

Abstract
Background:
Activation of the innate immune system may play a role in the development of alcohol use disorders (AUDs), which often originate with adolescent alcohol abuse. A key player in the innate immune system is microglia, the activation of which occurs along a spectrum from pro-inflammatory, or M1-like, to anti-inflammatory, or M2-like, phenotypes.
Methods:
Adolescent, male rats were gavaged with ethanol or isocaloric control diet every 8 hours for 4 days, then sacrificed at 0, 2, 7, and 14 days later. Microglia were isolated from the entorhinal cortex and hippocampus by Percoll gradient centrifugation, labeled with surface antigens for activation and analyzed by flow cytometry. Polarization states of microglia, defined as CD11b+CD45low cells, were determined by the expression of M1 surface markers, major histocompatibility complex (MHC) II, CD32, and CD86, and M2 surface marker, CD206 (mannose receptor). Cytokine gene expression was measured by reverse transcriptase polymerase chain reaction.
Results:
Isolated cells were a highly enriched population (>95% pure) of microglia/macrophages according to CD11b immunoreactivity. Ethanol rats showed the most dramatic increases in microglia activation markers CD11b and CD45, and M1 (MHC-II) and M2 (CD206) markers at T2, when additional M1 markers CD86 and CD32 were also increased. Surprisingly, pro-inflammatory gene expression of CCL2, IL-1ß, IL-6 and TNF-α, generally was decreased at all time points in ethanol rats except for IL-6 which was increased at T0 and TNF-α which was not changed at T0 in either region. Simultaneously, BDNF expression was increased at T2 and T7 while IGF1 and TGF-ß gene expression were decreased. Arginase was also increased at T0 in hippocampus, but not changed by alcohol otherwise.
Conclusions:
Altogether, these data support that microglia phenotype after alcohol dependence is not a simple M1 or M2 phenotype, though more indicators of an anti-inflammatory phenotype were observed. Determining microglia phenotype is critical for understanding their role in the development of AUDs.
Keywords: microglia, alcohol, adolescent, neuroimmune, cytokine, neuroinflammation, alcoholism, ethanol, flow cytometry
Introduction
Excessive alcohol consumption ranks as the #5 disease burden in 15-19-year-old adolescents (Gore et al., 2011). Drinking in adolescence remains unacceptably widespread: 10-40% of adolescents report frequent binge drinking defined as 4 or 5 drinks consumed across 2 h for women and men respectively that result in blood ethanol concentrations (BECs) greater than 80 mg/dl (SAMHSA, 2019). Adolescents, however, may drink much more than that with growing reports of extreme binge drinking, high blood ethanol concentrations, and rates of alcohol use disorders (AUDs) around 6% (Clark et al., 2002; Patrick and Terry-McElrath, 2019; van Hoof et al., 2011). Adolescent misuse of alcohol can lead to a greater likelihood of developing an AUD in adulthood and is more likely to result in brain damage than if drinking onset is delayed to adulthood (Grant, 1998; Crews et al., 2000).
The period of adolescence is a dynamic time in brain development that includes changes in gene expression, synaptic structure, myelination and neurotransmission in brain circuits that are critical for cognition, memory, emotion, and behavior (Spear, 2018). Adolescent alcohol exposure causes numerous changes in neuroimmune pathways, which may manifest as a range of consequences from altered neurogenesis and synaptic remodeling to volume loss in brain regions that are thought to drive the development of addiction behavior (reviewed in Crews et al., 2016; Guerri and Pascual, 2019; Spear, 2018). Microglia, acting as the brain’s resident immune cells, play critical roles in synaptic pruning and remodeling, especially during development (Tremblay et al., 2011). As the neuroimmune effects of alcohol are implicated in AUD pathogenesis in adolescence (Chastain and Sarkar, 2014; Crews et al., 2016), and some phenotypes of “activated” microglia drive neuroinflammation (Ransohoff and Perry, 2009), understanding the effect of alcohol on microglia phenotype is critical for elucidating their specific role in adolescent susceptibility to alcohol-induced neuropathology and development of AUDs.
A host of approaches across human and animal model studies show that microglia are activated by alcohol (Barton et al., 2017; He and Crews, 2008; Marshall et al., 2013; McClain et al., 2011; see also reviews by Chastain and Sarkar, 2014; Crews et al., 2016; Melbourne et al., 2019). Microglia acquire an activated morphology during alcohol exposure, indicated by process shortening and thickening in microglia in alcohol-treated rodents, as well as in tissues from humans with AUDs (Barton et al., 2017; He and Crews, 2008; Marshall et al., 2013; McClain et al., 2011). However, the activated state of microglial cells is far from a single phenotype. Similar to that described in macrophages in other organs, microglia activation occurs on a spectrum of activation states or phenotypes; the two most extreme phenotypes being the M1 proinflammatory/classically activated versus M2 anti-inflammatory/alternatively activated (Beynon and Walker, 2012; Cherry et al., 2014). The proinflammatory M1 phenotypes are thought to be detrimental to the brain through secondary neuronal toxicity via secretion of proinflammatory cytokines and chemokines as well as production of reactive oxygen species. The M2 phenotypes, however, likely promote tissue repair/remodeling and phagocytosis of cell debris through the secretion of anti-inflammatory cytokines and growth factors (Beynon and Walker, 2012; Cherry et al., 2014; Ransohoff and Perry, 2009).
While neuroimmune activation clearly plays a role in excessive alcohol consumption in animal models (Agrawal et al., 2011; Blednov et al., 2012; see also Mayfield and Harris, 2017, for review), the role of microglia specifically is not clear. Microglia have merely been implicated due to their role as the primary effector of the neuroimmune system coupled with the upregulation of proinflammatory gene expression; microglia are rarely examined specifically (Marshall et al., 2013; McClain et al., 2011; Melbourne et al., 2019; Peng et al., 2017). Indeed, the morphology of microglia in brains from AUD patients is ramified, not amoeboid as would be expected of fully pro-inflammatory microglia (Beynon and Walker, 2012; He and Crews, 2008). Across several groups, multiple animal models, and interdisciplinary approaches, the collective patterns of cytokine and growth factor protein expression, microglia surface marker expression, and hyper-ramified morphology support that alcohol exposure results in microglia that are more aligned with the M2-like “beneficial” end of the spectrum, at least in adult rats (Bell-Temin et al., 2013; Marshall et al., 2013; Peng et al., 2017; Zahr et al., 2010). However, how alcohol affects microglia phenotype in adolescent rats considering their enhanced susceptibility to alcohol-induced neurodegeneration (Crews et al., 2000) and potentially different neuroimmune reactivity than adult rats, is not known (Brenhouse and Schwarz, 2016; Crews et al., 2013; Doremus-Fitzwater et al., 2015; Pascual et al., 2007; Sharma et al., 2018). Therefore, in order to determine microglia phenotype and expression profiles in a model of an AUD in adolescent rats, we used Percoll gradient centrifugation followed by flow cytometry to assess microglia activation states, i.e. phenotypes, in the hippocampus and entorhinal cortices at 0, 2, 4, 7 and 14 days after a four-day alcohol binge in adolescent rats. Cytokine and regulatory molecular profiles in isolated microglia were then determined by real time RT-PCR.
Materials and Methods
Binge ethanol model:
All procedures were in accordance with the Guide for the Care and Use of Laboratory Animals and were approved by the University of Kentucky Institutional Animal Care and Use Committee prior to the start of experimentation. Forty-eight (n=24 control, n=24 ethanol) male, Sprague Dawley adolescent rats (P30 at arrival, Charles River Laboratories, Raleigh, NC) were run as a part of three cohorts where ~4% mortality due to gavage error occurred. Rats were pair-housed under a 12h light:dark cycle in an AALAC-accredited vivarium at the University of Kentucky. While no model perfectly mimics every aspect of AUDs, the Majchrowicz (1975) model of alcohol dependence was chosen for its oral/gastric route of exposure identical to the human condition, the production of high BECs characteristic of binge consumption, and most specifically to compare to past work in adult rats (Peng & Nixon, 2017). Rats had ad libitum food and water access except during the four days of binge treatment. Following two days of undisturbed acclimation, rats were handled for three days prior to experimentation. For the binge treatment, rats were intragastrically gavaged with ethanol diet (25% w/v ethanol in Vanilla Ensure Plus®, Abbott Laboratories, Abbott Park, IL; n=24) or an isocaloric control diet (42.75% w/v dextrose; n=24) every 8 hours for 4 days. Initially, ethanol rats were administered 5g/kg ethanol diet and subsequent doses were titrated based on the rat’s intoxication behavior which was scored using a behavioral scale modified from Majchrowicz as described (Morris et al., 2010, supplemental Table 1). For each dosing session, control rats received the average volume of diet that all ethanol rats received. Ninety minutes following the 6th dose of ethanol, tail bloods were taken for blood ethanol concentration (BEC) determination, measured on an AM1 Alcohol Analyzer (Analox, London, UK). At 10-26 hours after their last dose, ethanol rats were observed for 30 minutes of each hour and withdrawal behaviors scored as in previous studies (Morris et al., 2010; supplement Table 2). Tissue was obtained immediately after their last dose of ethanol or 2, 7, or 14 days later (e.g. T0, T2, etc.). One portion of cells were stained for flow cytometry as described below. Extra cells were pooled for RNA extraction and real-time RT-PCR were run in triplicate.
Isolation of microglia
As described previously (Peng et al., 2017) according to the original method of Frank (Frank et al., 2006), microglia were isolated from brain tissue via Percoll density gradient centrifugation. At one of four time points, rats were overdosed with sodium pentobarbital (Fatal-Plus®; Vortech Pharmaceuticals, Dearborn, MI), and perfused transcardially with 0.9% NaCl. Brains were excised and bilateral entorhinal cortices and hippocampi were dissected on ice. These regions were chosen as they are targets of alcohol neurotoxicity in the human condition and consistently damaged in this model (Crews et al., 2000; Kelso et al., 2011). For each region homogenates were prepared by finely mincing tissue with a scalpel, homogenizing in Dulbecco’s phosphate buffered saline (PBS), pH 7.4 with a Wheaton Tissue grinder (Thomas Scientific, Swedesboro, NJ), and further passing the homogenate through a 40 μm nylon cell strainer (VWR, Batavia, IL). Homogenates were then centrifuged for 6 min at 400 × g and cell pellets were resuspended in 2 ml 50% isotonic Percoll (GE Healthcare, Piscataway, NJ). Cells were gently applied to the top of a 70% Percoll layer with phosphate buffered saline (PBS) layered atop of the 50% Percoll layer. The cells/density gradient were centrifuged for 45 min at 1200 × g (minimum acceleration and brake) at 20°C. Microglia were collected from the intersection of the 50% and 70% Percoll phases as described (Frank et al., 2006; Peng et al., 2017).
Microglia staining and flow cytometry
Isolated microglia were suspended in an incubation buffer (50 μl; 1 × PBS + 0.1%BSA) for 30 min on ice then Fc receptors blocked with anti-CD32 (BD Bioscience, San Jose, CA). Fluorescent conjugated antibodies were applied on ice for 30 min in the dark to assess microglia purity (mouse anti-rat CD11b-FITC, BD Pharmingen, San Jose, CA; mouse anti-rat-CD45-APC, eBioscience, San Diego, CA) and state of activation (mouse anti-rat: MHC-II-PE, CD32-PE, CD86-PE; BD Bioscience, San Jose, CA). For CD206, cells were incubated in rabbit anti-rat CD206 then donkey anti-rabbit-PE secondary antibody (BD Bioscience, San Jose, CA). Following washes in PBS, cells were analyzed on an Attune Acoustic Focusing Cytometer (ABI, Carlsbad, CA) calibrated with commercially available beads prior to each run. Fluorescence spillover compensation values were generated from non-stained cell populations and single-color staining controls. Isotype controls were used to exclude the non-specific binding of antibodies. For each staining condition, 1 × 104 events were collected.
RNA isolation and real-time PCR.
Total RNA was extracted from isolated microglia/macrophages with TRIZOL Reagent (Life Technologies, Carlsbad, CA) and mirVana miRNA Isolation Kit (Life Technologies) following the manufacturer’s protocols. Real-time RT-PCR was conducted with Assays-on-Demand primers (Applied Biosystems Inc.), using a one-step quantitative Real-time RT-PCR system (Applied Biosystems Inc.). mRNA levels were standardized by comparison to glyceraldehyde-3-phosphate dehydrogenase (GAPDH). GAPDH was chosen as it is commonly used in alcohol-microglia studies for its stability across multiple alcohol models (e.g. Doremus-Fitzwater et al. 2015) and is unchanged in a 2-day binge model according to RNA-seq studies in isolated microglia (transcripts per million, unpublished observations). As with previous (Lan et al., 2012), data were analyzed utilizing the comparative threshold cycle method. Results were expressed as fold difference.
Statistical analyses
Data were compiled into excel and analyzed in Prism (v7, GraphPad Software, San Diego, CA). All data are reported as mean ± SEM. Alcohol model subject data, i.e. mean dose per day, mean withdrawal score, peak withdrawal score and BEC, were analyzed by one-way ANOVA. Data from flow cytometry and RT-PCR were compared using planned comparisons via T-test. Statistical significance was accepted at a p value <0.05.
Results
Binge data
Subject data for the various binge parameters are shown for each time point in Table 1. All binge parameters except one were statistically similar between groups and all values were similar to our previous report comparing withdrawal severity in adults versus adolescents in this model (Morris et al., 2010). Across the four day alcohol binge exposure, rats received a mean dose of 11.8 ± 1.1 g/kg/day ethanol. Peak blood ethanol concentrations were also similar between time points and averaged 404.4 ± 81.7 mg/dl, as measured on the third day of exposure. While BEC appeared higher than what we have observed in this model historically, the mean dose per day and range of BEC values were statistically similar to our previous reports (Marshall et al., 2013; Morris et al., 2010). Mean withdrawal in the T14 group was significantly less than the other time point groups. Withdrawal severity, however, has not correlated to microglia measures in our past work, reducing our concern about the impact of this one value (Marshall et al., 2013; McClain et al., 2011).
Table 1:
Binge Data
| Time Point (Days post E) |
N | BEC (mg/dL) | Peak Withdrawal |
Mean Withdrawal |
Dose (g/kg/day) |
|---|---|---|---|---|---|
| 0 | 6 | 408.4 ± 54.7 | n/a | n/a | 11.2 ± 1.3 |
| 2 | 8 | 388.5 ± 123.5 | 3.5 ± 0.3 | 1.9 ± 1.0 | 11.9 ± 0.9 |
| 7 | 6 | 431.0 ± 47.4 | 3.4 ± 0.3 | 1.1 ± 0.7 | 11.9 ± 1.4 |
| 14 | 4 | 390.0 ± 66.7 | 3.1 ± 0.4 | 0.9 ± 0.3* | 12.4 ± 0.5 |
BEC = Blood ethanol concentration
Increased expression of activation markers on the surface of microglia after 4-day binge alcohol exposure.
Myeloid cells were isolated from bilateral hippocampi and entorhinal cortices at 0, 2, 7 and 14 days after alcohol exposure. Time points were chosen accordance with our previous report of microglial activation in the four-day binge model in adult rats (Peng et al., 2017). As with prior reports using this method, isolated cells were highly enriched for microglia and macrophages and were suitable for immediate characterization ex vivo (Frank et al., 2006): isolated cells were >95% according to CD11b+ immunoreactivity, a microglia and macrophage antigen (Figure 1).
Figure 1. Flow cytometry analysis of Percoll-isolated microglia from 4-day binge alcohol exposure adolescent rats.

Male adolescent rats were gavaged with 25% (w/v) ethanol or isocaloric control diet every 8 hours for 4 days following a modified Majchrowicz protocol. Rats were sacrificed 2 days later and microglia were isolated from hippocampal and entorhinal cortex (E-Cortex) homogenates through Percoll density gradient centrifugation. Cells were labeled with microglia surface antigens and analyzed by flow cytometry. A. Isolated microglia exhibited highly uniform light scattering properties. Gate R1 circumscribes the cell population and excludes any remaining myelin debris or aggregates based on scatter characteristics. B. Both CD11b+CD45lo microglia (R5) and CD11b+CD45hi (R3) were evident after excluding debris, aggregates, and CD11b− cells from the analysis (R2 and R4). C-D. The relative frequencies of the CD11b+CD45hi cell populations varied between control and ethanol in hippocampus (C) and entorhinal cortex (D) after 4-day binge exposure. E-H. Mean fluorescent intensity (MFI) of CD11b and CD45 expression on CD11b+CD45lo microglia. Data presented show MFI of CD11b (E&F) and CD45 (G&H) on CD11b+CD45lo microglia isolated from control and alcohol rat hippocampus (E&G) and entorhinal cortex (F&H) as a percent of control. *p<0.05 versus respective control.
CD11b+ cells were divided into subpopulations of CD11b+CD45low “microglia” and a small subpopulation of CD11b+CD45high cells, which indicates fully activated CNS microglia/macrophages, infiltrating monocytes/macrophages, or neutrophils (Bedi et al., 2013). CD11b and CD45 are constitutively expressed by microglia though expression increases with activation (Marshall et al., 2013; Morioka et al., 1992). The frequency of CD11b+CD45high cells increased slightly but significantly in alcohol groups (3.56% ± 2.35 at T2) versus controls (0.92% ± 0.32 at T2) in hippocampus (Figure 1C) and entorhinal cortex in alcohol rats (1.95% ± 0.61 at T0, 2.32% ± 1.49 at T2) versus control rats (1.13% ± 0.17 at T0, 1.08% ± 0.23 at T2) (Figure 1D). Four-day binge alcohol exposure increased CD11b expression on microglia significantly in both hippocampus (Figure 1E) and entorhinal cortex at T2 (Figure 1F). Alcohol exposure also increased the expression of CD45 on CD11b+CD45low microglia in hippocampus (Figure 1G) at T0 and T2 and entorhinal cortex (Figure 1H) at T0, T2, and T7, all of which resolved to levels observed in controls by T14.
MHC II expression has been used to identify activated macrophage/microglia especially M1-polarized cells (David and Kroner, 2011). Therefore, to characterize the activation state of microglia after ethanol exposure, we analyzed the surface expression of MHC-II on the CD11b+CD45low population of microglia/macrophages isolated from the hippocampus or entorhinal cortex at 0, 2, 7 and 14 days after alcohol exposure. The majority of CD11b+CD45low microglia were negative for MHC-II expression. However, after 4-day binge alcohol exposure, the frequency of the MHC-II+ cells increased in both hippocampus and entorhinal cortex at T2 (from 3.04% in controls to 10.0% in EtOH, p=0.009 in hippocampus; from 1.87% to 6.39%, p=0.017 in entorhinal cortex; Figure 2A&B), all of which returned to control levels by T7 in hippocampus and by T14 in entorhinal cortex. The overall expression of MHC-II in CD11b+CD45high cells (activated microglia/macrophages) was much higher than CD11b+CD45low microglia and ethanol exposure did not affect the expression of MHC-II in the CD11b+CD45high cell population (data not shown). We also examined two other M1 markers, CD86 and CD32, on CD11b+CD45low microglia isolated from the hippocampus and entorhinal cortex at the T2 time point. The majority of CD11b+CD45low microglia were negative for CD86 expression though increased expression of CD86+ cells was observed in hippocampus (3.96% in controls to 8.31% in EtOH, p=0.062; Figure 2C) and entorhinal cortex of alcohol-exposed rats (3.07% in controls to 7.53% in EtOH, p=0.015; Figure 2E). We also observed increased expression of CD32+ cells in hippocampus (17.96% in controls to 46.89% in EtOH, p=0.008; Figure 2D); and in entorhinal cortex in alcohol animals (15.90% in controls to 51.58% in EtOH, p=0.007; Figure 2F).
Figure 2. Increased expression of MHC II, CD86, CD32 on CD11b+CD45lo cells after a four-day binge alcohol exposure.
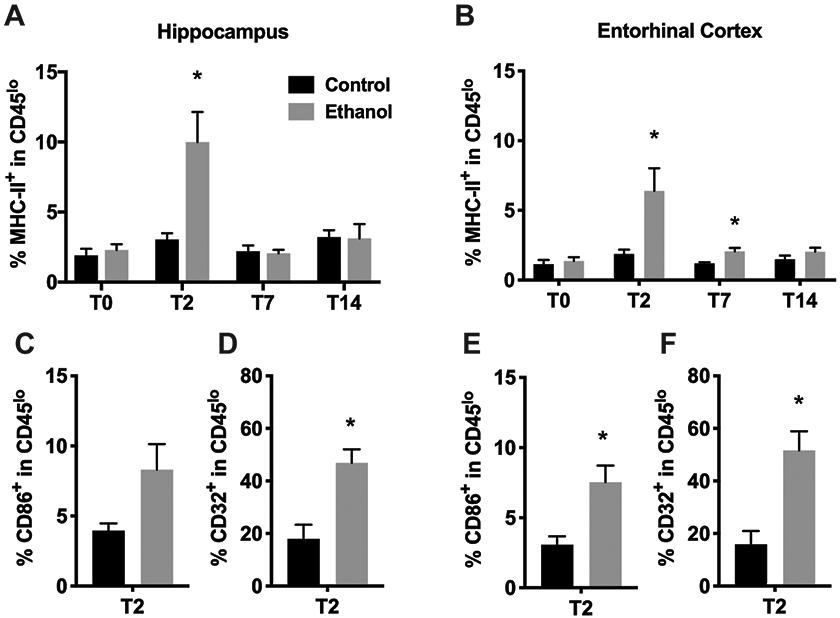
A-B. The percentage of MHC II+ cells within CD11b+CD45lo microglia varied significantly between control and alcohol-exposed rat hippocampus (A; left panels) and entorhinal cortex (B; right panels). C, E. The percentage of CD86+ cells within CD11b+CD45lo microglia at T2 varied significantly between control and alcohol-exposed rat hippocampus (C) and entorhinal cortex (D). D, F. The percentage of CD32+ cells within CD11b+CD45lo microglia at T2 varied significantly between control and alcohol-exposed rat hippocampus (D) and entorhinal cortex (F). * p<0.05 versus respective control.
To identify M2 microglia, the prototypical anti-inflammatory surface marker CD206 (macrophage mannose receptor 1), was used (Bedi et al., 2013; Beynon and Walker, 2012; Cherry et al., 2014). After alcohol exposure, the frequency of the CD206+ cells in CD11b+CD45low microglia increased at the T2 time point in hippocampus (3.72% in controls vs. 9.7% in EtOH, p=0.015; Figure 3A) and in entorhinal cortex of alcohol-exposed rats (3.09% in controls vs. 7.25% in EtOH, p=0.042; Figure 3B). The expression frequency of CD206+ cells in CD11b+CD45low microglia resolved to control levels by T7 in hippocampus. However, we still observed a slight increase in CD206+ cells in CD11b+CD45low microglia isolated from entorhinal cortex of alcohol rats at T7 (2.52% in controls to 4.63% in EtOH, p=0.014; Figure 3B) which resolved to control levels at T14. The overall expression of CD206 on CD11b+CD45high activated microglia/macrophages is much higher than CD11b+CD45low microglia although four day binge alcohol exposure did not affect CD206 expression on the CD11b+CD45high cell population (data not shown).
Figure 3. Increased expression of CD206 on CD11b+CD45lo cells after four-day binge alcohol exposure.
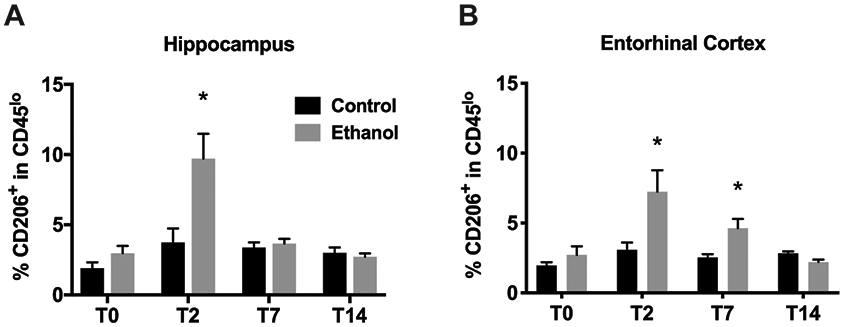
A-D. The percentage of CD206+ cells within CD11b+CD45lo microglia varied significantly between control and alcohol-exposed rat hippocampus (A) and entorhinal cortex (B). * p<0.05 versus respective control.
Cytokine and growth factor profile in isolated microglia after four-day binge exposure
Activated microglia have been shown to release pro-inflammatory or anti-inflammatory cytokines, chemokines and growth factors, and several studies have demonstrated that alcohol exposure induces an inflammatory response based on the assessment of pro-inflammatory factors both in situ and in cell culture models. However, anti-inflammatory factors are rarely examined. Therefore, in this study, we examined gene expression of a broader array of immune molecules important for determining microglia function in fresh Percoll-enriched microglia. The levels of mRNA encoding pro-inflammatory cytokine/chemokines TNF-α, CCL2, IL-1β, and IL-6, growth factors BDNF, IGF-1 and TGF-β and M2-like marker, arginase, were quantified by real-time RT-PCR. After binge exposure overall expression of pro-inflammatory genes (TNF-α, CCL2, IL-1β, and IL-6) was decreased significantly in microglia isolated from both hippocampus (Figure 4A-D) and entorhinal cortex (Figure 4 E-H) at both T2, T7, and T14 compared to control. Exceptions include a slight but not statistically significant decrease in IL-1ß and IL-6 in the hippocampus at T14. Strikingly, immediately after the last dose (T0) and notably while the animals were still intoxicated, IL-6 was increased over 3-fold in both hippocampus (Figure 4A) and entorhinal cortex (Figure 4G) while TNF-α was unchanged versus controls in both regions (Figure 4D, 4H). Interestingly, ethanol also decreased expression of anti-inflammatory cytokine TGF-β in microglia isolated from both hippocampus (Figure 5C) and entorhinal cortex (Figure 5G) at all time points examined. Simultaneously, BDNF expression was initially unchanged at T0 then increased significantly in microglia isolated from hippocampus (2.75 ± 0.06-fold compared to control microglia, p<0.01; Figure 5A) and entorhinal cortex (1.89 ± 0.63-fold compared to control microglia, p<0.01; Figure 5E) of alcohol rats at T2. Microglia isolated at T7 also showed increased BDNF expression after alcohol exposure (1.53 ± 0.06-fold, p<0.01 for hippocampus, 1.60 ± 0.03 folds, p<0.01 for entorhinal cortex) though the fold change was not as high as at T2, values which returned to control levels at T14. In contrast, a decrease in IGF1 expression was detected in microglia from both hippocampus (Figure 5B) and entorhinal cortex (Figure 5F) of alcohol-exposed rats at T2, T7 and T14, but only hippocampus at T0. Finally, arginase was increased over 4-fold (p<0.01) in microglia from hippocampus at T0 only (Figure 5D).
Figure 4. Expression profiles of proinflammatory cytokines in isolated microglia.
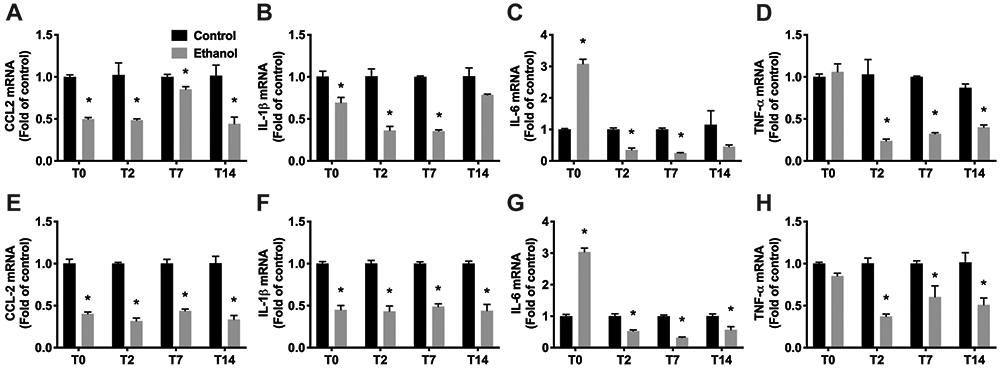
A-H. Total RNA was extracted from Percoll-enriched microglia from hippocampus (A-D) or entorhinal cortex (E-H) of control (n=8 pooled) and alcohol (n=8 pooled) group rats. mRNA expression was determined for CCL2 (A, E), IL-1β (B, F), IL-6 (C, G), TNF-α (D, H) by real-time RT-PCR. The results are expressed as folds of control rats. *p<0.05 versus respective control.
Figure 5. Expression profiles of growth factors in isolated microglia.
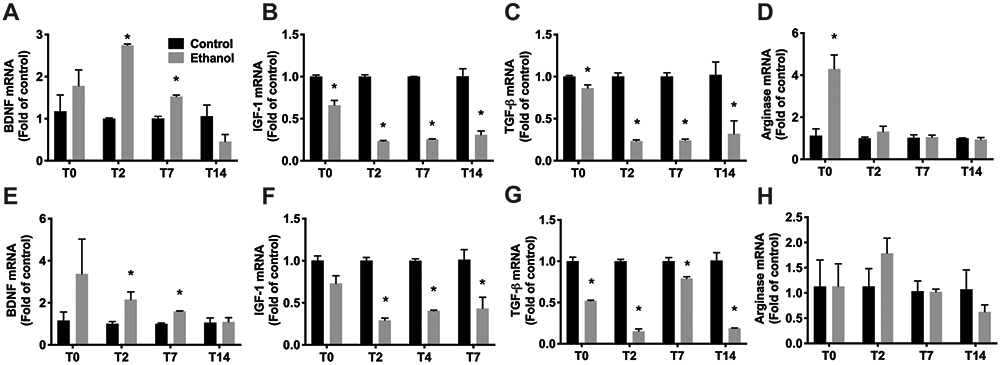
A-H. Total RNA was extracted from Percoll-enriched microglia from hippocampus (A-D) or entorhinal cortex (E-H) of control (n=8 pooled) and alcohol (n=8 pooled) group rats. mRNA expression was determined for BDNF (A, E), IGF-1 (B, F), TGF-β (C, G) and Arginase (D,H) by real-time RT-PCR. The results are expressed as folds of control rats. *p<0.05 versus respective control.
Discussion
Excessive alcohol consumption, the hallmark of an AUD, damages the brain (Crews and Nixon, 2009), however, the specific cellular mechanisms that drive these pathologies remain poorly understood. Neuroimmune activation, and specifically microglia activation, a central figure in the neuroimmune response under alcohol exposure and in secondary neurotoxic cascades in other neurodegenerative disorders, has logically been implicated in AUD pathogenesis (Chastain and Sarkar, 2014; Crews and Nixon, 2009; Mayfield and Harris, 2017). In this study, we evaluated the effects of 4-day binge alcohol exposure in adolescent rats on macrophage/microglia polarization by flow cytometry and real-time RT-PCR. Utilizing Percoll gradient centrifugation, microglia/macrophages were isolated and their polarization state was characterized by examining the expression of MHC-II, CD32, and CD86 as M1 surface markers versus CD206 as an M2 surface marker. We found that four-day binge alcohol exposure activated microglia according to significant increases in both M1 and M2 markers on microglia isolated from the hippocampus and entorhinal cortex, with the most dramatic effects observed at T2. While the timing of these major effects was generally similar to our previous report in adult male rats (Peng et al., 2017), some effects such as increased CD45 expression (as indicated by mean fluorescent intensity; MFI) and increased CD206+ microglia in entorhinal cortex persisted longer in adolescents, which may suggest an enduring activation state in adolescents versus adults although this study did not directly compare ages. Alcohol exposure in the four-day binge paradigm, significantly increased CD11b (complement receptor 3) expression on microglia isolated from both hippocampus and entorhinal cortex (Figure 1E-F), which aligns with our previous report of increased CD11b immunoreactivity in adult rats using immunohistochemistry (Marshall et al., 2016; Marshall et al., 2013). Data here support a transient upregulation of complement receptor 3, a common early step in neuroimmune activation, which we have observed to be persistently increased for months using immunohistochemistry (Marshall et al., 2016; Marshall et al., 2013). These data also align with reports of other microglial markers that are upregulated due to alcohol activating microglia (Barton et al., 2017; He and Crews, 2008; McClain et al., 2011; Sanchez-Alavez et al., 2019).
A variety of changes and interesting lack of effect of alcohol were observed for gene expression. During intoxication at T0, IL-6 gene expression is initially increased in microglia of both hippocampus and entorhinal cortices before dropping to below controls at almost all other timepoints. The alternative activation marker, arginase, was also only increased at T0 in hippocampal microglia and no other time point or regions. Intriguingly, four-day binge alcohol exposure decreased gene expression of pro-inflammatory cytokines TNF-α, CCL2, IL-1β, and IL-6 but also growth factors IGF-1 and TGF-β in microglia isolated from hippocampus and entorhinal cortex at 2- and 7-days post binge compared to controls. Simultaneously, increased BDNF gene expression was observed in microglia isolated at T2 and T7 compared to controls. Some changes in gene expression are consistent with our previous reports including no change in TNF-α protein expression in the hippocampus of adolescent rats at T2 (McClain et al., 2011) or at any time point in adult rats (Marshall et al., 2013), and a decreased in IL6 was observed at T2 for hippocampus only (Marshall et al., 2013). As gene expression does not always predict protein, BDNF protein expression was not changed in this same adolescent AUD model, though increased BDNF has been observed in adult alcohol models and correlates to microglia number (McClain et al., 2011; McClain et al., 2014; Marshall et al., 2016). Altogether, these effects on gene expression coupled with phenotypic surface marker expression support that four-day binge alcohol exposure does not polarize microglia towards purely M1-like or M2-like states, but along a spectrum, as has been suggested in other fields (Ransohoff, 2016). Microglia phenotypes may be more of a disease specific signature (Butovsky and Weiner, 2018), and as such these data support more of an anti-inflammatory state, which is consistent with a number of past reports in rat models (Barton et al., 2017; Bell-Temin et al., 2013; Marshall et al., 2013; Zahr et al., 2010). While these data support neuroimmune activating effects of alcohol, they do not necessarily support a role of microglia in pro-inflammatory effects in AUD models. Further, determining microglia phenotype is critical for understanding the specific role of these cells in adolescence, a critical window in innate immune system activation driven effects on adult behavior, such as the development of AUDs.
These data provide evidence that alcohol activates the neuroimmune system in rodent adolescent models of an AUD. Many groups equivocate alcohol activating microglia with “neuroinflammation,” but without examining the phenotype or phenotypic signature of microglia in AUD models, it is not clear what role microglia play as either cause or consequence of alcohol neurotoxicity (Melbourne et al., 2019). Many of the above results indicate a reparative or anti-inflammatory state, not a pro-inflammatory state of microglia after alcohol dependence. An over two-fold increase in the number of microglia expressing the M2-marker CD206 (mannose receptor), coupled with decreases in pro-inflammatory cytokine gene expression but increases in growth factor gene expression were observed in microglia isolated from the hippocampus and entorhinal cortex at 2 and 7 days after the last dose of alcohol (Fig 3-5). Considering the severity of this model, including past observations of degeneration (e.g. Crews et al., 2000), it is striking that limited pro-inflammatory indices are observed. Collectively, these measures support that microglia and/or their activation are driving a more reparative state during the first week after alcohol exposure. Alternatively, the lack of pro-inflammatory state could also reflect a blunted neuroimmune response, which has been suggested by rapid gene expression changes after an acute dose of alcohol (Doremus-Fitzwater et al., 2015). Acutely, it is well-accepted that alcohol blunts peripheral immune responses (e.g. Nelson et al., 1989). However, the four-day binge model, though the first ethanol experience, is a characterized by repeated binge-like exposure with intervals where BEC approaches zero, not a single acute exposure. Furthermore, the model results in cell death in corticolimbic regions that causes the release of the damage-associated molecular pattern (DAMP), High Mobility Group-Box 1 (HMGB1), which is thought to be secreted by degenerating neurons to elicit neuroimmune activation (Crews et al., 2016; Vetreno and Crews, 2012; Wang et al., 2015). Considering the HMGB1 release in this model and data presented above, a blunting of the immune response is not consistent with the neuroimmune activating events evident in this more chronic four-day exposure.
Several reports support a strong, causal tie between neuroimmune activation and excessive alcohol drinking (Agrawal et al., 2011; Blednov et al., 2012; Warden et al., 2020); see also for review (Mayfield and Harris, 2017), however, the role of microglia specifically in mediating alcohol-induced neurodegeneration in AUDs has been less clear (Melbourne et al., 2019; Walter and Crews, 2017). Most compelling, in human brain tissue, the morphology of microglia is not amoeboid, but ramified, which suggests that microglia are not cytotoxic M1-like or fully activated to a pro-inflammatory state (He and Crews, 2008). Furthermore, the time course of alcohol-induced cell death versus microglia activation does not support a causal role of microglia driving alcohol-induced neuronal toxicity in rat models (Crews et al., 2000; Kelso et al., 2011; Marshall et al., 2013). In this model, neurons die via necrosis (Obernier et al., 2002), which releases DAMPs such as HMGB1 (Wang et al., 2015) and likely ATP that activate microglia and neuroimmune signaling (Asatryan et al., 2018; Coleman et al., 2018; Crews et al., 2013; Gofman et al., 2014; Zou and Crews, 2014). ATP activated purinergic P2X4 or P2X7 receptors are endogenously expressed in microglia and known to modulate microglia activity (e.g. Monif et al., 2009). ATP activates P2X4Rs with lower (0.1 mM) concentrations, but is sufficient to trigger BDNF release (Ullman et al., 2008). P2X7 receptors are activated at higher (>1.0 mM) ATP concentrations, which triggers microglia migration, proliferation and modulates the release of pro-inflammatory cytokines such as IL-1ß and TNF-α (Suzuki et al., 2004; Monif et al., 2009; Clark et al., 2010). P2X4 or P2X7 receptors have been shown to regulate alcohol-induced responses in microglia (Gofman et al., 2014; Asatryan et al., 2018), which leads to the speculation that 4-day binge alcohol exposure may activate P2X4 receptors but not P2X7Rs, and thus increase BDNF but decrease proinflammatory cytokine IL-1ß, TNF-α expression. ATP release or purinergic receptors have not been examined in the binge model of an AUD, but these make for intriguing future directions for how alcohol activates microglia. Some groups have shown that alcohol acts directly on microglia through toll-like receptor 4 (TLR4; Alfonso-Loeches et al., 2010). However, astrocytes also have TLR4 receptors through which they become activated by alcohol (Blanco et al., 2005). Besides astrocytes and microglia, endothelial cells, oligodendrocytes, and neurons also secrete immune effectors in the brain. Indeed, astrocytes are highly activated in the hippocampus and entorhinal cortex of the current AUD model used (Kelso et al., 2011), therefore astrocytes may play a primary role in the pro-inflammatory actions of alcohol (Blanco et al., 2005). This point is why Percoll gradient centrifugation was used to isolate the entire microglia/macrophage population from regions of interest (hippocampus and entorhinal cortex), in order to provide cell-specific analysis of microglia phenotypic markers and function.
Percoll density gradient centrifugation allowed for rapid isolation of myeloid cells from brain tissue, which include microglia and may include brain parenchymal macrophages, as well as myeloid cells from circulating blood. While the initial saline perfusion excluded the majority of circulating myeloid cells in the brain vessels, a slight increase in the frequency of CD11b+CD45high cells in ethanol group versus control animals was observed (Figure 1C&D). CD11b+CD45high cells are thought to be endogenous CNS macrophages. According to studies of various brain injury models, post insult, CD11b+CD45high cells appear to be a mixed population of pro-inflammatory myeloid cell types, including infiltrating monocytes/macrophages, activated CNS parenchymal microglia/macrophages and potentially a few neutrophils and dendritic cells (Bedi et al., 2013). In this case, these CD11b+CD45high cells are likely activated CNS microglia/macrophages though there is no evidence of blood brain barrier compromise in the four-day binge model used (Marshall et al., 2013). In conditions with an established neuroinflammatory state, e.g. stroke, traumatic brain injury, viral, bacterial, or autoimmune diseases, activated immune cells in the blood are capable of crossing a compromised blood brain barrier (BBB) into the parenchyma. Although some evidence suggests that alcohol consumption may compromise the BBB (Haorah et al., 2005), little immune cell extravagation was observed under alcohol exposure (Peng et al., 2017) and no increases in IgG immunoreactivity have been reported (Marshall et al., 2013).
Alcohol has greater damaging effects on the adolescent brain (Crews et al., 2000) and the neuroimmune response is thought to play a role in adolescent alcohol drinking leading to greater severity of adult alcohol-related problems (Crews et al., 2013). This point immediately leads one to question whether there are developmental differences in microglia between adults and adolescents. Although direct comparisons are few, some groups have shown PND 28 to be very similar to adults while others describe subtle differences between mid-adolescence (PND 28-42) and adulthood (>PND 60; Brenhouse and Schwarz, 2016; Crews et al., 2019; Dubbelaar et al., 2018). Microglia number and distribution across the hippocampus peak during the second or third postnatal week, declining rapidly during the third and fourth week (Kim et al., 2015; Nikodemova et al., 2015), the latter of which corresponds to the early adolescence. Between PND 35-39 when we exposed rats to ethanol, there is only a modest decrease in microglia number versus adulthood (Kim et al., 2015). While microglia number in the hippocampus correlated to sensitivity to seizure triggers across the first postnatal weeks (Kim et al., 2015), several other relevant signaling factors and receptors also peak around adolescence which may better explain the sensitivity (reviewed in Spear, 2018). Importantly, withdrawal severity is remarkably similar in adolescents and adults in the Majchrowicz model and the timing of peak cell death at T0, prior to withdrawal, rules out alterations in microglia number driving differences through seizure severity (Morris et al., 2010). Further, microglia loss occurs across the 4-days in both adults and adolescents (Marshall, McClain et al., 2020). The Majchrowicz model in adolescent rats, however, has limitations as it models a more restricted population of adolescents, those with an AUD. This model was chosen to compare to past work in adult rats as the Majchrowicz model overcomes slight pharmacokinetic differences between adult and adolescent rats (Morris et al., 2010; Peng & Nixon, 2017).
Microglia in the adolescent brain may react more strongly than in adults (e.g. Figure 3, Brenhouse and Schwarz, 2016). Cytokine expression peaks in early adolescence at PND25 and drops across maturation to PND55, the pattern of which mirrors reports of increased reactivity of microglia in adolescents versus adults (Brenhouse and Schwarz, 2016). Adolescents may have increased neuroimmune activation, depending on the marker examined though effects vary by brain region, sex, and time examined (Sharma et al., 2018). In one of the few direct comparisons, CCL2 mRNA was increased more in adolescent rats versus adults, but the effect correlated to BEC which was higher in adolescents as they drank more ethanol (Harper et al., 2015). Ethanol pharmacokinetics differ slightly between adults versus adolescents which requires a model that can overcome these differences to produce similar BEC, one of the reasons why the Majchrowicz 4-day binge model was employed in the current study (Morris et al., 2010). It is of note that during development, microglia are much more likely to be “primed,” or become hyper-sensitive to subsequent insult (Ransohoff and Perry, 2009). Developmental susceptibility to priming is evident after early life stress, fetal infection, pain and opiate exposure but it is unclear the point at which adult reactivity is reached (Bilbo and Schwarz, 2009). Direct evidence of microglia priming shows that it continues through adolescence for drugs of abuse (Schwarz and Bilbo, 2013), and evidence of priming – the heightened response to subsequent insult - is clear in adult alcohol exposure (Marshall et al., 2016). Although demonstrating the increased reaction of microglia to a second insult is necessary to truly support that they are primed (as in Marshall et al., 2016 in adults), changes consistent with a low level of activation such as upregulation of complement receptor 3 (OX42), Iba-1 densitometry, and/or hyper-ramified morphology have been used to identify microglia that are likely primed (Ransohoff and Perry, 2009; Barton et al., 2017). To date, adolescent susceptibility to alcohol priming microglia has only been reported in preliminary form (Peng & Nixon, unpublished observations). It is of note that our observations of long-term, low levels of activation in microglia that are not fully M1 or pro-inflammatory, predict that microglia are primed by excessive alcohol exposure in adolescent models (McClain et al., 2011; Marshall et al., 2016; Peng et al., 2017). The implications of alcohol priming microglia in adolescence are concerning. Priming of microglia alters developmental trajectories for brain, behavior, and immune function (Bilbo and Schwarz, 2009; Ganguly and Brenhouse, 2015; Mouihate et al., 2010). Thus, microglia, even while merely in a primed state, can have a long-term impact on development, plasticity and behavior (Brenhouse and Schwarz, 2016).
In sum, alcohol exposure in the Majchrowicz four-day binge model has neuroimmune activating effects specifically on microglia, but not to a fully pro-inflammatory or M1-like phenotype even in regions known to be damaged by alcohol in adolescent male rats. Definitions of microglia phenotype and their relationship to function as well as dysfunction in disease are evolving (Ransohoff & Perry, 2009; Ransohoff, 2016; Butovsky and Weiner, 2018; Melbourne et al., 2019). While potential phenotypes are discussed according to the macrophage terminology of M1-like to the M2-like spectrum, alcohol - and perhaps the different phases of alcoholism - likely has its own disease signature (Butovsky and Weiner, 2018; Warden et al., 2020). These data highlight the complexity of microglia reactivity in disease: multiple phenotypes of microglia were observed and across the spectrum of M1 to M2-like markers. While these neuroimmune effects appear to produce a phenotype that is more M2-like and perhaps reparative microglia, these descriptors coincide with the definition of microglia being primed (Ransohoff and Perry, 2009). Priming microglia in the adolescent brain can have long term-detrimental effects on adult brain and behavior (Brenhouse and Schwarz, 2016). Alcohol activating or priming microglia may result in the derangement of neuroimmune signaling on neuronal activity, alter the trajectory of adolescent brain development or remove microglia from their homeostatic roles and impair plasticity (Melbourne et al., 2019; Tremblay et al., 2011). Thus, despite their putative reparative phenotype, much remains to be understood about the impact of primed microglia during adolescence on adult brain and behavior.
Supplementary Material
Acknowledgements:
We gratefully acknowledge assistance of Kevin Chen, Pharm.D. and Chelsea Geil Nickell, Ph.D. with production of the animal model and Jennifer Melbourne, Ph.D. for critical reading of the manuscript. This work was supported by NIAAA grants R01AA016959, R01AA025591 and R21 AA025563 and the University of Kentucky Department of Pharmaceutical Sciences.
Footnotes
Conflicts: The authors have no conflicts of interest in which to declare.
References
- Agrawal RG, Hewetson A, George CM, Syapin PJ & Bergeson SE 2011. Minocycline reduces ethanol drinking. Brain Behav Immun, 25 Suppl 1, S165–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alfonso-Loeches S, Pascual-Lucas M, Blanco AM, Sanchez-Vera I & Guerri C 2010. Pivotal role of TLR4 receptors in alcohol-induced neuroinflammation and brain damage. J Neurosci, 30, 8285–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asatryan L, Ostrovskaya O, Lieu D, & Davies DL 2018. Ethanol differentially modulates P2X4 and P2X7 receptor activity and function in BV2 microglial cells. Neuropharmacology, 128, 11–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barton EA, Baker C & Leasure JL 2017. Investigation of Sex Differences in the Microglial Response to Binge Ethanol and Exercise. Brain Sci, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedi SS, Smith P, Hetz RA, Xue H & Cox CS 2013. Immunomagnetic enrichment and flow cytometric characterization of mouse microglia. J Neurosci Methods, 219, 176–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell-Temin H, Zhang P, Chaput D, King MA, You M, Liu B & Stevens SM Jr. 2013. Quantitative proteomic characterization of ethanol-responsive pathways in rat microglial cells. J Proteome Res, 12, 2067–77. [DOI] [PubMed] [Google Scholar]
- Beynon SB & Walker FR 2012. Microglial activation in the injured and healthy brain: what are we really talking about? Practical and theoretical issues associated with the measurement of changes in microglial morphology. Neuroscience, 225, 162–71. [DOI] [PubMed] [Google Scholar]
- Bilbo SD & Schwarz JM 2009. Early-life programming of later-life brain and behavior: a critical role for the immune system. Front Behav Neurosci, 3, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanco AM, Valles SL, Pascual M & Guerri C 2005. Involvement of TLR4/type I IL-1 receptor signaling in the induction of inflammatory mediators and cell death induced by ethanol in cultured astrocytes. J Immunol, 175, 6893–9. [DOI] [PubMed] [Google Scholar]
- Blednov YA, Ponomarev I, Geil C, Bergeson S, Koob GF & Harris RA 2012. Neuroimmune regulation of alcohol consumption: behavioral validation of genes obtained from genomic studies. Addict Biol, 17, 108–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenhouse HC & Schwarz JM 2016. Immunoadolescence: Neuroimmune development and adolescent behavior. Neurosci Biobehav Rev, 70, 288–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butovsky O & Weiner HL 2018. Microglial signatures and their role in health and disease. Nat Rev Neurosci, 19, 622–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chastain LG & Sarkar DK 2014. Role of microglia in regulation of ethanol neurotoxic action. Int Rev Neurobiol, 118, 81–103. [DOI] [PubMed] [Google Scholar]
- Cherry JD, Olschowka JA & O'banion MK 2014. Neuroinflammation and M2 microglia: the good, the bad, and the inflamed. J Neuroinflammation, 11, 98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark AK, Staniland AA, Marchand F, Kaan TK, McMahon SB, & Malcangio M 2010. P2X7-dependent release of interleukin-1beta and nociception in the spinal cord following lipopolysaccharide. J Neurosci, 30, 573–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark DB, Bukstein O & Cornelius J 2002. Alcohol use disorders in adolescents: epidemiology, diagnosis, psychosocial interventions, and pharmacological treatment. Paediatr Drugs, 4, 493–502. [DOI] [PubMed] [Google Scholar]
- Coleman LG Jr., Zou J, Qin L & Crews FT 2018. HMGB1/IL-1beta complexes regulate neuroimmune responses in alcoholism. Brain Behav Immun, 72, 61–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crews FT, Braun CJ, Hoplight B, Switzer RC 3rd & Knapp DJ 2000. Binge ethanol consumption causes differential brain damage in young adolescent rats compared with adult rats. Alcohol Clin Exp Res, 24, 1712–23. [PubMed] [Google Scholar]
- Crews FT & Nixon K 2009. Mechanisms of neurodegeneration and regeneration in alcoholism. Alcohol Alcohol, 44, 115–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crews FT, Qin L, Sheedy D, Vetreno RP & Zou J 2013. High mobility group box 1/Toll-like receptor danger signaling increases brain neuroimmune activation in alcohol dependence. Biol Psychiatry, 73, 602–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crews FT, Robinson DL, Chandler LJ, Ehlers CL, Mulholland PJ, Pandey SC, Rodd ZA, Spear LP, Swartzwelder HS & Vetreno RP 2019. Mechanisms of Persistent Neurobiological Changes Following Adolescent Alcohol Exposure: NADIA Consortium Findings. Alcohol Clin Exp Res, 43, 1806–1822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crews FT, Vetreno RP, Broadwater MA & Robinson DL 2016. Adolescent Alcohol Exposure Persistently Impacts Adult Neurobiology and Behavior. Pharmacol Rev, 68, 1074–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David S & Kroner A 2011. Repertoire of microglial and macrophage responses after spinal cord injury. Nat Rev Neurosci, 12, 388–99. [DOI] [PubMed] [Google Scholar]
- Doremus-Fitzwater TL, Gano A, Paniccia JE & Deak T 2015. Male adolescent rats display blunted cytokine responses in the CNS after acute ethanol or lipopolysaccharide exposure. Physiol Behav, 148, 131–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubbelaar ML, Kracht L, Eggen BJL & Boddeke E 2018. The Kaleidoscope of Microglial Phenotypes. Front Immunol, 9, 1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank MG, Wieseler-Frank JL, Watkins LR & Maier SF 2006. Rapid isolation of highly enriched and quiescent microglia from adult rat hippocampus: immunophenotypic and functional characteristics. J Neurosci Methods, 151, 121–30. [DOI] [PubMed] [Google Scholar]
- Ganguly P & Brenhouse HC 2015. Broken or maladaptive? Altered trajectories in neuroinflammation and behavior after early life adversity. Dev Cogn Neurosci, 11, 18–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gofman L, Cenna JM, & Potula R 2014. P2X4 receptor regulates alcohol-induced responses in microglia. J Neuroimmune Pharmacol, 9, 668–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gore FM, Bloem PJ, Patton GC, Ferguson J, Joseph V, Coffey C, Sawyer SM & Mathers CD 2011. Global burden of disease in young people aged 10-24 years: a systematic analysis. Lancet, 377, 2093–102. [DOI] [PubMed] [Google Scholar]
- Grant BF 1998. The impact of a family history of alcoholism on the relationship between age at onset of alcohol use and DSM-IV alcohol dependence: results from the National Longitudinal Alcohol Epidemiologic Survey. Alcohol Health Res World, 22, 144–7. [PMC free article] [PubMed] [Google Scholar]
- Guerri C & Pascual M 2019. Impact of neuroimmune activation induced by alcohol or drug abuse on adolescent brain development. Int J Dev Neurosci, 77, 89–98. [DOI] [PubMed] [Google Scholar]
- Haorah J, Knipe B, Leibhart J, Ghorpade A & Persidsky Y 2005. Alcohol-induced oxidative stress in brain endothelial cells causes blood-brain barrier dysfunction. J Leukoc Biol, 78, 1223–32. [DOI] [PubMed] [Google Scholar]
- Harper KM, Knapp DJ & Breese GR 2015. Withdrawal from Chronic Alcohol Induces a Unique CCL2 mRNA Increase in Adolescent But Not Adult Brain--Relationship to Blood Alcohol Levels and Seizures. Alcohol Clin Exp Res, 39, 2375–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He J & Crews FT 2008. Increased MCP-1 and microglia in various regions of the human alcoholic brain. Exp Neurol, 210, 349–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelso ML, Liput DJ, Eaves DW & Nixon K 2011. Upregulated vimentin suggests new areas of neurodegeneration in a model of an alcohol use disorder. Neuroscience, 197, 381–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim I, Mlsna LM, Yoon S, Le B, Yu S, Xu D & Koh S 2015. A postnatal peak in microglial development in the mouse hippocampus is correlated with heightened sensitivity to seizure triggers. Brain Behav, 5, e00403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan X, Chen Q, Wang Y, Jia B, Sun L, Zheng J & Peng H 2012. TNF-alpha affects human cortical neural progenitor cell differentiation through the autocrine secretion of leukemia inhibitory factor. PLoS One, 7, e50783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall SA, Geil CR & Nixon K 2016. Prior Binge Ethanol Exposure Potentiates the Microglial Response in a Model of Alcohol-Induced Neurodegeneration. Brain Sci, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall SA, McClain JA, Kelso ML, Hopkins DM, Pauly JR & Nixon K 2013. Microglial activation is not equivalent to neuroinflammation in alcohol-induced neurodegeneration: The importance of microglia phenotype. Neurobiol Dis, 54, 239–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall SA †, McClain JA †, Wooden JI, & Nixon K 2020. Microglia Dystrophy Following Binge-Like Alcohol Exposure in Adolescent and Adult Male Rats. Front Neuroanat, 14: 52. †authors contributed equally. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayfield J & Harris RA 2017. The Neuroimmune Basis of Excessive Alcohol Consumption. Neuropsychopharmacology, 42, 376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClain JA, Morris SA, Deeny MA, Marshall SA, Hayes DM, Kiser ZM & Nixon K 2011. Adolescent binge alcohol exposure induces long-lasting partial activation of microglia. Brain Behav Immun, 25 Suppl 1, S120–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClain JA, Morris SA, Marshall SA & Nixon K 2014. Ectopic hippocampal neurogenesis in adolescent male rats following alcohol dependence. Addict Biol, 19, 687–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melbourne JK, Thompson KR, Peng H & Nixon K 2019. Its complicated: The relationship between alcohol and microglia in the search for novel pharmacotherapeutic targets for alcohol use disorders. Prog Mol Biol Transl Sci, 167, 179–221. [DOI] [PubMed] [Google Scholar]
- Monif M, Reid CA, Powell KL, Smart ML, Williams DA 2009. The P2X7 receptor drives microglial activation and proliferation: a trophic role for P2X7R pore. J Neurosci 29, 3781–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morioka T, Kalehua AN & Streit WJ 1992. Progressive expression of immunomolecules on microglial cells in rat dorsal hippocampus following transient forebrain ischemia. Acta Neuropathol, 83, 149–57. [DOI] [PubMed] [Google Scholar]
- Morris SA, Kelso ML, Liput DJ, Marshall SA & Nixon K 2010. Similar withdrawal severity in adolescents and adults in a rat model of alcohol dependence. Alcohol, 44, 89–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouihate A, Galic MA, Ellis SL, Spencer SJ, Tsutsui S & Pittman QJ 2010. Early life activation of toll-like receptor 4 reprograms neural anti-inflammatory pathways. J Neurosci, 30, 7975–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson S, Bagby GJ, Bainton BG & Summer WR 1989. The effects of acute and chronic alcoholism on tumor necrosis factor and the inflammatory response. J Infect Dis, 160, 422–9. [DOI] [PubMed] [Google Scholar]
- Nikodemova M, Kimyon RS, De I, Small AL, Collier LS & Watters JJ 2015. Microglial numbers attain adult levels after undergoing a rapid decrease in cell number in the third postnatal week. J Neuroimmunol, 278, 280–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obernier JA, Bouldin TW, & Crews FT 2002. Binge ethanol exposure in adult rats causes necrotic cell death. Alcohol Clin Exp Res, 26, 547–557. [PubMed] [Google Scholar]
- Pascual M, Blanco AM, Cauli O, Minarro J & Guerri C 2007. Intermittent ethanol exposure induces inflammatory brain damage and causes long-term behavioural alterations in adolescent rats. Eur J Neurosci, 25, 541–50. [DOI] [PubMed] [Google Scholar]
- Patrick ME & Terry-Mcelrath YM 2019. Prevalence of High-Intensity Drinking from Adolescence through Young Adulthood: National Data from 2016-2017. Subst Abuse, 13, 1178221818822976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng H, Nickell CRG, Chen KY, McClain JA & Nixon K 2017. Increased expression of M1 and M2 phenotypic markers in isolated microglia after four-day binge alcohol exposure in male rats. Alcohol, 62, 29–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ransohoff RM 2016. A polarizing question: do M1 and M2 microglia exist? Nat Neurosci, 19, 987–91. [DOI] [PubMed] [Google Scholar]
- Ransohoff RM & Perry VH 2009. Microglial physiology: unique stimuli, specialized responses. Annu Rev Immunol, 27, 119–45. [DOI] [PubMed] [Google Scholar]
- Sanchez-Alavez M, Nguyen W, Mori S, Wills DN, Otero D, Ehlers CL & Conti B 2019. Time course of microglia activation and brain and blood cytokine/chemokine levels following chronic ethanol exposure and protracted withdrawal in rats. Alcohol, 76, 37–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz JM & Bilbo SD 2013. Adolescent morphine exposure affects long-term microglial function and later-life relapse liability in a model of addiction. J Neurosci, 33, 961–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma R, Rooke J, Kolmogorova D, Melanson B, Mallet JF, Matar C, Schwarz J & Ismail N 2018. Sex differences in the peripheral and central immune responses following lipopolysaccharide treatment in pubertal and adult CD-1 mice. Int J Dev Neurosci, 71, 94–104. [DOI] [PubMed] [Google Scholar]
- Spear LP 2018. Effects of adolescent alcohol consumption on the brain and behaviour. Nat Rev Neurosci, 19, 197–214. [DOI] [PubMed] [Google Scholar]
- Substance Abuse and Mental Health Services Administration (SAMHSA), 2019. Key substance use and mental health indicators in the United States: Results from the 2018 National Survey on Drug Use and Health. (HHS Publication No. PEP19-5068, NSDUH Series H-54). Rockville, MD: Center for Behavioral Health Statistics and Quality, SAMHSA. Retrieved from https://www.samhsa.gov/data/ [Google Scholar]
- Suzuki T, Hide I, Ido K, Kohsaka S, Inoue K, Nakata Y 2004. Production and release of neuroprotective tumor necrosis factor by P2X7 receptor-activated microglia. J Neurosci, 24, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tremblay ME, Stevens B, Sierra A, Wake H, Bessis A & Nimmerjahn A 2011. The role of microglia in the healthy brain. J Neurosci, 31, 16064–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulmann L, Hatcher JP, Hughes JP, Chaumont S, Green PJ, Conquet F, Buell GN, Reeve AJ, Chessell IP, Rassendren F 2008. Up-regulation of P2X4 receptors in spinal microglia after peripheral nerve injury mediates BDNF release and neuropathic pain. J Neurosci, 28, 11263–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Hoof JJ, Van Der Lely N, Bouthoorn SH, Van Dalen WE & Pereira RR 2011. Adolescent alcohol intoxication in the dutch hospital departments of pediatrics: a 2-year comparison study. J Adolesc Health, 48, 212–4. [DOI] [PubMed] [Google Scholar]
- Vetreno RP & Crews FT 2012. Adolescent binge drinking increases expression of the danger signal receptor agonist HMGB1 and Toll-like receptors in the adult prefrontal cortex. Neuroscience, 226, 475–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter TJ & Crews FT 2017. Microglial depletion alters the brain neuroimmune response to acute binge ethanol withdrawal. J Neuroinflammation, 14, 86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Chu G, Yang Z, Sun Y, Zhou H, Li M, Shi J, Tian B, Zhang C & Meng X 2015. Ethanol directly induced HMGB1 release through NOX2/NLRP1 inflammasome in neuronal cells. Toxicology, 334, 104–10. [DOI] [PubMed] [Google Scholar]
- Warden AS, Wolfe SA, Khom S, Varodayan FP, Patel RR, Steinman MQ, Bajo M, Montgomery SE, Vlkolinsky R, Nadav T, Polis I, Roberts AJ, Mayfield RD, Harris RA, Roberto M 2020. Microglia Control Escalation of Drinking in Alcohol-Dependent Mice: Genomic and Synaptic Drivers. Biol Psychiatry. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahr NM, Luong R, Sullivan EV & Pfefferbaum A 2010. Measurement of Serum, Liver, and Brain Cytokine Induction, Thiamine Levels, and Hepatopathology in Rats Exposed to a 4-Day Alcohol Binge Protocol. Alcohol Clin Exp Res. 34, 1858–1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou JY & Crews FT 2014. Release of neuronal HMGB1 by ethanol through decreased HDAC activity activates brain neuroimmune signaling. PLoS One, 9, e87915. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.