

ABSTRACT
Although the blockade of the programmed cell death protein 1/programmed cell death ligand 1 (PD-1/PD-L1) pathway has become a promising treatment strategy for several types of cancers, the constitutive activation of c-Met in tumors may cause a low overall response rate to PD-1 inhibitors. Increasing evidence indicates that the dual inhibition of c-Met and PD-1 could improve the efficacy of anti-PD-1/PD-L1 monoclonal antibodies for tumor immunotherapy. In this study, we developed two bispecific single-chain diabodies targeting c-Met and PD-1 for the treatment of solid tumors based on protein homology modeling, and we identified that the binding affinity of diabody-mp to c-Met was 50-folds higher than that of diabody-pm. The results of in vitro studies revealed that both diabodies suppressed HGF-induced proliferation, migration, and invasion of tumor cells, inhibiting the activation of c-Met signaling by antagonizing HGF binding to c-Met. Moreover, they promoted T cell activation by blocking the PD-1 pathway, mediating tumor cellular cytotoxicity through T cell engagement. In vivo studies with mice models demonstrated that diabody-mp exhibited higher therapeutic efficacy than other structural antibodies, greatly enhancing the survival of c-Met-positive tumor-bearing mice compared to single or combined c-Met and PD-1 blockade therapy. Furthermore, diabody-mp, which had a higher c-Met binding affinity, showed better anti-tumoral activity than diabody-pm, which had a lower c-Met binding affinity. In conclusion, bispecific anti-PD-1/c-Met diabody-mp, with high c-Met-associated affinity, inhibited tumor growth by activating T cells, suggesting its therapeutic potential for c-Met-positive solid tumors.
KEYWORDS: PD-1, c-Met, bispecific antibodies, diabodies, immunotherapy, solid tumor
Introduction
Immunotherapy is a promising treatment that provides new hope for patients with advanced tumor that are not treatable with conventional treatment. Activated T cells induce immune tolerance by upregulating the expression of programmed cell death protein 1 (PD-1), a negative immune checkpoint receptor.1,2 Tumor cells overexpress programmed cell death ligand 1 (PD-L1), a ligand of PD-1, to facilitate immune evasion.3 Although blocking the PD-1/PD-L1 pathway could partially restore exhaustible T cells in preclinical models4 and PD-1-based monotherapies have shown remarkable clinical efficacy in some patients with multiple cancers, including melanoma, non-small cell lung cancer, and colorectal cancer, most patients experience poorly sustained clinical benefit and relapse.5 The objective response rate for nivolumab is 20% in patients with advanced hepatocellular carcinoma (HCC);6 therefore, there is an urgent need to develop strategies that can improve the response rate of PD-1 inhibitors. PD-1 inhibition combined with radiotherapy, chemotherapy, or targeted therapy could achieve enhanced anti-tumor effect; however, the strategy is associated with increased risk of side effects.
The c-Met tyrosine kinase receptor encoded by the MET proto-oncogene, also known as the hepatocyte growth factor (HGF) receptor, is activated in multiple malignancies through MET amplification, mutation, receptor overexpression, and/or a ligand-dependent mechanism.7 Dimerization and activation of c-Met receptor after binding with HGF would promote the proliferation, migration, and invasion of tumor cells.7–9 A previous preclinical study reported that abnormal activation of the c-Met signaling pathway is associated with poor clinical outcomes, and hence, c-Met-targeted therapy could be credible for cancer treatment.10 Although several c-Met tyrosine kinase inhibitors (TKIs) have been used clinically with positive therapeutic effects,11,12 HGF produced in the tumor microenvironment results in innate and acquired resistance to TKIs.13 An alternative approach to the blocking of the HGF/c-Met pathway is the development of therapeutic antibodies; however, it is difficult to produce effective antibodies against both HGF-dependent and HGF-independent c-Met signaling. Moreover, bivalent anti-c-Met antibodies may promote receptor dimerization and activation,14 and monovalent antibodies may lack potency.15
The combinations of immune checkpoint inhibitors and molecular targeted agents have shown great promise in recent years. Previous studies have reported that c-Met signaling plays immunosuppressive roles, such as the impairment of dendritic cell functions and the induction of T cell tolerance, which participate in immune regulation in tumors.16 The mutation of MET as a driver gene in tumors could cause markedly decreased clinical efficacy of PD-1 inhibition therapy. Data from several clinical trials showed that the deficiency of PD-L1 in tumors might limit the efficacy of anti-PD-1 monoclonal antibodies.17 However, most patients with MET exon 14-altered lung cancers expressing PD-L1 still have a low overall response rate to PD-1 inhibitors and a short median progression-free survival after treatment.18 In addition, it has been proposed that c-Met inhibitors promote tumor immune escape in HCC by stabilizing PD-L1.19 Therefore, dual blocking of c-Met and PD-1 as a feasible strategy could release or improve the efficacy of anti-PD-1/PD-L1 monoclonal antibodies (mAbs) for tumor immunotherapy. Several global clinical trials investigating the combination of PD-1/PD-L1 and c-Met inhibitors are in progress.20 In 2020, the US FDA accepted priority review applications for nivolumab in combination with cabozantinib in advanced renal cell carcinoma.21
Mounting evidence suggests that T cells could play a powerful role in the inhibition of tumor growth in solid tumor patients.22 Bispecific antibodies (bsAbs) with the specificity of two antibodies could simultaneously target two different antigens or epitopes, and hence, they could overcome MHC restriction to recruit T cells for the elimination of tumor cells.3,23 In 2009, the anti-EpCAM/CD3 catumaxomab, a trifunctional bsAb for the treatment of cancerous ascites, was the first bsAb to receive market approval.24 In 2014, blinatumomab was approved by the FDA for the treatment of patients with Philadelphia chromosome-negative precursor B cell acute lymphocytic leukemia.25 At present, popular tumor immunotherapies, such as bsAbs and CAR-T, have been approved for the treatment of hematological malignancies, but most of these therapies do not have the desired effect on solid tumors.
The design of monoclonal antibodies (mAbs) focuses on target selection, and it is also critical to determine the structural design of bsAbs. To achieve the treatment of solid tumors, we designed, expressed, and purified two bispecific anti-c-Met/PD-1 single-chain antibodies, also called diabodies,26 with different c-Met binding affinities, and we evaluated their anti-tumor activity in vivo and in vitro. Collectively, the results of our studies demonstrated the potential therapeutic benefits of diabodies for various c-Met-positive solid tumors.
Materials and methods
Cell lines
A549, MKN45, MHCC-97 H, and HEK293E cells were obtained from American Type Culture Collection, and CHO/hPD-1 EXD (extracellular domain) cells were generated in our laboratory. HEK293E cells were cultured in FreeStyle™293 Expression Medium (Gibco, NY, USA). The hPBMCs and T cells were cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS) (Gibco, NY, USA) and IL-2 (10 ng/mL) (Selleckchem, TX, USA). Other tumor cell lines were cultured in RPMI 1640 or Dulbecco’s modified Eagle’s medium (Sigma-Aldrich, MO, USA) supplemented with 10% FBS and 1% penicillin/streptomycin (Hyclone, Shanghai, China) at 37°C and 5% CO2.
Expression vector of diabodies construction
The coding sequences of the anti-c-Met and anti-PD-1 mAbs were created in previous studies in our laboratory. To successfully express the diabodies protein, the Kozak sequence and secretory signal peptide of human IgG were inserted after the CMV promoter of plasmid pCEP4, also called pCEP4-LC-SP. Next, using the previously synthesized anti-c-Met and anti-PD-1 mAbs sequence as templates, we cloned the VL and VH of the anti-c-Met and anti-PD-1 mAbs, respectively. The genes of diabody-mp and diabody-pm were cloned by overlap polymerase chain reaction (PCR). Finally, the genes of the diabodies were linked to the pCEP4-LC-SP plasmid linearized by restriction enzyme (NruI and Not I) digestion.
Expression and purification
For the production of diabodies, anti-PD-1 scFv, anti-c-Met scFv, anti-PD-1 mAb, anti-c-Met mAb, and anti-PD-1/c-Met scFv-Fc, a suspension of growing HEK293E cells at a final concentration of 106 cells/mL were transfected with plasmids by PEI (Polysciences, Shanghai, China). Tryptone N1 was added to the culture at a final concentration of 0.5%. After 7 days of culture in conical flasks gently shaken on the platform of an orbital incubator rotating at 125 rpm, 37°C, and 8% CO2, the supernatants were harvested and purified using His-tag (Roche, Mannheim, Germany) or Protein A Purification column (GenScript, Nanjing, China) by AKTA Explorer. Eluted fractions were analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis.
Flow cytometry
For cellular c-Met and PD-1 binding studies, MKN45 and CHO/hPD-1 EXD cells were harvested and resuspended in culture medium, respectively. The cells were added to a round bottomed 96-well plate (Coring Falcon, NY, USA) at 100 µL/well and incubated with the indicated concentrations of diabody-mp or diabody-pm in triplicate wells at 4°C for 1 hr. Next, the cells were incubated with anti-His-tag antibody-FITC (GenScript, Nanjing, China), followed by the analysis of the cells on Intellicyt iQuescreener. Data analysis was done by software FlowJo. The mean fluorescence intensity (MFI) of the cells was used to evaluate antibody affinity. When the MFI reaches half of its maximum value, the corresponding concentration value (EC50) of diabodies is the KD value of diabodies relative to c-Met or PD-1. For the T cell activation assay, the tumors were enzymatically digested using collagenase I, collagenase IV, and hyaluronidase (YEASEN, Shanghai, China) for 1 hr at 37°C. The TILs were separated by Percoll separation (Gibico, NY, USA) and incubated with antibodies (BioLegend, CA, USA) against FITC-anti-human CD4, FITC-anti-human CD8, and PE-anti-human CD69. For apoptosis detection, the tumor cells were plated in 12-well dishes and incubated overnight at 37°C and 5% CO2. The cells were then pre-treated with IFN-γ (10 ng/mL) (PeproTech, NJ, USA) for 24 hr, followed by co-culturing with activated T cells for 48 hr with or without 100 nM of diabody-mp or diabody-pm. The immunostaining of single cell suspensions was performed using an Annexin V-FITC/PI Apoptosis Detection Kit (Meilun, Dalian, China) according to the manufacturer’s instructions. Sample analysis was carried out using an Accuri C6 flow cytometer (BD Biosciences, CA, USA) and data analysis was performed using the Accuri software.
qRT-PCR
Cells were lysed and total RNA was extracted following standard laboratory protocols. The RNA was reverse-transcribed to complementary DNA using a 5× All-In-One RT MasterMix (Abm, Zhenjiang, China). Quantitative real-time PCR was performed using the ChamQTM Universal SYBR® qPCR Master Mix (Vazyme, Nanjing, China).
Enzyme linked immunosorbent assay
For the cell supernatants, MKN45 cells were seeded in a 96-well plate and cultured overnight at 37°C and 5% CO2. Next, the cells were pre-treated with IFN-γ (10 ng/mL) for 24 hr, after which the medium was removed. Upon the addition of diabody-mp or diabody-pm at the indicated concentrations, the cells were co-cultured with stimulated human T cells at 37°C for 48 hr. The cell supernatants were collected and analyzed using human IFN-γ and human IL-2 ELISA kits (MULTISCIENCES, Hangzhou, China). For tumor plasma, the tumors were dissociated in RPMI 1640 medium and then centrifuged to collect the supernatant. The supernatant was concentrated with Ultra-4 Centrifugal Filters-3 K MWCO (MerckMillipore, MA, USA). For blood plasma, blood was collected by cardiac puncture and centrifuged at 1500 g for 10 min. The supernatant was collected and analyzed using a human IFN-γ ELISA kit.
Luciferase assay
The Jurkat-PD-1+ cell line stably expressing hPD-1 was engineered by transfecting the cells with PCDH-CMV-MCS-EF1-GFP+Puro vector expressing PD-1, followed by selection with puromycin. The Jurkat-PD-1− cells, from which PD-1 has been knocked out, were engineered by transfecting the cells with the PX459 vector cloned into gRNA targeting PD-1, as described in Zheng’s protocol27 (Oligo1: 5′-CACCGCACGAAGCTCTCCGATGTGT-3′; Oligo2: 5′-AAACACACATCGGAGAGCTTCGTGC-3). The expression and knock-out of PD-1 were validated by western blotting and FACS. Jurkat-PD-1+ or Jurkat-PD-1− cells were co-transfected with pGL3-basic vector expressing luciferase regulated by the binding of NFAT with the IL-2 promoter and phRL-TK vector as a control plasmid. MKN45 cells were seeded in a 96-well plate and cultured overnight at 37°C and 5% CO2. After removing the medium, MKN45 cells were pre-treated with IFN-γ (10 ng/mL) for 24 hr. Jurkat-PD-1+/–NFAT cells were activated with PHA-L (2 µg/mL) and IL-2 (10 ng/mL) for 48 hr and then co-cultured with MKN45 cells upon the addition of 100 nM of diabody-mp, diabody-pm, or anti-PD-1 mAb at 37°C for 6 hr. Luciferase activity was determined using the Dual Luciferase Reporter Assay Kit (Vazyme, Nanjing, China).
Western blotting
A549 or MHCC-97 H cells were plated in 6-well plates and incubated overnight at 37°C and 5% CO2. The cells were incubated with 100 nM of diabody-mp, diabody-pm, anti-c-Met mAb, and JNJ-38877605 (Selleckchem, TX, USA) at 37°C for 2 hr. They were stimulated with or without 1 nM of HGF (GenScript, Nanjing, China) before collection and lysed with RIPA cell lysis buffer (Meilun, Dalian, China). Equal amounts of cell lysate were subjected to 10% SDS-PAGE. Immunoblotting was performed with primary antibodies against c-Met (ProteinTech, #25869-1-AP), Phospho-Met (Tyr1234/1235) (CST, #3077), Akt (CST, #4691), phospho-Akt (Ser473) (CST, #4060), p44/42 MAPK (CST, #4695), Phospho-p44/42 MAPK (Thr202/Tyr204) (CST, #4370), and GAPDH (CST, #5174) overnight at 4°C. HRP-conjugated secondary antibodies (CST, MA, USA) were incubated at room temperature for 1 hr.
Proliferation assay
A549 and MHCC-97 H cells were dispensed in 100 µL aliquots, seeded in 96-well plates, and allowed to adhere to the plates overnight. The cells were treated with HGF, followed by incubation with diabody-mp, diabody-pm, anti-c-Met mAb, and JNJ-38877605. They were further incubated in 10% Cell Counting Kit-8 solution (Meilun, Dalian, China) and diluted in culture medium for an additional 2 hr at 37°C. The absorbance at 450 nm was determined in each well.
Tumor cells migration and invasion
For cell migration detection, A549 and MHCC-97 H cells were plated in 6-well plates and incubated at 37°C and 5% CO2 for 24 hr. At > 90% cell confluence, the medium was removed and the cells were wounded with a tip. The detached cells were removed by washing the cells with a serum-free medium. Diabody-mp, diabody-pm, or JNJ-37788605 was added to serum-free medium with or without 1 nM HGF. The wounds were photographed at 0 hr, 24 hr, and 48 hr. For cell migration detection, 8-µm transwell inserts in 24-well plates (Coring Falcon, NY, USA) were coated with Matrigel basement membrane matrix (50 μL/well) (BD Biosciences, Shanghai, China). The cells were re-suspended in 200 μL serum-free RPMI 1640 medium and cultured in the upper chamber. RPMI 1640 medium (with 20% FBS) was added to the bottom wells. Diabody-mp, diabody-pm, or JNJ-37788605 was added to the medium in both inserts. The cells in the upper chamber were removed after 24 hr of incubation at 37°C. The migrated cells were fixed with 4% paraformaldehyde (PFA) and stained with 0.1% crystal violet solution for 30 min at room temperature. The wound area and the number of migrated cells were quantified using ImageJ software.
T cell-mediated tumor cell killing assay
A549, MHCC-97 H, or MKN45 cells were pre-treated with IFN-γ (10 ng/mL) for 24 hr and co-cultured in 12-well plates with either of the diabodies and with or without activated T cells for 48 hr. The wells were washed twice with PBS to remove the T cells. The surviving tumor cells were fixed with 4% PFA and stained with 0.1% crystal violet solution. They were quantified by a spectrometer at OD (570 nm), followed by elution with 33% acetic acid.
Mouse xenograft studies
All the animal experiments were in accordance with the ethical standards of the Department of Laboratory Animal Science of Fudan University, and this study was approved on March 6, 2019. BALB/c and NOD-SCID mice (6 weeks old) (Jihui, Shanghai, China) were raised and treated under specific-pathogen-free conditions. The mice were injected subcutaneously in the right flank with single-cell suspensions of MKN45 or MHCC-97 H cells (5 × 106). The immune system of mice was reconstructed by administering intraperitoneal injection of 107 hPBMCs every 2 weeks. Diabody-mp, diabody-pm, anti-PD-1 scFv and anti-c-Met scFv, anti-PD-1/c-Met scFv-Fc, anti-PD-1 mAb, capmatinib, and toripalimab were administered to the mice as indicated. The vehicle group was administered 0.5% methylcellulose supplemented with 1% polysorbate-80. Tumor volume was measured as indicated time and calculated as follows: volume (cm3) = (width) 2 × length × 0.5.
Patient and healthy donor material
All the experiments involving human samples were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards, based on informed consent. Tumor tissues were obtained from a patient with HCC who underwent surgery at Fudan University Shanghai Cancer Center. The samples were enzymatically digested using collagenase I, collagenase IV, and hyaluronidase (YEASEN, Shanghai, China) for 1 hr at 37°C. Single-cell suspensions were filtered with 70-µm filter units (Coring Falcon, NY, USA), and then incubated with diabody-mp or diabody-pm at 37°C for 48 hr. The hPBMCs were isolated from healthy individuals using Ficoll-Paque (GE Healthcare, Little Chalfont, UK). Autologous T cells were enriched using RosetteSep™ human T cell enrichment cocktail kits (STEMCELL technology, Canada). The hPBMCs were activated with PHA-L (2 µg/mL) (Sigma-Aldrich, MO, USA), and T cells were activated with 100 ng/mL of anti-human CD3 and anti-human CD28 antibodies (PeproTech, NJ, USA) for 48 hr.
Homology modeling
Similarity searches were conducted for the sequences of diabody-mp and diabody-pm in the DS database, and high score sequences were selected as templates based on E-values (< 1 × 10−5). After the selected template structures were loaded and aligned, we aligned the model sequence to the template sequence. Next, homology models were built using the MODELER program and selected according to PDF Total Energy or DOPE Score. Ramachandran plot was used to evaluate the structural rationality of the model after further energy optimization of the protein. The binding modes of diabody-mp or diabody-pm to c-Met were predicted using ZDOCK, a computational protein-protein docking program. Possible binding poses were searched and refined using the CHARMm-based optimization procedure RDOCK. The most reasonable binding pose was selected according to RDOCK scores on the basis of annotated CDR loops of both light and heavy chain of diabody-mp and diabody-pm in the Kabat numbering scheme.
Immunohistochemical and immunofluorescence staining
Tumors were removed from sacrificed mice and fixed in 4% PFA for 24 hr. The samples were embedded in paraffin and sectioned into slides. Individual slides were then incubated overnight at 4°C with primary antibodies against Ki67 (ProteinTech, #27309–1-AP), VEGFA (ProteinTech, #19003–1-AP), MMP9 (CST, #13667), and CD8 (ProteinTech, #66868-1-Ig). Tumors were removed from sacrificed mice, embedded into OCT blocks, and frozen for cryostat sections (8 μm thick). Cryostat sections were fixed with 4% PFA for 15 min at room temperature and blocked with a blocking solution for 30 min at room temperature. The samples were stained with primary antibodies against CD8 (ProteinTech, #66868-1-Ig), Granzyme B (ProteinTech, #13588-1-AP), and CoraLite488-conjugated goat anti-mouse (ProteinTech, #SA00013-1) and CoraLite594-conjugated Goat anti-rabbit (ProteinTech, #SA00013-4) secondary antibodies at room temperature for 1 hr. 4′,6-Diamidino-2-phenylindole (Dalian Meilun Biotechnology, Dalian, China) was used for nuclear staining, and the EVOS M5000 microscope was used for image analysis. Data were analyzed using ImageJ software.
Statistical analysis
Statistical analyses were conducted using GraphPad Prism 7. The data are presented as means ± SD; pairwise multiple comparisons were carried out using two-way ANOVA tests, unpaired t-tests, and log-rank (Mantel-Cox) test. A p-value less than 0.05 indicated statistical significance (***p < .001; **p < .01; *p < .05; ns: not significant p > .05).
Results
Design of single-chain PD-1 specific diabodies with high and low affinities to c-Met
We developed two bispecific single-chain diabodies targeting c-Met and PD-1 for solid tumor treatment (Figure 1a-b). On one hand, only light and heavy chain variable regions of anti-c-Met and anti-PD-1 mAbs, diabodies (55 kDa) may show improved pharmacokinetics and tissue penetration. On the other hand, the diabodies have different affinities with c-Met (diabody-pm [low, KD = 32.48 nM] and diabody-mp [high, KD = 0.63 nM]) (Figure 1c). Therefore, diabody-mp with superior target retention by optimization of c-Met-associated affinity may theoretically have higher drug exposure level in tumors. This improves the anti-tumor efficacy without the damage of normal tissues caused by long-term drug retention in the periphery.
Figure 1.
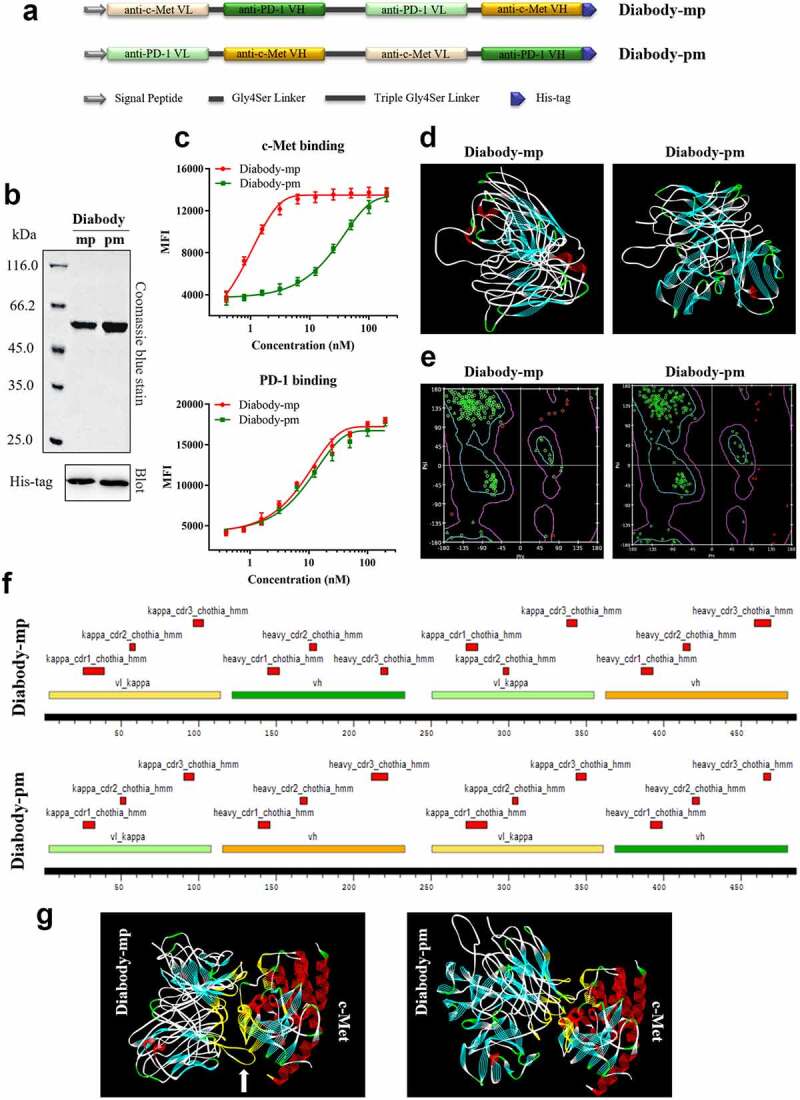
Development, homology modeling, and protein docking of diabodies with different c-Met binding affinities. a Schematics of the design of diabody-mp and diabody-pm. b Purified fractions were examined by sodium dodecyl sulfate polyacrylamide gel electrophoresis. Coomassie blue staining (top) and western blotting (bottom) were performed. c Binding titration of diabody-mp or diabody-pm with c-Met expressed on MKN45 cells and hPD-1 expressed on CHO cells. d The spatial conformation of diabody-mp and diabody-pm predicted by homology modeling were further optimized using the Discovery Studio. e Ramachandran plot was used to evaluate the structural rationality of the model. Amino acids with reasonable dihedral structure in the blue line were more than 95%. f CDR loops of light and heavy chain of diabody-mp and diabody-pm were annotated in Kabat numbering scheme and colored in red. VH and VL of anti-c-Met and anti-PD-1 frameworks were colored in yellow and green, respectively. g Putative binding modes of diabody-mp and diabody-pm to c-Met, conducted by using in silico modeling. The interaction interface is colored in yellow
Previous studies on the prediction of protein structure by homologous modeling found that diabodies28,29 could be developed into extremely flexible structures (Figure 1d) by exchanging the light and heavy chain variable regions of anti-c-Met and anti-PD-1 mAbs (Figure 1a). The Ramachandran plot showed high rationality of homology modeling of diabody-mp and diabody-pm (Figure 1e). Furthermore, we annotated the antibody sequence of the diabodies by labeling the complementary-determining region (CDR) using Discovery Studio, a computer protein simulation software (Figure 1f). Next, using the ZDOCK database to predict the binding mode of diabody-mp or diabody-pm with c-Met (PDB ID: 2RFS), we found that the binding details were quite different despite some common interaction interface, as diabody-mp has a relatively longer CDR loop (Figure 1g, white arrow). This result may shed light on the possible mechanism associated with the relatively high binding affinity of diabody-mp to c-Met.
Diabodies suppress HGF-induced proliferation, migration, and invasion of tumor cells
The c-Met tyrosine kinase receptor is activated through a ligand-dependent and/or ligand-independent mechanism.7,9 To detect the anti-c-Met effect of diabodies, we screened HGF-dependent A549 cells, HGF-independent MHCC-97 H cells with high c-Met expression, and MKN45 cells with low c-Met expression as c-Met-positive tumor cell lines for subsequent experiments (Figure 2a). Unlike anti-c-Met mAb and c-Met inhibitor JNJ-38877605, which played certain inhibitory roles in either HGF-dependent or HGF-independent settings, the diabodies significantly inhibited HGF-induced proliferation of both A549 and MHCC-97 H cells (Figure 2b). In addition, both diabodies inhibited HGF-induced migration and invasion of A549 and MHCC-97 H cells (Figure 2c-d). Together, our data illustrated that both diabody-mp and diabody-pm suppressed HGF-induced proliferation, migration, and invasion of tumor cells in vitro.
Figure 2.
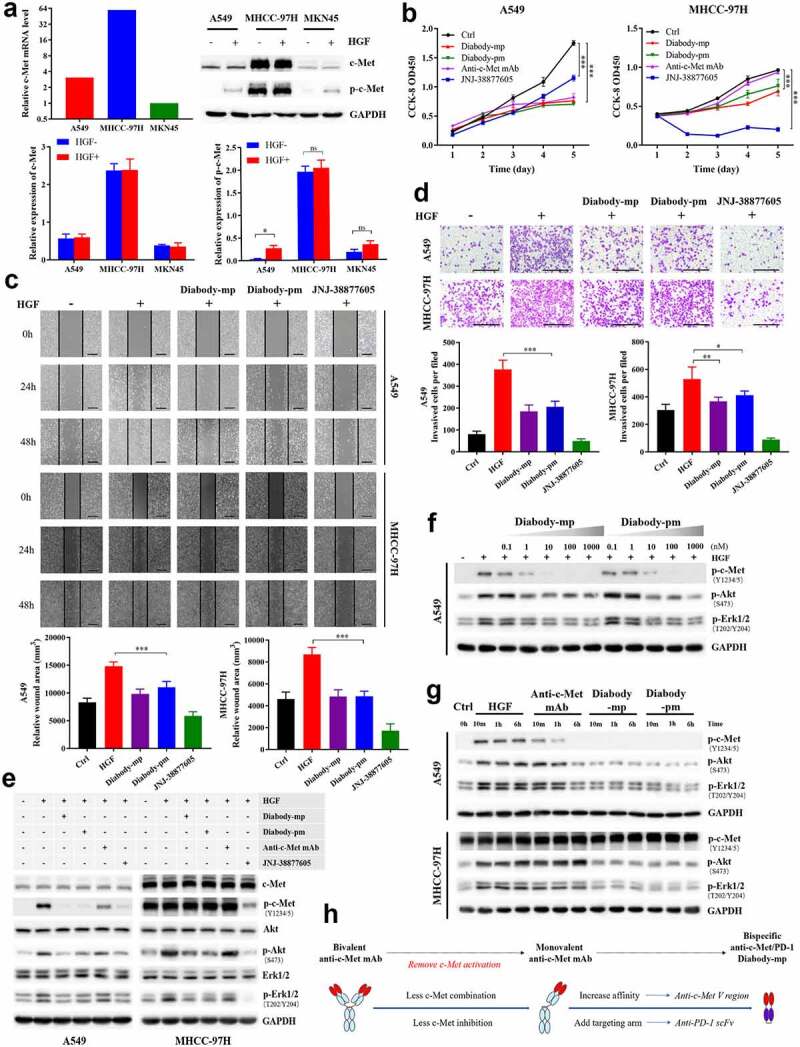
Diabodies inhibit the proliferation and movement of tumor cells and the activation of c-Met signaling by antagonizing HGF binding to c-Met. a The mRNA levels of c-Met detected by quantitative reverse transcription polymerase chain reaction in A549, MHCC-97 H, and MKN45 cells. Expression level of p-c-Met (Y1234/5) and total c-Met protein levels in A549, MHCC-97 H, and MKN45 cells stimulated with or without HGF (1 nM). b A549 or MHCC-97 H cells treated with 100 nM of diabody-mp, diabody-pm, anti-c-Met mAb, or JNJ-38877805 and stimulated with 1 nM HGF were subjected to a Cell Counting Kit-8 assay, and viable cells were measured on days 1–5. c Representative images of A549 cells and MHCC-97 H cells incubated with 100 nM of diabody-mp, diabody-pm, and JNJ-38877605 (positive control) from HGF-induced wound healing assay; scale bar, 400 µm. d Representative images of A549 and MHCC-97 H cells incubated with 100 nM of diabody-mp, diabody-pm, and JNJ-38877605 (positive control) from HGF-induced transwell assay; scale bar, 400 µm. e Western blot analysis of the activation of c-Met downstream molecules in A549 or MHCC-97 H cell lysates. Cells were treated with 100 nM of diabody-mp, diabody-pm, anti-c-Met mAb, and JNJ-38877605 for 2 hr and analyzed by immunoblotting with the indicated antibodies. HGF (1 nM) was added to stimulate cells at 37 ℃ for 15 min before sample collection. f Diabody-mp or diabody-pm inhibits HGF-induced c-Met signaling activation in a dose-dependent manner. A549 cells were incubated with the indicated concentration of diabody-mp or diabody-pm at 37 ℃ for 2 hr. g Western blot analysis of c-Met and other phosphorylated-targets in A549 and MHCC-97 H cells lysates. The cells were treated with HGF, anti-c-Met mAb, diabody-mp, and diabody-pm at indicated time point. h Design ideas of diabody-mp
Diabodies inhibit the activation of c-Met signaling by antagonizing HGF binding to c-Met
To further investigate the effect of the diabodies on cellular phosphor-c-Met, western blotting was used to quantify exogenous HGF (1 nM)-induced phospho-c-Met, and we analyzed the PI3K/Akt and MAPK signaling cascades triggered by the activation of c-Met tyrosine kinase in the lysates of A549 and MHCC-97 H cells. Both diabodies and JNJ-38877605 reduced HGF-induced c-Met phosphorylation in A549 cells, but anti-c-Met mAb was less effective in blocking HGF-induced c-Met phosphorylation (Figure 2e, left). Moreover, both diabodies showed dose-dependent inhibition of the c-Met signaling pathway in A549 cells (Figure 2f). In contrast to the HGF-dependent A549 cells, MHCC-97 H cells expressed high levels of constitutively phosphorylated c-Met. Although the inhibitory effect of the diabodies on phospho-c-Met was not significant, the diabodies similarly inhibited HGF-induced elevation of phospho-Akt and phospho-Erk (Figure 2e, right). Furthermore, treatment with the bivalent anti-c-Met mAb mimicked the activity of native HGF, which induced c-Met phosphorylation in both ligand-dependent A549 cells and ligand-independent MHCC-97 H cells (Figure 2g). Remarkably, the diabodies did not induce c-Met phosphorylation in the absence of HGF, which was consistent with our design ideas (Figure 2g-h). These results suggested that both diabodies inhibited the activation of c-Met signaling by antagonizing HGF binding to c-Met.
Diabodies promote T cell activation by blocking PD-1/PD-L1 axis
The binding of the TCR-CD3 complex on the effector T cells with the MHC-I complex on the tumor cells leads to the activation of effector cells.30 However, the interaction of PD-1 on the effector T cells with PD-L1 on the tumor cells causes T cell depletion and tumor immune escape.31 Herein, we engineered the Jurkat cell line that stably express or knockout PD-1 with luciferase expression regulated by the binding of the transcription factor NFAT with the promoter of IL-2. Furthermore, we measured the bioactivity of the diabodies by conducting a reporter gene assay with PHA-L-activated Jurkat cells expressing PD-1 (effector cells) and IFN-γ-induced tumor cells expressing PD-L1 (target cells).32 Both diabodies increased luciferase expression in Jurkat-PD-1+ cells, but not in Jurkat-PD-1− cells (Figure 3a), suggesting that they could promote target-specific activation of Jurkat cells by blocking PD-1/PD-L1 interaction.
Figure 3.
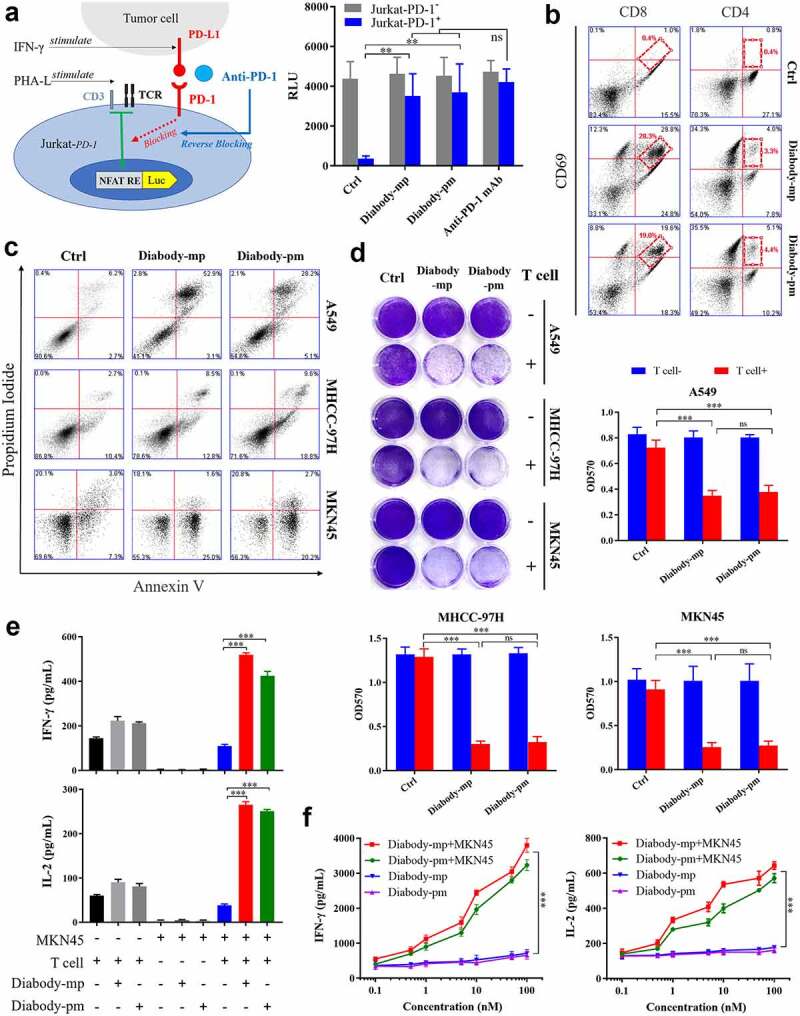
Diabodies block the PD-1 pathway, mediating the cellular cytotoxicity of c-Met-positive tumor cells through T cell engagement. a Schematic diagram of a reporter gene assay to measure the anti-PD-1 activity of diabody-mp or diabody-pm (left). The levels of luciferase activity were determined (right). b FACS quantification of CD69+-activated CD8+ T cells and tumor-infiltrating CD4+ effector T cells obtained from a patient with HCCs and incubated with 100 nM of diabody-mp, diabody-pm, and anti-PD-1 mAb for 48 hr after digestion with enzymes. c Apoptosis detection in tumor cells co-cultured with activated human T cells and treated with 100 nM of diabody-mp or diabody-pm for 48 hr at an E:T ratio of 10:1. The percent of positive apoptotic cells is shown. d A549, MHCC-97 H, or MKN45 cells co-cultured with activated human T cells at an E:T ratio of 10:1 upon the addition of 100 nM diabody-mp or diabody-pm for 48 hr, next subjected to crystal violet staining. e IFN-γ and IL-2 released in the presence of MKN45 cells and T cells at an E:T ratio of 10:1 upon the addition of 100 nM diabody-mp or diabody-pm, as measured by enzyme-linked immunosorbent assay (ELISA) kit after 48 hr. f IFN-γ and IL-2 released in the presence or absence of MKN45 cells at an E:T ratio of 64:1 upon the addition of diabody-mp or diabody-pm, as measured by ELISA kit after 48 hr
To determine the ability of the diabodies to activate T cells, a single-cell suspension was prepared from the fresh tumor tissue of a patient with HCC using the enzyme digestion method,33 and the suspension was incubated with the diabodies. An increase in the expression level of CD69 (the lymphocyte activating marker) was observed in tumor-infiltrating CD8+ and CD4+ T effector cells in the diabody-treated groups compared with that in the control group (Figure 3b), indicating that diabodies could promote the activation of CD8+ and CD4+ T cells in the tumor microenvironment. Together, these results demonstrated that diabody-targeted PD-1 could promote the activation of T cells and redirect the human immune system.
Diabodies mediate cellular cytotoxicity of HGF-dependent and HGF-independent tumor cells through T cell engagement
To evaluate the broad therapeutic potency of the diabodies in HGF-dependent and HGF-independent settings, we examined the non-small cell lung carcinoma cell line A549, HCC cell line MHCC-97 H, and gastric carcinoma cell line MKN45 as c-Met-expressing targets in an in vitro apoptosis assay. Human T cells as effectors were isolated by negative selection from the peripheral blood of healthy donors. PI and Annexin V FACS analyses revealed that both diabodies promoted the apoptosis of tumor cells with diverse c-Met aberration through T cell engagement (Figure 3c). Moreover, the diabodies induced human T cell-mediated killing (Figure 3d), which was likely due to the increased expression of cytokines IFN-γ and IL-2 (Figure 3e). Moreover, the diabodies induced dose-dependent secretion of IFN-γ and IL-2 after co-culturing with MKN45 cells (Figure 3f), suggesting that the diabody-induced activation of T cells can only occur in tumor cells. This also indicates the safety of the diabodies in normal cells. Accordingly, our results revealed that both diabodies induced T cell-mediated cellular cytotoxicity against c-Met-positive tumor by improving cytokine secretion.
Diabody-mp inhibits the growth of tumors with high and low expression of c-Met in vivo
No significant difference in anti-tumor activity was observed between both diabodies. Subsequently, to examine the therapeutic efficacy of two diabodies in NOD-SCID mice engrafted with human peripheral blood mononuclear cells (hPBMCs), we established transplant solid tumor models with low c-Met expression in MKN45 cells and high c-Met expression in MHCC-97 H cells. All the mice were injected once daily for 30 consecutive days with 1 mg/kg of diabody-mp or diabody-pm. Although the two distinct bispecific diabodies inhibited tumor growth in vivo, diabody-pm provided limited therapeutic benefits for low c-Met expression in the MKN45 tumor model, whereas diabody-mp therapy substantially decreased tumor burden in both high and low expression of c-Met tumors model. Moreover, all tumors progressed in the control mice treated with the vehicle (Figure 4a-c). In addition, diabody-mp suppressed spontaneous liver metastasis in the MKN45 tumor model, compared with the control group (Figure 4d). To explore the potential mechanisms associated with the anti-tumor activity of the diabodies, we assessed the activation of lymphocytes in an MKN45 tumor model and observed the upregulation of CD69 in CD8+ and CD4+ T cells. It is worth noting that diabody-mp induced higher CD69 expression in CD8+ T cells than diabody-pm (Figure 4e). Moreover, we identified that diabody-mp inhibited tumor proliferation, metastasis, and angiogenesis by inhibiting the expression of Ki67, MMP9, and VEGFA in tumor tissues (Figure 4f). However, diabody-pm did not significantly inhibit the expression of MMP9 and VEGFA (Figure 4f). In general, our observations in tumors models with both high and low expression of c-Met suggested that dual c-Met and PD-1 blockade by diabodies, especially diabody-mp, showed marked anti-tumor activity in vivo.
Figure 4.
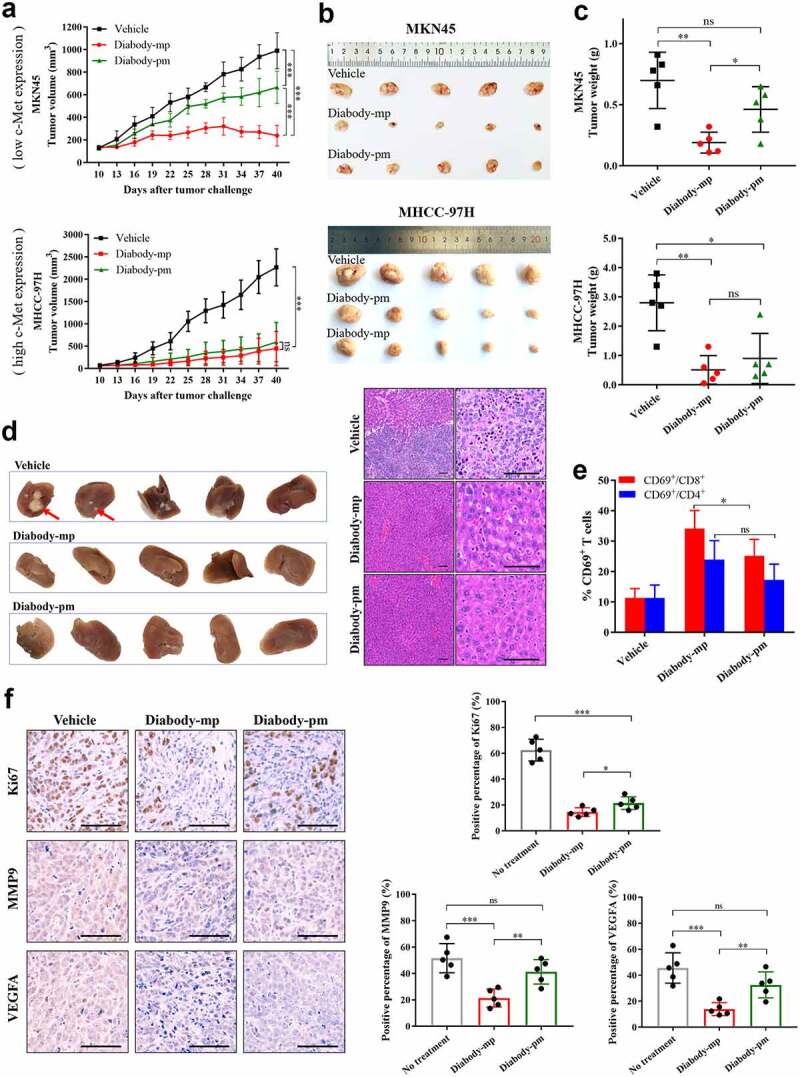
Diabody-mp inhibits the growth of tumor with high and low expression of c-Met in vivo. a Low c-Met-expressing MKN45 or high c-Met-expressing MHCC-97 H cells were subcutaneously transplanted into NOD-SCID mice, and the immune system was reconstructed with hPBMCs (intraperitoneal (i.p.) injection). Vehicle, diabody-mp (1 mg/kg), and diabody-pm (1 mg/kg) were administered once daily by i.p. injection starting from day 10 for a month. The tumor growth was monitored at the indicated times point. n = 5 mice per group. b The mice were sacrificed on day 40 and the tumors were removed and photographed. c Tumor tissues were weighed and plotted. d Liver removed from sacrificed mice bearing the MKN45 tumors were photographed (above), sectioned, and stained with hematoxylin and eosin (bottom); scale bar, 100 µm. e Percentage of activated CD4+ and CD8+ T cells from isolated TILs in MKN45 model based on CD69 expression. f Representative images of immunohistochemical staining of Ki67, MMP9, and VEGFA performed on MKN45 transplanted tumors treated as indicated; scale bar, 100 µm. The proportions of positive tumor cells were analyzed using ImageJ software
Diabody-mp exhibits stronger tumor inhibition than other structural antibodies
Furthermore, we evaluated if diabody-mp showed preferable therapeutic efficacy in vivo, compared with its IgG-like bispecific antibody counterparts (we focused on the scFv-Fc format, which is one of the most commonly used formats of bsAbs), the combination of its two antibodies functional region (anti-PD-1 scFv combined with anti-c-Met scFv), and anti-PD-1 mAb. The result showed diabody-mp exhibited stronger tumor growth inhibition in mice with subcutaneous MKN45 tumor transplants (Figure 5a). The diabody-mp-treated tumor had greater infiltration of CD8+ cytotoxic T lymphocytes than the other treated tumors (Figure 5b). In addition, diabody-mp increased the secretion of cytokine IFN-γ in the tumor microenvironment, but the level of IFN-γ in the peripheral blood was almost undetectable (Figure 5c), suggesting that diabody-mp activates the lymphocytes mainly in the tumor microenvironment. This indicates the safety and efficacy of diabody-mp and that it could effectively avoid cytokine storm generated by the activation of the systemic immune cells.
Figure 5.
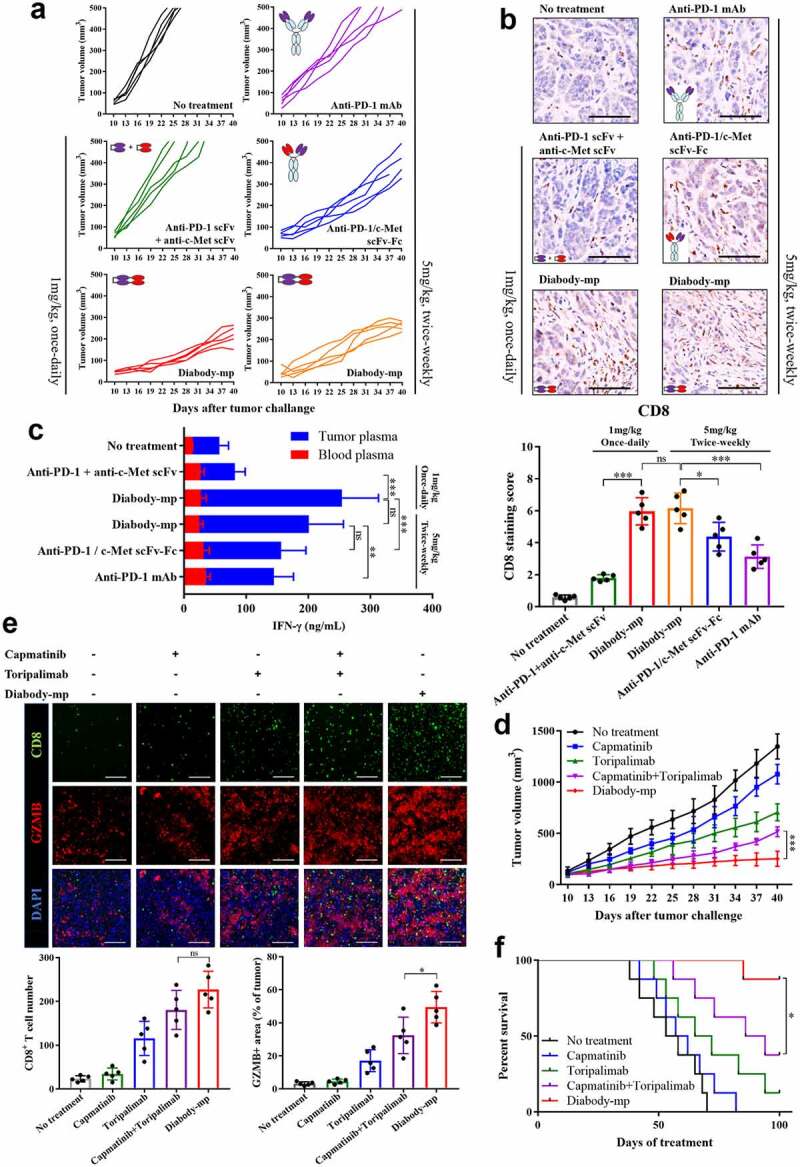
Diabody-mp exhibits improved tumor inhibition compared with other structural antibodies and single or combined c-Met and PD-1 blockade therapy. a Individual tumor growth curve. BALB/c mice were inoculated with MKN45 cells and treated as indicated for a month from day 10. The tumor growth was monitored at the indicated times point. n = 5 mice per group. b Representative images of immunohistochemical staining of CD8+ performed on MKN45 tumors treated as indicated; scale bar, 100 µm. c Quantification of IFN-γ secreted in the blood and tumor microenvironment of mice bearing the MKN45 tumors and treated as indicated. d Tumor growth curves of MKN45 gastric carcinoma in BALB/c mice treated with diabody-mp (1 mg/kg, once-daily, i.p.), capmatinib (10 mg/kg, once-daily, i.g.), toripalimab (10 mg/kg, once-weekly, i.v.) or capmatinib combined with toripalimab for a month. n = 8 mice per group. e Immunostaining of CD8 (CTL marker) and granzyme B (activity of T cell) in the MKN45 tumor sections; scale bar, 100 µm. f Mice survival curves were generated from MKN45-bearing mice. n = 8 mice per group
Diabody-mp shows superior anti-tumor efficacy than single or combined c-Met and PD-1 blockade therapy
We further compared the anti-tumor effects of single or combined c-Met and PD-1 blockade with diabody-mp in mouse MKN45 tumor models. The results showed that diabody-mp, but not single or combined capmatinib and toripalimab therapy, had the strongest tumor-inhibition effect (Figure 5d). We analyzed the tumor microenvironment by immunofluorescent detection, providing evidence that combined c-Met and PD-1 inhibition by diabody-mp exhibited higher therapeutic benefits than single or combined agents, possibly due to increased activation of CD8+ T cells (Figure 5e). Although the effect of diabody-mp on CD8+ T cell infiltration was slightly higher than that of combination therapy, there is no significant difference between two groups (Figure 5e). Moreover, diabody-mp prominently extended the survival of MKN45-bearing mice (Figure 5f), whereas combination with capmatinib and toripalimab had reduced effects, and the single agent did not show a considerable effect on the survival of the mice. Generally, these data demonstrated the superior anti-tumor activity of diabody-mp over that of single or combined c-Met and PD-1 blockade therapy in subcutaneous MKN45 gastric carcinoma mice models.
Discussion
Approximately 20–60% of solid tumors are poorly recognized by CD8+ T cells due to the deficiency of MHC-I, which is one of the principal reasons for tumor immune escape.34,35 Bispecific diabodies could overcome MHC restriction and recruit T cells to eliminate tumors. Moreover, unlike monovalent and bivalent anti-c-Met mAbs, diabodies do not activate c-Met receptor tyrosine kinase but induce the apoptosis of different c-Met-positive tumor cells through T cell engagement. Diabody-mp improves the anti-c-Met affinity by optimizing the c-Met variable region. The high c-Met binding affinity of diabody-mp may play a part in increasing the level of drug exposure in solid tumors by improving tissue permeability and tumor targeting.
Multiple factors determine the affinity and efficacy of antibodies, including the conformational flexibility of binding modules as well as the accessibility of epitopes.36 Early research proved that single mouse variable (V) domains could potentially target cryptic epitopes.37 Therefore, it is desirable to engineer anti-c-Met variable (V) domains and anti-PD-1 scFv fragment into diabody-mp with improved binding affinity to c-Met. The diabody provides a new c-Met-binding specificity, which is hardly formed by intact antibodies because their limited diversity of CDR loop lengths restricts antibody-antigen interaction.38,39
Inappropriate activation of cells expressing the Fc receptor could produce a massive level of cytokines and the associated toxic effects, which are undesirable for some antibodies, particularly bispecific antibodies that engage effector cells. Smaller recombinant antibody fragments without the Fc region as reliable alternatives retain the targeting specificity but possess other superior properties, such as improved avidity and tissue penetration. Bispecific diabodies, as intermediate-sized multivalent molecules (55 kDa) without the Fc region, are ideal tumor-targeting reagents, which could provide rapid tissue penetration and high target retention. Nevertheless, diabodies have short half-lives as a result of the defect in the Fc region, which imparts extended circulation. To maintain the effective concentration in vivo and ensure anti-tumor efficacy, diabodies need to be administered more frequently than traditional antibody drugs with long half-lives.
However, increasing clinical data have shown that the efficacy of immune checkpoint inhibitors with a half-life of only a few days is consistent with that of similar drugs with a half-life of several weeks. Target occupancy rate has been used as a pharmacokinetic biomarker for many therapeutic antibodies, one of which is the PD-1 inhibitor.40 There is no significant relationship between the half-life of anti-PD-1 antibody in the plasma and PD-1 occupancy on the T cell surface. More efficient and persistent procedures that can block PD-1/PD-L1 in vivo may provide improved therapeutic effects.41 In other words, as long as anti-PD-1 antibody occupies enough PD-1 on the T cell surface at the beginning of treatment, the anti-tumor effect is guaranteed regardless of the half-life of the PD-1 inhibitor in the plasma. Research has shown that most anti-PD-1 antibodies maintain a PD-1 occupancy rate of more than 70% for several weeks after a single injection;42,43 hence, their anti-tumor efficacy would not be affected by the half-life and frequency of medication. As shown in Figure 5a-b, there was almost no significant difference in the inhibition of tumor growth in mice administered diabody-mp once daily or twice weekly. Further related comparison and analysis could be carried out in the follow-up study.
Furthermore, immune checkpoint inhibitors with a long half-life may produce immune-related adverse reactions. It was recently reported that immune checkpoint inhibitors with a half-life of more than 15–20 days and a target binding rate of more than 70% after several months of treatment caused a series of severe side effects in clinical trials.44 Although the mechanism has not been fully elucidated, some researchers speculated that the severe side effects may be associated with the long half-life of such drugs, suggesting that diabodies with a short half-life might decrease the risk of adverse reactions. In mouse models, no obvious difference was observed between the diabody-treated groups and the control group in terms of activity, weight, and eating behavior. Moreover, hematoxylin and eosin staining of the liver and lung sections from the mice in the diabody-treated groups showed normal histological morphology without cell denaturation and inflammatory cell infiltration, suggesting no obvious organ toxicity. In addition, as shown in Figure 5c, diabody-mp increased the secretion of IFN-γ in the tumor microenvironment, but the level of IFN-γ in the peripheral blood was almost undetectable. This demonstrated that by activating lymphocytes mainly in the tumor microenvironment, diabody-mp could effectively avoid cytokine storm generated by the activation of the systemic immune cells.
Taken together, our results indicate that bispecific diabody-mp exhibited improved tumor growth inhibition, which greatly enhanced the survival of tumor-bearing mice compared with other structural antibodies and single or combined c-Met and PD-1 blockade therapy. The results of the in vitro studies indicate that there was no significant difference in the anti-tumor activities of the two diabodies. However, in vivo studies in mice models revealed that diabody-mp with higher c-Met binding affinity exhibited stronger anti-tumor activity than diabody-pm with lower c-Met binding affinity, suggesting the potential of diabody-mp in the treatment of c-Met-positive solid tumors.
Supplementary Material
Acknowledgments
We would like to thank Zhenhai Lin from Fudan University Shanghai Cancer Center for help in collection of liver cancer tissue. We would like to thank Editage (www.editage.cn) for English language editing.
Funding Statement
This work was supported in part by Science and Technology Commission of Shanghai Municipality 14431900100, in part by the National Key Research Project Bio-safety Key Technology Development Program 2016YFC1201501, in part by the National Natural Science Foundation of China, No. 31671228 and in part by the National Natural Science Foundation of China, No.81974314;National Natural Science Foundation of China [31671228];National Key Research Project Bio-safety Key Technology Development Program [2016YFC1201501];
Disclosure statement
No potential conflicts of interest were disclosed.
Author contributions
Min Yu, Huijie Wang, and Qingyun Yuan designed the study. Qingyun Yuan prepared all the materials and designed all the experiments. Qingyun Yuan, Weihua Hou, and Yuxiong Wang performed expression and purification of the antibodies. Qingyun Yuan and Xingxing Yuan assisted with all in vitro experiments. Qingyun Yuan and Qiaoyan Liang contributed to the in vivo experiments. Qingyun Yuan performed data collection, analysis, and interpretation. Qingyun Yuan wrote the first draft of the manuscript. All authors revised and approved the manuscript. Min Yu supervised the entire project.
Ethics approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards. All procedures performed in studies involving animals were in accordance with the ethical standards of the department of laboratory animal science of Fudan university and permitted on March 6, 2019.
Consent to participate
Informed consent was obtained from all individual participants included in the study.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website
References
- 1.Gubin MM, Zhang X, Schuster H, Caron E, Ward JP, Noguchi T, Ivanova Y, Hundal J, Arthur CD, Krebber W-J, et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens[J]. Nature. 2014;515(7528):577–14. doi: 10.1038/nature13988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Topalian SL, Drake CG, Pardoll DM.. Immune checkpoint blockade: a common denominator approach to cancer therapy[J]. Cancer Cell. 2015;27(4):450–461. doi: 10.1016/j.ccell.2015.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Taube JM, Anders RA, Young GD, Xu H, Sharma R, McMiller TL, Chen S, Klein AP, Pardoll DM, Topalian SL, et al. Colocalization of inflammatory response with B7-h1 expression in human melanocytic lesions supports an adaptive resistance mechanism of immune escape[J]. Sci Transl Med. 2012;4(127):127–137. doi: 10.1126/scitranslmed.3003689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Barber DL, Wherry EJ, Masopust D, Zhu B, Allison JP, Sharpe AH, Freeman GJ, Ahmed R. Restoring function in exhausted CD8 T cells during chronic viral infection[J]. Nature. 2006;439(7077):682–687. doi: 10.1038/nature04444. [DOI] [PubMed] [Google Scholar]
- 5.Ribas A, Wolchok JD. Cancer immunotherapy using checkpoint blockade[J]. Science. 2018;359(6382):1350–1355. doi: 10.1126/science.aar4060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Drew MP. The blockade of immune checkpoints in cancer immunotherapy[J]. Nat Rev Cancer. 2012;12(4):252–264. doi: 10.1038/nrc3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Trusolino L, Bertotti A, Comoglio PM. MET signalling: principles and functions in development, organ regeneration and cancer[J]. Nat Rev Mol Cell Biol. 2010;11(12):834–848. doi: 10.1038/nrm3012. [DOI] [PubMed] [Google Scholar]
- 8.Matsumoto K, Nakamura T. Hepatocyte growth factor and the Met system as a mediator of tumor–stromal interactions[J]. Int J Cancer. 2006;119(3):477–483. doi: 10.1002/ijc.21808. [DOI] [PubMed] [Google Scholar]
- 9.Tolbert WD, Daugherty-Holtrop J, Gherardi E, Vande Woude G, Xu HE. Structural basis for agonism and antagonism of hepatocyte growth factor[J]. Proc Natl Acad Sci U S A. 2010;107(30):13264–13269. doi: 10.1073/pnas.1005183107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gherardi E, Birchmeier W, Birchmeier C, Woude GV. Targeting MET in cancer: rationale and progress[J]. Nat Rev Cancer. 2012;12(2):89–103. doi: 10.1038/nrc3205. [DOI] [PubMed] [Google Scholar]
- 11.Drilon A, Clark JW, Weiss J, Ou SHI, Camidge DR, Solomon BJ, Otterson GA, Villaruz LC, Riely GJ, Heist RS, et al. Antitumor activity of crizotinib in lung cancers harboring a MET exon 14 alteration[J]. Nat Med. 2020;26(1):47–51. doi: 10.1038/s41591-019-0716-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Katayama R, Aoyama A, Yamori T, Qi J, Oh-hara T, Song Y, Engelman JA, Fujita N. Cytotoxic activity of tivantinib (ARQ 197) is not due solely to c-MET inhibition.[J]. Cancer Res. 2013;73(10):3087–3096. doi: 10.1158/0008-5472.CAN-12-3256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pennacchietti S, Cazzanti M, Bertotti A, Rideout WM, Han M, Gyuris J, Perera T, Comoglio PM, Trusolino L, Michieli P, et al. Microenvironment-derived HGF overcomes genetically determined sensitivity to anti-MET drugs.[J]. Cancer Res. 2014;74(22):6598–6609. doi: 10.1158/0008-5472.CAN-14-0761. [DOI] [PubMed] [Google Scholar]
- 14.Prat M, Crepaldi T, Pennacchietti S, Bussolino F, Comoglio PM. Agonistic monoclonal antibodies against the Met receptor dissect the biological responses to HGF.[J]. J Cell Sci. 1998;111:237–247. [DOI] [PubMed] [Google Scholar]
- 15.Pacchiana G, Chiriaco C, Stella MC, Petronzelli F, De Santis R, Galluzzo M, Carminati P, Comoglio PM, Michieli P, Vigna E, et al. Monovalency unleashes the full therapeutic potential of the DN-30 anti-Met antibody.[J]. J Biol Chem. 2010;285(46):36149–36157. doi: 10.1074/jbc.M110.134031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Benkhoucha M, Santiago-Raber M, Schneiter G, Chofflon M, Funakoshi H, Nakamura T, Lalive PH. Hepatocyte growth factor inhibits CNS autoimmunity by inducing tolerogenic dendritic cells and CD25+Foxp3+ regulatory T cells.[J]. Proc Natl Acad Sci U S A. 2010;107(14):6424–6429. doi: 10.1073/pnas.0912437107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Reck M, Rodríguez-Abreu D, Robinson AG, Hui R, Csőszi T, Fülöp A, Gottfried M, Peled N, Tafreshi A, Cuffe S, et al. Updated analysis of KEYNOTE-024: pembrolizumab versus platinum-based chemotherapy for advanced non-small-cell lung cancer with PD-L1 tumor proportion score of 50% or greater.[J]. J Clin Oncol. 2019;37(7):537–546. doi: 10.1200/JCO.18.00149. [DOI] [PubMed] [Google Scholar]
- 18.Sabari JK, Leonardi GC, Shu CA, Umeton R, Montecalvo J, Ni A, Chen R, Dienstag J, Mrad C, Bergagnini I, et al. PD-L1 expression, tumor mutational burden, and response to immunotherapy in patients with MET exon 14 altered lung cancers.[J]. Ann Oncol. 2018;29(10):2085–2091. doi: 10.1093/annonc/mdy334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li H, Li C, Li X, Ding Q, Guo L, Liu S, Liu C, Lai -C-C, Hsu J-M, Dong Q, et al. MET inhibitors promote liver tumor evasion of the immune response by stabilizing PDL1[J]. Gastroenterology. 2019;156(6):1849–1861. doi: 10.1053/j.gastro.2019.01.252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kudo M. Immuno-oncology therapy for hepatocellular carcinoma: current status and ongoing trials.[J]. Liver Cancer. 2019;8(4):221–238. doi: 10.1159/000501501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.U.S. food and drug administration accepts for priority review applications for OPDIVO® (nivolumab) in Combination with CABOMETYX® (cabozantinib) in advanced renal cell carcinoma[EB/OL]. https://www.businesswire.com/news/home/20201019005198/en.
- 22.Dudley ME, Rosenberg SA. Adoptive-cell-transfer therapy for the treatment of patients with cancer[J]. Nat Rev Cancer. 2003;3(9):666–675. doi: 10.1038/nrc1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li H, Er Saw P, Song E. Challenges and strategies for next-generation bispecific antibody-based antitumor therapeutics[J]. Cell Mol Immunol. 2020;17(5):451–461. doi: 10.1038/s41423-020-0417-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Topp MS, Gökbuget N, Zugmaier G, Klappers P, Stelljes M, Neumann S, Viardot A, Marks R, Diedrich H, Faul C, et al. Phase II trial of the anti-CD19 bispecific T cell-engager blinatumomab shows hematologic and molecular remissions in patients with relapsed or refractory B-precursor acute lymphoblastic leukemia.[J]. J Clin Oncol. 2014;32(36):4134–4140. doi: 10.1200/JCO.2014.56.3247. [DOI] [PubMed] [Google Scholar]
- 25.Baeuerle PA, Kufer P, Bargou R. BiTE: teaching antibodies to engage T-cells for cancer therapy.[J]. Curr Opin Mol Ther. 2009;11:22–30. [PubMed] [Google Scholar]
- 26.Fernandes RA, Su L, Nishiga Y, Ren J, Bhuiyan AM, Cheng N, Kuo CJ, Picton LK, Ohtsuki S, Majzner RG, et al. Immune receptor inhibition through enforced phosphatase recruitment[J]. Nature. 2020;586(7831):779–784. doi: 10.1038/s41586-020-2851-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR-Cas9 system.[J]. Nat Protoc. 2013;8(11):2281–2308. doi: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dolezal O, De Gori R, Walter M, Doughty L, Hattarki M, Hudson PJ, Kortt AA. Single-chain Fv multimers of the anti-neuraminidase antibody NC10: the residue at position 15 in the V(L) domain of the scFv-0 (V(L)-V(H)) molecule is primarily responsible for formation of a tetramer-trimer equilibrium.[J]. Protein Eng. 2003;16(1):47–56. doi: 10.1093/proeng/gzg006. [DOI] [PubMed] [Google Scholar]
- 29.Casey J L, Napier M P, King D J, et al . Tumour targeting of humanised cross-linked divalent-Fab’ antibody fragments: a clinical phase I/II study.[J]. Br J Cancer. 2002;86(9):1401–1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Smith-Garvin JE, Koretzky GA, Jordan MS. T Cell Activation[J]. Annu Rev Immunol. 2009;27:591–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zou W, Wolchok JD, Chen L. PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: mechanisms, response biomarkers, and combinations[J]. Sci Transl Med. 2016;8(328):324–328. doi: 10.1126/scitranslmed.aad7118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bellucci R, Martin A, Bommarito D, Wang K, Hansen SH, Freeman GJ, Ritz J. Interferon-γ-induced activation of JAK1 and JAK2 suppresses tumor cell susceptibility to NK cells through upregulation of PD-L1 expression.[J]. Oncoimmunology. 2015;4(6):e1008824. doi: 10.1080/2162402X.2015.1008824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rosenberg SA, Anderson WF, Blaese M, Hwu P, Yannelli JR, Yang JC, Topalian SL, Schwartzentruber DJ, Weber JS, Ettinghausen SE, et al. The development of gene therapy for the treatment of cancer[J]. Ann Surg. 1993;218(4):455–463. doi: 10.1097/00000658-199310000-00006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Seliger B, Cabrera T, Garrido F, Ferrone S. HLA class I antigen abnormalities and immune escape by malignant cells[J]. Semin Cancer Biol. 2002;12(1):3–13. doi: 10.1006/scbi.2001.0404. [DOI] [PubMed] [Google Scholar]
- 35.Garrido F, Aptsiauri N, Doorduijn EM, Garcia Lora AM, Van Hall T. The urgent need to recover MHC class I in cancers for effective immunotherapy[J]. Curr Opin Immunol. 2016;39:44–51. doi: 10.1016/j.coi.2015.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Holliger P, Hudson PJ. Engineered antibody fragments and the rise of single domains.[J]. Nat Biotechnol. 2005;23(9):1126–1136. doi: 10.1038/nbt1142. [DOI] [PubMed] [Google Scholar]
- 37.Ward ES, Güssow D, Griffiths AD, Jones PT, Winter G. Binding activities of a repertoire of single immunoglobulin variable domains secreted from Escherichia coli.[J]. Nature. 1989;341(6242):544–546. doi: 10.1038/341544a0. [DOI] [PubMed] [Google Scholar]
- 38.Cardoso RM, Zwick MB, Stanfield RL, Kunert R, Binley JM, Katinger H, Burton DR, Wilson IA. Broadly neutralizing anti-HIV antibody 4E10 recognizes a helical conformation of a highly conserved fusion-associated motif in gp41.[J]. Immunity. 2005;22(2):163–173. doi: 10.1016/j.immuni.2004.12.011. [DOI] [PubMed] [Google Scholar]
- 39.Hudson PJ, Souriau C. Engineered antibodies[J]. Nat Med. 2003;9(1):129–134. doi: 10.1038/nm0103-129. [DOI] [PubMed] [Google Scholar]
- 40.Liang M, Schwickart M, Schneider AK, Vainshtein I, Del Nagro C, Standifer N, Roskos LK. Receptor occupancy assessment by flow cytometry as a pharmacodynamic biomarker in biopharmaceutical development.[J]. Cytometry B Clin Cytom. 2016;90(2):117–127. doi: 10.1002/cyto.b.21259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Iwai Y, Ishida M, Tanaka Y, Okazaki T, Honjo T, Minato N. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade[J]. Proc Natl Acad Sci U S A. 2002;99(19):12293–12297. doi: 10.1073/pnas.192461099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Brahmer JR, Drake CG, Wollner I, Powderly JD, Picus J, Sharfman WH, Stankevich E, Pons A, Salay TM, McMiller TL, et al. Phase I study of single-agent anti-programmed death-1 (MDX-1106) in refractory solid tumors: safety, clinical activity, pharmacodynamics, and immunologic correlates.[J]. J Clin Oncol. 2010;28(19):3167–3175. doi: 10.1200/JCO.2009.26.7609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mo H, Huang J, Xu J, Chen X, Wu D, Qu D, Wang X, Lan B, Wang X, Xu J, et al. Safety, anti-tumour activity, and pharmacokinetics of fixed-dose SHR-1210, an anti-PD-1 antibody in advanced solid tumours: a dose-escalation, phase 1 study[J]. Br J Cancer. 2018;119(5):538–545. doi: 10.1038/s41416-018-0100-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sasikumar P, Shrimali R, Adurthi S, Ramachandra R, Satyam L, Dhudashiya A, Samiulla D, Sunilkumar KB, Ramachandra M. A novel peptide therapeutic targeting PD1 immune checkpoint with equipotent antagonism of both ligands and a potential for better management of immune-related adverse events[J]. J ImmunoTher Cancer. 2013;1(1suppl):O24. doi: 10.1186/2051-1426-1-S1-O24. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.