

Abstract
Immune checkpoint inhibitors (ICIs) therapy has been widely used to treat different human cancers, particularly advanced solid tumors. However, clinical studies have reported that ICI immunotherapy benefits only ~15% of colorectal cancer (CRC) patients, specifically those with tumors characterized by microsatellite instability (MSI), a molecular marker of defective DNA mismatch repair (dMMR). For the majority of CRC patients who carry proficient MMR (pMMR), ICIs have shown little clinical benefit. In this study, we examined the efficacy of sulindac to enhance the response of pMMR CRC to anti-PD-L1 immunotherapy. We utilized CT26 syngeneic mouse tumor model to compare the inhibitory effects of PD-L1 antibody (Ab), sulindac, and their combination on pMMR CRC tumor growth. We found that mice treated with combination therapy showed a significant reduction in tumor volume, along with increased infiltration of CD8+ T lymphocytes in the tumor tissues. We also demonstrated that sulindac could downregulate PD-L1 by blocking NF-κB signaling, which in turn led to a decrease in exosomal PD-L1. Notably, PD-L1 Ab can be bound and consumed by exosomal PD-L1 in the blood circulation. Therefore, in combination therapy, sulindac downregulating PD-L1 leads to increased availability of PD-L1 Ab, which potentially improves the overall efficacy of anti-PD-L1 therapy. We also show that low-dose sulindac does not appear to have a systemic inhibitory effect on prostaglandin E2 (PGE2). In conclusion, our findings provide unique insights into the mechanism of action and efficacy for sulindac as an immunomodulatory agent in combination with anti-PD-L1 therapy for the treatment of pMMR CRC.
Keywords: Colorectal cancer, Sulindac, Immunotherapy, PD-L1, MMR
Introduction:
Currently, colorectal cancer (CRC) remains the third most common cancer in both men and women, with an estimated 53,200 deaths per year (1). Although the overall survival of CRC patients has improved dramatically over the past few decades, the 5-year survival rate for individuals with stage IV CRC remains lower than 15% (1). Given this poor overall outcome, there is a greater need to develop more effective and safer treatment options for advanced and metastatic CRC. Recently, clinical immunotherapy with immune checkpoint inhibitors (ICIs), such as PD-1 and PD-L1 inhibitors, has shown remarkable inhibitory efficacy in a variety of cancers (2,3). The PD-1 cell receptor is a T-cell co-suppressor receptor that is expressed primarily on immune cells, such as T cells and macrophages (4,5); the ligand of PD-1, PD-L1, is mainly expressed on dendritic cells (DCs) and various types of tumor cells. Under human physiological cellular conditions, it appears that the interactions of PD-1 binding with PD-L1 leads to a protective mechanism against the development of auto-immune types of diseases in the host (6). However, when activated T lymphocytes recognize tumor cells, elevated PD-L1 on the surface of tumor cells can bind to PD-1 of T lymphocytes, blocking the attack of immune cells and resulting in a failure of immune surveillance (7). Therefore, the central concept of ICI immunotherapy is to block the direct binding between PD-1 and PD-L1 and restore the optimal surveillance and attack ability of immune cells on tumor cells (8).
CRC is characterized by a malignant transformation of cells involving a progressive accumulation of various genetic alterations, including chromosomal instability (CIN) and microsatellite instability (MSI) (9). The former CIN accounts for approximately 80% of sporadic CRCs with mutations in oncogenes and tumor suppressors, such as K-RAS, APC, and TP53 (10,11). Furthermore, the mechanism of MSI is characterized by elevated microsatellites with intragenic mutations of short and tandemly repeated DNA sequences, estimated to be present in approximately 15% of sporadic CRCs. In addition, MSI is regarded as a biomarker of deficient DNA mismatch repair (dMMR), an essential mechanism required for cellular repair of damaged DNA (9,11). Intriguingly, several clinical studies have reported that only CRC patients with dMMR, or highly microsatellite instability (MSI-H), responded to ICI immunotherapy, specifically anti-PD-1 therapy (12-14). Of significance, ICI immunotherapy dramatically improved overall survival (OS) and progression-free survival (PFS) in stage 4 CRC patients with dMMR/MSI-H, which accounts for less than 5% of all CRC cases. However, it did not appear to have a significant improvement in OS and PFS for those patients with proficient MMR (pMMR) and microsatellite stability (MSS) (15-17). Based on these clinical results, the FDA approved pembrolizumab (anti-PD-1), nivolumab (anti-PD-1), and ipilimumab (anti-CTLA-4) in combination with other agents for the treatment of dMMR/MSI-H metastatic CRC patients. Unfortunately, such options are only available for a very small group of patients with metastatic CRC.
We have a long-standing research interest in studying metastatic CRC, particularly examining the chemo-preventative effects of sulindac, an FDA-approved nonsteroidal anti-inflammatory drug (NSAID) that has been shown to inhibit CRC development and progression (18). In this study, we utilized low doses of sulindac in combination with PD-L1 antibody (Ab) to treat CT26 syngeneic mice, which are characterized as pMMR CRC (19). We showed that CT26 mice treated with the combination therapy [sulindac + PD-L1 Ab] displayed an enhanced immune response with subsequent tumor regression, compared to the monotherapy, in terms of PD-L1 Ab or sulindac alone. Of interest, infiltrating CD8+ T-cell aggregates were found in tumor tissues of mice treated with sulindac alone or in combination, inversely correlated with PD-L1 expression. Regarding the mechanism of action, we demonstrated that sulindac can downregulate PD-L1 gene expression by blocking NF-κB signaling. Thus, our findings provide evidence that sulindac acts as an immunomodulator to enhance the sensitivity of pMMR CRC tumors to anti-PD-L1 therapy.
Materials and Methods
Cell culture and reagents
The murine colon cancer cell line CT26 and the human colon cancer cell lines, Colo205, HT29, and Caco-2, were purchased from ATCC (Manassas VA, USA). We authenticate our cell lines at regular intervals of 12 months in addition to two authentications at the beginning and end of the project using the services provided by IDEXX BioAnalytics (Columbia, MO). The authentication includes short tandem repeat (STR) profiling, mycoplasma testing, and cross-species contamination checking. In this project, only early passages of the cell lines (<10) were utilized. Cell culture mediums were purchased from Thermo Fisher Scientific (Carlsbad CA, USA) and used for cell culture after mixing with 10% fetal bovine serum (Thermo Fisher Scientific). Cells were cultured at 37°C and in a 5% CO2 humidified incubator. Sulindac, CMC (Carboxymethylcellulose), and NF-κB inhibitor (Bay11-7082) were purchased from Sigma-Aldrich (St Louis MO, USA). CMC was used as the vehicle of sulindac for in vivo study. Mouse anti-PD-L1 antibody and isotype control rat IgG2b were purchased from BioXcell (West Lebanon NH, USA). Sulindac sulfide was purchased from Alfa Aesar (Haverhill MA, USA).
Animal study
BALB/c immunocompetent mice were purchased from Charles River Laboratories (Wilmington MA, USA). Animals were housed under specific pathogen-free conditions. All animal experiments were performed in accordance with a protocol approved by the Institutional Animal Care and Use Committee of Louisiana State University Health Sciences Center.
Orthotopic CRC mouse model with intrarectal (IR) injection was established as previously described (20). Briefly, after mice were anesthetized with 2.5% isoflurane, the anal canal was dilated with lubricated blunt-tipped forceps to expose the distal anal and rectal mucosa. Luciferase-labeled CT26 cells were injected into the distal posterior rectal submucosa, 1 to 2 mm above the anal canal, using a sterile 30-gauge removable needle on a 50-μL hamilton microliter syringe. A dissecting microscope was used to assist in the injection. We injected each mouse with 2 × 105 luciferase labeled CT26 cells in a volume of 10 μL.
Mice were monitored for tumor growth bi-weekly with the non-invasive IVIS™ Imaging System (PerkinElmer, Waltham MA, USA). In brief, mice were injected intraperitoneally with D-luciferin substrate (Promega, Madison WI, USA) in Dulbecco’s Phosphate Buffered Saline (DPBS) at 150 mg/kg. After 10 min, mice were anesthetized with 2.5% isoflurane and placed on a warm stage inside the light-tight camera box and continuously exposed to isoflurane. The exposure time was 1-5 sec. The level of light emitted from the bioluminescent CT26 cells was detected, integrated, digitized, and displayed by the IVIS™ Imaging System. The region of interest from displayed images was quantified as photons/second.
When orthotopic tumors were detectable with IVIS™, which was about 10 days after IR injection, twenty male and twenty female mice were randomly divided into four groups: (1) vehicle control treated with CMC+IgG2b; (2) sulindac (15 mg/kg, p.o., bid); (3) PD-L1 antibody (75 μg, i.p. M/W/F); (4) combination of sulindac and PD-L1 antibody (7.5 mg/kg p.o., bid + 37.5 μg, i.p. M/W/F). Both female and male mice in each group were used equally. Mice were euthanized on day 27 when some animals showed the symptoms of bowel obstruction. Blood samples were reserved for exosome isolation, and tumor tissue samples were collected and fixed in 10% buffered formalin for paraffin embedding.
Immunofluorescence staining
Immunofluorescence staining was performed on paraffin-embedded sections of CT26 tumor tissue and pMMR CRC lines (CT26, Colo205, HT29, and Caco-2). The paraffin-embedded sections were deparaffinized and rehydrated. After antigen retrieval and permeabilization, slides were blocked with 5% horse serum for 1 h at room temperature and then incubated with the primary antibodies at 4 °C overnight. To stain CD8+ T cells, we incubated tumor tissue slides with Alexa Fluor® 647 conjugated CD8a antibody (Biolegend, San Diego CA, USA) overnight. For PD-L1 and NF-κB staining, tumor tissue slides were incubated with primary mouse PD-L1 antibody (Abcam, Cambridge MA, USA) and primary NF-κB p65 antibody (Cell Signaling, Beverly MA, USA), followed by Alexa Fluor® 488 and 555 conjugated secondary antibody (Thermo Fisher Scientific, Worcester MA, USA) for 1 h at room temperature, respectively. For immunofluorescence staining of all four pMMR CRC cell lines, we seeded cells in flow dishes at 37°C overnight and then treated them with 10 μM Bay11-7082 for 2 h and 25 μM sulindac sulfide (SS) for 12 h. DMSO at 0.1% was used as vehicle control. Mouse TNF-α (Thermo Fisher Scientific) and human TNF-α (Millipore, Billerica MA, USA) at a concentration of 25 ng/mL were added to the cells for incubation of 20 min, respectively. Then, cells were fixed by 4 % formaldehyde (Alfa Aesar) for 10 min, followed by permeabilization with 1% Triton X-100 (Bio-Rad). After blocking with 1% BSA, cells were incubated with mouse PD-L1 antibody (Abcam), human PD-L1 antibody (Cell Signaling), and NF-κB p65 antibody (Cell Signaling) at 4°C overnight. After washing with PBS, cells were incubated with Alexa Fluor® 488 (PD-L1) or 555 (p65)-conjugated secondary antibody (Thermo Fisher Scientific) for 1 h at room temperature. Finally, after washing and staining with 5 ng/ml DAPI (Sigma-Aldrich), slides and cells were processed with Prolong® Diamond Antifade Mountant (Thermo Fisher Scientific) and analyzed by confocal microscopy (Nikon Eclipse Ti2, Tokyo, Japan). We used ImageJ v1.48 (21) to quantify the expression of PD-L1 and CD8+ T cell infiltration. For PD-L1, we calculated total corrected cell fluorescence (TCCF) using the formula: TCCF = integrated density - (area of selected cells × mean fluorescence of background readings). For CD8+ T cell infiltration, we calculated the percentage of CD8+ T cells in the tumor tissue using the formula of the number of CD8+T cells divided by the total number of surrounding tumor cells plus CD8+T cells.
Flow cytometry analysis
Referring to a published protocol (22), we analyzed the status of T cells in BALB/c splenocytes under different treatments in vitro using flow cytometry. First, we prepared single-cell suspensions from the spleens of BALB/c mice and performed mechanical dissociation. In brief, spleens were washed and minced in warm Dulbecco’s Modified Eagle Medium (DMEM) and minced, and then the clumps were removed by passing through a Corning® 40 μM cell strainer (Corning, NY, USA). The filtrate containing the single-cell suspensions was washed through DMEM and PBS and centrifuged at 300×g for 10 min at room temperature. Next, we seeded 1x106 cells per well in a 24 well plate and then treated with 1 μg/ml anti-mouse CD3e antibody (eBioscience, San Diego, CA, USA) and 1 μg/ml anti-mouse CD28 antibody (eBioscience) (positive control), 0.1% DMSO plus 200 nM IgG2b (vehicle control), 25 μM SS, 200 nM anti-mouse PD-L1 antibody (BioXcell), and a combination of 25 μM SS and 200 nM anti-mouse PD-L1 antibody for 72 h. After treatment, cells were blocked with Fc-blocked antibody (BD Bioscience, Sparks, MD, USA) at 4°C, followed by live-dead fixable viability stain 450 (BD Bioscience) for 20 min and flow cytometry antibodies for 30 min, including APC anti-mouse CD45 antibody (BD Bioscience), BV711 anti-mouse CD3e antibody (BD Bioscience), PE-CF594 anti-mouse CD8a antibody (BD Bioscience), and APC-Cy™7 anti-mouse CD4 antibody (BD Bioscience). Then, intracellular staining was performed using the BD GolgiStop™ (BD Bioscience) fixation and permeabilization solution kit. FITC anti-mouse granzyme B antibody (eBioscience) was incubated with the cells for 30 minutes. Cells were then washed once with PBS and fixed with 1% paraformaldehyde prior to analysis by flow cytometry.
Isolation and purification of exosomes from mouse plasma samples
Exosomes were extracted from mouse plasma samples and purified with the ExoQuick® ULTRA EV isolation kit, which was purchased from System Biosciences (Mountain View CA, USA). According to the manufacturer’s instruction, 125 μL of plasma was pre-treated with 1 μL of Thrombin to remove fibrin and then incubated with 33.5 μL of ExoQuick exosome precipitation solution for 30 min at 4°C. After centrifugation at 3,000 g for 10 min, the exosomes were concentrated at the bottom of the tubes and then resuspended with buffer. Finally, exosome lysates were prepared by adding RIPA buffer (Thermo Fisher Scientific) and analyzed by Western Blot.
Western blot assay
Membrane proteins were isolated with Mem-PER™ Plus Membrane Protein Extraction Kit (Thermo Fisher Scientific), and cytoplasmic proteins were extracted with NE-PER™ Nuclear and Cytoplasmic Extraction Kit (Thermo Fisher Scientific). We quantified the proteins with the DC Protein Assay kit (Bio-Rad, Hercules CA, USA). Denatured proteins were separated on a 10% SDS-PAGE gel and then transferred to PVDF filters (Bio-Rad). After blocking with 5% non-fat milk-TBST (Bio-Rad), the blots were incubated mouse PD-L1 antibody (Abcam), human PD-L1 antibody (Cell signaling), β-actin antibody (Cell signaling), CD63 polyclonal antibody (Abcam), or Calnexin polyclonal antibody (Abcam) at 4°C overnight, respectively. After washing 3 times with 1×TBST (Bio-Rad), the blots were incubated with peroxidase-linked secondary goat anti-rabbit IgG antibody (Bio-Rad), or goat-anti-mouse IgG antibody (Bio-Rad) were incubated for 1 h at room temperature. Finally, after incubation with SuperSignal West Pico PLUS chemiluminescent substrate (Thermo Scientific, Worcester, MA, USA), visual imaging was performed by VersaDoc™ Imaging System (Bio-Rad). Volume One software (Bio-Rad) was used for the relative quantification of blotted proteins.
Chromatin immunoprecipitation assay (ChIP)
EZ-Magna ChIP kit (Millipore, Billerica MA, USA) was purchased for ChIP analysis. We strictly followed the manufacturer’s instructions. In brief, 1 × 107 CT26 cells were treated with 25 ng/ml TNF-α for 20 min and then cross-linked with 1% formaldehyde (Sigma-Aldrich). After fixation and lysis, chromatin was ultrasonically sheared using pre-optimized conditions, followed by immunoprecipitated with NF-κB p65 antibody (Cell signaling) or goat anti-mouse IgG at 4°C overnight. After extraction and purification of DNA fragments from the pull-down complexes, PCR was implemented using a thermocycler in a total volume of 20 μL, and the program included the following steps: 94°C, 2 min; 30 cycles including denaturation at 94°C for 20 sec, annealing at 59°C for 30 sec, and extension at 72°C for 30 sec. Finally, PCR products were electrophoresed on a 2% agarose gel. The primer sequences are: Forward primer: 5’CACTTCCAGTTCGCAGAA 3’; Reverse primer: 5’ CAAGCAAATGACTCAGTTTTA 3’.
Enzyme-linked immunosorbent assay (ELISA)
Mouse plasma PGE2 was measured with Prostaglandin E2 Parameter Assay Kit (R&D Systems, Minneapolis MN, USA) following the manufacturer’s instruction. In brief, plasma samples (50 μL) were appropriately diluted and added to a 96 well polystyrene microplate coated with goat anti-mouse polyclonal antibodies. After incubation with 50 μL of mouse PGE2 monoclonal antibody for 1 h at room temperature, the PGE2 conjugate was added to each well and incubated for 2 h with gentle shaking. After washing, the substrate solution was added to each well and incubated for 30 min, and then the stop buffer was added to halt the color development reaction. Finally, the absorbance was measured at 450 nm with a microplate reader (Bio-Rad).
Statistical analysis
GraphPad Prism version 7.0 (San Diego CA, USA) and SAS (9.4, Cary NC, USA) were utilized for data analysis. Luciferase intensity data (photons/sec) acquired from IVIS imaging were exhibited with the mean plus and minus standard error of the mean (SEM), and one-way ANOVA along with Tukey’s method were used to analyze the difference of tumor size at the endpoint (day 27). Student’s t-test was used to evaluate the difference of tumor weight, PD-L1 expression, and infiltrating CD8+ T cells between two groups with Bonferonni adjustments. Simple linear regression was used to assess the percentage of CD8+ T cells and PD-L1 expression in tumor tissue, as well as the strength and direction of the linear correlation between exosomal PD-L1 expression and tumor weight. The goodness-of-fit measurement, R2 was calculated to measure the strength of the predictor in explaining the outcome, and the p-value of coefficients in the linear models was used to report the significance of the predictor.
Results:
Sulindac improves the efficacy of anti-PD-L1 therapy in vivo.
We established a syngeneic mouse model using well-established techniques, specifically intrarectal (IR) injection of luciferase-labeled murine CRC CT26 cells. We then treated each group of mice either with sulindac alone (15 mg/kg, p.o., bid), PD-L1 antibody alone (75 μg, i.p., M/W/F), or a combination of sulindac and PD-L1 antibody (7.5 mg/kg p.o., bid + 37.5 μg, i.p. M/W/F), respectively. Bioluminescence signals of labeled CT26 cells were measured twice per week using a non-invasive IVIS imaging system. We sacrificed mice on day 27 due to the development of intestinal obstruction in several mice. As shown in Figure 1a, there was no significant difference in tumor growth in mice treated with vehicle control, sulindac monotherapy, and anti-PD-L1 monotherapy. However, the mice treated with the combination of sulindac and PD-L1 Ab were found to have the lowest luciferase signal levels during treatment, which were significantly different from the other groups. Autopsy of each mouse revealed the orthotopic tumors from each group, which were fully resected, removed, and weighed. Figure 1b shows the average weight of CRC tumors at the endpoint. We found that the mice treated with the combination of sulindac and PD-L1 Ab had the smallest tumors compared to the vehicle controls, which in turn translated to the lowest tumor weight (p<0.05, t-test). We also measured mice the total body weight twice per week but did not find any significant differences between groups (Supplementary Figure 1), supporting the safety of our treatment regimens.
Figure 1. The combination of sulindac and PD-L1 antibody significantly inhibits tumor growth.

(a) Monitoring CT26 tumor orthotopic growth with bioluminescent imaging. Mice were screened with bioluminescent imaging (IVIS) twice per week after intrarectal (IR) injection. When orthotopic tumors were detectable with IVIS™, twenty male and twenty female mice were randomly divided into four groups: (1) vehicle control treated with CMC+IgG2b; (2) sulindac (15 mg/kg, p.o., bid); (3) PD-L1 antibody (75 μg, i.p. M/W/F); (4) combination with sulindac and PD-L1 antibody (7.5 mg/kg p.o., bid +37.5 μg, i.p. M/W/F). Both female and male mice were equally used in each group. Mice were sacrificed on day 27 when one mouse in the control group and two mice in the sulindac-treated group died from the bowel obstruction. Therefore, their final imaging results were not collected. Tumor sizes quantified by photons/s were compared and evaluated with one-way ANOVA along with Tukey’s method. *p<0.05, ** p<0.01, ***p<0.001. (b) Tumor weights of all mice at the endpoint. Differences between two groups were assessed by Student’s t-test, *p<0.05. Mice that died before the endpoint were not included in this analysis.
Sulindac increases the infiltration of CD8+ T lymphocytes in tumor tissues by downregulating the expression of the PD-L1 gene.
A recent study reported that CD8+ T lymphocytes mediated the inhibitory activity of sulindac upon tumor cell growth, specifically in the syngeneic breast cancer [4T1] animal model (23). We collected tumor tissues to analyze CD8+ T lymphocyte infiltration. Utilizing immunofluorescence imaging, we visualized infiltrating T lymphocytes (Green) and stained PD-L1 with Alexa Fluor® 488 conjugated secondary antibodies (Red). As shown in representative merged confocal images (Figure 2a; unmerged images are exhibited in Supplementary Figure 2), the combination group treated with sulindac and PD-L1 Ab were found to have the most infiltrating CD8+ T lymphocytes (Green) in the tumor tissues, compared to vehicle control and PD-L1 Ab treated mice. Mice treated with sulindac alone showed a 8-fold and 3-fold increase of infiltrating CD8+ T lymphocytes compared to vehicle control and PD-L1 Ab treated mice, respectively. These results support the notion that sulindac can facilitate T lymphocyte infiltration in pMMR CRC tumors. Intriguingly, when analyzing PD-L1 expression in the same set of tumor samples, we found that PD-L1 expression is inversely correlated with infiltrating CD8+ T lymphocytes in all groups, with the vehicle control and combination groups displaying a statistical significance (p<0.05, Figure 2b).
Figure 2. Sulindac increases the infiltration of CD8+ T lymphocytes in tumor tissues by down-regulating PD-L1 expression.

(a) Immunofluorescence imaging results show that sulindac increases the infiltration of CD8+ T lymphocytes in tumor tissues and downregulates PD-L1 expression. Treatments include vehicle control, sulindac, PD- L1 Ab, and sulindac+PD-L1 Ab. Red: PD-L1; Green: CD8+ T cells; Blue: DAPI. PD-L1 expression and CD8+ T cell infiltration were quantified by ImageJ (v1.48, NIH). The total corrected cell fluorescence (TCCF) of PD-L1 was calculated using the formula: [TCCF = integrated Density - (area of selected cell × mean fluorescence of background readings). For CD8+ T cell infiltration, we calculated the percentage of CD8+ T cells in tumor tissues using the formula of the number of CD8+ T cells divided by the total number of surrounding tumor cells plus CD8+ T cells. Mice that died before the endpoint or lacked visible tumor mass treatment were not included in this analysis. Differences between the two groups were evaluated by Student’s t-test, * p<0.05, ** p<0.01, *** p<0.001. Images were taken with a Nikon Eclipse Ti2 Laser Confocal Scanning Microscope. (b) Linear correlation of PD-L1 expression levels and infiltrating CD8+T cells between the two groups was calculated with Prism GraphPad 7.0. The goodness-of-fit measurement, R2 was calculated to measure the strength of the predictor in explaining the outcome, and the p-value of coefficients in the linear models was used to report the significance of the predictor. (c) Additive/synergistic effect of sulindac and PD-L1 Ab on activation of CD8+ T lymphocytes. Splenocytes were prepared from the spleens of BALB/c mice and mechanically dissociated and filtered. Then we treated the splenocytes with 1 μg/ml anti-mouse CD3e antibody and 1 μg/ml anti-mouse CD28 antibody (positive control), 0.1% DMSO plus 200nM IgG2b (vehicle control), 25 μM SS, 200 nM anti-mouse PD-L1 antibody, and a combination of 25 μM SS and 200 nM anti-mouse PD-L1 antibody for 72 h, respectively. After treatment, cells were incubated with flow cytometry antibodies as described in Materials and Methods prior to the analysis with flow cytometry. (d) Sulindac sulfide (SS) downregulates PD-L1 expression in pMMR CRC cells. CT26, Coloa205, HT29, and Caco-2 cells were treated with 25 μM SS and vehicle control (DMSO) for 48 h, followed by isolation of cell membrane and cytoplasm from cell lysis. PD-L1 expression was evaluated using Western Blot, and the intensity ratio between PD-L1 and β-actin was calculated using Quantity One software (Bio-Rad).
Although our results show that sulindac monotherapy facilitates CD8+T lymphocyte infiltration and reduces PD-L1 expression in tumor tissues, comparing the results of in vivo inhibitory activity, as shown in Figure 1b, it is unlikely that sulindac monotherapy exhibits compelling inhibition on tumor growth to the same extent of combination therapy. We speculated whether there were differences in the status of infiltrating CD8+T lymphocytes induced by sulindac monotherapy and combination therapy. Utilizing flow cytometry, we tested our hypothesis and found that anti-PD-L1 monotherapy significantly activated CD8+ T lymphocytes (49.80% GranzB+), compared to sulindac monotherapy (14.71% GranzB+); the combination therapy showed a synergistic effect of two agents (72.19% GranzB+), as shown in Figure 2c. A previous study reported that PD-L1 Ab or PD-1 Ab could increase mTOR signaling and granzyme B expression in virus-specific CD8+ T lymphocytes, resulting in faster clearance of lymphocytic choriomeningitis mammarenavirus (LCMV) infection (24). This study supports the notion that PD-L1 Ab can activate CD8+ T lymphocytes. Thus, we conclude that the synergistic effect of PD-L1 Ab and sulindac on CD8+ T lymphocyte activation resulted in the most significant inhibitory efficacy of the combination therapy.
To further investigate the relationship between sulindac and PD-L1 Ab, we determined the inhibitory effect of sulindac on PD-L1 expression using mouse CT26 cells, as well as human CRC Colo205, HT29, and Caco-2 cells. All of these cell lines were characterized as pMMR CRC models (19,25). After isolation of membrane and cytoplasmic proteins from all four cell lines, we compared the PD-L1 expression levels after the treatment of vehicle control and sulindac sulfide (SS), the active metabolite of sulindac using Western blot assay. As shown in Figure 2d, SS could effectively downregulate PD-L1 expression in the cell membrane and cytoplasm. Taken together, our results demonstrate that sulindac can facilitate the infiltration of CD8+ T lymphocytes in CRC tumors by downregulating PD-L1 expression.
Blockade of NF-κB signaling is associated with sulindac modulation of pMMR CRC response to anti-PD-L1 therapy.
We have previously demonstrated a novel mechanism by which sulindac inhibits tumor cell motility by blocking NF-κB signaling downregulation of selected oncogenic miRNAs (26). Recent studies have also reported the ability of NF-κB to regulate PD-L1 in different cancer cells (27-30). Since our in vivo data demonstrated that sulindac could downregulate PD-L1 expression in tumor tissues, we investigated if NF-κB signaling might also be involved in sulindac’s inhibitory effect on PD-L1 expression in pMMR CRC.
Figure 3a exhibited a putative binding site for NF-κB (GGGAAGTTCT) within the promoter region of the PD-L1 gene, as reported previously (31). We further determined the binding of NF-κB to the promoter of PD-L1 using the chromatin immunoprecipitation (ChIP) assay. In brief, NF-κB p65 antibodies were used to immunoprecipitate the sheared chromatin fractions extracted from CT26 cells. After purifying the DNA fragments from the pull-down complexes, we performed a PCR analysis with the designated primers to detect the target DNA fragments containing the putative NF-κB binding site (GGGAAGTTCT). As shown in Figure 3b, detected a band with the expected size using DNA templates extracted from the p65 immunoprecipitated complex. In addition, we employed a dual-luciferase reporter assay to confirm the ChIP results. In brief, wild-type or mutated promoter fragments of the mouse PDL-1 gene were cloned into pEZX-LvPG04 vectors, and the dual-reporter system used Gaussia Luciferase (GLuc) as the promoter reporter and Secreted Alkaline Phosphatase (SEAP) as an internal control for signal normalization. After transfection of these constructs into CT26 cells, we measured the signals of GLuc and SEAP and calculated the relative light unit (RLU) ratio. As shown in Supplementary Figure 3, the RLU of the mutated constructs is much lower than that of the wild-type constructs upon TNFα treatment or NF-κB/p65 co-transfection (p<0.05). Together, these results provide strong evidence that NF-κB binds directly to the promoter of PD-L1 in CT26 cells.
Figure 3: Blockade of NF-κB signaling is involved in sulindac modulation of pMMR/MSS CRC in response to anti-PD-L1 treatment.

(a) Schematic representation of the mouse PD-L1 promoter fragment containing a putative NF-κB p65 binding sequence (GGGAAGTTCT,−1028 to −1019). (b) ChIP assay results showing direct binding of NF- κB p65 to the PD-L1 promoter. CT26 cells were pre-treated with 25 ng/ml TNF-α for 20 min and immunoprecipitated by NF-κB p65 antibody or normal mouse IgG. The isolated DNA fragments were purified from the pull-down complexes and utilized as the templates for PCR amplification. The expected size of the PCR product containing the putative NF-κB p65 binding sequence is 253 bp. Input samples were derived from the starting chromatin that had been used for ChIP. (c) SS attenuates the induction of NF- κB signaling by TNFα. Four CRC cell lines were first treated with 10 μM Bay11-7082 for 2 h and 25 μM SS for 12 h, respectively. DMSO was used as vehicle control. Then, human TNF-α or mouse TNF-α at a concentration of 25 ng/ml was added to these cells and incubated for 20 min. Cells were fixed with 4 % formaldehyde for 10 min and then permeabilized with 1% Triton X-100. After blocking with 1% BSA, the cells were incubated with PD-L1 antibody and NF-κB p65 antibody at 4°C overnight. Then, cells were stained and imaged with confocal microscopy. (d) Sulindac downregulates PD-L1 expression by blocking NF-κB signaling in CT26 tumor issues. The same sample sets for Figure 2 were utilized in this analysis. Red: NF-κB p65; Green: PD- L1; Blue: DAPI. Images were captured by using a Nikon Eclipse Ti2 Laser Confocal Scanning Microscope.
Furthermore, we utilized immunofluorescence imaging to analyze the regulation of PD-L1 expression following sulindac treatment. Using our published protocol, TNFα and Bay11-7082 were included to stimulate and block NF-κB signaling, respectively (26). All four CRC cell lines were utilized in this study. As shown in Figure 3c and Supplementary Figure 4-7, TNFα treatment promoted translocation of NF-κB p65 into the nucleus, which in turn upregulated PD-L1 expression. Additionally, treatment with SS can effectively attenuate the induction of TNFα and blocked NF-κB signaling, which was comparable to that of the NF-κB inhibitor Bay11-7082. We also extended our study by examining NF-κB p65 and PD-L1 in tumor tissues that we analyzed above. As expected, mice treated with sulindac alone, or in combination with PD-L1 Ab, showed reduced NF-κB signaling in the nucleus along with PD-L1 downregulation (Figure 3d and Supplementary Figure 8), which is consistent with the in vitro results.
Sulindac inhibits exosomal PD-L1.
A recent study reported that exosomal PD-L1 could significantly affect the efficacy of PD-L1 Ab (32). After extracted exosomes from mouse plasma samples, we examined exosomal PD-L1 expression in mice from each treatment group. The exosome marker CD63 was used as the endogenous control to normalize the expression of PD-L1. As showed in Figure 4a, the expression of exosomal PD-L1 was decreased in all treated mice compared to the vehicle control group, and the lowest plasma exosomal PD-L1 expression was observed in the combination group. Supplementary Figure 9 exhibits the results of Western Blot and relative quantitation of exosomal PD-L1 in mice. We further analyzed the correlation between exosomal PD-L1 and tumor progression using a linear model. The values of tumor weight were used to indicate tumor progression. As shown in Figure 4b, only the combination group revealed a positive correlation, which was statistically significant (R2=0.81; p=0.01). Intriguingly, the PD-L1 Ab group showed a decrease in exosomal PD-L1, but a weak correlation to tumor weight (R2=0.23; p=0.23). This result implies that PD-L1 Ab may be competitively occupied by circulating exosomal PD-L1, leading to a lack of sufficient amount of PD-L1 Ab available to effectively block immune checkpoints by binding to PD-L1 on the surface of CRC tumor cells. In combination therapy, sulindac can downregulate PD-L1 expression, ultimately resulting in less PD-L1 being released into circulation via exosomes and relatively more PD-L1 Ab being available to approach CRC tumor cells, not only to bind to PD-L1 on the cell surface but also to activate CD8+ T cells, as shown in Figure 2c.
Figure 4: Sulindac inhibits exosomal PD-L1.
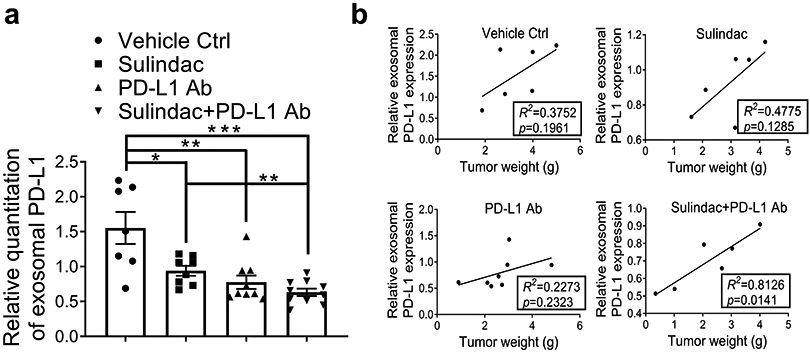
(a) Expression of exosomal PD-L1 was decreased in mice treated with all drugs compared to vehicle controls, and the combination of sulindac and PD-L1 Ab showed the lowest expression of exosomal PD-L1. We included only mice that were able to collect adequate blood samples for exosome isolation (Control group, n=7; Sulindac group, n=8; PD-L1 Ab group, n=9; Combination group, n=10). Exosomes were extracted from mouse plasma samples, purified with ExoQuick® ULTRA EV isolation kit, and lysed using RIPA buffer. Exosomal PD-L1 and CD63 were detected by Western blotting. Exosome marker CD63 was used as an endogenous control to normalize PD-L1 expression. Quantity One software (Bio-Rad) was used to calculate the intensity ratio between exosomal PD-L1 and CD63. (b) Linear correlation of exosomal PD-L1 expression levels and tumor weights between the two groups was calculated with Prism GraphPad 7.0. The goodness-of-fit measurement, R2 was calculated to measure the strength of the predictor in explaining the outcome, and the p-value of coefficients in the linear models was used to report the significance of the predictor.
Low doses of sulindac can effectively inhibit PD-L1 with no significant systematic toxicity.
The FDA-approved dose of sulindac for human use is 300-400 mg/day. Utilizing a published protocol(33), we calculated equivalent doses for the small animal study, which are 61.5-82.0 mg/kg. In this project, we only utilized doses of 15 and 7.5 mg/kg to treat mice, which are considered low doses of sulindac. To evaluate the systematic toxicity of sulindac, specifically the inhibition of PGE2, we utilized plasma samples collected from CT26 mice and assayed the concentration of PGE2 by ELISA. As shown in Figure 5, there was no significant difference between the vehicle control and the drug-treated mice. These results demonstrate that low-dose sulindac is a safe agent to modulate the response of pMMR CRC to ICI therapy, especially to anti-PD-L1 immunotherapy.
Figure 5. Low dose sulindac does not affect PGE2 at the systematic level.
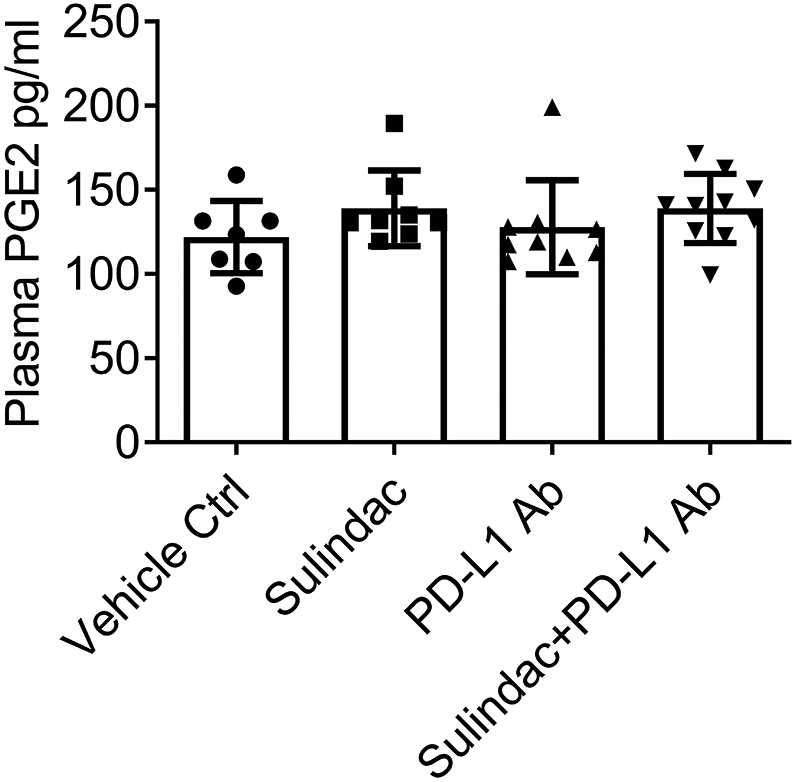
Plasma samples collected from mice were used to measure PGE2 levels using an ELISA assay. The same plasma sample sets for Figure 4 were utilized in this analysis. Plasma samples (50 μL) were appropriately diluted and added to a 96 well polystyrene microplate coated with goat anti-mouse polyclonal antibodies. After incubation with 50 μL of mouse PGE2 monoclonal antibody for 1 h at room temperature, the PGE2 conjugate was added to each well and incubated for 2 h with gentle shaking. After washing, the substrate solution was added to each well and incubated for up to 30 min, and then the stopping buffer was added to stop the color development reaction. Finally, the absorbance was measured at 450 nm using a microplate reader (Bio-Rad).
Discussion
The clinical benefit of checkpoint immunotherapy is well documented, with improvements in both long-term and overall survival seen primarily in the treatment of metastatic melanoma and lung cancer, and recently extended to metastatic triple-negative breast cancer (TNBC) (34). However, clinical studies have shown that the advantage of ICI immunotherapy for CRC is only confined to a small subset of patients with dMMR or highly microsatellite instability (MSI-H), representing less than 15% of CRC patients(12-14). Unfortunately, for most CRC patients carrying pMMR or MSI-L/MSS, current ICI regimens offer little clinical benefit in terms of patient survival.
Long-term use of NSAIDs has been shown to significantly reduce the incidence and risk of death from CRC and other cancers in both clinical and preclinical settings (35-39). In particular, sulindac is highly effective for treating pre-cancerous lesions in patients with familial adenomatous polyposis (FAP) and reducing the size and number of polyps by approximately 60-70% (18). Our previous studies have also exclusively investigated the anticancer activity of sulindac and several derivatives (26,40-45). In this study, we aimed to determine if sulindac could modulate the response to ICI immunotherapy of pMMR CRC. We chose CT26, a murine CRC cell line that was characterized as pMMR (19), to establish a syngeneic animal model using immunocompetent mice, which was shown by others to reduce immunogenicity to ICI treatment (46,47). After the intrarectal injection of luciferase-labeled CT26 cells, we treated mice with sulindac, PD-L1 Ab, and a combination of sulindac and PD-L1 Ab, respectively. Non-invasive bioluminescence imaging (IVIS) was utilized to monitor the orthotropic growth of CT26 tumors. Our results clearly demonstrate that the combination of sulindac and PD-L1 Ab was the most effective therapy in our observation, with a significant effect on tumor weight, size, and regression.
In view of a previous study reporting the involvement of CD8+ T lymphocytes in the anticancer activity of sulindac against breast cancer (23), we examined the infiltration of CD8+ T lymphocytes in tumors using immunofluorescence imaging. Our results showed a significant increase of infiltrating CD8+ T lymphocytes in the tumor tissues from the animals treated with sulindac monotherapy and combination therapy, with the latter group clearly exhibiting a more intensive infiltration of T cells. We also examined PD-L1 expression in the same tissue sets, given that previous studies have reported that elevated PD-L1 expression is associated with poor prognosis, reduced survival, and lymph node metastasis of CRC (48-50). Higher expression of PD-L1 in tumor cells has also been suggested to be one of the drivers of tumor immune escape (8). We found that PD-L1 expression was inversely correlated with the number of infiltrating CD8+ T lymphocytes, indicating that sulindac is able to target and inhibit PD-L1 expression in tumor cells, leading to the infiltration of CD8+ T lymphocytes. Of interest, we further demonstrated that sulindac and PD-L1 Ab have a synergistic effect on activation of CD8+T lymphocytes (GranzB+), supporting our in vivo results showing that the combination therapy has an improved inhibition on tumor growth, compared to the monotherapy.
Furthermore, we measured PD-L1 expression in the cytoplasm and cell membrane after sulindac treatment using four pMMR CRC cell lines, including CT26, Colo205, HT29, and Caco-2 cells. Our results demonstrate that sulindac can significantly downregulate intracellular and extracellular PD-L1 levels. We further explored the molecular mechanism by which sulindac inhibits PD-L1. Recently, others have reported that NF-κB is involved in the regulation of PD-L1 expression in cancer cells (27-30), and our previous studies have also shown that sulindac can block NF-κB signaling (26,43). Following our established protocol, we searched for NF-κB binding sequences and identified a putative locus in the PD-L1 gene promoter. Utilizing ChIP and luciferase reporter assays, we identified direct binding between NF-κB and the PD-L1 promoter. Recent studies have also reported that TNF-α can induce PD-L1 expression through the NF-κB pathway (30). Therefore, we used TNF-α as an inducer to stimulate NF-κB signaling and then determine whether sulindac treatment could effectively alter PD-L1 expression in all four pMMR CRC cell lines. As expected, our results support the notion that NF-κB signaling is a key mechanism by which sulindac downregulates PD-L1 through transcriptional regulation. We further examined the translocation of NF-κB in the tumor tissues collected from in vivo studies. Consistently, mice treated with sulindac, either alone or in combination with PD-L1 Ab, show a far less intensity of NF-κB signals within the cell nucleus, concomitant with downregulation of PD-L1.
Exosomes are 30-100 nm-sized membrane vesicles released by many cell types during physiological processes. Importantly, exosomes serve as a means of cell-cell communication because they can easily fuse with recipient cells and release their contents (51). Tumor cells typically produce far more exosomes than their normal non-cancerous counterparts, and their exosomes are often referred to as tumor-derived exosomes (TDEs) (52). TDEs contain key oncogenic elements, including a variety of miRNAs, mRNA, proteins, and lipids, which can initiate signaling pathways required for the tumor progression and metastatic processes (53). Recent studies have reported that exosomal PD-L1 is able to attenuate the inhibitory effect of PD-L1 Ab (32). Utilizing the blood samples collected from our in vivo studies, we analyzed exosomal PD-L1 in mice treated with sulindac [alone], PD-L1 Ab [alone], and the combination [sulindac+PD-L1 Ab]. We found that all treatment regimens significantly reduced exosomal PD-L1 expression, while the combination group is associated with the most reduction of exosomal PD-L1 and tumor weight. We further analyzed the correlation between exosomal PD-L1 expression levels and tumor progression in mice treated with monotherapy and combination therapy. The values of tumor weight were utilized as variables to indicate tumor progression.
Intriguingly, PD-L1 Ab treated mice showed a significant reduction in exosomal PD-L1, but it poorly correlated with tumor weights (R2=0.22; p=0.23). These results suggest that exosomal PD-L1 readily binds to and consumes PD-L1 Ab in circulation, leading to a reduction in PD-L1 Ab that can approach and target tumor cells. For the combination of sulindac and PD-L1 Ab, exosomal PD-L1 displayed a tight inverse correlation with tumor progression (R2=0.81; p=0.01), due to the ability of sulindac to downregulate PD-L1 at the transcriptional level, and less total intracellular PD-L1 could lead to less exosomal PD-L1 being secreted into the circulation. Ultimately, PD-L1 Ab in combination therapy can be relatively “enriched” around tumor cells compared to treatment with PD-L1 Ab monotherapy. Thus, although our results show that all treatments can reduce the levels of exosomal PD-L1 in circulation; however, the amount of “free” PD-L1 Ab that can engage tumor cells is the crucial parameter to interpret the difference of tumor inhibitory effect between different treatment arms, and our correlation analysis supports such a conclusion (Figure 4b). When excess “free” PD-L1 Ab reaches tumor cells, they can not only turn off immune checkpoints by binding to PD-L1 on the cell surface, but also activate CD8+ T lymphocytes to attack tumor cells.
Sulindac, an FDA-approved NSAID, is a non-selective cyclooxygenase-1 and −2 (COX-1 and COX-2) inhibitor. COX-2 inhibition can reduce the synthesis of prostaglandin 2 (PGE2). Therefore, long-term administration of NSAIDs, including higher doses of sulindac, is approached with particular caution regarding the possible human side effects associated with PGE2 reduction upon the gastrointestinal, renal, and cardiovascular systems (54). It has also been reported that COX-2 is capable of impairing tumor immunity and that inhibition of COX-2 has the potential to improve immunotherapeutic responses in a variety of tumor models (55-57). Therefore, if the systemic toxicity due to PGE2 inhibition is minimized, sulindac holds great promise as a very safe addition to ICI therapy. In our in vivo study, we used low doses of sulindac, which are equivalent to approximately ¼ of the FDA-approved dose for human usage. When testing PGE2 in the blood samples collected from mice, we did not see any significant difference in side effects or end-organ function among treatment groups, demonstrating that sulindac is relatively safe and effective at the low doses we chose.
We conclude that sulindac can effectively modulate the response of pMMR CRC to anti-PD-L1 immunotherapy. The mechanism of action involves sulindac downregulating PD-L1 through the blockade of NF-κB signaling. Given that pMMR CRC patients represent the majority of the CRC population but benefit little from current ICI regimens, our study provides a unique insight into the development of sulindac as part of a safe and effective combination immunotherapy for the treatment of CRC.
Supplementary Material
Acknowledgment:
This work was supported in part by the National Institutes of Health (NIH)-National Cancer Institute (NCI) grants (R01CA192395 and a pilot award of P20CA233374) to YX. We also appreciate Z. Liang and Y. Xun for their technical support.
Footnotes
The authors declare no potential conflicts of interest.
Reference:
- 1.Siegel RL, Miller KD, Goding Sauer A, Fedewa SA, Butterly LF, Anderson JC, et al. Colorectal cancer statistics, 2020. CA Cancer J Clin 2020 [DOI] [PubMed] [Google Scholar]
- 2.Xia L, Liu Y, Wang Y. PD-1/PD-L1 Blockade Therapy in Advanced Non-Small-Cell Lung Cancer: Current Status and Future Directions. Oncologist 2019;24:S31–S41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Munhoz RR, Postow MA. Clinical Development of PD-1 in Advanced Melanoma. Cancer J 2018;24:7–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pentcheva-Hoang T, Corse E, Allison JP. Negative regulators of T-cell activation: potential targets for therapeutic intervention in cancer, autoimmune disease, and persistent infections. Immunol Rev 2009;229:67–87 [DOI] [PubMed] [Google Scholar]
- 5.Carreno BM, Collins M. The B7 family of ligands and its receptors: new pathways for costimulation and inhibition of immune responses. Annu Rev Immunol 2002;20:29–53 [DOI] [PubMed] [Google Scholar]
- 6.Chen L, Han X. Anti-PD-1/PD-L1 therapy of human cancer: past, present, and future. J Clin Invest 2015;125:3384–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Blank C, Mackensen A. Contribution of the PD-L1/PD-1 pathway to T-cell exhaustion: an update on implications for chronic infections and tumor evasion. Cancer Immunol Immunother 2007;56:739–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Juneja VR, McGuire KA, Manguso RT, LaFleur MW, Collins N, Haining WN, et al. PD-L1 on tumor cells is sufficient for immune evasion in immunogenic tumors and inhibits CD8 T cell cytotoxicity. J Exp Med 2017;214:895–904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kreidieh M, Mukherji D, Temraz S, Shamseddine A. Expanding the Scope of Immunotherapy in Colorectal Cancer: Current Clinical Approaches and Future Directions. Biomed Res Int 2020;2020:9037217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell 1990;61:759–67 [DOI] [PubMed] [Google Scholar]
- 11.Fearon ER, Pierceall WE. The deleted in colorectal cancer (DCC) gene: a candidate tumour suppressor gene encoding a cell surface protein with similarity to neural cell adhesion molecules. Cancer Surv 1995;24:3–17 [PubMed] [Google Scholar]
- 12.Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N Engl J Med 2015;372:2509–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Papadopoulos N. Microsatellite instability (MSI) in non-colonic, non-HNPCC tumors: ‘instable’ evidence? Ann Oncol 1999;10:751–2 [DOI] [PubMed] [Google Scholar]
- 14.Vilar E, Tabernero J. Molecular dissection of microsatellite instable colorectal cancer. Cancer Discov 2013;3:502–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Le DT, Kavan P, Kim TW, Burge ME, Cutsem EV, Hara H, et al. KEYNOTE-164: Pembrolizumab for patients with advanced microsatellite instability high (MSI-H) colorectal cancer. Journal of Clinical Oncology 2018;36:3514- [Google Scholar]
- 16.Diaz L, Marabelle A, Kim TW, Geva R, Van Cutsem E, André T, et al. 386PEfficacy of pembrolizumab in phase 2 KEYNOTE-164 and KEYNOTE-158 studies of microsatellite instability high cancers. Annals of Oncology 2017;28 [Google Scholar]
- 17.Andre T, Lonardi S, Wong M, Lenz H-J, Gelsomino F, Aglietta M, et al. Nivolumab + ipilimumab combination in patients with DNA mismatch repair-deficient/microsatellite instability-high (dMMR/MSI-H) metastatic colorectal cancer (mCRC): First report of the full cohort from CheckMate-142. Journal of Clinical Oncology 2018;36:553- [Google Scholar]
- 18.Giardiello FM, Hamilton SR, Krush AJ, Piantadosi S, Hylind LM, Celano P, et al. Treatment of colonic and rectal adenomas with sulindac in familial adenomatous polyposis. N Engl J Med 1993;328:1313–6 [DOI] [PubMed] [Google Scholar]
- 19.Castle JC, Loewer M, Boegel S, de Graaf J, Bender C, Tadmor AD, et al. Immunomic, genomic and transcriptomic characterization of CT26 colorectal carcinoma. BMC Genomics 2014;15:190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hite N, Klinger A, Hellmers L, Maresh GA, Miller PE, Zhang X, et al. An Optimal Orthotopic Mouse Model for Human Colorectal Cancer Primary Tumor Growth and Spontaneous Metastasis. Dis Colon Rectum 2018;61:698–705 [DOI] [PubMed] [Google Scholar]
- 21.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods 2012;9:671–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Leelatian N, Doxie DB, Greenplate AR, Sinnaeve J, Ihrie RA, Irish JM. Preparing Viable Single Cells from Human Tissue and Tumors for Cytomic Analysis. Curr Protoc Mol Biol 2017;118:25C 1 1–C 1 3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yin T, Wang G, Ye T, Wang Y. Sulindac, a nonsteroidal anti-inflammatory drug, mediates breast cancer inhibition as an immune modulator. Sci Rep 2016;6:19534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ahn E, Araki K, Hashimoto M, Li W, Riley JL, Cheung J, et al. Role of PD-1 during effector CD8 T cell differentiation. Proc Natl Acad Sci U S A 2018;115:4749–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lengauer C, Kinzler KW, Vogelstein B. DNA methylation and genetic instability in colorectal cancer cells. Proc Natl Acad Sci U S A 1997;94:2545–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li X, Gao L, Cui Q, Gary BD, Dyess DL, Taylor W, et al. Sulindac inhibits tumor cell invasion by suppressing NF-kappaB-mediated transcription of microRNAs. Oncogene 2012;31:4979–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lim SO, Li CW, Xia W, Cha JH, Chan LC, Wu Y, et al. Deubiquitination and Stabilization of PD-L1 by CSN5. Cancer Cell 2016;30:925–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Asgarova A, Asgarov K, Godet Y, Peixoto P, Nadaradjane A, Boyer-Guittaut M, et al. PD-L1 expression is regulated by both DNA methylation and NF-kB during EMT signaling in non-small cell lung carcinoma. Oncoimmunology 2018;7:e1423170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jin X, Ding D, Yan Y, Li H, Wang B, Ma L, et al. Phosphorylated RB Promotes Cancer Immunity by Inhibiting NF-kappaB Activation and PD-L1 Expression. Mol Cell 2019;73:22–35 e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gowrishankar K, Gunatilake D, Gallagher SJ, Tiffen J, Rizos H, Hersey P. Inducible but not constitutive expression of PD-L1 in human melanoma cells is dependent on activation of NF-kappaB. PLoS One 2015;10:e0123410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Huang G, Wen Q, Zhao Y, Gao Q, Bai Y. NF-kappaB plays a key role in inducing CD274 expression in human monocytes after lipopolysaccharide treatment. PLoS One 2013;8:e61602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Poggio M, Hu T, Pai CC, Chu B, Belair CD, Chang A, et al. Suppression of Exosomal PD-L1 Induces Systemic Anti-tumor Immunity and Memory. Cell 2019;177:414–27 e13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nair AB, Jacob S. A simple practice guide for dose conversion between animals and human. J Basic Clin Pharm 2016;7:27–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mina LA, Lim S, Bahadur SW, Firoz AT. Immunotherapy for the Treatment of Breast Cancer: Emerging New Data. Breast Cancer (Dove Med Press) 2019;11:321–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Smalley W, Ray WA, Daugherty J, Griffin MR. Use of nonsteroidal anti-inflammatory drugs and incidence of colorectal cancer: a population-based study. Arch Intern Med 1999;159:161–6 [DOI] [PubMed] [Google Scholar]
- 36.Beazer-Barclay Y, Levy DB, Moser AR, Dove WF, Hamilton SR, Vogelstein B, et al. Sulindac suppresses tumorigenesis in the Min mouse. Carcinogenesis 1996;17:1757–60 [DOI] [PubMed] [Google Scholar]
- 37.Mahmoud NN, Boolbol SK, Dannenberg AJ, Mestre JR, Bilinski RT, Martucci C, et al. The sulfide metabolite of sulindac prevents tumors and restores enterocyte apoptosis in a murine model of familial adenomatous polyposis. Carcinogenesis 1998;19:87–91 [DOI] [PubMed] [Google Scholar]
- 38.Piazza GA, Alberts DS, Hixson LJ, Paranka NS, Li H, Finn T, et al. Sulindac sulfone inhibits azoxymethane-induced colon carcinogenesis in rats without reducing prostaglandin levels. Cancer Res 1997;57:2909–15 [PubMed] [Google Scholar]
- 39.Thompson HJ, Jiang C, Lu J, Mehta RG, Piazza GA, Paranka NS, et al. Sulfone metabolite of sulindac inhibits mammary carcinogenesis. Cancer Res 1997;57:267–71 [PubMed] [Google Scholar]
- 40.Yi B, Chang H, Ma R, Feng X, Li W, Piazza GA, et al. Inhibition of breast cancer cell motility with a non-cyclooxygenase inhibitory derivative of sulindac by suppressing TGFbeta/miR-21 signaling. Oncotarget 2016;7:7979–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li N, Xi Y, Tinsley HN, Gurpinar E, Gary BD, Zhu B, et al. Sulindac selectively inhibits colon tumor cell growth by activating the cGMP/PKG pathway to suppress Wnt/beta-catenin signaling. Mol Cancer Ther 2013;12:1848–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Whitt JD, Li N, Tinsley HN, Chen X, Zhang W, Li Y, et al. A novel sulindac derivative that potently suppresses colon tumor cell growth by inhibiting cGMP phosphodiesterase and beta-catenin transcriptional activity. Cancer Prev Res (Phila) 2012;5:822–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yi B, Piazza GA, Su X, Xi Y. MicroRNA and cancer chemoprevention. Cancer Prev Res (Phila) 2013;6:401–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xi Y. MicroRNA: A New Player for Cancer Chemoprevention. J Integr Oncol 2013;2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ma R, Yi B, Piazza GA, Xi Y. Mechanistic Role of MicroRNA in Cancer Chemoprevention by Nonsteroidal Anti-inflammatory Drugs. Curr Pharmacol Rep 2015;1:154–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yu G, Wu Y, Wang W, Xu J, Lv X, Cao X, et al. Low-dose decitabine enhances the effect of PD-1 blockade in colorectal cancer with microsatellite stability by re-modulating the tumor microenvironment. Cell Mol Immunol 2019;16:401–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yuzhakova DV, Shirmanova MV, Serebrovskaya EO, Lukyanov KA, Druzhkova IN, Shakhov BE, et al. CT26 murine colon carcinoma expressing the red fluorescent protein KillerRed as a highly immunogenic tumor model. J Biomed Opt 2015;20:88002. [DOI] [PubMed] [Google Scholar]
- 48.Shen Z, Gu L, Mao D, Chen M, Jin R. Clinicopathological and prognostic significance of PD-L1 expression in colorectal cancer: a systematic review and meta-analysis. World J Surg Oncol 2019;17:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shan T, Chen S, Wu T, Yang Y, Li S, Chen X. PD-L1 expression in colon cancer and its relationship with clinical prognosis. Int J Clin Exp Pathol 2019;12:1764–9 [PMC free article] [PubMed] [Google Scholar]
- 50.Li Y, He M, Zhou Y, Yang C, Wei S, Bian X, et al. The Prognostic and Clinicopathological Roles of PD-L1 Expression in Colorectal Cancer: A Systematic Review and Meta-Analysis. Front Pharmacol 2019;10:139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Marleau AM, Chen CS, Joyce JA, Tullis RH. Exosome removal as a therapeutic adjuvant in cancer. J Transl Med 2012;10:134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Steinbichler TB, Dudas J, Riechelmann H, Skvortsova, II. The role of exosomes in cancer metastasis. Semin Cancer Biol 2017;44:170–81 [DOI] [PubMed] [Google Scholar]
- 53.Kahlert C, Kalluri R. Exosomes in tumor microenvironment influence cancer progression and metastasis. J Mol Med (Berl) 2013;91:431–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mazhar D, Gillmore R, Waxman J. COX and cancer. QJM 2005;98:711–8 [DOI] [PubMed] [Google Scholar]
- 55.DeLong P, Tanaka T, Kruklitis R, Henry AC, Kapoor V, Kaiser LR, et al. Use of cyclooxygenase-2 inhibition to enhance the efficacy of immunotherapy. Cancer Res 2003;63:7845–52 [PubMed] [Google Scholar]
- 56.Veltman JD, Lambers ME, van Nimwegen M, Hendriks RW, Hoogsteden HC, Aerts JG, et al. COX-2 inhibition improves immunotherapy and is associated with decreased numbers of myeloid-derived suppressor cells in mesothelioma. Celecoxib influences MDSC function. BMC Cancer 2010;10:464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zelenay S, van der Veen AG, Bottcher JP, Snelgrove KJ, Rogers N, Acton SE, et al. Cyclooxygenase-Dependent Tumor Growth through Evasion of Immunity. Cell 2015;162:1257–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.