

Abstract
Biofluid-derived cell-free nucleic acids such as microRNAs (miRNAs) and circulating tumor-derived DNAs (ctDNAs) have emerged as promising disease biomarkers. Conventional detection of these biomarkers by digital PCR and next generation sequencing, although highly sensitive, requires time-consuming extraction and amplification steps that also increase the risk of sample loss and cross-contamination. To achieve the direct, rapid, and amplification-free detection of miRNAs and ctDNAs with near-perfect specificity and single-molecule level sensitivity, we herein designed a single-molecule kinetic fingerprinting assay, termed intramolecular single-molecule recognition through equilibrium Poisson sampling (iSiMREPS). iSiMREPS exploits a dynamic DNA nanosensor comprising a surface anchor and a pair of fluorescent detection probes: one probe captures a target molecule onto the surface, while the other transiently interrogates the target to generate kinetic fingerprints by intramolecular single-molecule Förster resonance energy transfer (smFRET) that are recorded by single-molecule fluorescence microscopy and identify the target after kinetic filtering and data analysis. We optimize the sensor design, use formamide to further accelerate the fingerprinting kinetics, and maximize sensitivity by removing non-target-bound probes using toehold-mediated strand displacement to reduce background. We show that iSiMREPS can detect, in as little as 10 seconds, two distinct, promising cancer biomarkers—miR-141 and a common EGFR exon 19 deletion—reaching a limit of detection (LOD) of ~3 fM and a mutant allele fraction among excess wild-type as low as 1 in 1 million, or 0.0001%. We anticipate that iSiMREPS will find utility in research and clinical diagnostics based on its features of rapid detection, high specificity, sensitivity, and generalizability.
Keywords: Single molecule fluorescence microscopy, smFRET, Kinetic fingerprinting, Nanosensor, Nucleic acids, Rapid biomarker detection
Graphical Abstract
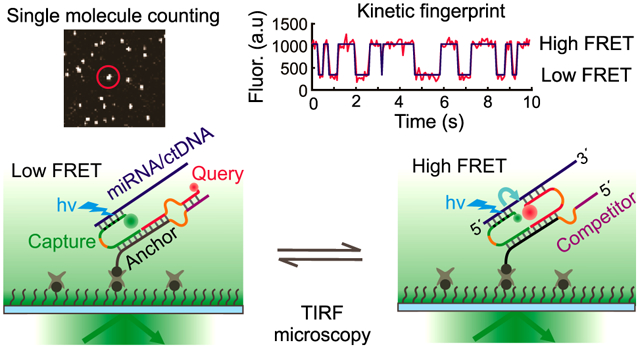
1. Introduction
Circulating cell-free nucleic acids (cfNAs) have emerged as promising diagnostic biomarkers for diseases such as cancer (Anfossi et al., 2018; Schwarzenbach et al., 2011). Among various cfNAs, microRNAs (miRNAs) are short non-coding RNAs with gene regulatory function and great potential as biomarkers due to high biofluid concentration and stability (Anfossi et al., 2018). Another important class of cfNAs comprises circulating, tumor-derived DNAs (ctDNAs), such as mutant copies of the epidermal growth factor receptor (EGFR) gene that are commonly found in the blood of some patients with non-small cell lung cancer (NSCLC) (Gazdar, 2009). Detecting abnormal levels of these biomarkers in biofluids (e.g., blood, urine) via non-invasive liquid biopsies has been a major area of clinical interest (Crowley et al., 2013; Heitzer et al., 2015), thus necessitating cfNA detection approaches that are rapid, highly specific, ultrasensitive, and robust for analyzing diverse biomarkers.
Advanced techniques such as next generation sequencing (NGS) for large-scale genome analysis (Schmitt et al., 2012) and digital PCR (Milbury et al., 2014) have arisen as gold standards for nucleic acid detection. Although they achieve high sensitivity for low abundance analytes and have sufficient specificity for allelic frequencies as low as 0.01% for ctDNA mutants, they require significant sample preparation, purification, enzymatic reactions, and amplification steps that are time-consuming and can introduce various errors and compromise assay performance when high specificity is necessary (Potapov and Ong, 2017). Several amplification-free methods (Cohen et al., 2017; Li et al., 2018; Wang and Zhang, 2015) for detecting cfNAs have been reported. However, the clinical utility of these techniques is constrained by upper limits on specificity imposed by the thermodynamics of nucleic acid binding (Zhang et al., 2012). Additionally, while a wide variety of innovative single molecule fluorescent based techniques (Akkilic et al., 2020; Ma et al., 2016) have been developed for ultrasensitive and rapid detection of nucleic acids utilizing non-enzymatic signal amplification steps, they suffer from lower specificity and dynamic range since many have a static readout that poorly distinguishes target-specific fluorescent signals from nonspecific background.
We recently reported an amplification-free single-molecule kinetic fingerprinting technique called Single Molecule Recognition through Equilibrium Poisson Sampling (SiMREPS) (Hayward et al., 2018; Johnson-Buck et al., 2015) for detection of miRNAs and ctDNAs from biofluids. SiMREPS uses single-molecule fluorescence microscopy to record the transient binding and dissociation of fluorescent probes to a surface-captured nucleic acid. Continuous imaging for ~10 minutes reveals repeated binding to individual captured molecules, yielding a time-resolved “kinetic fingerprint” that can be used to distinguish a target molecule from non-target molecules and non-specific background (Hayward et al., 2018; Johnson-Buck et al., 2015). While SiMREPS permits ultrahigh specificity, its sensitivity and speed are limited by the long acquisition times required to observe multiple probe binding events to each target molecule. While increasing fluorescent probe concentration would accelerate binding kinetics and permit faster acquisition times, the increased fluorescent background from diffusing probes would result in severe degradation of signal-to-noise in the measurement, effectively placing an upper limit on the speed of acquisition.
To overcome all of these limitations, we here developed a novel single-molecule Förster resonance energy transfer (smFRET)-based accelerated kinetic fingerprinting approach termed intramolecular SiMREPS, or iSiMREPS (Fig. 1). iSiMREPS introduces a dynamic DNA nanoscale sensor comprising a surface-immobilized anchor stably hybridized to a pair of fluorescent capture and query probes. The nanoscale intramolecular arrangement of the sensor creates a high local concentration of fluorescent probes. Thus, transient intramolecular interactions within the sensor result in rapid transitions between high- and low-FRET states in the presence of the correct target molecule, while showing almost no high-FRET signal in the absence of the target or in the presence of spurious targets. (Fig. 1). These FRET transitions reveal a characteristic kinetic fingerprint of the analyte that reduces false positives dramatically compared to a static readout because the signal must satisfy intensity thresholds and exhibit a specific kinetic signature that passes kinetic filtering criteria (Fig. 1). The rate of the transitions between high- and low-FRET states can be modulated by modifying sensor components and assay conditions and can be further increased by modifications that reduce thermodynamic stability of the probe-target complex (e.g. minimizing complementary base pairs, modifying the lengths of the sensor strands to facilitate transitions between both states, increasing temperature, or adding denaturant). Additionally, sensitivity can be improved by removing non-target-bound fluorescent probes via toehold-mediated strand displacement (TMSD) from invading strands, which lowers background fluorescence. Optimized iSiMREPS shows high sensitivity and ultrahigh specificity for detecting both a miRNA and a ctDNA in just 10 s acquisition per field of view (FOV), a 60-fold improvement over conventional (intermolecular) SiMREPS measurements, paving the way for dramatically accelerated molecular diagnostics.
Fig. 1.
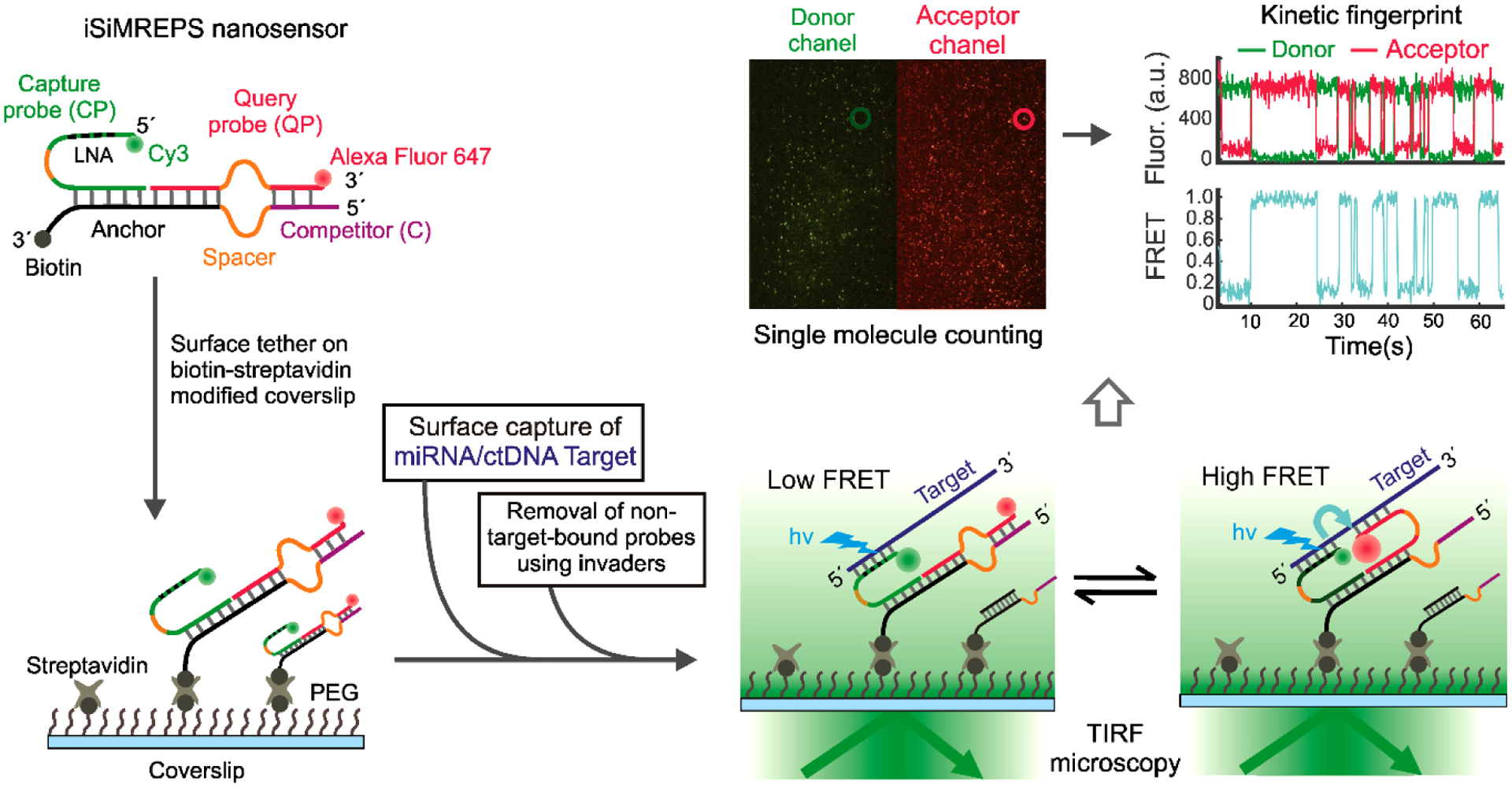
Schematic of iSiMREPS sensors for rapid kinetic fingerprinting of single nucleic acids. The iSiMREPS nanosensor consists of a biotinylated, surface-tethered anchor hybridized to a capture probe (CP) and a query probe (QP). The CP is labeled with a donor fluorophore (Cy3) and partially modified with locked nucleic acid residues for high affinity capture of a miRNA/ctDNA target molecule. The QP contains an acceptor fluorophore (Alexa Fluor 647) and transiently alternates between binding target and competitor sequences to generate high- and low-FRET signals, respectively. Transitions between FRET states are recorded by total internal reflection fluorescence (TIRF) microscopy, and FRET vs. time traces are analyzed computationally to count single biomarker molecules with high specificity.
2. Material and Methods
2.1. Oligonucleotides and reagents
All unmodified DNA oligonucleotides were purchased from Integrated DNA Technologies (IDT, www.idtdna.com) with standard desalting purification, unless otherwise noted. Biotinylated DNA oligonucleotides were purchased from IDT with polyacrylamide gel electrophoresis (PAGE) purification. Fluorescent query probes (QPs) with a 3′ Alexa Fluor 647 modification were purchased from IDT with high-performance liquid chromatography (HPLC) purification. Capture probes (CPs) that contained locked nucleic acid (LNA) residues were purchased either from IDT with a 5′Cy3 modification and HPLC purification or from Qiagen with a 5′amino modification with HPLC purification. The CPs from Qiagen were labeled with Cy3 monoreactive dye (GE Healthcare) and purified by ethanol precipitation (https://www.qiagen.com/us/resources). The miR-141 with a 5′-phosphate modification was purchased from IDT with HPLC purification. The double-stranded EGFR exon 19 deletion mutant DNA (EGFRΔexon_19) (COSMIC ID: COSM6223; c. 2235_2249del15 [p.E746_A750delELREA]) and wild type DNA (EGFRexon_19) substrates were prepared by annealing complementary single stranded oligonucleotides at 1 μM final concentration in annealing buffer (10 mM Tris-HCl, pH 8.0 at 25°C, 50 mM NaCl and 1 mM EDTA), heating at 95°C for 3 min, cooling to room temperature for 25 min, and finally holding at 4°C for 10 min before storage at −20°C for further use. All oligonucleotides’ sequences are shown in Tables S1 and S2.
2.2. Design of iSiMREPS probes
The intramolecular SiMREPS sensor design requires a stable complex of the anchor, capture probe (CP), and query probe (QP) that does not dissociate from the imaging surface in iSiMREPS assay conditions. The anchor contained a 12-nt segment rich in GC content (⩾75%) to have a melting temperature (Tm) of ~ 60 °C for stable hybridization with both CP and QP (Figs. 2A and 3A). The Tm between anchor and CP or QP was estimated by IDT OligoAnalyzer (https://www.idtdna.com/calc/analyzer) using the following parameters: target DNA concentration = 25 nM, NaCl = 600 mM, 25°C. All iSiMREPS sensors contained identical sequences in the anchor to stably hybridize with CP and QP. The CPs contained an 11- to 12-nt target-capturing sequence with 4 LNA residues (Tm = ~70°C, estimated using Qiagen web application) for high affinity and kinetically stable capturing of nucleic acid targets on the surface. All QPs used an 8-nt complementary segment for transient binding and dissociation with miR-141 (Tm = 30.2°C) (Table S3) or EGFRΔexon_19 (Tm = 23.9°C) (Table S4). The QPs also used a 6–7-nt complementary segment for transient binding with the competitor sequences extended from anchor for miR-141 (Tm = 7.5 to 18.1°C) (Table S3) and a 6–8-nt complementary section for EGFRΔexon_19 (Tm = 0 to 23.9°C) (Table S4). The Tm between query and target or competitor was estimated by IDT OligoAnalyzer as before, but with target RNA or DNA concentration = 1 μM. The discrimination between mutant (MUT) and wild-type (WT) DNA with a specific query probe was calculated using the web software NUPACK (Caltech: , 2007; Zadeh et al., 2011) and utilizing the following equation 1 (Zhang et al., 2012),
| (1) |
where Qmax,therm is the maximum theoretical discrimination, △△G0 is the difference in the Gibbs free energy of hybridization of a query with MUT and of the same query with WT DNA target. The detailed guidelines for designing SiMREPS query or fluorescent probes have been discussed elsewhere (Hayward et al., 2018; Johnson-Buck et al., 2019; Johnson-Buck et al., 2015).
Fig. 2.
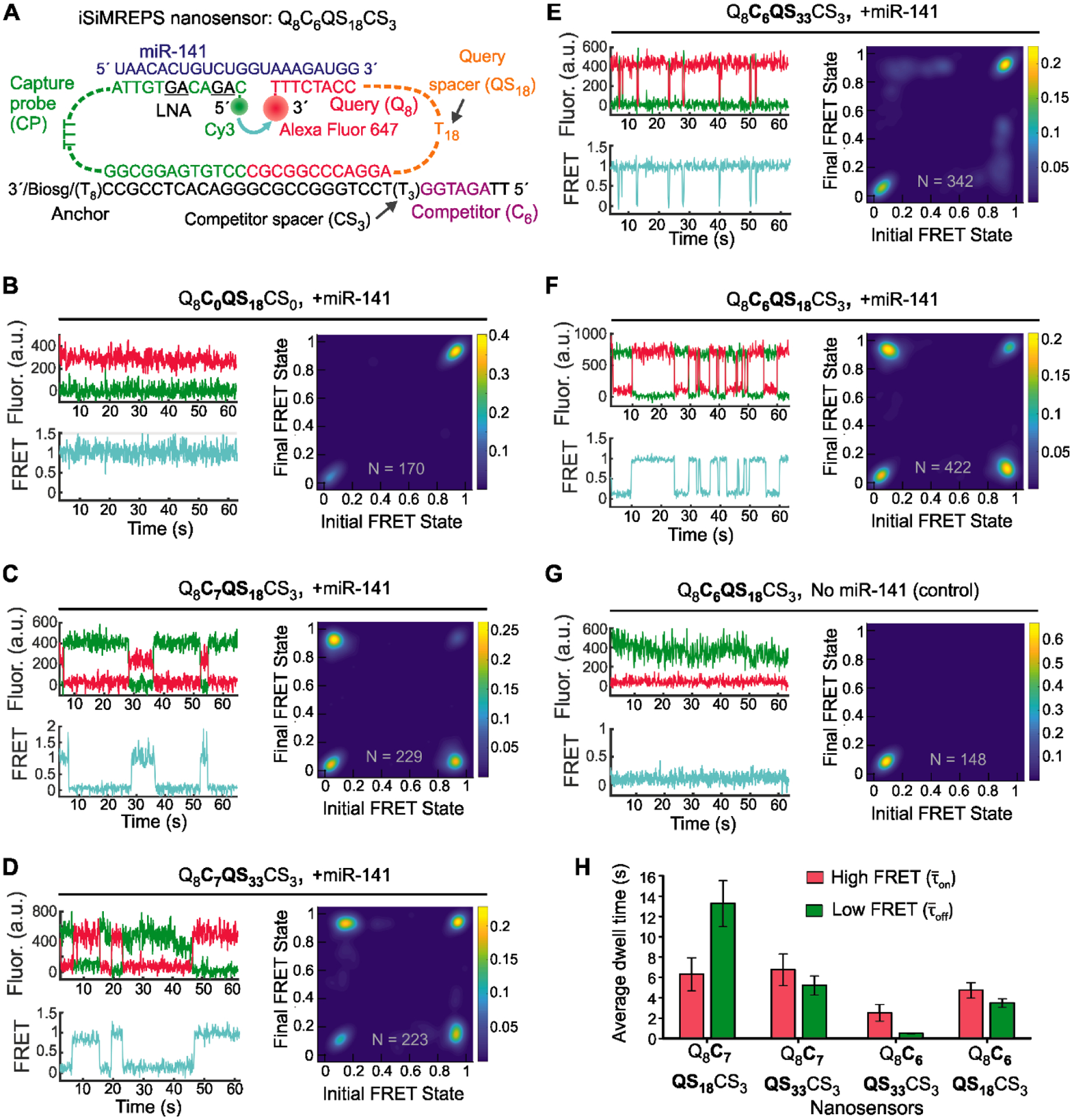
Design and optimization of iSiMREPS for detection of a miRNA. (A) Design of the optimized Q8C6QS18CS3 smFRET-based iSiMREPS sensor for detection of miR-141. The CP stably binds with the miRNA target with the assistance of locked nucleic acid residues (black and underlined) that increase the stability of the DNA-RNA duplex. The query (8 nt) switches between being bound to the 8 nt overhang of the target or to a 6 nt competitor sequence that extends from the anchor, resulting in dynamic kinetic smFRET fingerprints. (B-G) TODP plots and representative traces for different iSiMREPS sensor designs that have fixed query (8 nt), varying competitor (6 and 7 nt), fixed competitor spacer (3 nt), and varying query spacer (3, 18 and 33 nt) lengths in the presence of miR-141, as well as control without miR-141. The smFRET dynamics of each sensor is indicated. (H) The average dwell times of the high-FRET (red) and low-FRET (green) interactions for each sensor design. All data are presented as mean ± s.d., where n = 3 populations of a split data set for each condition.
Fig. 3.
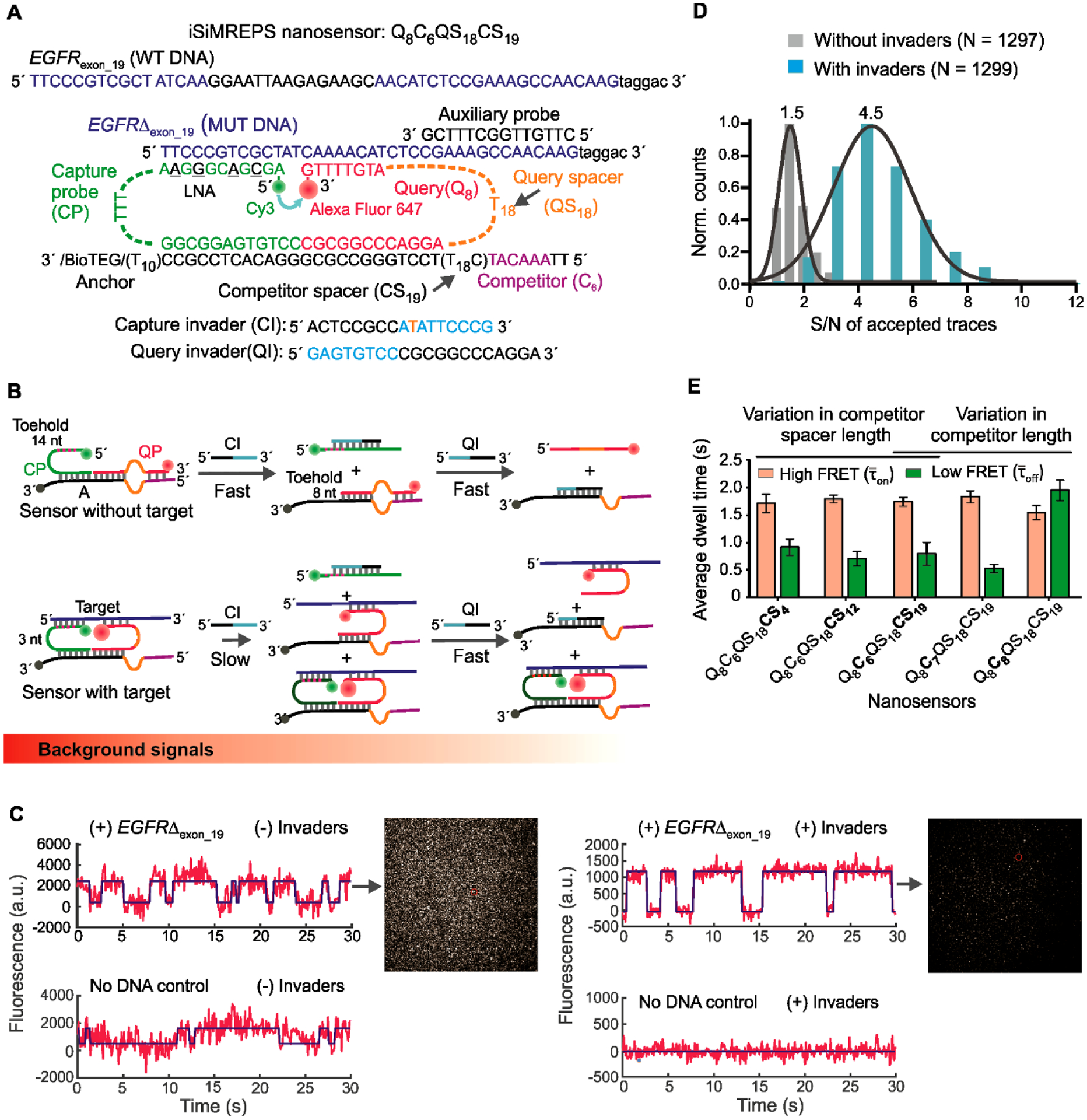
Design and optimization of iSiMREPS for detection of a ctDNA biomarker mutant DNA sequence. (A) Design of optimized smFRET-based iSiMREPS sensor for the detection of EGFR exon 19 deletion mutant DNA (EGFRΔexon_19). Two invaders, which are used to remove non-target-bound fluorescent probes from the surface, are also shown. (B) Schematic depiction of the removal of non-target-bound fluorescent probes (top) using CI and QI, and the much slower side reaction that removes target-bound probes (bottom). Each non-target-bound sensor has an exposed 9 nt toehold on the CP that binds with CI (cyan) and initiates the toehold displacement cascade. A 3 nt toehold on the CP in target-bound sensors can also bind with CI and ultimately prevent detection of a target molecule, but this reaction occurs much more slowly due to the shorter toehold. (C) Comparison of single-molecule FRET traces of iSiMREPS sensor in the presence (top) or absence (bottom) of the target sequence containing the EGFRΔexon_19. Background signals are significantly reduced with the application of invaders (right panel) compared to samples imaged without invader treatment (left panel). (D) Comparison of signal-to-noise (S/N) ratio with (cyan) and without (grey) invaders. (E) The average dwell times spent in the high-FRET (light red) and low-FRET (green) states for different iSiMREPS sensors designs. All data are presented as mean ± s.d., with n = 3 independent experiments.
2.3. Objective-TIRF iSiMREPS assay design
Detection experiments using the objective-TIRF microscope used sample cells made of cut pipette tips that were attached to a biotin-PEG and mPEG passivated glass coverslip (see Supplementary Information (SI)). The sample cells were first treated with 45 μL of 0.1–0.5 mg/mL streptavidin in T50 (10 mM Tris-HCl pH 8.0, 50 mM NaCl) for 10–20 min. The subsequent steps for this assay followed one of two procedures.
One procedure, along with a prism-TIRF protocol (see SI), was followed for the initial optimization of iSiMREPS assay parameters and conditions for detecting miR-141. The anchor, capture, query strands and miR-141 target were combined at 200, 225, 250 and 5 nM final concentrations, respectively in 4× PBS (Phosphate-buffered saline) (40 mM Na2HPO4, 7.2 mM KH2PO4, pH 7.4, 548 mM NaCl, 10.8 mM KCl), heated at 70°C for 7 min in a metal bath, and then cooled at room temperature for 25 min. Unless otherwise noted, all nucleic acid samples preparation were performed in GeneMate low-adhesion 1.7 mL micro centrifuge tubes in 4× PBS. The sensor was diluted 1000-fold, and 100 μL of the sensor solution was added to the cell for 45 min to tether the sensor on the surface via streptavidin-biotin affinity linkages. After removing non-surface-bound sensors and washing the cell 3 times with 4× PBS, a 100 μL solution of a pair of invader strands (see Table S1), each at 2 μM, was added to the cell and incubated for 20 min to remove non-target-bound fluorescent probes from the imaging surface. Next, the invaders solution was removed, the cell was washed 3 times with 4× PBS, and 200 μL imaging buffer containing oxygen scavenger system (OSS) consisting of 1 mM Trolox, 5 mM 3,4-dihydroxybenzoate, 50 nM protocatechuate dioxygenase, and the desired w/w% formamide in 4× PBS was added in the cell which was then imaged by TIRF microscopy.
The other procedure was followed for all experiments for detecting EGFRΔexon_19 as well as miR-141 quantification. Unless otherwise noted, a synthetic forward strand of EGFRΔexon_19 was used for optimizing the sensor parameters and assay conditions, while all experiments for quantifying concentration and determining sensitivity and specificity used duplex EGFRΔexon_19. The anchor, capture, and query strands for miR-141 or EGFRΔexon_19 was combined in a PCR tube at 400–500 nM final concentrations in 4× PBS, heated at 95°C for 3 min, 72°C for 7 min and 25°C for 25 min and 4°C for 10 min using a thermocycler to form a stable intramolecular complex. The sensor was then diluted to the desired concentration of 10 nM miR-141 sensor or 10–50 nM EGFRΔexon_19 sensor and 100 μL of the diluted sensor was added in the cell and incubated for 30 min to tether the sensor to the surface. Next, 100 μL of a solution containing either miR-141 or EGFRΔexon_19 target of the desired concentrations in 4× PBS was applied in the cell for 90 min for efficient capturing of the target by surface-tethered sensors. EGFRΔexon_19 target solution containing 100 nM of auxiliary probe and 2 μM of dT30 were heated at 90°C for 3 min in a metal block and cooled in a water bath at room temperature for 3 min prior to cell application. The non-target-bound probes were removed by invaders before imaging under an objective-type TIRF microscope in presence of OSS as outlined above.
2.4. Single-molecule fluorescence microscopy
iSiMREPS experiments were performed using either an Olympus IX-71 prism-type TIRF microscope equipped with a 60× water-immersion objective (Olympus UPLANAPO, 1.2NA) or an Olympus IX-81 objective-type TIRF microscope equipped with a 60× oil-immersion objective (APON 60XOTIRF, 1.49 NA) with CellTIRF and z-drift control modules. An ICCD (I-Pentamax, Princeton Instruments, MCP Gain 60) or sCMOS (Hamamatsu C13440-20CU) camera was used to record movies for the prism-TIRF while an EMCCD camera (IXon 897, Andor, EM gain 150) was used for the objective-TIRF. For recording smFRET signal, the Cy3-Alexa Fluor 647 fluorophore pairs were excited by light from a 532 nm laser at a power of 15–30 mW. For reliably detecting FRET signals with satisfactory S/N, an illumination intensity of ~50 W/cm2 is typically used, and the TIRF angle adjusted to achieve a calculated evanescent field penetration depth of ~70–85 nm. Two-channel images were recorded using a prism-TIRF microscope while only acceptor channel images were recorded using an objective-TIRF microscope. In prism-TIRF imaging, the signal integration time (exposure time) per frame was 100 ms, laser power was ~ 18 mW, and movies ranging from 1–15 minutes were collected to assess FRET behavior comprehensively. In objective-TIRF imaging, the exposure time per frame was 60–100 ms, and typically 200–600 movie frames were acquired per FOV.
2.5. Processing and analysis of objective-TIRF data
MATLAB scripts were used to identify areas of high average FRET acceptor intensity within each field of view, generate intensity-versus-time traces from these areas, and save these traces for further analysis. These traces were then analyzed using a two-state HMM (Bronson et al., 2009) algorithm to generate idealized (noise-less) intensity-versus-time traces to identify transitions between high- and low-FRET states. Thresholds of a minimum intensity of FRET transitions as well as a minimum S/N for the FRET signal were applied to each trace to distinguish genuine FRET transitions from baseline noise (Bronson et al., 2009) (Tables S6 and S7). Those traces passing the initial intensity and SNR thresholding were subjected to kinetic analysis to extract the number of FRET transitions per trace (Nb+d), the median dwell time in the high-FRET (τon, median), and low-FRET states (τoff, median), the intensity of the low-FRET (Ilow-FRET) and high-FRET (Ihigh-FRET) states, the longest individual dwell times in the high- and low-FRET states, and the coefficients of variation (CVs) of the dwell times in the high- and low-FRET states. These extracted parameters were subjected to minimum and maximum thresholding as indicated in Tables S6 and S7 to identify target-bound sensors based on their distinct kinetic and intensity behavior and to count the number of such target-bound sensors (“accepted counts”) observed in each movie. In addition, the cumulative frequencies of the dwell times in the high- and low-FRET states were fit to a single or double exponential function (see SI, and Figs. S4, S8 and S9) to obtain the average dwell time in each state and generate Nb+d histograms for each sensor. The Nb+d histograms and average dwell times were used to evaluate the sensor’s performance in terms of separation from background and capacity for rapid detection. The accepted counts were used for quantification and assessment of sensitivity. Prism-TIRF data was processed with similar considerations (See SI, and Table S5).
3. Results and Discussion
3.1. The general architecture and working principles of smFRET-based iSiMREPS nanosensors for nucleic acids
smFRET-based iSiMREPS for counting single nucleic acid molecules utilizes a surface-immobilized nanoscale sensor composed of three DNA oligonucleotides: a biotinylated anchor, a donor (Cy3)-labeled capture probe (CP), and an acceptor (Alexa Fluor 647)-labeled query probe (QP) (Fig. 1). The anchor stably binds the non-labeled ends of the CP and QP. The free end of the CP is partially modified with locked nucleic acid (LNA) residues, enabling high-affinity capture of the miRNA/ctDNA target molecule. The free end of the QP is designed to alternate between transient hybridization to the free end of the captured target nucleic acid molecule and a competitor (C) sequence that extends from the free end of the anchor. We introduced poly-deoxy-thymine (poly-dT) segments as spacers in the anchor, CP, and QP to introduce the flexibility necessary for the sensor to properly assemble and transition between target-bound and competitor-bound states.
In the optimized assay, iSiMREPS sensors composed of pre-hybridized anchor, CP, and QP are tethered first to a biotin-Bovine Serum Albumin (BSA)-passivated quartz slide (for prism-type total internal reflection fluorescence, or p-TIRF) or a PEG-passivated glass coverslip (for objective-type TIRF or o-TIRF) via streptavidin-biotin affinity linkages. The nucleic acid target molecules are then introduced into the solution above the surface. The target molecule binds strongly to the CP and thus become tethered to the surface. To minimize background signals before TIRF imaging, non-target-bound fluorescent probes are removed from the surface by toehold mediated strand displacement (TMSD) using a pair of capture and query invader strands (see below for detail).
In the presence of a target molecule, the QP alternates between transiently binding to the target and the competitor, yielding distinct FRET signatures depending on which sequence is bound. When the QP binds to the target, the donor and acceptor fluorophores are in close proximity, resulting in a high-FRET signal (Fig. 1). In contrast, when the QP dissociates from the target and/or binds to the competitor, the two fluorophores are far apart resulting in little to no FRET signal (Fig. 1). The repeated transitions between high- and low-FRET signals generate a characteristic kinetic fingerprint, permitting the accurate identification of single target nucleic acid molecules. Because these transitions occur much more rapidly than the transitions in conventional (intermolecular) SiMREPS, we anticipated that smFRET-based iSiMREPS should allow for faster and higher-confidence detection of nucleic acids through rapid fingerprint generation. In the following sections, we optimize this general design to detect two distinct nucleic acid biomarkers of disease.
3.2. Optimization of an iSiMREPS sensor design for detecting miRNA
An initial proof-of-concept iSiMREPS sensor (Fig. 2A) was designed to detect miR-141, a miRNA that has emerged as a biomarker for prostate cancer (Bryant et al., 2012; Mitchell et al., 2008). To develop iSiMREPS into an accelerated single-molecule kinetic fingerprinting technique, we initially tested several sensor designs (Fig. S1A–C) aiming for rapid smFRET transitions in the presence of target. To refer to different sensor designs, we use a QaCbQScCSd naming convention, where Q is the query sequence complementary to target, QS is the query spacer, C is the competitor sequence complementary to the query sequence, CS is the competitor spacer, and the letters a, b, c, and d are integers reflecting the number of nucleotides in each domain (Fig. 2A).
We initially omitted a competitor sequence (CS0) and instead used an 18-nt poly-dT query spacer (QS18) to introduce conformational flexibility (Murphy et al., 2004) in the QP and generate high- and low-FRET signals (Fig. S1A). p-TIRF characterization of Q8C0QS18CS0 in the presence of miR-141 showed clear smFRET signals, suggesting that the sensor hybridized successfully with miR-141 to induce a high-FRET state (Fig. 2B). However, the equilibrium FRET distribution overwhelmingly favored the high-FRET state (Fig. 2B), preventing the characterization of a kinetic fingerprint. This bias likely occurred due to the desired high local effective concentrations (Li et al., 2016) of the probes resulting in a high rate of query and target association. Consequently, while query-target dissociation events are expected, they appear too short-lived to be detected at the 6–100 ms time resolution achievable in smFRET. To disfavor the query-target interaction by increasing the entropic cost of hybridization, we increased the length of the query spacer from dT18 to dT33 (Fig. S1A). However, Q8C0QS33CS0 still heavily favored the high-FRET state (Fig. S1D).
We hypothesized that the addition of a competitor sequence could decrease the observed dwell times of the target bound (high-FRET) state by competing with the target and stabilizing the non-target-bound (low-FRET) state (Fig. 1). Therefore, in next design we introduced a 7-nt competitor sequence (C7) (Tm=18.1°C Table S3) to the anchor and used a 3-nt poly-dT as a competitor spacer (CS3) (Fig. S1B). Q8C7QS18CS3 showed repeated transitions between high- and low-FRET states that constituted a distinctive kinetic fingerprint in the presence of miR-141 (Fig. 2C). In fact, this design was somewhat biased towards the low-FRET state. This experiment demonstrated that the competitor sequence is required for iSiMREPS designs to exhibit measurable transitions between smFRET states.
To estimate the dwell times for high-(τon) and low-FRET (τoff) states each intensity-time trace was fit with a two-state Hidden Markov Model (HMM) (Bronson et al., 2009), and the dwell times of individual events were extracted (Fig. S2A). For each of the two states, the average dwell time was then calculated by fitting an exponential decay function to the cumulative frequency of the dwell time population (see SI and Fig. S2B–S2E). These FRET-state times encompass events where the query may have instantly re-bound to the same strand, and are thus are best described as the mean first passage times between bound states (with the unbound state serving as a short-lived intermediate) (Polizzi et al., 2016). The average dwell times for high- and low-FRET states for Q8C7QS18CS3 were 6.3 ± 1.6 s and 13.3 ± 2.2 s, respectively (Fig. 2H). Interestingly, was approximately 2-fold higher than (Fig. 2H) even though the query-competitor duplex was less thermodynamically stable (Tm = 18.1°C) than the query-target duplex (Tm = 30.2°C, see Table S3), suggesting that the geometry of the sensor causes the query to preferentially bind the competitor rather than the target. Together, these results suggested that single-molecule kinetic fingerprinting could be accelerated by fine-tuning sensor properties such as the thermodynamic stability of transient duplexes and the lengths of various spacers within the sensor. Considering the inverse relationship between the hybridization length of two oligonucleotides and the specificity of the interaction (Zhang et al., 2012), we opted to keep the query-target complementarity to 8 bp while tuning other parameters like spacer and competitor lengths to shorten τon and τoff and obtain more balanced distributions between FRET states.
To obtain a more balanced FRET distribution, we increased the query spacer from dT18 to dT33 (Fig. S1B). As expected, Q8C7QS33CS3 shifted towards the high-FRET state (Fig. 2D). was 5.2 ± 0.9 s, a reduction of approximately 60% compared to Q8C7QS18CS3. In contrast, there was no significant change in (Fig. 2H, left two panels). A possible explanation for this observation is that increasing the length of the query spacer also increases the entropic cost of query-competitor binding, reducing τoff. Consistent with this hypothesis, when we reduced the query spacer to 3 nt (Q8C7QS3CS3), a static low-FRET behavior was observed, presumably due to the minimized entropic cost of query-competitor binding (Fig. S1B and S1E).
To further reduce dwell times, we shortened the competitor sequence from C7 to C6 and tested three different iSiMREPS sensors with varying query spacer lengths of dT3, dT18, and dT33 (Fig. 2A and S1C). As with its C7 counterpart, Q8C6QS3CS3 did not show any high-FRET signal (Fig. S1F). Q8C6QS33CS3 showed a significant reduction in both τon and τoff compared to its C7 counterpart (Fig. 2E). However, was reduced substantially to 0.5 ± 0.1 s, and there was a strong dominance of the high-FRET state (Fig. 2H). We obtained the most promising results from Q8C6QS18CS3 (Fig. 2A), which showed near-parity between the high- and low-FRET states (Fig. 2F) with and (relative to Q8C7QS33CS3) of 4.7 ± 0.7 s and 3.5 ± 0.4 s, respectively (Fig. 2H). A control experiment confirmed that Q8C6QS18CS3 showed no high-FRET signal in the absence of miR-141 (Fig. 2G). Overall, the analysis of FRET behavior and dwell time suggests that as the query spacer (QSc) length is increased, and the competitor (Cb) length decreased, the query-target interaction is favored compared to query-competitor interaction. Since Q8C6QS18CS3 showed the best kinetic fingerprint, with short and well-balanced dwell times, this design was chosen for subsequent optimization and assay development for the rapid detection of miR-141.
3.3. Monte Carlo simulations rationalize the dependence of iSiMREPS sensor kinetics on spacer length
To better understand the effect of spacer length on iSiMREPS probe kinetics, we developed a coarse-grained Monte Carlo simulation model (see detail in SI). Our simulation results (Fig. S3) show that at very short spacer lengths, the distance between the target and query strands is large (i.e., pairing is inhibited) due to conformational rigidity of the stiff anchor duplex. Increasing spacer length up to 10 nt allows the target and query strands to interact without bending the anchor duplex. Beyond 10 nt, increasing the spacer length causes the target-query distance to gradually decrease due to the query strand’s increased radius of diffusion. By contrast, the query-competitor distance decreases monotonically across all spacer lengths. These findings (Fig. S3) are in qualitative agreement with the experimental results for detecting miR-141 using the 6-nt competitor (Fig. 2); the QS3 sensors showed that query-target interactions were unfavorable, while the QS18 sensors showed near-parity between the two states and the QS33 sensors favored query-target binding.
3.4. Optimization of an iSiMREPS sensor design for detecting ctDNA
To test for generality of the iSiMREPS approach, we next targeted a different class of nucleic acid biomarker: ctDNA. We chose an EGFR exon 19 deletion mutation DNA (EGFRΔexon_19) commonly found as fragmented ctDNA in biofluids of NSCLC patients (Fan et al., 2017). The optimized iSiMREPS sensor features the same fundamental components and architecture as the sensor design for miR-141 detection. However, it deals with the greater length and dsDNA nature of the ctDNA through two additional features. First, we added a short auxiliary probe that stably binds the extended 3’ end of the forward strand of the duplex mutant target DNA (Fig. 3A) to prevent reannealing of the complementary strand once melted during sample preparation. The auxiliary probe also aims to minimize any potential secondary structure of the target strand (Johnson-Buck et al., 2019).
Second, the DNA-based architecture of iSiMREPS sensors allows us to selectively remove the CP and QP of target-less sensors after target capture and before imaging (Fig. 1) to reduce background. To this end, we developed a two-step process that employs a pair of ssDNA “invaders” that selectively bind and disassemble target-less iSiMREPS sensors via TMSD, a strategy often displayed in dynamic DNA nanotechnology (Zhang and Seelig, 2011) (Fig. 3B). In the first step, a capture invader (CI) binds to a toehold exposed on the CP in the absence of target. Via TMSD, the CI disrupts the capture-anchor duplex to remove the CP from the surface (Fig. 3B, top panel). This first step reveals a second toehold, which is then bound by a query invader (QI) in the second step. The QI disrupts the query-anchor duplex to remove the QP and its fluorescent signal from the iSIMREPS sensor (Fig. 3B, top panel). Although these invaders are designed to work on non-target-bound probes to reduce background signals significantly, the spacer on the CP can also act as a toehold and there is a minor probability (Dunn et al., 2016) that this can lead to removal of probes from target-bound sensors (Fig. 3B, bottom panel).
We performed proof-of-concept studies for detecting EGFRΔexon_19 using a Q8C6QS18CS19 sensor (Fig. 3A) modelled after the optimized sensor for miR-141 (Fig. 2A). For this sensor, we used a longer competitor spacer (CS19 versus CS3) to further improve parity between the FRET states. We used a query specific to EGFRΔexon_19 (Tm = 23.9°C, Table S4) that was designed to maximize discrimination between EGFRΔexon_19 and the off-target wild type sequence (Fig. 3A), as predicted using NUPACK (Caltech: , 2007; Zadeh et al., 2011). For optimization of sensor designs, we used a synthetic forward strand of EGFRΔexon_19. A pair of CI and QI strands, as shown in Figure 3A, were designed to remove non-target-bound fluorescent probes from the surface (Fig. 3B). However, the initial design of CI contains a single mismatch in the spacer region to prevent the use of the capture spacer as a toehold (Fig. 3A).
To examine the performance of Q8C6QS18CS19 for detecting EGFRΔexon_19 and to assess the efficacy of the invader strands, the preassembled sensor was first tethered to the glass coverslip and the mutant DNA target was introduced to bind the sensor probes on the surface. Next the samples were (or were not) incubated with invaders and imaged with o-TIRF. We found that invader treatment significantly reduced background signal in single-molecule intensity-time traces, resulting in a 3-fold higher S/N ratio relative to samples that were not treated with invaders (Fig. 3C and 3D). Exponential fitting of dwell time distributions (see SI and Fig. S4C) showed a and of 1.7 ± 0.1 s and 0.8 ± 0.2 s respectively, indicating some bias for the high-FRET state (Fig. 3E). These and are shorter than those measured for miR-141 detection under similar salt concentration and temperature. This change likely arose because the query-mutant DNA duplex (Tm = 23.9°C) was less stable than query-miR-141 duplex (Tm = 30.2°C). Moreover, the presence of the extra 3´ sequences in the EGFRΔexon_19 target may destabilize the interaction with the query strand slightly by introducing more electrostatic repulsion from the nearby phosphates.
To modulate the dwell times of high-(τon) and low-FRET (τoff) states, we designed several additional iSiMREPS sensors. Firstly, we decreased the length of competitor spacer of Q8C6QS18CS19 to CS12 and CS4 (Fig. S5A). We expected that decreasing the CS length would 1) increase the rate of the query–competitor interactions because of higher local effective concentrations, and 2) increase τoff, making it more closely resemble the high-FRET state (τon). However, the results showed that varying the CS length had an insignificant effect on the dynamics of FRET transitions in iSiMREPS sensors (Figs. 3E and S5B). This result may be because of the relatively long QS (dT18) present in this series of designs introduced substantial flexibility to all constructs, thus undercutting attempts to finely tune effective local concentrations. Secondly, we ran experiments where we increased the length of competitors of Q8C6QS18CS19 to C7 and C8 to raise the thermodynamic stability of the query–competitor interaction (Table S4). Indeed, increasing the competitor length from 6- to 8-nt increased significantly (Fig. 3E), further confirming that competitor length is one of the most important parameters in iSiMREPS sensor design. Overall, the Q8C6QS18CSd = 4, 12, 19 design, where d is the number of nucleotides in CS, worked well for EGFRΔexon_19. Given the insignificant effect of CS length, Q8C6QS18CS19 was chosen for further assay optimization.
3.5. Denaturant (formamide)-assisted rapid detection of miRNA and ctDNA using iSiMREPS
Having optimized iSiMREPS designs for both miRNA and mutant DNA, we next sought to further accelerate sensor kinetics to increase the speed of kinetic fingerprinting. As a simple approach that maintains specificity, we chose to use the denaturant formamide, which is known to destabilize nucleic acid duplexes and decrease Tm by ~2.4–2.9°C /(mol L−1) (Blake and Delcourt, 1996). Due to the intramolecular assembly of the iSiMREPS sensor, we still expected fast association kinetics of the probes even in the presence of denaturant.
As predicted, adding formamide (10% v/v) to the imaging buffer resulted in intensity-time traces with much shorter τon and τoff for both miR-141 and EGFRΔexon_19 (Figs. 4A and 4D, left panels). With a standard acquisition time of 10 s per FOV and image processing (see Tables S6 and S7), histograms of the number of binding and dissociation events (Nb+d) for both miR-141 and EGFRΔexon_19 targets showed good separation from background with, but not without, 10% formamide (Figs. 4A and 4D, right panels).
Fig. 4.
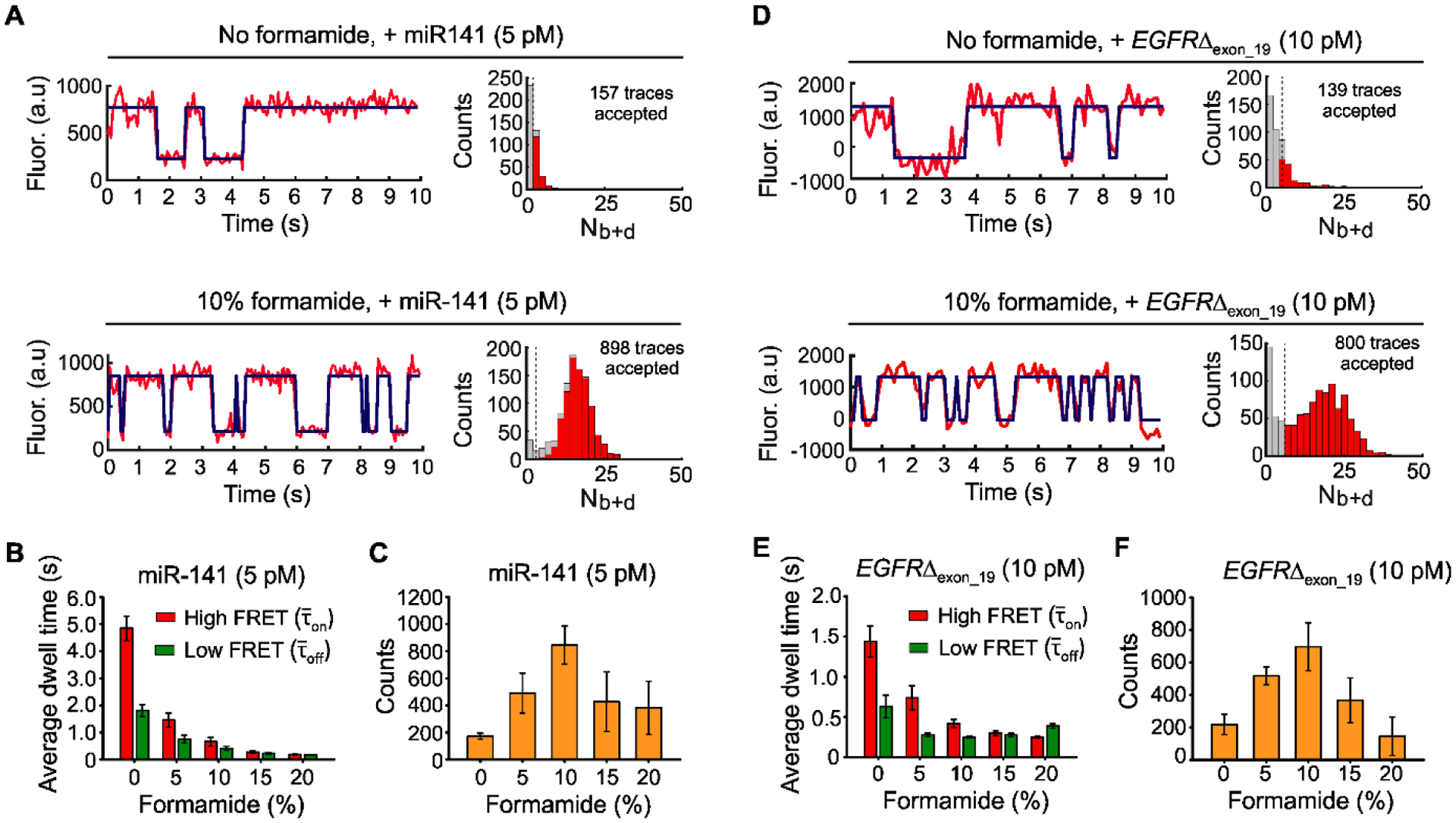
The effects of formamide on iSiMREPS sensors for rapid detection of miRNA and ctDNA. (A) Representative single- molecule kinetic fingerprints and histograms of the number of candidate molecules per FOV showing a given number of binding and dissociation events (Nb+d) after applying thresholds for FRET intensity, S/N, and dwell times of bound and unbound states in presence of 5 pM miR-141, without (top) and with 10% (v/v) formamide (bottom). The Q8C6QS18CS3 sensor as depicted in Figure 2A was used for this study and pre-treated with a capture invader (5´TCCGCCATATAACACTGTCTG 3´) and query invader (5´GAGTGTCCCGCGGCCCAGGA 3´) to remove non-target-bound sensors from coverslip before imaging under an objective-TIRF microscope. (B) The average dwell times for miR-141 bound state (high-FRET and non-bound state (low-FRET) as a function of formamide (0–20%, v/v). (C) The number of candidate miR-141 bound molecules per FOV as a function of formamide after applying an optimized kinetic parameter (see SI, and Table S6). (D) Representative single- molecule kinetic fingerprints and Nb+d histograms per FOV in presence of 10 pM EGFRΔexon_19 without (top) and with 10% formamide (bottom). Q8C6QS18CS19 sensor and invaders as depicted in Figure 3A were used for this study. (E) The and for EGFRΔexon_19 as a function of formamide (0–20%, v/v). (F) The number of candidate EGFRΔexon_19 bound molecules per FOV as a function of formamide after applying optimized kinetic parameters (see SI, and Table S7). All data are processed at a standard data acquisition of 10s. All data are presented as mean ± s.d., where n ⩾ 3 independent experiments.
Next, we varied the formamide volume fraction from 0% to 20% (v/v) to minimize data acquisition time while retaining sensor function and high sensitivity. The single molecule kinetic traces showed that the τon and τoff decreased with increasing formamide (Figs. S6A and S7A). The and gradually decreased with increasing formamide from 0–10% for both targets but stayed roughly constant from 10–20% for EGFRΔexon_19 and 15–20% for miR-141 (Figs. S8, S9, 4B and 4E). Specifically, shifting from 0% formamide to 10% formamide decreased and by factors of 7 and 4.5 respectively, for miR-141 (Fig. 4B) and 3.5 and 2.5, respectively, for EGFRΔexon_19 (Fig. 4E). The differences between the two sensors are consistent with the fact that DNA-RNA duplexes are more sensitive to destabilization by formamide than DNA-DNA duplexes (Casey and Davidson, 1977). The target bound signals separated well from background at ⩾10% formamide and poorly or inconsistently at 0 and 5 % formamide (Figs. S6B and S7B). The standard acquisition of ~ 10 s per FOV obtained in iSiMREPS as assisted by 10% formamide is approximately 60-times faster than intermolecular SiMREPS approaches (Hayward et al., 2018; Johnson-Buck et al., 2015).
We next evaluated the formamide dependence of sensitivity. For miR-141 and EGFRΔexon_19, the number of accepted traces per FOV (a measure of assay sensitivity) increased with increasing formamide up to 10%, then decreased at higher formamide concentrations (Fig. 4C and 4F). The accepted counts for 0 and 5% formamide likely underrepresented the number of true molecules because many target-bound sensors could not be effectively differentiated from the background in the 10 s data acquisition period (Figs. S6B and S7B). The lower number of counts observed in 15 and 20% formamide likely occurred because the reduced stability of the duplexes at these percentages decreases S/N and shortens some events to below the camera exposure time (Figs. S6C, S6D, S7C and S7D). We thus used 10% formamide during subsequent sensor optimization for maximizing sensitivity and specificity.
3.6. Use of invaders to increase the sensitivity of iSiMREPS
Our previous experiments showed that use of TMSD invaders significantly reduced background and improved S/N in single-molecule kinetic traces (Fig. 3). We next quantified the effect invaders have on the number of accepted counts per FOV. Firstly, we performed five experiments with the optimized EGFRΔexon_19 sensor, each pairing the identical query invader (QI) with one of five different capture invaders (CIs) shown in Figure 5A. These CIs have different toehold and pairing region lengths. Some contain mismatches to the spacer region of the CP, which are intended to mitigate undesired displacement of target-bound sensors. We also performed a control experiment without invaders. These experiments showed that all five CIs increase the number of detected counts per FOV and decrease the number of false positives in a control without mutant DNA (Fig. 5B). However, treatment with CIs that contain one or more mismatches (CI17, CI18, and CI21) with the capture probe’s 3 nt spacer showed more accepted traces and, surprisingly, improved S/N compared to treatment with fully complementary CIs (CI20 and CI15) (Figs. 5B, 5C, and S10). These results suggest that fully complementary CIs cause unwanted removal of target-bound probes. Overall, treatment with CI17 and QI performed the best, increasing the number of accepted traces ~4.5-fold compared to assays without invaders (Figs. 5B and 5C). This strategy was also tested and optimized for the detection of miR-141 (Fig. S11); the best-performing CI had a mismatch and exhibited a ~3.5-fold increase in accepted traces and improved S/N (Fig. S11).
Fig. 5.
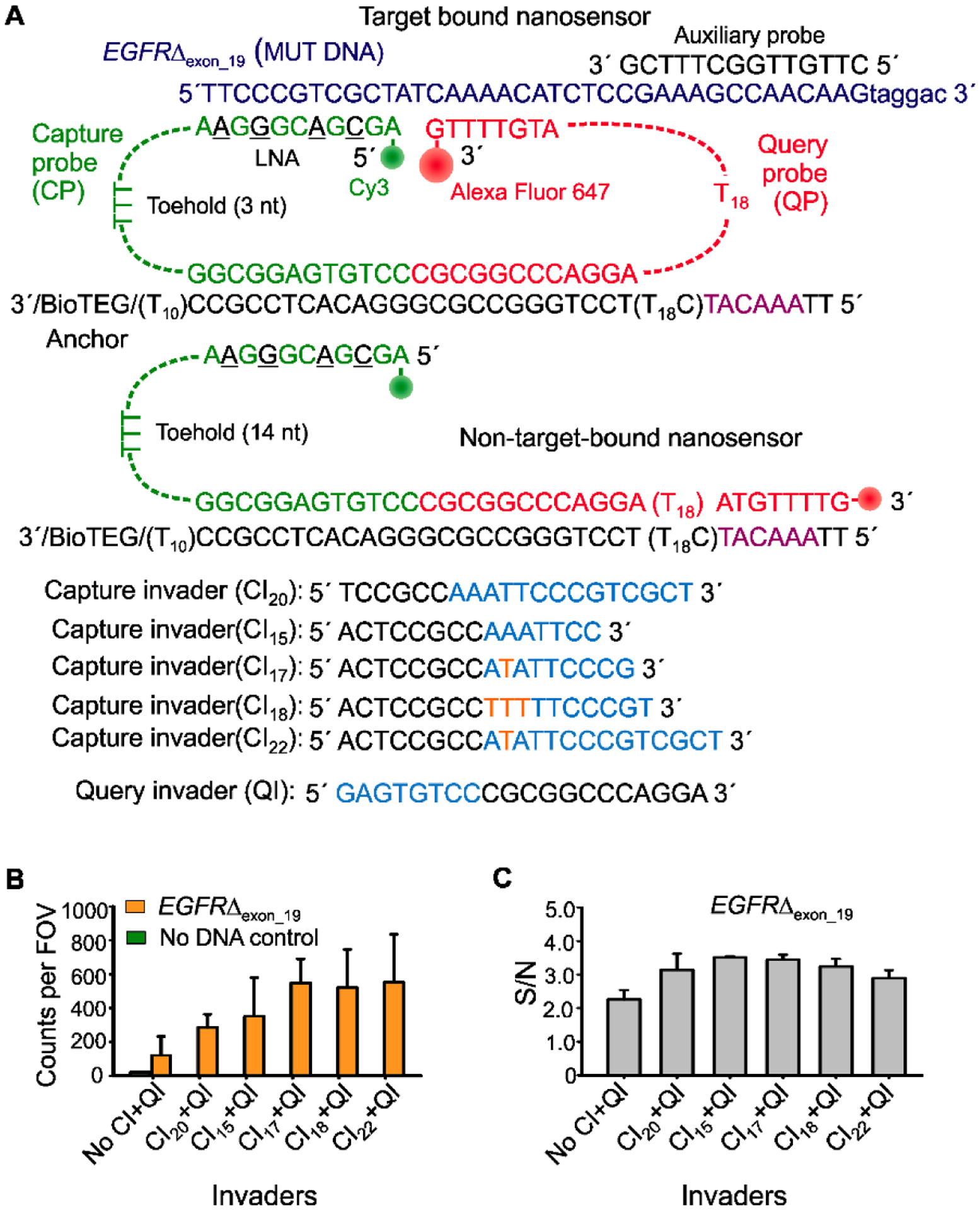
Optimization of the invaders for increased sensitivity of iSiMREPS assays for nucleic acids. (A) Schematic of target bound and non-target-bound iSiMREPS sensors, depicting the toehold available for invader binding as well as capture invaders (CIs) of variable lengths. Cyan segments of the invaders are complementary to the exposed toeholds, while orange sequences represent the nucleotides that are mismatched between invaders and toeholds in the CP. (B) Number of accepted counts per FOV in the presence of EGFRΔexon_19 after application of different CIs. (C) S/N ratio in the candidate target bound molecules after application of different CLs. All data are presented as the mean ± s.d. of n = independent experiments.
3.7. Sensitivity and specificity of detecting EGFR exon 19 deletion mutation DNA and miR-141
To further improve EGFRΔexon_19 detection sensitivity, we next optimized iSiMREPS preparation procedures and assay conditions (e.g., sensor concentration, invaders, target incubation time) (Fig. S12). Since EGFRΔexon_19 exists in double-stranded (ds) DNA form in biofluids, the target was thermally denatured at 90°C for 3 min and cooled at room temperature in the presence of an auxiliary probe that binds stably to the forward strand of mutant DNA (Fig. 3A). During this step, the poly-dT (dT30) was included in high molar excess as a carrier (Hayward et al., 2018). The auxiliary probe and dT30 help keep the capture region of the target DNA in an ssDNA form, permitting efficient and specific capture. Experiments using mutant ssDNA and dsDNA treated with the above denaturation steps showed similar results (Fig. 6A). The iSiMREPS assay for the EGFRΔexon_19 dsDNA was found to have a LOD of 3.2 fM in buffer (Fig. 6B) and a linear dynamic range spanning ~4 orders of magnitude (Fig. S13A), a ~1.5-fold improvement over conventional SiMREPS (Hayward et al., 2018). EGFRΔexon_19 detection in the presence and absence of a large (105–6-fold) excess of wild-type DNA showed 99.9996–99.9999% specificity for, permitting mutant detection at an allelic fraction of 0.001–0.0001% (Figs. 6C, S14 and Table S8). The miR-141 assay exhibited an LOD of 3.4 fM (Fig. 6D) with a dynamic range of approximately 3.2 orders of magnitude (Fig. S13B), a 1.2-fold improvement over conventional SiMREPS (Johnson-Buck et al., 2015),with similar sensitivity in one 60th of the time. Overall, iSiMREPS shows slightly lower sensitivity than some existing technologies for detecting nucleic acids like droplet digital PCR (Mestdagh et al., 2014; Milbury et al., 2014) and NGS (Postel et al., 2018). However, it exhibits superior specificity, comparable or better dynamic range than existing single-nucleic acid detection techniques (Gilboa et al., 2020) and achieves rapid detection with lower risk of cross-contamination, an elimination of sequence bias or inhibition due to enzymatic amplification, and a lack of need for purification, complex sample preparations or enzymatic reactions compared to existing techniques.
Fig. 6.
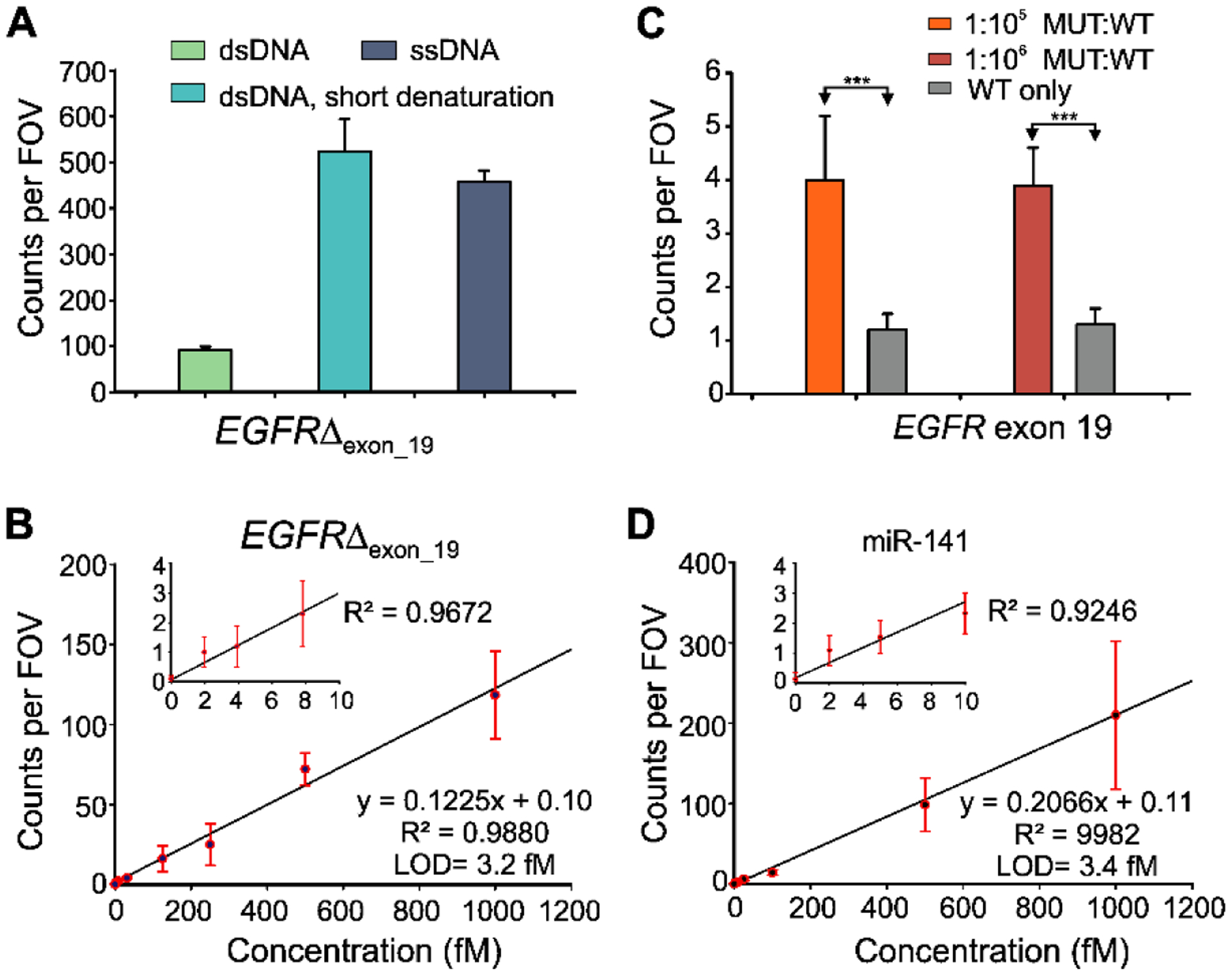
Standard curve and specificity of detecting EGFR exon 19 deletion (EGFRΔexon_19) and miR-141. (A) Effect of short thermal denaturation on the accepted counts of EGFRΔexon_19 duplex DNA (dsDNA). (B) Standard curve for EGFRΔexon_19 showing a LOD of 3.2 fM. Linear fits were constrained to a y-intercept of accepted counts at 0 fM. (C) Comparison of counts from low MUT allelic fraction and WT only conditions for determining specificity. Triple asterisks indicate the significant differences at 95% confidence levels as assessed using a two-tailed, unpaired t test and showed a specificity of 99.9996–99.9999% over the MUT fraction of 0.001–0.0001%. (D) Standard curve for miR-141 showing a LOD of approximately 3.4 fM. Linear fits were constrained to a y-intercept of accepted counts at 0 fM. All data are presented as mean ± s.d., where n ⩾ 3 independent experiments.
4. Conclusions
In this study, we developed a novel intramolecular smFRET-based kinetic fingerprinting technique (iSiMREPS) using a dynamic DNA nanosensor that enables rapid, highly sensitive and ultraspecific detection of diverse nucleic acid biomarkers. iSiMREPS’s intramolecular sensor design allows for accelerated kinetic fingerprinting by dramatically increasing the local effective concentration of probes and target. We optimized functional features of iSiMREPS such as the hybridization length of the query probe to the target and competitor, as well the length of the spacer in the query probe, to obtain sensor designs capable of quick fingerprint generation. We then demonstrated that the denaturant formamide further accelerates probe-target kinetics and, because iSiMREPS’s unique features permit sensors to retain high-affinity capture of single oligonucleotides under such conditions, fingerprint generation was achieved in 10 s per FOV. Moreover, the sensor design was amenable to a TMSD strategy that removed non-target bound sensors, nearly eliminating background noise. Yet, despite its several advances iSiMREPS is susceptible to fluorophore photobleaching and its sensitivity is, as with all surface-based assays, limited by the diffusion of the target molecules to the surface. In the future, sensitivity may be further improved by increasing surface concentration with a pre-concentration technique or by recording additional FOVs, a prospect possible because of the short acquisition times. Additionally, faster cameras with higher laser intensities or sensor designs that withstand higher formamide concentrations and/or temperatures may permit even faster detection speeds. Overall, iSiMREPS has demonstrated the analytical performance necessary for advanced applications in the liquid biopsy field with greater speed and potential for higher sample throughput. We anticipate that iSiMREPS can be further generalized for the rapid, highly sensitive and specific analysis of diverse analytes including nucleic acids, proteins, and small molecules in a spatially addressable microarray format.
Supplementary Material
Amplification-free single-molecule kinetic fingerprinting identifies diverse disease biomarkers
A dynamic DNA nanosensor generates kinetic fingerprints via intramolecular single-molecule FRET
iSiMREPS in 10 s detects biomarkers with a limit of detection of ~3 fM in 1,000,000-fold WT excess
Funding sources
This work was supported by National Institute of Health (NIH) [R21 CA204560 and R33 CA229023 to N.G.W. and M.T.]
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of competing interest
The University of Michigan has filed patent applications related to the SiMREPS technique on which M.T., A.J.-B. and N.G.W. are co-inventors. M.T., A.J.-B. and N.G.W. are co-founders of a startup company, aLight Sciences Inc., which seeks to commercialize this technology. A.J.-B. is an employee of aLight Sciences Inc. The remaining authors declare no competing interests.
Data availability
The data discussed in this publication will be made available upon request.
Appendix A. Supplementary data
Supplementary information includes Preparation of slides, coverslips, and sample cells, Prism-type TIRF iSiMREPS assay for miR-141, Cumulative frequency exponential fitting, Statistical mechanical simulations of sensors, Optimization of iSiMREPS assay conditions, Calculation of specificity of mutant DNA. Supplementary table S1–S8. Supplementary figures S1–S15.
References
- Akkilic N, Geschwindner S, Höök F, 2020. Biosens. Bioelectron 151, 9. [DOI] [PubMed] [Google Scholar]
- Anfossi S, Babayan A, Pantel K, Calin GA, 2018. Nat. Rev. Clin. Oncol 15, 541–563. [DOI] [PubMed] [Google Scholar]
- Blake RD, Delcourt SG, 1996. Nucleic Acids Res 24, 2095–2103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bronson JE, Fei J, Hofman JM, Gonzalez RL Jr., Wiggins CH, 2009. Biophys. J 97, 3196–3205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant RJ, Pawlowski T, Catto JW, Marsden G, Vessella RL, Rhees B, Kuslich C, Visakorpi T, Hamdy FC, 2012. Br. J. Cancer 106, 768–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caltech:, 2007.
- Casey J, Davidson N, 1977. Nucleic Acids Res 4, 1539–1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen L, Hartman MR, Amardey-Wellington A, Walt DR, 2017. Nucleic Acids Res 45, e137–e137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowley E, Di Nicolantonio F, Loupakis F, Bardelli A, 2013. Nat. Rev. Clin. Oncol 10, 472–484. [DOI] [PubMed] [Google Scholar]
- Dunn KE, Trefzer MA, Johnson S, Tyrrell AM, 2016. Sci. Rep 6, 29581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan G, Zhang K, Ding J, Li J, 2017. Oncotarget 8, 33922–33932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gazdar AF, 2009. Oncogene 1, 198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilboa T, Garden PM, Cohen L, 2020. Anal. Chim. Acta 1115, 61–85. [DOI] [PubMed] [Google Scholar]
- Hayward SL, Lund PE, Kang Q, Johnson-Buck A, Tewari M, Walter NG, 2018. J. Am. Chem. Soc 140, 11755–11762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heitzer E, Ulz P, Geigl JB, 2015. Clin. Chem 61, 112–123. [DOI] [PubMed] [Google Scholar]
- Johnson-Buck A, Li J, Tewari M, Walter NG, 2019. Methods 153, 3–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson-Buck A, Su X, Giraldez MD, Zhao M, Tewari M, Walter NG, 2015. Nat. Biotech 33, 730–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F, Tang Y, Traynor SM, Li X-F, Le XC, 2016. Anal. Chem 88, 8152–8157. [DOI] [PubMed] [Google Scholar]
- Li Z, Zhou X, Li L, Liu S, Wang C, Yu C, Su X, 2018. Anal. Chem 90, 6804–6810. [DOI] [PubMed] [Google Scholar]
- Ma F, Li Y, Tang B, Zhang C. y., 2016. Acc. Chem. Res 49, 1722–1730. [DOI] [PubMed] [Google Scholar]
- Mestdagh P, Hartmann N, Baeriswyl L, Andreasen D, Bernard N, Chen C, Cheo D, D’Andrade P, DeMayo M, Dennis L, Derveaux S, Feng Y, Fulmer-Smentek S, Gerstmayer B, Gouffon J, Grimley C, Lader E, Lee KY, Luo S, Mouritzen P, Narayanan A, Patel S, Peiffer S, Rüberg S, Schroth G, Schuster D, Shaffer JM, Shelton EJ, Silveria S, Ulmanella U, Veeramachaneni V, Staedtler F, Peters T, Guettouche T, Wong L, Vandesompele J, 2014. Nat. Methods 11, 809–815. [DOI] [PubMed] [Google Scholar]
- Milbury CA, Zhong Q, Lin J, Williams M, Olson J, Link DR, Hutchison B, 2014. Biomol. Detect. Quantif 1, 8–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell PS, Parkin RK, Kroh EM, Fritz BR, Wyman SK, Pogosova-Agadjanyan EL, Peterson A, Noteboom J, O’Briant KC, Allen A, Lin DW, Urban N, Drescher CW, Knudsen BS, Stirewalt DL, Gentleman R, Vessella RL, Nelson PS, Martin DB, Tewari M, 2008. Proc. Natl. Acad. Sci. U.S.A 105, 10513–10518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy MC, Rasnik I, Cheng W, Lohman TM, Ha T, 2004. Biophys. J 86, 2530–2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polizzi NF, Therien MJ, Beratan DN, 2016. Isr. J. Chem 56, 816–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Postel M, Roosen A, Laurent-Puig P, Taly V, Wang-Renault SF, 2018. Expert. Rev. Mol. Diagn 18, 7–17. [DOI] [PubMed] [Google Scholar]
- Potapov V, Ong JL, 2017. PLOS ONE 12, e0169774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt MW, Kennedy SR, Salk JJ, Fox EJ, Hiatt JB, Loeb LA, 2012. Proc. Natl. Acad. Sci. U.S.A 109, 14508–14513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarzenbach H, Hoon DS, Pantel K, 2011. Nat. Rev. Cancer 11, 426–437. [DOI] [PubMed] [Google Scholar]
- Wang JS, Zhang DY, 2015. Nat. Chem 7, 545–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zadeh JN, Steenberg CD, Bois JS, Wolfe BR, Pierce MB, Khan AR, Dirks RM, Pierce NA, 2011. J. Comput. Chem 32, 170–173. [DOI] [PubMed] [Google Scholar]
- Zhang DY, Chen SX, Yin P, 2012. Nat. Chem 4, 208–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang DY, Seelig G, 2011. Nat. Chem 3, 103–113. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.