient centrifugation (GE Healthcare Life Science) as described previously (10). Single-cell suspensions were stained and myeloid and lymphoid populations characterized by flow cytometry as in (13,33) and myeloid cells were stained as in (13,33). Briefly, monocytic immature myeloid cells (MO-IMC) were characterized as CD45+CD3−CD11bhiLy6ChiLy6G−F4/80−/lowI-A/E−/low, polymorphonuclear immature myeloid cells (PMN-IMC) were CD45+CD3−CD11bhi Ly6CintLy6G+F4/80−I-A/E−, Myeloid CD45+CD3−CD11bintLy6C−Ly6G−F4/80+ cells were further subsetted into F4/80highI-E/Ahigh (CD45+CD11b+Ly6G−Ly6C−F4/80highI-A/Ehigh) and F4/80lowI-A/Elow (CD45+CD11b+Ly6G−Ly6C−F4/80lowI-A/Elow).
Intracellular cytokine staining was performed following 4.5 hr stimulation with phorbol 12-myristate 13-acetate and ionomycin stimulation cocktail (eBioscience) in the presence of protein inhibitor cocktail (eBioscience). Cells were subsequently stained as referenced (13,24) with cell surface markers followed by fixation/permeabilization (Cytofix/Cytoperm, BD) enabling intracellular staining. Flow cytometry was performed using a LSR-FORTESSA cytometer (BD) and data were analyzed with FACSDiVa 6.1.3 software (BD FACSDiva Software, RRID:SCR_001456).
In the case of tumor tissues, for each mouse all colon tumors were pooled, minced in 5% FBS RPMI and enzymatically processed (400U/ml Liberase and 0.1 mg/ml DNAse1; Sigma Aldrich) for 15 min in a 37°C 5% CO2 incubator. Tissues pieces were then passed through a 70µM cell strainer to create a single cell suspension. Cells were then washed and counted before being stained for flow cytometry analysis.
Cell-sorting of colonic myeloid cells.
Single cell suspension of mid-proximal portions of the colons were obtained by enzymatic digestion as described above, and stained with CD45, Ly6C, Ly6G, F4/80 and I-A/E antibodies (13,33). CD45+CD11b+Ly6G−Ly6C−F4/80lowI-A/Elow and CD45+CD11b+Ly6G−Ly6C−F4/80highI-A/Ehigh were cell sorted using a BD FACSAria™ FUSION cell sorter. Cells were directly collected in RLT buffer of RNeasy micro kit (Qiagen Cat# 74004) for RNA extraction as described below.
Fixation of tissues for histology and immunohistochemistry
Colons for histology were flushed, bisected longitudinally, pinned flat and fixed using 10% buffered formalin for 48 hours at room temperature. Colons were then rolled into cassettes for standard paraffin-embedding. For samples in which the maintenance of mucus and/or mucosal/epithelial contact was desired, intact colons were fixed directly in methacarn (60% methanol, 30% chloroform, 10% acetic acid) for 48 hours at 4°C. Periodic acid-Schiff (PAS) staining of these tissues was carried out according to kit instructions (Millipore Cat# 101646). Other tissues such as spleen, liver, and mesenteric lymph nodes were fixed in formalin for 48 hours.
Histology
Inflammation scores for colons 7 days post-ETBF colonization were graded blindly by a board-certified pathologist as in (12). Briefly, scores for inflammation were: 0, normal; 1, mild increase in inflammatory cells and no mucosal epithelial changes (no proliferation or loss of crypt structure); 2, moderate increase in inflammatory cells and mild scattered mucosal epithelial proliferation with or without focal loss of crypt architecture; 3, moderate increase in inflammatory cells, diffuse or nearly diffuse (more than two sites) mucosal epithelial proliferation, and edema with or without focal loss of crypt architecture; 4, severe increase in inflammatory cells and marked consistent proliferation with extensive loss (more than two sites) of crypt architecture.
Immunohistochemistry
Immunostaining of colon sections was performed with the PowerVision kit according to the manufacturer’s protocol (Leica Biosystems). Briefly, slides were heated at 60°C for 10 min, deparaffinized and hydrated through xylene, graded ethyl alcohols, dH2O, dH2O with 20% Tween 20 (Sigma-Aldrich Cat# P-7949). After antigen retrieval, 25 minutes of steaming in Target Retrieval Solution (Dako Cat# S170084-2) using Black and Decker Handy Steamer Plus, sections were treated 5 minutes with Dual Blocking Solution (Dako Cat# S2003). Primary antibodies γH2AX (Cell Signaling Cat# 9718, 1:800) and MUC2 (Santa Cruz Biotechnology Cat# sc-15334, RRID:AB_2146667, 1:100) were incubated at room temperature for 45 minutes. Slides for CD8 and F4/80 staining were pre-blocked in DakoCytomation Biotin Blocking System (Dako Cat# X0590). Antibodies for CD8 (eBiosciences Cat# 14-0808, 1:800) and F4/80 (Bio-Rad Cat# MCAP497, RRID:AB_2335598 (formerly Serotec, MCAP497), 1:1000) were incubated at room temperature for 45 minutes, soaked an additional 45 min in PBS-Tween, and followed by mouse-adsorbed biotinylated anti-Rat IgG (Vector Laboratories Cat# BA-4001, RRID:AB_10015300, 1:500). For all, the secondary antibody used was the anti-rabbit IgG-reagent provided in the Powervision+PolyDAB kit (Leica Biosystems Cat# PV6119). DAB chromogen (Sigma-Aldrich Cat# D4293) was used for immunostaining visualization and sections were counterstained with Mayer’s hematoxylin.
Methyl CpG binding domain (MBD)-seq
DNA was isolated from tumors or epithelium using the QIAamp DNA mini kit (Qiagen Cat# 51306) following the manufacturer’s protocol. To identify differentially methylated regions (DMRs), MBD enrichment was performed on DNA from epithelium or individual tumors using Diagenode’s MethylCap kit. Libraries were prepared using Bio Scientific’s Methyl Sequencing kit. Single-end 75 bp sequencing was performed using an Illumina Nextseq. For this study, MBD-seq was performed on Min sham epithelium (N=1), Min ETBF tumor (N=1), BLM sham epithelium (N=3), BLM ETBF epithelium (N=3), and BLM ETBF tumors (N=5). Previous MBD-seq data from Maiuri et al. (14) was also included (N=3 Min sham epithelium, N=3 Min ETBF epithelium, N=5 Min ETBF tumors). Z-scores were calculated using a 500bp fixed-sized bin spanning CpG islands based on the distribution of coverage from uniquely mapped reads. Z-ratios were derived from the comparison of z-scores for the different sample types for the 500bp sized regions. Additional details are in Maiuri et al. (14).
RNA-seq
For cell-sorted MDSC and macrophages (mid-proximal regions from mice, WT N=3; BL N=3), RNA was isolated and DNase treated using the RNeasy micro kit (Qiagen Cat# 74004). Libraries were prepared with Nugen Ovation RNAseq system v2. Paired-end 150 bp sequencing was performed using an Illumina HiSeqX. Illumina’s CASAVA 1.8.4 was used to convert BCL files to FASTQ files using default parameters. Data was aligned to the mm10 reference genome.
For BLM and Min tumors and epithelium, RNA was isolated from epithelium (4 each sham Min distal and sham BLM mid-proximal) and tumors (4 ETBF Min distal and 5 ETBF BLM mid-proximal) using the RNeasy mini kit (Qiagen, 74104) and RNeasy micro kit (Qiagen Cat# 74004), respectively, and DNase-treated. Libraries were prepared with Illumina’s TruSeq Stranded mRNA library kit and were poly-A tail selected. Paired-end 75 bp sequencing was performed using an Illumina Nextseq. Raw fastq files reflecting single-end TruSeq Stranded RNA libraries were submitted to RSEM for stranded alignment (Bowtie2) using the GRCm38 reference genome masked for miscellaneous RNA (e.g. rRNA, lincRNA, snRNA etc.), mitochondrial genes, and ribosomal proteins.
For tumors from the PDL1 checkpoint experiment, RNA was isolated from tumors (BLM isotype: distal N=3, mid-proximal N=3; BLM PDL1: distal N=3, mid-proximal N=4; Min isotype: distal N=4; Min PDL1: distal N=4) and then DNase treated using the RNeasy micro kit. Libraries were generated using NEB Next Ultra RNA Library Prep Kit for Illumina (NEB, USA). Paired-end 150 bp sequencing was performed on an Illumina Novaseq 6000. Data was aligned to the GRCm38 reference genome.
RSEM quantified counts were submitted to DESeq2 (RRID:SCR_000154) for downstream normalization and differential expression analysis. p-values were calculated using the Wald test implemented in DESeq2 and adjusted p-values reflect an FDR correction for multiple hypothesis testing. CIBERSORT (RRID:SCR_016955) was run on RNA-seq profiles with default parameters and utilizing a mouse-specific immune cell gene signature matrix (ImmuCC; (47)). Gene Set Enrichment Analysis (GSEA) was performed on RNA-seq data using the GSEA software SeqGSEA (RRID:SCR_005724) and Hallmark, BioCarta Pathways (RRID:SCR_006917), and mouse MDSC gene sets (48), (49).
Anti-PDL1 therapy
Antibodies from Bristol-Myers Squibb were isotype control anti-DT 1D12 mIgG1 clone 4F7_RAS_Ab U2017010. AB107918.13 1469/Diptheria and anti-PDL1 PDL1 14D8 Chimeric_RAS_Ab_05 U20161214. AB294451.01 1105/B7-H1 Anti-mouse PDL1 and isotype controls were each given at 5 mg/kg dosages intraperitoneally (ip). Mice began treatment 7 days after ETBF colonization and antibodies were administered i.p. twice a week for a 6 week tumorigenesis terminal time point.
Data availability
Raw next-generation sequencing data utilized in these studies is available through NCBI under accession numbers: PRJNA687231, PRJNA687232, PRJNA687233, PRJNA687236 and SRP105286.
Supplementary Material
SIGNIFICANCE.
Colorectal cancers (CRCs) with BRAF mutations have distinct characteristics. We present evidence of specific CRC gene:microbial interactions where colonization with toxigenic bacteria drives tumorigenesis in BRAFV600ELgr5CreMin mice wherein tumors phenocopy aspects of human BRAF-mutated tumors and have a distinct interferon-gamma-dominant immune microenvironment uniquely responsive to immune checkpoint blockade.
Acknowledgements
We are deeply grateful for the contributions of Dr. David L. Huso in evaluating the pathology of this model.
Funding
Support for this research was provided from the following grants:
National Institute of Environmental Health Sciences, National Institutes of Health grants R01 ES023183 to HMOH and R01 ES011858 to SBB;
Bloomberg Philanthropies, Cancer Research UK (#C10674/A27140) and institutional resources from JHU SOM and DOM to CLS;
Swim Across America and National Cancer Institute, National Institutes of Health grant R01 CA203891 to FH;
We thank the Indiana University Center for Genomics and Bioinformatics and the Indiana Molecular Biology Institute for their assistance.
We thank the members of the Sidney Kimmel Comprehensive Cancer Center Experimental and Computational Genomics Core (supported by NCI grant P30CA006973) for their support with RNA-seq. We thank the Johns Hopkins University Oncology Tissue Services for tissue processing and slides (supported by NCI grant P30CA006973).
COI statement: CLS reports research funding from Bristol Myers Squibb and Janssen and International Patent Application No.: PCT/US19/20161. The other authors declare no potential conflicts of interest.
REFERENCES
- 1.Siegel RL, Miller KD, Goding Sauer A, Fedewa SA, Butterly LF, Anderson JC, et al. Colorectal cancer statistics, 2020. CA Cancer J Clin 2020;70(3):145–64 doi 10.3322/caac.21601. [DOI] [PubMed] [Google Scholar]
- 2.Sears CL, Garrett WS. Microbes, microbiota, and colon cancer. Cell host & microbe 2014;15(3):317–28 doi 10.1016/j.chom.2014.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen J, Domingue JC, Sears CL. Microbiota dysbiosis in select human cancers: Evidence of association and causality. Seminars in immunology 2017;32:25–34 doi 10.1016/j.smim.2017.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Garrett WS. The gut microbiota and colon cancer. Science 2019;364(6446):1133–5 doi 10.1126/science.aaw2367. [DOI] [PubMed] [Google Scholar]
- 5.Tuddenham S, Sears CL. The intestinal microbiome and health. Curr Opin Infect Dis 2015;28(5):464–70 doi 10.1097/QCO.0000000000000196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gao Z, Guo B, Gao R, Zhu Q, Qin H. Microbiota disbiosis is associated with colorectal cancer. Frontiers in microbiology 2015;6:20 doi 10.3389/fmicb.2015.00020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Boleij A, Hechenbleikner EM, Goodwin AC, Badani R, Stein EM, Lazarev MG , et al. The Bacteroides fragilis toxin gene is prevalent in the colon mucosa of colorectal cancer patients. Clin Infect Dis 2015;60(2):208–15 doi 10.1093/cid/ciu787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Toprak NU, Yagci A, Gulluoglu BM, Akin ML, Demirkalem P, Celenk T , et al. A possible role of Bacteroides fragilis enterotoxin in the aetiology of colorectal cancer. Clin Microbiol Infect 2006;12(8):782–6 doi CLM1494[pii] 10.1111/j.1469-0691.2006.01494.x. [DOI] [PubMed] [Google Scholar]
- 9.Purcell RV, Pearson J, Aitchison A, Dixon L, Frizelle FA, Keenan JI. Colonization with enterotoxigenic Bacteroides fragilis is associated with early-stage colorectal neoplasia. PLoS One 2017;12(2):e0171602 doi 10.1371/journal.pone.0171602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wu S, Rhee KJ, Albesiano E, Rabizadeh S, Wu X, Yen HR , et al. A human colonic commensal promotes colon tumorigenesis via activation of T helper type 17 T cell responses. Nat Med 2009;15(9):1016–22 doi 10.1038/nm.2015nm.2015[pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rajagopalan H, Bardelli A, Lengauer C, Kinzler KW, Vogelstein B, Velculescu VE. Tumorigenesis: RAF/RAS oncogenes and mismatch-repair status. Nature 2002;418(6901):934 doi 10.1038/418934a. [DOI] [PubMed] [Google Scholar]
- 12.Rhee KJ, Wu S, Wu X, Huso DL, Karim B, Franco AA , et al. Induction of persistent colitis by a human commensal, enterotoxigenic Bacteroides fragilis, in wild-type C57BL/6 mice. Infect Immun 2009;77(4):1708–18 doi 10.1128/IAI.00814-08IAI.00814-08[pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chung L, Orberg ET, Geis AL, Chan JL, Fu K, DeStefano Shields CE , et al. Bacteroides fragilis Toxin Coordinates a Pro-carcinogenic Inflammatory Cascade via Targeting of Colonic Epithelial Cells. Cell host & microbe 2018;23(3):421 doi 10.1016/j.chom.2018.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol 2009;9(3):162–74 doi 10.1038/nri2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bronte V, Serafini P, Mazzoni A, Segal DM, Zanovello P. L-arginine metabolism in myeloid cells controls T-lymphocyte functions. Trends in immunology 2003;24(6):302–6 doi 10.1016/s1471-4906(03)00132-7. [DOI] [PubMed] [Google Scholar]
- 16.Srivastava MK, Sinha P, Clements VK, Rodriguez P, Ostrand-Rosenberg S. Myeloid-derived suppressor cells inhibit T-cell activation by depleting cystine and cysteine. Cancer Res 2010;70(1):68–77 doi 10.1158/0008-5472.CAN-09-2587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ding L, Hayes MM, Photenhauer A, Eaton KA, Li Q, Ocadiz-Ruiz R , et al. Schlafen 4-expressing myeloid-derived suppressor cells are induced during murine gastric metaplasia. J Clin Invest 2016;126(8):2867–80 doi 10.1172/JCI82529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Groth C, Hu X, Weber R, Fleming V, Altevogt P, Utikal J , et al. Immunosuppression mediated by myeloid-derived suppressor cells (MDSCs) during tumour progression. Br J Cancer 2019;120(1):16–25 doi 10.1038/s41416-018-0333-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ostrand-Rosenberg S Myeloid-derived suppressor cells: more mechanisms for inhibiting antitumor immunity. Cancer Immunol Immunother 2010;59(10):1593–600 doi 10.1007/s00262-010-0855-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Whittaker Hawkins RF, Patenaude A, Dumas A, Jain R, Tesfagiorgis Y, Kerfoot S , et al. ICAM1+ neutrophils promote chronic inflammation via ASPRV1 in B cell-dependent autoimmune encephalomyelitis. JCI Insight 2017;2(23) doi 10.1172/jci.insight.96882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hanata N, Shoda H, Hatano H, Nagafuchi Y, Komai T, Okamura T , et al. Peptidylarginine Deiminase 4 Promotes the Renal Infiltration of Neutrophils and Exacerbates the TLR7 Agonist-Induced Lupus Mice. Front Immunol 2020;11:1095 doi 10.3389/fimmu.2020.01095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol 2012;12(4):253–68 doi 10.1038/nri3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Maiuri AR, Peng M, Sriramkumar S, Kamplain CM, DeStefano Shields CE, Sears CL , et al. Mismatch Repair Proteins Initiate Epigenetic Alterations during Inflammation-Driven Tumorigenesis. Cancer Res 2017;77(13):3467–78 doi 10.1158/0008-5472.CAN-17-0056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Maiuri AR, Savant SS, Podicheti R, Rusch DB, O’Hagan HM. DNA methyltransferase inhibition reduces inflammation-induced colon tumorigenesis. Epigenetics 2019;14(12):1209–23 doi 10.1080/15592294.2019.1634986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tao Y, Kang B, Petkovich DA, Bhandari YR, In J, Stein-O’Brien G , et al. Aging-like Spontaneous Epigenetic Silencing Facilitates Wnt Activation, Stemness, and Braf(V600E)-Induced Tumorigenesis. Cancer Cell 2019;35(2):315–28 e6 doi 10.1016/j.ccell.2019.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liberzon A, Birger C, Thorvaldsdottir H, Ghandi M, Mesirov JP, Tamayo P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst 2015;1(6):417–25 doi 10.1016/j.cels.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen B, Khodadoust MS, Liu CL, Newman AM, Alizadeh AA. Profiling Tumor Infiltrating Immune Cells with CIBERSORT. Methods Mol Biol 2018;1711:243–59 doi 10.1007/978-1-4939-7493-1_12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rouillard AD, Gundersen GW, Fernandez NF, Wang Z, Monteiro CD, McDermott MG , et al. The harmonizome: a collection of processed datasets gathered to serve and mine knowledge about genes and proteins. Database (Oxford) 2016;2016:baw100 doi 10.1093/database/baw100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Abraham SN, John AL. Mast cell-orchestrated immunity to pathogens. Nat Rev Immunol 2010;10(6):440–52 doi 10.1038/nri2782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hodges K, Kennedy L, Meng F, Alpini G, Francis H. Mast cells, disease and gastrointestinal cancer: A comprehensive review of recent findings. Transl Gastrointest Cancer 2012;1(2):138–50. [PMC free article] [PubMed] [Google Scholar]
- 31.Sommerfeld SD, Cherry C, Schwab RM, Chung L, Maestas DR Jr., Laffont P , et al. Interleukin-36gamma-producing macrophages drive IL-17-mediated fibrosis. Sci Immunol 2019;4(40) doi 10.1126/sciimmunol.aax4783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Moschen AR, Gerner RR, Wang J, Klepsch V, Adolph TE, Reider SJ , et al. Lipocalin 2 Protects from Inflammation and Tumorigenesis Associated with Gut Microbiota Alterations. Cell host & microbe 2016;19(4):455–69 doi 10.1016/j.chom.2016.03.007. [DOI] [PubMed] [Google Scholar]
- 33.Thiele Orberg E, Fan H, Tam AJ, Dejea CM, Destefano Shields CE, Wu S , et al. The myeloid immune signature of enterotoxigenic Bacteroides fragilis-induced murine colon tumorigenesis. Mucosal Immunol 2017;10(2):421–33 doi 10.1038/mi.2016.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Taube JM, Anders RA, Young GD, Xu H, Sharma R, McMiller TL , et al. Colocalization of inflammatory response with B7-h1 expression in human melanocytic lesions supports an adaptive resistance mechanism of immune escape. Sci Transl Med 2012;4(127):127ra37 doi 10.1126/scitranslmed.3003689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Taieb J, Le Malicot K, Shi Q, Penault Lorca F, Bouche O, Tabernero J , et al. Prognostic Value of BRAF and KRAS Mutations in MSI and MSS Stage III Colon Cancer. J Natl Cancer Inst 2017;109(5) doi 10.1093/jnci/djw272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rad R, Cadinanos J, Rad L, Varela I, Strong A, Kriegl L , et al. A genetic progression model of Braf(V600E)-induced intestinal tumorigenesis reveals targets for therapeutic intervention. Cancer Cell 2013;24(1):15–29 doi 10.1016/j.ccr.2013.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sakamoto N, Feng Y, Stolfi C, Kurosu Y, Green M, Lin J , et al. BRAFV600E cooperates with CDX2 inactivation to promote serrated colorectal tumorigenesis. eLife 2017;6 doi 10.7554/eLife.20331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tong K, Pellon-Cardenas O, Sirihorachai VR, Warder BN, Kothari OA, Perekatt AO , et al. Degree of Tissue Differentiation Dictates Susceptibility to BRAF-Driven Colorectal Cancer. Cell reports 2017;21(13):3833–45 doi 10.1016/j.celrep.2017.11.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dejea CM, Wick EC, Hechenbleikner EM, White JR, Mark Welch JL, Rossetti BJ , et al. Microbiota organization is a distinct feature of proximal colorectal cancers. Proc Natl Acad Sci U S A 2014;111(51):18321–6 doi 10.1073/pnas.1406199111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.O’Hagan HM, Wang W, Sen S, Destefano Shields C, Lee SS, Zhang YW , et al. Oxidative damage targets complexes containing DNA methyltransferases, SIRT1, and polycomb members to promoter CpG Islands. Cancer Cell 2011;20(5):606–19 doi 10.1016/j.ccr.2011.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ling A, Lundberg IV, Eklof V, Wikberg ML, Oberg A, Edin S , et al. The infiltration, and prognostic importance, of Th1 lymphocytes vary in molecular subgroups of colorectal cancer. The journal of pathology Clinical research 2016;2(1):21–31 doi 10.1002/cjp2.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Smith KN, Llosa NJ, Cottrell TR, Siegel N, Fan H, Suri P , et al. Persistent mutant oncogene specific T cells in two patients benefitting from anti-PD-1. J Immunother Cancer 2019;7(1):40 doi 10.1186/s40425-018-0492-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Grasso CS, Tsoi J, Onyshchenko M, Abril-Rodriguez G, Ross-Macdonald P, Wind-Rotolo M , et al. Conserved Interferon-gamma Signaling Drives Clinical Response to Immune Checkpoint Blockade Therapy in Melanoma. Cancer Cell 2020;38(4):500–15 e3 doi 10.1016/j.ccell.2020.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hugo W, Zaretsky JM, Sun L, Song C, Moreno BH, Hu-Lieskovan S , et al. Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell 2016;165(1):35–44 doi 10.1016/j.cell.2016.02.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Barker N, van Es JH, Kuipers J, Kujala P, van den Born M, Cozijnsen M , et al. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature 2007;449(7165):1003–7 doi 10.1038/nature06196. [DOI] [PubMed] [Google Scholar]
- 46.Ortega-Cava CF, Ishihara S, Rumi MA, Aziz MM, Kazumori H, Yuki T , et al. Epithelial toll-like receptor 5 is constitutively localized in the mouse cecum and exhibits distinctive down-regulation during experimental colitis. Clin Vaccine Immunol 2006;13(1):132–8 doi 10.1128/CVI.13.1.132-138.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen Z, Huang A, Sun J, Jiang T, Qin FX, Wu A. Inference of immune cell composition on the expression profiles of mouse tissue. Scientific reports 2017;7:40508 doi 10.1038/srep40508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA , et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 2005;102(43):15545–50 doi 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Alshetaiwi H, Pervolarakis N, McIntyre LL, Ma D, Nguyen Q, Rath JA , et al. Defining the emergence of myeloid-derived suppressor cells in breast cancer using single-cell transcriptomics. Sci Immunol 2020;5(44) doi 10.1126/sciimmunol.aay6017. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw next-generation sequencing data utilized in these studies is available through NCBI under accession numbers: PRJNA687231, PRJNA687232, PRJNA687233, PRJNA687236 and SRP105286.