

Abstract
Transient receptor potential vanilloid 4 (TRPV4) is a ubiquitously expressed polymodally activated ion channel. TRPV4 has been implicated in tumor progression; however, the cell-specific role of TRPV4 in tumor growth, angiogenesis and metastasis is unknown. Here, we generated endothelial-specific TRPV4 knockout (TRPV4ECKO) mice by crossing TRPV4 lox/lox mice with Tie2-Cre mice. Tumor growth and metastasis were significantly increased in a syngeneic Lewis lung carcinoma tumor model of TRPV4ECKO mice compared to TRPV4lox/lox mice. Multiphoton microscopy, dextran leakage and immunohistochemical analysis revealed increased tumor angiogenesis and metastasis that were correlated with aberrant leaky vessels (increased width and reduced pericyte and VE-cadherin coverage). Mechanistically, increases in VEGFR2, p-ERK and MMP-9 expression and DQ gelatinase activity were observed in the TRPV4ECKO mouse tumors. Our results demonstrated that endothelial TRPV4 is a critical modulator of vascular integrity and tumor angiogenesis and that deletion of TRPV4 promotes tumor angiogenesis, growth, and metastasis.
Keywords: Endothelial cell, metastasis, transient receptor potential vanilloid 4, tumor angiogenesis, vascular endothelial growth factor receptor 2
1. Introduction
TRPV4 is a polymodal cation channel activated by hypotonicity, temperature, shear stress, arachidonic acid, epoxyeicosatrienoic acid (EET), matrix stiffness and mechanical stretch in various cell types[1–9]. Recently, TRPV4 has been implicated in the growth and progression of breast, gastric, colon, and melanoma cancers[10–15]. In contrast to these findings, Peters et al. demonstrated that activation of TRPV4 induced oncosis and apoptosis of breast cancer cells in vitro and reduced tumor growth in vivo[16]. These findings suggest that either activation or inhibition of TRPV4 in tumor cells can differentially regulate tumor growth and metastasis of different tumors.
In addition to tumor cells, TRPV4 is expressed in cells of the tumor microenvironment. Notably, the mechanosensitive role of TRPV4 in endothelial cells (ECs) has attracted increased attention. In breast tumor-derived ECs but not in normal human ECs (HMVECs), TRPV4 was shown to regulate arachidonic acid-induced migration[17], implicating it in tumor angiogenesis. However, this study did not measure angiogenesis in vitro or in vivo. We recently demonstrated enhanced tumor growth in vivo in global TRPV4 knockout (KO) mice[1]. Nevertheless, it is not clear whether the enhanced tumor growth in the global TRPV4KO mice is due to the effects of TRPV4 on tumor cells themselves, tumor-associated ECs or both types of cells. To demonstrate the cell-specific role of TRPV4, we generated endothelial-specific TRPV4KO mice and investigated its role in tumor angiogenesis, growth, and metastasis.
2. Materials and Methods
2.1. Generation of endothelial-specific TRPV4 knockout mice
All animal (C57BL/6J background) experiments were approved by the Institutional Animal Care and Use Committees of Northeast Ohio Medical University (NEOMED). EC-specific TRPV4 knockout mice (TRPV4f/f-Tie2-Cre; TRPV4ECKO) were generated by a two-step process: in the first generation, TRPV4lox/lox mice[18] were crossed with Tie2-Cre mice, and the resulting heterozygous (Cre/Loxphet) mice were again crossed to generate homozygous TRPV4f/f Tie2-Cre mice. Littermates without Tie2–Cre were employed as controls. Genotyping was performed by PCR using ear punch/tail snip biopsies. Endothelial deletion of TRPV4 was further confirmed by measuring TRPV4 expression (RT-PCR) and calcium influx (calcium imaging) in primary ECs isolated from aortic explants.
2.2. Cell culture
Lewis lung carcinoma cells (LLCs) were cultured as described previously [2, 5]. Isolated ECs were characterized and cultured as previously described[19,1,20,21]. Briefly, ECs were isolated from the aorta in a two-step positive isolation process that utilized anti-CD31 antibody conjugated to magnetic beads and then anti-ICAM2 conjugated beads[22]. The EC phenotype was characterized by measuring a) the endothelial markers CD31 and VEGFR2, b) uptake of acetylated LDL and c) tube formation (2D angiogenesis) on Matrigel (Suppl. Fig. 1). Cells from passages 2–4 were employed to carry out all the experiments.
2.3. Syngeneic tumor model
LLCs injection and tumor growth assessment were performed as described previously[1,3].
2.4. RT-PCR and qPCR
RNA isolation, cDNA synthesis, RT-PCR, and qPCR were performed as previously described[23]. The following real-time mouse-specific primers were obtained from IDT Technologies (USA):
TRPV4 (F-5’-CAAGTACCCCGTGGTCTTCAT −3’; R: 5’-GTTCAACAGGAGCACGAAGG −3’),
VEGFR2 (F-5’-CTACAAGTGCTCGTACCGGG −3’; R-5’-TCGGTGATGTACACGATGCC-3’),
VEGF (F-5’-CAAACCTCACCAAAGCCAGC −3’; R-5’-GCGCTTTCGTTTTTGACCCT −3’),
MMP-9 (F −5’-GGTCTTCCCCAAAGACCTGAAA −3’; R-5’-AGAGACTGCTTCTCTCCCATCA −3’),
MMP-2 (F-5’-AACGGTCGGGAATACAGCAG −3’; R-5’-TGGTAAACAAGGCTTCATGGGG −3’),
GAPDH (F-5’-GGGTCCCAGCTTAGGTTCATC −3’, R-5’-ATCCGTTCACACCGACCTTC −3’) and
TIMP-2 (F-5’-AGAAGGAGGTGGATTCCGGG-3’, R-5’-AGAGGGGGCCGTGTAGATAAA-3’).
2.5. Immunohistochemistry for OCT-embedded tissues
Aorta, lungs, liver, and retina were extracted from the TRPV4lox/lox and TRPV4ECKO mice and frozen in OCT (Tissue Tek, USA,) or fixed in paraformaldehyde. Tissue sections (10 μm) were incubated with the following primary antibodies: mouse anti-CD31 (1:50; Invitrogen, Carlsbad, CA, USA) and rabbit anti-TRPV4 (1:50; Biorbyt, UK). After the samples were washed with PBS, the tissue sections were incubated with appropriate secondary antibodies coupled to Alexa Fluor-488 or Alexa Fluor-594 (Invitrogen) and mounted with DAPI Fluoromount (Vector Labs, USA). Retinal flat mounts were stained with Alexa Fluor-594-conjugated isolectin IB4. Images (x10, x20) were captured using either an Olympus IX81 or FV1000 confocal microscope and processed using NIH ImageJ software.
2.6. Western blot
Tumor tissues were lysed, and the protein content was measured by Western blotting. The membranes were incubated with primary antibodies against total VEGFR2 (1:1000; Cell Signaling, Danvers, MA, USA), total and phosphor-ERK1/2 (1:1000; Cell Signaling), HIF-1α (1:1000; Cell Signaling Technologies, USA) and GAPDH (1:5000; Cell Signaling Technologies, USA) followed by goat anti-rabbit (1:20,000) antibodies conjugated with horseradish peroxidase (Cell Signaling Technologies, USA). Clarity Western ECL Substrate (Bio-Rad, Hercules, CA, USA) was employed to detect the signals using a FluorChem M Simple Imager (Protein Simple, USA).
2.7. Calcium imaging
Calcium imaging was performed as previously described[19,1,2,23,20]. Cells were stimulated with the TRPV4 selective agonist GSK1016790A (GSK101; 100 nM, Sigma, USA).
2.8. Tumor vascular leakage/permeability
Tumor vascular leakage/permeability was assessed as previously described[1,3]. Briefly, dextran, Texas Red™, (3000 MW, Thermo Fisher Scientific, USA) was injected into tumor-bearing mice via tail vein injections. Tumors were extracted and fixed, and sections were imaged. Image processing and quantification were performed by using ImageJ software (National Institutes of Health). The results are shown as the mean fluorescence intensity.
2.9. Multiphoton microscopy
Mice bearing LLC tumors were anesthetized with isoflurane (2–3%) and injected with heparin (100 units/100 μl) via the inferior vena cava to prevent blood clotting. One hundred microliters of Alexa Fluor™ 594 isolectin GS-IB4 conjugate (1 μg/μl) (Molecular Probes, Thermo Fisher Scientific, USA) was injected via the inferior vena cava, and after 15 min, the mice were euthanized by Fatal-Plus solution. The mice were perfused with 10 ml of 10% formalin followed by 10 ml of PBS. Tumors were harvested, and deep vascular imaging (20x) was performed using either multiphoton or confocal microscopy. Image processing and quantification were performed by Fluoview Viewer software. The tortuosity index was measured by dividing the segmented line of the vessel by the distance between the start and end of the vessel using NIH ImageJ software.
2.10. Lung metastasis
The metastasis of LLCs to the lung tissue was monitored as previously described[3], and the total number of metastatic foci was calculated per lung. The data are reported as the total number of metastatic foci.
2.11. Immunohistochemistry of OCT-embedded tumors
Tumor immunohistochemistry was performed as previously described[1,3,21]. The following antibodies were employed: mouse-anti-CD31 (1:50; Invitrogen,USA), anti-CD45 (1:50; Cell Signaling Technologies, USA), rabbit anti- NG2 (1:200) (Millipore, USA), rabbit and mouse anti- VE-cadherin (1:100) (BD Biosciences, USA), rabbit anti-VEGFR2 (1:50; Cell Signaling Technologies, USA) and appropriate secondary antibodies coupled to Alexa Fluor-488 or Alexa Fluor-594 (Invitrogen, USA). The total number of CD31+ stained vessels and the number of vessels that colocalized with NG2/VE-cadherin/VEGFR2 were counted and are expressed as a percentage of the VEGFR2+ vessels (CD31+ + VEGFR2+/CD31+) and mural cell coverage of NG2 and vascular integrity with VE-cadherin. Images were captured using an Olympus IX81 microscope and processed using NIH ImageJ software.
2.12. In situ zymography
Frozen tumor sections of the TRPV4lox/lox and TRPV4ECKO mice were brought to room temperature and rinsed with distilled water. Tumor tissue sections were incubated with DQ-gelatin fluorescein conjugate (1 mg/ml; 1: 50 dilution; Thermo Fisher) in reaction buffer (50 mM Tris-HCl, 150 mM NaCl, 5 mM CaCl2; pH 7.6) at 37°C for 2 hrs. After incubation, slides were washed with Milli-Q water and then fixed in 4% paraformaldehyde solution for 10 min at room temperature. The slides were then washed with PBS three times and mounted with Fluoro-G containing DAPI. Confocal images were acquired at 40X, and the DQ-gelatin fluorescence intensity was quantified using ImageJ software.
2.13. Statistical analyses
Statistical analyses were carried out using either GraphPad Prism 5 or Microsoft Excel. Analyses were performed using Student’s t-test or Mann-Whitney tests. All values reported are the mean ± SEM. A value of *p < 0.05, **p < 0.01, or ***p < 0.001 was considered significant.
3. Results
3.1. Endothelial TRPV4 deletion enhances tumor growth and results in leaky and tortuous tumor vasculature
To determine the role of endothelial TRPV4 in tumor growth, we generated TRPV4ECKO mice by crossing TRPV4lox/lox mice with Tie2-Cre transgenic mice. First, we confirmed the TRPV4 deletion by genotyping using specific primers for Cre and loxp[18] and found deletion of TRPV4 in the TRPV4ECKO animals but not the TRPV4lox/lox animals (not shown). Immunohistochemical analysis of aortic cross sections showed TRPV4 staining in the endothelium (red; arrowheads in Fig. 1A) and smooth muscle layer (red) of the TRPV4lox/lox mice; this staining was colocalized with CD31 in the endothelium (yellow; arrows; Fig. 1A). Importantly, TRPV4 staining was absent in the endothelium of the TRPV4ECKO mouse aorta, showing no colocalization with CD31 (green; Fig. 1A), but was present in the smooth muscle layer (red), indicating specific deletion of TRPV4 in the endothelium. Further, we confirmed the deletion of TRPV4 in ECs isolated from the TRPV4lox/lox mice but not from the TRPV4ECKO mice using a two-step isolation protocol employing anti-CD31 and anti-ICAM2 magnetic beads. Next, we found that these isolated ECs expressed the cell surface receptor CD31 (Suppl. Fig. 1A) and VEGFR2 (Fig. 1 B), exhibited uptake of acetylated-LDL (Suppl. Fig. 1 B) and formed tubes on Matrigel (Suppl. Fig. 1C), confirming the typical EC functional phenotype. We next confirmed TRPV4 deletion in these ECs. First, RT-PCR analysis revealed that TRPV4 was expressed in ECs from the TRPV4lox/lox mice but not in ECs from the TRPV4ECKO mice (Fig. 1B). Finally, calcium imaging revealed that ECs from the TRPV4ECKO mice failed to induce Ca2+ influx when stimulated with the TRPV4 selective agonist GSK1016790A (Fig. 1C, D), confirming the functional deletion of TRPV4 in the endothelium. Importantly, immunohistochemical analysis of various organs revealed that there was no apparent vascular phenotype in the TRPV4ECKO mice compared to the TRPV4lox/lox mice (Suppl. Fig. 2).
Figure 1. Characterization of endothelial TRPV4 knockout (TRPV4ECKO) deletion in mice.

A) Immunohistochemistry shows the colocalization (yellow; arrows) of the endothelial marker CD31 (green) with TRPV4 (red; arrowheads) in the TRPV4lox/lox mouse aortae; this finding is absent in TRPV4ECKO mouse aortae (only green). Scale bar: 10 μm. Note that TRPV4 (red) is present only in the smooth muscle layer of the TRPV4ECKO mouse aortae. B) RT-PCR analysis of TRPV4, VEGFR2, and GAPDH expression in isolated mouse aortic endothelial cells from the TRPV4lox/lox and TRPV4ECKO mice. C) Representative average traces showing relative changes in cytosolic calcium in response to GSK1016790A (GSK101) in the Fluo-4/AM-loaded TRPV4lox/lox and TRPV4ECKO endothelial cells (n=64). The arrow represents the time when the cells were stimulated with GSK101 (F/F0 = ratio of normalized Fluo-4 fluorescence intensity relative to time). D) Quantitative analysis of cytosolic calcium influx induced by GSK101 in the TRPV4lox/lox and TRPV4ECKO endothelial cells. All data represented are the mean ± SEM from at least three independent experiments. Statistical significance is indicated by *** p≤0.001.
To confirm a direct role for endothelial TRPV4 in tumor angiogenesis in vivo, we induced syngeneic tumors in the TRPV4ECKO and TRPV4lox/lox mice by subcutaneously injecting mouse LLCs and monitored tumor growth for 21 days. We found that the tumors were palpable by 7 days and that there was a significant increase in tumor growth by 14 days. Tumor growth was 1.5-fold higher in the TRPV4ECKO mice than in the TRPV4lox/lox mice at 21 days (Fig. 2A, B). Multiphoton and confocal microscopy revealed that the tumors from the TRPV4ECKO mice exhibited abnormal angiogenesis, as shown by increased vessel density characterized by tortuous and larger vessels (Fig. 2C–F). Quantification of vessel tortuosity revealed a significant increase in the tumors from the TRPV4ECKO mice compared to the TRPV4lox/lox mice (Suppl. Fig. 3). Further, the TRPV4ECKO mouse tumors displayed vessels with reduced pericyte coverage (NG2/CD31) and vascular integrity (CD31/VE-cadherin) compared to the tumors from the TRPV4lox/lox mice (Fig. 3A, B). Finally, we found increased leakage of intravenously injected fluorescent dextran in the TRPV4ECKO mouse tumors compared to the TRPV4lox/lox mouse tumors (Fig. 3C). Tie-2 Cre may impact cells other than ECs, such as CD45-positive hemopoietic cells[24]. Therefore, we measured the recruitment of CD45-positive cells in the tumors and found no difference between the tumors in the TRPV4lox/lox and TRPV4ECKO groups (Suppl. Fig. 4). suggesting that the observed effects on tumor growth are mainly due to deletion of TRPV4 in ECs.
Figure 2. Deletion of endothelial TRPV4 channels promotes tumor growth and abnormal vasculature.
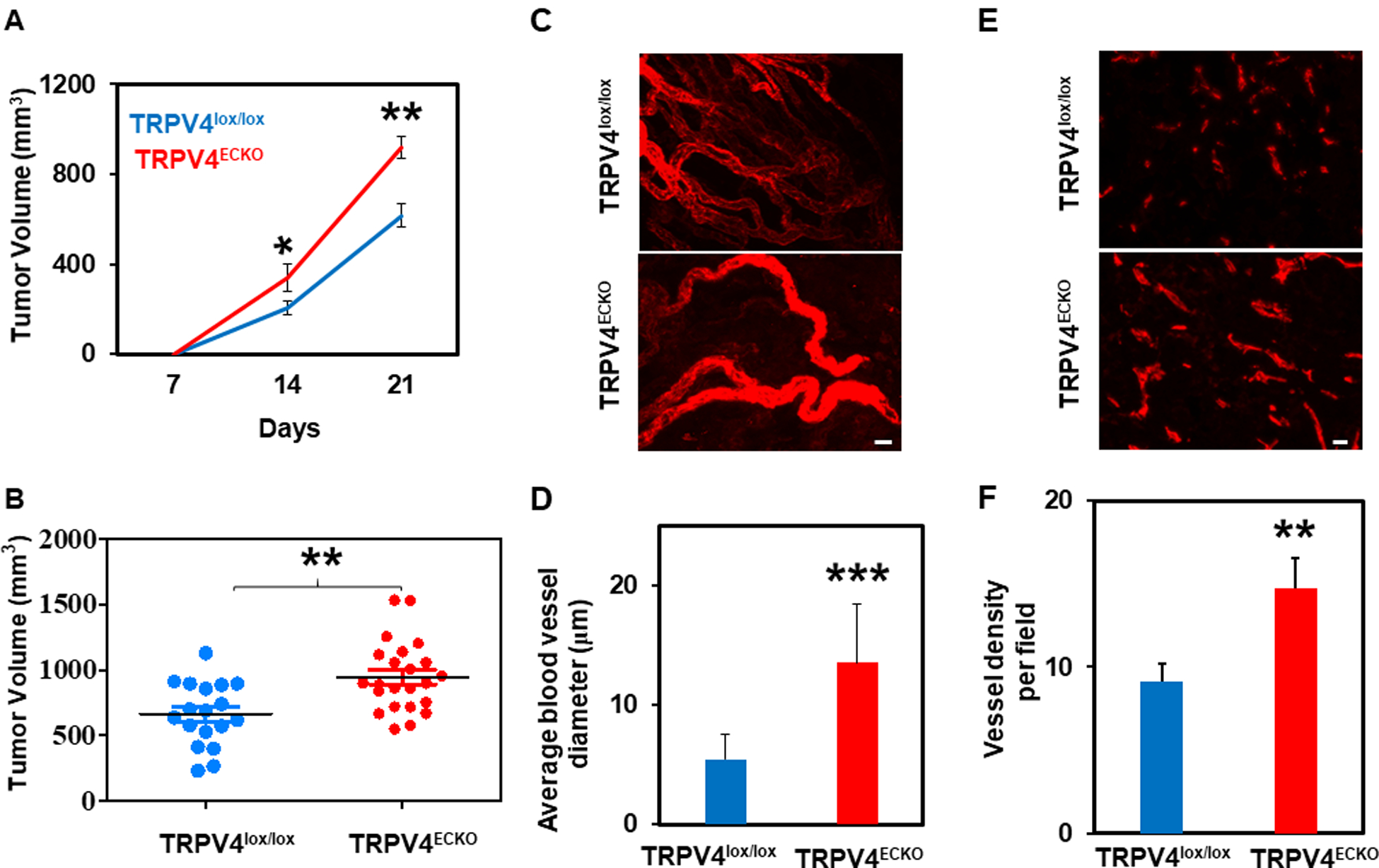
A) Tumor growth curves of the TRPV4lox/lox (n =9) and TRPV4ECKO (n =11) mice bearing Lewis lung carcinoma (LLC) cells. B) Tumor volumes of the TRPV4lox/lox and TRPV4ECKO mice at day 21. C-D) Multiphoton microscopy images of tumor vasculature from the TRPV4lox/lox and TRPV4ECKO mice (n=3) showing the blood vessel diameter. Mice were infused with Alexa Fluor 594-conjugated isolectin B4 prior to tumor extraction. Scale bar: 10 μm. E) Representative immunofluorescence images of tumor sections of the TRPV4lox/lox and TRPV4ECKO mice stained with an endothelial marker (CD31, red) (n=3). Scale bar: 10 μm. F) Quantification of vessel density from sections of the TRPV4lox/lox and TRPV4ECKO mouse tumors. All data are presented as the mean ± SEM, and statistical significance is indicated by * p≤0.05, **p≤0.01, and *** p≤0.001.
Figure 3. Endothelial TRPV4 channel deletion disrupts vascular integrity, leading to leaky vasculature.

A) Representative merged immunofluorescence images from tumor sections of the TRPV4lox/lox and TRPV4ECKO mice stained with an endothelial marker (CD31, red) and pericyte marker (NG2, green) (n=3). Scale bar: 10 μm. Quantification of pericyte coverage (NG2, pericytes in green/CD31, EC in red). B) Representative merged immunofluorescence images from the TRPV4lox/lox and TRPV4ECKO mouse tumor sections stained with an endothelial marker (CD31, red) and endothelial adherens junctional protein (VE-cadherin, green) (n=3). Scale bar: 10 μm. Quantification of VE-cadherin coverage (VE-cadherin, adherens junctional protein in green/CD31, ECs in red). C) Representative immunofluorescence images showing the leakage of Texas red dextran from the vasculature of the TRPV4lox/lox and TRPV4ECKO mouse tumors at day 21. Quantification of mean fluorescence intensity (MFI) (n=3). All data are presented as the mean ± SEM, and statistical significance is indicated by **p≤0.01 and *** p≤0.001.
3.2. Loss of endothelial TRPV4 increases the expression of VEGFR2 in tumors
To delineate the molecular mechanism underlying abnormal tumor angiogenesis in the TRPV4ECKO mice, we focused on the VEGFR2 pathway, which is downstream of TRPV4 in ECs[23]. Western blot analysis and immunohistochemistry revealed a significant increase in VEGFR2 expression and concomitant colocalization with CD31 in the tumors from the TRPV4ECKO group compared to the TRPV4lox/lox group (Fig. 4A, B). Further, we found significant upregulation of the p-ERK level in the tumors of the ECKO mice compared to the TRPV4lox/lox mice (Suppl Fig. 5). However, we found no difference in the expression of VEGF or HIF-1α between the tumors in the TRPV4ECKO group and the TRPV4lox/lox group (Suppl. Figs. 6 and 7).
Figure 4. Loss of endothelial TRPV4 increases VEGFR2 expression in LLC tumors.
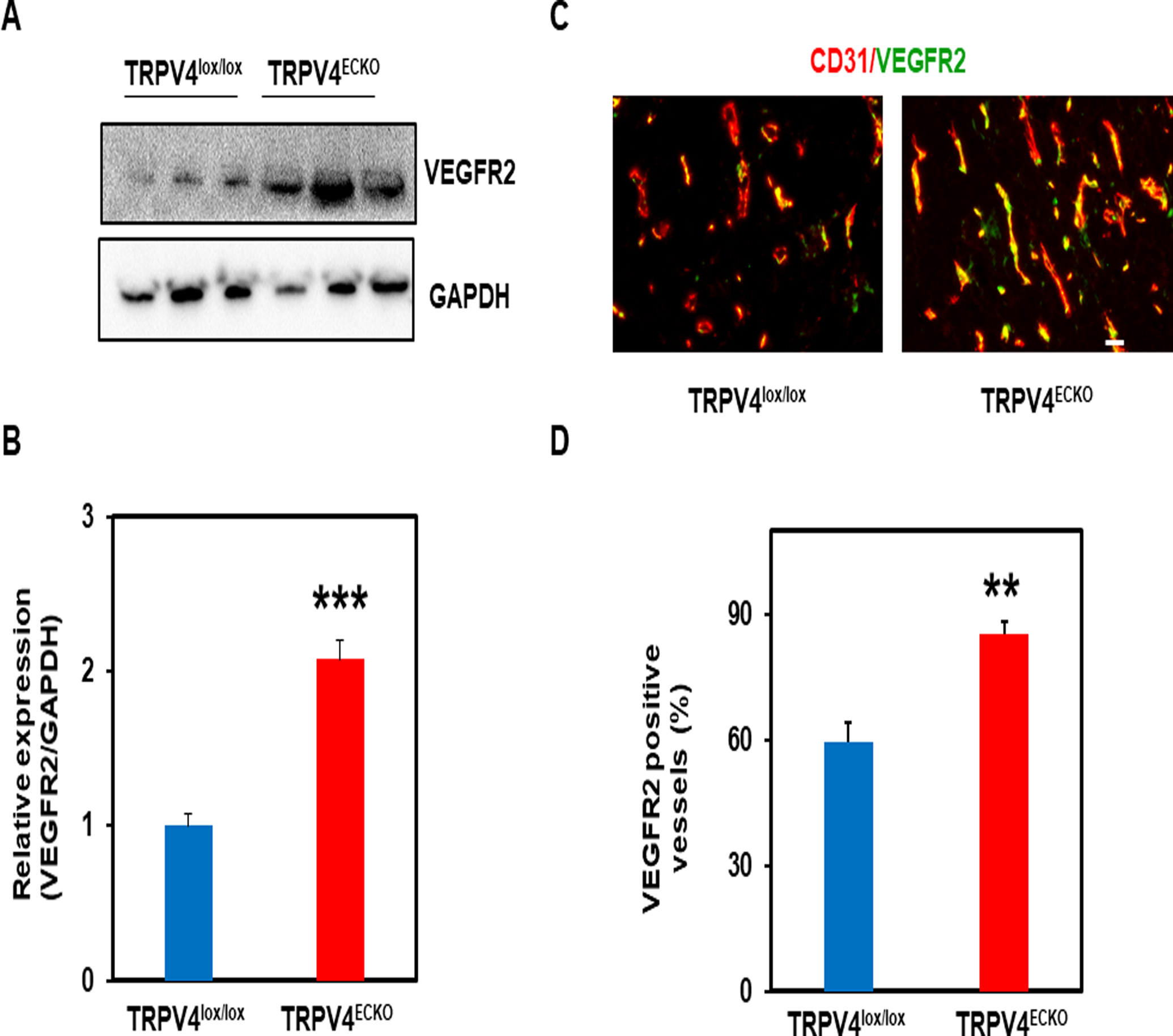
Western blot analysis (A) and quantification (B) of VEGFR2 expression in LLC tumors from the TRPV4lox/lox and TRPV4ECKO mice (n=3). C) Representative merged immunofluorescence images of tumor sections stained with endothelial markers (CD31, red) and VEGFR2 (green) from the TRPV4lox/lox and TRPV4ECKO mice. Scale bar: 10 μm. D) Quantification of VEGFR2-positive vessels in tumor sections from the TRPV4lox/lox and TRPV4ECKO mice (n=3). All data are presented as the mean ± SEM, and statistical significance is indicated by **p≤0.01 and ***p≤0.001.
3.3. TRPV4 deletion in the endothelium promotes lung metastasis
To explore the functional consequence of abnormal tumor angiogenesis (leaky vessels) in the TRPV4ECKO mice, we measured the metastasis of LLCs to the lung tissue. Consistent with our previous findings with the global TRPV4KO mice[3], lung metastasis was significantly higher in the TRPV4ECKO mice at 21 days than in the TRPV4lox/lox mice (Fig. 5A). Quantitative analysis revealed a significant increase (TRPV4lox/lox vs TRPV4ECKO; 8 vs 18; p<0.05) in the number of metastatic foci in the lungs of the TRPV4ECKO mice compared to those of the TRPV4lox/lox mice (Fig. 5B). Further, we found increased expression of matrix metalloproteinase-9 (Fig. 5C) but not matrix metalloproteinase-2 (Fig. 5D) in the TRPV4ECKO mouse tumors compared to the TRPV4lox/lox mouse tumors. Importantly, in situ zymography revealed increased DQ gelatinase activity in the TRPV4ECKO mouse tumors compared to the TRPV4lox/lox mouse tumors (Suppl. Fig. 8) with no difference in TIMP-2 expression (Suppl. Fig. 9).
Figure 5. TRPV4 deletion in the endothelium promotes lung metastasis via increased MMP-9 expression in tumors.

Representative H&E-stained images of lung sections (A) and quantification of metastatic foci (B) from the LLC tumor-bearing TRPV4lox/lox (n=9) and TRPV4ECKO mice (n=11). Scale bar: 200 μm. qPCR analysis of relative MMP-9 (C) and MMP-2 (D) expression in the TRPV4lox/lox and TRPV4ECKO mouse tumors (n=3). All data are presented as the mean ± SEM, and statistical significance is indicated by *p≤0.05 and ** p≤0.01, NS: nonsignificant.
4. Discussion
In the present study, we demonstrated that deletion of endothelial TRPV4 enhanced tumor progression compared to that of the TRPV4lox/lox mice. Tumors from the TRPV4ECKO mice exhibited increased vessel density characterized by large and tortuous vessels. Further, endothelial TRPV4 deletion produced leaky tumor vessels with a concomitant increase in metastasis to the lung compared with that of the TRPV4lox/lox mice. Ion channels have recently been identified as promising VEGF alternative therapeutic targets for tumor growth and metastasis. Among these channels, TRPV4 has attracted increased attention in the regulation of tumor endothelial function and angiogenesis[1,3,17,23]. However, TRPV4 in tumor cells has been shown to exhibit opposing effects on tumor growth and metastasis in different cancers[10–16]. These contrasting reports indicate that TRPV4 may have a tumor cell-independent role in inducing tumor growth and metastasis.
Tumors perfectly exploit the endothelium to induce tumor angiogenesis and metastasis by triggering the formation of abnormal, leaky vasculature. We previously demonstrated that TRPV4 expression is reduced in tumor ECs in vitro and that tumor growth and angiogenesis are increased in a subcutaneously implanted tumor in global TRPV4KO mice[1]. Although our studies demonstrated a role for TRPV4 in the regulation of endothelial function in vitro and vasodilation ex vivo[19,1,23,9,20], no studies have directly investigated the specific role of endothelial TRPV4 in tumor growth and metastasis in vivo. To confirm the specific role of endothelial TRPV4 in tumor angiogenesis and progression, we generated endothelial-specific knockout of TRPV4. To our knowledge, this is the first report to describe the role of endothelial TRPV4 in angiogenesis using endothelial-specific knockout of TRPV4. We did not observe any obvious vascular phenotype in the TRPV4ECKO mice because there was no change in the retinal vasculature compared to that of the TRPV4lox/lox mice. However, syngeneic tumor experiments revealed enhanced tumor growth and increased expression of p-ERK in the TRPV4ECKO mice compared to the TRPV4lox/lox mice. These findings agree with our previous findings from the global TRPV4KO mice[1,3] and tumor ECs[20] and confirm the endothelial-specific role of TRPV4 in tumor angiogenesis and growth. Multiphoton microscopy revealed large and convoluted vessels in the TRPV4ECKO tumors compared to the TRPV4lox/lox tumors, indicating abnormal vessel formation. Both NG2 and VE-cadherin staining, and Texas red dextran injections confirmed that loss of endothelial TRPV4 triggered vascular destabilization, leading to leaky vessels. In fact, these leaky vessels resulted in enhanced metastasis to the lung. VE-cadherin is critical for endothelial barrier integrity, which was shown to be compromised in the tumor vasculature[25,26]. A small molecule that directly potentiates VE-cadherin-mediated EC junctions induced tumor vascular normalization and reduced metastasis in mice[27].
Mechanistically, TRPV4 deletion in the endothelium appears to induce activation of the VEGF/VEGFR2 pathway. Both VEGF and VEGFR2 expression and activation have been shown to be enhanced during tumor angiogenesis, which has become the major target for cancer therapy[28]. We have recently demonstrated that downregulation or absence of TRPV4 in ECs in vitro induces translocation of VEGFR2 from the Golgi to the plasma membrane through YAP. Further, we found increased tyrosine phosphorylation of VEGFR2 at Y-1175[23]. Consistent with this observation, we found that the tumors from the TRPV4ECKO mice displayed increased expression of VEGFR2 and its downstream effector ERK1/2, indicating that deletion of endothelial TRPV4 augments the VEGF/VEGFR2 pathway. However, we did not find a significant difference in VEGF and HIF-1α expression between the tumors in the TRPV4ECKO and TRPV4lox/lox groups. We measured the VEGF and HIF-1α levels only at the end of the experiment (21 days) and cannot rule out the possibility that VEGF/HIF-1a expression may peak at earlier stages (7–14 days). Interestingly, we observed increased expression of matrix metalloproteinase-9 and DQ gelatinase activity in the TRPV4ECKO mouse tumors compared to the TRPV4lox/lox mouse tumors, suggesting that increased MMP-9 may be responsible for increased metastasis in the absence of endothelial TRPV4.
Our study demonstrates that endothelial TRPV4 suppresses tumor angiogenesis, tumor growth, vascular disruption, and metastasis, most likely by controlling VEGF/VEGFR2 localization/activation. These results indicate that angiogenesis is regulated by a tight coordination between mechanical and soluble (growth factor) signaling. Although the endothelium is continuously exposed to mechanical forces such as shear stress and cyclic strain, most studies on the endothelial role in tumor angiogenesis have focused on soluble factors such as VEGF. Previous work by Mammoto et al. demonstrated that ECM stiffness governs VEGFR2 gene promoter activity and expression via a mechanotranscriptional mechanism to stimulate angiogenesis[29]. However, the identity of the proximal mechanosensor(s) involved remains unknown. We recently demonstrated that the mechanosensitive ion channel TRPV4 regulates angiogenesis in an ECM stiffness-dependent manner in vivo[3]. Further, we showed that the absence or downregulation of TRPV4 in ECs induces membrane translocation and phosphorylation of VEGFR2 through the Rho/YAP pathway[23]. Taken together, these findings suggest that mechanical stiffness regulates angiogenesis through a mechanotranscriptional mechanism through both VEGFR2 expression and/or its activation, which is regulated by TRPV4. VEGF and its receptor VEGFR2 are critical for angiogenesis, yet direct targeting of the VEGF/VEGFR2 pathway yielded modest results in the clinic[30–33]. Nevertheless, most vascular normalization therapies still target growth factors such as VEGF and PDGF[34,28]. In conclusion, we propose that endothelial TRPV4 can be an alternate therapy for cancer instead of current strategies that inhibit angiogenesis by targeting VEGF and its receptors. Importantly, targeting endothelial TRPV4 could also offer a potential novel strategy for vascular normalization and cancer therapy.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health [NIH-(R15CA202847; R01HL119705 and R01HL148585 to CKT; R01 AI144115 and R15HL133918 to SP].
Footnotes
Conflict of Interest:
The authors declare no conflict of interest.
Data Availability Statement: The data to support the findings of the present study are available from the corresponding author upon reasonable request.
References:
- 1.Adapala RK, Thoppil RJ, Ghosh K, Cappelli HC, Dudley AC, Paruchuri S, Keshamouni V, Klagsbrun M, Meszaros JG, Chilian WM, Ingber DE, Thodeti CK (2016) Activation of mechanosensitive ion channel TRPV4 normalizes tumor vasculature and improves cancer therapy. Oncogene 35 (3):314–322. doi: 10.1038/onc.2015.83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Adapala RK, Thoppil RJ, Luther DJ, Paruchuri S, Meszaros JG, Chilian WM, Thodeti CK (2013) TRPV4 channels mediate cardiac fibroblast differentiation by integrating mechanical and soluble signals. Journal of molecular and cellular cardiology 54:45–52. doi: 10.1016/j.yjmcc.2012.10.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cappelli HC, Kanugula AK, Adapala RK, Amin V, Sharma P, Midha P, Paruchuri S, Thodeti CK (2019) Mechanosensitive TRPV4 channels stabilize VE-cadherin junctions to regulate tumor vascular integrity and metastasis. Cancer letters 442:15–20. doi: 10.1016/j.canlet.2018.07.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hartmannsgruber V, Heyken WT, Kacik M, Kaistha A, Grgic I, Harteneck C, Liedtke W, Hoyer J, Kohler R (2007) Arterial response to shear stress critically depends on endothelial TRPV4 expression. PLoS One 2 (9):e827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liedtke W (2005) TRPV4 plays an evolutionary conserved role in the transduction of osmotic and mechanical stimuli in live animals. J Physiol 567 (Pt 1):53–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Matthews BD, Thodeti CK, Tytell JD, Mammoto A, Overby DR, Ingber DE (2010) Ultra-rapid activation of TRPV4 ion channels by mechanical forces applied to cell surface beta1 integrins. Integr Biol (Camb) 2 (9):435–442. doi: 10.1039/c0ib00034e [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mendoza SA, Fang J, Gutterman DD, Wilcox DA, Bubolz AH, Li R, Suzuki M, Zhang DX (2010) TRPV4-mediated endothelial Ca2+ influx and vasodilation in response to shear stress. Am J Physiol Heart Circ Physiol 298 (2):H466–476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.O’Neil RG, Heller S (2005) The mechanosensitive nature of TRPV channels. Pflugers Archiv : European journal of physiology 451 (1):193–203. doi: 10.1007/s00424-005-1424-4 [DOI] [PubMed] [Google Scholar]
- 9.Thodeti CK, Matthews B, Ravi A, Mammoto A, Ghosh K, Bracha AL, Ingber DE (2009) TRPV4 channels mediate cyclic strain-induced endothelial cell reorientation through integrin-to-integrin signaling. Circ Res 104 (9):1123–1130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fang Y, Liu G, Xie C, Qian K, Lei X, Liu Q, Liu G, Cao Z, Fu J, Du H, Liu S, Huang S, Hu J, Xu X (2018) Pharmacological inhibition of TRPV4 channel suppresses malignant biological behavior of hepatocellular carcinoma via modulation of ERK signaling pathway. Biomedicine & pharmacotherapy = Biomedecine & pharmacotherapie 101:910–919. doi: 10.1016/j.biopha.2018.03.014 [DOI] [PubMed] [Google Scholar]
- 11.Lee WH, Choong LY, Jin TH, Mon NN, Chong S, Liew CS, Putti T, Lu SY, Harteneck C, Lim YP (2017) TRPV4 plays a role in breast cancer cell migration via Ca(2+)-dependent activation of AKT and downregulation of E-cadherin cell cortex protein. Oncogenesis 6 (5):e338. doi: 10.1038/oncsis.2017.39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lee WH, Choong LY, Mon NN, Lu S, Lin Q, Pang B, Yan B, Krishna VS, Singh H, Tan TZ, Thiery JP, Lim CT, Tan PB, Johansson M, Harteneck C, Lim YP (2016) TRPV4 Regulates Breast Cancer Cell Extravasation, Stiffness and Actin Cortex. Scientific reports 6:27903. doi: 10.1038/srep27903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu X, Zhang P, Xie C, Sham KWY, Ng SSM, Chen Y, Cheng CHK (2019) Activation of PTEN by inhibition of TRPV4 suppresses colon cancer development. Cell death & disease 10 (6):460. doi: 10.1038/s41419-019-1700-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Olivan-Viguera A, Garcia-Otin AL, Lozano-Gerona J, Abarca-Lachen E, Garcia-Malinis AJ, Hamilton KL, Gilaberte Y, Pueyo E, Kohler R (2018) Pharmacological activation of TRPV4 produces immediate cell damage and induction of apoptosis in human melanoma cells and HaCaT keratinocytes. PLoS One 13 (1):e0190307. doi: 10.1371/journal.pone.0190307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tang B, Wu J, Zhu MX, Sun X, Liu J, Xie R, Dong TX, Xiao Y, Carethers JM, Yang S, Dong H (2019) VPAC1 couples with TRPV4 channel to promote calcium-dependent gastric cancer progression via a novel autocrine mechanism. Oncogene 38 (20):3946–3961. doi: 10.1038/s41388-019-0709-6 [DOI] [PubMed] [Google Scholar]
- 16.Peters AA, Jamaludin SYN, Yapa K, Chalmers S, Wiegmans AP, Lim HF, Milevskiy MJG, Azimi I, Davis FM, Northwood KS, Pera E, Marcial DL, Dray E, Waterhouse NJ, Cabot PJ, Gonda TJ, Kenny PA, Brown MA, Khanna KK, Roberts-Thomson SJ, Monteith GR (2017) Oncosis and apoptosis induction by activation of an overexpressed ion channel in breast cancer cells. Oncogene 36 (46):6490–6500. doi: 10.1038/onc.2017.234 [DOI] [PubMed] [Google Scholar]
- 17.Fiorio Pla A, Ong HL, Cheng KT, Brossa A, Bussolati B, Lockwich T, Paria B, Munaron L, Ambudkar IS (2012) TRPV4 mediates tumor-derived endothelial cell migration via arachidonic acid-activated actin remodeling. Oncogene 31 (2):200–212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Moore C, Cevikbas F, Pasolli HA, Chen Y, Kong W, Kempkes C, Parekh P, Lee SH, Kontchou NA, Yeh I, Jokerst NM, Fuchs E, Steinhoff M, Liedtke WB (2013) UVB radiation generates sunburn pain and affects skin by activating epidermal TRPV4 ion channels and triggering endothelin-1 signaling. Proc Natl Acad Sci U S A 110 (34):E3225–3234. doi: 10.1073/pnas.1312933110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Adapala RK, Talasila PK, Bratz IN, Zhang DX, Suzuki M, Meszaros JG, Thodeti CK (2011) PKCalpha mediates acetylcholine-induced activation of TRPV4-dependent calcium influx in endothelial cells. Am J Physiol Heart Circ Physiol 301 (3):H757–765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Thoppil RJ, Adapala RK, Cappelli HC, Kondeti V, Dudley AC, Gary Meszaros J, Paruchuri S, Thodeti CK (2015) TRPV4 channel activation selectively inhibits tumor endothelial cell proliferation. Scientific reports 5:14257. doi: 10.1038/srep14257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Thoppil RJ, Cappelli HC, Adapala RK, Kanugula AK, Paruchuri S, Thodeti CK (2016) TRPV4 channels regulate tumor angiogenesis via modulation of Rho/Rho kinase pathway. Oncotarget 7 (18):25849–25861. doi: 10.18632/oncotarget.8405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang J, Niu N, Xu S, Jin ZG (2019) A simple protocol for isolating mouse lung endothelial cells. Scientific reports 9 (1):1458. doi: 10.1038/s41598-018-37130-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kanugula AK, Adapala RK, Midha P, Cappelli HC, Meszaros JG, Paruchuri S, Chilian WM, Thodeti CK (2019) Novel noncanonical regulation of soluble VEGF/VEGFR2 signaling by mechanosensitive ion channel TRPV4. FASEB journal : official publication of the Federation of American Societies for Experimental Biology 33 (1):195–203. doi: 10.1096/fj.201800509R [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tang Y, Harrington A, Yang X, Friesel RE, Liaw L (2010) The contribution of the Tie2+ lineage to primitive and definitive hematopoietic cells. Genesis 48 (9):563–567. doi: 10.1002/dvg.20654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bentley K, Franco CA, Philippides A, Blanco R, Dierkes M, Gebala V, Stanchi F, Jones M, Aspalter IM, Cagna G, Westrom S, Claesson-Welsh L, Vestweber D, Gerhardt H (2014) The role of differential VE-cadherin dynamics in cell rearrangement during angiogenesis. Nature cell biology 16 (4):309–321. doi: 10.1038/ncb2926 [DOI] [PubMed] [Google Scholar]
- 26.Le Guelte A, Dwyer J, Gavard J (2011) Jumping the barrier: VE-cadherin, VEGF and other angiogenic modifiers in cancer. Biology of the cell 103 (12):593–605. doi: 10.1042/BC20110069 [DOI] [PubMed] [Google Scholar]
- 27.Agrawal V, Maharjan S, Kim K, Kim NJ, Son J, Lee K, Choi HJ, Rho SS, Ahn S, Won MH, Ha SJ, Koh GY, Kim YM, Suh YG, Kwon YG (2014) Direct endothelial junction restoration results in significant tumor vascular normalization and metastasis inhibition in mice. Oncotarget 5 (9):2761–2777. doi: 10.18632/oncotarget.1942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Duda DG, Batchelor TT, Willett CG, Jain RK (2007) VEGF-targeted cancer therapy strategies: current progress, hurdles and future prospects. Trends Mol Med 13 (6):223–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mammoto A, Connor KM, Mammoto T, Yung CW, Huh D, Aderman CM, Mostoslavsky G, Smith LE, Ingber DE (2009) A mechanosensitive transcriptional mechanism that controls angiogenesis. Nature 457 (7233):1103–1108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Caporarello N, Lupo G, Olivieri M, Cristaldi M, Cambria MT, Salmeri M, Anfuso CD (2017) Classical VEGF, Notch and Ang signalling in cancer angiogenesis, alternative approaches and future directions (Review). Molecular medicine reports 16 (4):4393–4402. doi: 10.3892/mmr.2017.7179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Casanovas O, Hicklin DJ, Bergers G, Hanahan D (2005) Drug resistance by evasion of antiangiogenic targeting of VEGF signaling in late-stage pancreatic islet tumors. Cancer Cell 8 (4):299–309 [DOI] [PubMed] [Google Scholar]
- 32.Cuevas I, Boudreau N (2009) Managing tumor angiogenesis: lessons from VEGF-resistant tumors and wounds. Adv Cancer Res 103:25–42 [DOI] [PubMed] [Google Scholar]
- 33.Shibuya M (2006) Vascular endothelial growth factor (VEGF)-Receptor2: its biological functions, major signaling pathway, and specific ligand VEGF-E. Endothelium : journal of endothelial cell research 13 (2):63–69. doi: 10.1080/10623320600697955 [DOI] [PubMed] [Google Scholar]
- 34.di Tomaso E, London N, Fuja D, Logie J, Tyrrell JA, Kamoun W, Munn LL, Jain RK (2009) PDGF-C induces maturation of blood vessels in a model of glioblastoma and attenuates the response to anti-VEGF treatment. PLoS One 4 (4):e5123. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.