

Abstract
Non-enveloped viruses must create a transient membrane lesion to initiate infection by transferring their genomes into a target cell1. Rotaviruses offer a particularly favorable opportunity to visualize the mechanism for subviral particle delivery, the principal function of their outer-layer protein, VP42–4. We show here by electron cryomicroscopy (cryo-EM) that VP4, activated by cleavage to VP8* and VP5*, rearranges on the virion surface from an “upright” to a “reversed” conformation. The reversed structure projects an initially buried, “foot” domain outward into the host cell membrane to which the virion has attached. Analysis of cryo-tomograms of virus particles entering cells is consistent with this picture. We have stabilized with a disulfide mutant a likely intermediate in this transition. The results define molecular mechanisms for the first steps in viral membrane penetration and suggest similarities with mechanisms postulated for other viruses.
Rotaviruses and other non-enveloped, mammalian, double-strand RNA (dsRNA) viruses have evolved a mechanism to deliver a large (~700 Å diameter) macromolecular complex into the cytosol of a target cell. These viruses package segmented genomes in icosahedral capsids with multiple protein layers (Fig. 1a)2,3. The role of the outer layer of an infectious rotavirus particle (a “triple-layer” particle or TLP), is to insert the “double-layer” particle (DLP) that it surrounds into the cell to which the virus has attached. The DLP does not dissociate further; the polymerase and capping activities it contains instead transcribe the eleven genome segments, cap the mRNA products, and extrude the completed transcripts into the cytosol (Fig. 1b)2.
Fig. 1 |. Rotavirus entry and cryo-EM structures of the penetration protein in upright and reversed conformations.
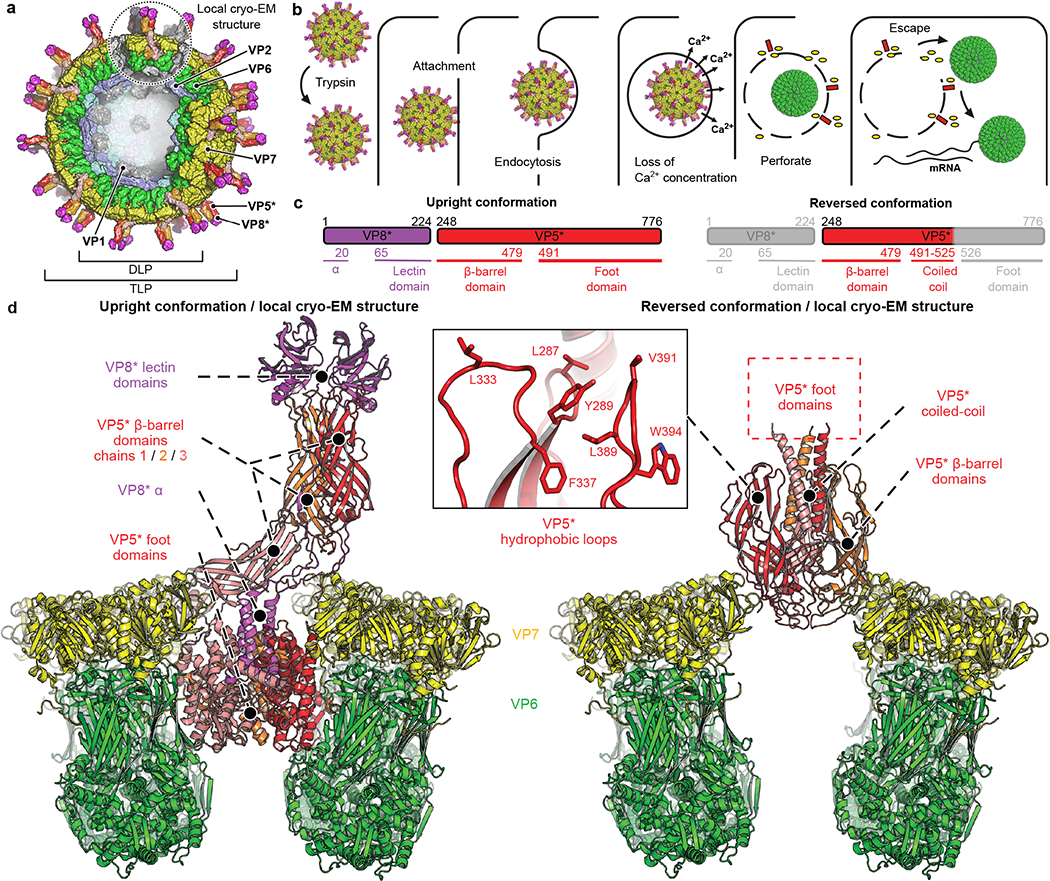
a, Overall structure of a rotavirus TLP. b, Schematic virus entry pathway derived from live-cell imaging experiments8,16,20. c, Domain organization of the VP8* and VP5* proteins in the upright (left) and reversed (right) conformations. Domains are labeled beneath the primary structure. Residue numbers indicate termini and domain boundaries; α, N-terminal α-helical segment of VP8*; regions in grey, not detected (disordered) in the reversed-conformation structure. d, Atomic models of upright (left) and reversed (right) conformations of VP5* (and VP8*) on the virion surface, from our cryo-EM reconstructions of wild-type recoated RRV TLPs. VP5*, red, orange and salmon; VP6, green; VP7, yellow; VP8*, magenta. Some VP6 and VP7 subunits omitted for clarity.
Previous studies have defined the functions of the two TLP outer-layer proteins, VP4 and VP7, in DLP delivery4–9. Infection requires cleavage of VP4, both in cell culture and in vivo, into an N-terminal fragment, VP8*, and a C-terminal fragment, VP5*. The former attaches the virus to its cellular receptor (in many cases, a glycolipid)10–14; the latter perforates the lipid bilayer of the vesicular membrane that surrounds it after initial uptake by the target cell (Fig. 1b). The function of VP7, a Ca2+-stabilized trimer15, is to anchor VP4 onto the virion surface and to respond to loss of Ca2+ at an early stage in infection by dissociating and releasing VP8* and VP5* from the DLP16.
On a virion, the VP4 “spike” is a trimer with an unusual, asymmetric conformation, both before and after the activating cleavage into VP8* and VP5*6,17. From one of the three VP8*s, the cleavage releases all but a short, N-terminal segment of VP8* (VP8* α: Fig. 1c). The two intact VP8* fragments associate with the bean-shaped β-barrel domains of the VP5* fragments from which cleavage has separated them, forming a twofold symmetric projection; the β-barrel domain of the third VP5* supports this projection, holding it away from the particle surface like a diagonal cantilever6. The VP8* lectin domains at the tip of the spike cover hydrophobic loops at one end of the two projecting VP5* β barrels with which they associate; the opposite ends of the projecting VP5* barrels similarly cover the hydrophobic loops of the third VP5* β barrel. The C-terminal regions of all three VP5* chains associate with threefold symmetry into the “foot” of the spike, clamped onto the particle surface by VP76.
Recombinant VP4 is monomeric; successive treatment with chymotrypsin and trypsin yields a trimeric product, designated VP5CT, from which VP8* has dissociated completely and from which most residues of the foot have been removed18. An α-helical coiled-coil at the center of VP5CT projects in the same direction as the hydrophobic loops on the β barrels -- a chain reversal with respect to the spike on a TLP. We have proposed that the transition, from the asymmetric, “upright” spike conformation seen on virions to the symmetric, “reversed” conformation seen in VP5CT, leads to perforation of a target membrane, when the transition is coupled to interaction of the β-barrel hydrophobic loops with the membrane lipid bilayer18. Mutations that reduce the hydrophobicity of those VP5* loops compromise infectivity and reduce in vitro liposome association of VP5*5.
The cryo-EM analysis described here shows that transition to the reversed conformation can occur spontaneously on the surface of the TLP, without VP7 dissociation (Fig. 1d), and that association of the hydrophobic loops with a target membrane will cause residues of the foot region to enter the membrane lipid bilayer. We infer that formation of an initial membrane lesion by interaction with the bilayer of the foot regions from one or more VP5* trimers is a decisive, early event in rotavirus penetration and DLP delivery.
Reversed VP5* on rotavirus particles
Pursuing an observation made initially with a human rotavirus vaccine strain, we determined the three-dimensional structures of rhesus rotavirus (RRV) (Supplementary Data 1–3) native TLPs and wild-type recoated TLPs (rcTLPs), prepared as described in Methods (Extended Data Fig. 1). We initially imposed icosahedral symmetry and then classified subparticles corresponding to VP4 positions (Extended Data Fig. 2). Classification of rcTLPs (without further alignment) yielded three distinct classes (Extended Data Figs. 3 and 4, Extended Data Table 1) -- one corresponding to the asymmetric upright spike (Fig. 1d, left), a second corresponding to a threefold symmetric reversed structure (Fig. 1d, right), and a third corresponding to empty vertices. Classification of spikes from native TLPs, treated the same way as rcTLPs, yielded classes identical to those from rcTLPs, but with a different distribution of particles among the classes (Extended Data Fig. 5). The threefold symmetric conformation represented a reversed structure not previously seen on rotavirus particles. When we omitted the 37 °C incubation, the TLPs had only upright VP4 spikes and empty positions without VP4.
Inspection of the reversed-conformation map showed that the VP5CT coordinates18 matched closely the reconstructed density. In VP5CT, three β-barrel domains surround a central, three-chain coiled-coil, formed by polypeptide-chain segments C-terminal to the β barrels (Fig. 1d, right). Hydrophobic loops at one end of the β barrel project in the same direction as the distal end of the coiled-coil. VP5CT, produced by proteolytic cleavage of recombinant VP4, terminates at the end of the coiled-coil; about 250 additional amino-acid residues on intact VP4 form the foot that anchors VP4 in the upright spike conformation onto the DLP (Fig. 1d, left). The VP5* in our rcTLPs was intact (Extended Data Fig. 1b), but the region beneath the VP5CT-like density, normally occupied by the foot, was completely empty in the reversed reconstructions (Fig. 1d, right). That is, as expected from the direction of the coiled-coil, the last ~250 amino acid residues of the VP4 polypeptide chain projected outwards. We could detect little or no density C-terminal to the coiled-coil in the reversed class map; we concluded that the foot residues must be substantially disordered. We have previously described the VP5CT conformation as a “folded back” structure and suggested that a conformational transition of the spike might involve folding back of the β-barrel domain toward the still-anchored foot8,18. In the reversed structure we have now visualized, coiled-coil formation has instead projected the foot-domain residues outward, while retaining the orientation of the β barrels (Fig. 1d and Extended Data Fig. 6a).
After the transition to the reversed conformation, VP5* does not dissociate, despite loss of interactions between the foot and the surrounding VP6 and VP7 trimers. The inward-facing end of each of the three VP5* β-barrels contacts a set of loops in the adjacent VP7 through a network of largely polar, side-chain interactions (Fig. 2a, right, and Extended Data Fig. 7). VP8* remains associated with the particle, even after the transition (Extended Data Fig. 8a)19. On a TLP, the foot domain of VP5* includes, along its threefold axis, the initial ~16 residues of the VP4 polypeptide chain, two of which then connect outwards to the VP8* lectin domains at the apex of the spike, while the third terminates at residue 27 (Fig. 2b, left). As the foot unfolds and projects outwards along the threefold axis of the spike, the three VP8* N-terminal segments must therefore remain trapped in the foot cavity, with the connections to the two lectin domains emerging through a channel at the VP5*-VP7 interface (Fig 2b, right). These emerging connections could in principle also participate in the VP5*-VP7 contacts.
Fig. 2 |. Interactions of the penetration protein domains VP5* and VP8* with VP7 capsid subunits.
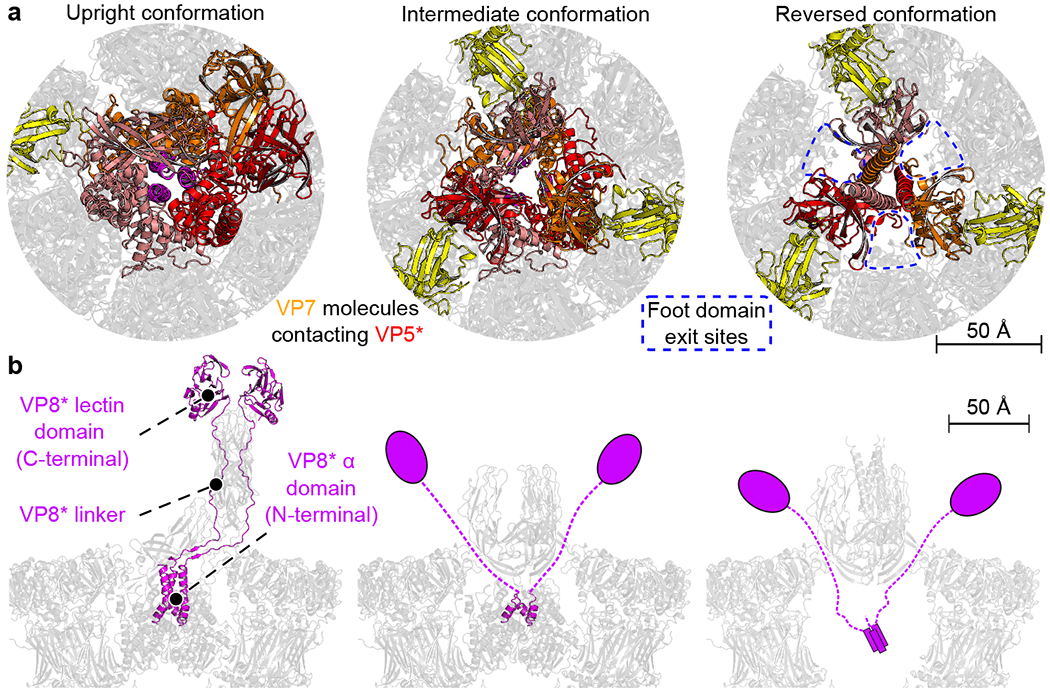
a, Top views of the upright (left), intermediate (middle), and reversed (right) conformations. VP5*, red, orange and salmon; VP7 chains interacting with VP5*, yellow. Close-up views of the interfaces are in Extended Data Fig. 7. Gaps between VP5* and VP7 shell molecules that allow for exit of VP5* foot domains are marked by blue dashed boxes. b, Side views of the upright (left), intermediate (middle) and reversed (right) conformations. VP8*, magenta. In the reversed structure (right), VP8* is shown schematically: no density for it was observed, but biochemical data shows its presence.
In an early cryo-EM study of rotavirus structure, Prasad and co-workers found that brief incubation of RRV at pH 11, followed by return to pH 8, caused an apparent collapse of the spikes into three, symmetrically disposed, protuberances19. We repeated their protocol with both native TLPs and wild-type VP4 rcTLPs and found that exposure to pH 11 had induced nearly complete transition to the reversed conformation described above (Extended Data Fig. 8b). Thus, the protuberances described in the earlier work were the three β-barrel domains seen at low resolution (see also Supplementary Discussion).
Trapping a potential intermediate
The conformational transition from upright (Fig. 1d, left) to reversed VP5* (Fig. 1d, right) might plausibly proceed in two steps -- rearrangement of the β-barrel domains into a threefold cluster followed by an eversion along the threefold axis of the rest of VP5*, in which coiled-coil formation thrusts the unfolded foot domain outwards (Fig. 2b). We screened a set of double cysteine mutants, designed to produce disulfide links within the foot and hence to stabilize its folded structure, and obtained the most robust recoating from a variant with cysteines at positions 567 and 590 in RRV VP4 (Fig. 3). Recoating DLPs with the mutant protein (along with wild-type VP7) yielded particles with infectivity 1600-fold lower than wild-type VP4 recoated controls (Fig. 3a).
Fig. 3 |. Infectivity and structure of TLPs recoated with mutant S567C-A590C penetration proteins.
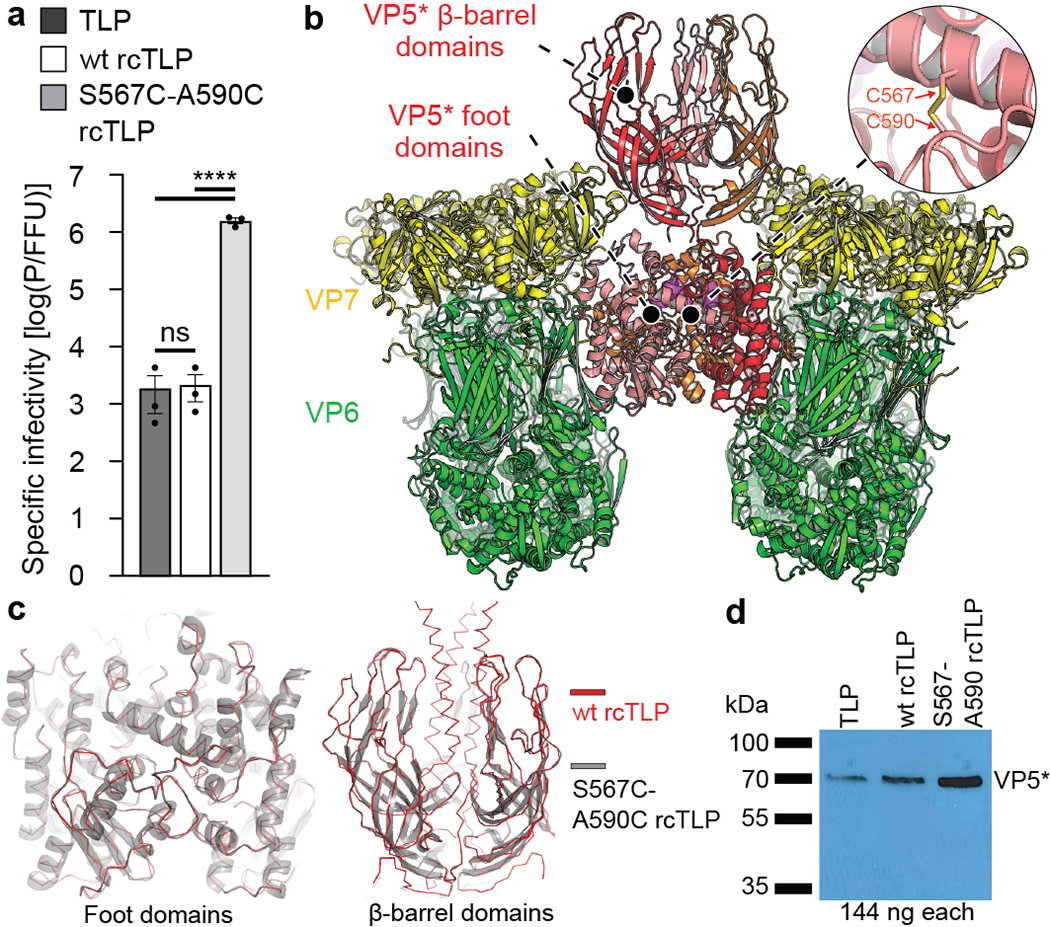
a, Particle to focus-forming unit (FFU) ratios of TLPs, wt rcTLPs and S567C-A590C rcTLPs determined as described in Methods. n = 3 biologically independent experiments. Error bars, mean ± s.d. Statistics: one-way ANOVA with Tukey post hoc test. p < 0.0001 for overall test (**** = p < 0.0001, ns = not significant). For source data, see Supplementary Data 4. b, Structure of the foot-locked intermediate obtained from cryo-EM analysis of S567C-A590C rcTLPs. VP5*, red, orange and salmon; VP6, green; VP7, yellow; VP8*, magenta. c, Left, structural comparison of the VP5* foot domains from the upright conformation of wt rcTLPs (red) and intermediate conformation of S567C-A590C rcTLPs (grey). Right, structural comparison of VP5* β-barrel domains from the reversed conformation of wt rcTLPs (red) and intermediate conformation of S567C-A590C rcTLPs (grey). d, Comparison of relative amounts of VP5* determined by Western blotting as described in Methods. Experiment repeated independently two times with similar results. For Western blot source data, see Supplementary Fig. 1.
We analyzed by cryo-EM 3,607 rcTLPs bearing S567C-A590C VP4 spikes. After initial icosahedral reconstruction, we classified the 216,420 VP4 positions (60 per virion) and found the mature spike conformation (same as Fig. 1d, left), empty positions without VP4, and a new class with three outward projecting β-barrel domains but no central, three-chain coiled coil (Fig. 3b). Instead, the foot VP5* appeared to be firmly in place, with little or no difference in structure, at the resolution we obtained, from the foot in wild-type rcTLPs (Figs. 3b, c). The anchored VP8* N-terminal segments were present, but clearly interpretable density did not extend beyond residue 16, where the three polypeptide chains encountered the inward-facing ends of the VP5* β barrels (Fig. 2b).
When we treated the S567C-A590C VP4 rcTLPs briefly at pH 11 and reneutralized, we found that most (69%) of the spikes were in the intermediate conformation (Extended Data Fig. 8c). The remaining 31% had converted to reversed trimers, probably because of inefficient disulfide formation.
Spike rearrangement during cell entry
In previous work, electron cryotomography (cryo-ET) of RRV entering at the rim of BSC-1 cells, where they were thin enough for imaging, showed that the spacing between particle and membrane at the virion-membrane interface could be either “loose” (~120 Å) or “tight” (~60 Å)8. VP4 projections spanned the gaps in both cases; those spanning the wider gap had about the length expected for the familiar “spike” conformation. We have now extracted from more than 100 recent tomographic data sets individual sub-tomograms of nearly fully engulfed particles and calculated icosahedral averages for each (Fig. 4a and Extended Data Fig. 9). The “loose” engulfment appears to correspond to the spike contacts expected for initial lectin contacts with glycolipid, even though the resolution of the icosahedral average is not sufficient to show the contacts directly. The “tight” engulfment corresponds closely to our expectation from Fig. 1d (right): the volume occupied by the foot in the unrearranged structure is empty and the gap between the VP7 surface and the membrane would place the VP5* hydrophobic loops at the surface of the bilayer, with the extruded foot apparently embedded in the membrane. We infer that the transformation we have detected on the surface of virions in vitro also occurs during cell entry as a key step in delivery of the DLP into the cytosol.
Fig. 4 |. Molecular rearrangements of the membrane-penetration protein during rotavirus entry.
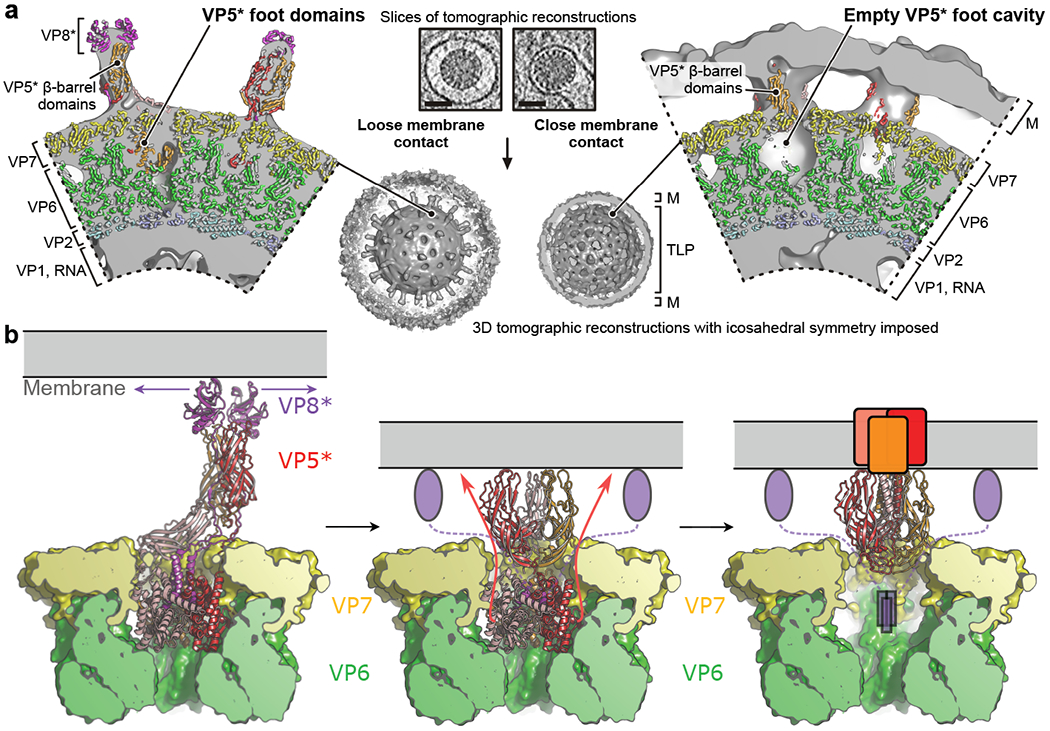
a, Cryo-ET reconstructions of RRV entering at the thin edge of BSC-1 cells. Tomographic analysis is shown for two viral particles that are representative for a “loose” (left) or “tight” (right) virion-membrane (M) interface, respectively. In the close-up views of the tomographic reconstructions, density is partially cut and shown in solid gray; protein subunits of positioned atomic models are shown in ribbon representation and colored as in Fig. 1. The scale bar in the tomographic slices (low-pass filtered and contrast-enhanced) is 50 nm. b, Model for VP5* and VP8* rearrangement and membrane-interaction; see Discussion for details.
Discussion
A transition of VP4 from upright to reversed conformation requires the following steps (Fig. 4b): separation of the VP8* lectin domains from the tips of the twofold clustered, VP5* β-barrel domains; outward flip of the third VP5* β barrel and formation of a β-barrel trimer; unfolding and outward projection of the foot domain by zippering of the central, three-strand, α-helical coiled coil. Formation of the observed β-barrel trimer requires that the lectin domains dissociate from the tips of the β barrels, but they remain tethered to the particle by the long N-terminal segment of VP8* (VP8* α + VP8* linker, Fig. 4b). Even after the foot has unfolded, the two complete VP8* fragments do not dissociate (Fig. 2b right and Extended Data Fig. 8a)19.
VP5* in its reversed conformation remains bound on the virion through contacts with three of the six VP7 subunits that surround it (Fig. 2a right and Extended Data Fig. 6a and 7); the same contacts are present in the structure of the “foot-locked” mutant (S567C-A590C VP4). A smooth transition from foot-locked to reversed VP5* is possible, because adjacent β barrels have no direct contact in the former structure, and unfolding and outward projection of the foot can occur without dissociation (Extended Data Fig. 6b). The foot-locked structure is therefore a plausible representation of an intermediate in the overall conformational change (see Supplementary Discussion).
The subtomogram averages in Fig. 4a show that the molecular rearrangements we see in vitro correspond to events occurring during virus entry. The absence of density in the foot cavity supports our proposed function for the conformational change. The very low infectivity of particles recoated with foot-locked VP4 indicates that complete rearrangement is necessary for virion penetration.
Figure 4b suggests how the findings reported here might map onto the sequence of events during cell entry derived from live-cell imaging experiments8,16,20. Steps in that cell-entry sequence are membrane interaction, invagination, engulfment, Ca2+ loss, VP4 and VP7 loss, DLP release (Fig. 1b). Membrane deformation requires more free energy than simple attachment; interaction of the VP5* hydrophobic loops with the membrane bilayer may be necessary for the observed “wrapping” (Fig. 4a center). The hydrophobic-loop interaction might initially occur through the two β-barrel domains that project in the spike conformation. Spontaneous fluctuations could generate local dissociation of the VP8* lectin domains (arrows in Fig. 4b, left), allowing the adjacent membrane to capture the hydrophobic loops of the two β-barrel domains that project in the spike conformation. Transition to a state resembling the foot-locked (intermediate) structure could then ensue (Fig. 4b, center), followed by unfurling of the foot, formation of the coiled-coil, and interaction of the foot with the target membrane (Fig. 4b, right).
During entry, at least two distinct regions of VP5* interact with the lipid bilayer of the target-cell membrane: the hydrophobic loops exposed by VP8* lectin-domain dissociation and the foot translocated outward by the conformational change described here. Determining how multiple local perturbations, each introduced by insertion of three foot regions into the target membrane, promote longer-range disruption (“perforation”: Fig. 1b) and DLP release will require high-resolution visualization of the inserted foot regions.
Like other non-enveloped animal viruses with dsDNA or dsRNA genomes, rotaviruses penetrate by delivering a modified but still assembled particle into the cytosol. The structures described here define more closely than yet achieved for other dsRNA or dsDNA viruses, how rotaviruses deliver a very large, macromolecular “payload” into a cell. The molecular description of the rotavirus delivery mechanism, and of those of other viruses that insert a modified virion or subviral particle into the cytosol, can inform efforts to design delivery vehicles for biologicals with cytosolic targets.
Methods
Data reporting
No statistical methods were used to predetermine sample size. The experiments were not randomized and the investigators were not blinded to allocation during experiments and outcome assessment.
Cells, plasmids and constructs
MA104 cells (ATCC) were cultured in M199 media (Invitrogen) supplemented with 25 mM HEPES and 10% HI-FBS (Invitrogen). BSC-1 cells (ATCC) were cultured in DMEM (Invitrogen) supplemented with 10% HI-FBS (Invitrogen). For VP4 and VP7 expression, full-length genomic sequences from rhesus rotavirus (G3 serotype, NCBI:txid444185) were amplified by PCR and cloned into pFastbac (Thermo Fisher Scientific) expression vector. Mutations were introduced by quick-change mutagenesis in DH10α cells (Thermo Fisher Scientific). Purified plasmid constructs were transfected into DH10-Bac cells (Thermo Fisher Scientific). Purified bacmids were transfected into SF9 cells (ATCC) grown in SF900 II SFM media supplemented with 1% pen-strep.
Rhesus rotavirus (RRV) TLP/DLP production and purification
MA104 cells were grown to confluency in 850-cm2 roller bottles, followed by exchange of medium to M199 supplemented with 1 μg/ml trypsin (Gibco). Cells were infected with trypsin activated RRV particles (G3 serotype) at a multiplicity of infection (MOI) of 0.1 and incubated for 24 h. Media and cell debris were collected and frozen for storage at −80 °C. Virus in thawed supernatants was concentrated by ultracentrifugation (225,500 g, 30 min), added to thawed cell debris pellets (which contain most of the virus), and extracted by sonication with Freon-113 as described21,22. The aqueous phases of three successive rounds of Freon extraction were combined, and TLPs were separated from DLPs by cesium chloride gradient centrifugation. TLPs were dialyzed twice against 2 liter of 20 mM Tris-HCl pH 8.0, 100 mM NaCl, 1 mM CaCl2 (TNC) and DLPs twice against 2 liter of 20 mM HEPES, pH 7.4, 100 mM NaCl (HN). Concentration of purified virus was determined by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) densitometry of VP6 bands against standards of DLP stocks ranging from 0.1 to 1.0 mg/ml.
Expression and purification of VP4 and VP7
Recombinant VP4 and VP7 were expressed in SF9 insect cells, using the Bac-to-Bac expression system as described previously23,24. In brief, insect cells at 2 million cells/ml were infected with baculovirus inoculum passaged three times in SF9 cells. For VP4, cells were harvested 72 h post infection and resuspended in 100 ml lysis buffer (75 mM Tris-HCl pH 8.0, 100 mM NaCl, 5 mM EDTA, 7.5 % glycerol). After freeze thawing, cells were lysed in a Dounce homogenizer and the homogenate cleared by centrifugation (70,409 g, 30 min). VP4 was purified from the cleared supernatant by ammonium sulfate precipitation followed by hydrophobic interaction chromatography (Phenyl FF column) and anion exchange chromatography (Q sepharose Column)25. Final purification was on a size exclusion chromatography column (S200) equilibrated in HNE (20 mM HEPES pH 7.3, 100 mM NaCl, 1 mM EDTA). Pure VP4 was concentrated to 5 mg/ml, flash frozen with liquid nitrogen, and stored at −80 °C. For VP7, cells were removed from media 72 h post infection and the supernatant was loaded onto a ConcavalinA resin. After elution with α-methyl-mannose, protein was adsorbed to a resin with immobilized M159 antibody26 (5 mg per ml of Protein G Sepharose resin) and eluted with HNE15. Fractions containing VP7 were pooled and concentrated to 3 mg/ml, flash frozen with liquid nitrogen, and stored at −80 °C.
Recoating of RRV DLPs
Recoating DLPs with VP4 and VP7 to form rcTLPs followed previously described protocols16. In brief, 1 M sodium acetate (pH 5.2) was added to a volume of DLP resulting in a final concentration of 100 mM sodium acetate. VP4 was added to a final concentration of 0.9 mg/ml (~33-fold excess) and the mixture incubated at room temperature for 1 h. VP7 was then added in 2.3-fold excess along with a final addition of sodium acetate and CaCl2 to maintain their respective concentrations of 100 mM and 1 mM. The mixture was incubated at room temperature for 1 h, and recoated particles were separated from excess components by ultracentrifugation (112,000 g, 30 min). Pellets were resuspended in 150 μl of TNC, additional TNC was added to a final volume of 1 ml, and centrifugation repeated. Final pellets were resuspended in 50 μl of TNC.
Infectivity assay of TLPs and rcTLPs
Titers and particles to focus-forming unit (FFU) ratios for TLP and rcTLPs were determined by infectious focus assays as previously described16, and specific infectivities were derived from concentration measurements based on densitometry of a Western blot using a primary antibody specific for VP6 (antibody 2B4, Santa Cruz Biotechnology, 1:4000 dilution). We determined the relative amounts of VP5* by Western blotting of 144 ng of purified particles with antibody HS227 (1:5000 dilution) in combination with horseradish-peroxidase (HRP)-coupled goat anti-mouse IgG (1:5000 dilution).
pH shift experiments with TLPs and rcTLPs
RRV particles were incubated with 5 μg/ml trypsin for 30 min at 37 °C and the reaction was quenched by addition of 1 mM phenylmethylsulfonyl fluoride (PMSF). pH induced conformational change was achieved by adding a 0.83 M N-cyclohexyl-3-aminopropanesulfonic acid (CAPS) buffer at pH 11.0 to a final concentration of 100 mM. As a control, we used 100 mM of Tris-HCl pH 8.0. Samples were incubated for 30 min at room temperature and neutralized to pH 8.0 by addition of 1 ml of TNC (20 mM Tris-HCl, 100 mM NaCl, 1 mM CaCl2). Viral particles were pelleted in a TLS 55 rotor (50,000 rpm, 1 h, 4 °C). Supernatants were removed and the pellets were resuspended in 50–80 μl TNC.
VP8* fractionation and detection by Western blotting
We digested 12 μg of TLPs with 5 μg/ml trypsin in a total volume of 12 μl for 30 min at 37 °C. Trypsin was quenched by adding 0.6 μl of aprotinin and 1.2 μl of PMSF to final concentrations of 2 μg/ml and 1 mM, respectively. We shifted the pH by adding 1.2 μl of 1 M CAPS buffer at pH 11.0 (or 1.2 μl of 1 M TRIS pH 8.0 for the control samples), incubated the samples for 30 min at room temperature, and re-neutralized them by adding 1 ml TNC (20 mM Tris-HCl pH 8.0, 100 mM NaCl, 1 mM CaCl2) or 1 ml TN2E (20 mM Tris-HCl pH 8.0, 100 mM NaCl, 2 mM EDTA). We monitored pH shift and re-neutralization by pH strips. Samples were then incubated for 30 min at 37 °C to promote uncoating of TLPs. TLPs and uncoated DLPs were then pelleted in TLS 55 rotor (50,000 rpm, 1 h, 4 °C), resuspended in 1 ml buffer and pelleted again (50,000 rpm, 1 h, 4 °C). We carefully removed the supernatants and resuspended the pellets in 50 μl either TNC or TN2E. We analyzed 10 μl of each sample by SDS-PAGE and silver staining, and also by SDS-PAGE and Western blotting with the VP8*-specific antibody HS127 (1:500 dilution of mouse HS1 ascitic fluid and 1:5000 dilution of HRP-coupled goat anti-mouse IgG).
Sample preparation for single particle electron cryomicroscopy (cryo-EM)
2.5 μl of RRV rcTLPs or TLPs at concentrations between 1 and 2 mg/ml were treated with 5 μg/ml trypsin at 37 °C for 30 min, after which trypsin was inactivated with PMSF at a final concentration of 1 mM. The trypsin-treated particles were then incubated with 2.5 μl TNC for a further 30 min at 37 °C, followed by application of 4 μl to a C-flat holey carbon grid (Electron Microscopy Science), blotting, and freezing with a CP3 plunge freezer (Gatan). The efficiency of the spike transition at pH 8.0 from upright to reversed was noticeably higher for rcTLPs than for authentic TLPs. We therefore used the former to generate samples for electron microscopy of pH 8.0 incubated particles. Because expression and purification of recombinant VP4 and VP7 involve quite different conditions than does virion propagation, we have not tried to track down the source of the difference. Previous structural studies show no significant structural differences, and specific infectivity of the rcTLPs is sometimes even higher than that of authentic virions.
Single particle cryo-EM and data processing
Vitrified grids were imaged with a Tecnai Polara F30 microscope (FEI/Thermo Fisher Scientific) operated at 300 kV and images recorded with a Gatan K2 direct electron detector. Movies of rotavirus particles were taken with an exposure time of 10 s and 0.2 s/frame (50 frames) using a physical pixel size of 1.231 Å in counting mode. Movies were motion corrected with MotionCor2 (5×5 patch alignment)28. We used Gautomatch for viral particle picking with template projections obtained with EMAN229 from a previous reconstruction30. For automatic particle picking, the angular sampling was 3° and the low-pass filter cutoff 40 Å. We determined the contrast transfer function (CTF) parameters using Gctf31 from total-summed images. We fitted defocus and astigmatism parameter at individual particle coordinates and used relion_preprocess32 for particle extraction. We aligned the rotavirus particle images and calculated reconstructions with cisTEM (refine3d version 1.01, reconstruct3d version 1.02)33 as described previously30. We imposed icosahedral symmetry in setting I2 for alignment and reconstruction and used a spherical shell mask for the reference with an inner radius of 222 Å, and an outer radius of 403 Å (corresponding to the VP2, VP6, and VP7 protein layers of the TLP). After the initial alignment, we used relion_motion_refine and relion_ctf_refine in order to obtain a polished particle stack and parameters for beam-tilt correction, respectively34, and carried out a second round of alignment with cisTEM. The Fourier shell correlation (FSC)-based resolution estimates for density within the spherical shell were 3.1 Å for the wild-type rcTLP reconstruction and 3.9 Å for the S567C-A590C rcTLP reconstruction (Extended Data Table 1).
Local reconstruction of VP4 structures
We used subparticle image analysis for local reconstructions and classification of VP4 structures (Extended Data Fig. 2). Similar to what we previously described for structure determination of the VP1 rotavirus polymerase30,35, we used signal subtraction of non-VP4 proteins (except a single VP7 trimer) with relion_project. We extracted subparticle images (corresponding to the location of VP4 proteins) based on the icosahedral alignment from the original polished, and signal-subtracted particle stacks using IMOD36. We used the programs refine3d (version 1.01) and reconstruct3D (version 1.02) from cisTEM for subparticle classification. We kept the subparticle alignment parameters constant during iterative classification (40–80 cycles); the high-resolution limit for classification was 8 Å; and we 3D-masked the references with a single mask encompassing volume of all VP4 conformations and a single adjacent VP7 trimer. Per-particle weighting in the reconstruction step was essentially turned off by setting the BSC parameter to 0.0. After classification, we calculated final maps for each class from non-signal-subtracted particle stacks and applied standard postprocessing steps. As classification reduced the number of particles per class compared to the icosahedral reconstruction (with 60-fold averaging), the final resolution of the local reconstructions was slightly lower, ranging from 3.3 to 4.3 Å resolution (Extended Data Table 1). FSC plots and local resolution analyses of the local reconstructions are shown in Extended Data Fig. 3. Density for the VP8* lectin and the dimeric VP5* β-barrel domains was poorly resolved in the upright spike conformation local reconstruction because of flexibility at the base of the dimeric spike projection (Extended Data Fig. 3a). We therefore improved this region of the map using alignment by classification. In this approach, we iteratively classified without alignment (6 classes, 5 Å high-resolution limit for classification, 80 cycles, 3D mask encompassing the VP8* lectin and VP5* β-barrel domains), superimposed the maps of the classes, and updated particle alignment parameters according to the 3D map alignment. The resulting 4 Å-resolution map allowed us to model the connection between the VP8* N terminus (located in the foot) and the peripheral VP8* lectin domains.
Structure modeling, refinement and validation
Our atomic models consist of the trimetric VP4 spike protein (VP5* and VP8*) surrounded by six trimers of VP6 and VP7 each (some of the VP6 and VP7 subunits are related by icosahedral symmetry of the virion). We initially placed subunits obtained from published structures with PDB-IDs 4V7Q6 and 1SLQ (in case of the VP5* reversed conformation)18 into our local reconstruction maps. We manually adjusted and completed the models using the programs O37 and Coot38, and used phenix.real_space_refine39 for structure refinement. We used standard stereochemical and B-factor restraints, as well as Ramachandran, rotamer, and secondary structure restraints. Residues included in the models are summarized in Extended Data Table 1. We validated the models with MolProbity40. Model statistics are in Extended Data Table 1. The refined structures show expected FSC values with the cryo-EM maps (Extended Data Fig. 3). Close-up views of density map sections are shown in Extended Data Fig. 4a. To locate structural changes between the upright and reversed structures, we calculated per residue Cα distances after subunit-vise superposition of V4, VP7, and VP6 subunits from the upright and reversed conformation structures (Extended Data Fig. 4b).
Sample preparation for electron cryotomography (cryo-ET)
BSC-1 cells, grown on gold grids and incubated with rotavirus TLPs, were plunge frozen for cryotomography as previously described8. In brief, Quantifoil gold grids (EMS Q250-AR2) with a holey carbon film (R2/2, 200 mesh) were glow discharged at −40 mA for 30 sec, coated with 0.1% poly-L-lysine hydrobromide (Sigma P1524-25mg), and sterilized with 70% ethanol for 10 min. BSC-1 cells were cultured on these grids in DMEM with 10% FBS and 1x Glutamax at 37 °C and 5% CO2 in a 35 mm diameter glass bottom dish (MatTek, P35G-1.5-14-C) for 24 h prior to inoculation. Grids were then washed by aspirating the medium from the dish and replaced it with MEM α (Gibco 41061-029) buffer at 37 °C. This process was repeated (with a total amount of about 2 ml buffer) until the buffer remained clear. Grids were then transferred to glass light microscope slides covered with Parafilm. Just before inoculation, the virus was activated using trypsin stock solution 50 μg/ml in TNC (pH 8.0) at 37 °C for 30 min. After diluting the activated virus 1:5 or 1:10 in MEM α, 5 μl of the solution were immediately added to the cells at 37 °C for inoculation. The grids were then incubated at 37 °C for a time between 5 and 45 min, before removing the buffer by blotted the grid from the edge with filter paper while simultaneously adding 5 μl of concentrated, BSA-coated 10 nm colloidal gold (Sigma-Aldrich)41. Grids were then blotted from the front with filter paper for 7–8 s and plunge-frozen in liquid ethane using a manual plunge-freezing device. The frozen grids were stored in liquid nitrogen.
cryo-ET and data processing
Vitrified grids were imaged using a Tecnai F30 transmission electron microscope (FEI/Thermo Fischer Scientific) equipped with a field emission gun operating at an accelerating voltage of 300 kV, GIF2000 post column energy filter (Gatan) operated in zero loss mode with a 20 eV slit width, and a 2k×2k charge-coupled device camera (Gatan). The cells were screened in low dose mode for areas of interest, i.e. areas that contained virus particles interacting with the edge of BSC-1 cells that were thin enough for cryo-ET imaging (up to 300 nm thick). In each area of interest, a single-axis tilt series was recorded in low dose mode using the microscope control software SerialEM42. The sample was tilted over a range of about −65° to +65° with 1.5–2.5° increments. The total accumulated electron dose per tilt series was limited to ∼100 e/Å2. The defocus was −6 μm to −8 μm and the pixel size was 1.077 nm or 0.986 nm, respectively. The software program IMOD43 was used for fiducial alignment of the tilt series images and tomogram reconstruction using weighted back projection. We analyzed virions from 90 raw cryo-tomograms by extracting virus-particle subtomograms with a box size of 150×150×150 pixels. We contrast-inverted and normalized the extracted viral particle subtomograms, and 3D-aligned them to a low-pass filtered reference using e2spt_classaverage.py and e2spt_extract_align.py from EMAN2 (version 2.2)44. At this stage, subtomographic viral densities were oriented in standard I4 icosahedral symmetry setting, which allowed us to i) calculate an icosahedral density average for each particle by just applying icosahedral symmetry, ii) extract sub-subtomograms at individual spikes positions, or iii) average selected subtomograms (individual viruses) or sub-subtomograms (individual VP4 trimer positions) (Fig. 4a and Extended Data Fig. 9).
Figure preparation
We prepared the figures with PyMOL (The PyMOL Molecular Graphics System, Version 2.1 Schrödinger, LLC), POV-Ray (www.povray.org), and matplotlib45. We retrieved rotavirus sequences from GenBank46 with BioPython47, aligned them with MAFFT48, and used ESPript49 to display the multiple sequence alignments of VP4 (Supplementary Data 1), VP7 (Supplementary Data 2), and VP6 (Supplementary Data 3). Serotypes and genotypes were compiled from published studies50–53.
Extended Data
Extended Data Fig. 1 |. Sample preparation and cryo-EM data collection.
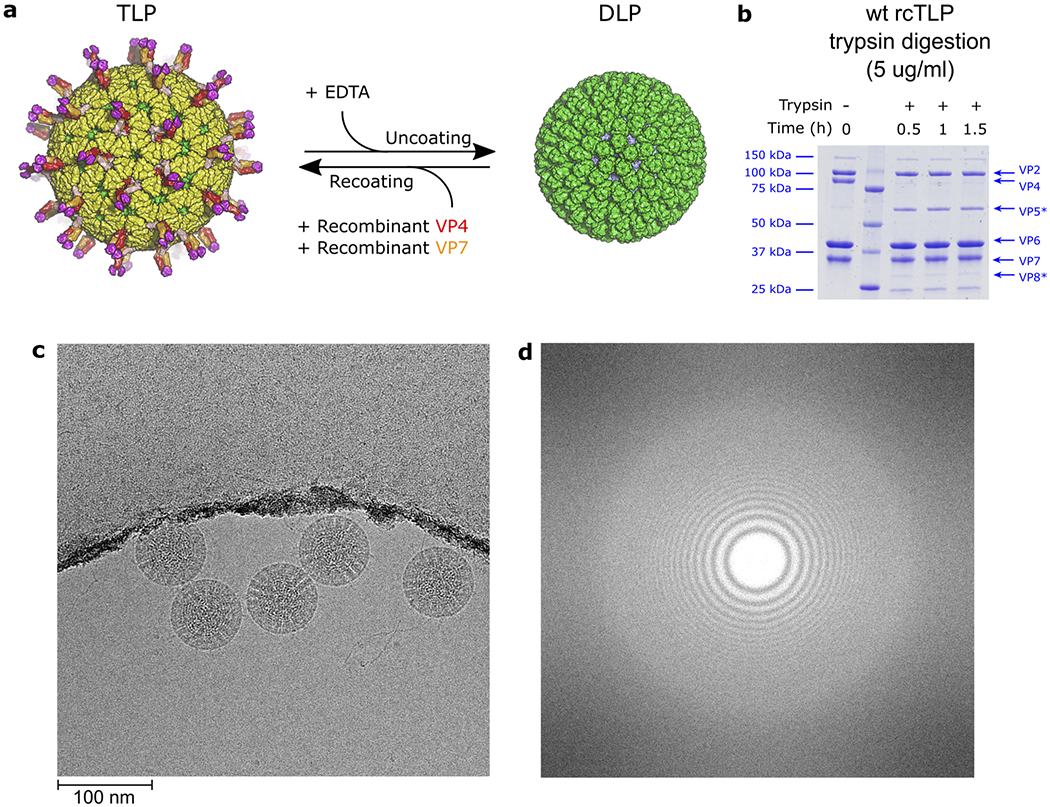
a, Schematic protocol for recoating of double-layer particles (DLPs) with recombinant VP4 and VP7. b, Time course of wild-type recoated triple-layer particles (wt rcTLPs) trypsin digestion with 5 μg/ml trypsin at 37 °C. Samples were analyzed after the shown incubation times by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). The experiment was repeated independently two times with similar results. For gel source data, see Supplementary Fig. 1. c, Representative micrograph (aligned and summed movie frames) of wt rcTLPs recorded with a Polara F30 electron microscope equipped with a K2 summit detector (magnification = 40650). The scale bar corresponds to 100 nm. d, Power spectrum of the micrograph shown in c.
Extended Data Fig. 2 |.
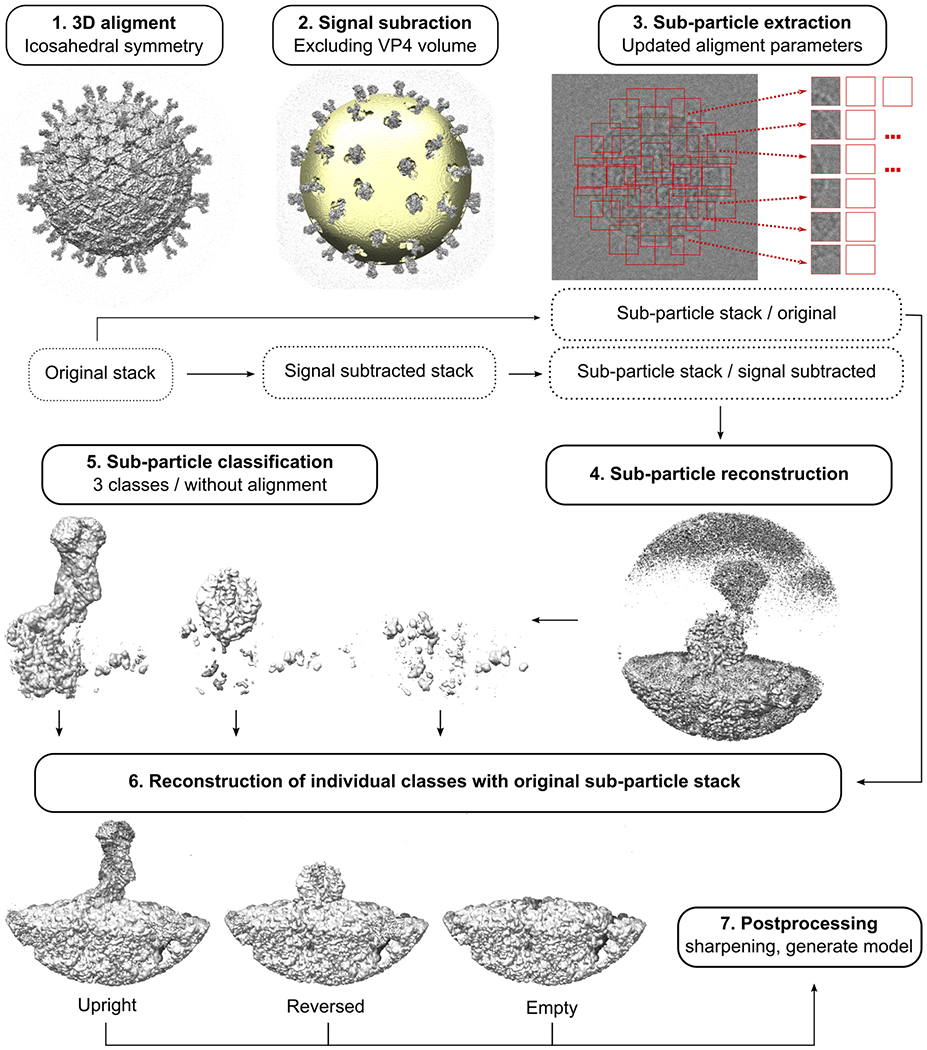
Data processing workflow for local reconstructions of rotavirus spike proteins.
Extended Data Fig. 3 |. Resolution analysis of the local cryo-EM reconstructions.
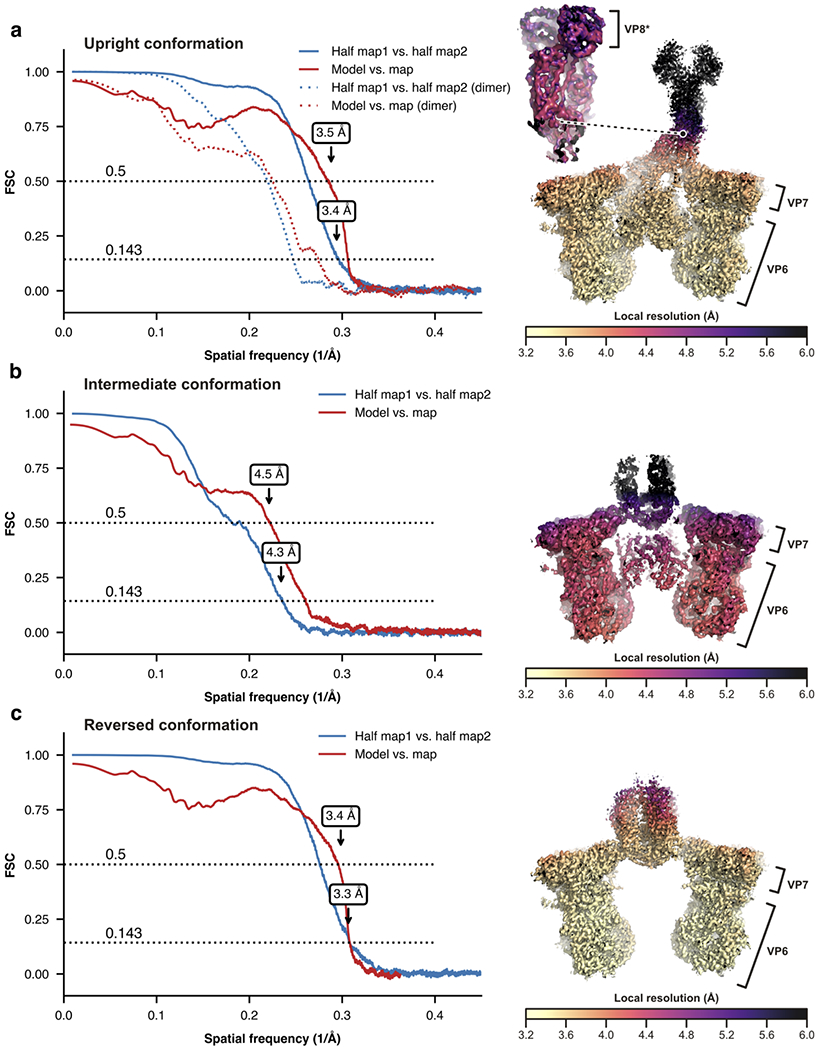
Left, Fourier shell correlation (FSC) curves for the reconstructions and refined models calculated with phenix.mtriage54. Correlations for the two half maps are shown as solid blue lines after applying a mask encompassing the models. Correlations between the refined model and final map are shown as red solid lines. Nominal resolution estimates at conventional FSC values are indicated by arrows. Right, local resolution of the reconstructions calculated with ResMap55. a, Upright conformation. Dashed lines (left) are the FSC analysis for the reconstruction of the distal VP5*/VP8* dimeric density that was obtained through alignment by classification (see Methods). The inset (right) shows the local resolution of this reconstruction. b, Intermediate conformation. c, Reversed conformation. For source data of the FCS plots, see Supplementary Data 5.
Extended Data Fig. 4 |. cryo-EM density and structure comparison.
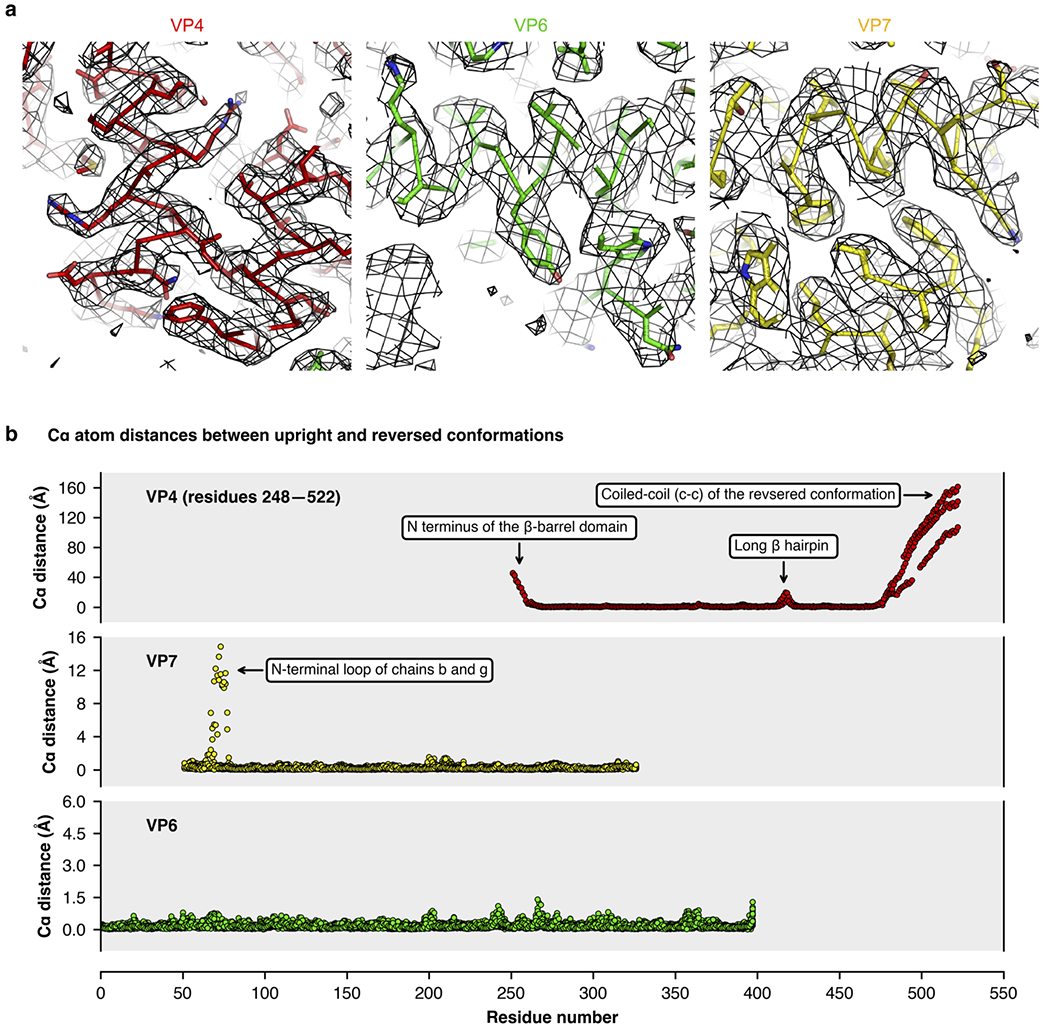
a, Close-up views of representative regions of the cryo-EM density maps obtained by local reconstruction. Density is shown as gray mesh; polypeptide-chain backbone as ribbon; side-chain atoms as sticks (carbon, black; nitrogen, blue; oxygen, red; sulfur, orange). b, Per-residue Cα distances after subunit-vise superposition of V4, VP7, and VP6 subunits from the upright and reversed conformation structures.
Extended Data Fig. 5 |. Comparison of penetration protein conformations on rcTLPs and native TLPs.
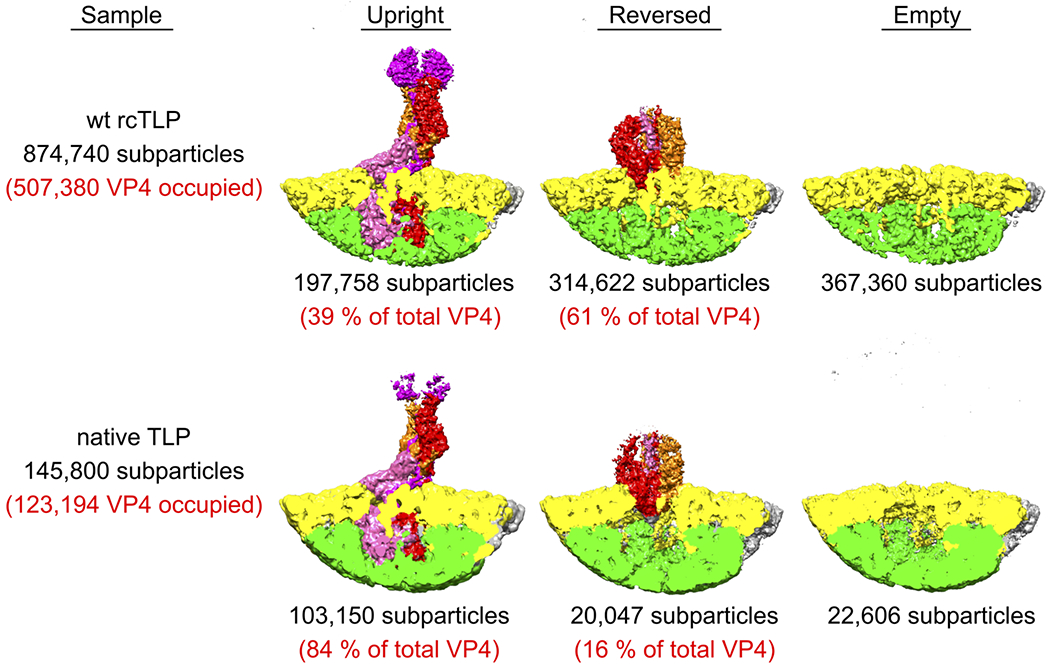
Relative subparticle amounts, and corresponding cryo-EM reconstructions obtained from rcTLPs (top row) and native TLPs (bottom row). Local spike reconstructions of rcTLPs were obtained from two cryo-EM samples prepared from two independent recoating reactions. Local spike reconstructions of native TLPs were obtained from one cryo-EM sample.
Extended Data Fig. 6 |. Rearrangements of the VP8* and VP5* penetration proteins during transition from upright to reversed conformation on the virion surface.
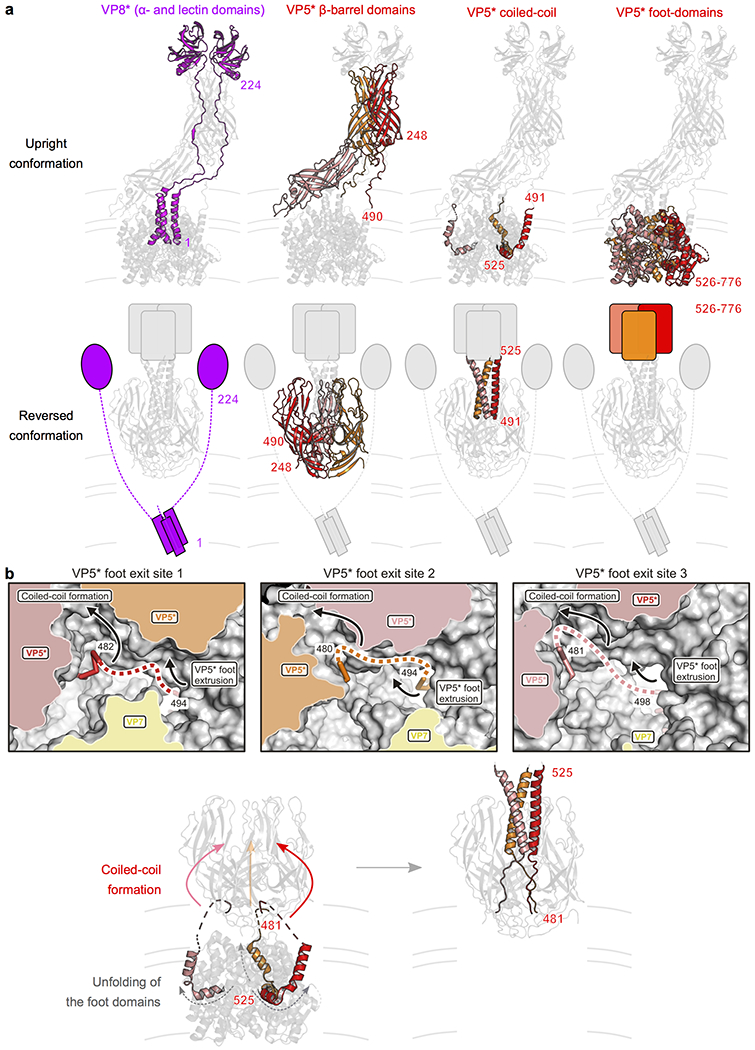
a, Distinct domains of the VP8* and VP5* spike proteins are colored separately to illustrate their conformational change during transition from upright (top row) to reversed (bottom row) conformation. Domains that were not observed in our cryo-EM maps because of flexible attachment are drawn schematically. VP8*, magenta; VP5*, red, orange and salmon. b, Formation of the trimeric coiled coil and extrusion of the foot domains. Top row, close-up views of the VP5* foot domain exit sites as observed in the intermediate conformation structure. A partially cut surface representation is shown. VP5*, red, orange and salmon; VP7, yellow. The last modeled residues of the VP5* β-barrel domains, 482 (chain 1), 480 (chain 2), and 481 (chain 3), are located on the outside. The connections to the first modeled VP5* foot domain residue, 494 (chain 1), 494 (chain 2), and 498 (chain 3), are indicated by dashed lines (fuzzy density in the cryo-EM map). Arrows indicate a suggested mechanism for foot domain reversal, involving zipping-up of the trimeric coiled-coil and unfolding and extrusion of foot domain residues. Bottom row, proposed transition from the intermediate structure (left) to the reversed structure by “zipping up” of the coiled coil and unfolding and extrusion of the foot domains.
Extended Data Fig. 7 |. Molecular details of the VP5*-VP7 interfaces for the upright and reversed conformations.
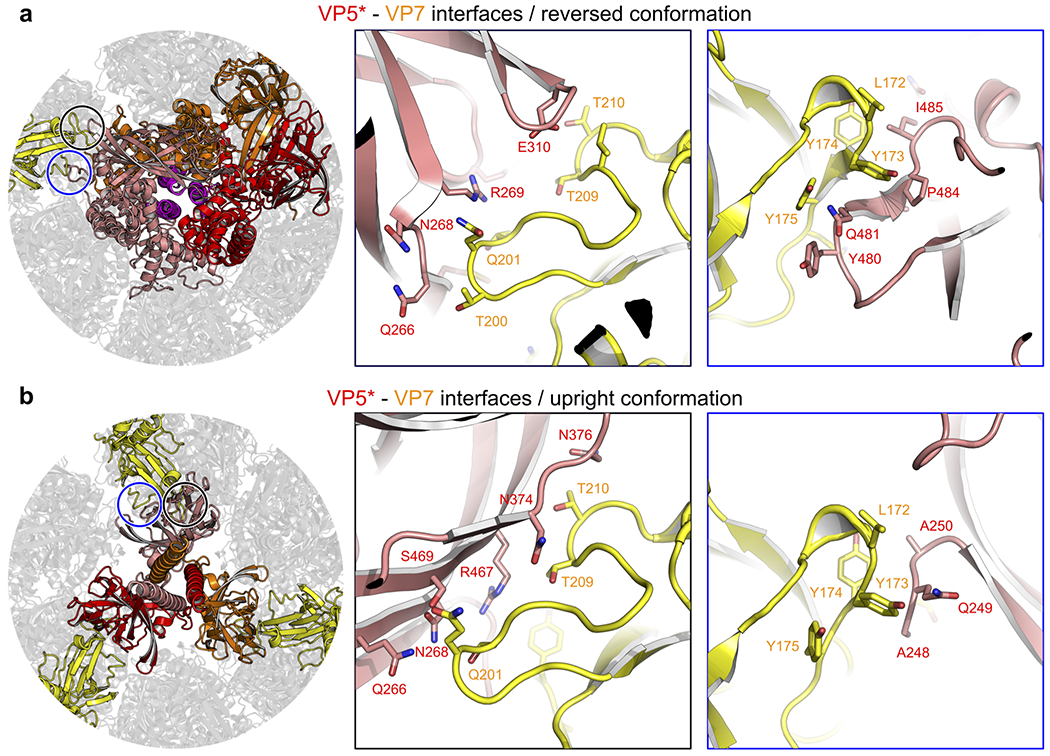
VP5*, salmon; VP7, yellow. a, VP5*-VP7 interface for the VP5* upright conformation. b, VP5*-VP7 interface for the VP5* reversed conformation. The following VP5* interface residues are conserved (Supplementary Data 1): N268, N376, R467, S469. The VP5* β-barrel N terminus (residues 248–250) is not strictly conserved, however, the interaction is based on main-chain hydrogen bonds and can likely be maintained for different side chains as well. The following VP7 interface residues are conserved (Supplementary Data 2): S201, T210, L172–Y175.
Extended Data Fig. 8 |. Inducing penetration protein reversal at alkaline pH and VP8* association with TLPs.
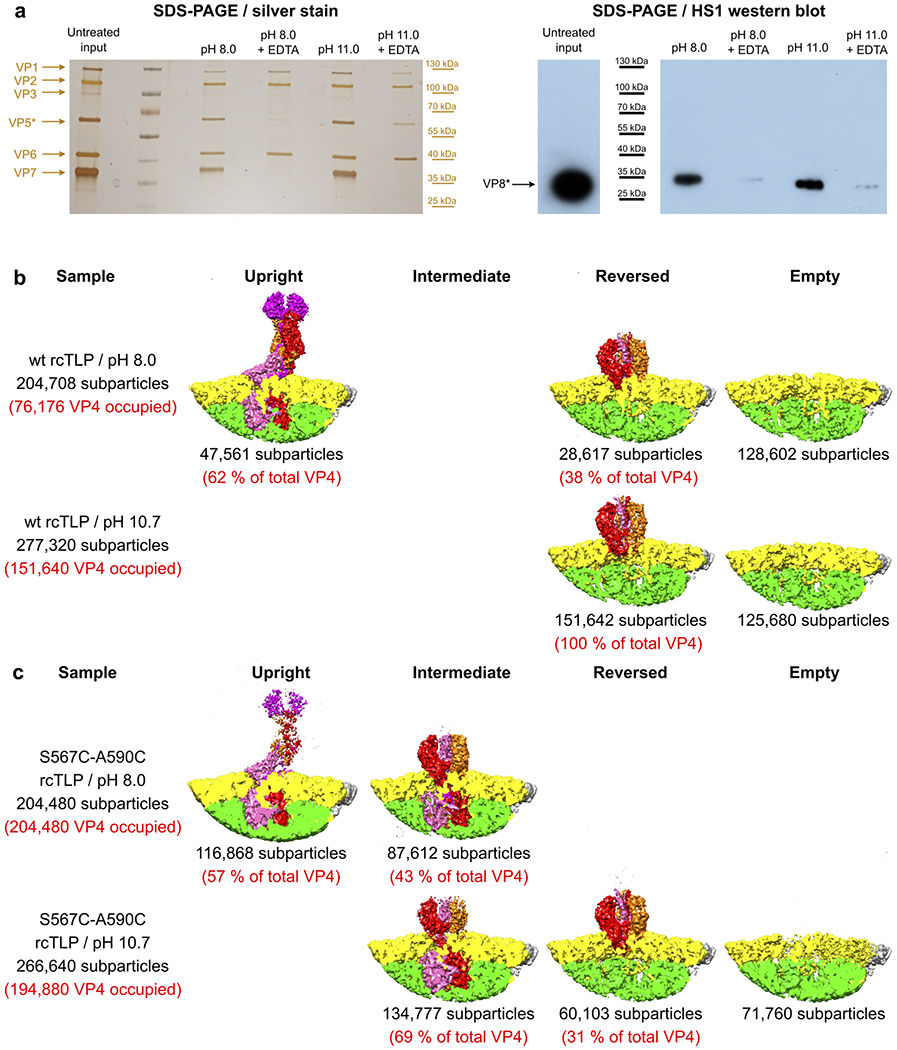
a, Analysis of rotavirus particles without (lanes 1 and 2) and with (lines 3 and 4) pH-induced conformational change of VP8*/VP5*, and without (lanes 1 and 3) and with (lanes 2 and 4) EDTA-induced uncoating. Pelleted fractions were analyzed by SDS-PAGE and silver staining (left) and by Western blotting with the VP8*-specific antibody HS127 (right) (see Methods). The experiment was repeated independently three times with similar results. For gel and Western blot source data, see Supplementary Fig. 1. b, Relative VP4 subparticle amounts, and corresponding cryo-EM reconstructions obtained from wild-type (wt) rcTLPs. c, Relative VP4 subparticle amounts, and corresponding cryo-EM reconstructions obtained from S567C-A590C rcTLPs. Recoating reactions for all samples were carried out at the same time and with the same VP7 and DLP stock solutions. All cryo-EM samples were prepared in the same blotting session.
Extended Data Fig. 9 |. Cryo-tomographic (cryo-ET) analysis of RRVs entering BSC-1 cells.
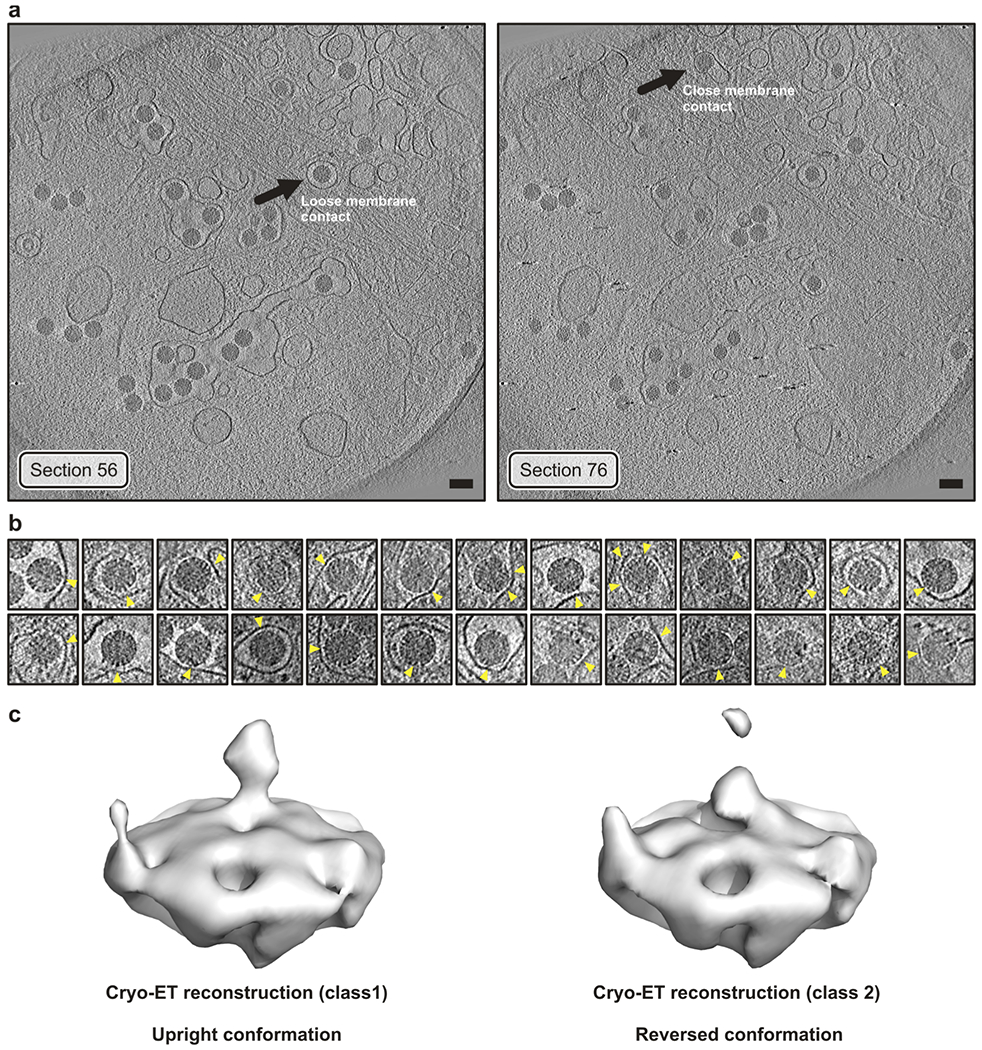
a, Sections of the tomogram from which the reconstructions (icosahedral average of single virion subtomograms) in Fig. 4a were obtained. Left, virus with “loose” membrane contact indicated by an arrow. Right, virus with “tight” membrane contact indicated by an arrow. Images were low-pass filtered and contrast enhanced for display. Scale bar corresponds to 100 nm. b, Tomographic slices of manually selected viruses (not including particle in panel a) from several tomograms with tight membrane contacts (yellow arrowheads). Images were low-pass filtered and contrast enhanced for display. Those particles were selected to detect VP4 reversed conformations in additional viruses (other than particle in Fig. 4a). c, Sub-subtomogram classification of VP4 positions extracted from the RRV particles shown in panel b yielded a class (left) with the upright conformation and a class (right) with the reversed conformation. VP4 positions were extracted from 26 selected particles chosen to have an easily identified region of close membrane contact. The particle shown in Fig. 4a was excluded from this selection. Note that the reconstructions shown in Fig. 4a are from single particles with imposed icosahedral symmetry, whereas we show here classified averaged individual volumes extracted at VP4 positions.
Extended Data Table 1 |.
Cryo-EM data collection and model statistics.
| Upright conformation | Reversed conformation | Intermediate conformation | |
|---|---|---|---|
| EMD-21955 PDB-ID 6WXE | EMD-21957 PDB-ID 6WXG | EMD-21956 PDB-ID 6WXF | |
| wt rcTLP | wt rcTLP | S567C-A590C rcTLP | |
| Data collection | |||
| Electron microscope | Polara | Polara | |
| Magnification | 40650 | 40650 | |
| Voltage (kV) | 300 | 300 | |
| Defocus range (μm)* | 1.0–2.5 | 1.0–3.0 | |
| Pixel size (Å) | 1.23 | 1.23 | |
| Icosahedral reconstruction | |||
| Number of images | 14432 | 3408 | |
| Box size (pixels) | 1024 | 1024 | |
| Symmetry imposed | I (setting I2) | I (setting I2) | |
| Map resolution (Å) † | 3.1 | 3.9 | |
| Local reconstruction | |||
| Number of images | 167147 | 252548 | 82008 |
| Box size (pixels) ‡ | 320 (264) | 320 (264) | 320 (264) |
| Symmetry imposed | C1 | C1 | C1 |
| Map resolution (Å) § | 3.4 | 3.3 | 4.3 |
| Sharpening B factor (Å2) | −124 | −125 | 0.0 |
| Model statistics | |||
| Modeled residues ∥ | |||
| VP4 chain 1 | 2–224, 250–596, 605–776 | 248–522 | 2–16,260–482, 496–596, 607–776 |
| VP4 chain 2 | 1–224, 250–482, 488–598, 606–776 | 248–522 | 2–16, 260–480, 494–596, 607-776 |
| VP4 chain 3 | 1–27, 261–494, 498–598, 606–776 | 248–522 | 2–16, 260–481, 498–594, 606–776 |
| VP7 chains a, h, q | 57–315 | 57–315 | 57–315 |
| VP7 chains b, g, i, j, m, p, r | 55–326 | 55–326 | 55–326 |
| VP7 chain c | 55–315 | 55–315 | 55–315 |
| VP7 chains d, f, k, I, n, o | 51–315 | 51–315 | 51–315 |
| VP7 chain e | 55–314 | 55–314 | 55–314 |
| VP6 | 1–397 | 1–397 | 1–397 |
| Sugars | 54 | 54 | 54 |
| Ca2+ ions | 18 | 18 | 18 |
| Refinement resolution (Å) | 3.4 | 3.3 | 4.3 |
| CC (mask) | 0.77 | 0.79 | 0.77 |
| Model composition | |||
| Non-hydrogen atoms | 111012 | 101709 | 107222 |
| Protein residues | 13952 | 12763 | 13466 |
| B factors (Å2) | |||
| Protein | 29.4 | 18.0 | 184 |
| R.m.s. deviations | |||
| Bond lengths (Å) | 0.004 | 0.004 | 0.004 |
| Bond angles (°) | 0.766 | 0.751 | 0.691 |
| Validation | |||
| MolProbity clash score | 3.8 | 3.8 | 3.3 |
| Poor rotamers (%) | 2.0 | 1.7 | 2.0 |
| Ramachandran plot | |||
| Favored (%) | 99.3 | 99.5 | 99.5 |
| Allowed (%) | 0.7 | 0.5 | 0.5 |
| Disallowed (%) | 0.0 | 0.0 | 0.0 |
Approximate range of underfocus.
Resolution where Fourier shell correlation (FSC) between half-maps drops below 0.143 after applying a spherical shell mask (inner radius = 222 Å, outer radius = 403 Å).
Pixel number in parenthesis used during intermediate processing steps.
Resolution where FSC between half-maps drops below 0.143 after applying a mask encompassing VP6, VP7, and VP4 (VP5* and VP8*).
Modeled amino-acid residues are given for each chain (full-length sequences: VP4, 1–776; VP7, 1–326; VP6, 1–397).
Supplementary Material
Acknowledgments
We thank Zongli Li, Sarah Sterling, Richard Walsh, and Shaun Rawson for assistance and guidance at the Harvard Medical School Cryo-EM Center for Structural Biology and the Harvard Medical School Molecular Electron Microscopy Suite, and Chen Xu for assistance at the Brandeis University cryo-EM facility. We thank Harry B. Greenberg for the kind gift of the HS1 and HS2 antibodies. The work was supported by NIH grant CA-13202 (to SCH). SCH is an Investigator in the Howard Hughes Medical Institute.
Footnotes
Data availability
We have deposited the maps of the cryo-EM reconstructions in the Electron Microscopy Data Bank (accession no. EMD-21955, upright conformation; EMD-21956, intermediate conformation; EMD-21957, reversed conformation), and the refined coordinates in the Protein Data Bank (PDB-IDs 6WXE, upright conformation; 6WXF, intermediate conformation; EMD-6WXG, reversed conformation). We obtained previously published rotavirus structures from the Protein Data Bank (PDB-IDs 4V7Q and 1SLQ). Rotavirus protein sequence used to prepare the sequence alignments shown in Supplementary Data 1–3 were retrieved from GenBank46. The accession identifiers for VP4 are: RRV, P12473; SA11, P12976; WA, P11193; S2, AQT31697; DS1, P11196; ST3, P11200; AU-1, P39033; 116E, Q09113; 69M, P26451; NCDV, P17465; UK, P12474; 993/83, Q08010; Gottfried, P23045; OSU, P11114; CH3, Q86184; L338, Q98636; EW, Q83450. The accession identifiers for VP7 are: RRV, P12473; SA11, P12976; WA, P11193; S2, AQT31697; DS1, P11196; ST3, P11200; AU-1, P39033; 116E, Q09113; 69M, P26451; NCDV, P17465; UK, P12474; 993/83, Q08010; Gottfried, P23045; OSU, P11114; CH3, Q86184; L338, Q98636; EW, Q83450. The accession identifiers for VP6 are: RRV, P12473; SA11, P12976; WA, P11193; S2, AQT31697; DS1, P11196; ST3, P11200; AU-1, P39033; 116E, Q09113; 69M, P26451; NCDV, P17465; UK, P12474; 993/83, Q08010; Gottfried, P23045; OSU, P11114; CH3, Q86184; L338, Q98636; EW, Q83450.
Competing interests
The authors declare no competing interests.
References
- 1.Harrison SC in Fields Virology (6th edition) (ed Knipe DM and Howley PM) 52–86 (Wolters Kluwer/Lippincott Williams and Wilkins, 2013). [Google Scholar]
- 2.Estes MK & Greenberg H in Fields Virology (6th edition) (ed Knipe DM and Howley PM) 1347–1401 (Wolters Kluwer/Lippincott Williams and Wilkins, 2013). [Google Scholar]
- 3.Trask SD, Ogden KM & Patton JT Interactions among capsid proteins orchestrate rotavirus particle functions. Curr. Opin. Virol 2, 373–379, doi: 10.1016/j.coviro.2012.04.005 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tihova M, Dryden KA, Bellamy AR, Greenberg HB & Yeager M Localization of membrane permeabilization and receptor binding sites on the VP4 hemagglutinin of rotavirus: implications for cell entry. J. Mol. Biol 314, 985–992, doi: 10.1006/jmbi.2000.5238 (2001). [DOI] [PubMed] [Google Scholar]
- 5.Kim IS, Trask SD, Babyonyshev M, Dormitzer PR & Harrison SC Effect of mutations in VP5 hydrophobic loops on rotavirus cell entry. J. Virol 84, 6200–6207, doi: 10.1128/JVI.02461-09 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Settembre EC, Chen JZ, Dormitzer PR, Grigorieff N & Harrison SC Atomic model of an infectious rotavirus particle. EMBO J. 30, 408–416, doi: 10.1038/emboj.2010.322 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Aoki ST et al. Structure of rotavirus outer-layer protein VP7 bound with a neutralizing Fab. Science 324, 1444–1447, doi: 10.1126/science.1170481 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Abdelhakim AH et al. Structural correlates of rotavirus cell entry. PLoS Pathog 10, e1004355, doi: 10.1371/journal.ppat.1004355 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Trask SD, Kim IS, Harrison SC & Dormitzer PR A rotavirus spike protein conformational intermediate binds lipid bilayers. J. Virol 84, 1764–1770, doi: 10.1128/JVI.01682-09 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dormitzer PR et al. Specificity and affinity of sialic acid binding by the rhesus rotavirus VP8* core. J. Virol 76, 10512–10517, doi: 10.1128/jvi.76.20.10512-10517.2002 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Delorme C et al. Glycosphingolipid binding specificities of rotavirus: identification of a sialic acid-binding epitope. J. Virol 75, 2276–2287, doi: 10.1128/JVI.75.5.2276-2287.2001 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Martinez MA, Lopez S, Arias CF & Isa P Gangliosides have a functional role during rotavirus cell entry. J. Virol 87, 1115–1122, doi: 10.1128/JVI.01964-12 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ramani S et al. The VP8* domain of neonatal rotavirus strain G10P[11] binds to type II precursor glycans. J. Virol 87, 7255–7264, doi: 10.1128/JVI.03518-12 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hu L et al. Cell attachment protein VP8* of a human rotavirus specifically interacts with A-type histo-blood group antigen. Nature 485, 256–259, doi: 10.1038/nature10996 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dormitzer PR, Greenberg HB & Harrison SC Purified recombinant rotavirus VP7 forms soluble, calcium-dependent trimers. Virology 277, 420–428, doi: 10.1006/viro.2000.0625 (2000). [DOI] [PubMed] [Google Scholar]
- 16.Salgado EN, Garcia Rodriguez B, Narayanaswamy N, Krishnan Y & Harrison SC Visualization of Calcium Ion Loss from Rotavirus during Cell Entry. J. Virol 92, doi: 10.1128/JVI.01327-18 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rodriguez JM et al. New insights into rotavirus entry machinery: stabilization of rotavirus spike conformation is independent of trypsin cleavage. PLoS Pathog 10, e1004157, doi: 10.1371/journal.ppat.1004157 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dormitzer PR, Nason EB, Prasad BV & Harrison SC Structural rearrangements in the membrane penetration protein of a non-enveloped virus. Nature 430, 1053–1058, doi: 10.1038/nature02836 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pesavento JB, Crawford SE, Roberts E, Estes MK & Prasad BV pH-induced conformational change of the rotavirus VP4 spike: implications for cell entry and antibody neutralization. J. Virol 79, 8572–8580, doi: 10.1128/JVI.79.13.8572-8580.2005 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Salgado EN, Upadhyayula S & Harrison SC Single-particle detection of transcription following rotavirus entry. J. Virol, doi: 10.1128/JVI.00651-17 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Smith RE, Zweerink HJ & Joklik WK Polypeptide components of virions, top component and cores of reovirus type 3. Virology 39, 791–810, doi: 10.1016/0042-6822(69)90017-8 (1969). [DOI] [PubMed] [Google Scholar]
- 22.Street JE, Croxson MC, Chadderton WF & Bellamy AR Sequence diversity of human rotavirus strains investigated by northern blot hybridization analysis. J. Virol 43, 369–378 (1982). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fiore L et al. Antigenicity, immunogenicity and passive protection induced by immunization of mice with baculovirus-expressed VP7 protein from rhesus rotavirus. J. Gen. Virol 76 (Pt 8), 1981–1988, doi: 10.1099/0022-1317-76-8-1981 (1995). [DOI] [PubMed] [Google Scholar]
- 24.Mackow ER, Barnett JW, Chan H & Greenberg HB The rhesus rotavirus outer capsid protein VP4 functions as a hemagglutinin and is antigenically conserved when expressed by a baculovirus recombinant. J. Virol 63, 1661–1668 (1989). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Trask SD & Dormitzer PR Assembly of highly infectious rotavirus particles recoated with recombinant outer capsid proteins. J. Virol 80, 11293–11304, doi: 10.1128/JVI.01346-06 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Greenberg HB et al. Production and preliminary characterization of monoclonal antibodies directed at two surface proteins of rhesus rotavirus. J. Virol 47, 267–275, doi: 10.1128/JVI.47.2.267-275.1983 (1983). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Padilla-Noriega L et al. Serologic analysis of human rotavirus serotypes P1A and P2 by using monoclonal antibodies. J. Clin. Microbiol 31, 622–628, doi: 10.1128/JCM.31.3.622-628.1993 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zheng SQ et al. MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 14, 331–332, doi: 10.1038/nmeth.4193 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bell JM, Chen M, Baldwin PR & Ludtke SJ High resolution single particle refinement in EMAN2.1. Methods 100, 25–34, doi: 10.1016/j.ymeth.2016.02.018 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jenni S et al. In situ Structure of Rotavirus VP1 RNA-Dependent RNA Polymerase. J. Mol. Biol 431, 3124–3138, doi: 10.1016/j.jmb.2019.06.016 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang K Gctf: Real-time CTF determination and correction. J. Struct. Biol 193, 1–12, doi: 10.1016/j.jsb.2015.11.003 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Scheres SH RELION: implementation of a Bayesian approach to cryo-EM structure determination. J. Struct. Biol 180, 519–530, doi: 10.1016/j.jsb.2012.09.006 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Grant T, Rohou A & Grigorieff N cisTEM, user-friendly software for single-particle image processing. Elife 7, doi: 10.7554/eLife.35383 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Scheres SH Beam-induced motion correction for sub-megadalton cryo-EM particles. Elife 3, e03665, doi: 10.7554/eLife.03665 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ding K et al. In situ structures of rotavirus polymerase in action and mechanism of mRNA transcription and release. Nat. Commun 10, 2216, doi: 10.1038/s41467-019-10236-7 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mastronarde DN & Held SR Automated tilt series alignment and tomographic reconstruction in IMOD. J. Struct. Biol 197, 102–113, doi: 10.1016/j.jsb.2016.07.011 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jones TA, Zou JY, Cowan SW & Kjeldgaard M Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr A 47, 110–119 (1991). [DOI] [PubMed] [Google Scholar]
- 38.Emsley P & Cowtan K Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr 60, 2126–2132, doi: 10.1107/S0907444904019158 (2004). [DOI] [PubMed] [Google Scholar]
- 39.Afonine PV et al. Real-space refinement in PHENIX for cryo-EM and crystallography. Acta Crystallogr D Struct Biol 74, 531–544, doi: 10.1107/S2059798318006551 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen VB et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr 66, 12–21, doi: 10.1107/S0907444909042073 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Iancu CV et al. Electron cryotomography sample preparation using the Vitrobot. Nat Protoc 1, 2813–2819, doi: 10.1038/nprot.2006.432 (2006). [DOI] [PubMed] [Google Scholar]
- 42.Mastronarde DN Automated electron microscope tomography using robust prediction of specimen movements. J. Struct. Biol 152, 36–51, doi: 10.1016/j.jsb.2005.07.007 (2005). [DOI] [PubMed] [Google Scholar]
- 43.Kremer JR, Mastronarde DN & McIntosh JR Computer visualization of three-dimensional image data using IMOD. J. Struct. Biol 116, 71–76, doi: 10.1006/jsbi.1996.0013 (1996). [DOI] [PubMed] [Google Scholar]
- 44.Galaz-Montoya JG, Flanagan J, Schmid MF & Ludtke SJ Single particle tomography in EMAN2. J. Struct. Biol 190, 279–290, doi: 10.1016/j.jsb.2015.04.016 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hunter JD Matplotlib: A 2D graphics environment. Comput. Sci. Eng 9, 90–95 (2007). [Google Scholar]
- 46.Benson DA et al. GenBank. Nucleic Acids Res. 46, D41–D47, doi: 10.1093/nar/gkx1094 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cock PJ et al. Biopython: freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics 25, 1422–1423, doi: 10.1093/bioinformatics/btp163 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Katoh K, Misawa K, Kuma K & Miyata T MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 30, 3059–3066, doi: 10.1093/nar/gkf436 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gouet P, Courcelle E, Stuart DI & Metoz F ESPript: analysis of multiple sequence alignments in PostScript. Bioinformatics 15, 305–308, doi: 10.1093/bioinformatics/15.4.305 (1999). [DOI] [PubMed] [Google Scholar]
- 50.Gorziglia M, Larralde G, Kapikian AZ & Chanock RM Antigenic relationships among human rotaviruses as determined by outer capsid protein VP4. Proc. Natl. Acad. Sci. U. S. A 87, 7155–7159, doi: 10.1073/pnas.87.18.7155 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Martella V et al. Molecular analysis of the VP7, VP4, VP6, NSP4, and NSP5/6 genes of a buffalo rotavirus strain: identification of the rare P[3] rhesus rotavirus-like VP4 gene allele. J. Clin. Microbiol 41, 5665–5675, doi: 10.1128/jcm.41.12.5665-5675.2003 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Matthijnssens J et al. Full genome-based classification of rotaviruses reveals a common origin between human Wa-Like and porcine rotavirus strains and human DS-1-like and bovine rotavirus strains. J. Virol 82, 3204–3219, doi: 10.1128/JVI.02257-07 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Patton JT Rotavirus diversity and evolution in the post-vaccine world. Discov. Med 13, 85–97 (2012). [PMC free article] [PubMed] [Google Scholar]
- 54.Afonine PV phenix.mtriage: a tool for analysis and validation of cryo-EM 3D reconstructions. Computational Crystallography Newsletter 8, 25 (2017). [Google Scholar]
- 55.Kucukelbir A, Sigworth FJ & Tagare HD Quantifying the local resolution of cryo-EM density maps. Nat. Methods 11, 63–65, doi: 10.1038/nmeth.2727 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.