

Abstract
The mechanisms behind the antitumor effects of exercise training (ExTr) are not fully understood. Using mouse models of established breast cancer (BC), we examined here the causal role of CD8+ T cells in the benefit acquired from ExTr in tumor control, as well as the ability of ExTr to improve immunotherapy responses. We implanted E0771, EMT6, MMTV-PyMT, and MCa-M3C BC cells orthotopically in wild-type or Cxcr3−/− female mice and initiated intensity-controlled ExTr sessions when tumors reached ~100 mm3. We characterized the tumor microenvironment (TME) using flow cytometry, transcriptome analysis, proteome array, ELISA, and immunohistochemistry. We used antibodies against CD8+ T cells for cell depletion. Treatment with immune checkpoint blockade (ICB) consisted of anti-PD-1 alone or in combination with anti-CTLA-4. ExTr delayed tumor growth and induced vessel normalization, demonstrated by increased pericyte coverage and perfusion, and decreased hypoxia. ExTr boosted CD8+ T-cell infiltration, with enhanced effector function. CD8+ T-cell depletion prevented the antitumor effect of ExTr. The recruitment of CD8+ T cells and the antitumor effects of ExTr were abrogated in Cxcr3−/− mice, supporting the causal role of the CXCL9/CXCL11-CXCR3 pathway. ExTr also sensitized ICB-refractory BCs to treatment. Our results indicate that ExTr can normalize the tumor vasculature, reprogram the immune TME, and enhance the antitumor activity mediated by CD8+ T cells via CXCR3, boosting ICB responses. Our findings and mechanistic insights provide a rationale for the clinical translation of ExTr to improve immunotherapy of BC.
Keywords: Exercise, Vascular normalization, Tumor microenvironment, Tumor immunology, Immune checkpoint blockade
Introduction
It is now recognized that exercise helps prevent cancer, reduces morbidity associated with cancer therapy, and enhances overall survival in patients with breast cancer (BC) (1-4). Because exercise is a homeostatic physiological intervention, it also affects the tumor microenvironment (TME) (5-10). However, how exercise modulates host immune responses against cancer or in combination with immune modulators is not fully understood.
Preclinical studies indicate that tumor perfusion and oxygenation increase from the very first exercise session (5,9,11). Because the tumor vasculature is immature (12) and lacks myogenic and autonomic regulation (5), any transient increase in mean arterial pressure, as seen during exercise, increases tumor perfusion (13). Beyond the acute increase in perfusion, exercise training (ExTr, accumulated bouts of exercise sessions) induces vessel normalization, thus enhancing tumor blood flow and oxygenation in subsequent resting conditions as well (6,7). As a consequence, the TME becomes less hypoxic, rendering tumors less aggressive (14). This may partly explain the association of ExTr with reduced tumor burden (6,8) and suggests ExTr as a form of vascular normalization therapy. More importantly, a normalized tumor vasculature and microenvironment reprograms the immunosuppressive TME and enhances antitumor immunity (15). An improved vascular network favors adaptive antitumor immunity by promoting trafficking and re-localization of tumor-infiltrating lymphocytes (TILs), leading to improved outcomes of various immunotherapies, including immune checkpoint blockade (ICB) (15-20).
Limited T-cell recruitment and function in the TME correlates with poor prognosis in BC (21,22). Among TILs, CD8+ T cells predict overall treatment response and overall survival in patients with BC (23-25). A varying degree of infiltrating CD8+ T cells express multiple immune checkpoints (e.g., co-expression of programmed cell death protein 1 (PD-1) and cytotoxic T-lymphocyte-associated protein 4 (CTLA-4)), which cause these cells to become exhausted (26). ICB mitigates CD8+ T-cell exhaustion by targeting these receptors and is approved for a subset of triple-negative BC patients (27). An abnormal TME, caused in part by hypoperfusion, results in the exclusion of CD8+ T cells and reduces the impact of immunotherapies such as ICB. We previously demonstrated that vascular normalization could increase immune cells’ infiltration into tumors, and multiple studies have confirmed these findings in several tumor models (15,17-19).
Here, we investigated the effect of ExTr on the tumor vasculature, TME, and TILs in established orthotopic BC models. We found that ExTr normalized the tumor vasculature, increased CD8+ T cells in primary BC tumors, and delayed tumor growth. Mechanistically, we demonstrated that the antitumor effect of ExTr was dependent on CD8+ T cells and that the CXCL9/11-CXCR3 pathway mediated the ExTr-induced tumoral infiltration of CD8+ T cells and the observed antitumor effect. We also demonstrated that ExTr reprogramed the TME, enhanced infiltration and function of CD8+ T cells, and sensitized refractory BCs to combined PD-1 and CTLA-4 blockade. These findings highlight the potential role of ExTr as a physiological strategy to revive antitumor immunity and improve the clinical outcomes of ICB therapy in BC.
Materials and Methods
Breast cancer cell lines.
E0771 (CH3 BioSystems) and EMT6 (ATCC) cells were cultured in RPMI-1640 medium with 10% FBS (Gibco, USA). MMTV-PyMT cells from transgenic breast cancer mouse models, as well as the MCa-M3C cell line (derived clusters from MMTV-PyMT tumors that spontaneously colonized the lungs; cells were isolated by Dr. Peigen Huang and established as a metastatic BC cell line) were cultured in DMEM medium with 10% FBS supplemented with 4.5 g/L glucose (Corning, USA). All cells were cultured at 37°C in a humidified incubator with 5% CO2. Before starting the study, the cells were expanded and frozen in single vials (containing 106 cells in 50% FBS / 10% DMSO, Sigma-Aldrich, USA), so vials from the same batch would be thawed anytime we started a new mice cohort. These cells were tested and authenticated (morphology check by microscopy, and antibodies for common viral contaminations, VRL Diagnostics, USA). The cell lines were also tested negative for mycoplasma. Aliquots were thawed and implanted in mice after 3 (no more than 5) passages within 5-7 days.
Preclinical tumor models of Breast Cancer.
Syngeneic, orthotopic breast tumors were established by injecting 50 μL of media containing BC cells in the 3rd mammary fat pad of 8-10-week-old female C57BL/6 (E0771, 2x105), FVB (MCa-M3C and MMTV-PyMT, 1x105), and Balb/c (EMT6, 2x105) mice. All these strains were bred and maintained in Cox-7 gnotobiotic animal facility. C57BL/6 mice (8 weeks old) were subjected to an ad libitum high-fat diet (HFD, cod. D12492, 60% fat) or low-fat diet (LFD, cod. D12450B 10% fat) (both from Research Diets, USA) starting 12 weeks before tumor implantation, and throughout the study. After 3 months of HFD/LFD, 106 E0771 cells were implanted in the 3rd mammary fat pad, as previously described in (14). Cxcr3−/− mice were obtained from Jackson Laboratory and were in the C57BL/6 background. For tumor implantation and subsequent collection, mice were anesthetized for 30 minutes using intramuscular injections of ketamine/xylazine (K/X; 90 mg / 9 mg per kg body weight; Patterson Vet Supply, USA). Tumor dimensions were initially measured every three days by both ultrasound (Vevo 2100, FUJIFILM VisualSonics) and calipers after treatment initiation. Tumor volume (in mm3) was estimated using the formula: tumor volume = (long axis) × (short axis)2 × π/6. When tumors reached ~100 mm3 (around 10 days for E0771 and 20 days for MCa-M3C), we started neoadjuvant intensity-controlled, moderate-to-vigorous aerobic exercise training (ExTr) treatment alone or in combination with immune checkpoint blockade (ICB) as described in the following sections. All animal procedures were carried out following the Public Health Service Policy on Humane Care of Laboratory Animals and approved by the Institutional Animal Care and Use Committee (IACUC) of Massachusetts General Hospital.
Exercise testing and prescription.
For patients with cancer, the current exercise recommendation follows the guidelines from the American College of Sports Medicine (ACSM) (28), which is endorsed by the American Cancer Society (ACS) (29) and similar to the World Health Organization (WHO) (30) for general health. It states that cancer survivors (after a cancer diagnosis) should engage in moderate-to-vigorous intensity (rate at which the exercise is performed) exercise 3-5 or more days per week for 30-60 minutes per session. The intensity should be expressed as relative to the maximum amount an individual can perform. The goal is to identify the body’s maximal capacity for uptake, transport, and use of oxygen, defined as the maximal oxygen uptake (VO2 max). ACSM considers moderate-intensity exercise ranging from 46-63% and vigorous 64-90% of VO2 max (28). In mice, similar physiological responses to graded exercise have been demonstrated (31,32). Thus, we measured the maximal oxygen capacity in mice to determine a proper prescription of the ExTr.
One week before tumor implantation, mice were subjected to acclimatization on a running treadmill (5-lane with shock grid, Panlab/Harvard Apparatus). The acclimatization consisted of running on the treadmill at a low velocity (10 m/min) for 10 minutes on three alternate days. Once mice were familiarized with the treadmill, we performed a maximal exercise test, which is correlated with and predicts VO2 max. The test started at 6 m/min with 3 m/min increments every 3 minutes until volitional exhaustion with 0° inclination. At the exhaustion point, mice were rapidly removed from the running treadmill, and the time (s), distance (m), and velocity (m/min) were recorded. Intensity-controlled, moderate-to-vigorous aerobic ExTr began when tumors reached ~100 mm3 and consisted of daily sessions at ~60% of maximal velocity in clusters of 5 mice per time (the number of lanes in the treadmill). In a cohort MMTV-PyMT tumor bearing mice, ExTr started when tumors were at a smaller size (~20 mm3). ExTr began with 30 minutes in the first session, with consecutive increments of 5 minutes until reaching 45 minutes per day, which was sustained throughout the ExTr protocol. Compliance was accessed by the percentage of completion of the prescribed ExTr program. We excluded mice that did not reach at least 75% of prescribed ExTr after randomization, representing ~20% of C57BL/6 mice. All FVB mice completed the study. A 7-day ExTr protocol was used for analyzing TME parameters, and a 14-day protocol was used for tumor growth endpoints.
Depletion of CD8+ T cells.
CD8+ T cells were depleted by administering intraperitoneal (i.p.) injections of 250 μg of anti-CD8β (53-5.8, BioXcell) or IgG. Concurrently with the ExTr protocol, the depletion protocol started when tumors reached 100 mm3 by injecting every 4 days (days 1, 5, 9 and 13) until tumors in the control group reached 1 cm3.
Immune checkpoint blockade treatment.
ICB with anti-PD-1 alone (200 μg, RMP 1-14, BioXcell), anti-PD-1 combined with anti-CTLA-4 (100 μg, 9D9 BioXcell), or IgG was administered i.p. and re-administered every 4 days (days 1, 5, 9 and 13, a total of 4 doses). ICB treatment started concomitantly with ExTr protocol when tumors had reached 100 mm3. The endpoint was determined by the time when the control group reached 1 cm3.
Flow cytometry analysis.
24 hours after the last exercise session, mice were anesthetized with K/X (90 mg / 9 mg per kg body weight), and breast tumors were harvested, minced and incubated for 1 hour at 37°C with DMEM media containing collagenase type 1A (1.5 mg/mL), hyaluronidase (1.5 mg/mL), and DNase (20 μg/mL). The digested sample was filtered through 70-μm strainers and centrifuged at 1200 rpm for 5 minutes. The supernatant was discarded, and cells were resuspended in 4 mL of ACK lysing buffer (Lonza, USA) for about 4 minutes. 6 mL of flow cytometry buffer (PBS with 3% BSA and 0.05% sodium azide) was first added, cells were then centrifuged, and the pellet (~2 million cells per panel) was resuspended (150μL). An aliquot from each sample was collected for counting cells in a hemocytometer. To detect cytokine production (IFNγ, granzyme B, and CXCL9), we additionally stimulated cells for 4 hours at 37°C with cell activation cocktail (PMA/Ionomycin and Brefeldin A, Biolegend, 1:100). Single-cell suspensions were incubated with Fc block (anti-CD16/CD32 antibody for 10 minutes at 4°C, Biolegend, 1:500) and then stained with cell surface-conjugated antibodies. The stained cells were washed and resuspended in 100μL of cold flow cytometry buffer (1% BSA, 5% FBS, 0.1% sodium azide in PBS; salts from Sigma-Aldrich, USA). Flow cytometry data were obtained using an LSRII flow cytometer (BD Biosciences) and analyzed with FlowJo software. The double/aggregated cells were gated out using forward scatter area (FSC-A) versus forward scatter width (FSC-W) and side scatter area (SSC-A) versus side scatter width (SSC-W). No viability dyes were used. Different fluorophores conjugated with the following mAb were used: CD45 (30-F11, Biolegend), TCRβ chain (H57-597, Biolegend), CD4 (RM4-5, Biolegend), FOXP3 (150D, Biolegend, 1:100), PD-L1 (10F.9G2, Biolegend), CD8a (53-6.7, Biolegend), PD-1 (29F.1A12, Biolegend), Tim3 (B8.2C12, Biolegend), CTLA4 (CD152, Biolegend), LAG3 (C9B7W, Biolegend), IFNγ (XMG1.2, Biolegend), Granzyme B (GB11, Biolegend), Ki67 (16A8, Biolegend), CXCR3 (173, Biolegend, 1:100), CD11b (M1/70, Biolegend), Ly6G (1A8, Biolegend), F4/80 (BM8, Biolegend), MHCII (M5/114.15.2, BD biosciences), CD206 (MMR, BD biosciences), NK1.1 (PK136, BD biosciences), and CXCL9 (MIG-2F5.5, Biolegend). Unless specified otherwise, all the antibodies were used at 1:200. Representative dot plots and gating strategies can be found in the supplementary information. Markers used to define cell populations are in their respective figures.
Vessel perfusion and hypoxia histology.
24 hours after the last exercise session, mice were injected with pimonidazole hydrochloride (60 mg/kg; Hyproxyprobe) i.p. 1 hour before sacrifice. Mice were then anesthetized, and biotinylated lectin (Vector Labs) was administered in the retro-orbital sinus (in 100 μL of 1 mg/mL) 5 minutes before breast tumor removal. Harvested tumors were then fixed in 4% formaldehyde in PBS for 6 hours and incubated with 30% sucrose in PBS overnight. Tumors were then mounted in optimal cutting temperature gel (Tissue-Tek). Transverse 20 μm tumor sections were stained with antibodies for CD31 (Millipore MAB1398Z, 1:100), Cy3-αSMA (Sigma C6198, 1:100), lectin (Strepavidin-Alexa546, Invitrogen S11225, 1:200), Pimonidazole (Hyproxyprobe HP6-100Kit antibody, 1:50), and CD8 (cell signaling 98941, 1:50). Lastly, they were counterstained with DAPI (Vector Labs, USA).
Histological image analysis.
Stained sections were imaged with a slide scanner (AxioScan Z1, Zeiss) for fluorescence quantification of mosaic images. Vessels (CD31) co-stained with lectin allowed the quantification of perfused vessel fraction (CD31+lectin+), whereas normalized vessels corresponded to CD31+αSMA+ co-stained vessels. Quantification of the hypoxia fraction was calculated by the percent area of tissue positive for pimonidazole. Unbiased image quantification was conducted using custom software designed in MATLAB (Mathworks, USA). Briefly, whole slide images were imported into MATLAB and first manually cleaned to ensure no imaging artifacts remained. The images were then individually thresholded. For each stain on each image, the mean [μ] and variance [σ2] for each background intensity was first calculated, and a threshold value of at least μ+4σ was used. Co-localization was then measured on channels of interest, namely between the channels of interest, namely between CD31 and lectin or CD31 and αSMA. For quantification of CD8+ T-cell density, 5-10 random fields were imaged for distribution of CD8 (ImageJ, NIH) and for acquiring representative images using a confocal microscope (Olympus) at 20x oil magnification. To avoid bias during the image acquisition, we took images in a blinded manner. The histological slides were named following a random numeric sequence with no group info given to the researcher, using DAPI channel to define a field of view (FOV) with no overlaps, no large necrotic areas, and avoiding tumor borders. We took at least 5 random qualified FOVs per slide and then, as many qualified FOVs as possible up to 10 FOVs per slide.
Bulk RNA-seq analysis.
mRNA was extracted from snap frozen fragments of E0771 tumors (n=5 mice per group, Control vs. ExTr) using the RNeasy Mini Kit (Qiagen). For this analysis, tumors were time-matched and size-matched to avoid potential confounding factors. The Massachusetts Institute of Technology BioMicro Center performed RNA-sequencing on extracted RNA samples. First, a Fragment Analyzer (Advanced Analytical Technologies Inc.) confirmed the quality of RNA samples (RIN 7.9-10). Illumina libraries were then prepared and indexed from ~250 ng of total RNA using the Kapa Hyperprep kit following the manufacturer’s recommendations (Roche). The libraries were confirmed using the Fragment Analyzer, quantified by qPCR, then pooled and sequenced on an Illumina NextSeq500 using 40+40 paired-end reads. The samples were demultiplexed by custom scripts, which allowed a single mismatch to the index sequencing. The raw FASTQ files were aligned using STAR (33) to the mouse Ensembl GRCm38 reference genome. The Bioconductor R package, GenomicAlignments (34), was used to generate gene read counts. DESeq2 (35) was used to normalize and determine the differentially expressed genes. Gene set enrichment analysis (GSEA)(36) was performed using GSEA software from the Broad Institute (37). Gene set enrichment network analysis was visualized with EnrichmentMap (36) in Cytoscape 3.0 v3.7.2 (https://cytoscape.org/cy3.html), using a cutoff of FDR < 0.10. Each node in the network corresponded to a gene set. The sizes of the nodes were proportional to the number of genes in the gene set. Gene sets with overlapping genes were organized as a network. Raw RNA-sequencing data of bulk E0771 mouse tumors performed in this study have been deposited in the NCBI Sequence Read Archive (SRA) under BioProject ID PRJNA689055.
RNA-seq data from human breast cancer patients from Ligibel et al. (38) was downloaded from the Gene Expression Omnibus (GEO) database (accession number GSE129508, from the study registered on the clinicaltrials.gov as NCT01516190). The authors stated that the participants obtained a medical clearance before participation, signed an informed consent, and that the study was approved by the Institutional Review Board (Dana-Farber/Harvard Cancer Center). We used STAR to align the raw FASTQ files to the human Ensembl GRCh38 reference genome, gene read counts were generated using GenomicAlignments, DESeq2 was used to normalize the raw counts (expressed by Volcano Plot and heat map), and GSEA was performed on the normalized counts.
Proteome cytokine array.
Homogenized fragments from snap frozen bulk tumors from both E0771 and MCa-M3C mouse models were used to profile cytokines using the Proteome Profiler Mouse Cytokine Array Kit (Panel A; R&D Systems), following the manufacturer’s protocol. 300 μg of protein pooled from n=6 mice per group (50 ¼g/mice). The array membranes (ThermoFischer, USA) were imaged (Kodak X-OMAT 2000A, USA), and the densitometry analysis was carried out using ImageJ (NIH, USA).
ELISAs.
Each E0771 and MCa-M3C tumors were homogenized in reagent diluent provided by the DuoSet ELISA (R&D Systems) and mechanically disrupted for protein extraction. 50 μg of protein were loaded in duplicate, and ELISAs for CXCL9/MIG (DY492), CXCL10/IP-10/CRG-2 (DY466), and CXCL11/I-TAC (DY572) and plates were read at 450nm with wavelength correction at 540nm (iMark, Bio-Rad, USA), following the manufacturer’s protocol. Absorbance values were interpolated from the standard values, and the final concentration expressed in ng/mg of protein.
Statistical analysis.
Whenever the presumptions of normality and homoscedasticity were achieved, we conducted Student’s t-test to compare two groups. One-way ANOVA was used for comparison among 3 or more groups, and two-way ANOVA was used for tumor growth analysis, followed by Tuckey’s post hoc test whenever necessary to identify specific group differences. In the other cases, we conducted a non-parametric analysis, comparing two groups by Mann-Whitney test and more than two groups by Kruskal-Wallis test. Correlation between variables was accessed by regression analysis. All statistical analyses were performed using GraphPad Prism 8 software. A P value ≤ 0.05 was considered significant. Data presented as mean ± SEM or median ± interquartile range (box plot and violin plot) and distribution (violin plot).
Results
ExTr delays tumor growth in established BCs
We first conducted an incremental running exercise test until exhaustion to define ExTr intensity for C57BL/6, FVB and Balb/c mice. Then, we implanted murine BCs orthotopically in syngeneic, immunocompetent mice [E0771 (C57BL/6), MCa-M3C (FVB), and EMT6 (Balb/c)]. When tumors reached ~100 mm3, mice were randomized to control (sedentary) or ExTr groups (Fig. 1A). The ExTr consisted of daily sessions of continuous moderate-to-vigorous intensity aerobic exercise on a running treadmill, progressing from 30 to 45 minutes per session (Supplementary Fig. S1A-C). We trained mice with the same relative exercise intensity (Supplementary Fig. S1D, ~60% of maximal running velocity). FVB mice displayed a higher maximal running velocity and better compliance with the ExTr program than C57BL/6 mice (Supplementary Fig. S1E-F). Therefore, total ExTr volume in FVB mice was higher than that in C57BL/6 mice. Using different models, we showed that ExTr delayed tumor growth in BC. Seven days of consecutive ExTr reduced tumor burden in the three models of BC (Fig. 1B–G, Supplementary Fig. S2A-F). Using a more traditional exercise intervention in preclinical models (i.e., 5 days of consecutive exercise for 2 weeks), we saw a similar tumor control induced by ExTr (Supplementary Fig. S2G-I). The consistent effect of ExTr on tumor growth also observed also on individual tumor growth and time to triple tumor volume (Supplementary Figs. S2C, S2F, S2I). After seven days of ExTr, tumor weight was ~30% smaller in ExTr than in control groups (Fig. 1E-G, Supplementary Fig. S2H). Similar antitumor effects of ExTr were found when started in smaller tumors (~20 mm3 on MMTV-PyMT in FVB, Supplementary Fig. S2J). ExTr also blunted obesity-induced accelerated tumor growth (Supplementary Fig. S2K). Interestingly, ExTr prevented cancer-induced chronic fatigue, as demonstrated by increased exercise capacity (Supplementary Fig. S3A-D), which is a biomarker of ExTr response and a predictor of overall survival in BC patients (39). More importantly, ExTr was associated with reduced recurrence rate and metastatic burden in the metastatic MCa-M3C model (Supplementary Fig. S4A-C).
Figure 1. Impact of exercise training on tumor progression in established breast tumors.
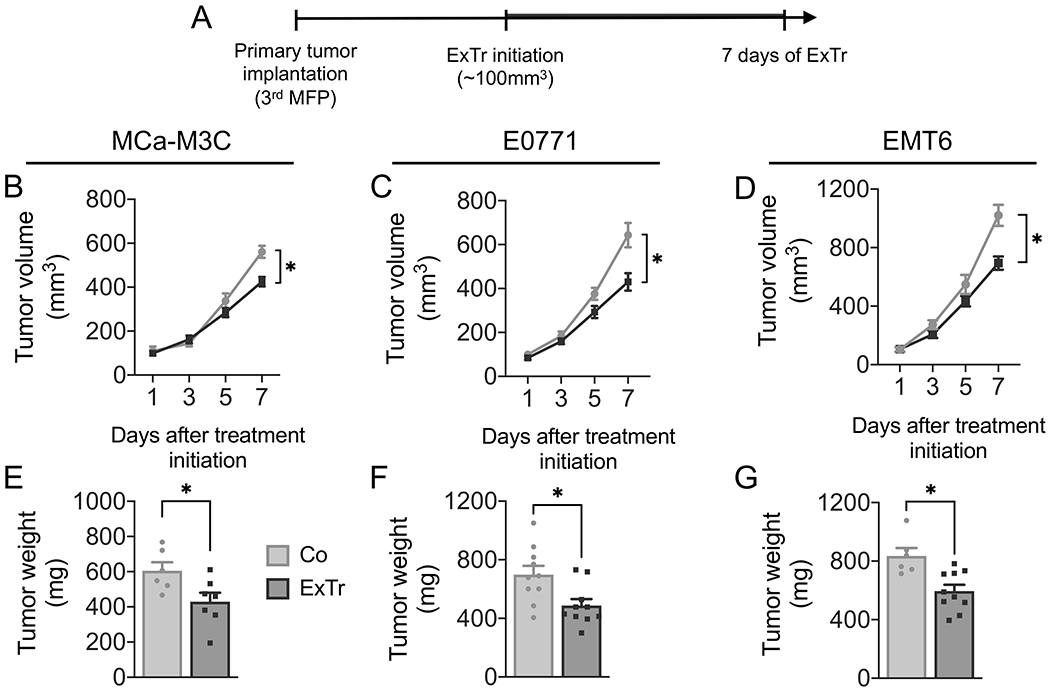
A, Experimental design of the study: MCa-M3C (in FVB), E0771 (in C57BL/6), and EMT6 (in Balb/c) tumors were implanted in the mammary fat pad (MFP) of immunocompetent female mice. When tumors reached ~100 mm3 in size, mice underwent intensity-controlled, moderate-to-vigorous aerobic exercise training (ExTr, 60% of maximal running velocity) or remained sedentary (Control) for 7 consecutive days. B–D, Tumor growth in (B) MCa-M3C (n=6-7), (C) E0771 (n=10), and (D) EMT6 (n=6-10) tumor models in response to 7 days of ExTr. E–G, Tumor weight after 7 days of ExTr the mouse BC models. Statistical comparison by two-way ANOVA (B-D) or unpaired Student’s t test (E-G). Data presented as mean ± SEM.
ExTr normalizes the tumor vasculature and TME
ExTr did not change vessel density (Fig. 2A-B) but promoted tumor vessels’ maturation. As expected, we found ExTr induced vessel normalization in both E0771 and MCa-M3C BC models. ExTr led to an increased percentage of vessels associated with αSMA+ perivascular cells (Fig. 2C–D), an indicator of vascular normalization. ExTr also increased the fraction of vessels that were perfused (Fig. 2E–F), indicating improved vascular function. More functional vessels resulted in reduced hypoxia (Fig. 2G–H), indicating normalization of TME.
Figure 2. Exercise training normalizes tumor blood vessels and reduces hypoxia.
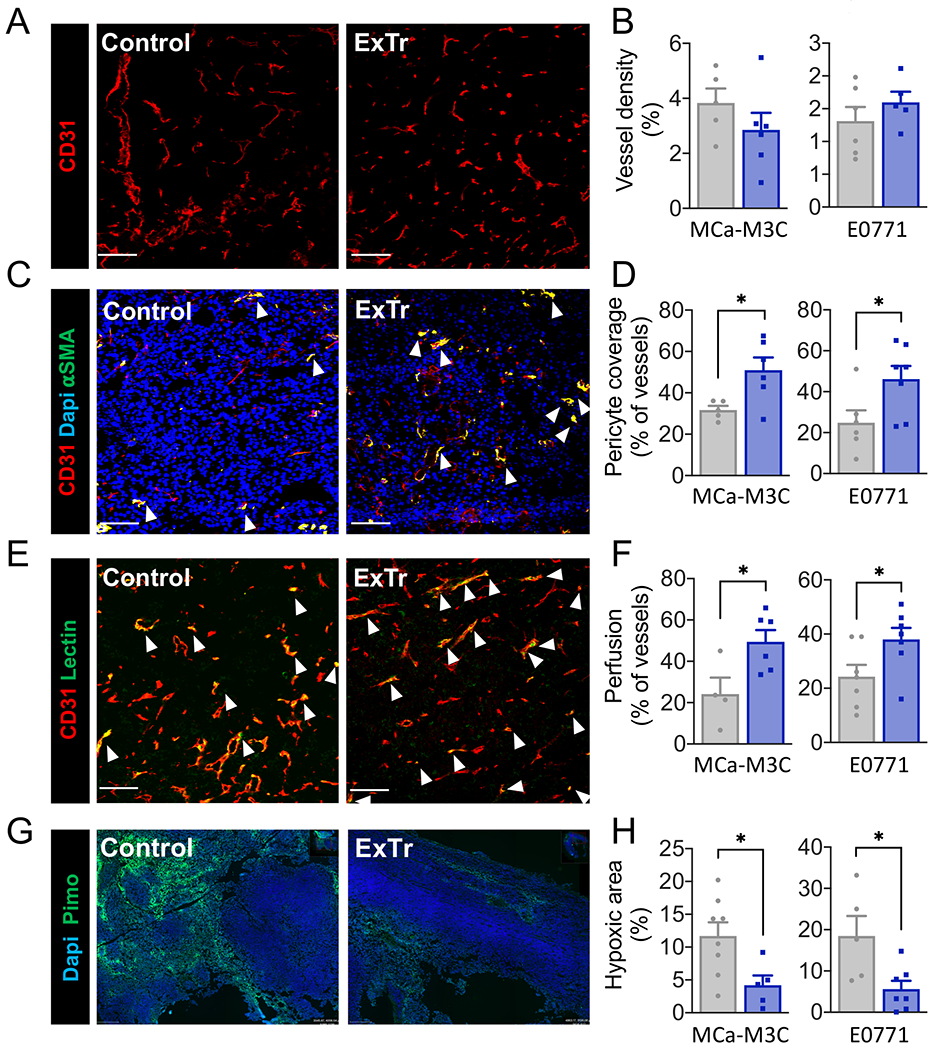
Immunofluorescence quantification was done in tumor sections to measure vessel normalization and hypoxia. Representative images from E0771 model (A, C, E and G) and corresponding quantitative data (B, D, E and H). A–B, Vessel density, indicated by fractional area (stained for CD31+). C–D, Vessel normalization, characterized by the fraction of vessels with pericyte coverage (CD31+αSMA+). E–F, Perfused vessels, expressed as the fraction of vessels (CD31+) also positive for lectin. G–H, Fractional hypoxic area. Red indicates vessels; Green indicates pericytes (αSMA, C), perfused vessels (lectin, E), or hypoxic area (pimonidazole, G); Blue indicates nucleus (Dapi, C and G). White arrows highlight co-staining. Scale bar, 100 μm. n=4-8 (MCa-M3C) and n=5-7 (E0771). Statistical differences between groups done using unpaired Student’s t test. Data presented as mean ± SEM.
ExTr reprograms the transcriptional signature of the TME
To determine potential mechanisms associated with the effects of ExTr on BCs, we performed unbiased RNA-seq analysis of E0771 tumor samples from Control versus ExTr mice (Fig. 3A-B, Supplementary Fig. S5). Gene Set Enrichment Analysis (GSEA) of the RNA-seq data indicated several pathways modulated by ExTr compared to the Control group (comprehensive list is in Supplementary Table S1). ExTr induced a metabolic reprogramming by reducing glucose uptake/insulin pathways and upregulated several gene sets associated with oxidative metabolism, such as oxidative phosphorylation (OXPHOS). It also enhanced gene expression linked to cytokine activation and downregulated several pathways involved with immunosuppression in BC (20,40,41), whose inhibition is demonstrated in the clinic to synergize with anti-PD-1 against solid tumors (42). This molecular reprogramming suggests a less glycolytic and less immunosuppressive tumor, which is consistent with vessel normalization.
Figure 3. Exercise training reprograms the transcription profile in the TME of breast tumors.
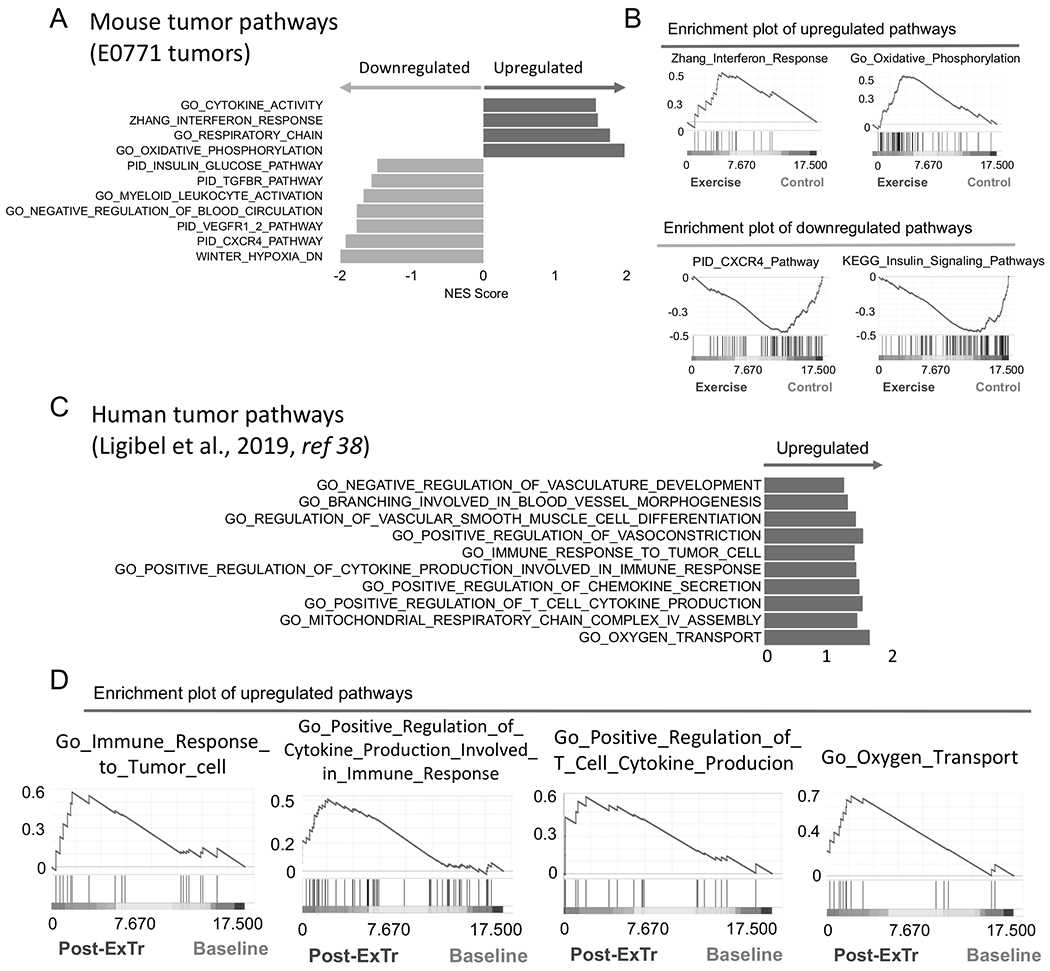
A–B, RNA-seq analysis of bulk tumors showing effects of ExTr signaling pathways. (A) Horizontal bars indicate NES (Normalized Enrichment Score) of pathways upregulated (positive NES, in dark color, right) or downregulated (negative NES, in light color, left) by ExTr, indicated by Gene Set Enrichment Analysis (GSEA). (B) Representative GSEA plots showing enhanced molecular signatures. E0771 tumors, collected after 7 days of ExTr. n=5 mice per group, FDR (false discovery rate) q-value<0.12, p<0.05. Comprehensive list of pathways modulated by ExTr are included in the Supplementary section (Supplementary Fig. S5, Supplementary Table S1). C–D, Gene signatures of pathways related to antitumor immunity and vessel normalization in patients in response to ExTr. Original data set from Ligibel et al. (38).
To test whether ExTr induced similar transcriptional signatures in the TME in BC patients, we analyzed the RNA-seq dataset previously published by Ligibel et al. (38). There were two groups of BC patients in the published study – exercise intervention (pre-ExTr and post-ExTr) and a mind-body control group (pre-mind and post-mind). We found via GSEA that in the ExTr group, there was upregulation of transcriptional pathways consistent with antitumor immune activity and vessel normalization in the BC patients (Fig. 3C–D), similar to what we found in mice. These results indicate that ExTr can improve antitumor immune activity and vessel normalization in not only mice, but also human BC patients.
ExTr increases the infiltration of CD8+ T cells into BC TME
We and others have shown that vessel normalization using anti-angiogenic therapies enhances trafficking of CD8+ T cells in murine BC models (17-20). Transcriptomic changes associated with ExTr supports a shift from immunosuppression to activation of antitumor immunity (Fig. 3). Therefore, we studied the phenotypic changes in the immune TME induced by ExTr using flow cytometry. Indeed, we found that ExTr increased the infiltration of CD8+ T cells into the BC TME (Fig. 4A-D, Supplementary Fig. S6A–B). We profiled the immune TME in BCs during ExTr and found that ExTr consistently boosted CD8+ T-cell infiltration, indicated by changes in relative (Fig. 4A) and absolute numbers (Fig. 4B) of CD8+ TILs. We also analyzed the intratumoral localization of CD8+ T cells with immunofluorescence. Consistent with flow cytometry analysis, we found that ExTr increased the intratumoral distribution of CD8+ T cells within the TME (Fig. 4C), indicated by the increased number of infiltrating CD8+ T cells per field (Fig. 4D). TILs are associated with favorable outcomes in BCs (22,24,43). We, therefore, determined the potential effects of ExTr in promoting the trafficking of TILs. Changes in tumor volume were inversely correlated with CD8+ TIL density (Supplementary Fig. S6C). The higher the increase in maximal aerobic exercise capacity (a biomarker of response to ExTr), the higher was the infiltration of CD8+ TILs (Supplementary Fig. S6D). On the other hand, no significant changes in conventional CD4+ (TCR+CD4+FOXP3−) or regulatory T cells (TCR+CD4+FOXP3+) were found (Fig. 4A). We did not see any changes in other cell populations potentially relevant to antitumor immunity (Supplementary Fig. S7), such as tumor-associated macrophages (TAMs, CD45+Ly6G−Ly6ClowCD11b+F4/80+), DCs (CD45+Ly6G−CD11blowCD11c+), or natural killer cells (NK, CD45+TCRβ−NK1.1+). Hence, we focused on the potential role of CD8+ T cells in mediating the antitumor effect of ExTr.
Figure 4. Exercise training improves infiltration and effector function of CD8+ T cells in breast tumors.
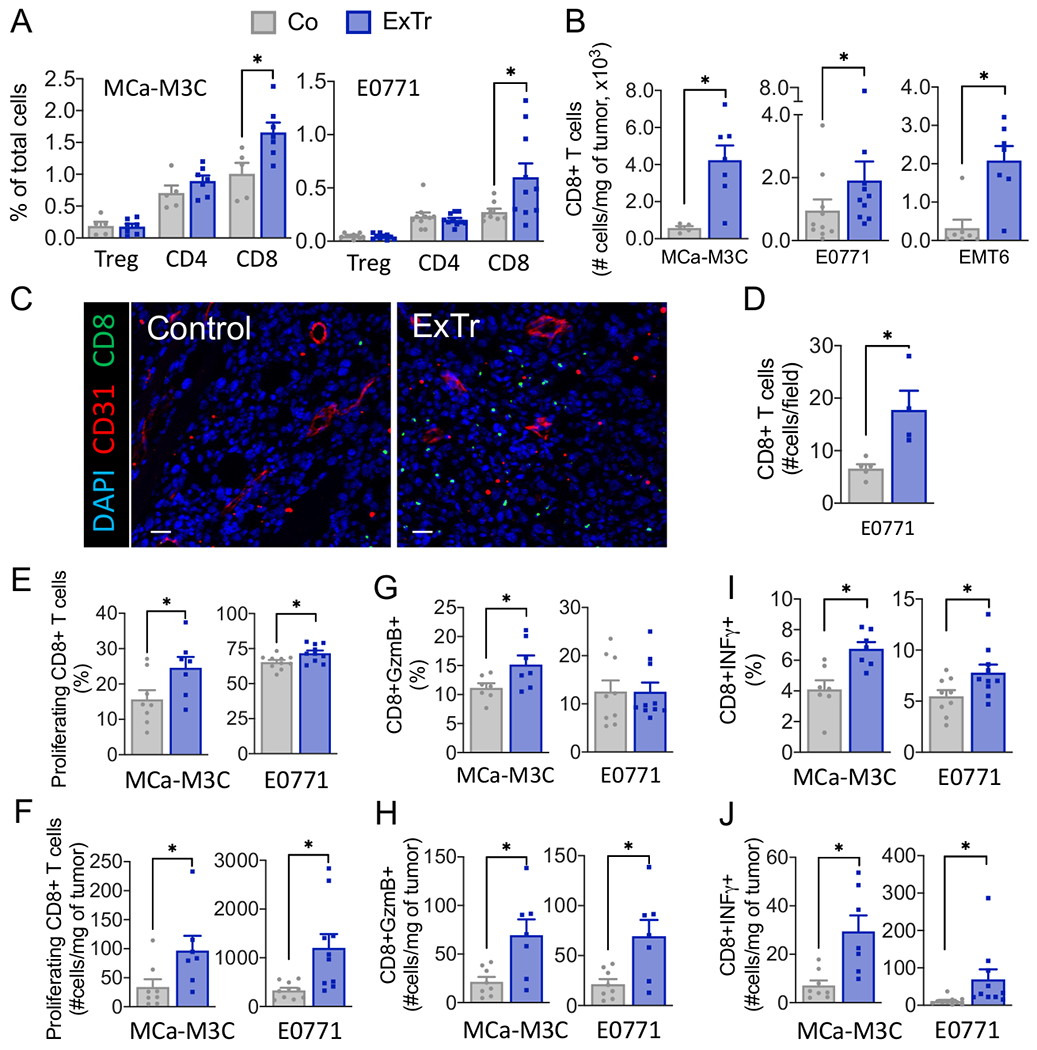
A, Flow cytometric analysis of tumor infiltrating TCR+CD4+FOXP3+ (Tregs), TCR+CD4+, and TCR+CD8+ T cells, assessed as percentage of total cells in breast tumors. B, Number of CD8 T cells in the three BC models. C, Representative immunofluorescence image [red indicates vessels (CD31+) and green indicates CD8+ T cells] and quantification of tumor immunohistochemistry images of CD8+ T cells per field. Scale bar, 25 μm (n=5 mice per group). D, Absolute number of CD8+ T cells per field. E–J, CD8+ T-cell functions. (E, H) Proliferating (Ki67+) CD8+ T cells. (F, I) Granzyme B+ and (G, J) IFNγ+ CD8+ T cells. For flow cytometry, 2 to 3 independent experiments were performed, and the results are representative of one single experiment (n=6-10 mice per group). Statistical differences comparing groups by unpaired Student’s t test. Data presented as mean ± SEM.
ExTr enhances the effector function of CD8+ T cells
We measured the immune phenotype of CD8+ T cells in BCs in response to ExTr. The proliferation marker Ki67 was elevated in ExTr groups (Fig. 4E–F). In addition, ExTr increased the fraction of effector CD8+ T cells expressing granzyme B (Fig. 4G–H and) and IFNγ (Fig. 4I–J). Because exhausted cells restrain the potential for antitumor effector function, we determined the expression of the checkpoint receptors PD-1, CTLA-4, TIM3, and LAG3 in CD8+ T cells (Supplementary Fig. S8A–B). We found ExTr reduced the amount of CD8+CTLA-4+ in MCa-M3C tumors and CD8+LAG3+ in E0771 tumors. ExTr also increased the number of non-immune cells expressing PD-L1 (Supplementary Fig. S8C). We then checked for the co-receptor expression (PD-1+CTLA-4+) in CD8+ T cells (Supplementary Fig. S8D), which is considered a marker of exhausted T cells (26). We found that ExTr significantly reduced CD8+ T cells showing an exhausted phenotype in E0771 and did not change the percentage in MCa-M3C (Supplementary Fig. S8E). However, ExTr significantly increased the total number of CD8+ T cells, contributing to the increased number of exhausted PD-1+CTLA-4+ T cells within the tumors (Supplementary Fig. S8F).
CD8+ T cells are necessary for the antitumor effect of ExTr
After characterizing the selective infiltration of CD8+ T cells, we examined the association of higher CD8+ T-cell infiltration with the antitumor effect of ExTr. We subjected mice to 14 consecutive days of ExTr when tumors in the Control group would have reached the maximum tumor size of ~1cm3. Treatment with depleting-antibodies targeting CD8+ T cells was started concurrently with ExTr when tumors were ~100 mm3 (Fig. 5A). We found that CD8+ T-cell depletion completely abrogated ExTr-induced tumor growth delay (Fig. 5B–C; individual values and time to triple tumor volume in Supplementary Fig. S9A-D; comparison between tumor growth curve from anti-CD8 and IgG in Supplementary Fig. S9E), indicating that CD8+ T cells’ tumor-infiltration is necessary for the antitumor effect of ExTr.
Figure 5. CD8+ T cells are required for the antitumor effect of exercise training in established breast tumors.
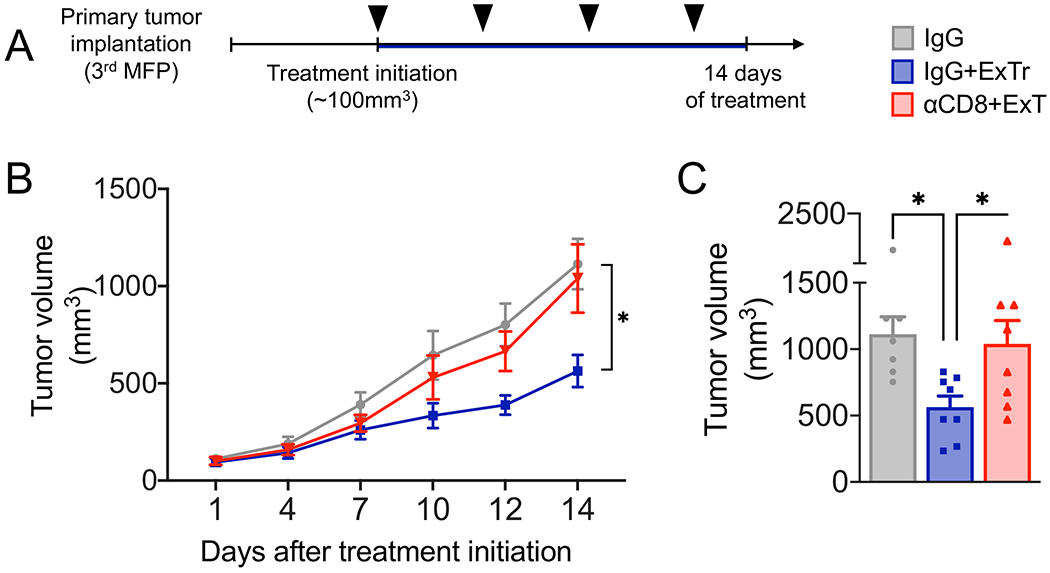
A, Experimental design of the study. When MCa-M3C tumors reached ~100 mm3, mice underwent daily ExTr (60% of maximal exercise velocity for 14 days) and treatment with anti-CD8β or isotype control IgG (250 μg, i.p., every 4 days; black arrow heads) or kept sedentary with IgG treatment. B, Effects of antibody-mediated CD8+ T-cell depletion (αCD8β) on the antitumor effect of ExTr as measured by tumor growth (B, Two-way ANOVA) and C, final tumor volume (one-way ANOVA), n=7-8 per group. Data presented as mean ± SEM.
CXCL9/11-CXCR3 pathway mediates the antitumor effect of ExTr
To dissect the mechanism of ExTr-induced CD8+ T-cell infiltration, we conducted a proteome profile array of bulk tumors, and identified CXCL9 and CXCL11 as potential candidates (Fig. 6A, Supplementary Fig. S10). We then quantified all three CXCR3 ligands (CXCL9, CXCL10, and CXCL11) by ELISA in the BC models. We found ExTr significantly increased CXCL9 in E0771 and CXCL11 in the MCa-M3C model (Fig. 6B–D). CXCL9 also showed a trend towards increase in MCa-M3C (Fig. 6B). There was no change in CXCL10 in both models (Fig. 6C). C57BL/6 mice do not express functional CXCL11. We also determined cellular CXCL9 expression and found several CXCL9-expressing cell populations (Supplementary Fig. S10A-J). CXCL9+ immune cells were predominantly macrophages (Supplementary Fig. S10H–I). It is possible that macrophages mediate beneficial effects of ExTr directly via CXCL9 expression and/or indirectly via vascular normalization (44-46). However, we could not identify a particular cell type that dominantly elevated CXCL9 expression with ExTr. CXCL9 and CXCL11 chemokines are specific ligands for the CXCR3 receptor. We also observed that ExTr increased the absolute number of CD8+ T cells expressing CXCR3 more than 3-fold in MCa-M3C (p<0.05) and 2-fold in the E0771 (p=0.07) tumor (Fig. 6E). Considering its role in the recruitment of CD8+ T cells into tumors (47-50), we asked whether CXCR3-deficiency would impair the antitumor effect of ExTr. To this end, we orthotopically implanted E0771 cells in Cxcr3−/− mice (C57BL/6 background), and, when tumors reached 100 mm3, we subjected them to ExTr for 11 consecutive days, when these tumors began to reach 1cm3. Loss of Cxcr3 completely abolished antitumor effects of ExTr, as assessed by tumor growth kinetics and final tumor weight at day 11 (Fig. 6F–G). The number of CD8+ T cells in E0771 tumors grown in Cxcr3−/− mice was 10-fold lower than that observed in wild-type mice after 7 days of ExTr (Fig. 6H). The frequency of CD8+ T cells was also lower in Cxcr3−/− mice (Fig. 6I). Majority of the CD8+ T cells expressed IFNγ+, and there was no difference between groups (Fig. 6J). ExTr induced a small but significant increase in CD8+ T cells in Cxcr3-KO mice, suggesting CXCR3-independent mechanisms of CD8+ T-cell recruitment (Fig. 6H–I). However, these CD8+ T cells were not enough to induce tumor control.
Figure 6. CXCL9/11-CXCR3 pathway is required for the CD8+ T cell-mediated antitumor effect of exercise training.
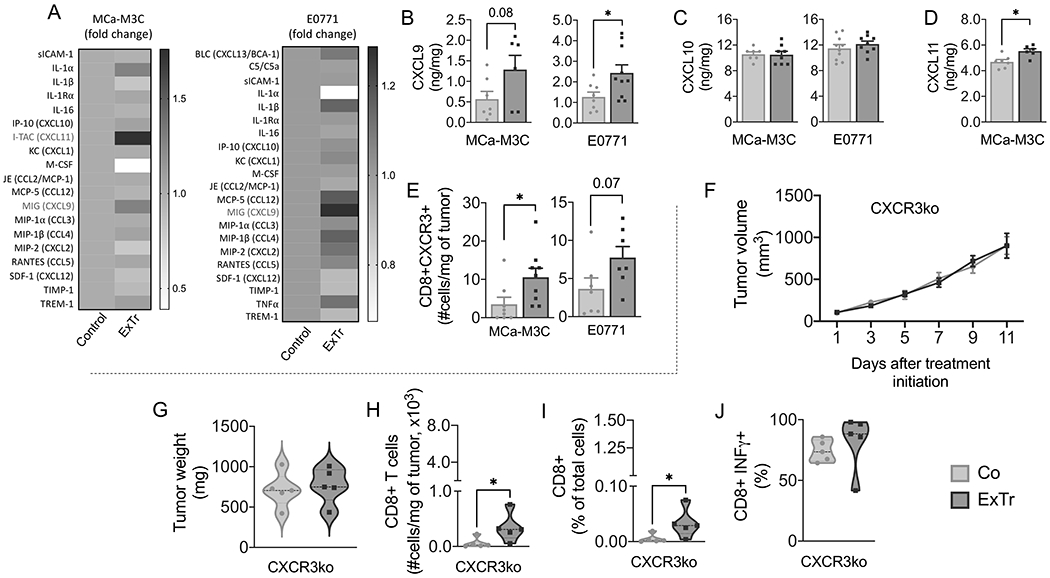
A, Heat map of cytokine array on MCa-M3C and E0771 tumors (pooled samples from n=6 mice per group). B–D, ELISA of chemokines (B) CXCL9, (C) CXCL10, and (D) CXCL10 in bulk tumors after 7 days of ExTr (n=6-10 per group, samples run in duplicate). E, Absolute number of CXCR3+CD8+ T cells for both MCa-M3C and E0771 tumor models (flow cytometry). (F-G) We implanted E0771 tumors in Cxcr3−/− mice (C57BL/6 background). When tumors reached ~100 mm3, mice started ExTr (daily sessions at 60% of maximal exercise velocity for 11 days) or were kept sedentary. (F) Tumor growth and (G) final tumor weight in Cxcr3−/− mice. (H) total and (I) relative tumor infiltration of CD8+ T cells in Cxcr3−/− mice. (J) CD8+ T-cell expression of IFNγ in both groups. n=5 mice/group. Statistical differences by comparing groups by unpaired Student’s t test (B-E), two-way ANOVA (F), and Mann-Whitney test (G-J). Data presented as mean ± SEM or median ± interquartile range and distribution (violin plot, G-J).
ExTr improves immune checkpoint blockade therapy in BC
ExTr increased perfusion, alleviated hypoxia, and enhanced the infiltration of CD8+ T cells in the TME. However, due to the increased total CD8+ T-cell recruitment in ExTr mice tumors, the number of exhausted CD8+ T cells also increased proportionally. Hence, we hypothesized that the combination of ExTr and ICB therapy would have a superior effect on BCs. When MCa-M3C tumors reached ~100 mm3 in size, we treated mice with either anti-PD-1 alone (Supplementary Fig. S11A–B) or anti-PD-1 in combination with anti-CTLA-4 (Fig. 7A, Supplementary Fig. S11C-G). During treatment, mice remained either sedentary or performed daily exercise (14-day ExTr protocol). We found no effect of ICB alone in the orthotopic BCs (Fig. 7B-C, Supplementary Fig. S11). On the other hand, the addition of ICB to ExTr exhibited superior BC tumor control, delaying tumor growth, reducing the final tumor volume, and further enhancing the antitumor effect of ExTr (Fig. 7B-C, Supplementary Fig. S11). Further analysis indicated similar infiltration of CD8+ T cells in both groups subjected to ExTr (Fig. 7D–G). ExTr also increased the total number of intratumoral granzyme B+CD8+ T cells with ExTr, although the fraction of granzyme B+CD8+ T cells was increased only in the ExTr+ICB group (Fig. 7E–H). Finally, ExTr and ICB seemed to have a synergistic effect in inducing an IFNγ+ response in CD8+ T cells, as both the relative and the absolute number of CD8+IFNγ+ T cells increased only with the combined treatment (Fig. 7F–I). ExTr also prevented cancer-induced fatigue in ICB-treated mice (Supplementary Fig. S11G). Altogether, we demonstrated that ExTr intervention in established tumors normalized blood vessels, increased CD8+ T-cell infiltration, and improved control of refractory BC tumors when combined with ICB.
Figure 7. Exercise training sensitizes breast cancer to immune checkpoint blockade.
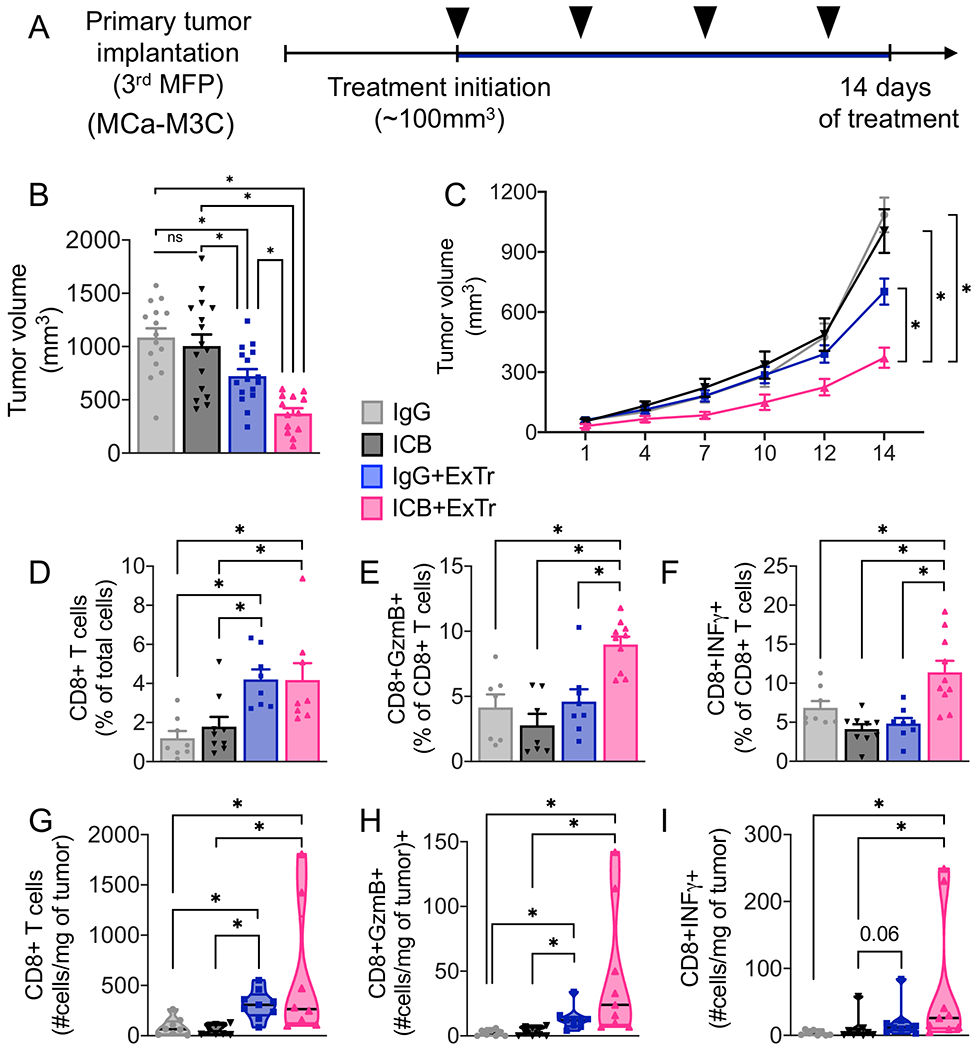
A, Experimental design of the study. When MCa-M3C tumors reached ~100 mm3, mice underwent ExTr (60% of Vmax ExTr, for 14 days) or kept sedentary and were treated with anti-PD-1 plus anti-CTLA-4 (ICB) or isotype control IgG (i.p., every 4 days; black arrow heads). B, ICB effects on tumor volume. C, The effect of ExTr on MCa-M3C tumor growth with ICB. D–I, TCR+CD8+ T-cell infiltration and function. (D) Relative and (G) absolute numbers of CD8+ T cells. (E) Relative and (H) absolute number of granzyme B+CD8+ T cells. (F) Relative and (I) absolute number of IFNγ+CD8+ T cells with ICB and ExTr. Statistical differences by (B, D-F) one-way or (C) Two-way ANOVA, or (G-I) Kruskal-Wallis test. n=7-16 mice per group, pooled data from 2-3 independent experiments. Data presented as mean ± SEM or median ± interquartile range and distribution (violin plot, G-I).
Discussion
ExTr is an emerging antitumor strategy (2,10), yet its potential to enhance immunotherapy in established tumors has not been reported. To study exercise in a therapeutic setting, we needed to define the exercise dose. Because most previous preclinical studies use ad libitum physical activity models (6,8,10), which do not properly capture human exercise behavior, we adapted a controlled exercise. We used a treadmill to precisely control the intensity and duration of exercise dose. Unlike the voluntary running model, in which mice can run 20 km per day (51), our treadmill model’s exercise volume is 10-20 times less than this. Thus, it better recapitulates ExTr in patients participating in clinically supervised ExTr programs. Using this clinically relevant ExTr protocol, we found that aerobic ExTr decreases tumor growth and reprograms the TME by increasing perfusion and decreasing hypoxia in established orthotopic BC models. To investigate the causal role of CD8+ T cells on ExTr-induced tumor growth delay, we used antibody-mediated CD8+ T-cell depletion. The depletion of CD8+ T cells abolished the antitumor effect of ExTr, confirming that the mechanism of antitumor activity from ExTr is CD8+ T cell-dependent.
Emerging data suggest that exercise dose plays a significant role in modulating tumor physiology (2,52). This is supported by clinical observations showing that even fulfillment of the recommended levels of physical activity (e.g., 150 minutes of accumulated moderate-intensity aerobic exercise most days of the week or 75 minutes of vigorous exercise every other day) reduces overall mortality in breast cancer (1,4). Although meeting the minimum guidelines for physical activity may improve survivorship in the long-term, it might not be sufficient to promote clinical benefits during cancer treatment. It seems only supervised ExTr, accounting for planned interventions considering both duration and intensity, has been proven beneficial (3,53-55). We found a significant variability of ExTr dose within preclinical models, even though it is usually assumed that the same ExTr velocity would fit all mouse strains and experimental conditions. Our study demonstrated that the relative exercise intensity for C57BL/6 versus FVB female mice are distinct. This may explain, at least in part, why previous preclinical studies failed to show antitumor activity under similar ExTr protocols (7,52,56). More importantly, the lack of proper control of exercise dose might account for the large variability and absence of significant results from exercise interventions in clinical trials (2).
The timing and type of ExTr intervention might also induce different TIL phenotypes. In voluntary running models conducted in the prevention setting (i.e., starting ad libitum run before implantation or appearance of tumors), NK cells (8) and CD8+ T cells (10) were previously reported to explain the antitumor effect of ExTr. However, because the host was already primed by the adaptations induced by ExTr, including the intrinsic metabolic capacity of CD8+ T cells (10), exercise might have enhanced antitumor capabilities even before a tumor’s existence. The problem is that, along with the much larger exercise volume that does not recapitulate human exercise behavior, the running wheel model does not provide a reliable preclinical model on the therapeutic ExTr either. This is particularly relevant considering that, although 3 out of 4 patients are willing to start an exercise program, nearly 60% of them do not follow an exercise routine (57).
Previous therapeutic ExTr interventions (i.e., starting treatment when tumors were established) on running treadmills could not demonstrate enhancement of cytotoxic NK nor CD8+ T cells (7,52,56). This is probably due to differences in exercise intensity. It is well established that both NK and CD8+ T cells increase in the peripheral blood in response to acute exercise (58). This occurs as a function of exercise-induced adrenergic activation (8,59), linked to exercise intensity. Hence, transient mobilization of immune cells might occur in response to every exercise session, thus favoring re-localization and immune surveillance of tumors. Naïve and memory subsets of CD8+ T cells are increased only beyond moderate-to-vigorous exercise intensity sessions (58). Therefore, if a certain threshold of exercise intensity is not achieved, one should not expect mobilization and redistribution of antitumor cells.
Consistent with previous preclinical reports (6,7), we showed that ExTr induces vessel normalization, characterized by a larger number of vessels with higher pericyte coverage, resulting in less tumoral hypoxia. To gain insight into changes induced by ExTr, we conducted an unbiased RNA-seq analysis of bulk RNA isolated from tumors to characterize the effects of ExTr on the TME. We found that ExTr modulated antitumor immune responses in preclinical models, as well as a transcriptional signature, consistent with the metabolic switch towards OXPHOS metabolism. This shift can occur as a consequence of enhanced tumor perfusion due to vessel normalization (60). We compared our results with the transcriptomic changes induced by a short-term ExTr in the TME in naïve-treated BC patients (38). In this study, the ExTr consisted of short-term (~1 month) supervised sessions of strength training twice a week, and patients were asked to perform aerobic exercise sessions on their own. It was not possible to categorize transcriptome signatures based on aerobic exercise dosing in this study. Nevertheless, we found a transcriptional TME reprogramming in patients consistent with improved antitumor immunity and vessel normalization, similar to what we observed in preclinical models of BC. Vessel normalization may occur at ExTr intensities lower than that employed in our study. For example, Schadler et al. (7) showed increased perfusion in subcutaneous tumors in C57BL/6 mice by ExTr at 12 m/min. This velocity would have represented ~35% of maximal running capacity for C57BL/6 mice in our study, instead of 60%. These authors did not observe changes in tumor growth or immune cell infiltration with the low-intensity ExTr. Although vessel normalization can be observed with low-intensity ExTr (7), a higher exercise dose may be required for mobilization and redistribution of immune cells (8,58,59). Thus, the synergism between induced vessel normalization and CD8-mediated tumor control seems to occur when sufficient intensity (i.e., from moderate-to-vigorous) of ExTr is reached.
ExTr causes a ubiquitous homeostatic physiological response, initiating several events that might contribute to the enhanced tumoral infiltration of CD8+ T cells. Tumors are predominantly glycolytic and with low glucose in the TME of aggressive tumors (61). Conversely, increased glycolytic rate and low perfusion/oxygenation contribute to intratumoral hypoxia and acidosis, which is alleviated with enhanced perfusion (12). Thus, a metabolic switch towards less hypoxic and more oxidative metabolism that favors CD8+ T-cell effector function (62) might have facilitated the enhanced CD8+ T cell effector function with ExTr. Also, CD8+ T cells cannot compete with cancer cells for the scarce intratumoral glucose availability, which further impairs CD8+ T cells’ density and activity (61). Therefore, by inducing vessel normalization and promoting a less hypoxic tumor, ExTr allows a more favorable metabolic TME to facilitate the trafficking of T cells and trigger antitumor effector function (63).
To understand mechanisms promoting enhanced CD8+ T cell trafficking, we found ExTr increases the concentration of the chemokines CXCL9 and CXCL11, but not CXCL10, in breast tumors. CXCL11 is not functional in C57BL/6 mice, so it was only quantified in the MCa-M3C tumor model in FVB mice (48). CXCL9/10/11 are ligands of the CXCR3 receptor involved in increased trafficking of CD8+CXCR3+ T cells to tumors (47,48,64). These chemokines are induced by IFNγ and were potentially triggered in response to the ExTr-mediated increase of IFNγ-producing effector CD8+ T cells, thereby inducing positive feedback to attract more CD8+ T cells to tumors. There is an association of IFNγ-secreting T cells and vessel normalization (18), and IFNγ promotes the enhanced expression of adhesion molecules in endothelial cells (65). These cells display a strong affinity to CD8+CXCR3+ T cells (66), further increasing their infiltration. ExTr antitumor effect was entirely abolished in Cxcr3−/− mice, demonstrating that the CXCR3 pathway participates in the antitumor effect of ExTr. Data suggest that CXCR3 is not essential for the early recruitment of CD8+ T cells into tumors (64). However, CXCR3 in CD8+ T cells is necessary to trigger initial effector responses in bystander CD8+ T cells (67), although not required for sustained IFNγ effector responses (68). Indeed, a large number of infiltrating CD8+ T cells expressed IFNγ in Cxcr3−/− mice. This may explain why CD8+CXCR3+ T cells are required for improved response to immunotherapy. Their ligands are reported to function both as biomarkers and inducers of response to anti-PD-1 alone (64) or in combination with anti-CTLA-4 (47). It also explains why Cxcr3−/− mice blunted the antitumor effect of ExTr. Future studies need to examine the inducers of activation and effector function of tumoral CD8+ T cells by ExTr.
The relevance of the CXCL9/10/11-CXCR3 pathway for recruiting and triggering cytotoxic T cells is well known (47-50,64). Several therapeutic approaches have been tested to leverage this pathway to attract more CD8+ T cells, including radiation, chemotherapy (50), and vaccination (49). Multimodal therapy increases ICB response, but it is associated with increased adverse events (69). Therefore, less toxic approaches are necessary. Our findings support ExTr as a safer strategy to stimulate the antitumor CXCL9/11-CXCR3 system in solid tumors.
ICB has revolutionized the treatment of multiple malignancies. However, its efficacy in BC has been modest (27,70). Lack of perfusion, tumoral hypoxia, and T-cell exclusion are considered significant resistance mechanisms (15,17-19). Several strategies have been proposed to alleviate immune exclusion and sensitize tumors to ICB (70). Here, we show that controlled ExTr can facilitate immune cell Infiltration into tumors and sensitize them to ICB. ExTr increases CD8+ T cells by reprogramming the TME towards a less aggressive phenotype, but a large fraction of CD8+ T cells remain expressing immune checkpoint receptors, suggesting these tumors would benefit from ICB therapy. Our work shows that ExTr and ICB synergistically enhance the activity of effector CD8+ T cells (i.e., a fraction of IFNγ- or granzyme B-expressing CD8+ T cells). Both CD8+ T-cell infiltration and IFNγ signaling signatures are indicative of benefit from combined anti-PD-1 and anti-CTLA-4 in patients with solid tumors (71) or anti-PD-L1 alone in BC (72). ExTr increased the number of cells expressing PD-L1, a biomarker of response for metastatic BC (25). Our data indicate that, supervised ExTr has the potential to unleash antitumor response from immunotherapy in the clinic.
Supplementary Material
Synopsis:
Optimized exercise therapy induces vessel normalization, boosts antitumor effector cell infiltration and function, and delays tumor growth in a CXCR3 pathway/CD8+ T cell-dependent manner. This results in sensitization of refractory breast cancer to immune checkpoint blockade.
Acknowledgments
We thank Dr. Joao Incio for preliminary discussions, Dr. Peigen Huang, Sylvie Roberge, and Carolyn Smith for technical support, Dr. Yao Sun, for experimental assistance, Drs. Patrik C. Anderson, Dan G. Duda, Gino B. Ferraro, and Timothy P. Padera for their helpful input on the manuscript.
Additional information.
This work was supported by Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP #2014/13690-8 and #2016/21320-1), in agreement with Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES), as visiting fellow from Instituto do Coração do Hospital das Clínicas da Faculdade de Medicina da Universidade de São Paulo (InCor-HCFMUSP), Sao Paulo, Brazil (ILG-S); Cancer Research Institute/ Merck Fellowship (NPT), by NCI grant #R25CA174650 through the CanCure Co-Op Program at Northeastern University in Boston, MA (HC); by International fellowship from the Alumni Association Sanjukai Kagawa University Faculty of Medicine, Overseas research Fellowship from the Japan Society for the Promotion of Science & KANAE foundation (KK). RKJ’s research is supported by NIH grants P01-CA080124, R35-CA197743, and R01-CA208205, and by the National Foundation for Cancer Research, Harvard Ludwig Cancer Center, Advanced Medical Research Foundation and Jane’s Trust Foundation.
Disclosure.
We disclose that RKJ received honorarium from Amgen and consultant fees from Chugai, Elpis, Ophthotech, Merck, SPARC, SynDevRx and XTuit. RKJ owns equity in Accurius, Enlight, and SynDevRx, and serves on the Boards of Trustees of Tekla Healthcare Investors, Tekla Life Sciences Investors, Tekla Healthcare Opportunities Fund and Tekla World Healthcare Fund. No funding or reagents from these companies were used in this study.
References
- 1.Holmes MD, Chen WY, Feskanich D, Kroenke CH, Colditz GA. Physical activity and survival after breast cancer diagnosis. JAMA 2005;293(20):2479–86 doi 10.1001/jama.293.20.2479. [DOI] [PubMed] [Google Scholar]
- 2.Iyengar NM, Jones LW. Development of Exercise as Interception Therapy for Cancer: A Review. JAMA Oncol 2019. doi 10.1001/jamaoncol.2019.2585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Williams PT. Significantly greater reduction in breast cancer mortality from post-diagnosis running than walking. Int J Cancer 2014;135(5):1195–202 doi 10.1002/ijc.28740. [DOI] [PubMed] [Google Scholar]
- 4.Cannioto RA, Hutson A, Dighe S, McCann W, McCann SE, Zirpoli GR, et al. Physical activity before, during and after chemotherapy for high-risk breast cancer: relationships with survival. J Natl Cancer Inst 2020. doi 10.1093/jnci/djaa046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McCullough DJ, Stabley JN, Siemann DW, Behnke BJ. Modulation of blood flow, hypoxia, and vascular function in orthotopic prostate tumors during exercise. J Natl Cancer Inst 2014;106(4):dju036 doi 10.1093/jnci/dju036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Betof AS, Lascola CD, Weitzel D, Landon C, Scarbrough PM, Devi GR, et al. Modulation of murine breast tumor vascularity, hypoxia and chemotherapeutic response by exercise. J Natl Cancer Inst 2015;107(5) doi 10.1093/jnci/djv040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schadler KL, Thomas NJ, Galie PA, Bhang DH, Roby KC, Addai P, et al. Tumor vessel normalization after aerobic exercise enhances chemotherapeutic efficacy. Oncotarget 2016;7(40):65429–40 doi 10.18632/oncotarget.11748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pedersen L, Idorn M, Olofsson GH, Lauenborg B, Nookaew I, Hansen RH, et al. Voluntary Running Suppresses Tumor Growth through Epinephrine- and IL-6-Dependent NK Cell Mobilization and Redistribution. Cell Metab 2016;23(3):554–62 doi 10.1016/j.cmet.2016.01.011. [DOI] [PubMed] [Google Scholar]
- 9.Miller A, Nace R, Ayala-Breton CC, Steele M, Bailey K, Peng KW, et al. Perfusion Pressure Is a Critical Determinant of the Intratumoral Extravasation of Oncolytic Viruses. Mol Ther 2016;24(2):306–17 doi 10.1038/mt.2015.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rundqvist H, Velica P, Barbieri L, Gameiro PA, Bargiela D, Gojkovic M, et al. Cytotoxic T-cells mediate exercise-induced reductions in tumor growth. Elife 2020;9 doi 10.7554/eLife.59996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Garcia E, Becker VG, McCullough DJ, Stabley JN, Gittemeier EM, Opoku-Acheampong AB, et al. Blood flow responses to mild-intensity exercise in ectopic vs. orthotopic prostate tumors; dependence upon host tissue hemodynamics and vascular reactivity. J Appl Physiol (1985) 2016;121(1):15–24 doi 10.1152/japplphysiol.00266.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jain RK. Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science 2005;307(5706):58–62 doi 10.1126/science.1104819. [DOI] [PubMed] [Google Scholar]
- 13.Zlotecki RA, Baxter LT, Boucher Y, Jain RK. Pharmacologic modification of tumor blood flow and interstitial fluid pressure in a human tumor xenograft: network analysis and mechanistic interpretation. Microvasc Res 1995;50(3):429–43 doi 10.1006/mvre.1995.1069. [DOI] [PubMed] [Google Scholar]
- 14.Incio J, Ligibel JA, McManus DT, Suboj P, Jung K, Kawaguchi K, et al. Obesity promotes resistance to anti-VEGF therapy in breast cancer by up-regulating IL-6 and potentially FGF-2. Sci Transl Med 2018;10(432):eaag0945 doi 10.1126/scitranslmed.aag0945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huang Y, Yuan J, Righi E, Kamoun WS, Ancukiewicz M, Nezivar J, et al. Vascular normalizing doses of antiangiogenic treatment reprogram the immunosuppressive tumor microenvironment and enhance immunotherapy. Proc Natl Acad Sci U S A 2012;109(43):17561–6 doi 10.1073/pnas.1215397109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ashcraft KA, Warner AB, Jones LW, Dewhirst MW. Exercise as Adjunct Therapy in Cancer. Semin Radiat Oncol 2019;29(1):16–24 doi 10.1016/j.semradonc.2018.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zheng X, Fang Z, Liu X, Deng S, Zhou P, Wang X, et al. Increased vessel perfusion predicts the efficacy of immune checkpoint blockade. J Clin Invest 2018;128(5):2104–15 doi 10.1172/JCI96582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tian L, Goldstein A, Wang H, Ching Lo H, Sun Kim I, Welte T, et al. Mutual regulation of tumour vessel normalization and immunostimulatory reprogramming. Nature 2017;544(7649):250–4 doi 10.1038/nature21724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schmittnaegel M, Rigamonti N, Kadioglu E, Cassara A, Wyser Rmili C, Kiialainen A, et al. Dual angiopoietin-2 and VEGFA inhibition elicits antitumor immunity that is enhanced by PD-1 checkpoint blockade. Sci Transl Med 2017;9(385) doi 10.1126/scitranslmed.aak9670. [DOI] [PubMed] [Google Scholar]
- 20.Chen IX, Chauhan VP, Posada J, Ng MR, Wu MW, Adstamongkonkul P, et al. Blocking CXCR4 alleviates desmoplasia, increases T-lymphocyte infiltration, and improves immunotherapy in metastatic breast cancer. Proc Natl Acad Sci U S A 2019;116(10):4558–66 doi 10.1073/pnas.1815515116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chung W, Eum HH, Lee HO, Lee KM, Lee HB, Kim KT, et al. Single-cell RNA-seq enables comprehensive tumour and immune cell profiling in primary breast cancer. Nature communications 2017;8:15081 doi 10.1038/ncomms15081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Denkert C, von Minckwitz G, Darb-Esfahani S, Lederer B, Heppner BI, Weber KE, et al. Tumour-infiltrating lymphocytes and prognosis in different subtypes of breast cancer: a pooled analysis of 3771 patients treated with neoadjuvant therapy. Lancet Oncol 2018;19(1):40–50 doi 10.1016/S1470-2045(17)30904-X. [DOI] [PubMed] [Google Scholar]
- 23.Liu S, Chen B, Burugu S, Leung S, Gao D, Virk S, et al. Role of Cytotoxic Tumor-Infiltrating Lymphocytes in Predicting Outcomes in Metastatic HER2-Positive Breast Cancer: A Secondary Analysis of a Randomized Clinical Trial. JAMA Oncol 2017;3(11):e172085 doi 10.1001/jamaoncol.2017.2085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang K, Shen T, Siegal GP, Wei S. The CD4/CD8 Ratio of Tumor-infiltrating Lymphocytes at the Tumor-host Interface Has Prognostic Value in Triple Negative Breast Cancer. Hum Pathol 2017;69:110–7 doi 10.1016/j.humpath.2017.09.012. [DOI] [PubMed] [Google Scholar]
- 25.Savas P, Virassamy B, Ye C, Salim A, Mintoff CP, Caramia F, et al. Single-cell profiling of breast cancer T cells reveals a tissue-resident memory subset associated with improved prognosis. Nat Med 2018;24(7):986–93 doi 10.1038/s41591-018-0078-7. [DOI] [PubMed] [Google Scholar]
- 26.Beavis PA, Henderson MA, Giuffrida L, Davenport AJ, Petley EV, House IG, et al. Dual PD-1 and CTLA-4 Checkpoint Blockade Promotes Antitumor Immune Responses through CD4(+)Foxp3(−) Cell-Mediated Modulation of CD103(+) Dendritic Cells. Cancer Immunol Res 2018;6(9):1069–81 doi 10.1158/2326-6066.CIR-18-0291. [DOI] [PubMed] [Google Scholar]
- 27.Zhang JQ, Plitas G. Immunotherapeutic strategies in breast cancer: A clinical update. J Surg Oncol 2020. doi 10.1002/jso.26287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.American College of Sports Medicine, Riebe D, Ehrman JK, Liguori G, Magal M. ACSM’s guidelines for exercise testing and prescription. Philadelphia: Wolters Kluwer; 2018. 472 pages p. [Google Scholar]
- 29.Rock CL, Doyle C, Demark-Wahnefried W, Meyerhardt J, Courneya KS, Schwartz AL, et al. Nutrition and physical activity guidelines for cancer survivors. CA Cancer J Clin 2012;62(4):243–74 doi 10.3322/caac.21142. [DOI] [PubMed] [Google Scholar]
- 30.Bull FC, Al-Ansari SS, Biddle S, Borodulin K, Buman MP, Cardon G, et al. World Health Organization 2020 guidelines on physical activity and sedentary behaviour. Br J Sports Med 2020;54(24):1451–62 doi 10.1136/bjsports-2020-102955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hoydal MA, Wisloff U, Kemi OJ, Ellingsen O. Running speed and maximal oxygen uptake in rats and mice: practical implications for exercise training. Eur J Cardiovasc Prev Rehabil 2007;14(6):753–60 doi 10.1097/HJR.0b013e3281eacef1. [DOI] [PubMed] [Google Scholar]
- 32.Gomes-Santos IL, Fernandes T, Couto GK, Ayres Ferreira-Filho JC, Cury Salemi VM, Fernandes FB, et al. Effects of Exercise Training on Circulating and Skeletal Muscle Renin-Angiotensin System in Chronic Heart Failure Rats. Plos One 2014;9(5) doi 10.1371/journal.pone.0098012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 2013;29(1):15–21 doi 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lawrence M, Huber W, Pages H, Aboyoun P, Carlson M, Gentleman R, et al. Software for computing and annotating genomic ranges. PLoS Comput Biol 2013;9(8):e1003118 doi 10.1371/journal.pcbi.1003118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 2014;15(12):550 doi 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Merico D, Isserlin R, Stueker O, Emili A, Bader GD. Enrichment map: a network-based method for gene-set enrichment visualization and interpretation. PLoS One 2010;5(11):e13984 doi 10.1371/journal.pone.0013984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 2005;102(43):15545–50 doi 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ligibel JA, Dillon D, Giobbie-Hurder A, McTiernan A, Frank E, Cornwell M, et al. Impact of a Pre-Operative Exercise Intervention on Breast Cancer Proliferation and Gene Expression: Results from the Pre-Operative Health and Body (PreHAB) Study. Clin Cancer Res 2019;25(17):5398–406 doi 10.1158/1078-0432.CCR-18-3143. [DOI] [PubMed] [Google Scholar]
- 39.Jones LW, Courneya KS, Mackey JR, Muss HB, Pituskin EN, Scott JM, et al. Cardiopulmonary function and age-related decline across the breast cancer survivorship continuum. J Clin Oncol 2012;30(20):2530–7 doi 10.1200/JCO.2011.39.9014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gunderson AJ, Yamazaki T, McCarty K, Fox N, Phillips M, Alice A, et al. TGFbeta suppresses CD8(+) T cell expression of CXCR3 and tumor trafficking. Nature communications 2020;11(1):1749 doi 10.1038/s41467-020-15404-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Peranzoni E, Lemoine J, Vimeux L, Feuillet V, Barrin S, Kantari-Mimoun C, et al. Macrophages impede CD8 T cells from reaching tumor cells and limit the efficacy of anti-PD-1 treatment. Proc Natl Acad Sci U S A 2018;115(17):E4041–E50 doi 10.1073/pnas.1720948115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bockorny B, Semenisty V, Macarulla T, Borazanci E, Wolpin BM, Stemmer SM, et al. BL-8040, a CXCR4 antagonist, in combination with pembrolizumab and chemotherapy for pancreatic cancer: the COMBAT trial. Nat Med 2020. doi 10.1038/s41591-020-0880-x. [DOI] [PubMed] [Google Scholar]
- 43.DeNardo DG, Brennan DJ, Rexhepaj E, Ruffell B, Shiao SL, Madden SF, et al. Leukocyte complexity predicts breast cancer survival and functionally regulates response to chemotherapy. Cancer Discov 2011;1(1):54–67 doi 10.1158/2159-8274.CD-10-0028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Huang Y, Goel S, Duda DG, Fukumura D, Jain RK. Vascular normalization as an emerging strategy to enhance cancer immunotherapy. Cancer Res 2013;73(10):2943–8 doi 10.1158/0008-5472.CAN-12-4354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rolny C, Mazzone M, Tugues S, Laoui D, Johansson I, Coulon C, et al. HRG inhibits tumor growth and metastasis by inducing macrophage polarization and vessel normalization through downregulation of PlGF. Cancer Cell 2011;19(1):31–44 doi S1535-6108(10)00474-5 [pii] 10.1016/j.ccr.2010.11.009. [DOI] [PubMed] [Google Scholar]
- 46.Chen P, Bonaldo P. Role of macrophage polarization in tumor angiogenesis and vessel normalization: implications for new anticancer therapies. Int Rev Cell Mol Biol 2013;301:1–35 doi 10.1016/B978-0-12-407704-1.00001-4. [DOI] [PubMed] [Google Scholar]
- 47.House IG, Savas P, Lai J, Chen AXY, Oliver AJ, Teo ZL, et al. Macrophage-Derived CXCL9 and CXCL10 Are Required for Antitumor Immune Responses Following Immune Checkpoint Blockade. Clin Cancer Res 2020;26(2):487–504 doi 10.1158/1078-0432.CCR-19-1868. [DOI] [PubMed] [Google Scholar]
- 48.Tokunaga R, Zhang W, Naseem M, Puccini A, Berger MD, Soni S, et al. CXCL9, CXCL10, CXCL11/CXCR3 axis for immune activation - A target for novel cancer therapy. Cancer Treat Rev 2018;63:40–7 doi 10.1016/j.ctrv.2017.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Moon EK, Wang LS, Bekdache K, Lynn RC, Lo A, Thorne SH, et al. Intra-tumoral delivery of CXCL11 via a vaccinia virus, but not by modified T cells, enhances the efficacy of adoptive T cell therapy and vaccines. Oncoimmunology 2018;7(3):e1395997 doi 10.1080/2162402X.2017.1395997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Luo R, Firat E, Gaedicke S, Guffart E, Watanabe T, Niedermann G. Cisplatin Facilitates Radiation-Induced Abscopal Effects in Conjunction with PD-1 Checkpoint Blockade Through CXCR3/CXCL10-Mediated T-cell Recruitment. Clin Cancer Res 2019;25(23):7243–55 doi 10.1158/1078-0432.CCR-19-1344. [DOI] [PubMed] [Google Scholar]
- 51.Manzanares G, Brito-da-Silva G, Gandra PG. Voluntary wheel running: patterns and physiological effects in mice. Braz J Med Biol Res 2018;52(1):e7830 doi 10.1590/1414-431X20187830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Colbert LH, Westerlind KC, Perkins SN, Haines DC, Berrigan D, Donehower LA, et al. Exercise effects on tumorigenesis in a p53-deficient mouse model of breast cancer. Med Sci Sports Exerc 2009;41(8):1597–605 doi 10.1249/MSS.0b013e31819f1f05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Park JH, Lee J, Oh M, Park H, Chae J, Kim DI, et al. The effect of oncologists’ exercise recommendations on the level of exercise and quality of life in survivors of breast and colorectal cancer: A randomized controlled trial. Cancer 2015;121(16):2740–8 doi 10.1002/cncr.29400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Buffart LM, Kalter J, Sweegers MG, Courneya KS, Newton RU, Aaronson NK, et al. Effects and moderators of exercise on quality of life and physical function in patients with cancer: An individual patient data meta-analysis of 34 RCTs. Cancer Treat Rev 2017;52:91–104 doi 10.1016/j.ctrv.2016.11.010. [DOI] [PubMed] [Google Scholar]
- 55.Sweegers MG, Altenburg TM, Chinapaw MJ, Kalter J, Verdonck-de Leeuw IM, Courneya KS, et al. Which exercise prescriptions improve quality of life and physical function in patients with cancer during and following treatment? A systematic review and meta-analysis of randomised controlled trials. Br J Sports Med 2018;52(8):505–13 doi 10.1136/bjsports-2017-097891. [DOI] [PubMed] [Google Scholar]
- 56.Glass OK, Bowie M, Fuller J, Darr D, Usary J, Boss K, et al. Differential response to exercise in claudin-low breast cancer. Oncotarget 2017;8(60):100989–1004 doi 10.18632/oncotarget.21054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Knowlton SE, O’Donnell EK, Horick N, Perez GK, Park E, Rabin J, et al. Moving forward on all fronts: impact, patterns, and barriers to exercise in cancer survivors and patients living with advanced disease. Support Care Cancer 2020;28(10):4979–88 doi 10.1007/s00520-020-05344-w. [DOI] [PubMed] [Google Scholar]
- 58.Campbell JP, Riddell NE, Burns VE, Turner M, van Zanten JJ, Drayson MT, et al. Acute exercise mobilises CD8+ T lymphocytes exhibiting an effector-memory phenotype. Brain Behav Immun 2009;23(6):767–75 doi 10.1016/j.bbi.2009.02.011. [DOI] [PubMed] [Google Scholar]
- 59.Graff RM, Kunz HE, Agha NH, Baker FL, Laughlin M, Bigley AB, et al. beta2-Adrenergic receptor signaling mediates the preferential mobilization of differentiated subsets of CD8+ T-cells, NK-cells and non-classical monocytes in response to acute exercise in humans. Brain Behav Immun 2018;74:143–53 doi 10.1016/j.bbi.2018.08.017. [DOI] [PubMed] [Google Scholar]
- 60.Navarro P, Bueno MJ, Zagorac I, Mondejar T, Sanchez J, Mouron S, et al. Targeting Tumor Mitochondrial Metabolism Overcomes Resistance to Antiangiogenics. Cell Rep 2016;15(12):2705–18 doi 10.1016/j.celrep.2016.05.052. [DOI] [PubMed] [Google Scholar]
- 61.Chang CH, Qiu J, O’Sullivan D, Buck MD, Noguchi T, Curtis JD, et al. Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell 2015;162(6):1229–41 doi 10.1016/j.cell.2015.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Calcinotto A, Filipazzi P, Grioni M, Iero M, De Milito A, Ricupito A, et al. Modulation of microenvironment acidity reverses anergy in human and murine tumor-infiltrating T lymphocytes. Cancer Res 2012;72(11):2746–56 doi 10.1158/0008-5472.CAN-11-1272. [DOI] [PubMed] [Google Scholar]
- 63.Weninger W, Crowley MA, Manjunath N, von Andrian UH. Migratory properties of naive, effector, and memory CD8(+) T cells. J Exp Med 2001;194(7):953–66 doi 10.1084/jem.194.7.953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chow MT, Ozga AJ, Servis RL, Frederick DT, Lo JA, Fisher DE, et al. Intratumoral Activity of the CXCR3 Chemokine System Is Required for the Efficacy of Anti-PD-1 Therapy. Immunity 2019;50(6):1498–512 e5 doi 10.1016/j.immuni.2019.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Melrose J, Tsurushita N, Liu G, Berg EL. IFN-gamma inhibits activation-induced expression of E- and P-selectin on endothelial cells. J Immunol 1998;161(5):2457–64. [PubMed] [Google Scholar]
- 66.Curbishley SM, Eksteen B, Gladue RP, Lalor P, Adams DH. CXCR 3 activation promotes lymphocyte transendothelial migration across human hepatic endothelium under fluid flow. Am J Pathol 2005;167(3):887–99 doi 10.1016/S0002-9440(10)62060-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Maurice NJ, McElrath MJ, Andersen-Nissen E, Frahm N, Prlic M. CXCR3 enables recruitment and site-specific bystander activation of memory CD8(+) T cells. Nature communications 2019;10(1):4987 doi 10.1038/s41467-019-12980-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hu JK, Kagari T, Clingan JM, Matloubian M. Expression of chemokine receptor CXCR3 on T cells affects the balance between effector and memory CD8 T-cell generation. Proc Natl Acad Sci U S A 2011;108(21):E118–27 doi 10.1073/pnas.1101881108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Martins F, Sofiya L, Sykiotis GP, Lamine F, Maillard M, Fraga M, et al. Adverse effects of immune-checkpoint inhibitors: epidemiology, management and surveillance. Nat Rev Clin Oncol 2019;16(9):563–80 doi 10.1038/s41571-019-0218-0. [DOI] [PubMed] [Google Scholar]
- 70.Adams S, Gatti-Mays ME, Kalinsky K, Korde LA, Sharon E, Amiri-Kordestani L, et al. Current Landscape of Immunotherapy in Breast Cancer: A Review. JAMA Oncol 2019. doi 10.1001/jamaoncol.2018.7147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Grasso CS, Tsoi J, Onyshchenko M, Abril-Rodriguez G, Ross-Macdonald P, Wind-Rotolo M, et al. Conserved Interferon-gamma Signaling Drives Clinical Response to Immune Checkpoint Blockade Therapy in Melanoma. Cancer Cell 2020;38(4):500–15 e3 doi 10.1016/j.ccell.2020.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sceneay J, Goreczny GJ, Wilson K, Morrow S, DeCristo MJ, Ubellacker JM, et al. Interferon Signaling Is Diminished with Age and Is Associated with Immune Checkpoint Blockade Efficacy in Triple-Negative Breast Cancer. Cancer Discov 2019;9(9):1208–27 doi 10.1158/2159-8290.CD-18-1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.