

Abstract
Steroid-resistant nephrotic syndrome (SRNS) is a genetically heterogeneous kidney disease that is the second most frequent cause of kidney failure in the first 2 decades of life. Despite the identification of mutations in more than 39 genes as causing SRNS, and the localization of its pathogenesis to glomerular podocytes, the disease mechanisms of SRNS remain poorly understood and no universally safe and effective therapy exists to treat patients with this condition. Recently, genetic research has identified a subgroup of SRNS patients whose kidney pathology is caused by primary coenzyme Q10 (CoQ10) deficiency due to recessive mutations in genes that encode proteins in the CoQ10 biosynthesis pathway. Clinical and preclinical studies show that primary CoQ10 deficiency may be responsive to treatment with CoQ10 supplements bypassing the biosynthesis defects. Coenzyme Q10 is an essential component of the mitochondrial respiratory chain, where it transports electrons from complexes I and II to complex III. Studies in yeast and mammalian model systems have recently identified the molecular functions of the individual CoQ10 biosynthesis complex proteins, validated these findings, and provided an impetus for developing therapeutic compounds to replenish CoQ10 levels in the tissues/organs and thus prevent the destruction of tissues due to mitochondrial OXPHOS deficiencies. In this review, we will summarize the clinical findings of the kidney pathophysiology of primary CoQ10 deficiencies and discuss recent advances in the development of therapies to counter CoQ10 deficiency in tissues.
Keywords: CoQ10, Ubiquinone, Steroid-resistant nephrotic syndrome, Mitochondriopathies
Introduction
Nephrotic syndrome (NS) is the most common glomerular disorder in individuals under 21 years, characterized by heavy proteinuria, hypoalbuminemia, edema, and hyperlipidemia. The estimated incidence of NS ranges from 2 to 16.9 per 100,000 children worldwide [1–4]. Based on pediatric patients’ response to treatment with steroids, the first-line therapy of NS, the syndrome is further classified as a steroid-sensitive nephrotic syndrome (SSNS) if patients achieve remission within 8–12 weeks of steroid therapy, or steroid-resistant nephrotic syndrome (SRNS) if patients continue to have proteinuria despite steroid administration [5, 6]. SSNS constitutes the predominant form of NS (~90%) and usually has a favorable long-term outcome. In contrast, SRNS which makes up ~ 10% of NS cases [7] has a poor prognosis, with 30–40% of patients developing kidney failure within 10 years [6], and is the second most frequent cause of kidney failure requiring kidney replacement therapy in individuals under 21 years of age [8].
In the majority of cases, the histological pattern of SRNS is characterized by a glomerular injury in the form of childhood-onset focal segmental glomerulosclerosis (FSGS), and less frequently by a developmental-onset diffuse mesangial sclerosis (DMS) [9]. At a cellular level, both types of glomerular histology represent impaired podocyte function and loss of podocyte differentiation. Podocytes are highly specialized epithelial cells that line the glomerular capillaries with whom they share the glomerular basement membrane and whose integrity plays an important role in ultrafiltration. Neighboring podocytes develop elaborate interdigitating foot processes, which form the slit diaphragm, a selective filtration barrier consisting of various cytoskeletal proteins. Formation and maintenance of the slit diaphragm is energy intensive, but critical for normal podocyte function. To compensate for the high energy demand it has been suggested that podocytes depend on mitochondrial oxidative phosphorylation, while glycolysis has been thought to be less relevant [10]. However, recent investigations using in vivo and in vitro models of podocyte-specific interference with mitochondrial metabolism have contrasted this view and demonstrated that anaerobic glycolysis plays the predominant role as the energy source in podocytes [11]. Clearly, further investigations are needed to establish the precise roles of either energy-generating pathway in the etiology of different glomerular diseases.
Over the last two decades, rapid progress in genetic sequencing technology and decrease in cost has led to a significant increase in the ability to identify the molecular causes of SRNS. To date, mutations in at least 50 genes are known to cause either autosomal recessive or dominant forms of SRNS [7, 12]. In addition to isolated glomerular disease, SRNS can occur as part of a syndromic disorder with extrarenal manifestations [13]. For excellent reviews on the genetics of SRNS and the use of genetic testing in SRNS, please see [7, 13]. Through improved understanding of the genetic causes of SRNS, it has been shown that any disruption of podocyte cell function or structure is central in the pathogenesis of disease. The heterogeneity of disease-causing mutations highlights the molecular complexity of SRNS pathology, but also has renewed a hope for improved pathway-based management of SRNS that is based on a molecular diagnosis instead of relying on broad histopathologic classification, something that has plagued past clinical trials and hindered the development of effective novel therapies [14]. Importantly, genetic testing of SRNS patients provides a greater prognostic ability to assess patient’s responsiveness to immunosuppressants and clinical outcomes [13]. This will also allow future trials to appropriately group SRNS patients in similar cohorts.
Significantly, recent clinical and preclinical studies have demonstrated that individuals with a primary coenzyme Q10 (CoQ10) deficiency, a rare cause of SRNS, may benefit from a treatment with dietary CoQ10 supplementation or its precursor analogs. Indeed, genetic testing has identified a small cohort of SRNS patients with mutations in genes encoding the CoQ10 biosynthesis pathway enzymes PDSS1, PDSS2, COQ2, COQ6, and COQ8B (ADCK4) [15–21]. Primary defects of CoQ10 are associated with decreased mitochondrial oxidative phosphorylation (OXPHOS) in cells and tissues with high energy requirement, including the glomerular podocytes and renal tubular cells. In addition to SRNS, primary CoQ10 deficiency is associated with several other extrarenal clinical phenotypes including (1) encephalomyopathy with seizures and ataxia [22], (2) a multisystem infantile form marked by encephalopathy, cardiomyopathy, and kidney failure [23], (3) a predominantly cerebellar form with ataxia and cerebellar atrophy [24], (4) Leigh syndrome with growth retardation [25], and (5) an isolated myopathic form [26]. Here, we will focus on the clinical findings associated with SRNS caused by causal mutations in the CoQ10 biosynthesis pathway and discuss recent advances in our understanding of the underlying molecular mechanisms.
CoQ10 biosynthesis
Coenzyme Q (CoQ) or ubiquinone is a lipid protein that is found in all cell membranes of eukaryotic organisms. The CoQ molecule consists of a hydrophilic redox-active benzoquinone ring and a hydrophobic polyisoprenoid tail that is embedded in the lipid bilayer of the cell membranes (Fig. 1). The length of the polyisoprenoid tail varies between species—the human isoform consists mostly of 10 isoprene residues (CoQ10), whereas the rodent isoform has 9 (CoQ9) and yeast has 6 residues (CoQ6). CoQ is synthesized de novo in each cell, and based on biochemical studies performed in rats, it is estimated that only ~ 2–4% of the dietary CoQ10 is taken up by the body [28]. In the same study, it was also shown that CoQ10 supplementation did not affect the endogenous CoQ9 biosynthesis [28]. While there may exist species-specific and formulation-specific differences in CoQ10 absorption, it is generally accepted that CoQ10 has low bioavailability in human tissue [29, 30]. In the human body, CoQ10 membrane concentration varies widely between different organs and between cell types within tissues. Its levels are highest in cells that harbor greater numbers of mitochondria and have high metabolic activity, such as podocytes in the kidney, cardiomyocytes, nerve cells in the brain, and myocytes in the skeletal muscle [29].
Fig. 1.
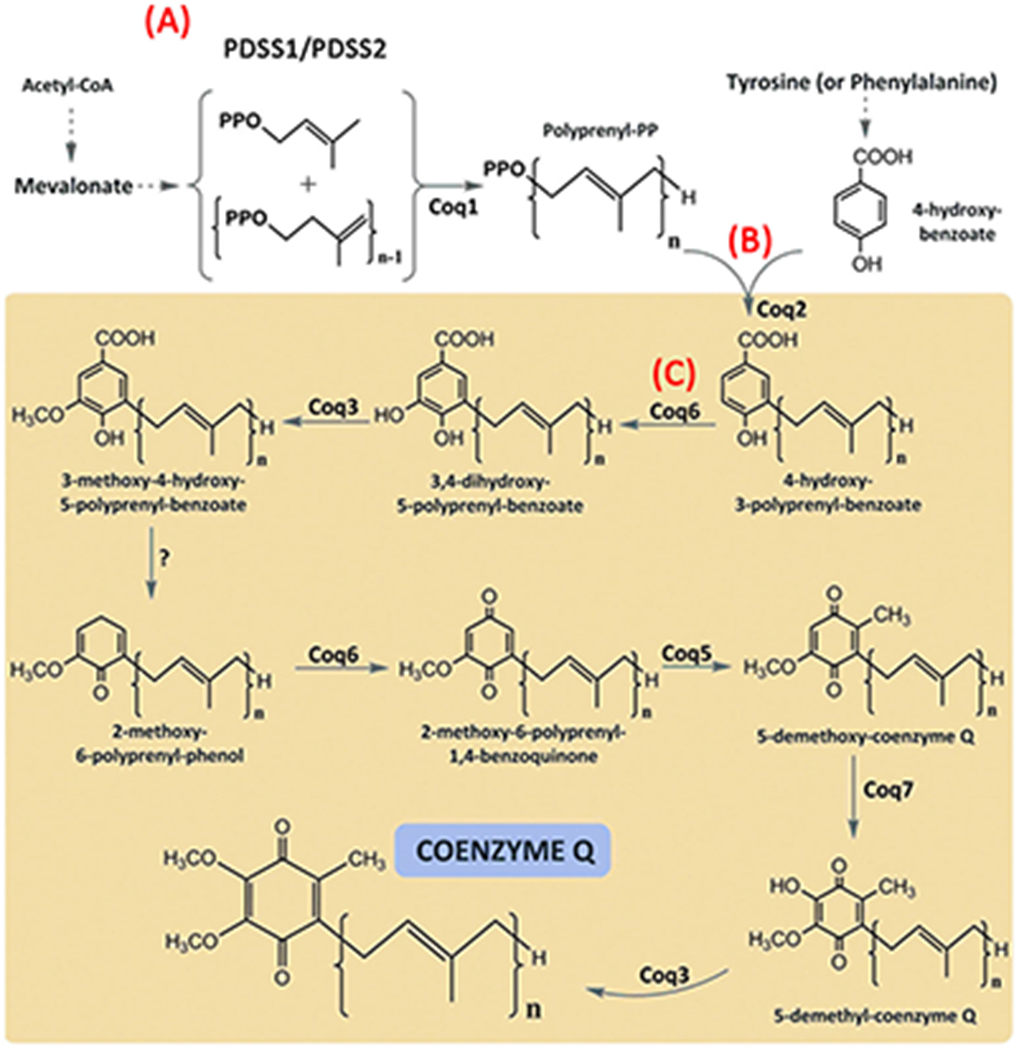
Schematic view of CoQ biosynthesis pathway in a eukaryotic cell. (A) The isoprene units are derived from the mevalonate pathway and polymerized by PDSS1/PDSS2 heterotetramer in the cytoplasm. (B) COQ2 links the polyprenylated tail and the qui-none head (4-hydroxybenzoate) in mitochondria. (C) The rest of the CoQ biosynthesis reactions take place in the matrix side of the mitochondrial inner membrane (yellow box), consisting of sequential modifications of the aromatic ring. Coq6 and Coq7 are hydroxylases; Coq3 andCoq5 are methylases; Coq4, Coq8, Coq9, and Coq10 are proteins with regulatory functions. Adapted from [27]; used with permission
In a eukaryotic cell, CoQ biosynthesis takes place in the mitochondrial inner membrane in a membrane-bound CoQ complex, which is a multienzyme aggregate, orthologous to the yeast CoQ biosynthetic complex named complex Q [31]. The constituent proteins of the complex are encoded by nuclear genes and their functions are highly conserved through evolution, enabling researchers to perform complementation studies in which human CoQ10 biosynthesis pathway genes are substituted for their yeast orthologs to examine their cellular function. In humans, the CoQ10 biosynthesis pathway is made of at least 16 enzymes (PDSS1, PDSS2, COQ2, COQ3, COQ4, COQ5, COQ6, COQ7, COQ8A, COQ8B, COQ9, COQ10A, COQ10B, FDX1L, FDXR, and ALDH3A1) [32, 33].
In mammalian cells, CoQ biosynthesis starts with the production of the redox-active benzoquinone ring—4-hydroxybenzoate (4HB)—from amino acids phenylalanine and tyrosine, and the formation of isoprene units from acetyl-CoA via the mevalonate pathway [31]. In the first CoQ biosynthetic step, the PDSS1/PDSS2 heterotetramer catalyzes the polymerization of the polyisoprenoid tail, which in humans contains 10 isoprene subunits (Fig. 1A). Subsequent to the hydrophobic tail formation, COQ2 catalyzes the condensation reaction by which 4HB is attached to the membrane-embedded polyisoprenoid tail (Fig. 1B). Once the head group is attached, the newly formed molecule is translocated to the CoQ complex where the p-hydroxybenzoate head group is further modified through hydroxylation, methylation, and decarboxylation reactions that are carried out by the individual peptides in the complex including COQ6 (Fig. 1C). Not all of the CoQ complex proteins are enzymes; for instance, COQ4, COQ5, and COQ9 are believed to be scaffolds that anchor the complex to the mitochondrial inner membrane, thus stabilizing the complex [31], whereas COQ8A and COQ8B enhance the catalytic activity of the complex and minimize the escape of intermediates [34]. Whether the polypeptides that form the CoQ complex localize to other cellular membranes besides the mitochondrial inner membrane remains controversial and warrants further research. For instance, COQ6, COQ7, and COQ9 have all been demonstrated to localize to the Golgi complex and to the cellular processes in cultured human podocytes in addition to mitochondria [17].
Functions of CoQ10
Most of the energy in a eukaryotic cell is generated in mitochondria through oxidative phosphorylation (OXPHOS) that uses oxygen and simple sugars to create adenosine triphosphate (ATP). CoQ10 has a central role in ATP production by transporting electrons within the mitochondrial respiratory chain from complex I (NADH-ubiquinone oxireductase) and complex II (succinate-ubiquinone oxireductase) to complex III (ubiquinol-cytochrome c reductase) after receiving electrons from NADH or succinate respectively [29]. In addition, CoQ10 mediates the transfer of electrons from the electron transport flavoprotein dehydrogenases family (ETFDH) enzymes, which are involved in fatty acid β-oxidation and branched-chain amino acid oxidation pathways, to complex III [35, 36]. The electron transfer by CoQ10 is facilitated by the 4HB ring within the CoQ10 molecule, which can oscillate between a fully reduced form (ubiquinol or CoQH2), after receiving 2 electrons and a fully oxidized form (ubiquinone or CoQ), after losing 2 electrons. Because of CoQ10’s essential function as an electron carrier in the mitochondrial respiratory chain, one of the major consequences of CoQ10 biosynthesis defects is impaired mitochondrial bioenergetics and reduced ATP synthesis [37].
In addition to its role in mitochondrial OXPHOS, CoQ10 in its oxidized form (ubiquinone) is a potent lipid-soluble antioxidant, protecting cellular membranes from lipid peroxidation by quenching reactive oxygen species (ROS) that form during mitochondrial respiration [38]. In agreement with this notion, increased ROS production and oxidative stress has been observed in the kidney tissue of Pdss2-knockout mice [39], as well as in the brains of Coq9-mutant mice [40]. Similarly, cultured human fibroblasts that were isolated from CoQ10-deficient patients with mutations in COQ2 displayed increased ROS production and oxidative damage [41]. Based on the biochemical evidence that CoQ10 acts as a membrane-bound antioxidant, it has been postulated that the tissue- and organ-specific phenotypes that are observed in human patients with primary CoQ10-deficiency, such as podocyte abnormalities, may be due to a selective vulnerability of certain cell types (i.e., podocytes) to increased levels of oxidative stress [42]. Indeed, the podocyte-specific kidney pathology of several COQ-gene mutations (COQ6, COQ8B) seems to provide support to this idea [17, 18].
An additional important biochemical role of CoQ10 in the cell is regulation of nucleotide biosynthesis through its function as an essential co-factor for mitochondrial dihydro-orotate dehydrogenase, an intermediate enzyme in de novo pyrimidine biosynthesis [43]. Evidence that this may be a clinically relevant pathway in CoQ10-deficient patients was provided by the demonstration that low pyrimidine levels and impaired cell growth in COQ2 patient fibroblasts could be reversed by supplementing the fibroblasts with uridine, a pyrimidine analog [44].
Genetic causes of SRNS due to CoQ10 biosynthesis deficiency
Primary CoQ10 deficiency was first associated with human disease in 1989 when Ogasahara et al. published a clinical report on two siblings who presented with recurrent rhabdomyolysis, associated with seizures and mental retardation, even though the molecular pathophysiology of the disease remained unknown [22]. It was not until 2006 when the molecular cause of CoQ10 deficiency was associated with a mutation in a gene involved in CoQ10 biosynthesis [45]. Within the last 15 years, numerous new cases of primary CoQ10 deficiency presenting with a broad spectrum of organ involvement, including SRNS and kidney tubulopathies among others, have been reported. Out of the 16 genes which are currently known to be involved in CoQ10 biosynthesis, mutations in 9 have been identified as causing human primary CoQ10 deficiency, while mutations in a subset of 5 of these have been reported to affect kidney development or contribute to the development of nephrotic syndrome (Table 1). Genes involved in the coenzyme Q10 biosynthesis pathway (PDSS2, COQ2, COQ6, and COQ8B/ADCK4) were found to be mutated in about 1–2.7% of SRNS cases [47, 65].
Table 1.
Coq10 biosynthesis pathway genes mutated in human SRNS
| Gene | Protein | Kidney phenotype | Extrarenal phenotype | CoQ10 dose | Outcome of CoQ10 treatment | References |
|---|---|---|---|---|---|---|
| PDSS1 | Prenyldiphosphate synthase subunit 1 | NS, tubular epithelial damage | Failure to thrive, developmental delay | − | − | [46] |
| PDSS2 | Prenyldiphosphate | SRNS | Leigh syndrome | 50 mg/day | No remission | [15] |
| synthase subunit 2 | SRNS | − | − | 47] | ||
| DMS | Encephalomyopathy, deafness, retinitis pigmentosa, hypertrophic cardiomyopathy | 20 mg/kd/day | 48] | |||
| COQ2 | Coenzyme Q2 4-hydroxybenzoate polyprenyltransferase | FSGS | Encephalomyopathy, nystagmus secondary to bilateral optic nerve atrophy, seizures, and developmental delay, right hemiplegia, myoclonus, swallowing difficulties | 30 mg/kg/day | 1 patient partial remission, 1 patient kidney failure | [45, 49] |
| NS | Seizures, liver failure, anemia, pancytopenia,insulin-dependent diabetes | − | − | [50] | ||
| FSGS, crescentic glomerulonephritis | Epileptic encephalomyopathy, hypotonia, psychomotor delay, optic nerve atrophy, | 30 mg/kg/day | No remission (kidney failure) | [16] | ||
| FSGS | − | − | [51] | |||
| COQ6 | Coenzyme Q6 monooxygenase | FSGS | Mild neurological symptoms | Unspecified | 2 patients no remission (kidney failure) | [52] |
| FSGS | 30 mg/kg/day | 1 patient remission, 1 patient partial remission, 1 patient kidney failure | [53] | |||
| FSGS | Neonatal diabetes, seizures, hypotonia, encephalopathy, respiratory failure, cortical and subcortical stroke-like lesions in the frontal, insular, and temporal regions with diffuse cerebral atrophy | 30 or 60 mg/kg/day | [54] | |||
| FSGS, DMS | SND, ataxia, seizures, facial dysmorphism, growth retardation, nephrolithiasis, white matter abnormalities | 15 or 30 mg/kg/day | Partial remission | [17] | ||
| FSGS | Tetralogy of Fallot | Unspecified | Remission | [55] | ||
| cFSGS | SND, mild muscle weakness in lower extremities, bilateral optic nerve atrophy, exotropia with nystagmus | − | − | [56] | ||
| MPGN | None | Unspecified | Kidney failure | [52] | ||
| FSGS, MCNS | None | 30 mg/kg/day | Remission | [57] | ||
| FSGS | SND | 20 mg/kg/day | Initiated after kidney transplant to prevent neurological damage | [58] | ||
| FSGS | SND, neurological impairment, optic atrophy | Idebenone 10 or 20 mg/kg/day | Enalapril treatment required for remission | [59] | ||
| COQ8B (ADCK4) | Coenzyme Q8B | FSGS, GS, cFSGS | Goiter, neurologic developmental delay, dilated cardiomyopathy, hypertension | − | − | [18] |
| cFSGS, GTL | Neurologic abnormalities, seizures, mild mental retardation, retinitis pigmentosa | 10 or 30 mg/kg/day | Partial remission | [21] | ||
| FSGS, mesangial proliferative glomerulonephritis | − | − | [60] | |||
| FSGS | Developmental delay | 15 or 30 mg/kg/day | 1 patient remission, 1 patient no remission | [61] | ||
| FSGS | Dysplastic ears, moderate hearing loss | − | − | [62] | ||
| FSGS | Seizure, pericardial effusion, hypertrophic cardiomyopathy, short stature, intellectual impairment, autism, hypothyroidism, pulmonary hypertension | 20 or 30 mg/kg/day | Partial remission | [19] | ||
| FSGS | 150 mg/kg/day | Partial remission | [63] | |||
| cFSGS, NOS | Medullary nephrocalcinosis, atrial septal defects, benign breast tumor, pancreatic pseudopapillary neoplasm | Cyclosporine | Remission | [64] |
FSGS, focal segmental glomerulosclerosis; cFSGS, collapsing focal segmental glomerulosclerosis; GS, global glomerulosclerosis; GTL, FSGS tip lesion variant; idebenone, hydrophilic short-chain coenzyme Q 10 analog; NS, nephrotic syndrome, DMS, diffuse mesangial sclerosis; MCNS, minimal change nephrotic syndrome; MPGN, membranoproliferative glomerulonephritis; NOS, not-otherwise-specified variant of FSGS; SND, sensorineural deafness; SRNS, steroid-resistant nephrotic syndrome; remission—no proteinuria at study endpoint; partial remission—proteinuria improved but remained above normal range based on study cut off; no remission—no improvement in proteinuria
PDSS1 nephropathy
PDSS1 encodes the prenyldiphosphate synthase subunit 1, which is the catalytic subunit of the PDSS1/PDSS2 heterotetramer that is responsible for the synthesis of the decaprenyl tail of coenzyme Q in the CoQ10 biosynthesis pathway (Fig. 1A). Mutations in PDSS1 are a rare cause of SRNS, and to date, only one patient presenting with SRNS in association with brain abnormalities and developmental delay, due to compound heterozygous mutations in PDSS1 and primary CoQ10 deficiency has been reported [46].
PDSS2 nephropathy
PDSS2 encodes the prenyldiphosphate synthase subunit 2, which is the regulatory subunit of the PDSS1/PDSS2 heterotetramer (Fig. 1A). Compared with PDSS1, PDSS2 deficiency causes a similar spectrum of multiorgan phenotypes, including neurological abnormalities. SRNS is more frequently associated with PDSS2 mutations. Mutations in PDSS2 were identified in a patient with Leigh syndrome, a severe neurological disorder characterized by progressive loss of mental and movement abilities, who subsequently developed an SRNS by 7 months of age [15]. Although CoQ10 treatment (50 mg/day) was initiated at 3 months of age, the patient showed no clinical improvement, likely because the severity of the neurological damage was irreversible at the time treatment was initiated. Using Fluidigm-targeted sequencing, Sadowski et al. identified two unrelated SRNS patients who harbored homozygous mutations in PDSS2; one of the individuals was diagnosed with SRNS at birth, whereas the other developed SRNS at 23 months of age and presented with cerebral palsy and intellectual disability [47]. Ivanyi et al. reported on a 7-month-old patient with SRNS along with encephalomyopathy, hypertrophic cardiomyopathy, and retinitis pigmentosa due to compound heterozygous mutations in PDSS2 [48]. The glomerular phenotype was identified as diffuse mesangial sclerosis (DMS) via histology. Although CoQ10 treatment was initiated shortly after admission, the patient showed no improvement and passed away at 8 months of age [48]. Although the patients described above carried a variety of deleterious alleles, no genotype to phenotype correlation has been established.
COQ2 nephropathy
COQ2 encodes the enzyme para-hydroxybenzoate-polyprenyl transferase that catalyzes the conjugation of the 4-hydroxybenzoate benzoquinone ring with the polyisoprenyl side chain [66], generating the first polyisoprenylated ring CoQ10 intermediate in the biosynthetic pathway (Fig. 1B). Using homozygosity mapping, Quinzii et al. identified a homozygous mutation in COQ2 as underlying CoQ10 deficiency in two siblings, one of whom presented with FSGS at 12 months and with progressive encephalomyopathy at 18 months, while his younger sister developed an isolated FSGS at 12 months of age [45, 67]. Although CoQ10 supplementation was started immediately after the diagnosis of CoQ10 deficiency in both siblings, the male showed no improvement since his neurological and kidney disease were already advanced, whereas his sister responded well to the treatment. After 50 months of therapy, she had demonstrated complete remission of proteinuria and did not show any signs of neurological abnormalities [49]. A rare case of isolated infantile SRNS with collapsing glomerulopathy caused by compound heterozygous mutations in COQ2 was also successfully treated with CoQ10 supplementation [16], whereas a homozygous frameshift mutation in COQ2 caused a rapid neonatal death due to multiorgan complications, including nephrotic syndrome in the affected patient [50]. While most of the known COQ2 mutations cause an early-onset multiorgan involvement, Gigante et al. identified homozygous COQ2 mutations in two cousins with adolescent-onset SRNS and mild neurological symptoms that were resolved after CoQ10 treatment [52]. Starr et al. report three individuals with compound heterozygous COQ2 mutations—two of the subjects, a 2-year-old Asian-American male presenting with focal mesangial sclerosis and collapsing glomerulopathy and a 4-year-old male of mixed European ancestry presenting with FSGS, were responsive to CoQ10 supplementation; while the advanced progression of the kidney disease in the third subject, a 12-year-old Caucasian male could not be mitigated by CoQ10 supplementation [53]. Eroglu et al. reported two case studies in which CoQ10 supplementation was initiated immediately after the diagnosis of insulin-dependent diabetes and proteinuria hours after birth (Patient 2) or 5 days after birth (patient 4) (patient designation given according to the original report) [54]. Both patients carried the same homozygous mutations in the COQ2 gene but were from two unrelated families. The patients responded initially well to CoQ10 supplementation by having normalized blood glucose and urine protein levels after 6 days or 1 month of treatment, respectively [54]. However, despite long-term CoQ10 treatment, the patients exhibited focal clonic seizures and neurological deterioration starting at the age of 2 (patient 2) or 3.5 (patient 4) months, which was complicated by respiratory difficulties, relapsing proteinuria, and sepsis (patient 2) or infections (patient 4) leading to death at 31 (patient 2) or 14 (patient 4) months of age [54]. Given the small number of COQ2 patients, it is difficult to establish clear genotype-phenotype correlation. However, the biochemical studies in COQ2 patient-derived fibroblasts and complementation assays in yeast using human COQ2 alleles have demonstrated that alleles with more damaging mutations are associated with more severe clinical symptoms, whereas the presence of at least one missense allele with reduced function (a hypomorphic allele) mitigates the pathology and results in isolated SRNS or adult-onset encephalopathy instead [68].
COQ6 nephropathy
COQ6 encodes the coenzyme Q6 monooxygenase, which is a component of the mitochondrial CoQ10 enzyme complex, where it catalyzes the hydroxylation of the ubiquinol head ring during CoQ10 biosynthesis (Fig. 1C). Heeringa et al. identified homozygous and compound heterozygous mutations in the COQ6 gene in seven families by homozygosity mapping as causing SRNS in humans [17]. In this cohort, COQ6 mutations were associated with sensorineural deafness (SND) in addition to the occurrence of SRNS. Kidney biopsies revealed that COQ6 kidney pathology ranges from diffuse mesangial sclerosis (DMS) to FSGS [17]. Importantly, treatment with oral COQ10 (15–30 mg/kg/d) early in the disease led to successful remission of the kidney disease in treated patients. Interestingly, Yildirim et al. report two siblings—a 7-year-old girl with SRNS and SND and her 10-year-old brother with isolated SND, as affected with homozygous COQ6 mutations, which were previously linked to SRNS and SND [17, 58]. This suggests that kidney involvement in COQ6 deficiency may be variable but also warrants careful follow-up of the patient to detect delayed onset of symptoms [58]. Stanczyk et al. identified compound heterozygous COQ6 mutations in a child from non-related parents, who presented isolated SRNS and responded well to 30 mg/kg/day CoQ10 treatment, with full remission 12 months after initiation of the treatment [57]. Although the most frequent glomerular phenotype in COQ6-deficiency is FSGS, a range of other histological patterns have been reported, including membranoproliferative glomerulonephritis (a phenotype usually associated with deficiency in the complement pathway) [52], DMS [17], collapsing FSGS [56], and minimal change disease [57], demonstrating high histological heterogeneity within COQ6 nephropathy.
COQ8B (ADCK4) nephropathy
COQ8B encodes the coenzyme Q8B (formerly known as ADCK4, aarF domain containing kinase 4), which localizes to the CoQ10 complex in the mitochondrial inner membrane. While the precise biochemical function of COQ8B is currently undefined, it has been shown to play a role in stabilizing the mitochondrial CoQ10 enzyme complex through its interaction with other CoQ10 complex peptides, such as COQ5/6/7 [18, 69]. Ashraf et al. described eight families with mutations in COQ8B as causing SRNS, with infrequent extrarenal manifestations including goiter, neurological developmental delay, and dilated cardiomyopathy [18]. To date, mutations in COQ8B causing FSGS with relatively rare extrarenal pathologies have been reported in 34 families from various ethnic backgrounds [19, 21, 61–63]. While the age of onset of the kidney clinical symptoms in COQ8B patients demonstrates great variability, the majority of them present in an adolescent population in contrast to the mostly neonatal and infantile onset of symptoms with mutations in other CoQ10 biosynthesis genes, such as COQ2d, PDSS1, and PDSS2. The reported kidney histology of COQ8B-deficient individuals includes FSGS, collapsing FSGS, global glomerulosclerosis, and mesangial proliferative glomerulonephritis [18, 19, 21, 60, 62–64, 70]. Treatment of asymptomatic individuals carrying COQ8B homozygous mutations with CoQ10 supplementation led to significant reduction (50–80%) of proteinuria within 6 weeks of treatment and partial remission was successfully maintained over several months [19, 21]. A few individuals with COQ8B nephropathy have also been reported to be either fully responsive to treatment with cyclosporin A [64] or have partial response to steroids or cyclosporin A [21]. Interestingly, complementation studies in ΔCOQ8 yeast mutants using human mutations revealed no genotype-phenotype correlation between the allele severity and mitochondrial respiratory deficiency, suggesting that COQ8B function might be redundant with other biosynthesis components in the CoQ10 pathway [71].
Genetic causes of non-SRNS kidney pathologies due to CoQ10 biosynthesis deficiency
COQ7 nephropathy
COQ7 encodes the 5-demethoxyubiquinone hydroxylase that catalyzes the hydroxylation of 2-polyprenyl-3-methyl-6-methoxy-1,4-benzoquinol (DMQH2), a critical step in CoQ10 biosynthesis. Mutations in COQ7 present with severe multiorgan phenotypes primarily affecting the nervous system [60, 72]. Currently, only one case has been reported with kidney involvement—the affected individual had multiple kidney cysts and a diffuse increase in kidney parenchymal echogenicity. However, the patient’s kidney function was unremarkable, and no glomerular pathology was noted [73].
COQ9 nephropathy
COQ9 encodes the coenzyme Q9, whose function in the CoQ10 complex is poorly studied. COQ9 interaction with COQ7 is required for COQ7 enzymatic activity. Mutations in human COQ9 lead to lethal multiorgan pathologies, which include kidney tubular dysfunction with no reported cases of glomerular involvement [74].
Model organisms of primary CoQ10 nephropathies
Much of our knowledge regarding the composition of the CoQ10 biosynthesis pathway and the function of the individual enzymes is derived from studies performed in various non-mammalian models, particularly the yeast Saccharomyces cerevisiae, which has been instrumental in helping to define the exact nature of the biochemical reactions and the order of the enzymatic steps involved in CoQ10 biosynthesis [34]. Moreover, biochemical characterization of the CoQ-synthome, the biosynthetic complex that produces CoQ in yeast, has inspired the prediction that an orthologous CoQ complex might exist in mammalian cells [31, 34]. Indeed, genetic studies have revealed a high level of evolutionary conservation in the CoQ10 biosynthesis pathway composition and structure between the yeast and mammalian cells, which has enabled functional testing of the human COQ alleles identified in primary CoQ10-deficient patients by performing genetic complementation assays in the yeast [34].
Even though the yeast has been invaluable in studying the biochemical properties of the CoQ10 biosynthesis pathway and the function of individual human alleles, a mammalian model is needed to investigate the pathophysiology of CoQ10 deficiency at the tissue and organ level and to assess potential therapies. The best studied mouse model of CoQ10-deficient nephropathy is the kidney disease, kd/kd mouse model, which arose spontaneously in an inbred mouse colony [75], and was subsequently identified as harboring a missense p.V117M amino acid substitution in the Pdss2 gene [76]. The mutant mice are healthy until 10 weeks of age, when proteinuria can be first detected and the kidneys develop glomerular sclerosis and tubular atrophy [75, 77]. Using a conditional allele of Pdss2, Peng et al. demonstrated that the kidney pathology in Pdss2 loss of function mice is specific to the glomerular podocytes, since deletion of Pdss2 from other kidney tissues (i.e., tubular epithelial cells), or from non-kidney tissues (monocytes or hepatocytes) did not result in overt pathology [78].
Recently, transgenic mouse models corresponding to other orthologous human CoQ10 biosynthesis genes have been developed, including two Coq9 mouse lines carrying p.Q95X and p.R239X nonsense mutations, respectively [79]. While these mice have been useful in studying the Coq9 pathophysiology in various organs, it appears that in contrast to the situation in humans, loss or reduction of CoQ9 cellular levels due to the lack of murine Coq9 does not cause kidney pathology in Coq9 mice, at least up to 18 months of age [79]. Recently, the kidney phenotype in mice with podocyte-specific depletion of Coq6 or Adck4 has been described [69, 80]. In both cases, the gene encoding the CoQ9 biosynthesis pathway peptide was inactivated in the glomerular podocytes by crossing Coq6flox/flox or Adck4flox/flox mice with Podocin-Cre mice. The resulting kidney clinical phenotypes in Coq6KO or Adck4KO mice were highly reminiscent of the human glomerular pathology of COQ6 or COQ8B patients, respectively [17, 69, 80, 81]. The transgenic mice developed progressive proteinuria by 5 months of age, which was accompanied by podocyte effacement and glomerular sclerosis. At the ultra-structural level, the podocytes contained mitochondria with abnormal morphology and increased size, indicative of defects in the OXPHOS pathway. Remarkably, the development of podocyte abnormalities and nephrotic syndrome were completely avoided by continuous treatment of the conditional knockout mice with a benzoate analog, 2,4-dihydroxybonzoic acid (2,4-diHB), in the drinking water. The treatment, which was initiated at the age of 3 months (Adck4KO) or 4 months (Coq6KO) prior to the onset of the histological and functional changes in the mutant mice, was shown to confer long-term protection from glomerular damage and proteinuria [69, 80]. These preclinical studies are the first to demonstrate successful prevention of glomerular pathology due to CoQ9-deficiency, by using a CoQ9 biosynthetic precursor analog as a therapeutic modality in a mouse model of SRNS. Of note, 2,4-diHB has been successfully used to counter senescence-associated mitochondrial defects and to extend the healthy life span in the Coq7/Mclk1 mouse model [82], as well as to increase CoQ9 biosynthesis in a Coq9R239X mouse model [79], indicating that analogs of CoQ9 precursors can be successfully used in bypass reactions to mitigate CoQ9 biosynthesis pathway defects. Indeed, it has been suggested that lower lipophilicity and higher bioavailability of the CoQ bypass compounds makes them more effective than CoQ supplementation in mitigating CoQ deficiency [82, 83].
Other analogs of 4-hydroxybenzoate, the natural CoQ10 head group precursor, that have been widely used in CoQ10 biosynthesis bypass experiments are vanillic acid and 3,4-dihydroxybenzoic acid (3,4-diHB) (Fig. 2); compounds whose beneficial effect to CoQ6 biosynthesis was first identified in yeast [84] and subsequently also confirmed in a variety of other CoQ-deficient organisms and cell lines (Table 2). All three analogs of 4-HB differ from the natural CoQ10 headgroup precursor by the presence of an additional hydroxyl group at either carbon 2 (2,4-dHB) or carbon 3 (3,4-dHB), or the presence of a methoxy group at carbon 3 (vanillic acid) of the benzoate ring, representing the intermediate steps of CoQ10 head group hydroxylation and methylation reactions, that are normally carried out by the specialized enzymes that constitute the CoQ10 biosynthesis pathway. This provides a rationale for using the bypass strategy to mitigate the functional deficiencies of COQ-enzymes in individuals with mutations in COQ-genes. Further studies are warranted to determine if the analogs of 4-hydroxybenzoate are useful alternatives or add-ons to CoQ10 supplementation in SRNS patients.
Fig. 2.
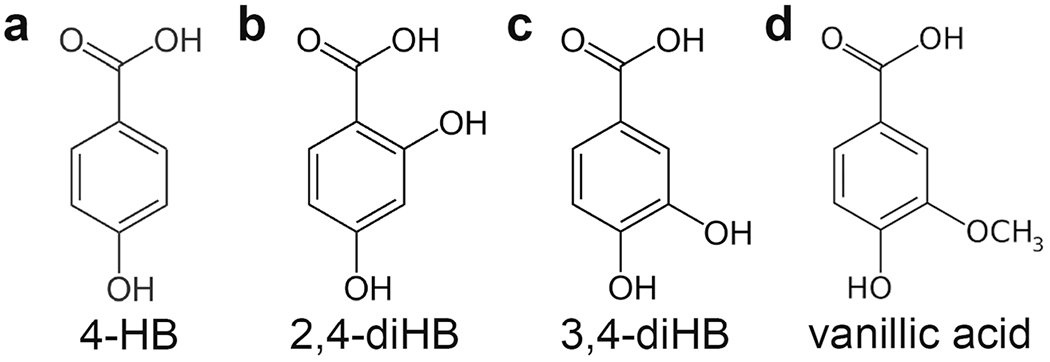
The structure of 4-HB and its analogs that have been successfully used in COQ bypass reactions. (A) 4-HB, 4-hydroxybenzoate. (B) 2,4-diHB, 2,4-dihydroxybenzoate. (C) 3,4-diHB, 3,4-dihydroxybenzoate. (D) Vanillic acid
Table 2.
Chemical analogs of 4-hydroxybenzoate that have been successfully used in CoQ biosynthesis bypass reactions
| Compound | Experimental model | Effect observed | Reference |
|---|---|---|---|
| Vanillic acid | S.cerevisiae Δcoq6 strain | Restoration of CoQ6 biosynthesis and mitochondrial respiration | [84] |
| S.cerevisiae Δcoq6 strain overexpressing human COQ6 mutations | Rescue of growth on non-fermentable carbon source | [85] | |
| Drosophila Coq2-RNAi | Partial rescue of nephrocyte slit diaphragm defects | [86] | |
| COQ6-deficient HEK293 cell line | Restoration of CoQ biosynthesis and ATP production | [87] | |
| 2,4-Dihydroxybenzoic | COQ7.V141E human fibroblasts | Increased CoQ cellular levels | [72] |
| acid | Coq7 knockout mouse model | Partial rescue of CoQ cellular levels, improved mitochondrial respiration, prolonged survival | [82] |
| Coq9.R239X mouse model | Increased CoQ biosynthesis | [79] | |
| COQ7.L111P and COQ7.V141E human fibroblasts | Restoration of CoQ biosynthesis | [88] | |
| COQ6-deficient HEK293 cell line | Restoration of CoQ biosynthesis and ATP production | [87] | |
| Coq6 podocyte-specific ko mouse model | Prevention of FSGS, prolonged survival | [80] | |
| Adck4 podocyte-specific ko mouse model | Prevention of FSGS, prolonged survival | [69] | |
| 3,4-Dihydroxybenzoic | S.cerevisiae Δcoq6 strain | Restoration of CoQ6 biosynthesis and respiration | [84] |
| acid | S.cerevisiae Δcoq6 strain overexpressing human COQ6 mutations | Rescue of growth on non-fermentable carbon source | [85] |
Coenzyme Q10 supplementation to target CoQ10 deficiency in humans
Management of SRNS is challenging, since patients do not respond to steroid treatment. However, patients with primary CoQ10 deficiency frequently respond to oral coenzyme Q10 supplementation [18, 19, 21, 49, 78, 89]. Long-term coenzyme Q10 supplementation to mitigate primary CoQ10 pathology was first used 20 years ago on two siblings with multiple respiratory chain dysfunctions, including mitochondrial encephalomyopathy, bilateral sensorineural deafness, and nephrotic syndrome [23]. The patients responded favorably to the treatment and showed improvement in the clinical picture of the neurological symptoms, while the kidney disease had unfortunately progressed to the terminal stage by the time the treatment was started [23]. More recent clinical case series and reports in which coenzyme Q10 supplementation was used on SRNS patients after the confirmation of a mutation in a CoQ10 biosynthesis pathway gene have demonstrated marked success in achieving sustained remission of proteinuria [17–19, 21, 53, 57, 61]. In general, it has emerged that patients with kidney pathology respond well to coenzyme Q10 supplementation if the treatment is initiated early before significant structural damage to the podocytes or nephron epithelium has occurred [49, 62].
Overall, it is evident that SRNS due to mutations in CoQ10 biosynthesis is a rare disease, comprising less than 1% of all SRNS cases [65]. While the gold standard for evaluating a new treatment is a large randomized control trial, the rarity of these pediatric diseases makes the feasibility of achieving robust evidence-based medicine difficult as compared with more common entities, such as cardiovascular disease or diabetes mellitus. Therefore, one has to rely on the clinical case series and case reports to guide new directions in clinical practice when it comes to rare childhood disease such as SRNS due to CoQ10 deficiency. In fact, there have been some smaller studies demonstrating that long-term CoQ10 supplementation is effective for those with ADCK4 mutations [19, 20]. Given the rarity of these diagnoses, well-controlled longitudinal studies have yet to be performed to determine whether CoQ10 supplementation preserves long-term kidney function and protects patients from progressing to chronic kidney disease. Yet, it is these small studies that will drive innovation and allow clinicians to pursue this novel treatment for other patients with deficiencies in the CoQ10 biosynthesis pathway.
An important unknown in coenzyme Q10 supplementation is the observed variability in CoQ10 efficacy in mitigating the tissue pathology between different organ systems. For instance, some clinical studies have reported a beneficial effect of oral coenzyme Q10 supplementation for neurologic conditions, but not for kidney dysfunction [19,49, 67]. Conversely, when a positive effect was observed on kidney function after supplementation with CoQ10, no improvement was observed in extrarenal manifestations, such as hypertrophic cardiomyopathy, pulmonary hypertension, body growth parameters, hypothyroidism, or neurological symptoms [19, 54]. Moreover, siblings with the same molecular diagnosis may not display similar response to CoQ10 supplementation [19]. It has been postulated that difference in CoQ10 efficacy may arise from a variation in individual metabolism [90], but may also be caused by the low bioavailability of CoQ10 due to its large molecular weight, high lipophilicity, and poor aqueous solubility [91]. Together, more research is required to investigate the biochemical and pathological mechanism of CoQ10 deficiency in order to develop rational therapeutic approaches in patients with different genetic mutations in the CoQ10 biosynthesis pathway.
Conclusions and future perspectives
Treatment of SRNS has remained an elusive target for nephrologists for over 40 years. Recent clinical findings and animal models have demonstrated that patients with SRNS caused by primary CoQ10 deficiency may benefit from coenzyme Q10 supplementation if treatment is started early, before irreversible tissue damage has occurred. Thus, it is important to establish a molecular diagnosis for SRNS patients and their family members to identify individuals who may be responsive to CoQ10 supplementation. Current protocols used to treat people with CoQ10 supplementation are anecdotal without established evidence-based guidelines, necessitating future studies to address the optimal formulation and dosing of CoQ10 in the appropriate cohort of SRNS patients. Preclinical studies in mice, yeast, and other model systems have identified and validated the use of several small molecules with improved bioavailability that ameliorate CoQ10 biosynthesis in eukaryotic cells with primary CoQ10 deficiency and thus may provide a novel avenue that may be more efficacious in treating this condition.
Funding
This study was supported by the National Institutes of Health to RA (DK115403, P30DK079307).
Footnotes
Compliance with ethical standards
Conflict of interest The authors declare that they have no conflict of interest.
Publisher′s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.El Bakkali L, Rodrigues Pereira R, Kuik DJ, Ket JC, van Wijk JA (2011) Nephrotic syndrome in The Netherlands: a population-based cohort study and a review of the literature. Pediatr Nephrol 26: 1241–1246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McKinney PA, Feltbower RG, Brocklebank JT, Fitzpatrick MM (2001) Time trends and ethnic patterns of childhood nephrotic syndrome in Yorkshire, UK. Pediatr Nephrol 16:1040–1044 [DOI] [PubMed] [Google Scholar]
- 3.Wyatt RJ, Marx MB, Kazee M, Holland NH (1982) Current estimates of the incidence of steroid responsive idiopathic nephrosis in Kentucky children 1–9 years of age. Int J Pediatr Nephrol 3:63–65 [PubMed] [Google Scholar]
- 4.Chanchlani R, Parekh RS (2016) Ethnic differences in childhood nephrotic syndrome. Front Pediatr 4:39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kim JS, Bellew CA, Silverstein DM, Aviles DH, Boineau FG, Vehaskari VM (2005) High incidence of initial and late steroid resistance in childhood nephrotic syndrome. Kidney Int 68:[ISP-Check]1275–1281. [DOI] [PubMed] [Google Scholar]
- 6.Benoit G, Machuca E, Antignac C (2010) Hereditary nephrotic syndrome: a systematic approach for genetic testing and a review of associated podocyte gene mutations. Pediatr Nephrol 25:[ISP-Check]1621–1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vivante A, Hildebrandt F (2016) Exploring the genetic basis of early-onset chronic kidney disease. Nat Rev Nephrol 12:133–146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Weaver DJ Jr, Somers MJG, Martz K, Mitsnefes MM (2017) Clinical outcomes and survival in pediatric patients initiating chronic dialysis: a report of the NAPRTCS registry. Pediatr Nephrol 32: 2319–2330 [DOI] [PubMed] [Google Scholar]
- 9.Trautmann A, Bodria M, Ozaltin F, Gheisari A, Melk A, Azocar M, Anarat A, Caliskan S, Emma F, Gellermann J, Oh J, Baskin E, Ksiazek J, Remuzzi G, Erdogan O, Akman S, Dusek J, Davitaia T, Ozkaya O, Papachristou F, Firszt-Adamczyk A, Urasinski T, Testa S, Krmar RT, Hyla-Klekot L, Pasini A, Ozcakar ZB, Sallay P, Cakar N, Galanti M, Terzic J, Aoun B, Caldas Afonso A, Szymanik-Grzelak H, Lipska BS, Schnaidt S, Schaefer F, PodoNet Consortium (2015) Spectrum of steroid-resistant and congenital nephrotic syndrome in children: the PodoNet registry cohort. Clin J Am Soc Nephrol 10:592–600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Abe Y, Sakairi T, Kajiyama H, Shrivastav S, Beeson C, Kopp JB (2010) Bioenergetic characterization of mouse podocytes. Am J Physiol Cell Physiol 299:C464–C476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brinkkoetter PT, Bork T, Salou S, Liang W, Mizi A, Ozel C, Koehler S, Hagmann HH, Ising C, Kuczkowski A, Schnyder S, Abed A, Schermer B, Benzing T, Kretz O, Puelles VG, Lagies S, Schlimpert M, Kammerer B, Handschin C, Schell C, Huber TB (2019) Anaerobic glycolysis maintains the glomerular filtration barrier independent of mitochondrial metabolism and dynamics. Cell Rep 27(1551–1566):e1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cil O, Perwad F (2018) Monogenic causes of proteinuria in children. Front Med (Lausanne) 5:55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Preston R, Stuart HM, Lennon R (2019) Genetic testing in steroid-resistant nephrotic syndrome: why, who, when and how? Pediatr Nephrol 34:195–210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.De Vriese AS, Sethi S, Nath KA, Glassock RJ, Fervenza FC (2018) Differentiating primary, genetic, and secondary FSGS in adults: a clinicopathologic approach. J Am Soc Nephrol 29:759–774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lopez LC, Schuelke M, Quinzii CM, Kanki T, Rodenburg RJ, Naini A, Dimauro S, Hirano M (2006) Leigh syndrome with nephropathy and CoQ10 deficiency due to decaprenyl diphosphate synthase subunit 2 (PDSS2) mutations. Am J Hum Genet 79: 1125–1129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Diomedi-Camassei F, Di Giandomenico S, Santorelli FM, Caridi G, Piemonte F, Montini G, Ghiggeri GM, Murer L, Barisoni L, Pastore A, Muda AO, Valente ML, Bertini E, Emma F (2007) COQ2 nephropathy: a newly described inherited mitochondriopathy with primary renal involvement. J Am Soc Nephrol 18:2773–2780 [DOI] [PubMed] [Google Scholar]
- 17.Heeringa SF, Chernin G, Chaki M, Zhou W, Sloan AJ, Ji Z, Xie LX, Salviati L, Hurd TW, Vega-Warner V, Killen PD, Raphael Y, Ashraf S, Ovunc B, Schoeb DS, McLaughlin HM, Airik R, Vlangos CN, Gbadegesin R, Hinkes B, Saisawat P, Trevisson E, Doimo M, Casarin A, Pertegato V, Giorgi G, Prokisch H, Rotig A, Nurnberg G, Becker C, Wang S, Ozaltin F, Topaloglu R, Bakkaloglu A, Bakkaloglu SA, Muller D, Beissert A, Mir S, Berdeli A, Varpizen S, Zenker M, Matejas V, Santos-Ocana C, Navas P, Kusakabe T, Kispert A, Akman S, Soliman NA, Krick S, Mundel P, Reiser J, Nurnberg P, Clarke CF, Wiggins RC, Faul C, Hildebrandt F (2011) COQ6 mutations in human patients produce nephrotic syndrome with sensorineural deafness. J Clin Invest 121: 2013–2024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ashraf S, Gee HY, Woerner S, Xie LX, Vega-Warner V, Lovric S, Fang H, Song X, Cattran DC, Avila-Casado C, Paterson AD, Nitschke P, Bole-Feysot C, Cochat P, Esteve-Rudd J, Haberberger B, Allen SJ, Zhou W, Airik R, Otto EA, Barua M, Al-Hamed MH, Kari JA, Evans J, Bierzynska A, Saleem MA, Bockenhauer D, Kleta R, El Desoky S, Hacihamdioglu DO, Gok F, Washburn J, Wiggins RC, Choi M, Lifton RP, Levy S, Han Z, Salviati L, Prokisch H, Williams DS, Pollak M, Clarke CF, Pei Y, Antignac C, Hildebrandt F (2013) ADCK4 mutations promote steroid-resistant nephrotic syndrome through CoQ10 biosynthesis disruption. J Clin Invest 123:5179–5189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Atmaca M, Gulhan B, Korkmaz E, Inozu M, Soylemezoglu O, Candan C, Bayazit AK, Elmaci AM, Parmaksiz G, Duzova A, Besbas N, Topaloglu R, Ozaltin F (2017) Follow-up results of patients with ADCK4 mutations and the efficacy of CoQ10 treatment. Pediatr Nephrol 32:1369–1375 [DOI] [PubMed] [Google Scholar]
- 20.Atmaca M, Gulhan B, Atayar E, Bayazit AK, Candan C, Arici M, Topaloglu R, Ozaltin F (2019) Long-term follow-up results of patients with ADCK4 mutations who have been diagnosed in the asymptomatic period: effects of early initiation of CoQ10 supplementation. Turk J Pediatr 61:657–663 [DOI] [PubMed] [Google Scholar]
- 21.Korkmaz E, Lipska-Zietkiewicz BS, Boyer O, Gribouval O, Fourrage C, Tabatabaei M, Schnaidt S, Gucer S, Kaymaz F, Arici M, Dinckan A, Mir S, Bayazit AK, Emre S, Balat A, Rees L, Shroff R, Bergmann C, Mourani C, Antignac C, Ozaltin F, Schaefer F, PodoNet Consortium (2016) ADCK4-associated glomerulopathy causes adolescence-onset FSGS. J Am Soc Nephrol 27:63–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ogasahara S, Engel AG, Frens D, Mack D (1989) Muscle coenzyme Q deficiency in familial mitochondrial encephalomyopathy. Proc Natl Acad Sci U S A 86:2379–2382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rotig A, Appelkvist EL, Geromel V, Chretien D, Kadhom N, Edery P, Lebideau M, Dallner G, Munnich A, Ernster L, Rustin P (2000) Quinone-responsive multiple respiratory-chain dysfunction due to widespread coenzyme Q10 deficiency. Lancet 356:391–395 [DOI] [PubMed] [Google Scholar]
- 24.Lamperti C, Naini A, Hirano M, De Vivo DC, Bertini E, Servidei S, Valeriani M, Lynch D, Banwell B, Berg M, Dubrovsky T, Chiriboga C, Angelini C, Pegoraro E, DiMauro S (2003) Cerebellar ataxia and coenzyme Q10 deficiency. Neurology 60: 1206–1208 [DOI] [PubMed] [Google Scholar]
- 25.Van Maldergem L, Trijbels F, DiMauro S, Sindelar PJ, Musumeci O, Janssen A, Delberghe X, Martin JJ, Gillerot Y (2002) Coenzyme Q-responsive Leigh’s encephalopathy in two sisters. Ann Neurol 52:750–754 [DOI] [PubMed] [Google Scholar]
- 26.Lalani SR, Vladutiu GD, Plunkett K, Lotze TE, Adesina AM, Scaglia F (2005) Isolated mitochondrial myopathy associated with muscle coenzyme Q10 deficiency. Arch Neurol 62:317–320 [DOI] [PubMed] [Google Scholar]
- 27.Alcazar-Fabra M, Navas P, Brea-Calvo G (2016) Coenzyme Q biosynthesis and its role in the respiratory chain structure. Biochim Biophys Acta 1857:1073–1078 [DOI] [PubMed] [Google Scholar]
- 28.Zhang Y, Aberg F, Appelkvist EL, Dallner G, Ernster L (1995) Uptake of dietary coenzyme Q supplement is limited in rats. J Nutr 125:446–453 [DOI] [PubMed] [Google Scholar]
- 29.Turunen M, Olsson J, Dallner G (2004) Metabolism and function of coenzyme Q. Biochim Biophys Acta 1660:171–199 [DOI] [PubMed] [Google Scholar]
- 30.Bhagavan HN, Chopra RK (2007) Plasma coenzyme Q10 response to oral ingestion of coenzyme Q10 formulations. Mitochondrion 7(Suppl):S78–S88 [DOI] [PubMed] [Google Scholar]
- 31.Stefely JA, Pagliarini DJ (2017) Biochemistry of mitochondrial coenzyme Q biosynthesis. Trends Biochem Sci 42:824–843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Desbats MA, Lunardi G, Doimo M, Trevisson E, Salviati L (2015) Genetic bases and clinical manifestations of coenzyme Q10 (CoQ 10) deficiency. J Inherit Metab Dis 38:145–156 [DOI] [PubMed] [Google Scholar]
- 33.Payet LA, Leroux M, Willison JC, Kihara A, Pelosi L, Pierrel F (2016) Mechanistic details of early steps in coenzyme Q biosynthesis pathway in yeast. Cell Chem Biol 23:1241–1250 [DOI] [PubMed] [Google Scholar]
- 34.Awad AM, Bradley MC, Fernandez-Del-Rio L, Nag A, Tsui HS, Clarke CF (2018) Coenzyme Q10 deficiencies: pathways in yeast and humans. Essays Biochem 62:361–376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bentinger M, Tekle M, Dallner G (2010) Coenzyme Q-biosynthesis and functions. Biochem Biophys Res Commun 396: 74–79 [DOI] [PubMed] [Google Scholar]
- 36.Watmough NJ, Frerman FE (2010) The electron transfer flavoprotein: ubiquinone oxidoreductases. Biochim Biophys Acta 1797: 1910–1916 [DOI] [PubMed] [Google Scholar]
- 37.Lopez LC, Luna-Sanchez M, Garcia-Corzo L, Quinzii CM, Hirano M (2014) Pathomechanisms in coenzyme q10-deficient human fibroblasts. Mol Syndromol 5:163–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bentinger M, Brismar K, Dallner G (2007) The antioxidant role of coenzyme Q. Mitochondrion 7(Suppl):S41–S50 [DOI] [PubMed] [Google Scholar]
- 39.Quinzii CM, Garone C, Emmanuele V, Tadesse S, Krishna S, Dorado B, Hirano M (2013) Tissue-specific oxidative stress and loss of mitochondria in CoQ-deficient Pdss2 mutant mice. FASEB J 27:612–621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Garcia-Corzo L, Luna-Sanchez M, Doerrier C, Garcia JA, Guaras A, Acin-Perez R, Bullejos-Peregrin J, Lopez A, Escames G, Enriquez JA, Acuna-Castroviejo D, Lopez LC (2013) Dysfunctional Coq9 protein causes predominant encephalomyopathy associated with CoQ deficiency. Hum Mol Genet 22:[ISP-Check]1233–1248. [DOI] [PubMed] [Google Scholar]
- 41.Quinzii CM, Lopez LC, Von-Moltke J, Naini A, Krishna S, Schuelke M, Salviati L, Navas P, DiMauro S, Hirano M (2008) Respiratory chain dysfunction and oxidative stress correlate with severity of primary CoQ10 deficiency. FASEB J 22:1874–1885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ziosi M, Di Meo I, Kleiner G, Gao XH, Barca E, Sanchez-Quintero MJ, Tadesse S, Jiang H, Qiao C, Rodenburg RJ, Scalais E, Schuelke M, Willard B, Hatzoglou M, Tiranti V, Quinzii CM (2017) Coenzyme Q deficiency causes impairment of the sulfide oxidation pathway. EMBO Mol Med 9:96–111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ernster L, Dallner G (1995) Biochemical, physiological and medical aspects of ubiquinone function. Biochim Biophys Acta 1271: 195–204 [DOI] [PubMed] [Google Scholar]
- 44.Lopez-Martin JM, Salviati L, Trevisson E, Montini G, DiMauro S, Quinzii C, Hirano M, Rodriguez-Hernandez A, Cordero MD, Sanchez-Alcazar JA, Santos-Ocana C, Navas P (2007) Missense mutation of the COQ2 gene causes defects of bioenergetics and de novo pyrimidine synthesis. Hum Mol Genet 16:1091–1097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Quinzii C, Naini A, Salviati L, Trevisson E, Navas P, Dimauro S, Hirano M (2006) A mutation in para-hydroxybenzoate-polyprenyl transferase (COQ2) causes primary coenzyme Q10 deficiency. Am J Hum Genet 78:345–349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vasta V, Merritt JL 2nd, Saneto RP, Hahn SH (2012) Next-generation sequencing for mitochondrial diseases: a wide diagnostic spectrum. Pediatr Int 54:585–601 [DOI] [PubMed] [Google Scholar]
- 47.Sadowski CE, Lovric S, Ashraf S, Pabst WL, Gee HY, Kohl S, Engelmann S, Vega-Warner V, Fang H, Halbritter J, Somers MJ, Tan W, Shril S, Fessi I, Lifton RP, Bockenhauer D, El-Desoky S, Kari JA, Zenker M, Kemper MJ, Mueller D, Fathy HM, Soliman NA, Group SS, Hildebrandt F (2015) A single-gene cause in 29.5% of cases of steroid-resistant nephrotic syndrome. J Am Soc Nephrol 26:1279–1289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ivanyi B, Racz GZ, Gal P, Brinyiczki K, Bodi I, Kalmar T, Maroti Z, Bereczki C (2018) Diffuse mesangial sclerosis in a PDSS2 mutation-induced coenzyme Q10 deficiency. Pediatr Nephrol 33: 439–446 [DOI] [PubMed] [Google Scholar]
- 49.Montini G, Malaventura C, Salviati L (2008) Early coenzyme Q10 supplementation in primary coenzyme Q10 deficiency. N Engl J Med 358:2849–2850 [DOI] [PubMed] [Google Scholar]
- 50.Mollet J, Giurgea I, Schlemmer D, Dallner G, Chretien D, Delahodde A, Bacq D, de Lonlay P, Munnich A, Rotig A (2007) Prenyldiphosphate synthase, subunit 1 (PDSS1) and OH-benzoate polyprenyltransferase (COQ2) mutations in ubiquinone deficiency and oxidative phosphorylation disorders. J Clin Invest 117:[ISP-Check]765–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.McCarthy HJ, Bierzynska A, Wherlock M, Ognjanovic M, Kerecuk L, Hegde S, Feather S, Gilbert RD, Krischock L, Jones C, Sinha MD, Webb NJ, Christian M, Williams MM, Marks S, Koziell A, Welsh GI, Saleem MA, RADAR the UK SRNS Study Group (2013) Simultaneous sequencing of 24 genes associated with steroid-resistant nephrotic syndrome. Clin J Am Soc Nephrol 8:637–648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gigante M, Diella S, Santangelo L, Trevisson E, Acosta MJ, Amatruda M, Finzi G, Caridi G, Murer L, Accetturo M, Ranieri E, Ghiggeri GM, Giordano M, Grandaliano G, Salviati L, Gesualdo L (2017) Further phenotypic heterogeneity of CoQ10 deficiency associated with steroid resistant nephrotic syndrome and novel COQ2 and COQ6 variants. Clin Genet 92:224–226 [DOI] [PubMed] [Google Scholar]
- 53.Starr MC, Chang IJ, Finn LS, Sun A, Larson AA, Goebel J, Hanevold C, Thies J, Van Hove JLK, Hingorani SR, Lam C (2018) COQ2 nephropathy: a treatable cause of nephrotic syndrome in children. Pediatr Nephrol 33:1257–1261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Eroglu FK, Ozaltin F, Gonc N, Nalcacioglu H, Ozcakar ZB, Yalnizoglu D, Gucer S, Orhan D, Eminoglu FT, Gocmen R, Alikasifoglu A, Topaloglu R, Duzova A (2018) Response to early coenzyme Q10 supplementation is not sustained in CoQ10 deficiency caused by CoQ2 mutation. Pediatr Neurol 88:71–74 [DOI] [PubMed] [Google Scholar]
- 55.Nakanishi K, Okamoto T, Nozu K, Hara S, Sato Y, Hayashi A, Takahashi T, Nagano C, Sakakibara N, Horinouchi T, Fujimura J, Minamikawa S, Yamamura T, Rossanti R, Nagase H, Kaito H, Ariga T, Iijima K (2019) Pair analysis and custom array CGH can detect a small copy number variation in COQ6 gene. Clin Exp Nephrol 23:669–675 [DOI] [PubMed] [Google Scholar]
- 56.Park E, Ahn YH, Kang HG, Yoo KH, Won NH, Lee KB, Moon KC, Seong MW, Gwon TR, Park SS, Cheong HI (2017) COQ6 mutations in children with steroid-resistant focal segmental glomerulosclerosis and Sensorineural hearing loss. Am J Kidney Dis 70:139–144 [DOI] [PubMed] [Google Scholar]
- 57.Stanczyk M, Balasz-Chmielewska I, Lipska-Zietkiewicz B, Tkaczyk M (2018) CoQ 10-related sustained remission of proteinuria in a child with COQ6 glomerulopathy-a case report. Pediatr Nephrol 33:2383–2387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yuruk Yildirim Z, Toksoy G, Uyguner O, Nayir A, Yavuz S, Altunoglu U, Turkkan ON, Sevinc B, Gokcay G, Kurkcu Gunes D, Kiyak A, Yilmaz A (2020) Primary coenzyme Q10 Deficiency-6 (COQ10D6): two siblings with variable expressivity of the renal phenotype. Eur J Med Genet 63:103621. [DOI] [PubMed] [Google Scholar]
- 59.Justine Perrin R, Rousset-Rouviere C, Garaix F, Cano A, Conrath J, Boyer O, Tsimaratos M (2020) COQ6 mutation in patients with nephrotic syndrome, sensorineural deafness, and optic atrophy. JIMD Rep 54:37–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang F, Zhang Y, Mao J, Yu Z, Yi Z, Yu L, Sun J, Wei X, Ding F, Zhang H, Xiao H, Yao Y, Tan W, Lovric S, Ding J, Hildebrandt F (2017) Spectrum of mutations in Chinese children with steroid-resistant nephrotic syndrome. Pediatr Nephrol 32:1181–1192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Feng C, Wang Q, Wang J, Liu F, Shen H, Fu H, Mao J (2017) Coenzyme Q10 supplementation therapy for 2 children with proteinuria renal disease and ADCK4 mutation: case reports and literature review. Medicine (Baltimore) 96:e8880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lolin K, Chiodini BD, Hennaut E, Adams B, Dahan K, Ismaili K (2017) Early-onset of ADCK4 glomerulopathy with renal failure: a case report. BMC Med Genet 18:28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yang J, Yang Y, Hu Z (2018) A novel ADCK4 mutation in a Chinese family with ADCK4-associated glomerulopathy. Biochem Biophys Res Commun 506:444–449 [DOI] [PubMed] [Google Scholar]
- 64.Park E, Kang HG, Choi YH, Lee KB, Moon KC, Jeong HJ, Nagata M, Cheong HI (2017) Focal segmental glomerulosclerosis and medullary nephrocalcinosis in children with ADCK4 mutations. Pediatr Nephrol 32:1547–1554 [DOI] [PubMed] [Google Scholar]
- 65.Warejko JK, Tan W, Daga A, Schapiro D, Lawson JA, Shril S, Lovric S, Ashraf S, Rao J, Hermle T, Jobst-Schwan T, Widmeier E, Majmundar AJ, Schneider R, Gee HY, Schmidt JM, Vivante A, van der Ven AT, Ityel H, Chen J, Sadowski CE, Kohl S, Pabst WL, Nakayama M, Somers MJG, Rodig NM, Daouk G, Baum M, Stein DR, Ferguson MA, Traum AZ, Soliman NA, Kari JA, El Desoky S, Fathy H, Zenker M, Bakkaloglu SA, Muller D, Noyan A, Ozaltin F, Cadnapaphornchai MA, Hashmi S, Hopcian J, Kopp JB, Benador N, Bockenhauer D, Bogdanovic R, Stajic N, Chernin G, Ettenger R, Fehrenbach H, Kemper M, Munarriz RL, Podracka L, Buscher R, Serdaroglu E, Tasic V, Mane S, Lifton RP, Braun DA, Hildebrandt F (2018) Whole exome sequencing of patients with steroid-resistant nephrotic syndrome. Clin J Am Soc Nephrol 13:53–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Forsgren M, Attersand A, Lake S, Grunler J, Swiezewska E, Dallner G, Climent I (2004) Isolation and functional expression of human COQ2, a gene encoding a polyprenyl transferase involved in the synthesis of CoQ. Biochem J 382:519–526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Salviati L, Sacconi S, Murer L, Zacchello G, Franceschini L, Laverda AM, Basso G, Quinzii C, Angelini C, Hirano M, Naini AB, Navas P, DiMauro S, Montini G (2005) Infantile encephalomyopathy and nephropathy with CoQ10 deficiency: a CoQ10-responsive condition. Neurology 65:606–608 [DOI] [PubMed] [Google Scholar]
- 68.Desbats MA, Morbidoni V, Silic-Benussi M, Doimo M, Ciminale V, Cassina M, Sacconi S, Hirano M, Basso G, Pierrel F, Navas P, Salviati L, Trevisson E (2016) The COQ2 genotype predicts the severity of coenzyme Q10 deficiency. Hum Mol Genet 25:[ISP-Check]4256–4265. [DOI] [PubMed] [Google Scholar]
- 69.Widmeier E, Yu S, Nag A, Chung YW, Nakayama M, Fernandez-Del-Rio L, Hugo H, Schapiro D, Buerger F, Choi WI, Helmstadter M, Kim JW, Ryu JH, Lee MG, Clarke CF, Hildebrandt F, Gee HY (2020) ADCK4 deficiency destabilizes the coenzyme Q complex, which is rescued by 2,4-dihydroxybenzoic acid treatment. J Am Soc Nephrol 31:1191–1211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wu L, Zhang Q, Mo W, Feng J, Li S, Li J, Liu T, Xu S, Wang W, Lu X, Yu Q, Chen K, Xia Y, Lu J, Xu L, Zhou Y, Fan X, Guo C (2017) Quercetin prevents hepatic fibrosis by inhibiting hepatic stellate cell activation and reducing autophagy via the TGF-beta1/ Smads and PI3K/Akt pathways. Sci Rep 7:9289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Vazquez Fonseca L, Doimo M, Calderan C, Desbats MA, Acosta MJ, Cerqua C, Cassina M, Ashraf S, Hildebrandt F, Sartori G, Navas P, Trevisson E, Salviati L (2018) Mutations in COQ8B (ADCK4) found in patients with steroid-resistant nephrotic syndrome alter COQ8B function. Hum Mutat 39:406–414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Freyer C, Stranneheim H, Naess K, Mourier A, Felser A, Maffezzini C, Lesko N, Bruhn H, Engvall M, Wibom R, Barbaro M, Hinze Y, Magnusson M, Andeer R, Zetterstrom RH, von Dobeln U, Wredenberg A, Wedell A (2015) Rescue of primary ubiquinone deficiency due to a novel COQ7 defect using 2,4-dihydroxybensoic acid. J Med Genet 52:779–783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kwong AK, Chiu AT, Tsang MH, Lun KS, Rodenburg RJT, Smeitink J, Chung BH, Fung CW (2019) A fatal case of COQ7-associated primary coenzyme Q10 deficiency. JIMD Rep 47:23–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Duncan AJ, Bitner-Glindzicz M, Meunier B, Costello H, Hargreaves IP, Lopez LC, Hirano M, Quinzii CM, Sadowski MI, Hardy J, Singleton A, Clayton PT, Rahman S (2009) A nonsense mutation in COQ9 causes autosomal-recessive neonatal-onset primary coenzyme Q10 deficiency: a potentially treatable form of mitochondrial disease. Am J Hum Genet 84:558–566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lyon MF, Hulse EV (1971) An inherited kidney disease of mice resembling human nephronophthisis. J Med Genet 8:41–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Peng M, Jarett L, Meade R, Madaio MP, Hancock WW, George AL Jr, Neilson EG, Gasser DL (2004) Mutant prenyltransferaselike mitochondrial protein (PLMP) and mitochondrial abnormalities in kd/kd mice. Kidney Int 66:20–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Barisoni L, Madaio MP, Eraso M, Gasser DL, Nelson PJ (2005) The kd/kd mouse is a model of collapsing glomerulopathy. J Am Soc Nephrol 16:2847–2851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Peng M, Falk MJ, Haase VH, King R, Polyak E, Selak M, Yudkoff M, Hancock WW, Meade R, Saiki R, Lunceford AL, Clarke CF, Gasser DL (2008) Primary coenzyme Q deficiency in Pdss2 mutant mice causes isolated renal disease. PLoS Genet 4:e1000061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Luna-Sanchez M, Diaz-Casado E, Barca E, Tejada MA, Montilla-Garcia A, Cobos EJ, Escames G, Acuna-Castroviejo D, Quinzii CM, Lopez LC (2015) The clinical heterogeneity of coenzyme Q10 deficiency results from genotypic differences in the Coq9 gene. EMBO Mol Med 7:670–687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Widmeier E, Airik M, Hugo H, Schapiro D, Wedel J, Ghosh CC, Nakayama M, Schneider R, Awad AM, Nag A, Cho J, Schueler M, Clarke CF, Airik R, Hildebrandt F (2019) Treatment with 2,4-dihydroxybenzoic acid prevents FSGS progression and renal fibrosis in podocyte-specific Coq6 knockout mice. J Am Soc Nephrol 30:393–405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Gee HY, Otto EA, Hurd TW, Ashraf S, Chaki M, Cluckey A, Vega-Warner V, Saisawat P, Diaz KA, Fang H, Kohl S, Allen SJ, Airik R, Zhou W, Ramaswami G, Janssen S, Fu C, Innis JL, Weber S, Vester U, Davis EE, Katsanis N, Fathy HM, Jeck N, Klaus G, Nayir A, Rahim KA, Al Attrach I, Al Hassoun I, Ozturk S, Drozdz D, Helmchen U, O’Toole JF, Attanasio M, Lewis RA, Nurnberg G, Nurnberg P, Washburn J, MacDonald J, Innis JW, Levy S, Hildebrandt F (2014) Whole-exome resequencing distinguishes cystic kidney diseases from phenocopies in renal ciliopathies. Kidney Int 85:880–887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wang Y, Oxer D, Hekimi S (2015) Mitochondrial function and lifespan of mice with controlled ubiquinone biosynthesis. Nat Commun 6:6393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hargreaves IP (2014) Coenzyme Q10 as a therapy for mitochondrial disease. Int J Biochem Cell Biol 49:105–111 [DOI] [PubMed] [Google Scholar]
- 84.Ozeir M, Muhlenhoff U, Webert H, Lill R, Fontecave M, Pierrel F (2011) Coenzyme Q biosynthesis: Coq6 is required for the C5-hydroxylation reaction and substrate analogs rescue Coq6 deficiency. Chem Biol 18:1134–1142 [DOI] [PubMed] [Google Scholar]
- 85.Doimo M, Trevisson E, Airik R, Bergdoll M, Santos-Ocana C, Hildebrandt F, Navas P, Pierrel F, Salviati L (2014) Effect of vanillic acid on COQ6 mutants identified in patients with coenzyme Q10 deficiency. Biochim Biophys Acta 1842:1–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hermle T, Braun DA, Helmstadter M, Huber TB, Hildebrandt F (2017) Modeling monogenic human nephrotic syndrome in the Drosophila garland cell nephrocyte. J Am Soc Nephrol 28:[ISP-Check]1521–1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Acosta Lopez MJ, Trevisson E, Canton M, Vazquez-Fonseca L, Morbidoni V, Baschiera E, Frasson C, Pelosi L, Rascalou B, Desbats MA, Alcazar-Fabra M, Rios JJ, Sanchez-Garcia A, Basso G, Navas P, Pierrel F, Brea-Calvo G, Salviati L (2019) Vanillic acid restores coenzyme Q biosynthesis and ATP production in human cells lacking COQ6. Oxidative Med Cell Longev 2019:3904905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wang Y, Smith C, Parboosingh JS, Khan A, Innes M, Hekimi S (2017) Pathogenicity of two COQ7 mutations and responses to 2,4-dihydroxybenzoate bypass treatment. J Cell Mol Med 21:[ISP-Check]2329–2343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Sobreira C, Hirano M, Shanske S, Keller RK, Haller RG, Davidson E, Santorelli FM, Miranda AF, Bonilla E, Mojon DS, Barreira AA, King MP, DiMauro S (1997) Mitochondrial encephalomyopathy with coenzyme Q10 deficiency. Neurology 48:1238–1243 [DOI] [PubMed] [Google Scholar]
- 90.Lopez-Lluch G, Del Pozo-Cruz J, Sanchez-Cuesta A, Cortes-Rodriguez AB, Navas P (2019) Bioavailability of coenzyme Q10 supplements depends on carrier lipids and solubilization. Nutrition 57:133–140 [DOI] [PubMed] [Google Scholar]
- 91.Zaki NM (2016) Strategies for oral delivery and mitochondrial targeting of CoQ10. Drug Deliv 23:1868–1881 [DOI] [PubMed] [Google Scholar]