

Abstract
Biguanides are a class of anti-diabetic drugs that includes phenformin and metformin, however, the former was withdrawn from approval in many countries due to its toxicity. Findings from retrospective epidemiological studies in diabetic populations and pre-clinical laboratory models have demonstrated that biguanides possess anti-tumor activities that suggest their repurposing for cancer prevention and treatment. However, a better understanding of how these biguanides behave as anti-tumor agents is needed to guide their improved applications in cancer therapy, spurring increased interest in their pharmacology. Here, we present evidence for proposed mechanisms of action related to their anti-tumor activity, including their effects on central carbon metabolism in cancer cells and immune-modulating activity, and then review progress on biguanide repurposing in cancer therapeutics and the possible re-evaluation of phenformin as a cancer therapeutic agent.
Keywords: Metformin, Phenformin, AMPK, Cancer cell Metabolism, Immunometabolism, Cancer Immunotherapy
Biguanides, from diabetes to cancer
Biguanides are a class of anti-diabetic drugs originating from compounds identified in the French lilac, a plant recognized for its hypoglycemic properties since medieval times [1]. One biguanide, phenformin (N-Phenethylbiguanide), was prescribed to treat type 2 diabetes starting in the 1950s but was withdrawn from use in the late 1970s in the United States due to the risk of lactic acidosis. In 1995, metformin (dimethylbiguanide), another biguanide with less adverse side-effects, was approved in the United States and has become the most widely prescribed drug for diabetes [1]. Evidence from retrospective population-based studies revealed that metformin use was associated with reduced risk of cancer and decreased cancer-related mortality in diabetic patients compared to those not using metformin [2, 3]. Preclinical studies of both metformin and phenformin in cancer cell culture and mouse tumor models have demonstrated that both biguanides possess antitumor activity, and this has spurred attempts to repurpose them for cancer prevention and treatment. Several recent reviews on metformin have summarized what is known about its mechanisms of actions under different contexts [4–6]. Here, we focus on progress in repurposing biguanides for cancer therapy. We discuss metabolic determinants of biguanide sensitivity, adaptation to biguanide exposure, associated strategies towards targeting metabolic heterogeneity, effects of biguanides on the tumor immune microenvironment (TIME), and progress in testing biguanides in cancer clinical trials.
Molecular targets of biguanides underlying their anti-tumor activities
A broadly accepted mechanism of action to explain biguanides’ anti-tumor activities involves their inhibition of mitochondrial complex I (Complex I) (Figure 1). Metformin can inhibit the proliferation of cancer cells in vitro and reduce xenograft tumor growth, and these effects can be rescued by the expression of the metformin-resistant yeast-derived Complex I NADH dehydrogenase subunit, NDI1 [7]. Similarly, ectopic expression of NDI1 in cancer cells with Complex I mutations blocked the ability of phenformin to impair proliferation and oxygen consumption [8]. These findings highlight the importance of Complex I as a major target for biguanides in reducing tumor burden. Inhibition of Complex I led to an increase in the intracellular adenosine monophosphate (AMP) to adenosine triphosphate (ATP) ratio, which indirectly activates AMP-activated protein kinase (AMPK). AMPK modulates the expression and activities of various metabolic enzymes and regulators in pathways that are critical for cancer cell growth and proliferation. AMPK-dependent inhibition of mammalian target of rapamycin (mTOR) signaling is perhaps the most studied anti-neoplastic mechanism for biguanides [1,9]. Inhibition of Complex I by metformin is also associated with other metabolic changes, such as depletion of tricarboxylic acid (TCA) cycle metabolites [10] and modulation of hypoxia-inducible factor-1a (HIF-1α) activity [7]. Metformin may also trigger epigenetic changes by modulating the levels of critical substrates for key histone modification enzymes [11–13].
Figure 1. Molecular targets of biguanides that underlie their anti-tumor activities.
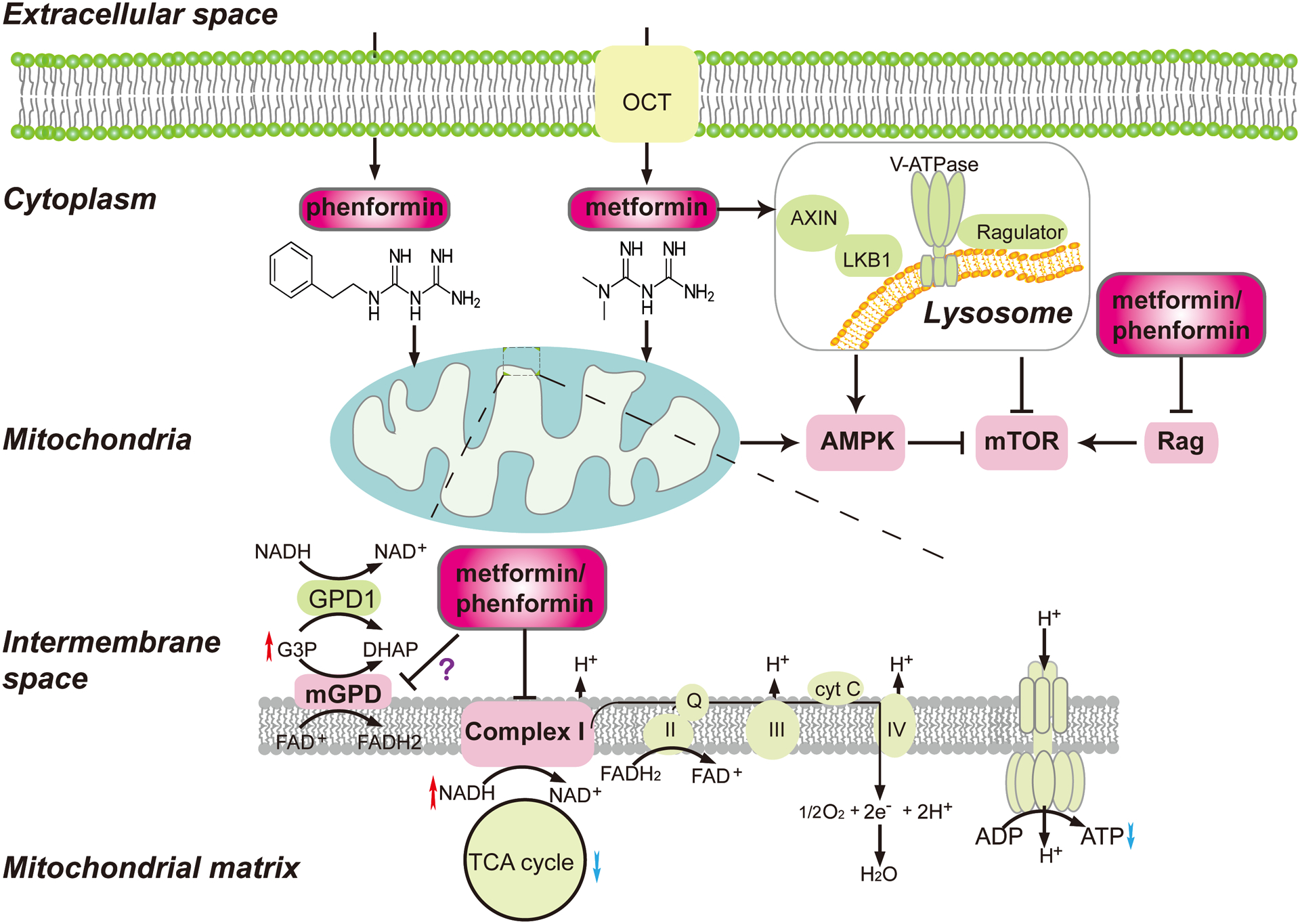
Biguanides enter cells either directly (phenformin) or via organic cation transporters (OCTs) (metformin), and target mitochondrial complex I, AMPK, mTOR and/or mGPD. Inhibition of mitochondrial complex I by biguanides triggers an increase in the AMP/ADP:ATP ratio, leading to activation of AMPK and subsequent attenuation of mTOR signaling. Biguanides also inhibit mGPD, leading to perturbation of the G3P shuttle and the accumulation of NADH and G3P. Finally, biguanides may activate AMPK and inhibit mTOR independent of mitochondrial complex I status by inducing the formation of v-ATPase-Ragulator-AXIN/LKB1-AMPK complex on lysosomal membranes.
Biguanides may also activate AMPK and inhibit mTOR signaling via complex I-independent mechanisms (Figure 1). Metformin induced the formation of v-ATPase-Ragulator-AXIN/LKB1-AMPK complex on lysosomal membranes, leading to AMPK activation [14]. The formation of this complex led to dissociation of Ragulator from mTORC1, resulting in mTOR inhibition [14]. In addition, biguanides modulated the activities of Rag GTPases [15, 16], including inhibiting RagC nuclear-cytoplasmic shuttling [15, 16] thus reducing mTORC1 signaling in an AMPK-independent manner.
Mitochondrial glycerophosphate dehydrogenase (mGPD), is another potential mediator of biguanides’ anti-tumor activities. mGPD and cytoplasmic GPD1 are major constituents of the glycerol-3-phosphate (G3P) shuttle, which allows reducing equivalents generated in the cytosol to contribute to ATP synthesis in the mitochondria and plays a significant role in regulating redox potential in the cell. mGPD has been suggested to be a target of metformin to inhibit hepatic gluconeogenesis and alleviate type 2 diabetes in an earlier study [17]. More recently, phenformin showed inhibition of mGPD activity in sonic hedgehog driven medulloblastoma cells, leading to alterations in cytoplasmic redox potential and increased NADH levels [18] (Figure 1). This increase in NADH levels promoted the interaction between the NADH-dependent co-repressor C-terminal binding protein 2 (CtBP2) and Gli1. Knockdown of CtBP2 abolished the effects of phenformin on inhibiting Hedgehog transcriptional output and Hedgehog-dependent tumor growth in nude mice [18]. Moreover, low expression levels of GPD1 correlated with poor responses to metformin in 15 cell lines of various cancer types [19]. Treatment of cells with metformin overexpressing GPD1 resulted in the accumulation of G3P, and G3P itself inhibited oxygen consumption rate and mitochondrial ATP generation in these cancer cells and enhanced the inhibitory effects of metformin on cell proliferation and xenografted tumor growth in nude mice [19]. However, evidence supporting direct effects of biguanide on the G3P shuttle are linked to their anti-tumor activities are still missing, and more studies are needed to determine if this plays a definitive role.
Phenformin has been found to possess more potent anti-tumor activities than metformin in preclinical studies [20–23]. Compared to metformin, phenformin is more lipophilic and therefore more passively cell-permeable, making it less dependent on active transport, while metformin requires a family of organic cation transporters (OCTs) to enter cells [24]. Many cancer and immune cells express inadequate levels of OCTs for efficient metformin uptake, likely limiting its anti-tumor activity to specific cellular lineages. For example, melanoma cell lines express much lower levels of OCT2 than the MDA-MB-468 breast cancer cell line [21] and over-expression of OCT2 in melanoma cells increased their response to metformin treatment while knockdown of OCT2 in MDA-MB-468 decreased their sensitivity [21]. OCT1 and OCT3 have also been reported to function as metformin transporters in cancer cells [25,26]. Taken together, while biguanides may act on multiple intracellular targets to confer their anti-tumor activities, their targeting differences and efficacy may be dictated by pharmacodynamic differences in cell permeability based on OCT expression.
Response to biguanides in the context of tumor metabolic heterogeneity
Consistent with Complex I being a major cellular target of biguanides, perturbations of mitochondrial oxidative phosphorylation (OXPHOS) correlate with the ability of these drugs to affect cellular responses. Increases in cellular susceptibility to biguanides is dependent on either high dependence on OXPHOS or mitochondrial dysfunction. For example, in triple-negative breast cancer (TNBC) the heme-binding transcriptional repressor, BTB and CNC homology 1 (BACH1) suppresses the transcription of electron transport chain (ETC) genes needed for OXPHOS function. Both degradation of BACH1 by hemin or BACH1 knockdown resulted in increased oxygen consumption rates and sensitized cancers to metformin treatment in cell culture and xenograft mouse models [27]. Deficiency of Liver kinase B1 (LKB1), a main upstream activator of AMPK, in non-small cell lung cancer cells (NSCLC) also correlated with increased sensitivity to phenformin, probably due to the accumulation of defective mitochondria resulting from aberrant AMPK-ULK1 mitophagy signaling [20]. In addition, mutations in mitochondrial genomic DNA may also serve as potential biomarkers for biguanides sensitivity in tumors. Cancer cells with mitochondrial DNA mutations in core Complex I subunits are unable to upregulate OXPHOS under low glucose conditions and are more sensitive to phenformin treatment when grown in xenografted tumors [8]. These studies demonstrate that OXPHOS alterations in cancer cells likely constitute a major determinant of biguanide sensitivity.
Given the genetic underpinning of many of these alterations, an important consideration is that mitochondrial OXPHOS status is likely to be heterogeneous across individual tumors, which may influence tumor sensitivity to biguanides. It is well established that such phenotypic heterogeneity contributes to drug resistance [28]. Specific sub-populations of cancer cells including cancer stem cells, slow-cycling cells or drug-resistant subclones have increased reliance on mitochondrial OXPHOS for survival and are more sensitive to mitochondria inhibitors. In a KRAS-driven pancreatic ductal adenocarcinoma cancer mouse model, a subpopulation of dormant tumor cells that survived KRAS oncogene ablation exhibited increased mitochondrial mass and higher oxygen consumption rate, which made them more sensitive to the mitochondrial complex V inhibitor, oligomycin. This subpopulation was responsible for tumor relapse, and treatment with oligomycin prolonged the overall survival of mice bearing regressed tumors upon KRAS “reactivation” [29]. In addition, leukemic stem cells that survived tyrosine kinase inhibitor (TKI) imatinib treatment exhibit increased oxygen consumption rates and contribute higher glucose flux to the TCA cycle [30].
In melanoma, a slow-cycling subpopulation marked by high expression of the H3K4 histone lysine demethylase KDM5B (aka JARID1B) [31] can dynamically interconvert with the rapidly proliferating tumor bulk of KDM5Blow cells. KDM5Bhigh cells possess higher self-renewal capacity in vitro and are critical for long-term maintenance of melanoma tumors in mouse models [31]. KDM5Bhigh cells are resistant to a broad panel of anticancer drugs, including cisplatin, bortezomib, and temozolomide [32]. In addition, this slow-cycling subpopulation of cells in BRAFV600E mutant melanoma is resistant to BRAF inhibitors (BRAFi), such as vemurafenib in contrast to their faster cycling counterparts [21, 32]. However, phenformin selectively targeted this KDM5Bhigh subpopulation of BRAF mutant melanoma cells [21] and the combination of phenformin and BRAFi synergistically inhibited cell viability in cultured melanoma cells, and induced tumor regression in xenograft models and a BRAFV600E-driven mouse model of melanoma [21]. Similar selective effects of phenformin on KDM5Bhigh subpopulations of NRAS-mutant and NF1-mutant melanoma cells have also been observed [33], supporting a vulnerability of such subpopulations of cancer cells to phenformin.
Biguanides also enhance the efficacy of chemotherapy or other therapeutic interventions in additional cancer mouse models by targeting cancer stem cells or quiescent cells. Metformin selectively eliminated breast cancer stem cells in several breast cancer cell lines [34, 35]. Combination of metformin and the widely used chemotherapeutic anthracycline, doxorubicin, significantly reduced tumor relapse in a breast cancer xenograft mouse model [34]. In a follow-up study, the ability of metformin to eliminate cancer stem cells in combination with doxorubicin was associated with the inhibition of an inflammatory feedback loop including NF-κB and STAT3 activation in the stem cell population [36]. Consistently, the combination of metformin with doxorubicin was effective in targeting xenografts derived from several inflammatory prostate and melanoma cell lines and failed in targeting those derived from non-inflammatory cell lines [36]. In addition, metformin assisted in eradicating patient-derived orthotopic pancreatic tumors in mice by eliminating the noncycling subpopulation, while the glutaminase inhibitor, BPTES killed the rapidly proliferating subpopulation [37]. These findings suggest that targeting OXPHOS with biguanides may be effective in limiting the emergence of drug-resistant clones and contributes to more durable clinically responses to targeted therapies or chemotherapies in cancer (Figure 2). Understanding the intra-tumor metabolic heterogeneity will provide important insights into the determinants of biguanide sensitivity and facilitate the translation of these basic findings into the clinic.
Figure 2. Biguanides enhance the efficacy of chemotherapy or targeted therapy to prevent tumor relapse by targeting tumor heterogeneity.
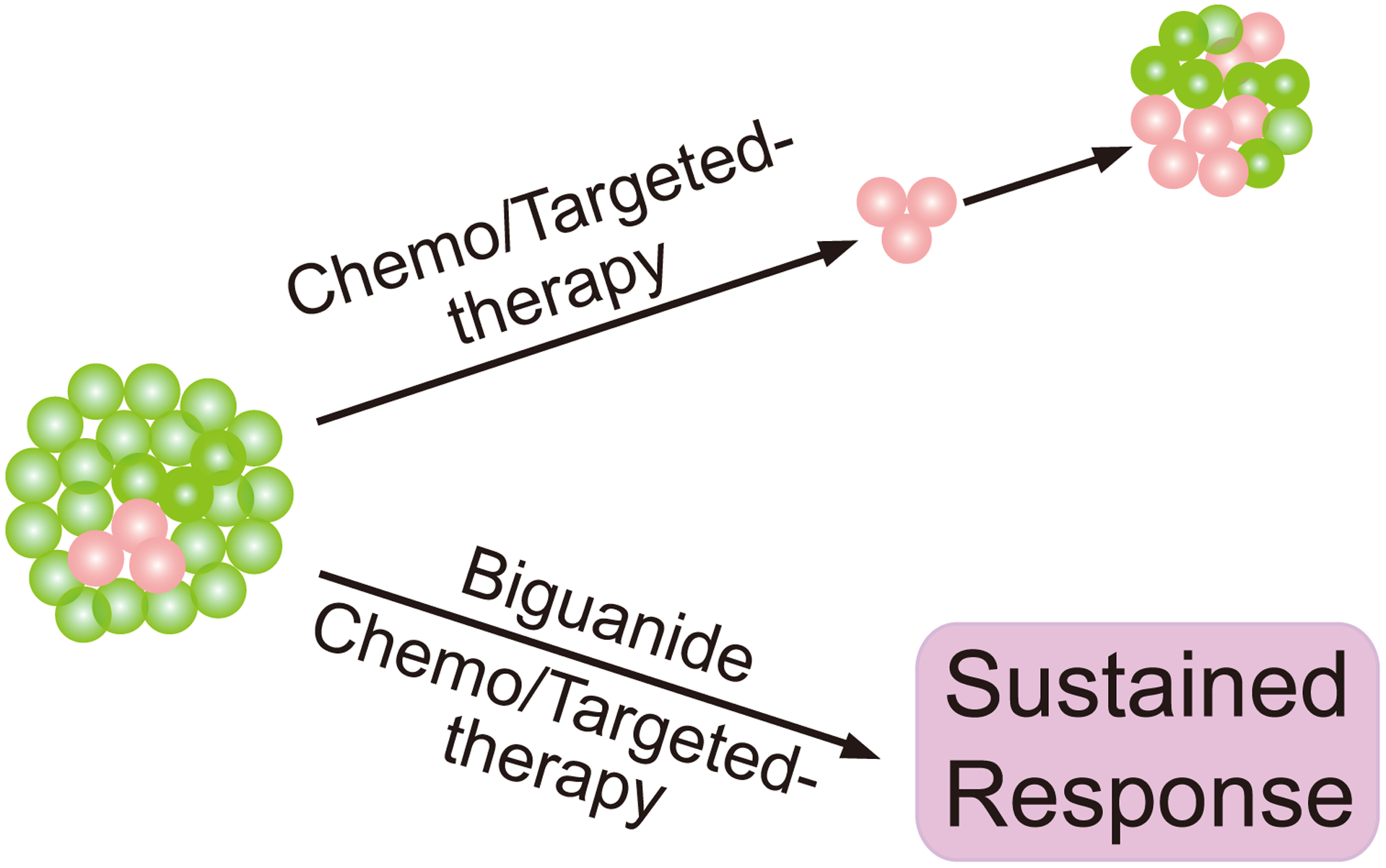
Tumor heterogeneity contributes to drug resistance and the use of biguanides prevents tumor relapse by targeting certain OXPHOS-addicting sub-populations of cancer cells within the tumor that are resistant to conventional therapies such as cancer stem cells or slow-cycling cell types (indicated in red).
Targeting compensatory metabolic pathways to enhance the efficacy of biguanides
When cancer cells are treated with biguanides, they usually undergo extensive metabolic reprogramming to cope with the drug-induced metabolic stress (Figure 3). Targeting these metabolic adaptations has shown promise for enhancing the efficacy of biguanides in recent preclinical studies.
Figure 3. Cancer cells undergo extensive metabolic reprogramming to compensate for metabolic stress induced by biguanides treatment.
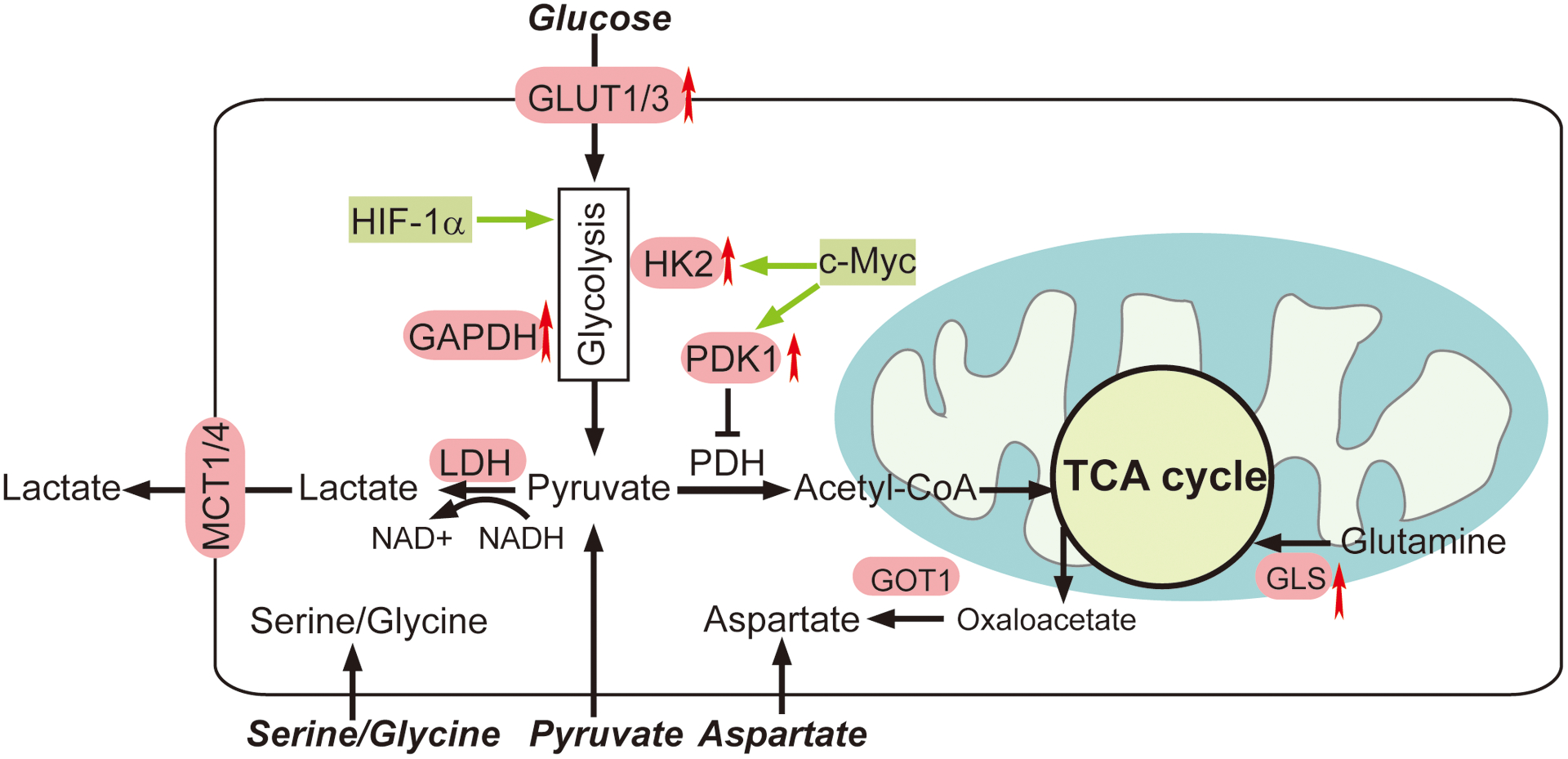
Metabolic adaptations include glycolysis, aspartate biosynthesis, glutamine metabolism and serine synthesis. Recent preclinical studies have suggested that targeting these metabolic adaptations could enhance the efficacy of biguanides in cancer.
Glycolysis
Alterations in glycolysis can influence the impact the effects of biguanides on cancer cells. Glucose deprivation sensitizes cancer cells to treatment by biguanides [7, 8], strongly supporting a role for glucose metabolism in the compensatory response to the action of biguanides on mitochondria metabolism. In contrast to high-glucose conditions, in glucose-limiting condition, metformin-treated HeyA8 cells recapitulate most of the metabolic changes in metformin-responsive mouse and human tumor tissues including decreases in TCA cycle metabolites [38]. Dynamic fluoro-deoxy-D-glucose positron emission tomography-computed tomography (FDG-PET-CT) scans of patients with treatment-naive primary breast cancer showed that exposure of metformin increased FDG signals in primary breast tumors, indicating an increased utilization of glucose within the tumor parenchyma [39]. This increase in glycolytic flux is most likely triggered by biguanide-mediated inhibition of OXPHOS and can occur via various mechanisms, such as upregulation of glucose transporter 1(GLUT1) or glucose transporter 3 (GLUT3) (Figure 3) [40–43]. For example, phenformin treatment induced NBR2 lncRNA expression and subsequent GLUT1 upregulation in several cancer cell lines, serving as an adaptive response by transporting more glucose into cells. Knockdown of GLUT1 sensitized these cancer cells to phenformin-induced apoptosis [40]. In addition, inhibition of GLUT1 expression by an mTOR inhibitor MLN0218 enhanced the efficacy of phenformin in KRAS/LKB1–mutant human NSCLC cell lines and in NSCLC genetically engineered models (GEM) [41]. Conversely, overexpression of GLUT3 rendered several biguanide-sensitive cancer cell lines resistant to phenformin under low glucose condition [8].
Alterations in the expression of genes encoding glycolytic enzymes may also influence the response to biguanide treatment. A subset of diffuse large B cell lymphomas (DLBCL) patients with low expression levels of glyceraldehyde 3-phosphate dehydrogenase (GAPDH) were sensitive to a combination therapy consisting of L-asparaginase, mTOR inhibitor, and metformin, whereas overexpression of GAPDH in the GAPDH-low DLBCL cell line or animal models reduced their sensitivities to biguanides by increasing glycolysis (Figure 3) [44]. Key transcriptional regulators of glycolytic enzymes, such as HIF-1α and c-Myc (Figure 3), may also participate in the adaptive response to biguanides. Hyperglycemia counters the sensitivity of biguanides in ovarian cancer models, probably through increased c-Myc expression and subsequent induction of glycolytic enzymes hexokinase 2 (HK2) and pyruvate dehydrogenase kinase 1 (PDK1) (Figure 3) [43]. These recent findings strongly support that up-regulation of glycolysis can act as an adaptive response to render cells resistant to biguanide treatment.
Effects of pharmacologic inhibitors that disrupt glucose transporters or glycolysis flux in combination with biguanides have been investigated extensively in various tumor types. Inhibition of HIF-1α by PX-478 induced synthetic lethality with biguanides in human acute lymphoblastic leukemia (ALL) and chronic lymphocytic leukemia (CLL) cells and lymphoma xenografts [42]. Dichloroacetate, an inhibitor of the glycolysis enzyme PDK, decreased lactic acidosis induced by biguanides, enhanced their effects on cell death induction in glioma stem cells and prolonged the survival of mice bearing xenograft tumors [45]. Oxamate, an inhibitor of lactate dehydrogenase (LDH) which converts pyruvate to lactate, overcame phenformin resistance in CT26 tumor bearing mice [46]. Moreover, inhibition of the monocarboxylate transporter (MCT) family (Figure 3), which induces the accumulation of lactate in cells, enhanced the anti-tumor activity of biguanides. Zinc finger-based ablation of MCT4, together with the MCT1 inhibitor AZD3965 and phenformin, suppressed the growth of LS174T colorectal adenocarcinoma xenograft tumors in nude mice [47]. Similarly, treatment with AZD3965 in combination with phenformin strongly inhibited proliferation of Raji Burkitt lymphoma cells with low MCT4 expression [48]. Dual inhibition of MCT1/MCT4 by syrosingopine, an inhibitor of the vesicular monoamine transporters used as an antihypertensive drug, exerted synthetic lethal effects with metformin in various cancer cell lines, likely through NAD+ depletion [49]. These findings strongly suggest that the combination of biguanides together with glycolytic inhibitors could be more potent than biguanides alone.
Aspartate metabolism
Aspartate is a critical amino acid precursor for nucleotide synthesis and is therefore important for cell proliferation [50, 51]. The biosynthesis of aspartate requires the electron acceptor NAD+ provided from the ETC. When complex I is inhibited by biguanide treatment, NAD+ regeneration from NADH is restricted, thus impairing the aspartate synthesis [50, 51]. Supplementation of exogenous electron acceptors including pyruvate or a-ketobutyrate rescues proliferation in such respiration-deficient cells [50–52]. Pyruvate withdrawal from culture medium conferred sensitivity to biguanide across different cancer cell lines [50, 52], while aspartate supplementation enabled the proliferation of Cytochrome B mutant 143B Cybrid Cells [51]. Moreover, the loss of glutamic-oxaloacetic transaminase 1 (GOT1), a cytosolic aspartate aminotransferase that catalyzes the interconversion between aspartate and oxaloacetate (Figure 3) rendered Jurkat cells more sensitive to phenformin. When the ETC is inhibited, GOT1 becomes essential for cell survival by converting oxalocacetate to aspartate in the cytosol, which compensates for the decrease of aspartate production in mitochondrial caused by phenformin [50].
Aspartate was also identified as a limiting nutrient for tumor growth, as it can neither be produced from asparagine due to the lack of asparaginase activity in cancer cells, nor be taken up by cells due to its poor cell permeability [53]. Heterologous expression of guinea pig asparaginase 1 (gpASNase1) supplied aspartate to tumor cells in nude mice, and gpASNase1 expression in 143B cancer cells conferred resistance to metformin when grown as xenografted tumors in nude mice. These findings indicate that reducing the availability of aspartate could increase the efficacy of biguanides.
Glutamine metabolism
Reductive carboxylation of glutamine to form citrate is a major pathway for cancer cells with dysfunctional mitochondria to produce TCA cycle intermediates and lipids (Figure 3). When treated with metformin, several prostate cancer cell lines and the transgenic adenocarcinoma mouse prostate (TRAMP) model have increased dependency on reductive glutamine metabolism that is accompanied by a metformin-induced decrease in glucose oxidation [54]. Inhibition of glutaminase by compound 968 or BPTES in combination with metformin further attenuated cell proliferation, whereas the increase of glutamine flux by upregulating mTOR, reduced sensitivity to metformin [54]. Similarly, a glutaminase inhibitor, CB839 potentiated the anti-tumor activities of metformin in osteosarcoma cell lines and an orthotopic xenograft mouse model [55]. 13C-glutamine tracing analysis illustrated that CB839 further decreased the levels of TCA metabolites triggered by metformin, suggesting an increased dependence on the anaplerotic utilization of glutamine in cancer cells under metformin treatment [55].
Serine metabolism
Serine is a precursor for the biosynthesis of nucleic acids, proteins, and lipids. Serine flux may increase to compensate for biguanide-induced decreases in OXPHOS (Figure 3) [56]. Conversely, serine withdrawal from media enhanced the inhibition of metformin on cell viability across multiple cancer cell lines, including NSCLC and colorectal cancer [56]. In addition, dietary restriction of serine and glycine enhanced the anti-tumor efficacy of phenformin in C57BL/6 mice bearing MC38 allografts [56]. Mechanistically, serine deprivation antagonized the effects of metformin by upregulating the levels of glycolytic metabolites and downregulating the levels of TCA metabolites [56]. Therefore, targeting serine synthesis may represent an alternative pathway to boost the anti-tumor actions of biguanides.
Effects of Biguanides on the tumor immune microenvironment
In addition to targeting cancer cells, biguanides target immune cells in the tumor microenvironment, such as CD8+ T cells, regulatory T cells (Tregs), myeloid-derived suppressor cells (MDSCs), and tumor associated macrophages (TAMs), which may contribute to anti-tumor activities of biguanides (Figure 4). Understanding the effects of biguanides on the TIME will provide new insights into how metabolic interventions can be used to augment anti-tumor immunity and improve the efficacy of cancer immunotherapies.
Figure 4. Modulation of anti-tumor immunity by biguanides.
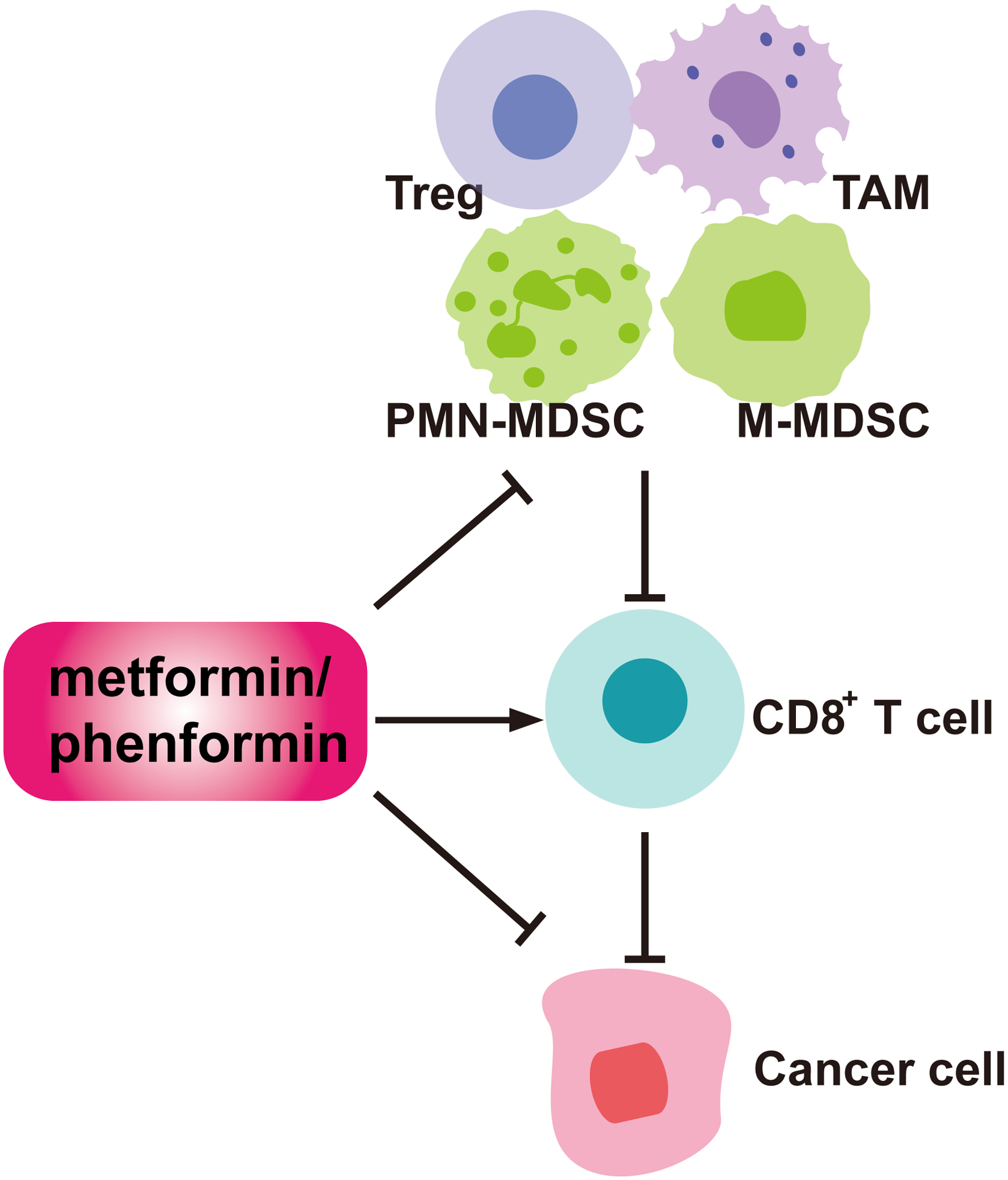
In the tumor immune microenvironment, biguanides can derepress the functionality of cytotoxic CD8+ T cells. In addition, they can inhibit the recruitment or activities of immunosuppressive populations including PMN-MDSCs, M-MDSCs, TAMs, and Tregs.
Biguanides can alter the function of CD8+ T cells in TIME, both directly and indirectly. Metformin directly promoted CD8+ memory T cell generation and enhances protective immunity in C57BL/6 mouse inoculated with EL4-Ova tumors [57]. Metformin also enhances tumor infiltration of CD8+ T cells, protects them from apoptosis and promotes IL-2, TNFα, and IFNγ cytokine production in mouse models [58]. Further, antibody depletion of CD8+ cells abolished the ability of metformin to inhibit tumor growth in this model, supporting a role for CD8+-dependent anti-tumor immunity [58].
Metformin may also increase the ability of CD8+ T cells to kill cancer cells through indirect mechanisms. Metformin reduced intratumoral hypoxia in B16 melanoma tumors in part by inhibiting cancer cell oxygen consumption, thus alleviating intra-tumoral CD8+ T cell suppression [59]. As a consequence, metformin augmented the effects of anti-PD-1 therapy, which relieves the suppression of CD8+ T cell by blocking the PD-1/PD-L1 immune checkpoint pathway, in these tumor models [59]. More recently, metformin-induced AMPK activation in cancer cells was suggested to enhance cytotoxic T cell immune response by altering the PD-1 ligand, PD-L1 glycosylation and trafficking [60]. Moreover, the combination of metformin and anti-CTLA4 therapy, another immune inhibitory checkpoint receptor that interferes with T cell receptor co-stimulation, suppressed tumor growth in several syngeneic mouse models [60]. It was proposed that AMPKα regulated PD-L1 glycosylation by directly phosphorylating S195 of PD-L1, which is in the extracellular/luminal side of PD-L1 [60]. This mechanism appears controversial as it remains unclear how AMPKα localizes to the luminal side of ER to mediate this phosphorylation. These studies suggest that metformin promotes cytotoxic CD8+ T cell function in certain mouse models. However, this may not be true in other settings. For example, phenformin decreased the production of IFNɣ by CD8+ effector T cells in vitro [61]. In addition, phenformin did not appear to affect the infiltration of CD8+ cytotoxic T cells into spontaneous tumors in the BRAF/PTEN GEM model [62]. Further studies are required to clarify the effects of biguanides on CD8+ T cells and elucidate the underlying mechanisms affecting T cell phenotypes.
In addition to CD8+ T cells, biguanides also act on Tregs, which suppress cytotoxic T cell effector functions needed for tumor elimination. Metformin administration decreased the infiltration of Tregs into tumors grown from several cancer cell lines [63] and in a C57BL/6 mouse model with inducible esophageal squamous-cell carcinoma [64]. Metformin impaired the differentiation of CD4+ naive T cells into inducible Tregs in vitro by decreasing mTORC1-dependent expression of Foxp3 [63].
Biguanides also target MDSCs, which constitute another major tumor-protective immune population within the TIME. MDSCs are immature myeloid cells that expand under both normal inflammation and in cancer [65]. MDSCs exert potent immunosuppressive activities towards T and natural killer (NK) cells through multiple mechanisms, including expression of arginase-1, inducible nitric oxide synthase and release of ROS. MDSCs can be broadly divided into two main populations, polymorphonuclear-, PMN- (aka granulocytic (G-)) and monocytic (M-) MDSCs. Phenformin selectively reduces PMN-MDSCs in the spleens of immune-competent mice bearing allograft melanoma tumors and induced apoptosis in PMN-MDSCs derived from bone marrow co-cultured with tumor cells in vitro [62]. MDSCs isolated from tumor-bearing mice treated with phenformin exhibited reduced suppressive activities towards CD8+ T cells [62]. Consequently, phenformin enhanced the efficacy of PD-1 immune checkpoint blockade in a BRAF/PTEN melanoma GEM model. Like phenformin, metformin treatment blocked the suppressive function of human MDSCs from ovarian cancer patients by downregulating the expression of the ectonucleotidases, CD39 and CD73 on both PMN- and M- MDSC subsets, in an AMPK- and HIF1α-dependent manner [66]. Similar inhibitory effects of metformin and phenformin on MDSCs have since been confirmed in several other mouse models [67–69].
TAMs contribute to an immunosuppressive tumor microenvironment that is favorable for cancer development. Low-dose metformin increased the infiltration of tumor-suppressive (CD11c+) macrophage and decreased the infiltration of tumor-promoting (CD163+) macrophages in human esophageal squamous cell carcinoma [64]. Metformin reduced IL-13-driven M2-like polarization of macrophage in vitro, which was partially abolished by AMPKα1 RNAi-knockdown [70]. Metformin also inhibited metastasis to the lungs from subcutaneously injected Lewis lung cancer cell tumors in C57BL/6 mice by blocking the accumulation of M2-like TAMs in the tumor parenchyma [70]. Similar inhibition of macrophage polarization towards M2 phenotype by metformin in the TIME has been observed in several other mouse models [71–74]. However, evidence supporting a direct effect of metformin on macrophages was lacking in these studies.
Taken together, biguanides produce immunomodulatory effects in both CD8+ T cells and other major groups of immunosuppressive cells in the TIME. Key questions regarding the contribution of each major immune cell type to the anti-tumor activities of biguanides and the collective effects of biguanides on anti-tumor immunity remain to be answered.
Clinical effort on repurposing biguanides for cancer treatment
Based on the evidence from preclinical studies in exploring the anti-tumor actions of biguanides, multiple interventional clinical trials have been conducted to investigate the effects of metformin alone or in combination with other drugs in non-diabetic patients with various cancers. Although metformin reduced the level of the Ki67 proliferation marker in tumor tissue in breast, prostate and endometrial cancers [75–79], no significant clinical benefit has been demonstrated for metformin monotherapy in overall survival and progression free survival among cancer patients [80–83].
Several clinical trials exploring the combination of metformin with chemo- or molecularly targeted therapies have been recently reported or are currently ongoing (Table 1). In a phase 1 clinical trial on 21 patients with advanced cancers, metformin in combination with the mTOR inhibitor Temsirolimus was well tolerated and showed modest efficacy [84]. In a phase 2 trial, metformin did not improve the overall survival of patients with advanced pancreatic cancer receiving gemcitabine and erlotinib with metformin at the 6 months primary endpoint [85]. However, metformin produced a significant benefit in progression-free survival in patients with advanced non-squamous NSCLC receiving chemotherapeutic drugs (carboplatin, paclitaxel) and the vascular endothelial growth factor (VEGF) inhibitor, bevacizumab [86]. In another phase 2 trial, metformin combined with EGFR kinase inhibitors improved progression free survival, objective response and overall survival rates in patients with epidermal growth factor receptor (EGFR)–mutated lung adenocarcinoma than EGFR inhibitors alone [87]. These two studies support potential benefits of metformin in combination with molecular targeted therapies in patients with lung cancer. Since LKB1 is frequently mutated in NSCLCs, and LKB1 deficiency correlated with increased sensitivity to biguanides in preclinical studies [20], it would be interesting to investigate if this sensitivity can be detected in clinical studies. Future mechanistic and biomarker studies may help to guide patient selection and improve therapeutic strategies.
Table 1.
Clinical trials on biguanides in combination with molecularly targeted therapy in cancer.
| Trial name (ID) | Study design | Treatment setting | Standard arm | Experimental arm | Patients | Primary endpoints | Status |
|---|---|---|---|---|---|---|---|
| (NCT01650506)I | Phase 1, open label | BC | Erlotinib +M | 8 | MTD | Completed | |
| CGMT(NCT01864681)II | Phase 2, randomized, double masking | NSCLC | Gefitinib + placebo | Gefitinib +M | 224 | PFS | Completed |
| GEM(NCT01210911)III | Phase 2, randomized, quadruple masking | Pancreatic cancer | Gefitinib + Erlotinib +Placebo | Gefitinib + Erlotinib +M | 120 | Survival | Completed |
| (NCT01087983)IV | Phase 1, non-randomized, open label | AC | Lapatinib+ Sirolimus | 111 | MTD | Completed | |
| Lapatinib +M | |||||||
| (NCT01477060)V | Phase 2, randomized, open label | MBC | Arm A: Lapatinib | 32 | PFS | Terminated | |
| Arm B: M | |||||||
| Arm C: Lapatinib +M | |||||||
| (NCT04428086)VI | Phase 1, randomized, open label | ST, Adults | Arm 1: Apatinib + Rosuvastatin | 36 | Cmax, AUC | Not yet recruiting | |
| Arm 2: Apatinib +M | |||||||
| (NCT03071705)VII | Phase 2, randomized, double masking | NSCLC | TKI | TKI+M | 120 | OS | Completed |
| (NCT02495103)VIII | Phase 1 & 2, non-randomized, open label | HLRCC, SDH-RCC, RCC | phase 1: Vandetanib+M | 7 | MTD | Completed | |
| phase 2: Vandetanib+M | ORR | ||||||
| (NCT02672488)IX | Phase 2, randomized, open label | HCC | Sorafenib | Sorafenib +M | 82 | OS | Unknown |
| (NCT01578551)X | Phase 2, randomized, open label | adenocarcin oma | Paclitaxel + Carboplatin + Bevacizumab | Paclitaxel+ Carboplatin+ Bevacizumab+ M | 25 | PFS | Terminated |
| (NCT02143050)XI | Phase 1, open label | Melanoma | Dabrafenib + Trametinib +M | 53 | Toxicity | Unknown | |
| Phase 2, open label | CRR | ||||||
| (NCT01638676)XII | Phase 1& 2, open label | Melanoma | Vemurafenib+M | 55 | AE | Unknown | |
| METALLICA(NCT04300790)XIII | Phase 2, randomized, multicenter, two cohort, open label | BC | CohortA: Normal fasting glycemia (M+ Fulvestrant + Alpelisib) CohortB: Fasting glycemia (M + Fulvestrant + Alpelisib) |
68 | % of patients with G3–4 HG | Not yet recruiting | |
| (NCT03006172)XIV | Phase 1, non-randomized, open label | BC, ST | Stage I, Arm A: GDC-0077 | 256 | Toxicity | Recruiting | |
| Stage I, Arm B: GDC-0077 + Palbociclib + Letrozole | |||||||
| Stage I, Arm C: GDC-0077 + Letrozole | |||||||
| Stage II, Arm B: GDC-0077 + Palbociclib + Letrozole | Phase 2 Dose of GDC-0077, AE | ||||||
| Stage II, Arm C: GDC-0077 + Letrozole | |||||||
| Stage II, Arm D: GDC-0077 + Fulvestrant | |||||||
| Stage II, Arm E: GDC-0077 + Palbociclib + Fulvestrant | |||||||
| Stage II, Arm F: GDC-0077 + Palbociclib + Fulvestrant + M | |||||||
| Stage II, Arm G: GDC-0077 + Trastuzumab + Pertuzumab | |||||||
| (NCT03829020)XV | Phase 1, open label | Recurrent/Refractory PCM | M+ Nelfinavir + Bortezomib | 36 | MTD | Recruiting | |
| INSIDE(NCT03151772)XVI | Early phase 1, non-randomized, open label | GBM | M | 3 | Bioavailabilty | Recruiting | |
| Disulfiram | Bioavailabilty | ||||||
| IMPACT(NCT03378297)XVII | Early Phase 1, randomized, open label | OC | Arm: no intervention | 143 | Changes in the expression of biomarkers | recruiting | |
| Arm: M | |||||||
| Arm: Acetylsalicylic acid | |||||||
| Arm: Olaparib | |||||||
| Arm: Letrozol | |||||||
| (NCT02948283)XVIII | Phase 1, open label | Relapsed/Refractory MM or CLL | M+ Ritonavir | 3 | safety, tolerability, feasibility | Active, not recruiting | |
| (NCT01430351)XIX | Phase 1, non-randomized, open label | GBM | Arm 1 (TMZ) | 144 | Incidence of toxicities | Active, not recruiting | |
| Arm 2 (TMZ, memantine hydrochloride) | |||||||
| Arm 3 (TMZ, mefloquine) | |||||||
| Arm 4 (TMZ, M) | |||||||
| Arm 5 (TMZ, memantine hydrochloride, mefloquine) | |||||||
| Arm 6 (TMZ, memantine hydrochloride, M) | |||||||
| Arm 7 (TMZ, mefloquine, M) | |||||||
| Arm 8 (TMZ, memantine hydrochloride, M, mefloquine) | |||||||
| MACIST(NCT02496741)XX | Phase 1b, open label | Glioma; CCA; CHS | M+ chloroquine | 15 | MTD | Completed | |
| ENDOLA(NCT02755844)XXI | Phase 1 &2, open label | Recurrent EC | Olaparib, M+ metronomic cyclophosphamide | 35 | RP2D of combined therapy | Active, not recruiting | |
| HERMET(NCT03238495)XXII | Phase 2, randomized, open label | HER2+ BC | Chemotherapy | Chemotherapy+ M | 100 | pCR | Recruiting |
| met-HEReMYTA (NCT02488564)XXIII | Phase 2, open label | HER2+ BC | Liposomal doxorubicin +Docetaxel+Trastuzumab+M | 49 | pCR | Completed | |
| DLBCL(NCT02531308)XXIV | Phase 2, open label | DLBCL | R-CHOP + M | 5 | PFS | Terminated | |
| (NCT01797523)XXV | Phase 2, open label | EC | Letrozole + M + RAD001 | 62 | CBR | Active, not recruiting | |
| (NCT01627067)XXVI | Phase 2, open label | BC | Everolimus + Exemestane + M | 23 | PFS | Terminated | |
| (NCT02917629)XXVII | Phase 2, randomized, double masking | Stage I-IV Oral Cavity or OPC Undergoing Definitive Treatment | Placebo | ACTOplus met XR | 13 | Ki-67 expression in tumor tissue | Terminated |
| (NCT01529593)XXVIII | Phase 1, open label | Advanced Cancers | Temsirolimus + M | 87 | MTD | Active, not recruiting | |
| (NCT03017833)XXIX | Phase 1, open label | Advanced/Metastatic Relapsed/Refractory Cancers | M + Sapanisertib | 50 | AE, Clinical and laboratory values, Vital sign measurements, GI symptoms, incidence of neurotoxicity | Recruiting | |
| (NCT02048384)XXX | Phase 1, randomized, open label | Metastatic PAC | M | M+ Rapamycin | 22 | Safety, feasibility | Completed |
| (NCT02874430)XXXI | Phase 2, open label | Localized BC or UC | M+Doxycycline | 46 | % of stromal cells expressing CAV1 | Recruiting | |
| (NCT03026517)XXXII | Phase 1, open label | Melanoma | Dabrafenib +Trametinib + Phenformin | 40 | DFS | Active, not recruiting | |
As of Sep 1, 2020
AC: advanced cancer, AE: adverse event, AUC: Area Under the Plasma Concentration-Time Curve, CAV1: Caveolin-1, CBR: Clinical Benefit Rate, CCA: Cholangiocarcinoma, CHS: Chondrosarcoma, CLL: Chronic Lymphocytic Leukemia, Cmax: maximum observed concentration, CRR: clinical response rate, DFS: Disease-free survival, DLBCL: Diffuse Large B-Cell Lymphoma, EC: Endometrial Cancer, GBM: glioblastoma, HCC: Hepatocellular Carcinoma, HG: hyperglycemia, HNSCC: Head and Neck Squamous Cell Carcinoma, M: metformin, MBC: metastatic breast cancer, MM: Multiple Myeloma, MTD: Maximum Tolerated Dose, NSCLC: non-small cell lung cancer, OC: ovarian cancer, ORR: overall response rate, OS: overall survival, OPC: Oropharynx Cancer, PAC: Pancreatic Adenocarcinoma, PCM: Plasma Cell Myeloma, pCR: Pathologic complete response, PFS: progression free survival, RP2D: Recommended phase 2 trial dose, ST: solid tumor, TKI: erlotinib, afatinib, gefitinib, TMZ: temozolomide, TNBC, triple negative breast cancer, UC: Uterine Cancer
With the promise of immune checkpoint inhibitors (ICI) in cancer immunotherapy and the support of preclinical studies [59, 60, 62], several clinical investigations on the combination of biguanides with anti-CTLA4 (Ipilimumab) or anti-PD-1 (Sintilimab, Nivolumab, Pembrolizumab) have been initiated in patients with various solid tumors (Table 2). So far, only retrospective clinical studies on the combination of metformin and ICI have been reported. In a retrospective study, clinical outcomes were improved in patients with non-small-cell lung cancer who received ICIs in combination with metformin [88]. In a retrospective melanoma study, several treatment-related outcomes, including objective response rate, disease control rate, overall survival and progression free survival improved in patients receiving ICIs in combination with metformin compared to those receiving ICIs alone, although without significance due to small sample size [89].
Table 2.
Clinical trials on biguanides in combination with immunotherapy in cancer.
| Trial name (ID) | Study design | Treatment setting | Standard arm | Experimental arm | Patients | Primary endpoints | Status |
|---|---|---|---|---|---|---|---|
| (NCT03994744)XXXIII | Phase 2, open label | SCLC | Sintilimab+M | 68 | ORR, safety | recruiting | |
| SMART(NCT03874000)XXXIV | Phase 2, open label | NSCLC | Sintilimab + M | 43 | ORR | recruiting | |
| (NCT03048500)XXXV | Phase 2, open label | Stage III-IV NSCLC | M+ Nivolumab | 17 | ORR | recruiting | |
| (NCT03800602)XXXVI | Phase 2, open label | Treatment Refractory MSSCC | Nivolumab+ M | 24 | ORR | recruiting | |
| (NCT03618654)XXXVII | Phase 1, randomized, open label | HNSCC | Durvalumab | Durvalumab + M | 38 | Immune cell polarization (Th1/Th2; M1/M2) | recruiting |
| (NCT03311308)XXXVIII | Phase 1, non-randomized, open label | Advanced Melanoma | Pembrolizumab | Pembrolizumab +M | 30 | Ki-67 proliferation index in T cell | recruiting |
| (NCT04114136)XXXIX | Phase 2, randomized, open label | ST | Nivolumab or Pembrozilumab | Nivolumab or Pembrozilumab +M | 108 | Best overall response | Not yet recruiting |
| Nivolumab or Pembrozilumab+Rosiglitazone | |||||||
| (UMIN000028405)XL | Phase 1b, non-randomized, open label | Refractory/Recurrent ST | Nivolumb + M | 39 | Safety, Pharmacokinetics property of metformin | No longer recruiting | |
As of Sep 1, 2020
CC: Colorectal Cancer, HNSCC: Head and Neck Squamous Cell Carcinoma, M: metformin, NSCLC: non-small cell lung cancer, ORR: overall response rate, SCLC: small cell lung cancer, ST: solid tumor, Th, T helper cell.
The outcome of various clinical studies of metformin in cancer patients are underwhelming, in contrast to promising results from preclinical studies on the anti-tumor activities of metformin. One possibility for the discrepancy between preclinical and clinical studies might be due to timing of treatment in animal models. In animal studies that claimed anti-tumor efficacy of metformin, metformin administration was usually initiated at the time of tumor inoculation or when tumor size was small, which does not necessarily reflect clinical settings, and the same efficacy may not be replicated for large tumors. For instance, metformin enhanced the efficacy of anti-PD-1 in C57BL/6 mouse bearing with B16 or MC38 tumors when the treatment was initiated at Day 5 after tumor inoculation [59]. However, metformin administrated when the tumor size exceeded 10 mm2 failed to exert synergistic effects with anti-PD-1 [59].
A potential solution to improve the clinical benefits of metformin in cancer treatment is to use a more potent biguanide, phenformin. The incidence of lactate acidosis associated phenformin use is high compared to metformin, which led to its discontinuation as a diabetic drug in most countries. However, phenformin possesses relatively low toxicity (64 cases of lactate acidosis per 100,000 patient years) [90] compared to other cancer treatments. Phenformin, but not metformin, enhanced the efficacy of BRAF inhibitors in suppressing the proliferation of BRAF-mutant melanoma cells in vitro and inhibiting BRAF-driven tumor growth in mouse models [21]. For the BRAF/PTEN GEM model experiment in the same study, administration of phenformin, but not metformin, when tumor size reached ~100 mm3, caused significant tumor regression when combined with the BRAFi, vemurafenib [21]. Based on these findings, a phase 1 clinical trial of phenformin with the dabrafenib/trametinib combination in patients with BRAF-mutated melanoma is currently underway (NCT03026517). If safety of phenformin is confirmed in this trial, combinations of phenformin with other targeted or immuno- therapies that were supported by preclinical animal model studies, such as phenformin-anti-PD-1 combination [62], could be explored. IM156, a novel biguanide derived from phenformin [91], has also been evaluated as a monotherapy in a basket Phase 1 clinical trials on 22 patients with various types of advanced cancers (NCT03272256) [91, 92]. The results showed that it was well tolerated and decreased tumor growth rates in 3 patients.
Concluding Remarks
Mitochondrial complex I, AMPK, mTOR, and mGPD have all been suggested as molecular targets for mediating the anti-tumor activities of biguanides. Consistent with mitochondrial complex I serving as a main target, biguanides preferentially target subpopulations of slower growing cancer cells that are more reliant on OXPHOS for survival, creating therapeutic opportunities for combining biguanides with drugs that act on rapidly cycling populations to exert greater tumor clearance. Targeting compensatory metabolic pathways needed for cancer cells to survive metabolic disruptions may also improve biguanide efficacy in combination. In addition, biguanides exert effects on the TIME, with the potential to influence immune recognition and elimination of the tumor. Interestingly, biguanides can also affect host pathophysiology by modulating gut microbiota [93–95], raising the possibility that biguanides may also indirectly impact patient response to cancer therapies. Therefore, it is unknown as to whether biguanides affect direct targets within cancer cells, the tumor microenvironment, or commensal microbiota, being mindful that none of these putative effects are mutually exclusive. Finally, the pharmacodynamic properties of phenformin suggests that it may be preferred over metformin for applications within cancer therapy, as metformin has shown limited efficacy in recent clinical trials possibly due to its more limited pharmacodynamic properties. While the toxicity profile of phenformin is not ideal as a long-term maintenance therapy for type 2 diabetes patients, it is well within the limits for treating cancer. Incorporation of preclinical and clinical studies of biguanides, including novel derivatives, will help to elucidate biomarkers that predict therapeutic efficacy, define proper patient cohorts with sensitivity to biguanides, and guide clinical trials of both mono- and combination therapies in cancer (see Outstanding Questions).
Supplementary Material
Acknowledgements
Research on biguanides in Dr. Zheng’s laboratory has been supported by NIH (R00 CA133245, R01 CA166717 and R21 CA227588), Melanoma Research Alliance, and Worldwide Cancer Research. We apologize for not being able to include all relevant literature due to the space limitation.
References:
- 1.Pollak M (2010) Metformin and other biguanides in oncology: advancing the research agenda. Cancer prevention research 3, 1060–1065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Evans JM, et al. (2005) Metformin and reduced risk of cancer in diabetic patients. Bmj 330, 1304–1305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Morales DR and Morris AD (2015) Metformin in cancer treatment and prevention. Annual review of medicine 66, 17–29 [DOI] [PubMed] [Google Scholar]
- 4.Vancura A, et al. (2018) Metformin as an Anticancer Agent. Trends in pharmacological sciences 39, 867–878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kulkarni AS, et al. (2020) Benefits of Metformin in Attenuating the Hallmarks of Aging. Cell metabolism [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rena G, et al. (2017) The mechanisms of action of metformin. Diabetologia 60, 1577–1585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wheaton WW, et al. (2014) Metformin inhibits mitochondrial complex I of cancer cells to reduce tumorigenesis. eLife 3, e02242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Birsoy K, et al. (2014) Metabolic determinants of cancer cell sensitivity to glucose limitation and biguanides. Nature 508, 108–112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Van Nostrand JL, et al. (2020) AMPK regulation of Raptor and TSC2 mediate metformin effects on transcriptional control of anabolism and inflammation. Genes & development 34, 1330–1344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Janzer A, et al. (2014) Metformin and phenformin deplete tricarboxylic acid cycle and glycolytic intermediates during cell transformation and NTPs in cancer stem cells. Proceedings of the National Academy of Sciences of the United States of America 111, 10574–10579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cuyas E, et al. (2018) Metformin regulates global DNA methylation via mitochondrial one-carbon metabolism. Oncogene 37, 963–970 [DOI] [PubMed] [Google Scholar]
- 12.Cuyas E, et al. (2018) Metformin directly targets the H3K27me3 demethylase KDM6A/UTX. Aging cell 17, e12772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Galdieri L, et al. (2016) Activation of AMP-activated Protein Kinase by Metformin Induces Protein Acetylation in Prostate and Ovarian Cancer Cells. The Journal of biological chemistry 291, 25154–25166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang CS, et al. (2016) Metformin Activates AMPK through the Lysosomal Pathway. Cell metabolism 24, 521–522 [DOI] [PubMed] [Google Scholar]
- 15.Kalender A, et al. (2010) Metformin, independent of AMPK, inhibits mTORC1 in a rag GTPase-dependent manner. Cell metabolism 11, 390–401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wu L, et al. (2016) An Ancient, Unified Mechanism for Metformin Growth Inhibition in C. elegans and Cancer. Cell 167, 1705–1718 e1713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Madiraju AK, et al. (2014) Metformin suppresses gluconeogenesis by inhibiting mitochondrial glycerophosphate dehydrogenase. Nature 510, 542–546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Di Magno L, et al. (2020) Phenformin Inhibits Hedgehog-Dependent Tumor Growth through a Complex I-Independent Redox/Corepressor Module. Cell reports 30, 1735–1752 e1737 [DOI] [PubMed] [Google Scholar]
- 19.Xie J, et al. (2020) GPD1 Enhances the Anticancer Effects of Metformin by Synergistically Increasing Total Cellular Glycerol-3-Phosphate. Cancer research 80, 2150–2162 [DOI] [PubMed] [Google Scholar]
- 20.Shackelford DB, et al. (2013) LKB1 inactivation dictates therapeutic response of non-small cell lung cancer to the metabolism drug phenformin. Cancer cell 23, 143–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yuan P, et al. (2013) Phenformin enhances the therapeutic benefit of BRAF(V600E) inhibition in melanoma. Proceedings of the National Academy of Sciences of the United States of America 110, 18226–18231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huang X, et al. (2008) Important role of the LKB1-AMPK pathway in suppressing tumorigenesis in PTEN-deficient mice. The Biochemical journal 412, 211–221 [DOI] [PubMed] [Google Scholar]
- 23.Vara-Ciruelos D, et al. (2019) Phenformin, But Not Metformin, Delays Development of T Cell Acute Lymphoblastic Leukemia/Lymphoma via Cell-Autonomous AMPK Activation. Cell reports 27, 690–698 e694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Daugan M, et al. (2016) Metformin: An anti-diabetic drug to fight cancer. Pharmacological research 113, 675–685 [DOI] [PubMed] [Google Scholar]
- 25.Vasan K, et al. (2020) Mitochondrial Metabolism as a Target for Cancer Therapy. Cell metabolism 32, 341–352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Madera D, et al. (2015) Prevention of tumor growth driven by PIK3CA and HPV oncogenes by targeting mTOR signaling with metformin in oral squamous carcinomas expressing OCT3. Cancer prevention research 8, 197–207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee J, et al. (2019) Effective breast cancer combination therapy targeting BACH1 and mitochondrial metabolism. Nature 568, 254–258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Turajlic S, et al. (2019) Resolving genetic heterogeneity in cancer. Nature reviews. Genetics 20, 404–416 [DOI] [PubMed] [Google Scholar]
- 29.Viale A, et al. (2014) Oncogene ablation-resistant pancreatic cancer cells depend on mitochondrial function. Nature 514, 628–632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kuntz EM, et al. (2017) Targeting mitochondrial oxidative phosphorylation eradicates therapy-resistant chronic myeloid leukemia stem cells. Nature medicine 23, 1234–1240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Roesch A, et al. (2010) A temporarily distinct subpopulation of slow-cycling melanoma cells is required for continuous tumor growth. Cell 141, 583–594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Roesch A, et al. (2013) Overcoming intrinsic multidrug resistance in melanoma by blocking the mitochondrial respiratory chain of slow-cycling JARID1B(high) cells. Cancer cell 23, 811–825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Trousil S, et al. (2017) Phenformin Enhances the Efficacy of ERK Inhibition in NF1-Mutant Melanoma. The Journal of investigative dermatology 137, 1135–1143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hirsch HA, et al. (2009) Metformin selectively targets cancer stem cells, and acts together with chemotherapy to block tumor growth and prolong remission. Cancer research 69, 7507–7511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee KM, et al. (2017) MYC and MCL1 Cooperatively Promote Chemotherapy-Resistant Breast Cancer Stem Cells via Regulation of Mitochondrial Oxidative Phosphorylation. Cell metabolism 26, 633–647 e637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hirsch HA, et al. (2013) Metformin inhibits the inflammatory response associated with cellular transformation and cancer stem cell growth. Proceedings of the National Academy of Sciences of the United States of America 110, 972–977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Elgogary A, et al. (2016) Combination therapy with BPTES nanoparticles and metformin targets the metabolic heterogeneity of pancreatic cancer. Proceedings of the National Academy of Sciences of the United States of America 113, E5328–5336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu X, et al. (2016) Metformin Targets Central Carbon Metabolism and Reveals Mitochondrial Requirements in Human Cancers. Cell metabolism 24, 728–739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lord SR, et al. (2018) Integrated Pharmacodynamic Analysis Identifies Two Metabolic Adaption Pathways to Metformin in Breast Cancer. Cell metabolism 28, 679–688 e674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu X and Gan B (2016) lncRNA NBR2 modulates cancer cell sensitivity to phenformin through GLUT1. Cell cycle 15, 3471–3481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Momcilovic M, et al. (2015) Heightening Energetic Stress Selectively Targets LKB1-Deficient Non-Small Cell Lung Cancers. Cancer research 75, 4910–4922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Khan H, et al. (2019) Metabolic Rewiring in Response to Biguanides Is Mediated by mROS/HIF-1a in Malignant Lymphocytes. Cell reports 29, 3009–3018 e3004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Litchfield LM, et al. (2015) Hyperglycemia-induced metabolic compensation inhibits metformin sensitivity in ovarian cancer. Oncotarget 6, 23548–23560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chiche J, et al. (2019) GAPDH Expression Predicts the Response to R-CHOP, the Tumor Metabolic Status, and the Response of DLBCL Patients to Metabolic Inhibitors. Cell metabolism 29, 1243–1257 e1210 [DOI] [PubMed] [Google Scholar]
- 45.Jiang W, et al. (2016) Repurposing phenformin for the targeting of glioma stem cells and the treatment of glioblastoma. Oncotarget 7, 56456–56470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Miskimins WK, et al. (2014) Synergistic anti-cancer effect of phenformin and oxamate. PloS one 9, e85576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Marchiq I, et al. (2015) Genetic disruption of lactate/H+ symporters (MCTs) and their subunit CD147/BASIGIN sensitizes glycolytic tumor cells to phenformin. Cancer research 75, 171–180 [DOI] [PubMed] [Google Scholar]
- 48.Noble RA, et al. (2017) Inhibition of monocarboxyate transporter 1 by AZD3965 as a novel therapeutic approach for diffuse large B-cell lymphoma and Burkitt lymphoma. Haematologica 102, 1247–1257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Benjamin D, et al. (2018) Dual Inhibition of the Lactate Transporters MCT1 and MCT4 Is Synthetic Lethal with Metformin due to NAD+ Depletion in Cancer Cells. Cell reports 25, 3047–3058 e3044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Birsoy K, et al. (2015) An Essential Role of the Mitochondrial Electron Transport Chain in Cell Proliferation Is to Enable Aspartate Synthesis. Cell 162, 540–551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sullivan LB, et al. (2015) Supporting Aspartate Biosynthesis Is an Essential Function of Respiration in Proliferating Cells. Cell 162, 552–563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gui DY, et al. (2016) Environment Dictates Dependence on Mitochondrial Complex I for NAD+ and Aspartate Production and Determines Cancer Cell Sensitivity to Metformin. Cell metabolism 24, 716–727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sullivan LB, et al. (2018) Aspartate is an endogenous metabolic limitation for tumour growth. Nature cell biology 20, 782–788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fendt SM, et al. (2013) Metformin decreases glucose oxidation and increases the dependency of prostate cancer cells on reductive glutamine metabolism. Cancer research 73, 4429–4438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ren L, et al. (2020) Glutaminase-1 (GLS1) inhibition limits metastatic progression in osteosarcoma. Cancer & metabolism 8, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gravel SP, et al. (2014) Serine deprivation enhances antineoplastic activity of biguanides. Cancer research 74, 7521–7533 [DOI] [PubMed] [Google Scholar]
- 57.Pearce EL, et al. (2009) Enhancing CD8 T-cell memory by modulating fatty acid metabolism. Nature 460, 103–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Eikawa S, et al. (2015) Immune-mediated antitumor effect by type 2 diabetes drug, metformin. Proceedings of the National Academy of Sciences of the United States of America 112, 1809–1814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Scharping NE, et al. (2017) Efficacy of PD-1 Blockade Is Potentiated by Metformin-Induced Reduction of Tumor Hypoxia. Cancer immunology research 5, 9–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cha JH, et al. (2018) Metformin Promotes Antitumor Immunity via Endoplasmic-Reticulum-Associated Degradation of PD-L1. Molecular cell 71, 606–620 e607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Blagih J, et al. (2015) The energy sensor AMPK regulates T cell metabolic adaptation and effector responses in vivo. Immunity 42, 41–54 [DOI] [PubMed] [Google Scholar]
- 62.Kim SH, et al. (2017) Phenformin Inhibits Myeloid-Derived Suppressor Cells and Enhances the Anti-Tumor Activity of PD-1 Blockade in Melanoma. The Journal of investigative dermatology 137, 1740–1748 [DOI] [PubMed] [Google Scholar]
- 63.Kunisada Y, et al. (2017) Attenuation of CD4(+)CD25(+) Regulatory T Cells in the Tumor Microenvironment by Metformin, a Type 2 Diabetes Drug. EBioMedicine 25, 154–164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wang S, et al. (2020) Low-Dose Metformin Reprograms the Tumor Immune Microenvironment in Human Esophageal Cancer: Results of a Phase II Clinical Trial. Clinical cancer research : an official journal of the American Association for Cancer Research [DOI] [PubMed] [Google Scholar]
- 65.Kumar V, et al. (2016) The Nature of Myeloid-Derived Suppressor Cells in the Tumor Microenvironment. Trends in immunology 37, 208–220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Li L, et al. (2018) Metformin-Induced Reduction of CD39 and CD73 Blocks Myeloid-Derived Suppressor Cell Activity in Patients with Ovarian Cancer. Cancer research 78, 1779–1791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Baumann T, et al. (2020) Regulatory myeloid cells paralyze T cells through cell-cell transfer of the metabolite methylglyoxal. Nature immunology 21, 555–566 [DOI] [PubMed] [Google Scholar]
- 68.Uehara T, et al. (2019) Metformin induces CD11b+-cell-mediated growth inhibition of an osteosarcoma: implications for metabolic reprogramming of myeloid cells and anti-tumor effects. International immunology 31, 187–198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Xu P, et al. (2019) Metformin inhibits the function of granulocytic myeloid-derived suppressor cells in tumor-bearing mice. Biomedicine & pharmacotherapy = Biomedecine & pharmacotherapie 120, 109458. [DOI] [PubMed] [Google Scholar]
- 70.Ding L, et al. (2015) Metformin prevents cancer metastasis by inhibiting M2-like polarization of tumor associated macrophages. Oncotarget 6, 36441–36455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Giles ED, et al. (2018) Metformin inhibits stromal aromatase expression and tumor progression in a rodent model of postmenopausal breast cancer. Breast cancer research : BCR 20, 50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Liu Q, et al. (2018) Metformin Inhibits Prostate Cancer Progression by Targeting Tumor-Associated Inflammatory Infiltration. Clinical cancer research : an official journal of the American Association for Cancer Research 24, 5622–5634 [DOI] [PubMed] [Google Scholar]
- 73.Ma Q, et al. (2019) PlGF signaling and macrophage repolarization contribute to the anti-neoplastic effect of metformin. European journal of pharmacology 863, 172696. [DOI] [PubMed] [Google Scholar]
- 74.Sloot YJE, et al. (2019) Effect of PTEN inactivating germline mutations on innate immune cell function and thyroid cancer-induced macrophages in patients with PTEN hamartoma tumor syndrome. Oncogene 38, 3743–3755 [DOI] [PubMed] [Google Scholar]
- 75.DeCensi A, et al. (2014) Differential effects of metformin on breast cancer proliferation according to markers of insulin resistance and tumor subtype in a randomized presurgical trial. Breast cancer research and treatment 148, 81–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Joshua AM, et al. (2014) A pilot ‘window of opportunity’ neoadjuvant study of metformin in localised prostate cancer. Prostate cancer and prostatic diseases 17, 252–258 [DOI] [PubMed] [Google Scholar]
- 77.Laskov I, et al. (2014) Anti-diabetic doses of metformin decrease proliferation markers in tumors of patients with endometrial cancer. Gynecologic oncology 134, 607–614 [DOI] [PubMed] [Google Scholar]
- 78.Niraula S, et al. (2012) Metformin in early breast cancer: a prospective window of opportunity neoadjuvant study. Breast cancer research and treatment 135, 821–830 [DOI] [PubMed] [Google Scholar]
- 79.Hadad SM, et al. (2015) Evidence for biological effects of metformin in operable breast cancer: biomarker analysis in a pre-operative window of opportunity randomized trial. Breast cancer research and treatment 150, 149–155 [DOI] [PubMed] [Google Scholar]
- 80.Kalinsky K, et al. (2014) Presurgical trial of metformin in overweight and obese patients with newly diagnosed breast cancer. Cancer investigation 32, 150–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Mark M, et al. (2019) Impact of Addition of Metformin to Abiraterone in Metastatic Castration-Resistant Prostate Cancer Patients With Disease Progressing While Receiving Abiraterone Treatment (MetAb-Pro): Phase 2 Pilot Study. Clinical genitourinary cancer 17, e323–e328 [DOI] [PubMed] [Google Scholar]
- 82.Montaudie H, et al. (2017) Metformin monotherapy in melanoma: a pilot, open-label, prospective, and multicentric study indicates no benefit. Pigment cell & melanoma research 30, 378–380 [DOI] [PubMed] [Google Scholar]
- 83.Pimentel I, et al. (2019) A phase II randomized clinical trial of the effect of metformin versus placebo on progression-free survival in women with metastatic breast cancer receiving standard chemotherapy. Breast 48, 17–23 [DOI] [PubMed] [Google Scholar]
- 84.Khawaja MR, et al. (2016) Phase I dose escalation study of temsirolimus in combination with metformin in patients with advanced/refractory cancers. Cancer chemotherapy and pharmacology 77, 973–977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kordes S, et al. (2015) Metformin in patients with advanced pancreatic cancer: a double-blind, randomised, placebo-controlled phase 2 trial. The Lancet. Oncology 16, 839–847 [DOI] [PubMed] [Google Scholar]
- 86.Marrone KA, et al. (2018) A Randomized Phase II Study of Metformin plus Paclitaxel/Carboplatin/Bevacizumab in Patients with Chemotherapy-Naive Advanced or Metastatic Nonsquamous Non-Small Cell Lung Cancer. The oncologist 23, 859–865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Arrieta O, et al. (2019) Effect of Metformin Plus Tyrosine Kinase Inhibitors Compared With Tyrosine Kinase Inhibitors Alone in Patients With Epidermal Growth Factor Receptor-Mutated Lung Adenocarcinoma: A Phase 2 Randomized Clinical Trial. JAMA oncology, e192553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Afzal MZ, et al. (2019) Clinical outcomes in non-small-cell lung cancer patients receiving concurrent metformin and immune checkpoint inhibitors. Lung cancer management 8, LMT11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Afzal MZ, et al. (2018) Efficacy of metformin in combination with immune checkpoint inhibitors (anti-PD-1/anti-CTLA-4) in metastatic malignant melanoma. Journal for immunotherapy of cancer 6, 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Stang M, et al. (1999) Incidence of lactic acidosis in metformin users. Diabetes care 22, 925–927 [DOI] [PubMed] [Google Scholar]
- 91.Izreig S, et al. (2020) Repression of LKB1 by miR-17 approximately 92 Sensitizes MYC-Dependent Lymphoma to Biguanide Treatment. Cell reports. Medicine 1, 100014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Rha SY, et al. (2020) Phase I study of IM156, a novel potent biguanide oxidative phosphorylation (OXPHOS) inhibitor, in patients with advanced solid tumors. Journal of Clinical Oncology 38, 3590–3590 [Google Scholar]
- 93.Cabreiro F, et al. (2013) Metformin retards aging in C. elegans by altering microbial folate and methionine metabolism. Cell 153, 228–239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Pryor R, et al. (2019) Host-Microbe-Drug-Nutrient Screen Identifies Bacterial Effectors of Metformin Therapy. Cell 178, 1299–1312 e1229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Sun L, et al. (2018) Gut microbiota and intestinal FXR mediate the clinical benefits of metformin. Nature medicine 24, 1919–1929 [DOI] [PMC free article] [PubMed] [Google Scholar]
Resources
- I. https://clinicaltrials.gov/ct2/show/NCT01650506.
- II. https://clinicaltrials.gov/ct2/show/NCT01864681.
- III. https://clinicaltrials.gov/ct2/show/NCT01210911.
- IV. https://clinicaltrials.gov/ct2/show/NCT01087983.
- V. https://clinicaltrials.gov/ct2/show/NCT01477060.
- VI. https://clinicaltrials.gov/ct2/show/NCT04428086.
- VII. https://clinicaltrials.gov/ct2/show/NCT03071705.
- VIII. https://clinicaltrials.gov/ct2/show/NCT02495103.
- IX. https://clinicaltrials.gov/ct2/show/NCT02672488.
- X. https://clinicaltrials.gov/ct2/show/NCT01578551.
- XI. https://clinicaltrials.gov/ct2/show/NCT02143050.
- XII. https://clinicaltrials.gov/ct2/show/NCT01638676.
- XIII. https://clinicaltrials.gov/ct2/show/NCT04300790.
- XIV. https://clinicaltrials.gov/ct2/show/NCT03006172.
- XV. https://clinicaltrials.gov/ct2/show/NCT03829020.
- XVI. https://clinicaltrials.gov/ct2/show/NCT03151772.
- XVII. https://clinicaltrials.gov/ct2/show/NCT03378297.
- XVIII. https://clinicaltrials.gov/ct2/show/NCT02948283.
- XIX. https://clinicaltrials.gov/ct2/show/NCT01430351.
- XX. https://clinicaltrials.gov/ct2/show/NCT02496741.
- XXI. https://clinicaltrials.gov/ct2/show/NCT02755844.
- XXII. https://clinicaltrials.gov/ct2/show/NCT03238495.
- XXIII. https://clinicaltrials.gov/ct2/show/NCT02488564.
- XXIV. https://clinicaltrials.gov/ct2/show/NCT02531308.
- XXV. https://clinicaltrials.gov/ct2/show/NCT01797523.
- XXVI. https://clinicaltrials.gov/ct2/show/NCT01627067.
- XXVII. https://clinicaltrials.gov/ct2/show/NCT02917629.
- XXVIII. https://clinicaltrials.gov/ct2/show/NCT01529593.
- XXIX. https://clinicaltrials.gov/ct2/show/NCT03017833.
- XXX. https://clinicaltrials.gov/ct2/show/NCT02048384.
- XXXI. https://clinicaltrials.gov/ct2/show/NCT02874430.
- XXXII. https://clinicaltrials.gov/ct2/show/NCT03026517.
- XXXIII. https://clinicaltrials.gov/ct2/show/NCT03994744.
- XXXIV. https://clinicaltrials.gov/ct2/show/NCT03874000.
- XXXV. https://clinicaltrials.gov/ct2/show/NCT03048500.
- XXXVI. https://clinicaltrials.gov/ct2/show/NCT03800602.
- XXXVII. https://clinicaltrials.gov/ct2/show/NCT03618654.
- XXXVIII. https://clinicaltrials.gov/ct2/show/NCT03311308.
- XXXIX. https://clinicaltrials.gov/ct2/show/NCT04114136.
- XL. https://upload.umin.ac.jp/cgi-open-bin/ctr_e/ctr_view.cgi?recptno=R000031915.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.