

Abstract
Objective –
Niacin therapy fails to reduce cardiovascular events in statin-treated subjects even though it increases plasma HDL cholesterol (HDL-C) and decreases LDL-C and triglyceride levels. To investigate potential mechanisms for this lack of cardioprotection, we quantified the HDL proteome of subjects in two niacin clinical trials: the Carotid Plaque Composition (CPC) study and the HDL Proteomics substudy of the Atherothrombosis Intervention in Metabolic Syndrome with Low HDL/High Triglycerides (AIM-HIGH) trial.
Approach and Results –
Using targeted proteomics, we quantified levels of 31 HDL proteins from 124 CPC subjects and 120 AIM-HIGH subjects. The samples were obtained at baseline and after 1 year of statin monotherapy or niacin-statin combination therapy. Compared with statin monotherapy, niacin-statin combination therapy did not reduce HDL-associated apolipoproteins APOC1, APOC2, APOC3 and APOC4, despite significantly lowering triglycerides. In contrast, niacin markedly elevated HDL-associated phospholipid transfer protein (PLTP), clusterin (CLU), and haptoglobin/haptoglobin related proteins (HP/HPR) (P≤0.0001 for each) in both the CPC and AIM-HIGH cohorts.
Conclusions –
The addition of niacin to statin therapy resulted in elevated levels of multiple HDL proteins linked to increased atherosclerotic risk, which might have compromised the cardioprotective effects associated with higher HDL-C levels and lower levels of LDL-C and triglycerides.
Keywords: atherosclerosis, HDL proteome, cardioprotection, lipid metabolism
Graphical Abstract
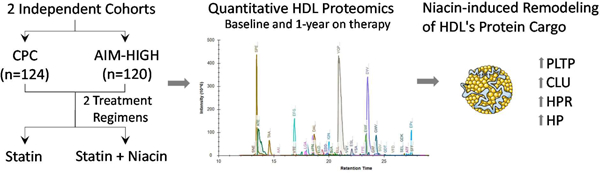
Introduction
Pharmacological elevation of HDL cholesterol (HDL-C) with niacin or cholesteryl ester transfer protein (CETP) inhibitors in combination with a statin have failed to reduce cardiovascular diseases (CVD) in randomized clinical trials.1–3 These observations have been widely interpreted to indicate that HDL is not in the causal pathway for atherosclerosis in humans, at least in statin-treated subjects. However, both niacin and CETP inhibitors also reduce LDL-C in statin-treated subjects; niacin also markedly reduces apolipoprotein B (APOB) and triglyceride levels. Thus, it remains unclear why these interventions failed to lower CVD risk. Indeed, in the only study of an HDL-C targeted drug (anacetrapib) that demonstrated cardioprotection, the effect was attributed to the reduction in LDL-C rather than HDL-C elevation.4 Therefore, it is critical to identify metrics distinct from HDL-C that define HDL’s proposed roles in cardioprotection.5
Two such proposed metrics are HDL’s cholesterol efflux capacity (CEC) and the concentration of HDL particles, termed HDL particle number (HDL-P). We previously showed that statin monotherapy modestly increased HDL-C but failed to increase HDL-P (as quantified by calibrated ion mobility analysis) or HDL CEC.6 Niacin and statin combination therapy markedly increased HDL-C but only modestly increased HDL-P and macrophage CEC. Moreover, combination therapy failed to improve ABCA1-specific CEC, the key first step in reverse cholesterol transport from macrophages.6 These observations support the proposal that HDL-C is a poor index of HDL-P and CEC.
Using shotgun proteomics, we demonstrated that HDL carries a family of 48 proteins implicated in lipid metabolism, inflammation, complement regulation, and regulation of proteolysis.7 Subsequent studies from multiple laboratories have demonstrated that over 90 proteins are reliably detected in HDL8 and that alterations in HDL protein levels may be linked to HDL’s cardioprotective effects.9, 10 We recently employed two targeted MS/MS methods—selected reaction monitoring and parallel reaction monitoring—to quantify HDL’s protein cargo.11, 12 Targeted proteomic approaches are suitable for quantification in a clinical setting and, when used with appropriate controls, are specific, reliable, and reproducible.13
In the current study, we examine the impact of statin monotherapy and niacin-statin combination therapy on HDL-C, LDL-C, triglycerides, and the HDL proteome. First, we used parallel reaction monitoring on HDL isolated from subjects enrolled in the Carotid Plaque Composition (CPC) study to identify candidate proteins altered by the lipid-lowering interventions. Second, we confirmed our observations by using selected reaction monitoring on HDL isolated from subjects enrolled in the Atherothrombosis Intervention in Metabolic Syndrome with Low HDL/High Triglycerides (AIM-HIGH) study. We show that adding niacin to statin therapy increases the abundance of proteins linked to atherogenesis, suggesting a possible explanation for why niacin therapy fails to provide cardioprotection.
Methods
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Study design
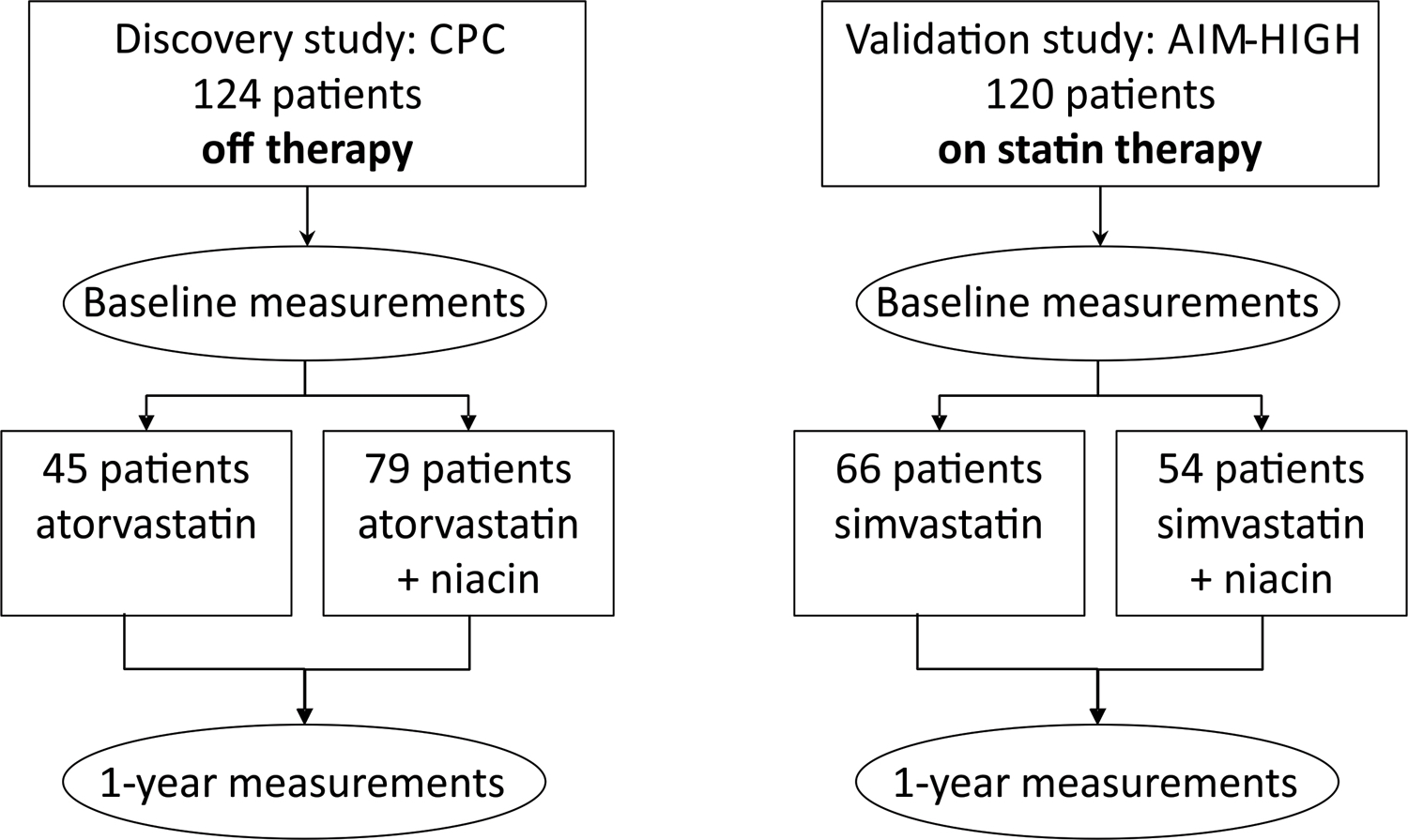
We quantified HDL protein levels in two cohorts, a discovery and a validation cohort. The discovery cohort was a subset of 124 subjects from the CPC study.14 Entry criteria for the CPC study were (i) established cardiovascular disease (defined as >50% stenosis or three 30% obstructive coronary lesions on coronary angiography, post myocardial infarction (MI), post coronary artery bypass graft, or carotid stenosis >15% by ultrasound) and (ii) APOB level ≥120 mg/dL. Male subjects were <67 years old; females were <70 years, and all subjects were lipid-lowering therapy naïve. All the subjects that were eligible for the CPC study, signed a consent and were statin-naïve were include in this work. Subjects were randomized to (i) atorvastatin (10–80 mg/day, n=45), (ii) atorvastatin and extended-release niacin (2 g/d, n=36), (iii) atorvastatin, niacin, and colesevelam (3.8 g/d, n=43). HDL-C levels in subjects with normal triglyceride levels are little affected by colesevelam, a bile acid-binding resin.15
Because we compared treatment-induced changes in HDL-C with other HDL metrics, and because all subjects in our study had triglyceride levels<230 mg/dL on entry into the trial, we pre-specified that subjects should be divided into two groups for analysis: statin treatment alone (monotherapy, n=45) or niacin-statin treatment (combining the statin/niacin group with the statin/niacin/colesevelam group, total n=79). Subgroup analysis confirmed that colesevelam treatment did not significantly affect the impact of niacin therapy on HDL-C.
The validation cohort was a subset of subjects from the HDL Proteomics substudy of AIM-HIGH.3 Subjects that completed at least 2 MRI examinations and had blood samples available at baseline and at 1 year were included in this work. Eligible subjects were 45 years of age or older and had established cardiovascular disease, which was defined as clinically documented stable coronary heart disease, cerebrovascular or carotid disease, or peripheral arterial disease. All subjects in AIM-HIGH had “atherogenic dyslipidemia” defined as: (i) HDL-C<40 mg/dL for men; <50 mg/dL for women; (ii) elevated triglyceride levels of 150 to 400 mg/dL; (iii) LDL-C levels <180 mg/dL in subjects who were not taking a statin at entry. More than 90% of the AIM-HIGH subjects were on statin therapy on entry into the trial. After a run-in period, the subjects were randomized into two treatment groups. Patients were randomized into the statin only group (simvastatin plus a placebo that contained a small dose (50 mg) of immediate-release niacin in each 0.5 g or 1 g tablet, blind the subjects and study personnel) and the niacin-statin combination group (niacin at a dose of 1.5 to 2 g per day plus simvastatin). In both groups, the dose of simvastatin was adjusted according to an algorithm specified in the protocol to achieve and maintain LDL-C between 40 and 80 mg/dL during treatment. Subjects in both groups could receive ezetimibe, at a dose of 10 mg per day, to achieve the target LDL-C levels. In both cohorts, venous blood was collected at entry into the trial and after 1 year on therapy. Freshly prepared EDTA plasma was stored at −80°C until analysis.
Importantly, the CPC subjects were not using any lipid-lowering drugs on entry into the trial. In contrast, all subjects in the AIM-HIGH were on statin monotherapy at entry.
Laboratory tests.
Fasting plasma concentrations of triglycerides, total cholesterol, HDL-C, LDL-C, apolipoprotein A-I (APOA1), and APOB were measured at Northwest Lipid Research Laboratories using validated biochemical and immunological methods.
Protein nomenclature.
Protein symbols (all capital letters, not italicized) are based on gene symbols as recommended by the Human Genome Organization (HUGO) Gene Nomenclature Committee (HGNC).16
HDL isolation.
Plasma was quickly thawed at 37°C and subjected to sequential ultracentrifugation to isolate HDL (density 1.063–1.210 g/mL) as previously described.17 Total protein concentration in HDL was measured using the Bradford assay, with albumin as the standard. Quality control plasma was included randomly in each isolation rotor to control for pre-analytical variability.
Sample workup.
For both the CPC and AIM-HIGH studies, samples from baseline and 1-year on therapy were randomized without discrimination regarding pre or post-treatment and processed at the same time. Ultracentrifugation was performed in batches. A control of pooled plasma from apparently healthy volunteers was included in the analyses to confirm that we reliably detected and quantified the proteins selected for quantification. Randomization was continued for protein digestion and MS/MS analyses.
HDL proteolytic digestion.
Isolated HDL (10 μg protein) was diluted with 0.2% RapiGest (Waters, Milford, MA) in 100 mM ammonium bicarbonate, reduced with dithiothreitol, alkylated with iodoacetamide, and digested with two additions of trypsin (1:20, w/w HDL protein; Promega, Madison, WI) for 4 h at 37°C and a second addition (1:20, w/w HDL protein) for overnight incubation at 37°C. After acidic hydrolysis of the RapiGest with 0.5% trifluoroacetic acid, samples were dried and stored at −20°C until mass spectrometric (MS) analysis. Digestion was performed in 96-well plates.
Discovery study: targeted mass spectrometric analyses of CPC cohort.
Digested HDL proteins were quantified by parallel reaction monitoring, a powerful approach for unbiased analysis of complex protein complexes.11, 12, 18 Details are provided in Supplemental Material.
Validation study: targeted mass spectrometric analyses of AIM-HIGH subjects.
Digested HDL proteins were quantified by selected reaction monitoring, which has been demonstrated to quantify precisely the HDL proteome.19 We previously showed that using parallel reaction monitoring or selected reaction monitoring to quantify the HDL proteome yields very similar results.11 Details are provided in Supplemental Material.
Statistical analyses.
Categorical variables are presented as percentages, and continuous variables as means and standard deviations (baseline characteristics) or as medians and interquartile ranges for proteins and other variables with skewed distributions (triglycerides).
For each treatment, differences in protein amounts between baseline and on-treatment were initially evaluated by the Wilcoxon signed rank test. To account for multiple comparison testing, P values obtained from the Wilcoxon signed rank test were corrected using the method of Benjamini and Hochberg.20–22 This step-up method controls the false discovery rate (FDR) and assumes a non-negative correlation. A corrected P value threshold is calculated based on a false discovery rate (FDR) of 5%, and only proteins above the corrected P value are considered significantly different.21, 22 To compare changes in the amounts of HDL protein caused by combination therapy (niacin plus statin) and statin monotherapy, we applied the Wilcoxon rank-sum test to the absolute changes from baseline to on-treatment values and used Benjamini and Hochberg correction for multiple comparisons20–22 as described above. Analyses were performed with STATA software, version 13.
Results
Study design, baseline characteristics, and workflow.
We tested the hypothesis that statins and niacin exert distinct effects on HDL’s proteome by analyzing HDL from two independent populations: CPC and AIM-HIGH subjects (Fig. 1). HDL was isolated from freshly thawed plasma (stored at −80°C after collection) by ultracentrifugation and its protein content digested with trypsin. Data from previous studies11, 12, 18 were used to select 31 HDL proteins for relative quantification. Two to five peptides from each targeted protein were then measured by tandem mass spectrometry. All statistical analyses were controlled for multiple comparisons.
Figure 1.
Workflow.
Baseline characteristics of the study participants were reported previously.3, 6 For the CPC study, we analyzed HDL isolated from i) 45 subjects assigned to monotherapy with atorvastatin, and ii) 79 subjects assigned to combination therapy (atorvastatin and niacin, ± colesevelam) (Table 1 and Fig. 1). For the AIM-HIGH study, we analyzed HDL from i) 66 subjects randomized to simvastatin, and ii) 54 subjects assigned to simvastatin plus niacin (Table 1 and Fig. 1).
Table 1.
Baseline characteristics of CPC and AIM-HIGH subjects.
| CPC | AIM-HIGH | |||||
|---|---|---|---|---|---|---|
| Study treatment | Atorvastatin | Atorvastatin + Niacin | P value | Simvastatin | Simvastatin + Niacin | P value |
| N | 45 | 79 | 66 | 54 | ||
| Age, y | 55±9 | 55±8 | 0.84 | 61±9 | 60±8 | 0.62 |
| Sex, % female | 38 | 31 | 0.49 | 25 | 17 | 0.31 |
| Cholesterol, mg/dL | 235±38 | 244±43 | 0.27 | 149±35 | 143±27 | 0.32 |
| Triglycerides*, mg/dL | 170 (102; 221) | 151 (114; 228) | 0.76 | 145 (128; 162) | 143 (124; 206) | 0.25 |
| LDL-C, mg/dL | 161±33 | 168±43 | 0.40 | 79±30 | 75±22 | 0.37 |
| HDL-C, mg/dL | 43±14 | 41±11 | 0.40 | 34±6 | 34±6 | 0.44 |
| Glucose, mg/dL | 102±17 | 98±14 | 0.20 | 106±2 | 106±2 | 0.88 |
Values are means and standard deviations (unpaired t-test) or * medians and interquartile ranges (Wilcoxon rank-sum test).
In both analyses, the subjects on monotherapy or combination therapy had similar ages, percentages of female subjects, smoking status, and lipid levels (Table 1). On entry into the studies, patients enrolled in AIM-HIGH were on statin therapy, while CPC subjects were not taking any cholesterol-lowering medication. This likely explains why the AIM-HIGH subjects had lower levels of total and LDL-C cholesterol than the CPC subjects.
Changes in lipid and glucose levels after therapy.
After 1 year on therapy, the median reduction in LDL-C levels was 42% in the CPC subjects on statin monotherapy (−66 mg/dL, P<0.0001) compared with 51% (−85 mg/dL, P<0.0001) in the subjects on combination therapy (Table 2). The degree of reduction was significantly greater in the subjects treated with the combination therapy (P=0.0015). LDL-C levels in the AIM-HIGH subjects, who were taking a statin when they entered the trial, did not differ significantly after 1 year on either therapy.
Table 2.
Median percentage changes in lipids and glucose after 1 year of statin or niacin-statin therapy in CPC and AIM-HIGH subjects.
| Study | Variable | Statin (%) | P Value | Statin + Niacin (%) | P Value | Statin vs. Statin + Niacin P Value |
|---|---|---|---|---|---|---|
| CPC | Triglycerides | −27 (−45; −4) | 0.0004 | −46 (−59; −24) | <0.0001 | 0.0015 |
| LDL-C | −42 (48; −30) | <0.0001 | −51 (−60; −41) | <0.0001 | 0.0016 | |
| HDL-C | 10 (0; 20) | 0.0001 | 26 (16; 43) | <0.0001 | <0.0001 | |
| Glucose | 2 (4; 8) | 0.334 | 6 (−1; 11) | 0.0001 | 0.0887 | |
| AIM-HIGH | Triglycerides | −6 (−27; 17) | 0.423 | −23 (−43; −5) | 0.0001 | 0.0023 |
| LDL-C | −11 (−22; 13) | 0.0784 | −10 (−27; 13) | 0.106 | 0.996 | |
| HDL-C | 11 (2; 23) | <0.0001 | 21 (11; 36) | <0.0001 | 0.0013 | |
| Glucose | 6 (−3; 10) | 0.00130 | 9 (2; 14) | <0.0001 | 0.103 |
Values are median (interquartile range) of percentage changes from baseline. P values for percentage change after 1 year of therapy are from the Wilcoxon matched-pairs signed rank test. P values for the changes between monotherapy and combination therapy are from the Wilcoxon rank-sum test.
Median triglyceride levels in the CPC subjects were significantly reduced after 1 year on either statin therapy (−27%, −36 mg/dL, P=0.0004) or statin/niacin (−46%, −90 mg/dL, P< 0.0001) (Table 2). The combination therapy lowered triglycerides more effectively than did the monotherapy (P=0.0015). Triglyceride levels in AIM-HIGH subjects did not change significantly on statin therapy, but they were 23% lower (−34 mg/dL, P=0.0023) after 1 year on the combination therapy, therefore combination therapy was more effective than monotherapy in lowering triglycerides (P=0.0023).
Median HDL-C levels increased by 10% in the CPC subjects treated with statin monotherapy (4 mg/dL, P=0.0001), while they increased by 26% in the subjects on combination therapy (12 mg/dL, P<0.0001) (Table 2). The increase in HDL-C levels on combination therapy was significantly greater than on monotherapy (P<0.0001). In AIM-HIGH subjects, monotherapy and the combination therapy produced similar increases in HDL-C: 11% (4 mg/dL, P<0.0001) and 21% (12 mg/dL, P<0.0001), respectively. As in the CPC trial, combination therapy was more effective than monotherapy at raising HDL-C in AIM-HIGH (P=0.0013).
Median plasma glucose levels did not differ significantly in either group after 1 year of statin monotherapy (Table 2). In contrast, they increased by 6% in the CPC subjects (P=0.0001) and 9% in AIM-HIGH subjects (P<0.0001) after 1 year of combination therapy. However, neither increase was significantly higher when comparing with monotherapy (P=0.089 and 0.101, respectively for CPC and AIM-HIGH. Taken together, these observations are consistent with previous studies showing that statin monotherapy lowers LDL-C while combination therapy of statin and niacin further reduces LDL-C while increasing HDL-C and decreasing triglyceride levels.3, 23
Statin monotherapy in the statin-naive subjects in the CPC study reduced apolipoprotein Cs (APOCs) in HDL.
The CPC subjects were not on lipid-lowering therapy when the entered the study. In this cohort, levels of 10 of 31 HDL proteins differed significantly after 1 year of statin monotherapy (Fig. 2). Levels of the apolipoproteins APOC1, APOC2, APOC3, and APOC4 in HDL were reduced by at least 15% (P< 0.0001 for each), as were levels of apolipoprotein E (APOE, 21% reduction, P<0.0001), apolipoprotein M (APOM, −13%, P<0.0001), apolipoprotein L1 (APOL1, −4%, P=0.0006), and cholesterol ester transfer protein (CETP, −23%, P<0.0001). In contrast, only one protein—phospholipid transfer protein (PLTP)—was elevated after 1 year of treatment with atorvastatin (11% increase, P=0.0025). All analyses were corrected for multiple comparisons (FDR 5%, cut-off P=0.004).
Figure 2. Effects of lipid-altering therapies on the HDL proteome in the CPC study.
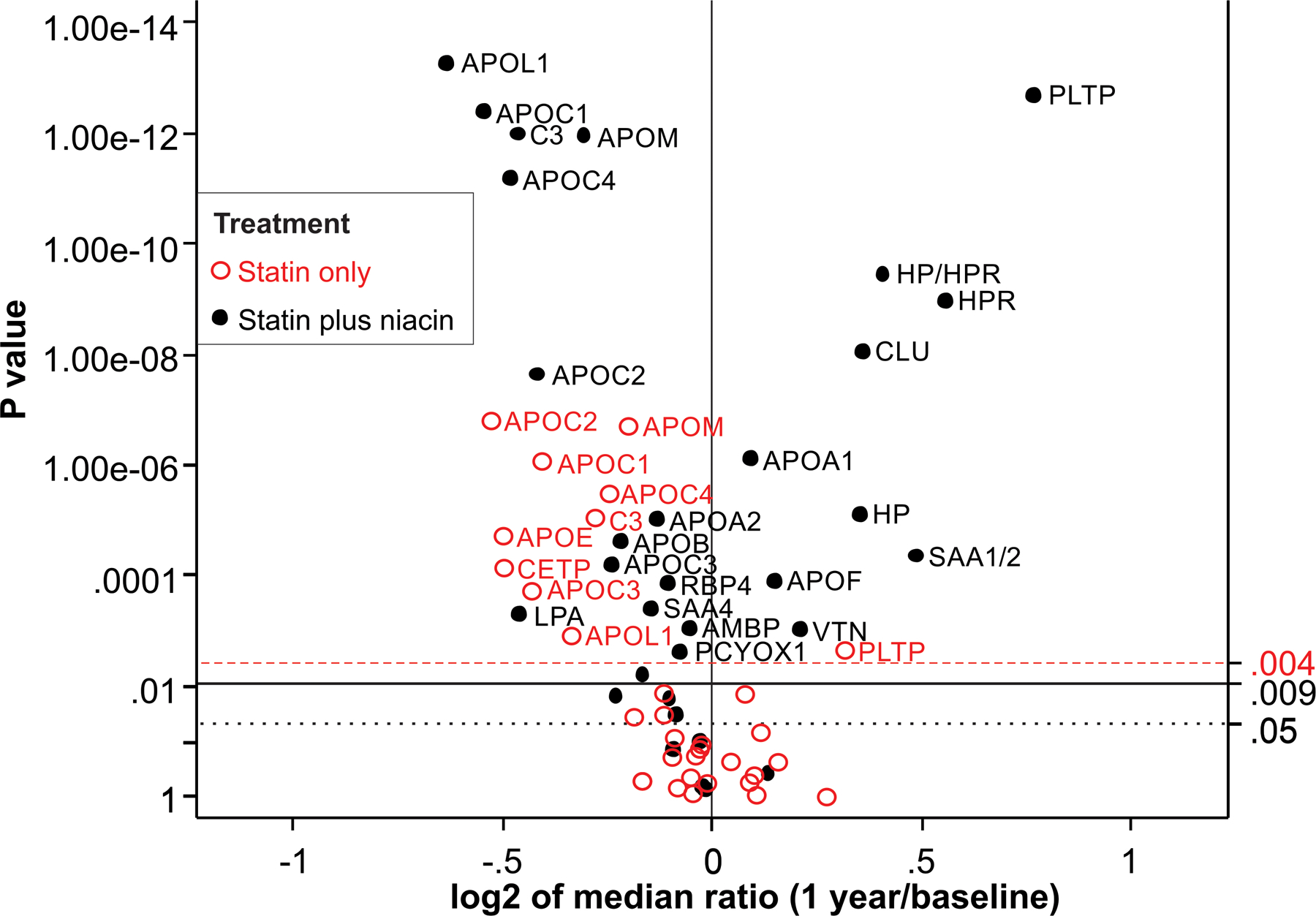
All CPC subjects were off all lipid-lowering drugs when they entered the study. HDL proteins were quantified by parallel reaction monitoring before and after 1 year on statin or niacin-statin therapy. For each protein, the P value from the Wilcoxon signed rank test is plotted against the log2 fold median change between 1-year on therapy and baseline. Proteins overexpressed after 1 year on therapy are displayed to the right of the value 0 on the x-axis, while underexpressed proteins are to the left. After we controlled for multiple comparison testing, only proteins situated above the P value of 0.004 for statin treatment and 0.009 for combination therapy (niacin plus statin) on the y-axis were considered significantly different between 1-year on therapy and baseline. The dashed (red) horizontal line shows the corrected critical P value threshold (P=0.004) for statin treatment. The solid horizontal line shows the corrected overall critical P value threshold (P=0.009) for statin plus niacin treatment. The dotted (black) horizontal line shows the uncorrected overall critical P value of 0.05.
The reduction in APOC1 and APOC3 levels in HDL correlated strongly with the reduction in plasma triglycerides levels (ρ=0.52, P=0.0002 and ρ=0.56, P=0.0001, respectively). Smaller but significant correlations were observed with APOC4 and APOC2 (ρ=0.42, P=0.0044 and ρ=0.29, P=0.05, respectively).
Combination niacin-statin therapy extensively remodels the HDL proteome.
In the CPC study, levels of 22 of 31 measured HDL proteins (71%) differed significantly (after correction for multiple comparisons) between baseline and 1 year of combination therapy (Fig. 2). Combination therapy significantly reduced levels of those proteins reduced by statin monotherapy (all APOCs at least 15% reduction, P<0.0001, APOE, - 11%, P=0.0062, APOM, −19% P<0.0001, and APOL1, −36%, P<0.0001). Although the niacin/statin-treated subjects in the CPC trial had markedly lower triglyceride levels than the subjects treated only with a statin, APOCs levels in HDL were similar in the two groups.
HDL isolated after combination therapy contained lower levels of apolipoprotein (a) (LPA, −23%, P<0.0007) and APOB (−18%, P<0.0001). Lipoprotein (a), which contains LPA covalently bound to APOB, is denser than LDL and can be isolated by ultracentrifugation in the HDL density range. It is known that niacin reduces lipoprotein(a) levels in blood.24
In contrast to monotherapy, which significantly increased the level of only one HDL protein (PLTP), niacin-statin combination therapy elevated levels of 8 HDL proteins (Fig. 2). Five of the 8 proteins increased in relative abundance by more than 25% (PLTP; clusterin [CLU]; haptoglobin [HP], haptoglobin-related protein [HPR], and serum amyloid A1/2 [SAA1/2], P<0.0001 for all). It is important to note that the amino acid sequences of HP and HPR are 91% homologous.25 For quantification of those proteins, we measured levels of one peptide found in both HP and HPR (termed HP/HPR, 32% increase) along with one peptide unique to HP (27% increase) and one unique to HPR (47% increase; details in Supplemental Methods).
We next determined which changes in HDL protein levels differed between treatment groups after 1 year of monotherapy or combination therapy (Fig. 3). Compared to statin monotherapy niacin-statin treatment increased levels of PLTP, CLU, HP/HPR, and APOF (all P<0.0001) in HDL. In contrast, levels of APOL1 (P<0.0001), complement component 3 (C3, P=0.003), and LPA (P=0.001) were decreased by niacin-statin treatment.
Figure 3. Changes in HDL protein amounts after 1-year on statin or niacin-statin therapy for CPC study subjects.
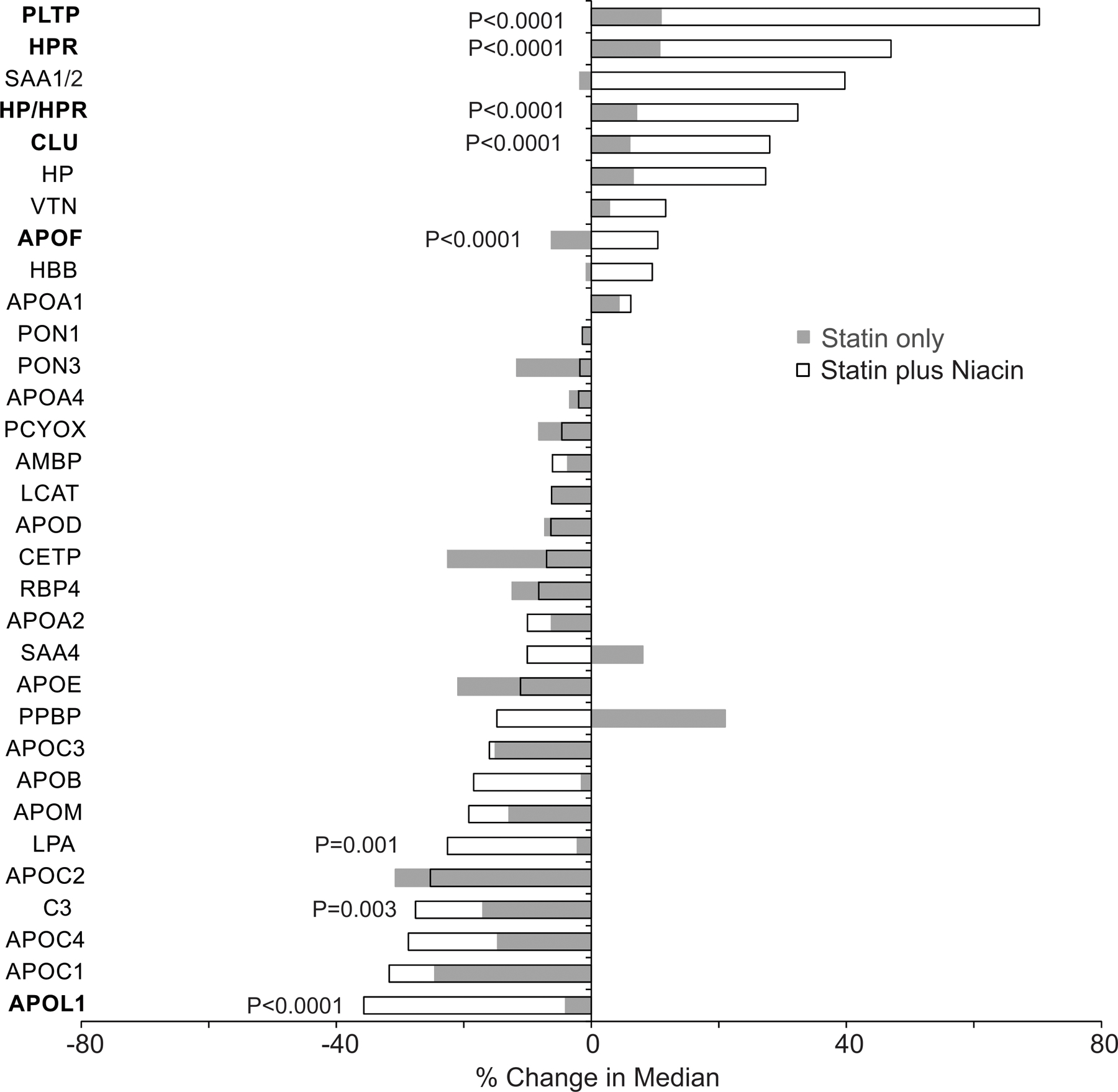
The differences in the changes between the therapies were compared using the Wilcoxon rank-sum test. P values were corrected for multiple comparisons using the method of Benjamini and Hochberg. P values of proteins with significant changes between therapies are shown. Proteins with P ≤0.0001 are in bold.
Remodeling of the HDL proteome by niacin-statin therapy in the CPC was recapitulated in AIM-HIGH.
AIM-HIGH study tested the hypothesis that adding niacin to statin therapy would further reduce the risk of cardiovascular events. In contrast to the CPC subjects, the AIM-HIGH subjects were already taking a statin when they entered the study. Consistent with this, the relative abundance of only two HDL proteins (CETP, −15%, P=0.0004 and serum paraoxonase/lactonase 3 [PON3], 11%, P=0.0004) differed after 1 year of statin monotherapy (Fig. 4). These differences may be due to change on statin type and dose regimen, as well as to stricter control of LDL-C levels of those patients.
Figure 4. Effect of niacin therapy on the HDL proteome in the AIM-HIGH study.
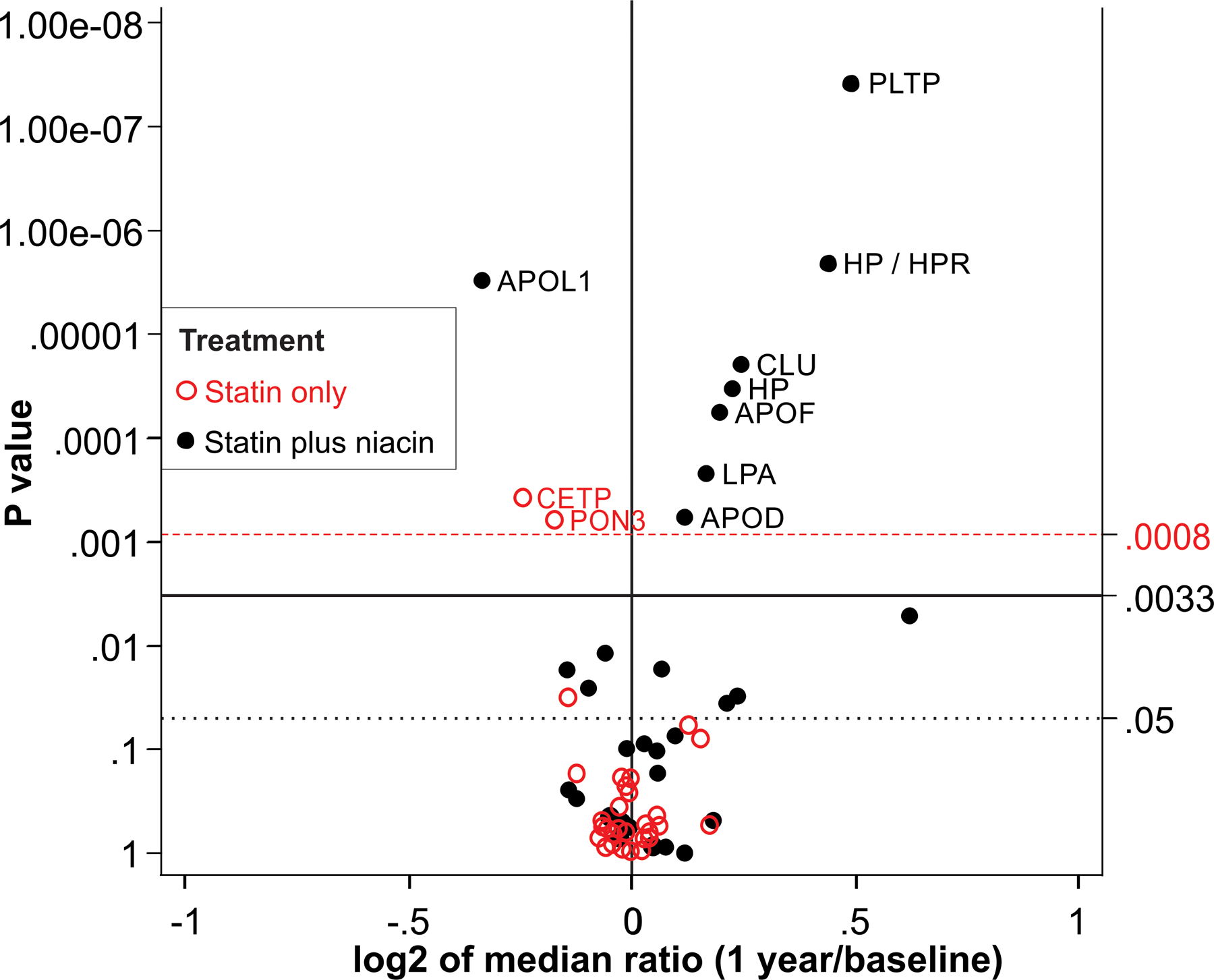
All subjects were on statin monotherapy on entry into the study. HDL proteins were quantified by selected reaction monitoring before and after 1 year on statin or niacin-statin therapies. For each protein, the P value from the Wilcoxon signed rank test is plotted against the log2 fold median change between 1-year on therapy and baseline. Proteins overexpressed after one year on therapy are displayed to the right of the value 0 on the x-axis, while underexpressed proteins are to the left. After we controlled for multiple comparison testing, only proteins situated above the P value of 0.004 for statin treatment and 0.009 for combination therapy (niacin plus statin) on the y-axis were considered significantly different between 1-year on therapy and baseline. The dashed (red) horizontal line shows the corrected critical P value threshold (P= 0.008) for statin treatment. The solid horizontal line shows the corrected overall critical P value threshold (P= 0.033) for statin plus niacin treatment. The dotted (black) horizontal line shows the uncorrected overall critical P value of 0.05.
As in the CPC study, 1 year of niacin-statin combination therapy in the AIM-HIGH markedly remodeled the HDL proteome (Fig. 4). HDL of subjects on combination therapy exhibited greater than 40% increases in levels of PLTP and HPR (P<0.0001 for each). The joint peptide for HP/HPR increased by 35%, while the HP unique peptide increased by 17% (P< 0.0001, for both). Levels of CLU (18%, P<0.0001), APOF (15% P<0.0001), LPA (12%, P=0.0002), and APOD (9%, P=0.0006) also increased significantly. As in CPC, combination therapy reduced APOL1 levels (−21%, P<0.0001). We next determined which HDL proteins differed in relative abundance after 1 year of monotherapy or combination therapy (Fig. 5). In agreement with the CPC study, levels of PLTP (P<0.0001), CLU (P=0.0002), HP/HPR (P<0.0001), and APOF (P=0.0006) where higher in HDL isolated from subjects on combination therapy than in that from subjects on statin monotherapy. Combination therapy also elevated PON3 (P<0.0001) and CETP (P=0.0034) levels in HDL when compared with statin-only therapy. APOL1 levels were lower after combination therapy than after simvastatin monotherapy (P=0.0003).
Figure 5. Changes in HDL protein amounts after 1-year on niacin therapy for AIM-HIGH study subjects.
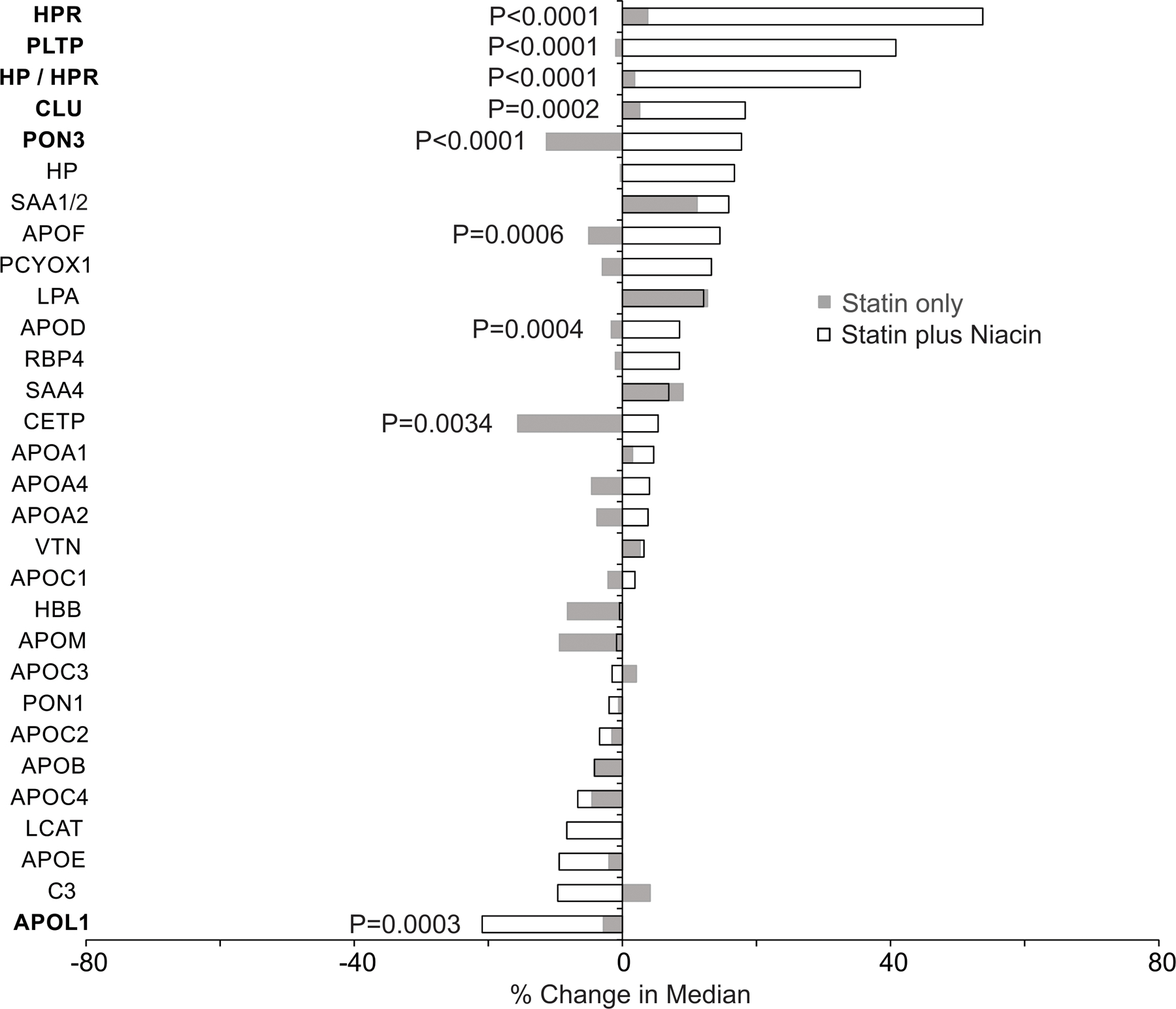
The differences in change between treatments (maintenance of statin monotherapy or addition of niacin to statin therapy) were compared using the Wilcoxon rank-sum test. P values were corrected for multiple comparisons using the method of Benjamini and Hochberg. P values of proteins with significant changes between therapies are shown. Proteins with P ≤0.0003 are in bold.
In previous studies, CLU levels in HDL correlated positively with plasma HDL-C levels and negatively with triglyceride levels.26 In the CPC subjects, we found similar correlations of HDL CLU levels with both HDL-C (ρ=0.54 at baseline and ρ=0.58 at 1 year, each P<0.001) and triglycerides (ρ=−0.31 at baseline [P=0.0004] and ρ=−0.54 [P<0.001]).
Two proteins, LPA (P=0.001) and complement C3 (C3, P=0.003), were significantly reduced by combination therapy relative to monotherapy in the CPC but not the AIM-HIGH studies. Also, three proteins, apolipoprotein D (APOD, P=0.0004), PON3 (P<0.0001), and CETP (P=0.0034) were increased by combination therapy only in AIM-HIGH study. The differences in the protein levels in the two studies are likely due differences in study design. All CPC subjects were statin-naïve, while AIM-HIGH patients were on statin therapy on entry into the trial.
Discussion
Although niacin induces a favorable lipid profile by increasing HDL-C and reducing LDL-C, APOB, Lp(a) and triglyceride levels, it fails to reduce cardiovascular risk in statin-treated subjects.3, 27–29 We tested the hypothesis that adding niacin to statin therapy might promote atherogenesis by altering HDL’s protein composition. Using quantitative mass spectrometric analyses of samples from two independent cohorts (CPC and AIM-HIGH), we demonstrated large (>25%), highly significant (P≤0.0001) increases in levels of four HDL proteins: PLTP, CLU, and HP and HPR. All of those proteins have been linked to an increased risk of atherosclerosis. In contrast, levels of only one HDL protein—APOL1—significantly decreased in both cohorts.
Niacin treatment elevated median PLTP levels in HDL by ~70%. PLTP, which belongs to the lipid transfer/lipopolysaccharide binding protein gene family, binds and transfers various lipids, including phospholipids, cholesterol, α-tocopherol, diacylglycerides, and lipopolysaccharides.30–33
In hypercholesterolemic mice, PLTP-deficiency reduced atherosclerotic lesion size.34 Overexpressing PLTP in mice heterozygous for the LDL receptor increased susceptibility to atherosclerosis35, as also happened in rabbits expressing human PLTP.36 PLTP expression markedly increased levels of small pre-β HDL particles as well as α-HDL in mice expressing human APOA1.37 In humans, two single nucleotide polymorphisms linked to lower PLTP transcription and activity associated with a greater number of HDL particles, smaller HDL size, and decreased risk of CVD.38 In another study, a tagging single nucleotide polymorphism in the PLTP region associated with elevated PLTP activity and carotid artery disease.39 In the Framingham Heart Study, PLTP activity associated positively with increased baseline plasma levels of PLTP, which also associated with increased risk of all-cause mortality in diabetic male subjects.40
In plasma, PLTP forms a complex with CLU.41 Like PLTP, CLU (also known as APOJ) in HDL was markedly elevated in both of our cohorts on niacin therapy. Circulating CLU predominantly associates with HDL, and it is enriched in the dense HDL3 subfraction.42 CLU is implicated in vascular injury and the regulation of smooth muscle cell proliferation and migration in vitro and in vivo.43, 44 Apoe−/− mice deficient in CLU are protected from atherosclerosis.45 Also, CLU levels are elevated in serum of patients with CVD.46 These observations suggest that CLU promotes vascular injury and atherogenesis.
After 1 year of treatment, levels of both, haptoglobin (HP) and haptoglobin-related protein (HPR) were higher in the HDL of both CPC and AIM-HIGH subjects on niacin-statin therapy. HP is an acute phase response protein47 that contributes to inflammation, insulin resistance, diabetes, and obesity,48 all of which associate with increased CVD risk. High levels of plasma HP predict an increased risk of developing type 2 diabetes.49 Importantly, HP haplotypes predict CVD risk in subjects with type 1 diabetes.50, 51
Only one protein, APOL1, was decreased in the HDL of niacin-treated subjects in both cohorts of subjects. APOL1, together with HRP, mediates the human innate immune response to African trypanosomes.52 Two APOL1 alleles are strongly linked to the risk of renal disease in African Americans, but neither allele associates with CVD risk in that ethnic group.53
Niacin therapy is a well-established risk factor for diabetes. However, in both the CPC and AIM-HIGH cohorts, only small changes in plasma glucose levels were detected after 1 year on niacin-statin combination therapy. Moreover, glucose levels were similar in both cohorts after monotherapy or combination therapy, suggesting that niacin-induced diabetes was unlikely to have contributed to the changes we observed in the HDL proteome.
We also found that statin monotherapy remodeled the HDL proteome in the CPC cohort, with major reductions in APOC1, APOC2, APOC3, APOC4, and APOE, with reductions in APOC3 and APOC1 correlating with a reduction in plasma triglyceride levels. As CPC study was not designed to evaluate statins effect on HDL proteome (i.e. no placebo group for the statin treated subjects), we cannot exclude the reductions in APOCs were caused by factors unrelated to the treatment. Because all subjects in AIM-HIGH were on statin therapy when they entered the study, we were unable to determine the effect on statin monotherapy on the HDL proteome in that cohort. Importantly, there were no significant differences in levels of APOCs between subjects on monotherapy and combination therapy, despite a significant reduction in triglycerides levels, indicating that levels of APOCs did not associate with the lower triglyceride levels seen in the niacin-treated subjects. Total plasma APOC3 levels were not measured in the present study and it is possible that niacin reduces APOC3 levels in APOB-containing lipoproteins in statin-treated subjects, but that HDL-associated APOC3 levels remain unchanged. APOC3 in HDL strongly associated with incident CVD risk in four different human cohorts 54, raising the possibility that niacin’s failure to further reduce APOC3 levels in HDL in statin-treated subjects contributed to its lack of cardiovascular benefit in those subjects.
Our study has several strengths. First, we analyzed samples from two independent cohorts enrolled in large cardiovascular outcome studies that compared the impact of niacin therapy on CVD risk in subjects treated with two different statins. Both studies detected no CVD benefit from niacin therapy despite favorable changes in LDL-C, HDL-C, and triglyceride levels. Second, we found that niacin therapy induced consistent, highly significant differences in levels of four HDL proteins in both studies. Third, we used different mass spectrometric methods to quantify HDL protein levels in the two cohorts, indicating that the results were analytically robust. Finally, in the AIM HIGH study subjects were on statin therapy on entry into the trial. We observed relatively small changes in relative abundance of only two HDL proteins, CETP and PON3, after 1 year of statin monotherapy in those subjects, demonstrating that MS/MS quantification of protein abundance in HDL is highly reproducible.
Our study also has several limitations. First, ultracentrifugation may have altered HDL’s protein composition.55 We choose ultracentrifugation to isolate HDL because it has been widely used for both clinical and translational studies, and because we have carefully standardized and validated the method in our laboratory.11, 12, 18, 56 Second, alterations in levels of proteins in plasma and/or other lipoproteins might have contributed to CVD risk in the statin-treated subjects. In future studies, it will be important to quantify potentially atherogenic proteins in HDL, other lipoproteins, and plasma. Third, we cannot assume that the assumption of randomization is correct for the statin-only group in the CPC cohort because of the relatively small number of subjects (n=45). However, we note that the major, highly significant different expression levels of the atherogenic proteins found in HDL of subjects treated with niacin were observed in both the CPC and Aim-High studies. Finally, some niacin-statin treated subjects in CPC were also treated with colesevelam, and some statin-treated subjects in the AIM-HIGH were also treated with ezetimibe. However, remarkably similar changes in the HDL proteome were observed in both the CPC and AIM-HIGH cohorts, indicating that these factors were unlikely to have confounded the analyses.
In conclusion, in two independent cohorts of subjects treated with two different statins, we found consistent, highly significant changes in levels of five HDL proteins when niacin was added to statin therapy. Four of the five proteins increased, and all four have been linked to an increased risk of CVD. These findings raise the possibility that niacin-induced changes in the HDL proteome, and perhaps similar changes in plasma and/or other lipoproteins, contribute to niacin’s failure to reduce CVD risk in statin-treated subjects.
Supplementary Material
Highlights section.
The HDL proteome was quantified in two independent clinical trials to identify potential mechanisms for the failure of niacin to reduce cardiovascular events in statin-treated subjects, even though it increases HDL-C and decreases LDL-C and triglyceride levels.
Statin monotherapy reduced the levels of HDL proteins strongly linked to triglyceride metabolism, including apolipoproteins APOC1, APOC2, APOC3, and APOC4.
Niacin therapy markedly elevated HDL-associated phospholipid transfer protein, clusterin, haptoglobin, and haptoglobin-related proteins in statin treated subjects, all of which are strongly linked to increased atherosclerotic risk in humans.
Acknowledgments
Sources of Funding
This work was supported by awards from the National Institutes of Health (R01HL149685, R01HL089504, R01HL63895, P01HL151328, P01HL092969, R35HL150754), American Heart Association award #15CVGPSD27260197, and Fundação de Amparo a Pesquisa do Estado de Sao Paulo (FAPESP 2016/00696-3).
Nonstandard Abbreviations and Acronyms
- HDL-C
high-density lipoprotein cholesterol
- LDL-C
low-density lipoprotein cholesterol
- CPC
Carotid Plaque Composition
- AIM-HIGH
Atherothrombosis Intervention in Metabolic Syndrome with Low HDL/High Triglycerides
- APOC1
apolipoprotein C1
- APOC2
apolipoprotein C2
- APOC3
apolipoprotein C3
- APOC4
apolipoprotein C4
- PLTP
phospholipid transfer protein
- CLU
clusterin
- HP
haptoglobin
- HPR
haptoglobin related protein
- CETP
cholesteryl ester transfer protein
- CVD
cardiovascular diseases
- APOB
apolipoprotein B
- CEC
cholesterol efflux capacity
- HDL-P
HDL particle number
- FDR
false discovery rate
- APOE
apolipoprotein E
- APOM
apolipoprotein M
- APOL1
apolipoprotein L1
- LPA
apolipoprotein (a)
- CLU
clusterin
- SAA1/2
serum amyloid A1/2
- APOF
apolipoprotein F
- C3
complement component 3
- PON3
serum paraoxonase/lactonase 3
- APOD
apolipoprotein D
Footnotes
Clinical Trials Registration – CPC - https://www.clinicaltrials.gov/ct2/show/NCT00715273. Identifier: NCT00715273. AIM-HIGH (HDL Proteomics) - https://clinicaltrials.gov/ct2/show/NCT00880178. Identifier: NCT00880178. AIM-HIGH - https://clinicaltrials.gov/ct2/show/NCT00120289. Identifier: NCT00120289.
Disclosures
Dr. Xue-Qiao Zhao received research grants from AstraZeneca, DelCore, Novartis. Dr. Tomas Vaisar received honoraria from AstraZeneca. Other authors have nothing to disclosure.
References
- 1.Rader DJ, Tall AR. The not-so-simple HDL story: Is it time to revise the HDL cholesterol hypothesis? Nat Med. 2012;18:1344–1346. [DOI] [PubMed] [Google Scholar]
- 2.Schwartz GG, Olsson AG, Abt M, et al. Effects of dalcetrapib in patients with a recent acute coronary syndrome. New Engl J Med. 2012;367:2089–2099. [DOI] [PubMed] [Google Scholar]
- 3.Boden WE, Probstfield JL, Anderson T, Chaitman BR, Desvignes-Nickens P, Koprowicz K, McBride R, Teo K, Weintraub W. Niacin in patients with low HDL cholesterol levels receiving intensive statin therapy. New Engl J Med. 2011;365:2255–2267. [DOI] [PubMed] [Google Scholar]
- 4.Bowman L, Hopewell JC, Chen F, Wallendszus K, Stevens W, Collins R, Wiviott SD, Cannon CP, Braunwald E, Sammons E, Landray MJ. Effects of Anacetrapib in Patients with Atherosclerotic Vascular Disease. N Engl J Med. 2017;377:1217–1227. [DOI] [PubMed] [Google Scholar]
- 5.Ronsein GE, Heinecke JW. Time to ditch HDL-C as a measure of HDL function? Curr Opin Lipidol. 2017;28:414–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ronsein GE, Hutchins PM, Isquith D, Vaisar T, Zhao XQ, Heinecke JW. Niacin therapy increases high-density lipoprotein particles and total cholesterol efflux capacity but not ABCA1-specific cholesterol efflux in statin-treated subjects. Arterioscler Thromb Vasc Biol. 2016;36:404–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vaisar T, Pennathur S, Green PS, et al. Shotgun proteomics implicates protease inhibition and complement activation in the antiinflammatory properties of HDL. J Clin Invest. 2007;117:746–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shah AS, Tan L, Long JL, Davidson WS. Proteomic diversity of high density lipoproteins: our emerging understanding of its importance in lipid transport and beyond. J Lipid Res. 2013;54:2575–2585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gourgari E, Ma J, Playford MP, Mehta NN, Goldman R, Remaley AT, Gordon SM. Proteomic alterations of HDL in youth with type 1 diabetes and their associations with glycemic control: a case-control study. Cardiovasc Diabetol. 2019;18:43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vaisar T, Tang C, Babenko I, Hutchins P, Wimberger J, Suffredini AF, Heinecke JW. Inflammatory remodeling of the HDL proteome impairs cholesterol efflux capacity. J Lipid Res. 2015;56:1519–1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ronsein GE, Pamir N, von Haller PD, Kim DS, Oda MN, Jarvik GP, Vaisar T, Heinecke JW. Parallel reaction monitoring (PRM) and selected reaction monitoring (SRM) exhibit comparable linearity, dynamic range and precision for targeted quantitative HDL proteomics. J Proteomics. 2015;113:388–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ronsein GE, Reyes-Soffer G, He Y, Oda M, Ginsberg H, Heinecke JW. Targeted proteomics identifies paraoxonase/arylesterase 1 (PON1) and apolipoprotein Cs as potential risk factors for hypoalphalipoproteinemia in diabetic subjects treated with fenofibrate and rosiglitazone. Mol Cell Proteomics. 2016;15:1083–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Carr SA, Abbatiello SE, Ackermann BL, et al. Targeted Peptide Measurements in Biology and Medicine: Best Practices for Mass Spectrometry-based Assay Development Using a Fit-for-Purpose Approach. Mol Cell Proteomics. 2014;13:907–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhao XQ, Phan BA, Chu B, Bray F, Moore AB, Polissar NL, Dodge JT Jr., Lee CD, Hatsukami TS, Yuan C. Testing the hypothesis of atherosclerotic plaque lipid depletion during lipid therapy by magnetic resonance imaging: study design of Carotid Plaque Composition Study. Am Heart J. 2007;154:239–246. [DOI] [PubMed] [Google Scholar]
- 15.Out C, Groen AK, Brufau G. Bile acid sequestrants: more than simple resins. Curr Opin Lipidol. 2012;23:43–55. [DOI] [PubMed] [Google Scholar]
- 16.Yates B, Braschi B, Gray KA, Seal RL, Tweedie S, Bruford EA. Genenames.org: the HGNC and VGNC resources in 2017. Nucleic Acids Res. 2017;45:D619–d625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mendez A, Oram J, Bierman E. Protein kinase C as a mediator of high density lipoprotein receptor- dependent efflux of intracellular cholesterol. J Biol Chem. 1991;266:10104–10111. [PubMed] [Google Scholar]
- 18.Silva ARM, Toyoshima MTK, Passarelli M, Di Mascio P, Ronsein GE. Comparing Data-Independent Acquisition and Parallel Reaction Monitoring in Their Abilities To Differentiate High-Density Lipoprotein Subclasses. J Proteome Res. 2020;19:248–259. [DOI] [PubMed] [Google Scholar]
- 19.Hoofnagle AN, Becker JO, Oda MN, Cavigiolio G, Mayer P, Vaisar T. Multiple-reaction monitoring-mass spectrometric assays can accurately measure the relative protein abundance in complex mixtures. Clin Chem. 2012;58:777–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Simes RJ. An improved Bonferroni procedure for multiple tests of significance. Biometrika. 1986;73:751–754. [Google Scholar]
- 21.Benjamini Y, Hochberg Y. Controlling the false discovery rate - a practical and powerful approach to multiple testing. J R Statist Soc B. 1995;57:289–300. [Google Scholar]
- 22.Benjamini Y, Yekutieli D. The control of the false discovery rate in multiple testing under dependency. Ann Statist. 2001;29:1165–1188. [Google Scholar]
- 23.Baigent C, Keech A, Kearney PM, Blackwell L, Buck G, Pollicino C, Kirby A, Sourjina T, Peto R, Collins R, Simes R. Efficacy and safety of cholesterol-lowering treatment: prospective meta-analysis of data from 90,056 participants in 14 randomised trials of statins. Lancet. 2005;366:1267–1278. [DOI] [PubMed] [Google Scholar]
- 24.Nordestgaard BG, Chapman MJ, Ray K, et al. Lipoprotein(a) as a cardiovascular risk factor: current status. Eur Heart J. 2010;31:2844–2853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nielsen MJ, Petersen SV, Jacobsen C, Oxvig C, Rees D, Moller HJ, Moestrup SK. Haptoglobin-related protein is a high-affinity hemoglobin-binding plasma protein. Blood. 2006;108:2846–2849. [DOI] [PubMed] [Google Scholar]
- 26.Hoofnagle AN, Wu M, Gosmanova AK, Becker JO, Wijsman EM, Brunzell JD, Kahn SE, Knopp RH, Lyons TJ, Heinecke JW. Low clusterin levels in high-density lipoprotein associate with insulin resistance, obesity, and dyslipoproteinemia. Arterioscler Thromb Vasc Biol. 2010;30:2528–2534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Landray MJ, Haynes R, Hopewell JC, Parish S, Aung T, Tomson J, Wallendszus K, Craig M, Jiang L, Collins R, Armitage J. Effects of extended-release niacin with laropiprant in high-risk patients. N Engl J Med. 2014;371:203–212. [DOI] [PubMed] [Google Scholar]
- 28.Garg A, Sharma A, Krishnamoorthy P, Garg J, Virmani D, Sharma T, Stefanini G, Kostis JB, Mukherjee D, Sikorskaya E. Role of Niacin in Current Clinical Practice: A Systematic Review. Am J Med. 2017;130:173–187. [DOI] [PubMed] [Google Scholar]
- 29.Schandelmaier S, Briel M, Saccilotto R, Olu KK, Arpagaus A, Hemkens LG, Nordmann AJ. Niacin for primary and secondary prevention of cardiovascular events. The Cochrane database of systematic reviews. 2017;6:Cd009744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tall AR, Krumholz S, Olivecrona T, Deckelbaum RJ. Plasma phospholipid transfer protein enhances transfer and exchange of phospholipids between very low density lipoproteins and high density lipoproteins during lipolysis. J Lipid Res. 1985;26:842–851. [PubMed] [Google Scholar]
- 31.Nishida HI, Klock DG, Guo Z, Jakstys BP, Nishida T. Phospholipid transfer protein can transform reconstituted discoidal HDL into vesicular structures. Biochim Biophys Acta. 1997;1349:222–232. [DOI] [PubMed] [Google Scholar]
- 32.Rao R, Albers JJ, Wolfbauer G, Pownall HJ. Molecular and macromolecular specificity of human plasma phospholipid transfer protein. Biochemistry. 1997;36:3645–3653. [DOI] [PubMed] [Google Scholar]
- 33.Hailman E, Albers JJ, Wolfbauer G, Tu AY, Wright SD. Neutralization and transfer of lipopolysaccharide by phospholipid transfer protein. J Biol Chem. 1996;271:12172–12178. [DOI] [PubMed] [Google Scholar]
- 34.Jiang XC, Qin S, Qiao C, Kawano K, Lin M, Skold A, Xiao X, Tall AR. Apolipoprotein B secretion and atherosclerosis are decreased in mice with phospholipid-transfer protein deficiency. Nat Med. 2001;7:847–852. [DOI] [PubMed] [Google Scholar]
- 35.van Haperen R, van Tol A, van Gent T, Scheek L, Visser P, van der Kamp A, Grosveld F, de Crom R. Increased risk of atherosclerosis by elevated plasma levels of phospholipid transfer protein. J Biol Chem. 2002;277:48938–48943. [DOI] [PubMed] [Google Scholar]
- 36.Masson D, Deckert V, Gautier T, et al. Worsening of diet-induced atherosclerosis in a new model of transgenic rabbit expressing the human plasma phospholipid transfer protein. Arterioscler Thromb Vasc Biol. 2011;31:766–774. [DOI] [PubMed] [Google Scholar]
- 37.Jiang X, Francone OL, Bruce C, Milne R, Mar J, Walsh A, Breslow JL, Tall AR. Increased prebeta-high density lipoprotein, apolipoprotein AI, and phospholipid in mice expressing the human phospholipid transfer protein and human apolipoprotein AI transgenes. J Clin Invest. 1996;98:2373–2380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vergeer M, Boekholdt SM, Sandhu MS, et al. Genetic variation at the phospholipid transfer protein locus affects its activity and high-density lipoprotein size and is a novel marker of cardiovascular disease susceptibility. Circulation. 2010;122:470–477. [DOI] [PubMed] [Google Scholar]
- 39.Jarvik GP, Rajagopalan R, Rosenthal EA, Wolfbauer G, McKinstry L, Vaze A, Brunzell J, Motulsky AG, Nickerson DA, Heagerty PJ, Wijsman EM, Albers JJ. Genetic and nongenetic sources of variation in phospholipid transfer protein activity. J Lipid Res. 2010;51:983–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cavusoglu E, Marmur JD, Chhabra S, Hojjati MR, Yanamadala S, Chopra V, Eng C, Jiang XC. Elevated baseline plasma phospholipid protein (PLTP) levels are an independent predictor of long-term all-cause mortality in patients with diabetes mellitus and known or suspected coronary artery disease. Atherosclerosis. 2015;239:503–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cheung MC, Vaisar T, Han X, Heinecke JW, Albers JJ. Phospholipid transfer protein in human plasma associates with proteins linked to immunity and inflammation. Biochemistry. 2010;49:7314–7322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Davidson WS, Silva RA, Chantepie S, Lagor WR, Chapman MJ, Kontush A. Proteomic analysis of defined HDL subpopulations reveals particle-specific protein clusters: relevance to antioxidative function. Arterioscler Thromb Vasc Biol. 2009;29:870–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Miyata M, Biro S, Kaieda H, Eto H, Orihara K, Kihara T, Obata H, Matsushita N, Matsuyama T, Tei C. Apolipoprotein J/clusterin is induced in vascular smooth muscle cells after vascular injury. Circulation. 2001;104:1407–1412. [DOI] [PubMed] [Google Scholar]
- 44.Millis AJ, Luciani M, McCue HM, Rosenberg ME, Moulson CL. Clusterin regulates vascular smooth muscle cell nodule formation and migration. Journal of cellular physiology. 2001;186:210–219. [DOI] [PubMed] [Google Scholar]
- 45.Hamada N, Miyata M, Eto H, Ikeda Y, Shirasawa T, Akasaki Y, Miyauchi T, Furusho Y, Nagaki A, Aronow BJ, Tei C. Loss of clusterin limits atherosclerosis in apolipoprotein E-deficient mice via reduced expression of Egr-1 and TNF-α. J Atheroscler Thromb. 2011;18:209–216. [DOI] [PubMed] [Google Scholar]
- 46.Trougakos IP, Poulakou M, Stathatos M, Chalikia A, Melidonis A, Gonos ES. Serum levels of the senescence biomarker clusterin/apolipoprotein J increase significantly in diabetes type II and during development of coronary heart disease or at myocardial infarction. Experimental gerontology. 2002;37:1175–1187. [DOI] [PubMed] [Google Scholar]
- 47.Oliviero S, Cortese R. The human haptoglobin gene promoter: interleukin-6-responsive elements interact with a DNA-binding protein induced by interleukin-6. The EMBO journal. 1989;8:1145–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Maffei M, Barone I, Scabia G, Santini F. The Multifaceted Haptoglobin in the Context of Adipose Tissue and Metabolism. Endocrine reviews. 2016;37:403–416. [DOI] [PubMed] [Google Scholar]
- 49.Schmidt MI, Duncan BB, Sharrett AR, Lindberg G, Savage PJ, Offenbacher S, Azambuja MI, Tracy RP, Heiss G. Markers of inflammation and prediction of diabetes mellitus in adults (Atherosclerosis Risk in Communities study): a cohort study. Lancet. 1999;353:1649–1652. [DOI] [PubMed] [Google Scholar]
- 50.Costacou T, Ferrell RE, Orchard TJ. Haptoglobin genotype: a determinant of cardiovascular complication risk in type 1 diabetes. Diabetes. 2008;57:1702–1706. [DOI] [PubMed] [Google Scholar]
- 51.Levy AP, Hochberg I, Jablonski K, Resnick HE, Lee ET, Best L, Howard BV. Haptoglobin phenotype is an independent risk factor for cardiovascular disease in individuals with diabetes: The Strong Heart Study. J Am Coll Cardiol. 2002;40:1984–1990. [DOI] [PubMed] [Google Scholar]
- 52.Smith AB, Esko JD, Hajduk SL. Killing of trypanosomes by the human haptoglobin-related protein. Science. 1995;268:284–286. [DOI] [PubMed] [Google Scholar]
- 53.Bajaj A, Susztak K, Damrauer SM. APOL1 and Cardiovascular Disease: A Story in Evolution. Arterioscler Thromb Vasc Biol. 2017;37:1587–1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jensen MK, Aroner SA, Mukamal KJ, Furtado JD, Post WS, Tsai MY, Tjonneland A, Polak JF, Rimm EB, Overvad K, McClelland RL, Sacks FM. High-Density Lipoprotein Subspecies Defined by Presence of Apolipoprotein C-III and Incident Coronary Heart Disease in Four Cohorts. Circulation. 2018;137:1364–1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.McVicar JP, Kunitake ST, Hamilton RL, Kane JP. Characteristics of human lipoproteins isolated by selected-affinity immunosorption of apolipoprotein A-I. Proc Natl Acad Sci U S A. 1984;81:1356–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ronsein GE, Vaisar T. Deepening our understanding of HDL proteome. Expert Rev Proteomics. 2019;16:749–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.