

Abstract
Background
India accounts for 20% of the global retinoblastoma (RB) burden. However, the existing data on RB1 gene germline mutations and its influence on clinical decisions is minimally explored.
Methods
Fifty children with RB underwent complete clinical examination and appropriate multidisciplinary management. Screening of germline RB1 gene mutations was performed through next-generation sequencing and Multiplex Ligation-dependent Probe Amplification (MLPA) analysis. The mutation and non-mutation groups were compared for clinical parameters especially severity, progression and recurrence.
Results
Twenty-nine patients had bilateral RB (BLRB) and 21 had unilateral RB (ULRB). The genetic analysis revealed 20 RB1 variations in 29 probands, inclusive of 3 novel mutations, known 16 mutations and heterozygous whole gene deletions. The mutation detection rate (MDR) was 86.2% in BLRB and 19% in ULRB. Associations of disease recurrence (p = 0.021), progression (p = 0.000) and higher percentage of optic nerve invasion, subretinal seeds and high-risk pathological factors were observed in the mutation group. Clinical management was influenced by the presence of germline mutations, particularly while deciding on enucleation, frequency of periodic follow up and radiotherapy.
Conclusions
We identified novel RB1 mutations, and our mutation detection rate was on par with the previous global studies. In our study, genetic results influenced clinical management and we suggest that it should be an essential and integral component of RB-care in India and elsewhere.
Background
Retinoblastoma (RB) (OMIM#180200) is the commonest childhood intraocular tumor, with a global estimated annual incidence of 1 in 15,000–20,000 live births [1]. India accounts for the highest global burden having one out of every five RB children with an estimated annual incidence of 1500 RB children [2–4]. RB occurs due to the two-hit hypothesis of Knudson, which is because of loss-of-function of the tumour suppressor RB1gene, owing to homozygous allelic mutations, loss of heterozygosity mechanism or gene silencing [5]. RB1 is a nuclear phosphoprotein, essential for G1/S check point during the cell cycle regulation, while in a dephosphorylated state binds to mitotic agents like E2F, viral particles and other factors, but releases them during mitosis when phosphorylated. RB1 gene is located on chromosome band 13q14.2, consisting of 27 exons, which encodes a 4.7 kb mRNA. So far, 1748 unique RB1 variants in 3366 individuals have been identified and summarized in the Leiden Open Variation Database (LOVD) [6]. Most of the RB1 mutations are unique and found in exon, splicing introns and untranslated regions [5–8]. Interestingly, RB1 exon deletions are seen not only in RB but also less frequently in breast cancer, osteosarcoma and lung cancer.
Usually, in any given population, there are more children with unilateral RB (ULRB-60%) than bilateral (BLRB-40%) and a clinician has to be noted that a majority of those with BLRB and a small proportion of those with ULRB might have germline RB1 mutations, who may need genetic screening [2]. Genetic screening could play a vital role in management of RB which could influence various crucial clinical management decisions [7].
Unless genetic testing is available, the minority of unilateral hereditary cases, fail to get the desirable clinical management decisions and frequent clinical surveillance. Hereditary RB tends to be early in onset, bilateral and multifocal, hence needs continuous surveillance for effective management. All cases with mutation, as mentioned earlier, have a lifetime risk for osteosarcoma, soft tissue sarcoma, malignant melanoma or multiple brain tumours. Hence they need lifelong follow-ups, as opposed to sporadic cases, which may not have genetic predisposition. Between 1905 and 2005 about 199 RB survivors were retrospectively analysed for second primary tumours (SPT) and found that 44 of them developed SPT [9]. Any form of radiation for investigation (like X-ray, CT scan) or treatment has to be preferably avoided in all germline cases, due to probable increased risk of second malignancies. Besides North America and Germany, RB1 mutations have been reported from various populations around the world like, Argentina [10], Brazil [11], China [12, 13], Colombia [14], Ecuador [15], Egypt [16], India [17–21], Iran [22], Israel [23, 24], Italy [25], Korea [26], Netherlands [27], Spain [28, 29], Malaysia [30], Mexico [31], Morocco [32], New Zealand [33], Pakistan [34], Swiss [35], Tunisia [36] Singapore [37] Thailand [38] and United Kingdom [39]. Out of five earlier studies from India, stratifying genetic tests is an option suggested by Thirumalairaj et al. [40].
Though enormous number of studies are available on RB1 gene mutations across the globe, including India, there is limited information on how the genetic result could influence clinical management outcomes. Hence, we undertook this study to describe and correlate the genetic and clinical parameters of 50 RB patients from India. We also examined the opportunities and challenges in clinical decisions which were influenced through RB1 gene screening in a developing country scenario.
Methods
Patient recruitment and clinical examination
Fifty (48 unrelated and two related siblings) RB patients (aged 0.2–5.3 years) with various clinical presentations, from the Department of Paediatric Ophthalmology, Narayana Nethralaya, Bangalore, India were recruited from June 2014 to Feb 2015. Among these twenty-nine were BLRB and twenty-one had ULRB. A complete clinical examination was carried out under general anaesthesia which included dilated retinal evaluation, imaging of retina using wide field fundus camera (Retcam), measurement of intraocular pressure, anterior segment evaluation by handheld slit lamp. Also, magnetic resonance imaging (MRI) of the orbits and brain, B scan ultrasonography of the eye, cerebrospinal fluid analysis and bone marrow analysis were performed when indicated. The clinical disease was classified as per the AJCC TNM classification for RB, as well as the International Classification of Intraocular Retinoblastoma [41]. The study was approved by the Institutional Ethical Committee, which followed the Tenets of the Declaration of Helsinki. After ascertaining pedigree and written informed parental consent, five ml of blood sample was obtained in EDTA coated vacutainer tubes from patients (during examination under anaesthesia) for genetic analysis. For clinical analysis the cohort was divided into two groups—those with and without RB1 mutations.
DNA isolation, NGS target sequencing of RB1 gene analysis
Nucleospin Blood XL kit (Macherey–Nagel)—About 5 of peripheral blood from the child is collected and 500 μl of proteinase K and PBS are added for lysis of RBCs. Then 10 ml of buffer BQ1 is added a shaken vigorously for 2 min and incubated at 56 °C for 15 min. Then add 10 ml of 96–100% ethanol and vortex for 10 s for lysate formation. Take Nucleospin Blood XL column and add 15 ml of the lysate and centrifuge at 5000 rpm for 3 min. Discard the flow-through and repeat the last step. Add 7.5 ml Buffer BQ2 and centrifuge at 5000 rpm for 2 min and repeat the step for 20 min. Insert the column into new collection tube and add 750 μl of prewarmed 70 °C Elution Buffer BE and incubate at room temperature for 16 h. The last step may be repeated and when centrifuged at 5000 rpm for 5 min, highly pure genomic DNA elutes through the silica membrane.
Genomic DNA was used for targeted gene capture using a custom capture kit. Briefly, 1ug of DNA was subjected to fragmentation resulting in an average size of 150 bp followed by end repair, adenylation, adaptor ligation and amplification to obtain whole genome libraries using the Kapa DNA library preparation kit v2.14. These libraries were then hybridized to biotinylated probes (NimbleGen, Roche) specific to RB1gene for 72 h and extracted using streptavidin beads, washed and normalized. The libraries were then sequenced to mean > 80–100× coverage on Illumina sequencing platform (HiSeq 2500). The sequences obtained are aligned to human reference genome (GRCh37/hg19) using BWA program [42, 43] and analyzed using Picard and GATK-Lite tool kit [44, 45] to identify variants relevant to the clinical indication. Annotations of the variants were performed against the Ensembl release 75 gene model [46]. Clinically relevant mutations were annotated using published variants in literature and a set of variant databases including ClinVar, OMIM, GWAS, HGMD and SwissVar [47–54].
Multiplex ligation-dependent probe amplification (MLPA) analysis
In order to detect large deletions/duplications in the RB1 gene, we performed Multiplex Ligation-dependent Probe Amplification (MLPA). SALSA MLPA kit P047 RB1 (Amsterdam, Netherlands) was used as per manufacturer’s recommendations.
Statistical analysis
Multivariate analysis for genotype phenotype correlation was done using Pearson Chi square test, SPSS software. Clinical factors like sub retinal seeds, optic nerve invasion, pathological high-risk factors (HRF), tumour recurrence, tumor resistance to treatment, need for 2nd line drugs like topotecan and need for radiotherapy were analysed in the mutation versus no mutation groups.
Results
Of 50 RB patients, 29 had BLRB (average age at presentation of 1.8 years) and 21 had ULRB (average age at presentation of 2.3 years). A family history of RB was observed in two patients. In the BLRB group, 25 out of 29 probands (86.2%) had a germline mutation whereas in the ULRB group, 4 out of 21 (19%) had a mutation. NGS and MLPA analyses revealed total of 20 RB1 gene variations in 29 probands, inclusive of three novel mutations (3 probands, 6%—c.1050-8_1050-2delTTATTTA) (intronic splice variant) (ClinVar ID: SCV001571344.1), Q444P (ClinVar ID: SCV001571345.1) and S567P (ClinVar ID: SCV001571346.1)), previously reported 16 mutations (22 probands: 44%) and heterozygous deletion of whole RB1 gene (3 BLRB, 1 ULRB, 8%). The types of mutations were, non-sense being the maximum [13], followed by missense [7], splice site [4], whole gene deletions [4]. One proband had frameshift (Table 1; Fig. 1).
Table 1.
Type of genetic abnormalities
| S. nos. | Genetic abnormality | Number of mutations | Number of patients | Component of novel mutation | No of pts with novel mutation | Unilateral /bilateral |
|---|---|---|---|---|---|---|
| 1 | Whole gene deletion | NA | 4 | NA | NA | 1:3 |
| 2 | Missense mutation | 7 | 7 | 2 | 2 | 3:4 |
| 3 | Frame shift | 1 | 1 | NIL | NIL | 0:1 |
| 4 | Splice site | 3 | 4 | 1 | 1 | 0:4 |
| 5 | Non-SENSE | 10 | 13 | NIL | NIL | 0:13 |
| Total | 19 | 29 | 3 | 7 | 4:25 |
Fig. 1.

Represents the mutations identified in RB patients distributed across the RB protein structure
Genotype to clinical analysis revealed that there was no direct correlation between age of presentation and disease severity between the groups. Clinical features, age of presentation and high-risk features like optic nerve invasion in the groups have been listed in Table 2. Mutation group had more patients with increased severity requiring enucleation (95.23%), optic nerve invasion (64.7%), sub-retinal seeds (68%) and pathological high-risk factors (73.9%). The disease severity factors like average clinical TNM and pathological TNM were stratified as per the mutation type (splice site, missense, termination and whole gene deletion) and the findings are listed in Table 3. In the current cohort, splice site mutation had the highest average clinical and pathological TNM, as well as the youngest average age of enucleation. Disease recurrence and disease progression correlated significantly with mutation group (p = 0.021 and p = 0.000 respectively). Notably, of the total 10 recurrences in the current cohort, 9 patients had the mutation (Table 4). The mutation detection rate (MDR) was 86.2% in BLRB (25 out of 29) and 19% in ULRB (4 out of 21), which was better than many other global studies and comparable to some of the recent robust ones (Table 5).
Table 2.
Clinical presentation
| Mutation | No mutation | |
|---|---|---|
| Average age of diagnosis | 1.82 years | 2.08 years |
| Need for enucleation | 23 of 29(79.31%) | 20 of 21(95.23%) |
| Average age of enucleation | 2.08 years* | 2.05 years # |
| Optic nerve invasion | 11 of 17 (64.7%) | 8 of 19(42.1%) |
| Sub retinal seeds | 17 OF 25(68%) | 4 OF 20(20%) |
| Pathological high risk factors | 17 of 23(73.9%) | 9 of 19 (47.36%) |
*Average age of enucleation calculated after excluding 1 patient who was enucleated at 13.77 years
#Includes one pt who presented at 5.13 years and was not enucleated. BL detected early and therefore managed early compared to UL which is late
Table 3.
Correlating mutation versus clinical disease severity
| Splice site mutation | Missenese mutation | Termination | Whole gene deletion | |
|---|---|---|---|---|
| Total | 4 | 7 | 14 | 4 |
| U:B | 0:04 | 3:04 | 0:14 | 1:03 |
| Age (years) | 1.45 | 1.81 | 1.28 | 4.09 |
| Enucleation | 4 (100%) | 7 (100%) | 10 (71.4%) | 3 (75%) |
| Average age at enucleation (years) | 1.83 | 1.95 | 2.02 | 1.98 * |
| Avg pathological TNM | 2.6 | 2.14 | 1.85 | 2.3 |
| Avg clinical TNM | 3.3 | 2.7 | 2.38 | 2.75 |
*Average age of enucleation calculated after excluding 1 patient who was enucleated at 13.77 years
Table 4.
Correlation of genotype with high risk phenotype
| Phenotypic features | Mutation present n (%) | No mutation n (%) | Pearson Chi-square tests | |
|---|---|---|---|---|
| Chi sq | Significance (p value) | |||
| Optic nerve invasion | ||||
| Y | 11 | 8 | 1.83 | 0.575 |
| N | 6 | 11 | ||
| Recurrence | ||||
| Y | 9 | 1 | 5.25 | 0.021 |
| N | 20* | 20 | ||
| Progression | ||||
| Y | 16 | 1 | 13.79 | 0.000 |
| N | 13 | 20 | ||
| Need for topotecan | ||||
| Y | 7 | 1 | 3.40 | 0.065 |
| N | 22 | 20 | ||
| Need for radiotherapy | ||||
| Y | 3 | 2 | 0.091 | 0.923 |
| N | 26 | 19 | ||
*Of the 20 without recurrence and with mutation, 7 had disease progression and 13 had ‘none’
Table 5.
Mutation detection rates in unilateral and bilateral RB patient groups studies across the globe
| S. nos | Author | Country | Type of mutations | Mutation detection rate BLRB (%) | Mutation detection rate ULRB | Year of study |
|---|---|---|---|---|---|---|
| 02 | Mohd Khalid, M.K., et al | Malaysia | Nonsense, Frame shift, Splice site and De-novo origin | 100 | 25% | 2015 |
| 05 | Grotta et al. | Italy | Point mutations, Frame shift, Large deletions | 96.5 | 22% | 2015 |
| 09 | Chen, Z., et al | USA | Nonsense, Splice, Frameshift | 97 | 18% | 2014 |
| 07 | Price et al | United Kingdom | Point mutation, deletions, missense, splice site mutations | 96 | 9.5% | 2014 |
| 10 | Seo, S.H., et al | Korea | Missense, nonsense, frameshift and splice | 94.5 | None | 2013 |
| 11 | Ottaviani, D., et al | Argentina | Nonsense, frameshift, missense, deletions | 94 | – | 2013 |
| 08 | Dommering, C.J., et al | Netherland | Nonsense, frameshift, splice, large indel, missense, chromosomal deletions and promoter | 92 | 10% | 2014 |
| 01 | Frenkal.Set al | France | Stop codon, Splice site and large deletions | 90 | 19.8% | 2016 |
| 15 | Macias, M., et al | Mexico | Nonsense, Splice, Frameshift | 76.9 | 34.8% | 2008 |
| 16 | Abouzeid et al | Switzerland | Nonsense, frameshift, missense, deletions | 73 | 10.7% | 2007 |
| 03 | Zhang, L., et al | China | Nonsense, Splice, Frameshift | 65 | 35% | 2015 |
| 06 | Devarajan et al | India | Nonsense, Frame shift, Splice site and Denovo origin | 63 | 37% | 2015 |
| 04 | Kalsoom, S., et al | Pakistan | Null mutation, deletions, missense, splice site mutations | 45.7 | 54.3% | 2015 |
| 12 | Barbosa, R.H., et al | Brasil | Nonsense, Splice, Frameshift | 42.2 | 56.3% | 2013 |
| 14 | Abidi et al., | Morocco | Duplication, Deletion, Splice, Frameshift | 40 | None | 2011 |
| 17 | Choy et al | Hong kong & China | Nonsense, Splice, Frameshift | 38 | 19% | 2002 |
| 13 | Ahani et al | Iran | Missense, frameshift and splice site | 16.6 | 18.2% | 2013 |
| 14 | Present study—Himika, Malaichamy, et al | India | Missense, frameshift, gene deletions | 86.2 | 19% | 2020 |
A nonsense mutation, c. 233G > A (p.W78Ter) was identified in two unrelated patients with bilateral RB. The novel nucleotide changes include two missense substitutions—c.1699 T > C (p.S567P), c.1331A > C (p.Q444P) and one splice site variation (c.1050-8_1050-2delTTATTTA). Bioinformatics prediction analysis of SIFT, PolyPhen–2, Provean, showed that the missense substitutions (p.S567P, p.Q444P) had deleterious effect which may affect the functional properties of the protein and both the missense variations are present in the retinoblastoma-associated protein A domain of the RB1 protein. The three novel mutations were in BLRB patients. One of the BLRB patients, who presented at 2.5 years of age, had a p.W78X mutation and he was diagnosed to have pinealoma and was a case of trilateral RB. Another proband, presented with bilateral disease at 1.5 years with a positive family history. The father had regressed tumour, both the daughter and father carried the same familial mutation, c.1789C > T (Q597Ter). Interestingly, the half sibling of this proband, who was of the same father, presented at the age of 2.3 years with BLRB and had the same mutation c.1789 C > T (Q597Ter). Another interesting aspect was the varied clinical spectrum presentation and outcome of our four BLRB probands who all had the same termination mutation c.1333C > T (p.R445Ter) (Fig. 2).
Fig. 2.
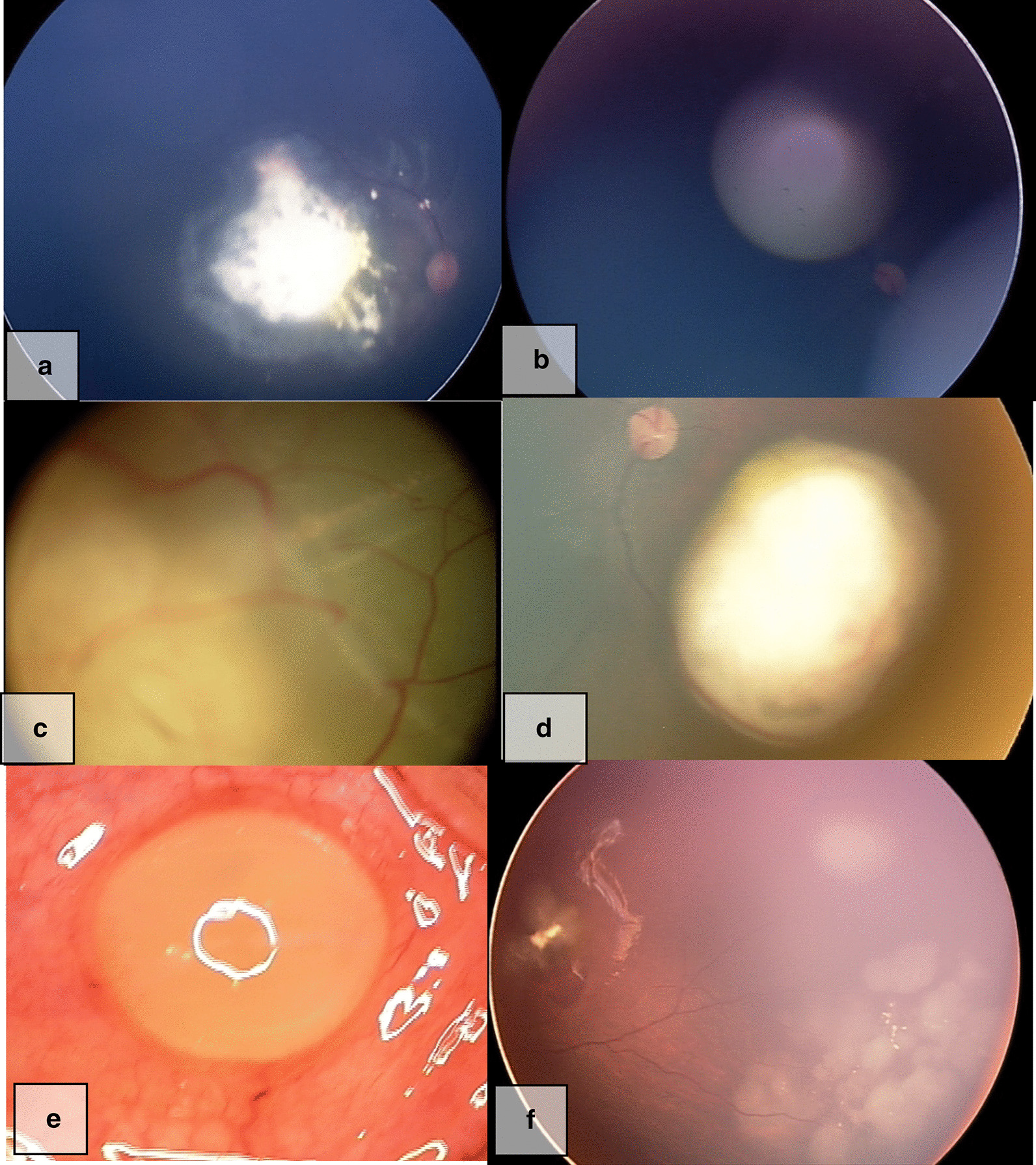
Variable phenotype of same genotype. Case75 a, b had mild disease in both eyes, bilateral globe salvage successful. Case 55. c One eye had severe disease needing enucleation, while other eye d had mild disease with successful globe salvage. Case 9 e one eye extensive tumor needing enucleation. Later the other eye f developed tumor which was nonresponsive to Rx and eventually needed enucleation
Discussion
In the current era of cancer-care—NGS/MLPA techniques have revolutionised the genetic diagnostic scenario of RB-care globally and also selectively in India [22–26]. Incorporating genetic testing as part of RB-care has significant advantages—these opportunities and challenges are highlighted in the current study. For example, our four ULRB cases who would have otherwise not been monitored closely post treatment completion with the mutation, were switched to 3–6 monthly surveillance, like any other BLRB patient with an RB1 mutation in our study cohort. Genetic test as a prognostic marker has been applied in medulloblastoma, paediatric gliomas [55, 56] and breast cancer [56]. However, in comparison, clinical adoption of RB genetic diagnostics is poor amongst the clinicians in India and other developing countries.
The mutation detection rates across countries in BLRB varied from 100 to 16.6% and in ULRB from 56.3 to 9.5% (Table 5), the wide variation could be due to various reasons inclusive of the fact that the studies were performed prior to highly sensitive NGS/MLPA tests era. Price et al., in United Kingdom studied 403 unrelated patients, 209 blood and 194 tumour samples and identified 533 variations, including RB1 gene mutations [39]. In Netherlands, 529 RB patients were screened with a 92% detection rate in BLRB and 10% in ULRB [27]. In the largest mutation meta-analysis of 932 RB patients, it was found that globally the most frequent mutations reported were R320X (nearly 50 times), R579X (nearly 40 times) and R251X (nearly 30 times) [57]. All the studies uniformly found deletions, duplications, missense, nonsense, splice and frameshift mutations, once again establishing that RB1 gene has no hotspot (10–13, 16, 17, 20–24, 26, 30–32, 34–36, 59–62). In our study, we found 20 RB1 gene variations in 29 probands (79%), inclusive of three novel mutations, 16 previously reported mutations, four heterozygous deletions of the whole RB1 gene. We had one case each of frameshift and commonly reported R251X, R320X mutations and it is to be noted that those with the arginine/termination mutations have a risk to develop SPT [15]. In our study, we identified mutations in 86% of BLRB patients and 19% in ULRB—which is comparable to other global studies, however we could not find any mutation in 4 BLRB patients and this could be because of various reasons including mosaicism and somatic MYCN gene mutations, which we did not study. Mosaicism is a tricky issue in RB diagnostics and prenatal genetic counselling, hence may go unnoticed suggests Rushlow et al. [58–62].
RB1 gross alterations were found in 15% of 433 BLRB and 6.5% of 262 ULRB patients—these patients developed fewer tumours compared to those with null mutations and interestingly, those with cytogenetic or sub-microscopic whole gene deletions often had ULRB, however all those with gross deletions with one breakpoint inside the RB1 gene had BLRB [63]. Notably, in our cohort all cases of ULRB, irrespective of their mutation type, had optic nerve invasion and were severe enough to warrant enucleation. Prior knowledge of mutation may influence enucleation decisions in the subset of ULRB patients, who all had the mutation, the other eye is also ‘at risk’ and must be treated potentially as a ‘bilateral’ case. In the four c.1333C > T (p.R445Ter) BLRB patients, three had disease progression despite treatment, in one bilateral globe salvage was successful by using plaque brachytherapy, two needed unilateral enucleation and one case needed bilateral enucleation due to progressive disease unresponsive to multimodality treatment (Fig. 2). The variable clinical phenotype and response to treatment despite the same mutation, could be due to epigenetic molecular events in the tumor [64]. In pineal cyst, a pre-malignant form of pinealoblastoma, BLRB is more common than ULRB where germline mutations are invariably identified [65] and we had a patient with pinealoma, trilateral RB who had the pW78X mutation.
In our study the mutation group had statistically significant progression, recurrence and higher percentage of optic nerve invasion, subretinal seeds and high-risk pathological factors but lower percentage of enucleation compared to the non-mutation group. Radiotherapy is contraindicated in patients with germline mutations and this valuable information could help the clinician to modify treatment options. There are studies describing ill effects of radiation on RB, which however do not have the mutation data [66].
Testing the RB1 gene for mutation is a challenging task, owing to its size, heterogeneity of mutations (with 200 reported), lack of hotspot and the variable intronic lengths [67]. Three patients in our cohort were exclusively referred for mutation analysis from other centres, envisaging the fact that clinical management of RB is well addressed, however the same level of care does not exist for genetic testing uniformly across RB care in India. This is despite established RB guidelines specifying the role of genetic testing in RB care [8]. Centres for RB care without a genetic support, must be aware of this need and should sensitize the family on the role and usefulness of genetic testing and also inform them of the additional cost of care to the family which is usually not covered by insurance (68).
There are many limitations of our study, we did not analyse tumor DNA samples and hence we did not detect somatic mutations especially in non-hereditary retinoblastoma [37]. Also, the conclusions made in the study were based on not performing chromosomal studies for large deletion, the study also had a small sample size, with short follow-up period and failure to detect mutations in few BLRB patients. In addition, the ULRB cases had a very low detection rate compared to other robust similar studies.
Conclusion
In summary, 50 RB patients were screened for RB1 mutations using targeted NGS and MLPA methodologies, which found detection rates on par with most global studies. Comparing case-wise genetic findings with various clinical parameters and mutations found that there were clinical phenotypic and allelic heterogeneities. The mutation group had a higher clinical risk of recurrence, which influenced clinical management. RB1 mutation screening is an important tool in RB-care globally, including developing countries.
Abbreviations
- RB
Retinoblastoma
- RB1
Retinoblastoma gene
- NGS
Next generation sequencing
- MLPA
Multiplex ligation dependent probe amplification
- ULRB
Unilateral retinoblastoma
- BLRB
Bilateral retinoblastoma
- MDR
Mutation detection rate
- AJCC
American Joint Committee on Cancer
- TNM
Tumor node metastasis
- DNA
Deoxyribo nucleic acid
- OMIM
Online Mendelian inheritance in man
- GWAS
Genome Wide Association Study
- HGMD
Human gene mutation database
- SPSS
Statistical package for the social sciences
- LOVD
Leiden open variation database
- BWA
Burrows wheeler aligner
- EDTA
Ethylenediaminetetraacetic acid
- LOH
Loss of heterozygosity
- SPT
Secondary primary tumors
- CAP
Certified analytics professional
Authors' contributions
HG: Writing up clinical part of the manuscript, phenotype and genotype analysis. SM: Writing up the manuscript, laboratory work and analysis. AM: Clinical analysis of the patients, reviewing the manuscript. SM: laboratory critical analysis, guidance, reviewing the manuscript. NJ: laboratory critical analysis, reviewing the manuscript. VS: laboratory analysis, writing up the manuscript. AG: laboratory work supervision, genetic counselling, reviewing the manuscript. AG: laboratory critical analysis, reviewing the manuscript. SS: laboratory critical analysis, reviewing the manuscript. SS: laboratory critical analysis, overall guidance, reviewing the manuscript. VR: laboratory critical analysis, overall guidance, reviewing the manuscript. GK: Overall guidance, genetic counselling, reviewing the manuscript. All authors read and approved the final manuscript.
Funding
Source of funding- SciGenom Research Foundation. The funding body have role in study design, collection, analysis, and interpretation of data.
Data availability
The sequencing datasets generated and analyzed during the current study are available in the NCBI Sequence Read Archive (SRA) repository (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA725658) under the BioProject ID PRJNA725658. The ClinVar IDs for the three RB1 variants: NM_000321.3(RB1):c.10508_10502del,NM_000321.3(RB1):c.1331A > C(p.Gln444Pro),NM_000321.3(RB1):c.1699 T > C(p.Ser567Po)are SCV001571344.1,SCV001571345.1,SCV001571346.1 respectively. Further informations on the current study are available from corresponding author on reasonable request.
Declarations
Ethics approval and consent
The ethics approval and consent for the study have been approved by the Institutional Ethics Committee of Narayana Nethralaya (EC Ref No: C/2013/03/02) and the study have been performed in accordance with the ethical standards as laid down in the 1964 Declaration of Helsinki and its later amendments or comparable ethical standards.
Consent for publication
Written informed parental consent were obtained for publication
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Himika Gupta and Sivasankar Malaichamy have contributed equally to this work
References
- 1.Broaddus E, Topham A, Singh AD. Incidence of retinoblastoma in the USA: 1975–2004. Br J Ophthalmol. 2009;93(1):21–23. doi: 10.1136/bjo.2008.138750. [DOI] [PubMed] [Google Scholar]
- 2.Lohmann D, Gallie B, Dommering C, Gauthier-Villars M. Clinical utility gene card for: retinoblastoma. Eur J Hum Genet. 2011;19(3):3. doi: 10.1038/ejhg.2010.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dimaras H. Retinoblastoma genetics in India: from research to implementation. Indian J Ophthalmol. 2015;63(3):219–226. doi: 10.4103/0301-4738.156917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vogel F. Genetics and mutation rate of retinoblastoma (glioma retinae), with general remarks on methods of determining mutation rate in humans. Z Mensch Vererb Konstitutionsl. 1954;32(4):308–336. [PubMed] [Google Scholar]
- 5.Knudson AJ. Mutation and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci USA. 1971;68(4):820–823. doi: 10.1073/pnas.68.4.820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lohman D. http://rb1-lovd.d-lohmann.de/.
- 7.Afshar AR, Pekmezci M, Bloomer MM, Cadenas NJ, Stevers M, Banerjee A, et al. Next-generation sequencing of retinoblastoma identifies pathogenic alterations beyond rb1 inactivation that correlate with aggressive histopathologic features. Ophthalmology. 2020;127(6):804–813. doi: 10.1016/j.ophtha.2019.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gallie B. National retinoblastoma strategy canadian guidelines for care. Can J Ophthalmol. 2009;44(SUPPL):2. doi: 10.3129/i09-194. [DOI] [PubMed] [Google Scholar]
- 9.Dommering CJ, Marees T, van der Hout AH, Imhof SM, Meijers-Heijboer H, Ringens PJ, et al. RB1 mutations and second primary malignancies after hereditary retinoblastoma. Fam Cancer. 2012;11(2):225–233. doi: 10.1007/s10689-011-9505-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ottaviani D, Parma D, Giliberto F, Ferrer M, Fandino A, Davila MT, et al. Spectrum of RB1 mutations in argentine patients: 20-years experience in the molecular diagnosis of retinoblastoma. Ophthalmic Genet. 2013;34(4):189–198. doi: 10.3109/13816810.2012.755553. [DOI] [PubMed] [Google Scholar]
- 11.Barbosa RH, Aguiar FC, Silva MF, Costa RA, Vargas FR, Lucena E, et al. Screening of RB1 alterations in Brazilian patients with retinoblastoma and relatives with retinoma: phenotypic and genotypic associations. Invest Ophthalmol Vis Sci. 2013;54(5):3184–3194. doi: 10.1167/iovs.13-11686. [DOI] [PubMed] [Google Scholar]
- 12.Zhang L, Jia R, Zhao J, Fan J, Zhou Y, Han B, et al. Novel mutations in the RB1 gene from Chinese families with a history of retinoblastoma. Tumour Biol. 2015;36(4):2409–2420. doi: 10.1007/s13277-014-2851-7. [DOI] [PubMed] [Google Scholar]
- 13.Choy KW, Pang CP, Yu CB, Wong HL, Ng JS, Fan DS, et al. Loss of heterozygosity and mutations are the major mechanisms of RB1 gene inactivation in Chinese with sporadic retinoblastoma. Hum Mutat. 2002;20(5):408. doi: 10.1002/humu.9077. [DOI] [PubMed] [Google Scholar]
- 14.Serrano ML, Yunis JJ. Identification of three new mutations in the RB1 gene in patients with sporadic retinoblastoma in Colombia. Biomedica. 2013;33(1):53–61. doi: 10.1590/S0120-41572013000100007. [DOI] [PubMed] [Google Scholar]
- 15.Leone PE, Vega ME, Jervis P, Pestana A, Alonso J, Paz-y-Mino C. Two new mutations and three novel polymorphisms in the RB1 gene in Ecuadorian patients. J Hum Genet. 2003;48(12):639–641. doi: 10.1007/s10038-003-0092-5. [DOI] [PubMed] [Google Scholar]
- 16.Mohammed AM, Kamel AK, Hammad SA, Afifi HH, El Sanabary Z, El Din ME. Constitutional retinoblastoma gene deletion in Egyptian patients. World J Pediatr. 2009;5(3):222–225. doi: 10.1007/s12519-009-0042-1. [DOI] [PubMed] [Google Scholar]
- 17.Devarajan B, Prakash L, Kannan TR, Abraham AA, Kim U, Muthukkaruppan V, et al. Targeted next generation sequencing of RB1 gene for the molecular diagnosis of Retinoblastoma. BMC Cancer. 2015;15:320. doi: 10.1186/s12885-015-1340-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Joseph B, Raman R, Uthra S, Jagadeesan M, Ganesh A, Paul PG, et al. Genotype-phenotype correlation analysis in retinoblastoma patients from India. Asian Pac J Cancer Prev. 2006;7(4):619–622. [PubMed] [Google Scholar]
- 19.Kumaramanickavel G, Joseph B, Narayana K, Natesh S, Mamatha G, Shanmugam MP, et al. Molecular-genetic analysis of two cases with retinoblastoma: benefits for disease management. J Genet. 2003;82(1–2):39–44. doi: 10.1007/BF02715880. [DOI] [PubMed] [Google Scholar]
- 20.Kiran VS, Kannabiran C, Chakravarthi K, Vemuganti GK, Honavar SG. Mutational screening of the RB1 gene in Indian patients with retinoblastoma reveals eight novel and several recurrent mutations. Hum Mutat. 2003;22(4):339. doi: 10.1002/humu.9181. [DOI] [PubMed] [Google Scholar]
- 21.JayaSingh AM, Pandian AJ, Mallipatna AC, Khetan V, Sripriya S, Kapoor S, Agarwal S, Sankaran S, Katragadda S, Veeramachaneni V, Hariharan R, Subramanian K, Mannan AU. Next-generation sequencing-based method shows increased mutation detection sensitivity in an Indian retinoblastoma cohort. Mol Vis. 2016;22:1036–1047. [PMC free article] [PubMed] [Google Scholar]
- 22.Ahani A, Akbari MT, Saliminejad K, Behnam B, Akhondi MM, Vosoogh P, et al. Screening for large rearrangements of the RB1 gene in Iranian patients with retinoblastoma using multiplex ligation-dependent probe amplification. Mol Vis. 2013;19:454–462. [PMC free article] [PubMed] [Google Scholar]
- 23.Sagi M, Frenkel A, Eilat A, Weinberg N, Frenkel S, Pe'er J, et al. Genetic screening in patients with Retinoblastoma in Israel. Fam Cancer. 2015;14(3):471–480. doi: 10.1007/s10689-015-9794-z. [DOI] [PubMed] [Google Scholar]
- 24.Frenkel S, Zloto O, Sagi M, Fraenkel A, Pe'er J. Genotype-phenotype correlation in the presentation of retinoblastoma among 149 patients. Exp Eye Res. 2016;146:313–317. doi: 10.1016/j.exer.2016.04.002. [DOI] [PubMed] [Google Scholar]
- 25.Grotta S, D'Elia G, Scavelli R, Genovese S, Surace C, Sirleto P, et al. Advantages of a next generation sequencing targeted approach for the molecular diagnosis of retinoblastoma. BMC Cancer. 2015;15:841. doi: 10.1186/s12885-015-1854-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Seo SH, Ahn HS, Yu YS, Kang HJ, Park KD, Cho SI, et al. Mutation spectrum of RB1 gene in Korean bilateral retinoblastoma patients using direct sequencing and gene dosage analysis. Clin Genet. 2013;83(5):494–496. doi: 10.1111/j.1399-0004.2012.01954.x. [DOI] [PubMed] [Google Scholar]
- 27.Dommering CJ, Mol BM, Moll AC, Burton M, Cloos J, Dorsman JC, et al. RB1 mutation spectrum in a comprehensive nationwide cohort of retinoblastoma patients. J Med Genet. 2014;51(6):366–374. doi: 10.1136/jmedgenet-2014-102264. [DOI] [PubMed] [Google Scholar]
- 28.Cuevas-Cuerda D, Salas-Trejo D. Evaluation after five years of the cancer genetic counselling programme of Valencian Community (Eastern Spain) Fam Cancer. 2014;13(2):301–309. doi: 10.1007/s10689-013-9693-0. [DOI] [PubMed] [Google Scholar]
- 29.Alonso J, Garcia-Miguel P, Abelairas J, Mendiola M, Sarret E, Vendrell MT, et al. Spectrum of germline RB1 gene mutations in Spanish retinoblastoma patients: phenotypic and molecular epidemiological implications. Hum Mutat. 2001;17(5):412–422. doi: 10.1002/humu.1117. [DOI] [PubMed] [Google Scholar]
- 30.Mohd Khalid MK, Yakob Y, Md Yasin R, Wee Teik K, Siew CG, Rahmat J, et al. Spectrum of germ-line RB1 gene mutations in Malaysian patients with retinoblastoma. Mol Vis. 2015;21:1185–1190. [PMC free article] [PubMed] [Google Scholar]
- 31.Macias M, Dean M, Atkinson A, Jimenez-Morales S, Garcia-Vazquez FJ, Saldana-Alvarez Y, et al. Spectrum of RB1 gene mutations and loss of heterozygosity in Mexican patients with retinoblastoma: identification of six novel mutations. Cancer Biomark. 2008;4(2):93–99. doi: 10.3233/CBM-2008-4205. [DOI] [PubMed] [Google Scholar]
- 32.Abidi O, Knari S, Sefri H, Charif M, Senechal A, Hamel C, et al. Mutational analysis of the RB1 gene in Moroccan patients with retinoblastoma. Mol Vis. 2011;17:3541–3547. [PMC free article] [PubMed] [Google Scholar]
- 33.Sheck LH, Ng YS, Watson M, Vincent AL. Clinical findings and molecular diagnosis of retinoblastoma in older children. Ophthalmic Genet. 2013;34(4):238–242. doi: 10.3109/13816810.2012.752015. [DOI] [PubMed] [Google Scholar]
- 34.Kalsoom S, Wasim M, Afzal S, Shahzad MS, Ramzan S, Awan AR, et al. Alterations in the RB1 gene in Pakistani patients with retinoblastoma using direct sequencing analysis. Mol Vis. 2015;21:1085–1092. [PMC free article] [PubMed] [Google Scholar]
- 35.Abouzeid H, Munier FL, Thonney F, Schorderet DF. Ten novel RB1 gene mutations in patients with retinoblastoma. Mol Vis. 2007;13:1740–1745. [PubMed] [Google Scholar]
- 36.Ayari-Jeridi H, Moran K, Chebbi A, Bouguila H, Abbes I, Charradi K, et al. Mutation spectrum of RB1 gene in unilateral retinoblastoma cases from Tunisia and correlations with clinical features. PLoS ONE. 2015;10(1):e0116615. doi: 10.1371/journal.pone.0116615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tomar S, Sethi R, Sundar G, Quah TC, Quah BL. Mutation spectrum of RB1 mutations in retinoblastoma cases from Singapore with implications for genetic management and counselling. PLoS ONE. 2017;12(6):e0178776. doi: 10.1371/journal.pone.0178776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rojanaporn D, Boontawon T, Chareonsirisuthigul T, Thanapanpanich O, Attaseth T, Saengwimol D, et al. Spectrum of germline RB1 mutations and clinical manifestations in retinoblastoma patients from Thailand. Mol Vis. 2018;24:778–788. [PMC free article] [PubMed] [Google Scholar]
- 39.Price EA, Price K, Kolkiewicz K, Hack S, Reddy MA, Hungerford JL, et al. Spectrum of RB1 mutations identified in 403 retinoblastoma patients. J Med Genet. 2014;51(3):208–214. doi: 10.1136/jmedgenet-2013-101821. [DOI] [PubMed] [Google Scholar]
- 40.Thirumalairaj K, Abraham A, Devarajan B, Gaikwad N, Kim U, Muthukkaruppan V, Vanniarajan A. A stepwise strategy for rapid and cost-effective RB1 screening in Indian retinoblastoma patients. J Hum Genet. 2015;60(9):547–552. doi: 10.1038/jhg.2015.62. [DOI] [PubMed] [Google Scholar]
- 41.AJCC. From the AJCC cancer staging manual. Cancer Staging Handbook. 2004; 6th edition. New York: Springer, pp. 13–5.
- 42.Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010;26(5):589–595. doi: 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Meyer LR, Zweig AS, Hinrichs AS, Karolchik D, Kuhn RM, Wong M, et al. The UCSC Genome Browser database: extensions and updates 2013. Nucleic Acids Res. 2013;41:D64–D69. doi: 10.1093/nar/gks1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20(9):1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25(16):2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zerbino DR, Achuthan P, Akanni W, Amode MR, Barrell D, Bhai J, et al. Ensembl 2018. Nucleic Acids Res. 2018;46(D1):D754–D761. doi: 10.1093/nar/gkx1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hamosh A, Scott AF, Amberger JS, Bocchini CA, McKusick VA. Online Mendelian Inheritance in Man (OMIM), a knowledgebase of human genes and genetic disorders. Nucleic Acids Res. 2005;33:D514–D517. doi: 10.1093/nar/gki033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Landrum MJ, Lee JM, Benson M, Brown G, Chao C, Chitipiralla S, et al. ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res. 2016;44(D1):D862–D868. doi: 10.1093/nar/gkv1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Amberger JS, Bocchini CA, Schiettecatte F, Scott AF, Hamosh A. OMIMorg: Online Mendelian Inheritance in Man (OMIM(R)), an online catalog of human genes and genetic disorders. Nucleic Acids Res. 2015;43:D789–D798. doi: 10.1093/nar/gku1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.MacArthur J, Bowler E, Cerezo M, Gil L, Hall P, Hastings E, et al. The new NHGRI-EBI Catalog of published genome-wide association studies (GWAS Catalog) Nucleic Acids Res. 2017;45(D1):D896–D901. doi: 10.1093/nar/gkw1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Stenson PD, Ball EV, Mort M, Phillips AD, Shiel JA, Thomas NS, et al. Human gene mutation database (HGMD): 2003 update. Hum Mutat. 2003;21(6):577–581. doi: 10.1002/humu.10212. [DOI] [PubMed] [Google Scholar]
- 52.Welter D, MacArthur J, Morales J, Burdett T, Hall P, Junkins H, et al. The NHGRI GWAS Catalog, a curated resource of SNP-trait associations. Nucleic Acids Res. 2014;42:D1001–D1006. doi: 10.1093/nar/gkt1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Stenson PD, Mort M, Ball EV, Howells K, Phillips AD, Thomas NS, et al. The human gene mutation database: 2008 update. Genome Med. 2009;1(1):13. doi: 10.1186/gm13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mottaz A, David FP, Veuthey AL, Yip YL. Easy retrieval of single amino-acid polymorphisms and phenotype information using SwissVar. Bioinformatics. 2010;26(6):851–852. doi: 10.1093/bioinformatics/btq028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kunder RJR, et al. Real-time PCR assay based on the differential expression of microRNAs and protein-coding genes for molecular classification of formalin-fixed paraffin embedded medulloblastomas. Neuro Oncol. 2013;15(12):1644–1651. doi: 10.1093/neuonc/not123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mistry MZN, et al. BRAF mutation and CDKN2A deletion define a clinically distinct subgroup of childhood secondary high-grade glioma. J Clin Oncol. 2015;33(9):1015–1022. doi: 10.1200/JCO.2014.58.3922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Valverde JR, Alonso J, Palacios I, Pestana A. RB1 gene mutation up-date, a meta-analysis based on 932 reported mutations available in a searchable database. BMC Genet. 2005;6:53. doi: 10.1186/1471-2156-6-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rushlow D, Piovesan B, Zhang K, Prigoda-Lee NL, Marchong MN, Clark RD, et al. Detection of mosaic RB1 mutations in families with retinoblastoma. Hum Mutat. 2009;30(5):842–851. doi: 10.1002/humu.20940. [DOI] [PubMed] [Google Scholar]
- 59.Amitrano S, Marozza A, Somma S, Imperatore V, Hadjistilianou T, De Francesco S, et al. Next generation sequencing in sporadic retinoblastoma patients reveals somatic mosaicism. Eur J Hum Genet. 2015;23(11):1523–1530. doi: 10.1038/ejhg.2015.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chen Z, Moran K, Richards-Yutz J, Toorens E, Gerhart D, Ganguly T, et al. Enhanced sensitivity for detection of low-level germline mosaic RB1 mutations in sporadic retinoblastoma cases using deep semiconductor sequencing. Hum Mutat. 2014;35(3):384–391. doi: 10.1002/humu.22488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.D'Elia G, Grotta S, Del Bufalo F, De Ioris MA, Surace C, Sirleto P, et al. Two novel cases of trilateral retinoblastoma: genetics and review of the literature. Cancer Genet. 2013;206(11):398–401. doi: 10.1016/j.cancergen.2013.11.001. [DOI] [PubMed] [Google Scholar]
- 62.Castera L, Gauthier-Villars M, Dehainault C, Michaux D, Benachi A, Lumbroso-Le Rouic L, et al. Mosaicism in clinical practice exemplified by prenatal diagnosis in retinoblastoma. Prenat Diagn. 2011;31(11):1106–1108. doi: 10.1002/pd.2837. [DOI] [PubMed] [Google Scholar]
- 63.Albrecht P, Ansperger-Rescher B, Schuler A, Zeschnigk M, Gallie B, Lohmann DR. Spectrum of gross deletions and insertions in the RB1 gene in patients with retinoblastoma and association with phenotypic expression. Hum Mutat. 2005;26(5):437–445. doi: 10.1002/humu.20234. [DOI] [PubMed] [Google Scholar]
- 64.Benavente CA, Dyer MA. Genetics and epigenetics of human retinoblastoma. Annu Rev Pathol. 2015;10:547–562. doi: 10.1146/annurev-pathol-012414-040259. [DOI] [PubMed] [Google Scholar]
- 65.RuizDelRio N, AbelairasGomez JM, AlonsoGarciadelaRosa FJ, PeraltaCalvo JM, deLasHerasMartin A. Trilateral retinoblastoma: correlation between the genetic anomalies of the RB1 gene and the presence of pineal gland cysts. Arch Soc Esp Oftalmol. 2014;89(1):4–9. doi: 10.1016/j.oftal.2013.07.006. [DOI] [PubMed] [Google Scholar]
- 66.Kleinerman RA, Tarone RE, Abramson DH, Seddon JM, Stovall M, Li FP, Fraumeni JF. Risk of new cancers after radiotherapy in long-term survivors of retinoblastoma: an extended follow-up. J Clin Oncol. 2020;23(10):2272. doi: 10.1200/JCO.2005.05.054. [DOI] [PubMed] [Google Scholar]
- 67.Lohmann DR GB. Retinoblastoma. GeneReviews; https://www.ncbinlmnihgov/books/NBK1452/. 2013.
- 68.Cohen JG, Dryja TP, Davis KB, Diller LR, Li FP. RB1 genetic testing as a clinical service: a follow-up study. Med Pediatr Oncol. 2001;37(4):372–378. doi: 10.1002/mpo.1213. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The sequencing datasets generated and analyzed during the current study are available in the NCBI Sequence Read Archive (SRA) repository (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA725658) under the BioProject ID PRJNA725658. The ClinVar IDs for the three RB1 variants: NM_000321.3(RB1):c.10508_10502del,NM_000321.3(RB1):c.1331A > C(p.Gln444Pro),NM_000321.3(RB1):c.1699 T > C(p.Ser567Po)are SCV001571344.1,SCV001571345.1,SCV001571346.1 respectively. Further informations on the current study are available from corresponding author on reasonable request.