

Abstract

11,12-Dihydrodibenzo[c,g]-1,2-diazocines have been established as a viable alternative to azobenzene for photoswitching, in particular, as they show an inverted switching behavior: the ground state is the Z isomer. In this paper, we present an improved method to obtain dibenzodiazocine and its derivatives from the respective 2-nitrotoluenes in two reaction steps, each proceeding in minutes. This fast access to a variety of derivatives permitted the study of substitution effects on the synthesis and on the photochemical properties. With biochemical applications in mind, methanol was chosen as a protic solvent system for the photochemical investigations. In contrast to the azobenzene system, none of the tested substitution patterns resulted in more efficient switching or in significantly prolonged half-lives, showing that the system is dominated by the ring strain.
1. Introduction
Photoswitches have become very useful tools for a wide range of applications, e.g., in pharmacology,1 biochemistry,2 nanotechnology,3 and catalytic chemistry,4 because they permit a reagent-free change of conformation with high temporal and spatial control. The probably most popular photochromic molecules are azobenzenes, as they typically offer good switching reversibility together with high photostabilities.2,5,6 In this class of molecules, the thermodynamically stable configuration is the E state, in which the backbone is almost rod-like, while in the Z configuration. it is adopting a V shape, which is typically used to relocate covalently attached moieties.3,4,7−9 The time frame, in which experiments can be performed with this photoactivated state, is limited by spontaneous relaxation, the half-life of which is typically in the range of seconds to hours depending on the exact structure of the system, the solvent, and certainly the temperature.5 Due to the considerable overlap of the spectra of the E and Z states, typically no complete transformation to the Z state can be attained but rather photostationary states (PSS), in which 5% or more material remains in the E configuration.6 While this is not much of a problem, if the Z state is the biologically active one, it significantly blurs the results of the switching event if the E state is the one to be observed.
An interesting alternative is 11,12-dihydrodibenzo[c,g]-1,2-diazocine, 1a (Scheme 1), which is derived from azobenzene by formally inserting an ethylene bridge between the 2-positions at the phenyl rings. This ethylene bridge provides so much strain on the system that the Z state becomes the ground state but not enough to inhibit the formation of the E state by excitation. Although first described in 1910,10 its photochemistry was only recently explored by Herges and Temps.11,12 Due to its distortion and the resulting shifts in the absorption spectra, 1a can be isomerized almost quantitatively (92%) by visible light with high quantum yield and a half-life of about 4.5 h (in hexane at 28.5 °C).11 These features gained the interest of the community, resulting in a series of publications.13−22 Nevertheless, the number of reported derivatives remains limited and typically only small yields were reported. Basically, two synthetic routes are commonly followed: the reduction of 2,2′-dinitrobibenzyls (2)10,17,23−25 and the oxidation of 2,2′-diaminobibenzyls,14,26 both of which result in the formation of the N=N bond. An interesting new and efficient alternative approach has recently been reported by Staubitz et al., who coupled hydrazine derivatives, which already bear the N–N bond, with 2,2′-diiodobibenzyls to obtain a series of new diazocine derivatives.27 A very extensive publication by Trauner et al. describes a series of mostly nonsymmetrical diazocine derivatives and their photochemical properties.28 As with the nonsymmetrical azobenzene derivatives,5,29,30 most of the nonsymmetrical diazocines show significantly lowered thermal stability as determined by the half-life times.
Scheme 1. (a) Switching Behavior of 11,12-Dihydrodibenzo[c,g]-1,2-diazocine 1a, for which the Z Configuration is the Ground State; (b) Strategies Reported in the Literature to Convert 2,2′-Dinitrobibenzyl (2a) to 1a.

i = Pb, base, MeOH; ii = Ph3P, MoCl2O2, THF or PCl3, C6H6; iii = Zn, Ba(OH)2, EtOH; iv = HgO, EtOH or O2, CuCl2, NaOH, MeOH; v = glucose, NaOH, EtOH, H2O or Pb in a ball mill.
In this project, we wanted to study the effect of different substituents in the series of symmetrical dibenzodiazocines to understand the impact of substitution on the photochemical properties and on the respective syntheses. In the parent azobenzene series, strategies have been reported that stabilize the photoexcited state, e.g., by introduction of four halogen atoms or methoxy groups at the ortho positions.30−32 This kind of substitution also shifts the absorption of the respective isomers into the visible range, which is very advantageous for the application in biological systems.33 It would be very useful to also attain similar effects in the diazocine series.
2. Results and Discussion
As the symmetric 2,2′-dinitrobibenzyls (2) can be very easily obtained by oxidative coupling of the respective 2-nitrotoluenes (3),24,34 many derivatives of which are readily accessible, we decided to use the reductive ring closure as an entrance to the respective class of diazocines. We consider this advantageous over the oxidative coupling of 2,2′-diaminobibenzyls, as the latter are often obtained from the nitro derivatives by hydrogenation or comparable reduction strategies. For the lead compound, 1a, three different routes for the reductive ring closure have been established (Scheme 1): the direct reduction,17,22,24,25,35 the path via the corresponding azoxy compound 4a,15,36 and the one via the hydrazine derivative 5a.16,22−24,35 Typical challenges of all the published synthetic strategies are the fair to poor yields, the long reaction times of several hours up to days, or the equipment, which typically cannot be found in organic labs, such as ball mills. Also, often heavy metal reagents are employed, which might interfere with biological systems even in trace amounts.
To our surprise, we found that the same reagent, which is frequently employed for the formation of the acyclic azobenzenes from the respective nitrobenzenes, lithium tetrahydridoaluminate,37,38 can be successfully employed for the direct synthesis of diazocine (1a) too. When using standard conditions, 1a was obtained in a yield of 28%, setting the basis for further investigations. As this approach led to the formation of significant amounts of azoxybenzene derivative 4a as a by-product, we tried to increase the number of hydride equivalents successively. At about 20 equiv, the formation of 4a could basically be suppressed, but a major side-product now was hydrazobenzene 5a. In contrast to the azoxybenzene derivative 4a, a convenient protocol exists for the efficient conversion of the hydrazobenzene 5a into the diazocine 1a.24 Thus, by exposing the organic phase after the work-up to air in the presence of Cu2+, the yield of 1a could be maximized. We also tried to moderate the reactivity of the LiAlH4 by replacing some of the hydrogen atoms with other ligands like alkoxy groups.39,40 In addition, alternative complex hydrides were tested, which have been reported for the dimerization of nitroarenes to the analogous azobenzenes.41−45 The results are summarized in Table 1.
Table 1. Optimization of the Synthesis of Diazocine 1a Starting from 2,2′-Dinitrobibenzyl 2a.
| reducing agent/mol L–1a | molar ratio to 2 | T/°Cb | time/minc | isol. yield/% | |
|---|---|---|---|---|---|
| LiAlH4 | 2.4 | 6.25 | RT | 80 | 28 |
| LiAlH4 | 2.4 | 15 | RT | 10 | 44 |
| LiAlH4 | 0.2 | 20 | RT | 10 | 53 |
| LiAlH4 | 2.4 | 20 | RT | 10 | 58 |
| LiAlH4 | 2.4 | 20 | 40 | 10 | 41 |
| LiAlH4 | 2.4 | 20 | 0 | 70 | 58 |
| LiAlH4 | 2.4 | 20 | –78 | 170 | - |
| LiAlH4d | 2.4 | 20 | RT | 10 | 22 |
| LiAlH(OMe)3 | 1.7 | 15 | RT | 230 | 48 |
| LiAlH(OMe)3 | 0.9 | 80 | RT | 230 | 33 |
| LiAlH(OMe)3 | 0.2 | 25 | RT | 230 | 40 |
| LiBHEt3 | 1.0 | 10 | RT | 230 | -e |
| LiAlH4·NMP | 0.5 | 20 | RT | 230 | 6 |
| LiAlH2(OtBu)2 | 0.5 | 20 | RT | 230 | 11 |
| LiAlH(StBu)3 | 0.6 | 20 | RT | 230 | - |
| LiAlH(SEt)3 | 0.6 | 20 | RT | 50 | -e |
| LiBH4 | 0.6 | 3 | RT | 230 | - |
Solution in THF.
RT = room temperature.
After the indicated reaction time, a 10 min reaction time for the reoxidation of the hydrazine side product followed.
Inverted addition.
Some formation of respective 2,2′-diaminobibenzyl.
LiAlH4 at a high concentration (2.4 mol L–1, concentration of the commercial reagent) and room temperature or 0 °C provided 1a with a yield of 58%, which is the same as the highest yields reported in the literature for the reductive coupling.24 When the reaction was run at 0 °C, it was completed after 80 min, while at room temperature, it took only 20 min, including the reoxidation time. This is a significant acceleration as compared to all the literature-known reductive ring closures, which take at least several hours. Of the alternative complex hydrides, only the trimethoxy derivative, LiAlH(OMe)3, resulted in acceptable yields.
This efficient method was extended to the synthesis of several derivatives in order to understand the width of applicability and substituent effects. A particular focus was put onto molecules, which can be integrated into biochemical systems via suitable linker groups. In contrast to many previous publications,16,19,27,28 we decided to use the hydroxyl group rather than the amino group, as we were planning to open a window into carbohydrate chemistry, where the typical functional groups are hydroxyl groups. Consequently, a number of protected OH groups, such as methoxymethoxy (OMOM) or benzyloxy (OBn), were tested together with other substituents, which are known to significantly alter the switching behavior in the acyclic azobenzene series.5
To this end, a series of 2-nitrotoluene derivatives 3b–n was oxidatively coupled to form the respective 2,2′-dinitrobibenzyls 2b–n (Scheme 2), see the Supporting Information for details. For the coupling, we basically used the established routine24 with the exception of lowering the reaction temperature to −78 °C, because this provided more reproducible results. The yields for the coupling steps are summarized in Table 2.
Scheme 2. Synthesis of the Symmetrical Dibenzodiazocines 1a–n with Different Functional Groups in Varying Substitution Patterns by Oxidative Coupling of the 2-Nitrotoluenes 3a–n Followed by Reductive Ring Closure with LiAlH4.

For indicating the position, we chose Roman numerals to avoid confusion with the IUPAC nomenclature or other assignments used in this publication.
Table 2. Synthetic Yields and Photochemical Parameters for the Dibenzodiazocines 1a–n Obtained by Reductive Ring Closure with Complex Metal Hydridesc.
| derivative with R = | yield bibenzyl 2/% | yield diazocine 1/% | τ1/2/h | % E @ PSS385 | |
|---|---|---|---|---|---|
| H | a | 60 | 58 | 5.30 ± 0.07 | 82 |
| i-F | b | 74 | 38 | 7.18 ± 0.02 | 78 |
| i-Cl | c | 80 | 26 | 10.5 ± 0.2 | 75 |
| i-Br | d | 64 | 20a | 10.8 ± 0.1 | 73 |
| i-CF3 | e | 79 | 33b | 8.44 ± 0.05 | 77 |
| i-NMe2 | f | 65 | 28 | 14.3 ± 0.2 | 28 |
| ii-OMe | g | 14 | 10 | 2.64 ± 0.03 | 31 |
| ii-OMOM | h | 21 | 22 | 2.21 ± 0.02 | 25 |
| ii-OBn | i | 57 | 3 | 2.64 ± 0.06 | 45 |
| ii-F | j | 71 | 44 | 10.2 ± 0.1 | 41 |
| iii-OMe | k | 25 | 20b | 8.6 ± 0.2 | 35 |
| iii-OMOM | l | 36 | 12 | 11.3 ± 0.2 | 37 |
| iv-F | m | 64 | 17a | 14.1 ± 0.2 | 49 |
| iv-Me | n | 44 | 6 | 20.0 ± 0.2 | 72 |
reduction with LiAlH(OMe)3
These compounds could not be completely purified, see Supp. Info. for the spectra. Nevertheless, the impurities turned out not be photoactive
Thermal half-life of the E isomers and their fraction at PSS at λ = 385 nm were determined at 23 °C by UV/vis spectroscopy in MeOH and 1H NMR in CD3OD, respectively.
As can be seen, the yields directly correlate with the substituents of the 2-nitrotoluene derivatives: electron-withdrawing groups such as −CF3 or −Cl increase the yields, presumably through the increased acidity of the methyl groups, while electron-donating groups typically lower the yields of the 2,2′-dinitrobibenzyls. A notable exception is 2f, in which the ortho dimethylamino group might stabilize the intermediate potassium salt by complexation.
The different 2,2′-dinitrobibenzyl derivatives 2b–n were subjected to the optimized ring-closure conditions, which in most cases, successfully yielded the respective dibenzodiazocins. Again, clear effects of the substituents could be observed: while the compounds with electron-withdrawing groups, such as −CF3 or halogen, generally resulted higher yields (up to 44%), the yields for the compounds with electron-donating groups were lower. A number of special cases have to be discussed: (1) The bromo derivative 2d lost its bromine atoms when treated with LiAlH4, resulting in the formation of the parent compound 1a. This could be suppressed by using LiAlH(OMe)3 instead, which worked satisfactory in the screening study (see Table 1) and is known to tolerate more functional groups than LiAlH4.41 (2) The benzyloxy compound 3i became partly cleaved by the hydride ions. In this case, the unwanted side reaction could not be suppressed by the use of the methoxylated aluminate so that the yield never exceeded 3%. (3) In the case of the ortho methyl compound 2n, the yields presumably are low because of the steric hindrance at the nitrogen atoms hampering the ring-closure reaction. (4) A similar effect might play a role for the ortho fluorine derivative 2 m, for which additionally, the formation of significant amounts of the respective 2,2′-diaminobibenzyl was observed. In this case, using LiAlH(OMe)3 could minimize the over-reduction and a yield of 17% diazocine 1m was obtained.
With this series of symmetrical dibenzodiazocines at hand, we set out to explore the photochemical properties. The thermal stability of the excited state and the photoconvertibility are important parameters, in particular, if molecular switches are to be included into larger systems. For many applications, an optimal photoswitch should be quantitatively converted from one state to the other by irradiation. Therefore, the n+ → π* transitions in the absorption spectra of the E and the Z isomers should be energetically well separated. To minimize uncontrolled back-switching during the experiments, the thermal half-life τ1/2 should be as long as possible.32 As τ1/2 significantly depends on the environment, in particular the solvent, it is advisable to determine its value under conditions as close as possible to the latter applications. For the use in biological systems, aqueous buffers would be ideal, but as the simple dibenzodiazocines are not soluble in water, we rather chose methanol as the model solvent for the photochemical experiments.
We first recorded the UV spectra for the ground state, Z-configured isomers, and then the ones of the PSS, in which a dynamic equilibrium between the E and the Z isomers is established. For the excitation of the system, we chose to use light of 385 nm, as in most of the derivatives, including the parent compound 1a, the absorption of the Z state at this wavelength is significantly higher than for the E state, favoring a maximal switching. Representative spectra are shown in Figure 1; the spectra of the other diazocines are depicted in the Supporting Information. The solutions in the PSS were then characterized by 1H NMR spectroscopy, where the signals of the two isomers are separated and can be individually integrated to evaluate the composition of the binary mixtures (see Table 2). In a series of separate experiments, the mixtures were permitted to relax thermally (at 23 °C) to the ground state while being observed by UV/vis spectroscopy. In all cases, clean isosbestic points were obtained, confirming the direct switching of the isomers without any intermediate states. The kinetic data were evaluated quantitatively by determination of the absorbances at 480 nm, which is close to the maximum of the n+ → π* transition of the E isomers. The respective semilogarithmic plots are shown in the Supporting information and yielded the τ1/2 for the different derivatives.
Figure 1.
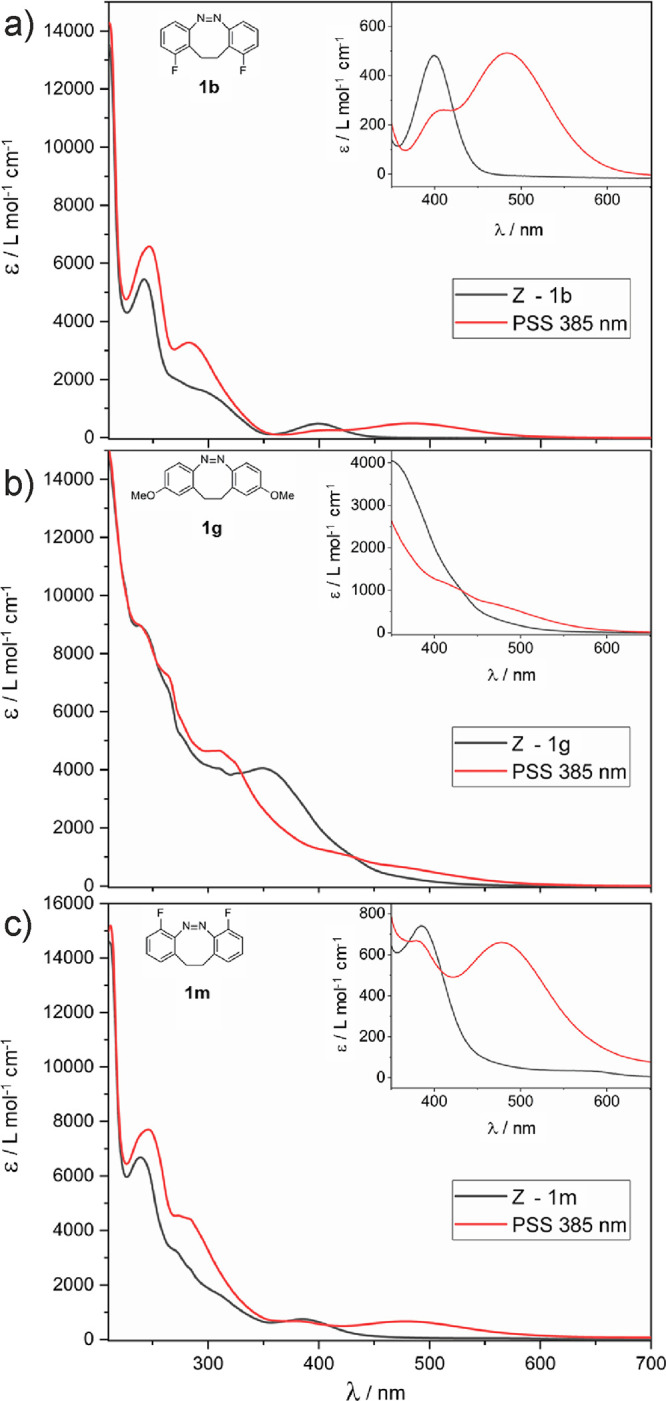
Absorption spectra of representative dibenzodiazocines at the ground state (Z isomer; black) and at the photostationary state (PSS; red) at 385 nm, recorded in MeOH. (A) Spectra of 1,10-difluoro-11,12-dihydrodibenzo[c,g]-1,2-diazocine 1b. (b) Spectra of 2,9-dimethoxy-11,12-dihydrodibenzo[c,g]-1,2-diazocine 1g. (c) Spectra of 4,7-difluoro-11,12-dihydrodibenzo[c,g]-1,2-diazocine 1m. Insets show the n+ → π* transitions.
It turns out that the substituents have significant impact on the spectra of both states. In the nonsubstituted 11,12-dihydrodibenzo[c,g]-1,2-diazocine 1a, the absorption maxima have been reported to be well separated, which is the basis for the excellent performance of the system. As a first trend, it has to be noted that the peak separation in methanol is less pronounced than in the aprotic solvents used in most previous studies. As a result, the fraction of the E isomer in the PSS is limited to 82% (Table 2, first entry). The same is true for all the other derivatives tested in this study and presumably is a general trend that needs to be accounted for in other protic solvents, such as water.
In the following, we discuss the effect of the substitutions in the four possible positions with the different functional groups. In the i position, which is meta to the azo group (see Scheme 2 for numbering), the separation and intensity of the absorption bands basically stays the same as in the parent compound 1a for most substituents. This goes in line with previous observations in the azobenzene system, the properties of which are not affected much by meta-substituents.5 Indeed, the −F group in the position i even induced a slight increase in the molar extinction of the n+ → π* absorption of the Z isomers while rendering the spectrum of the PSS almost unchanged (Figure 1a). In contrast to this, introducing −NMe2 groups into the same position led to a significant reduction of the intensity of the respective band basically to the point that the extinction maxima for both isomers became the same. This certainly also resulted in a decreased switching effect, as in the PSS only 28% of the molecules were converted to the E form. We believe that this is caused by the very strong +M effect of this group, but we cannot exclude at this stage that protonation by the solvent might play a role as well. A weakening of the double-bond character of the N=N bond can be excluded in this case, as the half-life of the E isomer is clearly longer (14.3 h) than the average life time in this series.
In the ii position, para to the azo group, the presence of substituents with +M effect is generally detrimental (1g–j). While for the case of the fluorine atoms, the effects are limited (spectra are similar to the ones of 1a, τ1/2 ≈ 10.2 h with 41% of E in the PSS), and the spectra of the alkoxy derivatives become almost featureless, as the clear maxima disappear for both, the Z and the E states (Figure 1b). This observation is in agreement with the situation for azobenzenes, where alkoxy groups in the para position lead to deteriorated switching and significantly reduced half-lives (by factors of up to 106), which generally is attributed to the weakening of the N=N double-bond character.46,47 As expected, this effect is less pronounced in the position iii, where the half-life of the E isomers rises to about 10 h again, although the fractions of E isomers in the PSS remain low (∼35%, 1k,l). This observation is meaningful, as the ii and iii positions are sterically equivalent in the dibenzodiazocine system, which is not the case in the azobenzene system.
Of basic interest are also the substitutions at the iv position, the only available ortho position to the azo group in this system. As mentioned before, this kind of substitution results in the azobenzene system in significantly prolonged half-lives and in red-shifted maxima in the spectra.30−32 In our case, the fluorine derivative 1m again shows spectra similar to the one of the lead compound 1a, only with a significantly increased shoulder at 400 nm for the E state (see Figure 1c), which limits the fraction of the E isomer in the PSS to 49%. The situation is even worse for the methyl derivative, for which neither the E nor the Z states show a maximum around 400 nm. Although no positive electronic effect was found, these two derivatives show the longest half-lives, with 14.1 h for 1m and 20 h for 1n, demonstrating a significant steric effect, although much smaller than the one found in the azobenzene series.27−30
3. Conclusions
As the use of lithium tetrahydridoaluminate significantly simplifies the ring-closure reaction of 2,2′-dinitrobibenzyls; we now have a facile route at hand to access dibenzodiazocines in two steps starting from the respective 2-nitrotoluenes. As both reactions can be run on the time scale of minutes and in homogeneous solution, this is the fastest way to this highly interesting class of photoswitches. For the lead compound 1a, the achievable yield is the same as the best ones previously published for the reductive ring closure. For the substituted derivatives, lower yields were attained, although the derivatives with electron-withdrawing groups show yields up to 44%. In the case of functional groups that become easily cleaved off by LiAlH4, a milder alternative was found in LiAlH(OMe)3.
The photochemical properties of the new diazocines in methanolic solution were investigated. Similar to acyclic azobenzenes, the variation of the substituents enables the tuning of the photochemistry of diazocines. Halogenation in the meta position to the N=N moiety leads to an increased stability of the excited state, whereas a NMe2 group at the same site results in an even more stable E isomer but leads to a lower photoconversion. A significant stabilization of the E state could also be attained by substitution in the ortho position. In general, the effects of the substitution on the photochemical properties are much smaller in the diazocine system than for the azobenzenes, e.g., the half-life can only be increased by a factor of four, not by several orders of magnitude. None of the substituents had an advantageous effect on the photoconversion efficiency: the value of 82% for 1a, which is lower than the one found in aprotic solvents (92%), could not be exceeded by any derivative.
For future incorporation into larger (bio)molecular systems via oxygen-containing groups, substitution at the meta position (iii) seems to be advisable, as substituents in the para position seem to be generally detrimental. The optimized synthetic procedure presented here and the determined impact of substituents on the photochemistry of 11,12-dihydrodibenzo[c,g]-1,2-diazocine will facilitate their application in further biochemical, pharmacological, and chemical studies.
4. Experimental Part
4.1. General Information
4.1.1. NMR Spectroscopy
The following NMR spectrometers were used in this study: Bruker AV400 and Bruker DRX600. All measurements were performed at room temperature in deuterated chloroform (CDCl3). For calibration, the signal of residual CHCl3 was employed. Chemical shifts refer to the δ scale in parts per million (ppm). Coupling constants J are given in Hertz (Hz).
4.1.2. Mass Spectrometry
Electrospray ionization mass spectrometry (ESI-MS) spectra were measured on a Thermo Fisher Surveyor MSQ. All high-resolution mass spectrometry (HRMS) spectra were recorded on a Thermo Fisher Scientific LTQ Orbitrap XL.
4.1.3. UV–Vis Spectroscopy
The UV–vis spectra were recorded in a 1.0 cm quartz cuvette using methanol (≥99.8%, HiPerSolv CHROMANORM, VWR) as a solvent.
The thermal half-life of the E isomers was determined with an Ocean Optics USB4000 detector, which is connected via optical fibers to a CVH100 cuvette holder from Thorlabs and to a DH-mini light source (Ocean Optics). The optical setup was controlled by PHITS (photoswitch irradiator test suite), which was programmed by the Heckel group at the University of Frankfurt.
All other UV–vis spectra were recorded on a Specord S 600 (Analytic Jena).
4.1.4. Light Sources
The samples for the determination of the thermal half-life were irradiated by an M385L2 LED (385 nm; 700 mA) from Thorlabs run with a DC2100 LED driver (Thorlabs).
The evaluation of the photoconvertibility was performed via NMR spectroscopy after irradiation with a custom-made 3 W LED (370 nm–380 nm; 750 mA) mounted on a starboard from Avonec.
4.1.5. Chemicals and Column Chromatography
All reactions except the synthesis of 6-(dimethylamino)-2-nitrotoluene were carried out under a nitrogen atmosphere using absolute solvents. DCM was dried over activated 4 Å molecular sieves. THF was first dried with Na and benzophenone and then distilled. LiAlH4 suspension in THF (2.40 M) was received from Albemarle.
Silica gel 60 (Macherey–Nagel) had a particle size of 0.063–0.2 mm. TLC plates with a layer thickness of 0.2 mm with a fluorescence indicator from Macherey–Nagel were used.
4.2. Syntheses
4.2.1. Syntheses of 2-Nitrotoluene Derivatives 3
4.2.1.1. 6-(Dimethylamino)-2-nitrotoluene 3f
The synthesis of 6-(dimethylamino)-2-nitrotoluene was performed according to Otevrel and Bobal.48 To a solution of formaldehyde (37%, 17.0 mL, 120 mmol) and sulfuric acid (20%, 27 mL, 99 mmol) in THF (33 mL) were alternatingly added a solution of 2-methyl-3-nitroaniline (2.17 g, 13.0 mmol) in THF (67 mL) and NaBH4 (6.23 g, 184 mmol). After 2 h of stirring at room temperature, the reaction mixture was transferred to a saturated solution of K2CO3 and then extracted with EtOAc. The combined organic layers were dried over MgSO4 and the solvent was removed in vacuum. The product could be isolated quantitatively (2.34 g) as yellow oil.
1H NMR (400 MHz, CDCl3) δ: 7.49 (dd, J = 6.6, 2.7 Hz, 1H), 7.31–7.23 (m, 2H), 2.77 (s, 6H), 2.46 (s, 3H).
ESI-MS: 181.21 [M + H]+
HRMS: calcd, 181.09770; found, 181.09722 [M + H]+
4.2.1.2. 4- and 5-(Methoxymethoxy)-2-nitrotoluene 3h,l
The introduction of the methoxymethyl group was performed according to Fuji et al.(49)
5-(Methoxymethoxy)-2-nitrotoluene3h: To a solution of 3-methyl-4-nitrophenol (10.18 g, 65.45 mmol) in dichloromethane (400 mL) were added first dimethoxymethane (29 mL, 0.33 mol) and then P2O5 (75 g, 0.53 mol). The reaction mixture was stirred at RT overnight and then decanted. The organic layer was washed with KOH solution and water. The combined organic layers were dried over MgSO4 and the solvent was removed in vacuum. The product could be isolated in a yield of 11.5 g (89%) as brown oil. 1H NMR (600 MHz, CDCl3) δ: 7.66 (d, J = 2.6 Hz, 1H), 7.23 (d, J = 8.5 Hz, 1H), 7.18 (dd, J = 8.5, 2.6 Hz, 1H), 5.19 (s, 2H), 3.48 (s, 3H), 2.52 (s, 3H). 13C NMR (101 MHz, CDCl3) δ: 160.8, 143.2, 137.0, 127.5, 119.5, 114.1, 94.3, 56.5, 21.7. ESI-MS: 197.93 [M + H]+, 196.24 [M-H]−. HRMS: calcd, 198.07608; found, 198.07599 [M + H]+
4-(Methoxymethoxy)-2-nitrotoluene 3l: 4-Methyl-3-nitrophenol was transformed analogously using the procedure described above. The product was isolated in a yield of 5.08 g (79%) as beige solid. 1H NMR (400 MHz, CDCl3) δ: 7.66 (d, J = 2.5 Hz, 1H), 7.23 (d, J = 8.5 Hz, 1H), 7.18 (dd, J = 8.5, 2.5 Hz, 1H), 5.19 (s, 2H), 3.48 (s, 3H), 2.53 (s, 3H). 13C NMR (101 MHz, CDCl3) δ: 155.8, 149.8, 133.6, 126.7, 121.6, 112.4, 94.8, 56.4, 19.8. ESI-MS: 197.94 [M + H]+. HRMS: calcd, 198.07608; found, 198.07609 [M + H]+
4.2.2. General Synthetic Procedure for 2,2′-Dinitrobibenzyls
The dimerization of the 2-nitrotoluenes to the corresponding bibenzyls was performed according to Moormann et al.24 In contrast to the literature, the best yields were observed at a reaction temperature of −78 °C.
At −78 °C, tBuOK solution in THF (1.05 equiv; 1.60 M) was slowly added to a solution of the 2-nitrotoluene derivative in THF (0.2 mM). After stirring for 5 min., Br2 (1 equiv) was added dropwise at −78 °C and the mixture was stirred again for 5 min. Then, the cooling was removed and the reaction was stopped by adding saturated aqueous Na2SO3 solution. The phases were separated and the aqueous one was extracted with DCM. The organic solvent was removed in vacuum and the raw product was further purified. Experimental details, purification, yields, and the spectroscopic data of compounds 2a–n can be found in the Supporting Information.
4.2.3. General Procedure for the Reductive Ring Closure to Dibenzodiazocines 1a–n
The corresponding bibenzyl 2 was slowly added to a suspension of LiAlH4 in THF (2.4 M, 80 equiv H–) at room temperature. After 10 min of stirring, the reaction was stopped by the slow addition of a THF/water mixture (9/1) until the gas evolution ceased. The resulting suspension was filtered and the residue was washed with acetone. CuCl2·2H2O (0.1 equiv) was added to the filtrate and compressed air was passed through the solution for 10 min. The solvent was removed in vacuum and the raw product was further purified. Experimental details, purification, yields, and the spectroscopic data of compounds 2a–n can be found in the Supporting Information.
Acknowledgments
We thank Prof. Alexander Heckel, University of Frankfurt, for giving us access to his photochemical setup for the determination of the thermal half-life of the diazocins 1a–m. The generous donation of LiAlH4 solution in THF as well as potassium t-butanolate by Albemarle is very much appreciated.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.1c02524.
It contains copies of the UV/vis, 1H, and 13C NMR spectra of relevant products as well as the experimental kinetic plots for the thermal relaxation (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Hüll K.; Morstein J.; Trauner D. In Vivo Photopharmacology. Chem. Rev. 2018, 118, 10710–10747. 10.1021/acs.chemrev.8b00037. [DOI] [PubMed] [Google Scholar]
- Lubbe A.; Szymanski W.; Feringa B. L. Recent Developments in Reversible Photoregulation of Oligonucleotide Structure and Function. Chem. Soc. Rev. 2017, 46, 1052–1079. 10.1039/C6CS00461J. [DOI] [PubMed] [Google Scholar]
- Wang L.; Li Q. Photochromism into Nanosystems: Towards Lighting up the Future Nanoworld. Chem. Soc. Rev. 2018, 47, 1044–1097. 10.1039/C7CS00630F. [DOI] [PubMed] [Google Scholar]
- Stoll R. S.; Hecht S. Artificial Light-Gated Catalyst Systems. Angew. Chem., Int. Ed. 2010, 49, 5054–5075. 10.1002/anie.201000146. [DOI] [PubMed] [Google Scholar]
- Bandara H. M. D.; Burdette S. C. Photoisomerization in Different Classes of Azobenzene. Chem. Soc. Rev. 2012, 41, 1809–1825. 10.1039/C1CS15179G. [DOI] [PubMed] [Google Scholar]
- Beharry A. A.; Woolley G. A. Azobenzene Photoswitches for Biomolecules. Chem. Soc. Rev. 2011, 40, 4422–4437. 10.1039/c1cs15023e. [DOI] [PubMed] [Google Scholar]
- Baroncini M.; Silvi S.; Credi A. Photo- and Redox-Driven Artificial Molecular Motors. Chem. Rev. 2020, 120, 200–268. 10.1021/acs.chemrev.9b00291. [DOI] [PubMed] [Google Scholar]
- Weber T.; Chandrasekaran V.; Stamer I.; Thygesen M. B.; Terfort A.; Lindhorst T. K. Switching of bacterial adhesion to a glycosylated surface by reversible reorientation of the carbohydrate ligand. Angew. Chemie Int. Ed. 2014, 53, 14583–14586. 10.1002/anie.201409808. [DOI] [PubMed] [Google Scholar]
- Chandrasekaran V.; Lindhorst T. K. Sweet Switches: Azobenzene Glycoconjugates Synthesized by Click Chemistry. Chem. Commun. 2012, 48, 7519–7521. 10.1039/c2cc33542e. [DOI] [PubMed] [Google Scholar]
- Duval H. Recherches sur la Benzidination. III. Recherches dans la série du diphényléthane. Bull. Soc. Chim. Fr. 1910, 7, 727–732. [Google Scholar]
- Siewertsen R.; Neumann H.; Buchheim-Stehn B.; Herges R.; Näther C.; Renth F.; Temps F. Highly Efficient Reversible Z–E Photoisomerization of a Bridged Azobenzene with Visible Light through Resolved S1 (nπ*) Absorption Bands. J. Am. Chem. Soc. 2009, 131, 15594–15595. 10.1021/ja906547d. [DOI] [PubMed] [Google Scholar]
- Siewertsen R.; Schönborn J. B.; Hartke B.; Renth F.; Temps F. Superior Z→E and E→Z Photoswitching Dynamics of Dihydrodibenzodiazocine, a Bridged Azobenzene, by S1 (nπ*) Excitation at λ = 387 and 490 nm. Phys. Chem. Chem. Phys. 2011, 13, 1054–1063. 10.1039/C0CP01148G. [DOI] [PubMed] [Google Scholar]
- Thapaliya E. R.; Zhao J.; Ellis-Davies G. C. R. Locked-Azobenzene: Testing the Scope of a Unique Photoswitchable Scaffold for Cell Physiology. ACS Chem. Neurosci. 2019, 10, 2481–2488. 10.1021/acschemneuro.8b00734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabré G.; Garrido-Charles A.; González-Lafont À.; Moormann W.; Langbehn D.; Egea D.; Lluch J. M.; Herges R.; Alibés R.; Busqué F.; Gorostiza P.; Hernando J. Synthetic Photoswitchable Neurotransmitters Based on Bridged Azobenzenes. Org. Lett. 2019, 21, 3780–3784. 10.1021/acs.orglett.9b01222. [DOI] [PubMed] [Google Scholar]
- Eljabu F.; Dhruval J.; Yan H. Incorporation of Cyclic Azobenzene into Oligodeoxynucleotides for the Photo-Regulation of DNA Hybridization. Bioorg. Med. Chem. Lett. 2015, 25, 5594–5596. 10.1016/j.bmcl.2015.10.043. [DOI] [PubMed] [Google Scholar]
- Samanta S.; Qin C.; Lough A. J.; Woolley G. A. Bidirectional Photocontrol of Peptide Conformation with a Bridged Azobenzene Derivative. Angew. Chem., Int. Ed. 2012, 51, 6452–6455. 10.1002/anie.201202383. [DOI] [PubMed] [Google Scholar]
- Li S.; Han G.; Zhang W. Concise Synthesis of Photoresponsive Polyureas Containing Bridged Azobenzenes as Visible-Light-Driven Actuators and Reversible Photopatterning. Macromolecules 2018, 51, 4290–4297. 10.1021/acs.macromol.8b00687. [DOI] [Google Scholar]
- Zhu Q.; Wang S.; Chen P. Diazocine Derivatives: A Family of Azobenzenes for Photochromism with Highly Enhanced Turn-On Fluorescence. Org. Lett. 2019, 21, 4025–4029. 10.1021/acs.orglett.9b01215. [DOI] [PubMed] [Google Scholar]
- Löw R.; Rusch T.; Röhricht F.; Magnussen O.; Herges R. Diazocine-Functionalized TATA Platforms. Beilstein J. Org. Chem. 2019, 15, 1485–1490. 10.3762/bjoc.15.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moormann W.; Langbehn D.; Herges R. Synthesis of functionalized diazocines for application as building blocks in photo- and mechanoresponsive materials. Beilstein J. Org. Chem. 2019, 15, 727–732. 10.3762/bjoc.15.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moormann W.; Tellkamp T.; Stadler E.; Röhricht F.; Näther C.; Puttreddy R.; Rissanen K.; Gescheidt G.; Herges R. Efficient Conversion of Light to Chemical Energy: Directional, Chiral Photoswitches with Very High Quantum Yields. Angew. Chem., Int. Ed. 2020, 59, 15081–15086. 10.1002/anie.202005361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tellkamp T.; Shen J.; Okamoto Y.; Herges R. Diazocines on Molecular Platforms. Eur. J. Org. Chem. 2014, 2014, 5456–5461. 10.1002/ejoc.201402541. [DOI] [Google Scholar]
- Paudler W. W.; Zeiler A. G. Diazocine chemistry. VI. Aromaticity of 5,6-dihydrodibenzo[b,f][1,2]diazocine. J. Org. Chem. 1969, 34, 3237–3239. 10.1021/jo01263a004. [DOI] [Google Scholar]
- Moormann W.; Langbehn D.; Herges R. Solvent-Free Synthesis of Diazocine. Synthesis 2017, 49, 3471–3475. 10.1055/s-0036-1590685. [DOI] [Google Scholar]
- Sell H.; Näther C.; Herges R. Amino-Substituted Diazocines as Pincer-Type Photochromic Switches. Beilstein J. Org. Chem. 2013, 9, 1–7. 10.3762/bjoc.9.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J.; He J.; Zhi C.; Luo B.; Li X.; Pan Y.; Cao X.; Gu H. Highly Efficient Synthesis of Azos Catalyzed by the Common Metal Copper (0) Through Oxidative Coupling Reactions. RSC Adv. 2014, 4, 16607–16611. 10.1039/c4ra00749b. [DOI] [Google Scholar]
- Li S.; Eleya N.; Staubitz A. Cross-Coupling Strategy for the Synthesis of Diazocines. Org. Lett. 2020, 22, 1624–1627. 10.1021/acs.orglett.0c00122. [DOI] [PubMed] [Google Scholar]
- Maier M. S.; Hüll K.; Reynders M.; Matsuura B. S.; Leippe P.; Ko T.; Schäffer L.; Trauner D. Oxidative Approach Enables Efficient Access to Cyclic Azobenzenes. J. Am. Chem. Soc. 2019, 141, 17295–17304. 10.1021/jacs.9b08794. [DOI] [PubMed] [Google Scholar]
- Dokić J.; Gothe M.; Wirth J.; Peters M. V.; Schwarz J.; Hecht S.; Saalfrank P. Quantum Chemical Investigation of Thermal Cis-to-Trans Isomerization of Azobenzene Derivatives: Substituent Effects, Solvent Effects, and Comparison to Experimental Data. J. Phys. Chem. A 2009, 113, 6763–6773. 10.1021/jp9021344. [DOI] [PubMed] [Google Scholar]
- Dong M.; Babalhavaeji A.; Samanta S.; Beharry A. A.; Woolley G. A. Red-Shifting Azobenzene Photoswitches for in Vivo Use. Acc. Chem. Res. 2015, 48, 2662–2670. 10.1021/acs.accounts.5b00270. [DOI] [PubMed] [Google Scholar]
- Beharry A. A.; Sadovski O.; Woolley G. A. Azobenzene Photoswitching without Ultraviolet Light. J. Am. Chem. Soc. 2011, 133, 19684–19687. 10.1021/ja209239m. [DOI] [PubMed] [Google Scholar]
- Bléger D.; Schwarz J.; Brouwer A. M.; Hecht S. o-Fluoroazobenzenes as Readily Synthesized Photoswitches Offering Nearly Quantitative Two-Way Isomerization with Visible Light. J. Am. Chem. Soc. 2012, 134, 20597–20600. 10.1021/ja310323y. [DOI] [PubMed] [Google Scholar]
- Benov L. Photodynamic Therapy: Current Status and Future Directions. Med. Princ. Pract. 2015, 24, 14–28. 10.1159/000362416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enyedy I. J.; Ling Y.; Nacro K.; Tomita Y.; Wu X.; Cao Y.; Guo R.; Li B.; Zhu X.; Huang Y.; Long Y. Q.; Roller P. P.; Yang D.; Wang S. Discovery of Small-Molecule Inhibitors of Bcl-2 through Structure-Based Computer Screening. J. Med. Chem. 2001, 44, 4313–4324. 10.1021/jm010016f. [DOI] [PubMed] [Google Scholar]
- Deo C.; Bogliotti N.; Métivier R.; Retailleau P.; Xie J. A Visible-Light-Triggered Conformational Diastereomer Photoswitch in a Bridged Azobenzene. Chem. – Eur. J. 2016, 22, 9092–9096. 10.1002/chem.201601400. [DOI] [PubMed] [Google Scholar]
- Joshi D. K.; Mitchell M. J.; Bruce D.; Lough A. J.; Yan H. Synthesis of Cyclic Azobenzene Analogues. Tetrahedron 2012, 68, 8670–8676. 10.1016/j.tet.2012.06.007. [DOI] [Google Scholar]
- Nystrom R. F.; Brown W. G. Reduction of Organic Compounds by Lithium Aluminum Hydride. III. Halides, Quinones, Miscellaneous Nitrogen Compounds1. J. Am. Chem. Soc. 1948, 70, 3738–3740. 10.1021/ja01191a057. [DOI] [PubMed] [Google Scholar]
- Merino E. Synthesis of Azobenzenes: the Coloured Pieces of Molecular Materials. Chem. Soc. Rev. 2011, 40, 3835–3853. 10.1039/c0cs00183j. [DOI] [PubMed] [Google Scholar]
- Málek J. Reductions by Metal Alkoxyaluminum Hydrides. Org. React. 2004, 34, 1–99. 10.1002/0471264180.or034.01. [DOI] [Google Scholar]
- Málek J. Reductions by Metal Alkoxyaluminum Hydrides. Part II. Carboxylic Acids and Derivatives, Nitrogen Compounds, and Sulfur Compounds. Org. React. 2004, 36, 249–334. 10.1002/0471264180.or036.03. [DOI] [Google Scholar]
- Brown H. C.; Weissman P. M. Selective Reductions. VII. Reaction of Lithium Trimethoxyaluminohydride with Selected Organic Compounds Containing Representative Functional Groups1. J. Am. Chem. Soc. 1965, 87, 5614–5620. 10.1021/ja00952a018. [DOI] [Google Scholar]
- Gaylord N. G. Reduction with Complex Metal Hydrides. J. Chem. Educ. 1957, 34, 367–374. 10.1021/ed034p367. [DOI] [Google Scholar]
- Brown H. C.; Kim S. C.; Krishnamurthy S. Selective Reductions. 26. Lithium Triethylborohydride as an Exceptionally Powerful and Selective Reducing Agent in Organic Synthesis. Exploration of the Reactions with Selected Organic Compounds Containing Representative Functional Groups. J. Org. Chem. 1980, 45, 1–12. 10.1021/jo01289a001. [DOI] [Google Scholar]
- Cha J. S.; Yu S. J. Reaction of Lithium Tris(tert-butylthiolato)hydridoaluminate with Selected Organic Compounds Containing Representative Functional Groups. J. Inclusion Phenom. Macrocyclic Chem. 2009, 65, 7–13. 10.1007/s10847-009-9628-4. [DOI] [Google Scholar]
- Fuller J. C.; Stangeland E. L.; Jackson T. C.; Singaram B. Lithium Aluminum Hydride-N-Methylpyrrolidine Complex. 1. Synthesis and Reactivity of Lithium Aluminum Hydride-N-Methylpyrrolidine Complex. An Air and Thermally Stable Reducing Agent Derived from Lithium Aluminum Hydride. Tetrahedron Lett. 1994, 35, 1515–1518. 10.1016/S0040-4039(00)76746-3. [DOI] [Google Scholar]
- Nishimura N.; Sueyoshi T.; Yamanaka H.; Imai E.; Yamamoto S.; Hasegawa S. Thermal Cis-to-Trans Isomerization of Substituted Azobenzenes II. Substituent and Solvent Effects. Bull. Chem. Soc. Jpn. 1976, 49, 1381–1387. 10.1246/bcsj.49.1381. [DOI] [Google Scholar]
- We wish to thank one of the reviewers of the manuscript for pointing out that the increased flexibility of the eight-membered ring becomes also visible in the 1H NMR spectra as coalescence of the methylene signals into one broad singlet.
- Otevrel J.; Bobal P. Diamine-Tethered Bis(thiourea) Organocatalyst for Asymmetric Henry Reaction. J. Org. Chem. 2017, 82, 8342–8358. 10.1021/acs.joc.7b00079. [DOI] [PubMed] [Google Scholar]
- Fuji K.; Nakano S.; Fujita E. An Improved Method for Methoxymethylation of Alcohols under Mild Acidic Conditions. Synthesis 1975, 4, 276–277. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.