

Abstract
Autophagy is a catabolic intracellular nutrient scavenging pathway triggered by nutrient deprivation and stress that captures and degrades intracellular proteins and organelles in lysosomes. The breakdown products are then recycled into metabolic pathways to sustain survival. Organelle turnover by autophagy contributes to quality control and suppresses inflammation. Autophagy is upregulated in many cancers and supports their growth, survival and malignancy in a tumor cell-autonomous fashion. Host autophagy also promotes tumor growth by maintaining a supply of essential nutrients and suppressing innate and adaptive anti-tumor immune responses. Autophagy is also upregulated in response to cancer therapy and confers treatment resistance. Thus, autophagy is a cancer vulnerability and autophagy inhibition is under investigation as a novel therapeutic approach.
Keywords: Autophagy, Cancer, T cells, Interferon, Immune Response, Metabolism
Regulation and Function of Autophagy
Autophagy is induced by nutrient deprivation and stress to capture, degrade and recycle intracellular proteins and organelles in lysosomes (Figure 1) [1, 2]. The master regulator of cell growth, Mammalian Target of Rapamycin (mTOR), inhibits autophagy, whereas the major sensor of energy limitation, AMP-activated protein kinase (AMPK), activates autophagy. In turn, stress responsive pathways such as those controlled by Hypoxia-inducible factors (HIFs) activate autophagy. Integration of these positive and negative regulators coordinates the control of autophagic degradation with the intracellular and extracellular environment to maintain homeostasis [3].
Figure 1. Cancer cells activate autophagy to promote survival.
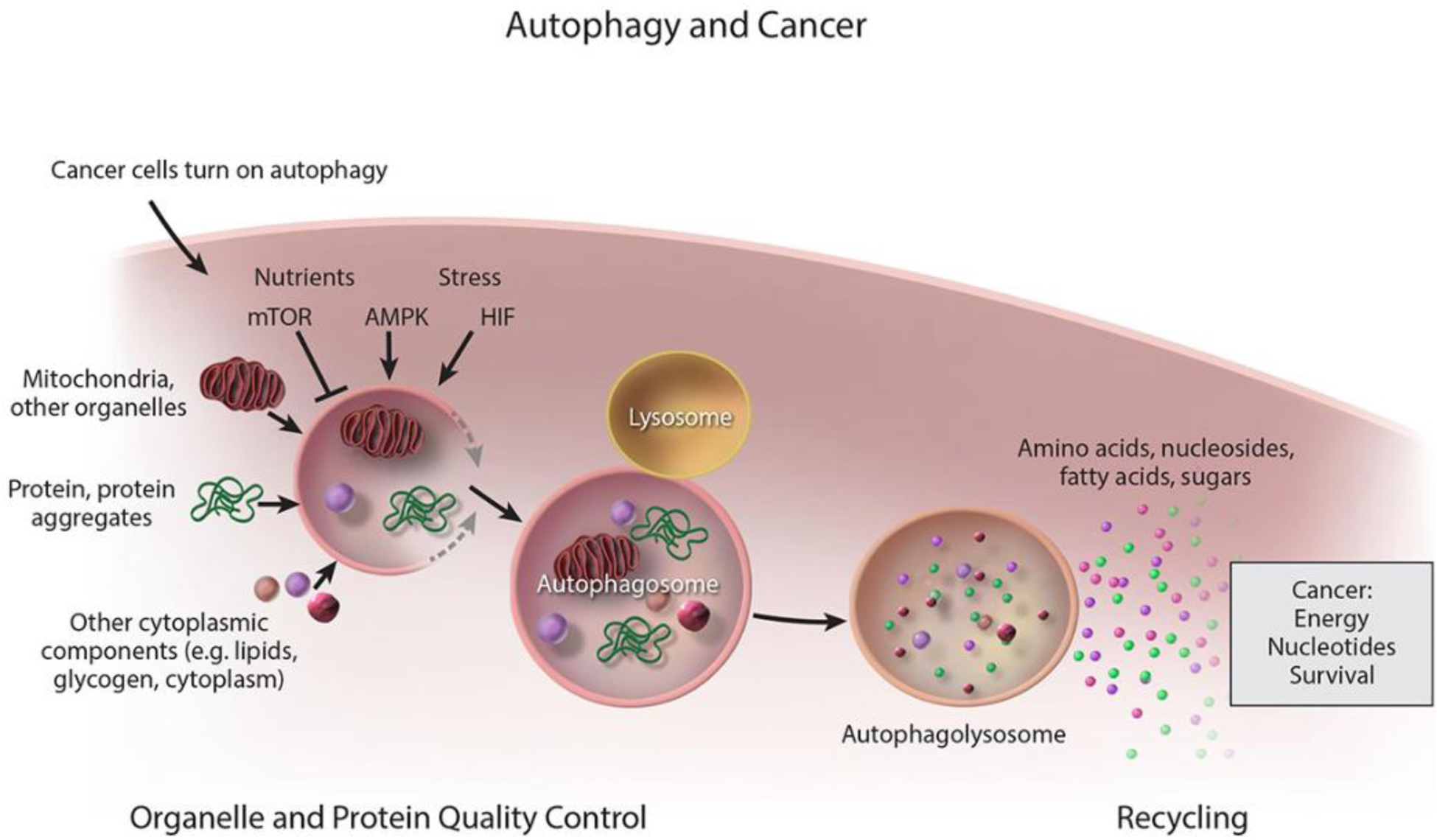
General scheme for the capture and degradation of autophagy cargo in both normal and cancer cells. Cancer cells upregulate autophagy and use the breakdown products to sustain nucleotide pools and energy homeostasis and thereby their growth and survival.
The main functions of autophagic degradation are sustaining metabolism through recycling and provision of metabolites, controlling protein and organelle quality, and eliminating intracellular bacteria. The essential autophagy gene(s) (Atg) orchestrate the initiation of double membrane autophagosome formation and membrane expansion, while cargo adaptors attached to the membrane bind and capture cargo such as proteins and organelles [4, 5]. Autophagosomes then fuse with lysosomes where the cargo is degraded, and the breakdown products are released into the cytoplasm where they can be used as substrates for metabolism and biosynthesis (Figure 1). Roughly half of cellular proteins are selectively targeted for degradation by autophagy during starvation, as such, autophagy is a robust mechanism for intracellular protein turnover [6]. In this way, autophagy ensures cellular homeostasis during interruptions in nutrient availability.
Protein and organelle quality control through autophagy is achieved by their selective degradation. Damaged proteins and organelles are identified and tagged with ubiquitin and eliminated, thereby preventing their accumulation which is toxic [4, 5]. A major characteristic of lack of autophagy function is the dramatic accumulation of damaged organelles, particularly mitochondria and also protein aggregates. Failure to degrade autophagy substrates is destructive as it is pro-inflammatory and can lead to overt disruption of cellular function in post-mitotic cells. Defects in autophagy lead to surprising selective tissue damage and disease, and understanding the underlying mechanisms is key to modulating autophagy as a therapeutic approach [3].
Role of Autophagy in the Adult Mice
Genetic approaches inactivating Atg genes in adult mice have revealed many important functions of autophagy in vivo. These functions center around sustaining metabolism, mounting a protective response to stress, and suppressing inflammation. When considering targeting a specific pathway or function in cancer it is essential to also understand the consequences to normal tissues to establish whether there is a therapeutic window where cancer cells will be preferentially sensitive. It is also important to understand the potential consequences of inhibiting a pathway in normal tissues to be aware of on-target toxicities. Finally, cancer therapy resistance is a major obstacle to successful treatment and knowing the possible resistance mechanisms that can emerge is useful.
Metabolism.
Constitutive autophagy deficiency in mice causes neonatal lethality, and various tissue-specific models of autophagy deficiency have revealed dependencies [5]. However, a global view of the role of autophagy in adult mice is clear with conditional, whole body deletions of essential autophagy genes [2, 7]. For example, deletion of Atg7 in adult mice predisposes to death from infection due to failure to eliminate intracellular Streptococcus, but most mice live for two to three months, eventually dying from neurodegeneration [8]. Deleted mice also display systemic metabolic impairment (cachexia) and inflammation. Tissue damage is selective with brain, liver, white adipose tissue (WAT) and muscle being most impacted [8] suggesting that loss of autophagy-mediated recycling at the cellular level creates a systemic metabolic imbalance that amplifies dependency on extracellular nutrients supplied from dedicated nutrient stores (e.g. WAT, muscle) thereby driving cachexia [8]. Why some tissues are more dependent on autophagy than others may be related to differences in metabolism, but recent findings reveal that this is caused in some cases by differential activation of stress response and adaptation pathways.
Fasting.
One salient feature of autophagy preserved though evolution from yeast to mammals is enabling survival to nutrient deprivation. Although genetically switching off Atg7 or Atg5 in adult mice permits survival for several months, fasting is lethal [8, 9]. Upon fasting, autophagy-deficient mice rapidly deplete stores of liver glycogen and WAT and activate muscle wasting indicative of cachexia [8]. Despite elevated mobilization of dedicated nutrient stores, circulating glucose levels plummet and mice die from hypoglycemia that is rescued by glucose supplementation [8]. Thus, autophagy is required for fasting glucose homeostasis, underscoring the important role of autophagy in sustaining cellular and systemic mammalian metabolism in response to nutrient limitation.
Inflammation.
The induction of inflammation upon conditional deficiency in autophagy likely resides from the failure to clear damaged proteins, organelles and bacteria that triggers an innate immune response [8]. The failure to remove autophagy substrates, particularly mitochondria that can release their DNA, for example, may be one of the triggers of inflammation [10]. Failure to clear dead cells through Microtubule-associated protein 1A/1B-light chain 3 (LC3)-mediated phagocytosis (LAP), which uses a subset of the autophagy machinery including Atg7 also contributes to inflammation [11].
Integration of Autophagy with Stress Response Pathways
Autophagy enables adaptation to stress exemplified by that fatal response of autophagy-deficient mice to fasting [8]. Accelerated mobilization of dedicated nutrient stores can temporarily compensate for loss of autophagy in the short term during fasting as one metabolic compensation mechanism [8]. Determining how autophagy functionally interacts with known stress response and compensatory pathways can provide insight into its global role in mammalian physiology.
p53.
Autophagy suppresses the activation of p53, a transcription factor and an essential mediator of response to DNA damage and other forms of stress, suggesting that autophagy loss potentiates p53 activation and the deleterious consequences therefrom (growth arrest, apoptosis, senescence, etc.), contributing to the phenotype of autophagy deficient mice [12]. Indeed, p53-deficiency prolongs survival of Atg7-deficient adult mice by delaying neurodegeneration [9, 13]. p53 deficiency also prolongs the survival of autophagy-deficient mice to fasting, which is consistent with a role for a potent p53 stress response in neurons [9, 13]. These findings demonstrate that autophagy is a negative regulator of the p53 stress response pathway in vivo. p53 also activates autophagy providing a negative feedback loop to limit p53 activation (Figure 2) [14].
Figure 2. Functional interaction of autophagy and stress response pathways.
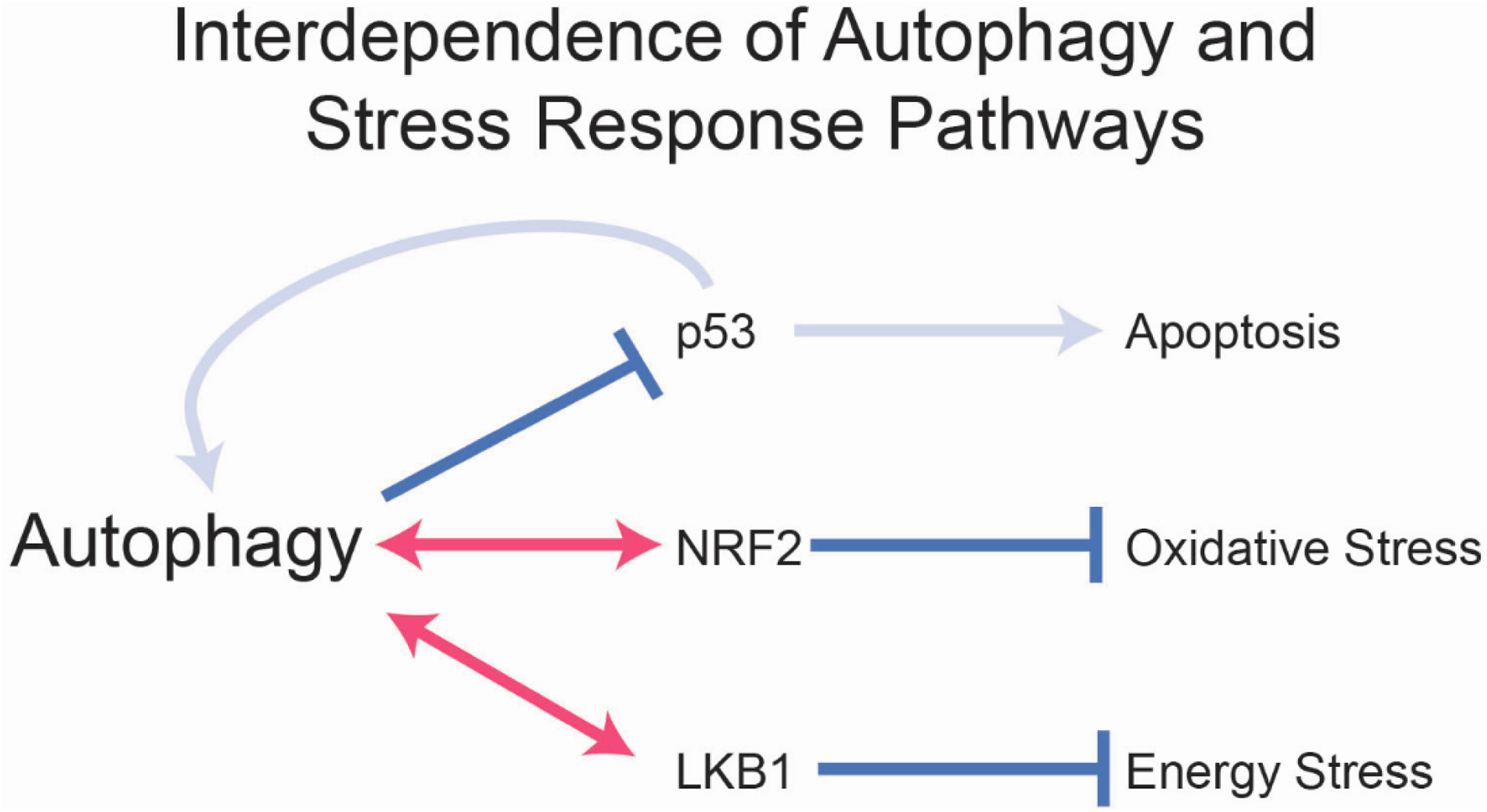
Autophagy suppresses p53 and thereby apoptosis and neurodegeneration. p53 in turn activates autophagy providing a negative feedback loop. Autophagy and NRF2 functionally complement each other to suppress oxidative stress and loss of both is synthetically lethal in vivo. Autophagy and LKB1 functionally complement each other and loss of both is synthetically lethal in vivo. Pink arrows indicate synthetic lethality.
Nuclear factor erythroid 2-related factor 2 (Nrf2).
The selective clearance of damaged mitochondria through mitophagy implicates autophagy as a mitigator of oxidative stress [10]. NRF2 is a transcription factor and master regulator of the response to oxidative stress where it provides adaptation and protection [15]. In cells, loss of autophagy directly activates NRF2 by causing accumulation of the autophagy substrate Sequestosome-1 (SQSTM1/p62) that binds and inhibits the NRF2 inhibitor Kelch-like ECH-associated protein 1 (KEAP1) [16, 17]. NRF2 mobilization is a protective adaptation to autophagy inactivation [18]. Moreover, conditional deletion of Atg7 in mice that lack Nrf2 is lethal, directly demonstrating the protective role this pathway plays in response to failure of autophagy (Figure 2) [9]. This synthetic lethality of Atg7 and Nrf2 is singularly displayed in the intestine with death arising from villus destruction and loss of barrier function [9]. Nrf2 can compensate to some degree for autophagy deficiency in the intestine, and it remains to be determined what aspects of Nrf2 function are required. In mouse models of pancreas cancer Nrf2-mediated induction of micropinocytosis compensates for loss of autophagy indicating that extracellular nutrient scavenging by micropinocytosis can compensate for loss of intracellular nutrient scavenging by autophagy [19]. Whether this is a compensatory mechanism in normal tissues is not known.
Liver kinase B1 (Stk11/Lkb1).
LKB1 is a kinase and master regulator of the response to energy crisis in part through activation of the energy deficit sensor AMPK. In turn, AMPK promotes energy conserving catabolic functions to facilitate protective adaptation that includes induction of autophagy (Figure 1) [20, 21]. As autophagy is essential to support metabolism and energy homeostasis, one can envision how deficiency in Lkb1 and loss of the response to energy crisis is detrimental in the absence of autophagy. Indeed, co-deleting Atg7 and Lkb1 in adult mice is more lethal than deleting either gene alone due to specific loss of intestinal barrier function (Figure 2) [22]. The synthetic lethality of Atg7 and Lkb1 in vivo suggests that LKB1 promotes metabolic adaptation, possibly through AMPK that enables tolerance to loss of autophagy. Note that one of the metabolic adaptations and protective mechanisms induced by AMPK is autophagy, which cannot be induced in Atg7-deficient mice, therefore protection derives from an autophagy-independent function. Thus, autophagy loss can be tolerated in mice as long as the Lkb1 function is intact, which allows compensatory energy stress adaptation mechanisms protect from that autophagy loss in a tissue-specific fashion. It will be of interest to determine how LKB1 and possibly AMPK compensate for autophagy deficiency.
Autophagy Promotes Cancer
Tumor Cell Autonomous Regulation of Autophagy
Unlike most normal cells and tissues where autophagy is active only at a low level in the fed state and induced by stress, autophagy is commonly and constitutively upregulated in cancer cells even in the presence of nutrients where it promotes tumor growth, survival and malignancy [1, 2]. These findings are clear in genetically engineered mouse models (GEMMs) for cancer where an essential autophagy gene is deleted at the onset of tumorigenesis specifically in tumor cells. Tumors generated in this way accumulate vast levels of autophagy substrates, most notably mitochondria, commonly resulting an oncocytic histology [23]. While there is a clear spectrum of autophagy addiction in different cancer models, this is seen in mouse models of Kras-driven lung and pancreas cancer, Braf-driven melanoma and lung cancer, and prostate, intestine and hereditary breast cancers [1, 2, 7].
Lkb1-deficient lung cancers are especially addicted to autophagy, consistent with the synthetic lethality of Atg7 and Lkb1 in mice, suggesting that inactivation of bioenergetic checkpoints in cancer is one means to cause autophagy addiction [24]. Similarly, tumors with p53 intact are more autophagy addicted, consistent with autophagy directly suppressing p53 activation in the brain with deleterious consequences including cell death [2, 12]. In the cancer setting; however, autophagy deficiency activates the p53 tumor suppressive function to limit tumor growth. In models of pancreas cancer autophagy deficiency promotes the benign pre-cancer state while blocking progression to malignancy in part by activating p53 [2]. These findings raised the question as to the biochemical nature of autophagy addiction in cancer.
Autophagy Regulates Tumor Cell Metabolism.
A key question is how does autophagy promote tumorigenesis at the biochemical level? As providing substrates to sustain metabolism is a main feature of autophagy, a comparison of the metabolism of isogenic autophagy-intact and -deficient lung cancer cell lines revealed the metabolic vulnerabilities caused by autophagy deficiency. Autophagy deficient tumor cells are sensitive to nutrient deprivation due to failure to degrade macromolecules and provide substrates to the tricarboxylic acid (TCA) cycle, which limits aspartate production (Figure 3A) [25]. Lack of sufficient aspartate limits do novo nucleotide synthesis, causing fatal nucleotide depletion, a phenotype rescued by nucleoside supplementation [25]. Autophagy deficiency also creates an energy crisis likely due to lack of TCA cycle support for the Electron Transport Chain (ETC) and ATP production (Figure 3A). Although there may be additional means by which autophagy promotes lung tumor metabolism, nucleotides are commonly identified as limiting in cancer and are the basis of the success of antagonistic nucleos(t)ide analogs and anti-folates in cancer chemotherapy [26].
Figure 3. Tumor cell autonomous and non-autonomous mechanisms by which autophagy promotes tumor metabolism.
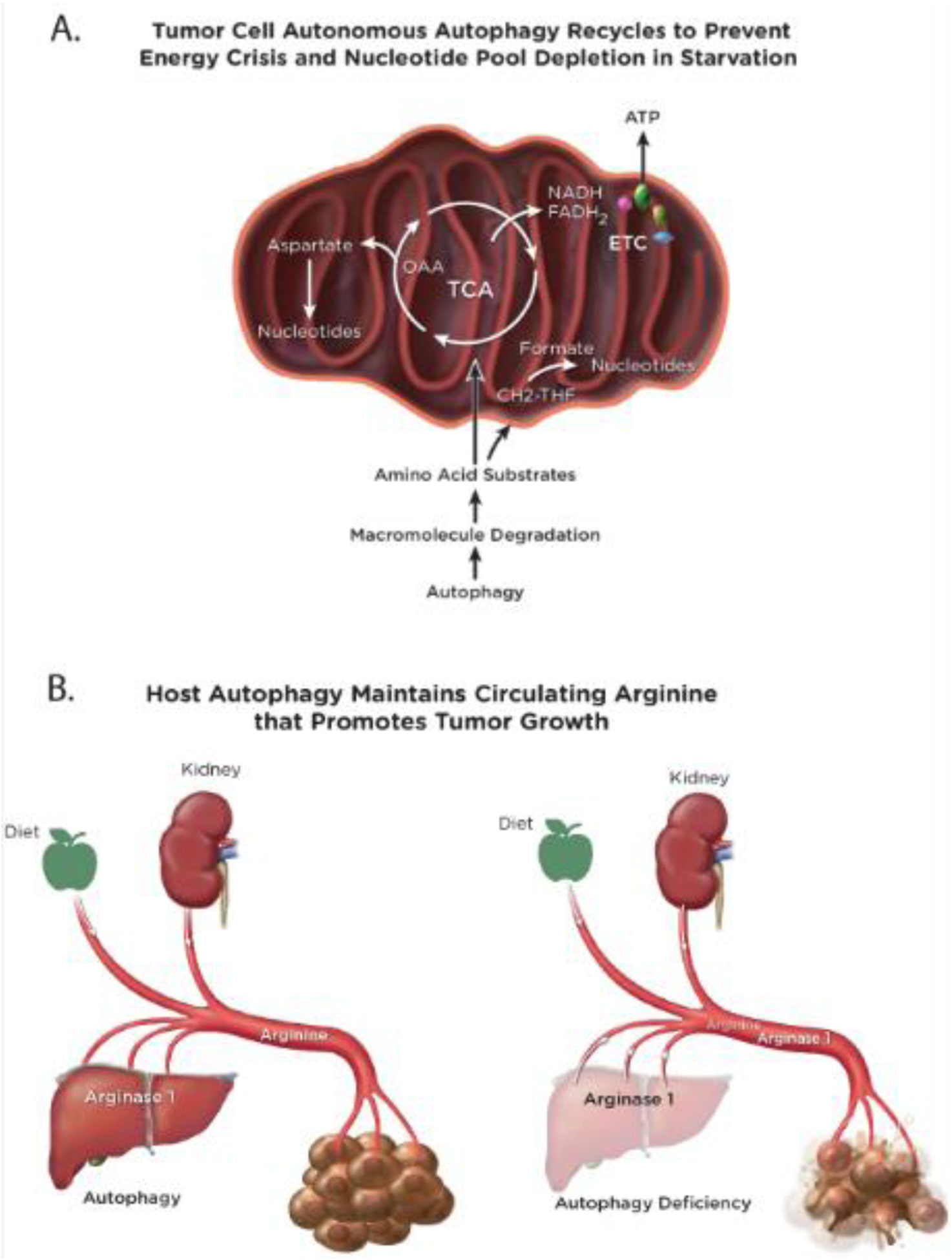
(A), In tumor cells autophagy recycles macromolecules to sustain the supply of substrates to the TCA cycle when nutrients are limiting, which enables continued nucleotide synthesis and energy homeostasis. (B), Host autophagy maintains circulating arginine levels by preventing the release of Arginase 1 from hepatocytes. Arginine is an essential tumor nutrient and its loss from the circulation in autophagy-deficient mice blocks tumor growth.
The metabolic role of autophagy in cancer cells may depend on cancer type. Metabolic analysis of pancreas cancer cell lines with and without autophagy revealed specific depletion of the cysteine pool and not that for the other amino acids [27]. Autophagy is required for plasma membrane localization of the cystine transporter SLC7A11, which gets diverted to lysosomes with loss of autophagy [27]. Whether this mechanism is specific to pancreas cancer or a general property of cancers more broadly is not known.
Selective Autophagy Dependency of Cancer.
Autophagy has a role in supporting the metabolism and survival of both normal tissues and cancer, which raised the question, are tumors more dependent on autophagy than normal tissues sufficient to provide a therapeutic window? This is a critical question as any autophagy inhibition approach to cancer therapy will most likely be given systemically, as such, selective sensitivity of tumors to autophagy inhibition is critical. Note that the GEMMs used to model cancer described above only tested the functional consequence of autophagy deficiency specifically within tumor cells. By engineering GEMMs for lung and pancreas cancer where genetic inactivation of autophagy is induced in mice with established cancer addressed this question. Indeed, switching off autophagy through conditional, systemic deletion of Atg7 in adult mice with established lung cancer causes dramatic tumor regression prior to onset of damage to host tissues [8]. Moreover, switching autophagy off through induced systemic expression of a dominant-negative version of Atg4b in mice with established pancreas cancer causes remarkable anti-tumor activity prior to detrimental consequences to normal tissues [28]. The above studies revealed the selective autophagy dependency of tumors.
Host Autophagy Promotes Tumor Growth
The pronounced anti-tumor activity of conditional, systemic autophagy ablation in mice with cancer was far greater that what was expected from studies with tumor cell-specific autophagy ablation, suggesting that host autophagy also plays a role in tumor promotion [8, 28]. To test this hypothesis, autophagy-proficient and -deficient adult host mice generated by conditional, systemic deletion of either Atg7 or Atg5 were assessed for the capacity to support tumor growth. While a variety of autophagy-proficient tumors grow on wild-type autophagy-proficient host mice, the same autophagy-proficient tumors commonly fail to grow on autophagy-deficient host mice [29]. Thus, host autophagy as well as tumor cell autonomous autophagy promote tumor growth, but by what mechanism?
As autophagy sustains metabolism, it is reasonable to hypothesize that host autophagy provides essential tumor nutrients through the circulation, the absence of which in autophagy-deficient host mice prevents tumor growth. Metabolic profiling of autophagy-proficient and -deficient adult mice through conditional, systemic deletion of either Atg7 or Atg5 revealed a number of altered circulating metabolites, the most striking of which is a substantial reduction of serum arginine levels with loss of autophagy [8, 29]. Arginine is essential for mTOR activation and tumors are commonly arginine auxotrophs due to silencing of genes essential for de-novo synthesis of arginine [30]. Proteomic analysis of serum revealed that autophagy-deficient mice have elevated levels of serum Arginase I (ARGI) the enzyme that degrades arginine [29]. In autophagy-deficient mice, ARGI is released from damaged liver hepatocytes into the circulation where it degrades arginine required for tumor growth, which is partly rescued by dietary supplementation with arginine [29]. Thus, one mechanism by which host autophagy promotes tumor growth is through sustaining an essential tumor nutrient, arginine (Figure 3B). Whether this is due to the requirement for arginine to sustain mTOR activity or remedy arginine auxotrophy, or both, is not known. Nonetheless, strategies to therapeutically target arginine by inducing its enzymatic degradation in cancer are ongoing [31].
Whereas circulating arginine maintained by autophagy is important for growth of a variety of cancers, pancreas cancer has a robust stromal component that impairs vasculature creating a nutrient poor environment. How do tumor cells meet their metabolic needs in this restrictive environment? Analysis of pancreas tumor and stromal cell metabolism revealed that autophagy in the stromal cells is required for the secretion of alanine, which is taken up by the tumor cells and used to support the TCA cycle [32]. Thus, autophagy promotes tumor growth at a distance (e.g. from the liver by preventing Arginase I release) [29] and locally (e.g. stromal alanine secretion) [32] to drive tumor metabolism and tumorigenesis.
Autophagy-Mediated Immune Evasion
Autophagy suppresses the innate immune response and inflammation [33] suggesting that autophagy may thereby suppress the adaptive immune response and contribute to tumor immune evasion. Unfortunately, the GEMMs for cancer that have been used to genetically dissect the role of autophagy in cancer in vivo are not optimal for assessing the capacity for tumor killing by T cells. GEMMs for cancer bypass the normal period of mutagenesis that occurs in human cancer that leads to oncogene activation and tumor suppressor gene inactivation since the oncogenic events occur instead through recombinase-mediated alterations in specific targeted genes. Hence, the resulting GEMM tumors lack the high truncal mutation burden and neoantigen load important for tumor recognition and killing by T cells and response to immune checkpoint inhibition [34] . As such, syngeneic, high mutation burden, antigenic mouse tumor cell lines transplanted on host mice with an intact immune system or assessed in vitro for tumor cell killing by T cells are necessary to address whether or not autophagy modulates an anti-tumor T-cell response. With these approaches, a series of recent studies demonstrate that inactivation of autophagy in either the tumor cells or the host, promotes cytokine production and activates tumor killing by T cells, with tumor growth being restored by depletion of T cells.
Autophagy Suppresses Activation of the Innate Immune Response.
One of the main functions of autophagy is suppressing activation of the innate immune response, and the induction of inflammation upon conditional deficiency in autophagy likely resides from the failure to clear damaged proteins, organelles and bacteria that triggers an innate immune response. The triggers of innate immunity, Damage-Associated Molecular Patterns and Pathogen-Associated Molecular Patterns (DAMPs and PAMPs) are autophagy substrates [33]. The elimination of DAMPs and PAMPs by their capture in autophagosomes and degradation is an important part of immune homeostasis to prevent exposure of damaged membranes and other organelle components that can trigger Toll-like receptor (TLR) signaling. Autophagy eliminates damaged mitochondria thereby preventing the release of mitochondrial DNA and the activation of the cytosolic DNA sensing mechanism (Cyclic GMP-AMP Synthase [cGAS] and Stimulator of Interferon Genes [STING]), type I Interferon-α (IFNα), and inflammasome activation. Autophagy also eliminates intracellular bacteria that would otherwise activate TLR signaling. The innate immune machinery is also degraded by autophagy indicating that autophagy is important for down regulating innate immune responses once they are triggered [6]. Cells with autophagy inactivated upregulate production and secretion of pro-inflammatory cytokines (e.g. type I and II interferons and tumor necrosis factor-α [TNFα]). Mice with deleted for essential autophagy genes display induction of proinflammatory cytokines (e.g. type I and II interferons, TNFα, chemokine (C-C motif) ligand 2 [CCL2], C-X-C motif chemokine ligand 10 [CXCL10]) [8, 35]. It is likely that some of the damage induced in normal tissues in autophagy-deficient mice is due to unchecked innate immune activation and this may also underlie human diseases associated with compromised function of essential autophagy genes such as Crohn’s disease that is a risk factor for colon cancer. Although the pro-inflammatory tumor microenvironment of autophagy-deficient primary tumors suppresses their growth it may be the cause of expansion of premalignant desmoplastic stroma in models of pancreas cancer [2, 7], and enhancement of metastasis in models of breast cancer [36–39].
Autophagy Suppresses Tumor Killing by T cells.
The innate immune response is a potent activator of the adaptive immune response. As autophagy suppresses innate immune responses, does this play a role in anti-tumor immunity, and if so how? In vitro experiments reveal evidence that autophagy can suppress the adaptive immune response directed at tumor cells. An in vitro genetic screen identified autophagy as a resistance mechanism to intrinsic killing by T cells [40]. Autophagy also protects from T cell killing through TNFα-induced cell death in vitro [41].
In vivo experiments demonstrated that autophagy suppresses the ability of T cells to kill tumor cells, and this correlates with less type II Interferon gamma (Ifnγ) expression [42]. Conversely, inactivation of autophagy in mice promotes the recognition and rejection of a variety of antigenic tumors by T cells [35, 43]. Autophagy is an important inhibitor of the innate and adaptive immune responses that creates a permissive environment for tumor immune evasion and growth.
Mechanisms of Autophagy-mediated Immune Evasion
A series of elegant in vivo experiments revealed several mechanisms by which autophagy suppresses an anti-tumor T cell response and how autophagy inhibition can be used to enhance tumor rejection by the immune system.
Inhibition of Antigen Presentation by Autophagy.
Pancreas cancers that are dominated by activating mutation in Kras have high levels of autophagy that promotes growth, survival and malignancy by sustaining metabolism via both tumor cell intrinsic and extrinsic mechanisms. Pancreas cancers also commonly suppress expression of the major histocompatibility complex class-I (MHC-I) and as a result have defective antigen presentation that prevents both tumor killing by T cells and the effectiveness of immunotherapy. MHC-I is recognized by the autophagy cargo receptor encoded by the Neighbor of BRCA1 gene product (NBR1), which targets it for degradation in lysosomes [43]. By eliminating MHC-I on the cell surface, autophagy prevents T cell-mediated tumor killing [43].
Hepatic Autophagy Immune Tolerance (HAIT).
Autophagy-deficient host mice are more efficient at rejecting antigenic tumors than autophagy-proficient mice, which is dependent on T cells [35]. In stark contrast to tumors grown on autophagy-proficient hosts, tumors grown on autophagy-deficient hosts show enhanced immune infiltration and gene expression signatures of activated type I and II interferon pathways. More specifically, the gene expression signature for STING activation is prominent in tumors grown on autophagy-deficient hosts. Genetic inactivation of both Sting and Atg7 in host mice restores tumor growth, demonstrating that innate immune activation via STING by loss of autophagy promotes tumor rejection [35]. Gene expression profiling of tumors growing on autophagy-proficient and -deficient hosts also revealed potent induction of the expression signature for Ifnγ in T cells specifically in tumors grown on autophagy-deficient hosts. Genetic deficiency in both Ifnγ and Atg7 restores defective tumor growth, demonstrating the essential role of Ifnγ in tumor cell killing by T cells induced by loss of host autophagy (Figure 4) [35]. Antigen presentation is also required for tumor rejection on autophagy-deficient hosts as downregulating β−2 microglobulin (a IFNγ -inducible gene) restores tumor growth [35]. Autophagy inactivation in mice produces an inflammatory response in the liver, suggesting that suppressing liver inflammation may contribute to immune evasion through autophagy. Specific deletion of autophagy in the liver hepatocytes is sufficient to promote tumor rejection that is dependent on both T cells and Ifnγ [35]. Thus, liver autophagy suppresses innate immune activation and thereby T cell activation and anti-tumor activity. HAIT may be overcome by inhibiting autophagy to enhance tumor rejection (Figure 4). Determining the exact means by which loss of autophagy activates STING and triggers type I and II interferon pathway activation in hepatocytes to activate T cells remains to be determined.
Figure 4. Autophagy suppresses innate and adaptive immune responses to promote tumor immune tolerance.
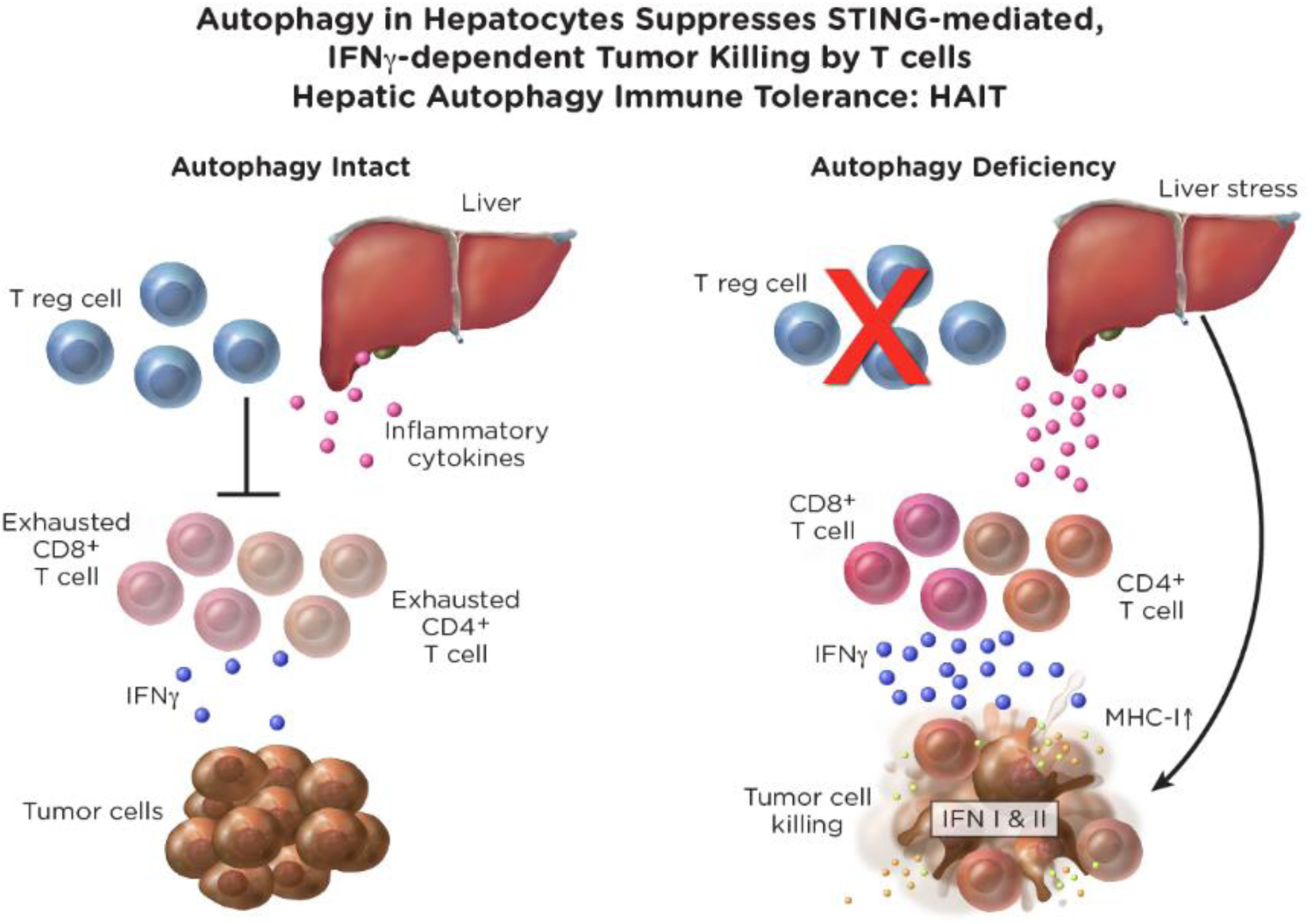
Liver autophagy suppresses STING and IFNγ activation to prevent tumor killing by T cells in MHC-I-dependent fashion. In pancreas cancer autophagy suppresses cell surface expression of MHC-I to promote immune evasion.
LAP.
A subset of autophagy pathway components, especially the conjugation machinery (Atg5, 7, 12, and 16) plays an additional role in the removal of dead cells by phagocytosis using a single membrane conjugated with the autophagy cargo receptor LC3 by a process called LAP [11]. Macrophages are professional phagocytes that remove dead cells, eliminating DAMPs and PAMPs, which suppresses inflammation [44]. Specifically inhibiting LAP but not autophagy in macrophages activates tumor killing by T cells. LAP mediated tumor killing requires STING and the type-I interferon response [45], suggesting that failure to eliminate dead cells and presumably their DNA through LAP in macrophages activates STING and type I interferon signaling to boost an anti-tumor T cell response. Interestingly HAIT is independent of LAP [35], indicating that inhibiting shared components for both LAP and autophagy (e.g. ATG7) will promote inflammation by two overlapping mechanisms to enhance an anti-tumor T cell response.
Autophagy Inhibition Enhances Response to Targeted Therapy and to Immune Checkpoint Blockade.
In Ras- and Braf-driven cancers autophagy is induced by MAP kinase pathway inhibition and promotes treatment resistance, and coordinate inhibition of autophagy with chloroquine analogs, which block autophagic cargo degradation in lysosomes [46], enhances treatment response [47–50]. In breast cancer autophagy deficiency promotes radiation sensitivity through mitochondrial DNA release and STING activation [51]. More compelling is the prospect that autophagy inhibition enhances response to immunotherapy. Small molecule inhibition of vacuolar protein sorting 34 (VPS34), which blocks autophagosome formation, promotes immune infiltration and enhances response to immune checkpoint blockade [52]. In melanoma inhibition of palmitoyl-protein thioesterase 1 (PTP1) in lysosomes, which interferes with lysosome function does the same [53]. Autophagy inhibition with chloroquine enhances the anti-tumor activity of immune checkpoint blockade in pancreas cancer by restoring cell surface MHC-I expression [43]. Thus. Autophagy inhibition may be additionally useful in preventing treatment resistance to targeted therapy as well as immunotherapy.
Concluding Remarks
Autophagy inhibition can induce selective tumor cell killing by tumor cell intrinsic and extrinsic mechanisms. Recycling by autophagy sustains tumor intrinsic metabolic function and host autophagy maintains essential tumor nutrients in the circulation and tumor microenvironment. Autophagy suppresses innate and adaptive immune responses to facilitate immune evasion and immunotherapy resistance. By suppressing type I and II interferon signaling and preventing cell surface expression of MHC-I , autophagy prevents tumor killing by T cells. These findings provide a novel opportunity to promote tumor rejection by activating the anti-tumor immune response by inhibiting autophagy and to broaden success of immunotherapies by coordinately inhibiting autophagy. Further work is required to determine how to best inhibit the autophagy pathway, eliminate essential tumor nutrients, and to defeat tumor immune tolerance conferred by autophagy to promote treatment response (see Outstanding Questions).
Outstanding Questions.
How does autophagy suppress p53?
How do NRF2 and LKB1 compensate for loss of autophagy?
What are other mechanisms that confer tissue-specific autophagy dependence?
Are there essential tumor nutrients supplied by autophagy beyond arginine and alanine?
What is the best way to inhibit autophagy for cancer therapy?
How can autophagy dependence be assessed in human cancers?
How can the efficacy of autophagy inhibition be maximized, and the predicted toxicities be minimized for cancer therapy?
Are there other essential tumor nutrients supplied by autophagy?
How does autophagy inactivation trigger the innate immune response?
Will autophagy inhibition suppress primary tumor growth at the expense of promoting metastasis?
Can we mimic tumor rejection by targeting mechanisms downstream of autophagy loss?
What are the best therapy combinations with autophagy inhibitors?
Can autophagy inhibition prevent emergence of resistance to immunotherapy?
Highlights.
Cancers upregulate autophagy to sustain metabolism and survival in a tumor-cell autonomous fashion
Autophagy sustains the essential tumor nutrients arginine and alanine
Autophagy mitigates stress and is functionally interdependent with p53, NRF2 and LKB1 stress response pathways
Host autophagy promotes tumor growth by providing essential tumor nutrients in a tumor-cell non-autonomous fashion
Autophagy promotes immune tolerance by suppressing and T cell activation
Acknowledgments
EW is supported by the NIH grants R01 CA243547, CA188096, CA163591, and the Ludwig Princeton Branch, Ludwig Institute for Cancer Research
EL is supported by the NIH grant R01 CA243547
JYG is supported by NIH grant R01CA237347-01A1, ACS grant 134036-RSG-19-165-01-TBG, the GO2 Foundation for Lung Cancer, and the New Jersey Health Foundation.
Glossary
- AMP-activated protein kinase (AMPK)
Kinase that provides energy stress adaptation
- Arginase I (ARGI)
Enzyme that degrades the amino acid arginine
- Essential autophagy gene (Atg)
Gene whose expression is essential for the autophagy pathway
- Chemokine (C-C motif) ligand 2 [CCL2]
Inflammatory cytokine
- C-X-C motif chemokine ligand 10 [CXCL10]
Inflammatory cytokine
- Cyclic GMP-AMP Synthase [cGAS]
Sensor of cytoplasmic DNA and STING activator
- Stimulator of Interferon Genes [STING]
In response to cGAS activates type I interferon signaling
- Damage-Associated Molecular Patterns and Pathogen-Associated Molecular Patterns (DAMPs and PAMPs)
Activators of the innate immune response to damage and infection
- Electron Transport Chain (ETC)
Enzyme complexes that function to pump protons out of mitochondria to create a membrane potential that drives ATP and energy production
- Essential autophagy gene (Atg)
Gene essential for function of the autophagy pathway
- Genetically engineered mouse model (GEMM)
Mice with engineered mutations that cause cancer
- Hepatic Autophagy Immune Tolerance (HAIT)
Liver inflammatory response blocked by autophagy that promotes immune evasion and tumor growth
- Hypoxia-inducible factors (HIFs)
Transcription factors induced by and protect from oxygen deprivation that activate autophagy
- LC3-mediated phagocytosis (LAP)
Phagocytic process that requires a subset of essential autophagy genes to engulf and degrade dead cells and suppress inflammation
- Liver kinase B1 (Stk11/Lkb1)
Kinase that responds to and protects from energy stress upstream of AMPK
- Major histocompatibility complex class-I (MHC-I)
Cell surface complex that presents antigens for immune recognition
- Mammalian Target of Rapamycin (mTOR)
Kinase and nutrient responsive master regulator of cell growth that inhibits autophagy
- Microtubule-associated protein 1A/1B-light chain 3 (LC3)
Autophagosome membrane bound autophagy cargo receptor
- Neighbor of BRCA1 gene product (NBR1)
Autophagy cargo receptor
- NRF2 inhibitor Kelch-like ECH-associated protein 1 (KEAP1)
Binds to and inhibits NRF2
- Nuclear factor erythroid 2-related factor 2 (NEF2)
Transcription factor and master regulator of the oxidative stress response
- Sequestosome-1 (SQSTM1/p62)
Autophagy cargo receptor
- Toll-like receptor (TLR)
Signaling complexes that are activated by DAMPs and PAMPs
- Tricarboxylic acid (TCA) cycle
Enzyme complexes that oxidize acetyl-CoA derived from carbohydrates, fats, and proteins to generate energy and building blocks
- Tumor necrosis factor-α [TNFα]
Inflammatory cytokine
- Type I Interferon-α (IFNα)
Cytokine that activates transcription in response to triggers of the innate immune response
- Type II Interferon gamma (IFNγ)
Cytokine critical for immune activation and defense from viral infection
- White adipose tissue (WAT)
White fat tissue
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures: EW is a founder of Vescor Therapeutics and a stockholder in Forma Therapeutics.
References
- 1.Amaravadi R et al. (2016) Recent insights into the function of autophagy in cancer. Genes Dev 30 (17), 1913–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kimmelman AC and White E (2017) Autophagy and Tumor Metabolism. Cell Metab 25 (5), 1037–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mizushima N and Levine B (2020) Autophagy in Human Diseases. N Engl J Med 383 (16), 1564–1576. [DOI] [PubMed] [Google Scholar]
- 4.Mizushima N (2020) The ATG conjugation systems in autophagy. Curr Opin Cell Biol 63, 1–10. [DOI] [PubMed] [Google Scholar]
- 5.Morishita H and Mizushima N (2019) Diverse Cellular Roles of Autophagy. Annu Rev Cell Dev Biol 35, 453–475. [DOI] [PubMed] [Google Scholar]
- 6.Mathew R et al. (2014) Functional role of autophagy-mediated proteome remodeling in cell survival signaling and innate immunity. Mol Cell 55 (6), 916–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Poillet-Perez L and White E (2019) Role of tumor and host autophagy in cancer metabolism. Genes Dev 33 (11–12), 610–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Karsli-Uzunbas G et al. (2014) Autophagy is required for glucose homeostasis and lung tumor maintenance. Cancer Discov 4 (8), 914–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yang Y et al. (2020) Autophagy promotes mammalian survival by suppressing oxidative stress and p53. Genes Dev 34 (9–10), 688–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Newman LE and Shadel GS (2018) Pink1/Parkin link inflammation, mitochondrial stress, and neurodegeneration. J Cell Biol 217 (10), 3327–3329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Heckmann BL and Green DR (2019) LC3-associated phagocytosis at a glance. J Cell Sci 132 (5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.White E (2016) Autophagy and p53. Cold Spring Harb Perspect Med 6 (4), a026120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang Y and White E (2020) Autophagy suppresses TRP53/p53 and oxidative stress to enable mammalian survival. Autophagy 16 (7), 1355–1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kenzelmann Broz D et al. (2013) Global genomic profiling reveals an extensive p53-regulated autophagy program contributing to key p53 responses. Genes Dev 27 (9), 1016–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Taguchi K and Yamamoto M (2020) The KEAP1-NRF2 System as a Molecular Target of Cancer Treatment. Cancers (Basel) 13 (1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Komatsu M et al. (2010) The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat Cell Biol 12 (3), 213–23. [DOI] [PubMed] [Google Scholar]
- 17.Lau A et al. (2010) A noncanonical mechanism of Nrf2 activation by autophagy deficiency: direct interaction between Keap1 and p62. Mol Cell Biol 30 (13), 3275–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Towers CG et al. (2020) Autophagy and cancer: Modulation of cell death pathways and cancer cell adaptations. J Cell Biol 219 (1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Su H et al. (2021) Cancer cells escape autophagy inhibition via NRF2-induced macropinocytosis. Cancer Cell. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Herzig S and Shaw RJ (2018) AMPK: guardian of metabolism and mitochondrial homeostasis. Nat Rev Mol Cell Biol 19 (2), 121–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lin SC and Hardie DG (2018) AMPK: Sensing Glucose as well as Cellular Energy Status. Cell Metab 27 (2), 299–313. [DOI] [PubMed] [Google Scholar]
- 22.Khayati K et al. (2020) Autophagy compensates for Lkb1 loss to maintain adult mice homeostasis and survival. Elife 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guo JY et al. (2013) Autophagy suppresses progression of K-ras-induced lung tumors to oncocytomas and maintains lipid homeostasis. Genes Dev 27 (13), 1447–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bhatt V et al. (2019) Autophagy modulates lipid metabolism to maintain metabolic flexibility for Lkb1-deficient Kras-driven lung tumorigenesis. Genes Dev 33 (3–4), 150–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Guo JY et al. (2016) Autophagy provides metabolic substrates to maintain energy charge and nucleotide pools in Ras-driven lung cancer cells. Genes Dev 30 (15), 1704–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tsesmetzis N et al. (2018) Nucleobase and Nucleoside Analogues: Resistance and Re-Sensitisation at the Level of Pharmacokinetics, Pharmacodynamics and Metabolism. Cancers (Basel) 10 (7). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mukhopadhyay S et al. (2021) Autophagy is required for proper cysteine homeostasis in pancreatic cancer through regulation of SLC7A11. Proc Natl Acad Sci U S A 118 (6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yang A et al. (2018) Autophagy Sustains Pancreatic Cancer Growth through Both Cell-Autonomous and Nonautonomous Mechanisms. Cancer Discov 8 (3), 276–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Poillet-Perez L et al. (2018) Autophagy maintains tumour growth through circulating arginine. Nature 563 (7732), 569–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Keshet R et al. (2018) Rewiring urea cycle metabolism in cancer to support anabolism. Nat Rev Cancer 18 (10), 634–645. [DOI] [PubMed] [Google Scholar]
- 31.Fultang L et al. (2016) Molecular basis and current strategies of therapeutic arginine depletion for cancer. Int J Cancer 139 (3), 501–9. [DOI] [PubMed] [Google Scholar]
- 32.Sousa CM et al. (2016) Pancreatic stellate cells support tumour metabolism through autophagic alanine secretion. Nature 536 (7617), 479–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Deretic V and Levine B (2018) Autophagy balances inflammation in innate immunity. Autophagy 14 (2), 243–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ganesan S and Mehnert J (2020) Biomarkers for Response to Immune Checkpoint Blockade. Annual Review of Cancer Biology 4 (1), 331–351. [Google Scholar]
- 35.Poillet-Perez L et al. (2020) Autophagy promotes growth of tumors with high mutational burden by inhibiting a T-cell immune response. Nature Cancer 1 (9), 923–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Aqbi HF et al. (2018) Autophagy-deficient breast cancer shows early tumor recurrence and escape from dormancy. Oncotarget 9 (31), 22113–22122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.La Belle Flynn A et al. (2019) Autophagy inhibition elicits emergence from metastatic dormancy by inducing and stabilizing Pfkfb3 expression. Nat Commun 10 (1), 3668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Marsh T et al. (2020) Autophagic Degradation of NBR1 Restricts Metastatic Outgrowth during Mammary Tumor Progression. Dev Cell 52 (5), 591–604 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vera-Ramirez L et al. (2018) Autophagy promotes the survival of dormant breast cancer cells and metastatic tumour recurrence. Nat Commun 9 (1), 1944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lawson KA et al. (2020) Functional genomic landscape of cancer-intrinsic evasion of killing by T cells. Nature 586 (7827), 120–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhu XG et al. (2021) Functional Genomics In Vivo Reveal Metabolic Dependencies of Pancreatic Cancer Cells. Cell Metab 33 (1), 211–221 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.DeVorkin L et al. (2019) Autophagy Regulation of Metabolism Is Required for CD8(+) T Cell Anti-tumor Immunity. Cell Rep 27 (2), 502–513 e5. [DOI] [PubMed] [Google Scholar]
- 43.Yamamoto K et al. (2020) Autophagy promotes immune evasion of pancreatic cancer by degrading MHC-I. Nature 581 (7806), 100–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Martinez J et al. (2016) Noncanonical autophagy inhibits the autoinflammatory, lupus-like response to dying cells. Nature 533 (7601), 115–9. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 45.Cunha LD et al. (2018) LC3-Associated Phagocytosis in Myeloid Cells Promotes Tumor Immune Tolerance. Cell 175 (2), 429–441 e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Amaravadi RK et al. (2019) Targeting Autophagy in Cancer: Recent Advances and Future Directions. Cancer Discov 9 (9), 1167–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bryant KL et al. (2019) Combination of ERK and autophagy inhibition as a treatment approach for pancreatic cancer. Nat Med 25 (4), 628–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kinsey CG et al. (2019) Protective autophagy elicited by RAF-->MEK-->ERK inhibition suggests a treatment strategy for RAS-driven cancers. Nat Med 25 (4), 620–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lee CS et al. (2019) MAP kinase and autophagy pathways cooperate to maintain RAS mutant cancer cell survival. Proc Natl Acad Sci U S A 116 (10), 4508–4517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.White E (2019) Blockade of RAF and autophagy is the one-two punch to take out Ras. Proc Natl Acad Sci U S A 116 (10), 3965–3967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yamazaki T et al. (2020) Mitochondrial DNA drives abscopal responses to radiation that are inhibited by autophagy. Nat Immunol 21 (10), 1160–1171. [DOI] [PubMed] [Google Scholar]
- 52.Noman MZ et al. (2020) Inhibition of Vps34 reprograms cold into hot inflamed tumors and improves anti-PD-1/PD-L1 immunotherapy. Sci Adv 6 (18), eaax7881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sharma G et al. (2020) PPT1 inhibition enhances the antitumor activity of anti-PD-1 antibody in melanoma. JCI Insight 5 (17). [DOI] [PMC free article] [PubMed] [Google Scholar]