

Abstract
Bioorthogonal activation of prodrugs provides a strategy for on-demand on-site production of therapeutics. Intracellular activation provides a strategy to localize therapeutics, potentially minimizing off-target effects. To this end, nanoparticles embedded with transition metal catalysts (nanozymes) were engineered to generate either ‘hard’ irreversible or ‘soft’ reversible coronas in serum. The hard corona induced nanozyme aggregation, effectively inhibiting nanozyme activity, whereas only modest loss of activity was observed with the non-aggregating soft corona nanozymes. In both cases complete activity was restored by treatment with proteases. Intracellular activity mirrored this reactivation: endogenous proteases in the endosome provided intracellular activation of both nanozymes. The role of intracellular proteases in nanozyme reactivation was verified through treatment of the cells with protease inhibitors, which prevented reactivation. This study demonstrates the use of intracellular proteolysis as a strategy for localization of therapeutic generation to within cells.
Keywords: Stimuli response, Endogenous activation, Bioorthogonal chemistry, Nanozymes, Proteolysis
Graphical Abstract
Bioorthogonal chemistry is a versatile strategy for in situ generation of imaging and therapeutic agents in living system using abiologic chemical processes.1–3 Bioorthogonal catalysis via transition metal catalysts (TMCs) provides access to processes that cannot be accomplished by natural enzymes.4–18 Loading of TMCs into nanomaterials provides nanozymes that feature enhanced stability,19 biocompatibility20, 21 and solubility.4, 22, 23 Through appropriate choice of nanomaterial scaffold these nanozymes can also be engineered to provide complex kinetic behavior analogous to enzymes24, 25 as well as the ability to localize in therapeutically important targets including bacterial biofilms,26 cells,27,28,29 and tissues.30–32
Intracellular activation of therapeutics provides a key strategy for therapeutic localization, with concomitant decreases in required dosing and off-target effects.33 Endogenous activation provides a key strategy for intracellular activation, relying on intrinsic cellular features such as reduced endosomal pH, increased thiol levels and intracellular enzymes.34–37 Enzymes, in particular, are attractive candidates for activating therapeutics due to their high efficiency and specificity.38, 39
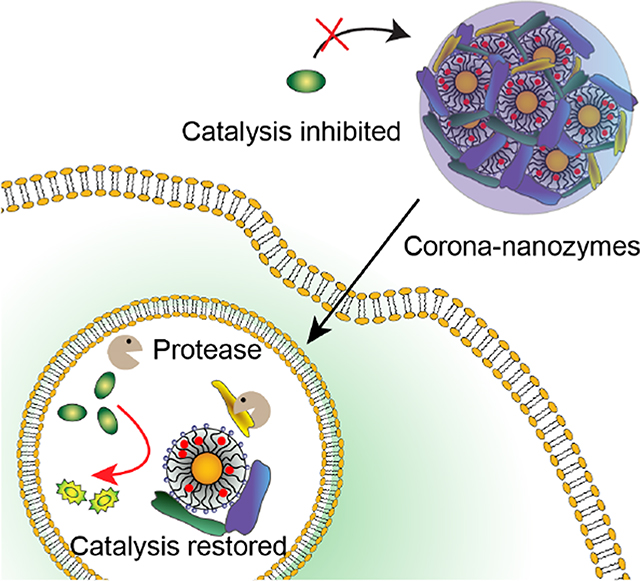
In recent studies we have demonstrated that gold nanoparticles could readily encapsulate TMCs into their monolayer, providing biorthogonal nanozymes.23, 26, 30 These studies used nanozymes featuring monolayers engineered to resist hard (irreversible) corona formation that retained intracellular activity for extended periods.40, 41 We hypothesized that engineering the nanoparticle surface to form a hard corona would result in nanozyme inactivation through aggregation and steric blocking of the nanozyme active sites (Figure 1). This inhibition could then be reversed through intracellular proteolysis by endogenous proteases found in endosomes, including the cathepsins. The above hypotheses were tested by generating nanozymes engineered to form either a hard (NZ1) or soft corona (NZ2) in serum. Nanozymes with hard coronas (Corona-NZ1) were almost completely deactivated, with activity restored in solution through proteolysis. In contrast, nanoparticles engineered to generate a soft corona (Corona-NZ2) were only minimally inhibited, with activity restored through proteolysis. Treatment of cells with inactive hard corona nanozymes provided activity only inside cells, whereas soft corona nanozymes were active both extra- and intracellularly. The observed activation of Corona-NZ1 and Corona-NZ2 arose from intracellular proteolysis, as evidenced by lack of reactivation when cells were incubated with protease inhibitors. Overall, this study demonstrates that the protein corona can be used to enable specific intracellular activation of nanozymes, providing a versatile strategy for on-demand generation of imaging and therapeutic agents.
Figure 1.
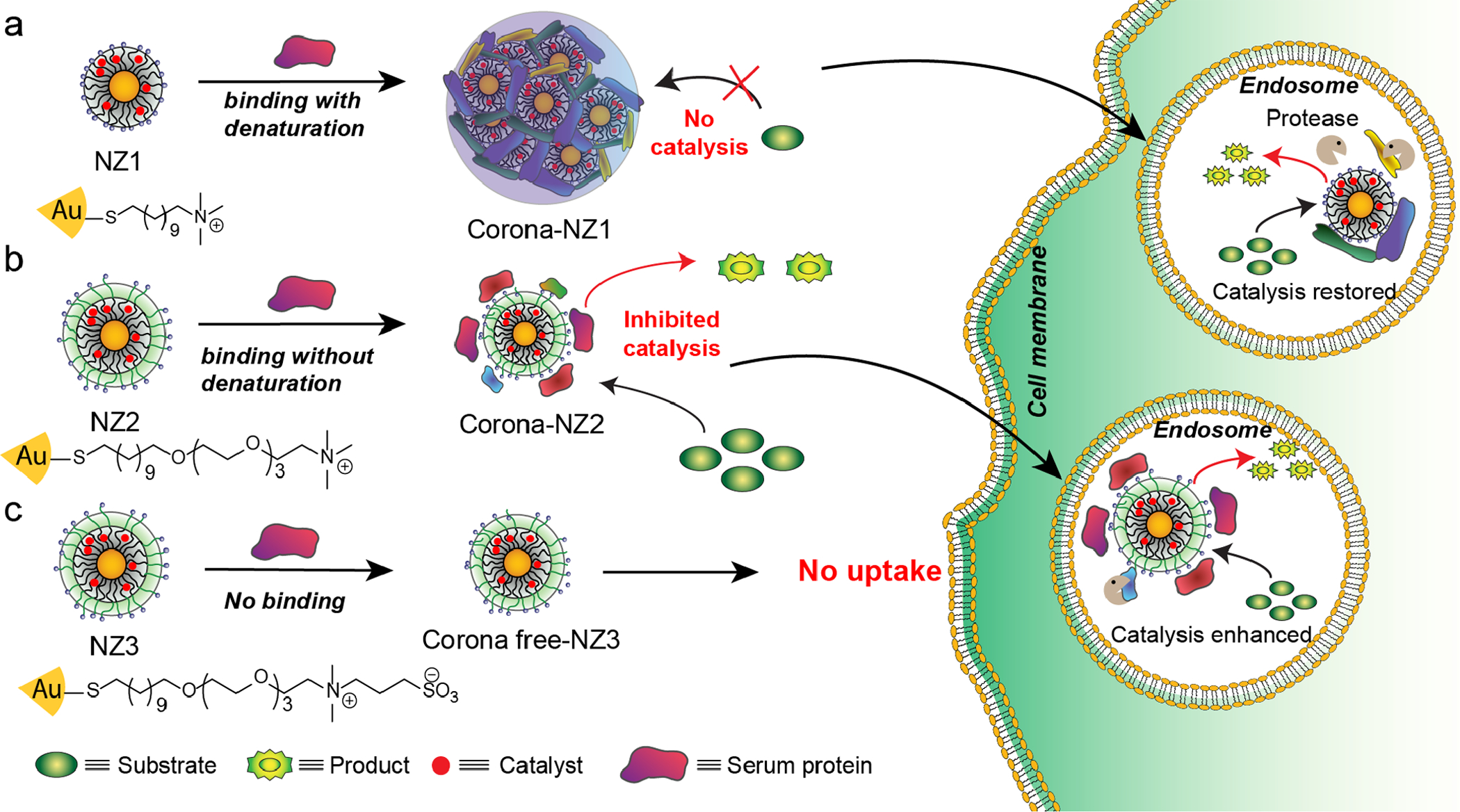
Strategy for intracellular activation of bioorthogonal nanozymes through endosomal proteolysis of the protein corona on 2nm core gold nanoparticles: 1) Hard corona effectively inhibits catalytic activity of NZ1, and the soft corona partial reduces catalytic activity of NZ2. 2) After cellular uptake, both cationic nanozymes (NZ1 and NZ2) activities were restored by endogenous proteases. 3) Corona-free nanozymes (NZ3) showed high catalytic activity only extracellularly due to the low cellular uptake.
Results and Discussion
The nature of the protein corona42–44 is determined by ligand design on nanomaterials.45, 46 For instance, the zwitterionic sulfobetaine terminal group has been shown to have ‘stealth’ properties, providing corona-free NPs in serum condition.45 In contrast, positively charged quaternary ammonium terminal groups interact with proteins to form protein coronas on NPs.46 The nature of this corona can be controlled by ligand design, with the incorporation of appropriate functionality allowing selection of hard or soft corona formation.41 As an example, AuNPs with exposed hydrophobic surfaces (e.g. NP1) lead to protein denaturation and irreversible protein adsorption (hard corona), rendering proteins readily susceptible to proteolysis.40, 41 In contrast, AuNPs with tetra(ethylene glycol) (TEG) spacers (e.g. NP2) prevent hydrophobic interactions between proteins and hydrophobic alkane chain in the monolayer of AuNPs, forming soft coronas with the retention of protein structure with concomitantly slower proteolysis.41
We hypothesized that this ability to control corona formation could be used to regulate intracellular activation of nanozymes through endosomal proteolysis of the protein corona. This control was demonstrated using functionalized AuNPs (2 nm core in diameter) as scaffolds for encapsulating transition metal catalysts (TMCs, Figure 1) to generate nanozymes. These nanozymes could be engineered to provide catalysis exclusively inside cells through hard corona formation (NZ1), intracellularly and extracellularly through soft corona generation, (NZ2), and exclusively extracellularly using a ‘stealth’ NP (NZ3) not uptaken by cells.46
Bioorthogonal nanozymes in aqueous solution (NZ1–3) were prepared by encapsulating a highly reactive ruthenium-based catalyst ([CpRu(8HQ)(allyl)PF6,Cp = cyclopentadienyl,8HQ = 8-hydroxy-quinolinate]47 (synthesis and characterization shown in Figure S1–3) into the monolayer of NP1–3 through nanoprecipitation followed by ultrafiltration to remove excess catalyst (Figure 1). Transmission electron microscopy (TEM) and dynamic light scattering (DLS) verified that the size of AuNPs and nanozymes were essentially the same, indicating that there was no aggregation of nanozymes after encapsulation of TMCs (Figure S4–7). The amount of catalyst encapsulation in nanozymes was quantified using inductively coupled plasma mass spectrometry (ICP-MS) by tracking 101Ru and 197Au. Similar Ru/AuNP ratios were obtained for each of the nanozymes, with ~6 Ru catalysts per nanozyme. (Table S1).
The catalytic activity of nanozymes was evaluated by monitoring Rhodamine 110 (Rho110) fluorescent intensity generated from non-fluorescent substrate pro-Rhodamine (pro-Rho: bis-N, N’-allyloxycarbonyl Rhodamine 110, synthesis shown in Figure S8) (Figure 2a). Addition of nanozymes to the pro-Rho solution (pH=7.4) resulted in rapid increase in fluorescence, indicating that the Ru catalyst retained its catalytic activity post-encapsulation (Figure 2b). We calculated the reaction rate of each nanozymes based on a calibration curve using Rhodamine 110 (Rho110) (Figure S9). We found each of the nanozymes had essentially identical catalytic activity in phosphate buffered saline (PBS) solution (Figure 2c).
Figure 2.
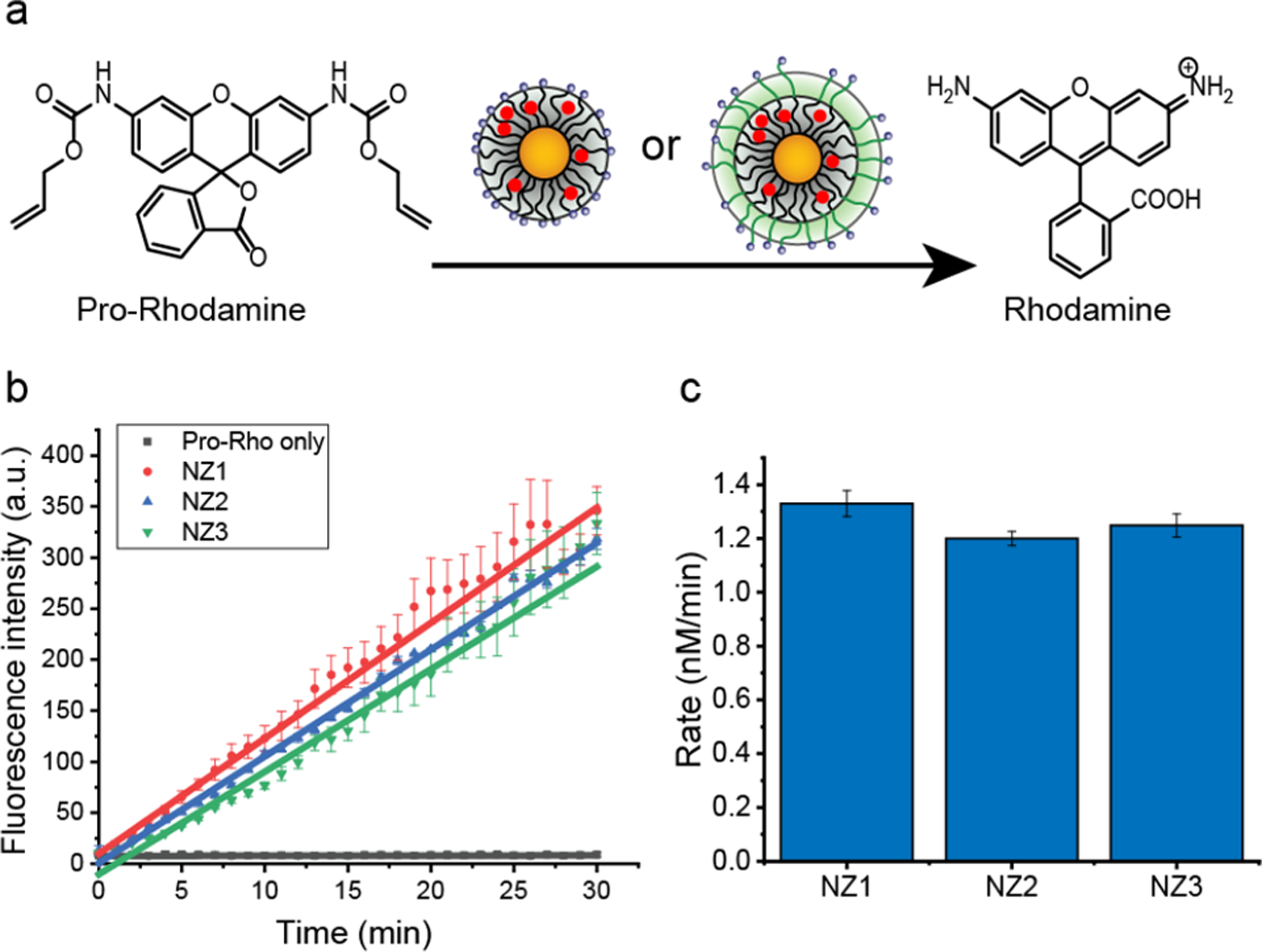
a. Ru-based nanozymes catalyze the deprotection of non-fluorescent pro-Rho to green fluorescent Rhodamine. b. Kinetic studies of nanozymes (200 nM) converting pro-Rho (10 μM) to fluorescent product in phosphate buffered saline (PBS, pH 7.4) solution at 37 °C. The average fluorescence was measured from three independent replicates. Nnanozymes (NZ1, NZ2 and NZ3) had similar catalytic activity. c. Rate of activation of pro-Rho by nanozymes in PBS solution based on the fluorescent calibration curve of Rhodamine (Figure S9).
We next characterized protein corona formation on the nanozymes in serum. Positively charged nanozymes are expected to rapidly absorb serum proteins, forming protein coronas.46 Corona formation was studied in 1 % serum, the highest concentration allowing DLS measurement.45 After 2 h incubation at 37 °C, the hydrodynamic diameter of the cationic nanozymes (NZ1 and NZ2) increased significantly (Figure 3a and b), while no significant change was observed in the zwitterionic nanozyme (NZ3) (Figure 3. c). For NZ1, the mean diameter increased from 5 nm to ~40 nm (Figure 3. a), indicating moderate aggregation. For NZ2, the size increased from ~9 nm to ~15 nm, indicating the formation of a protein corona with minimal aggregation (Figure 3b). DLS and TEM images confirmed the formation of protein corona around nanozymes (Figure S10 and Figure S11). Circular dichroism (CD) results verified that NZ1 induced partial conformational change of protein while NZ2 and NZ3 retained the original protein conformation (Figure 3d).
Figure 3.
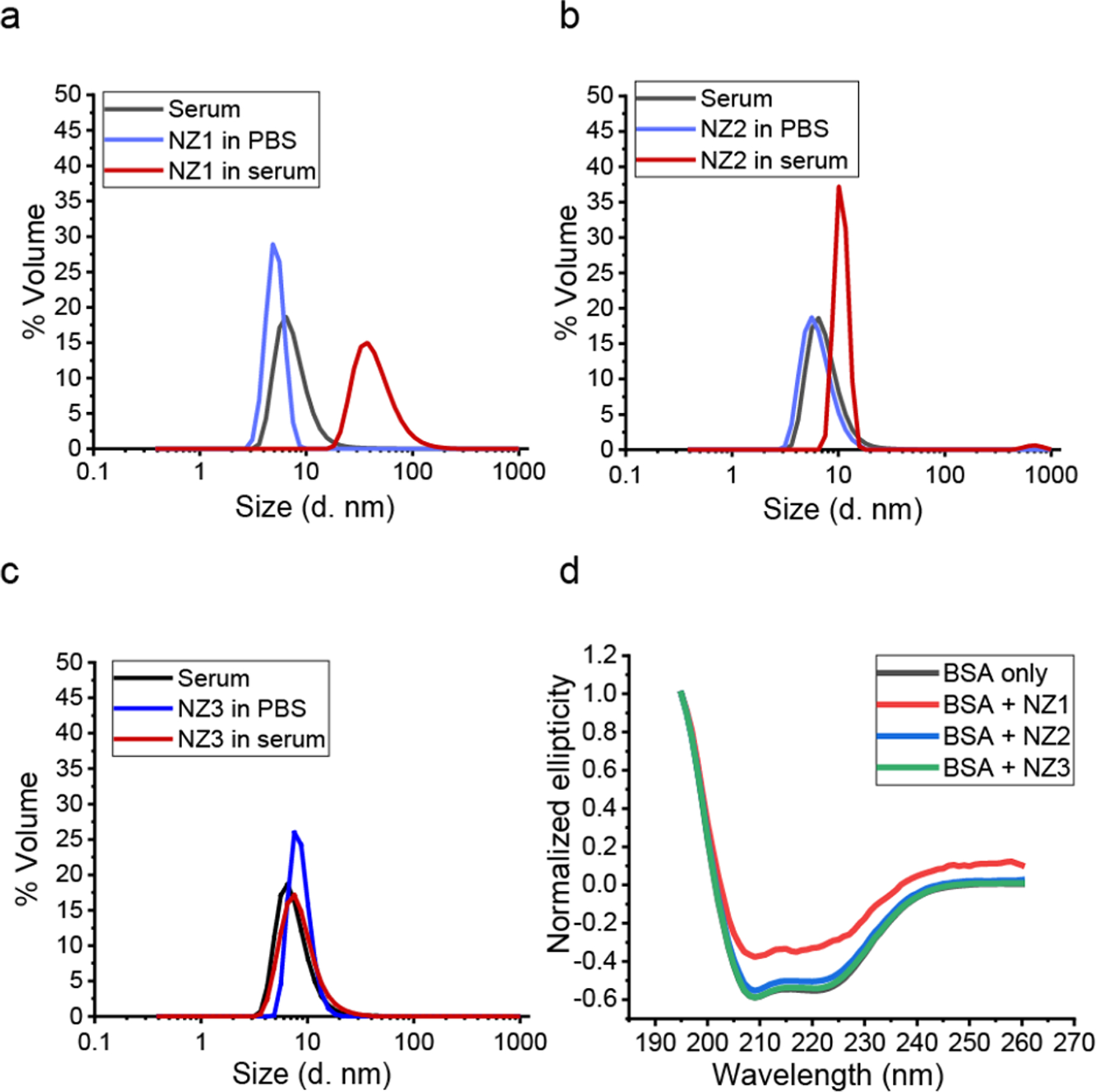
a. b. and c. Size distribution of nanozymes (200 nM) in PBS and 1% serum PBS solution was determined by DLS after 2h incubation under 37 °C. The result showed the measurement from three independent replicates. It indicated that NZ2 and serum protein formed corona-like structure while NZ1 further formed assemblies which caused the shift of size significantly. As a control, NZ3 showed corona free property. d. CD spectrum of BSA with nanozymes. NZ1 cause partial BSA conformational changes, while NZ2 and NZ3 have no effect to denature BSA proteins.
We next evaluated the catalytic efficiency of nanozymes under serum conditions. Nanozymes were pre-incubated in 1% serum at 37 °C for different times (0 h, 0.5 h, 1h and 2 h) and then transferred to the pro-Rho solution. Kinetic studies (Figure 4 a, b and c) indicated that upon increasing the duration of incubation in serum, the catalytic activity of NZ1 decreased significantly (ca. 10-fold decrease) whereas, NZ2 exhibited only a slight decrease (ca. 40% change) (Figure 4d). As expected, zwitterionic NZ3 shown no significant change in the catalytic activity in 1% serum due to its stealth property (Figure 4d). Overall, formation of a hard corona and nanozyme aggregation on NZ1 acted as a supramolecular gate that blocked the access of substrates to the catalyst. However, the soft corona around NZ2 allowed for the slow diffusion of substrates to the catalyst, thereby enabling the retention of catalytic activity. Corona free NZ3, as predicted was not affected by protein corona formation.
Figure 4.
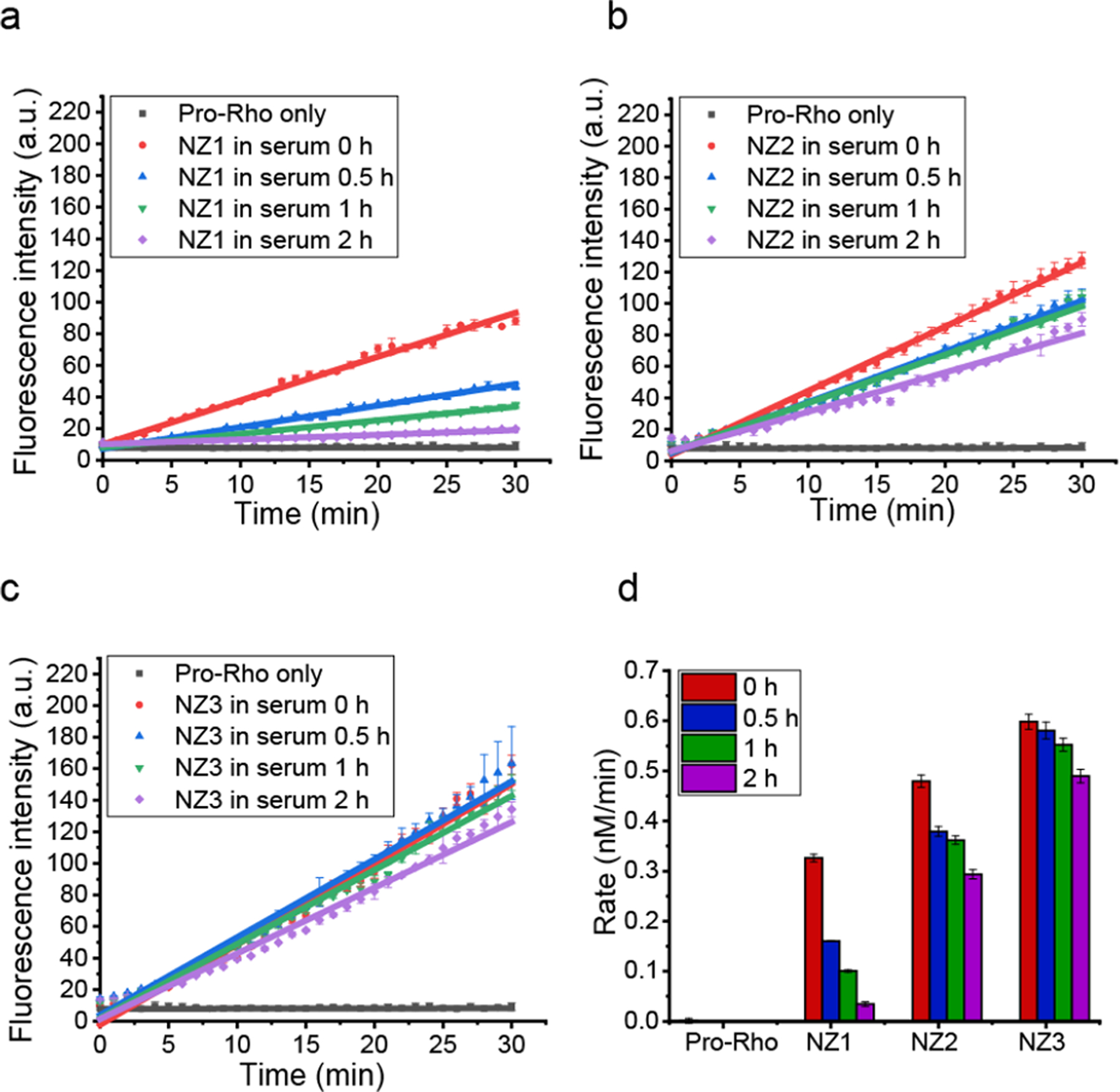
a. b. and c. Kinetic studies of nanozymes (200 nM) in 1% serum with different pre-incubation time at 37 °C. The average fluorescence was measured from three independent replicates. After incubation, NZ1 lost almost all catalytic activity, NZ2 showed a modest lost in efficiency while NZ3 remained similar activity. d. Catalytic activity of pro-Rho by nanozymes after incubation with 1% serum (based on fluorescent calibration curve of Rhodamine in Figure S9).
We next studied the ability of proteolysis to restore catalysis of serum-inhibited NZ1 and NZ2 (Corona-NZ1 and Corona-NZ2) through the removal of protein corona (Figure 5a). After 2 h pre-incubation of nanozymes with 1% serum, trypsin was directly added to corona-nanozyme solutions and further incubated for another 0.5 h. As shown in Figure 5b, a turn-on response of Corona-NZ1 (hard corona) was observed upon the addition of trypsin, as detected through a significant increase (~9-fold) in rate of fluorogenesis. Almost complete reactivation was also observed for Corona-NZ2 (soft corona) (Figure 5c). The rate of activation of pro-Rho was shown in Figure 5d, based on the fluorescent calibration curve of Rhodamine. A significant decrease of corona-particle assemblies was observed by DLS and TEM for both NZ1 and NZ2 (Figure S10 and Figure S12), consistent with proteolysis of the protein corona resulted in restoration of the catalytic activity of both Corona-NZ1 and Corona-NZ2.
Figure 5.
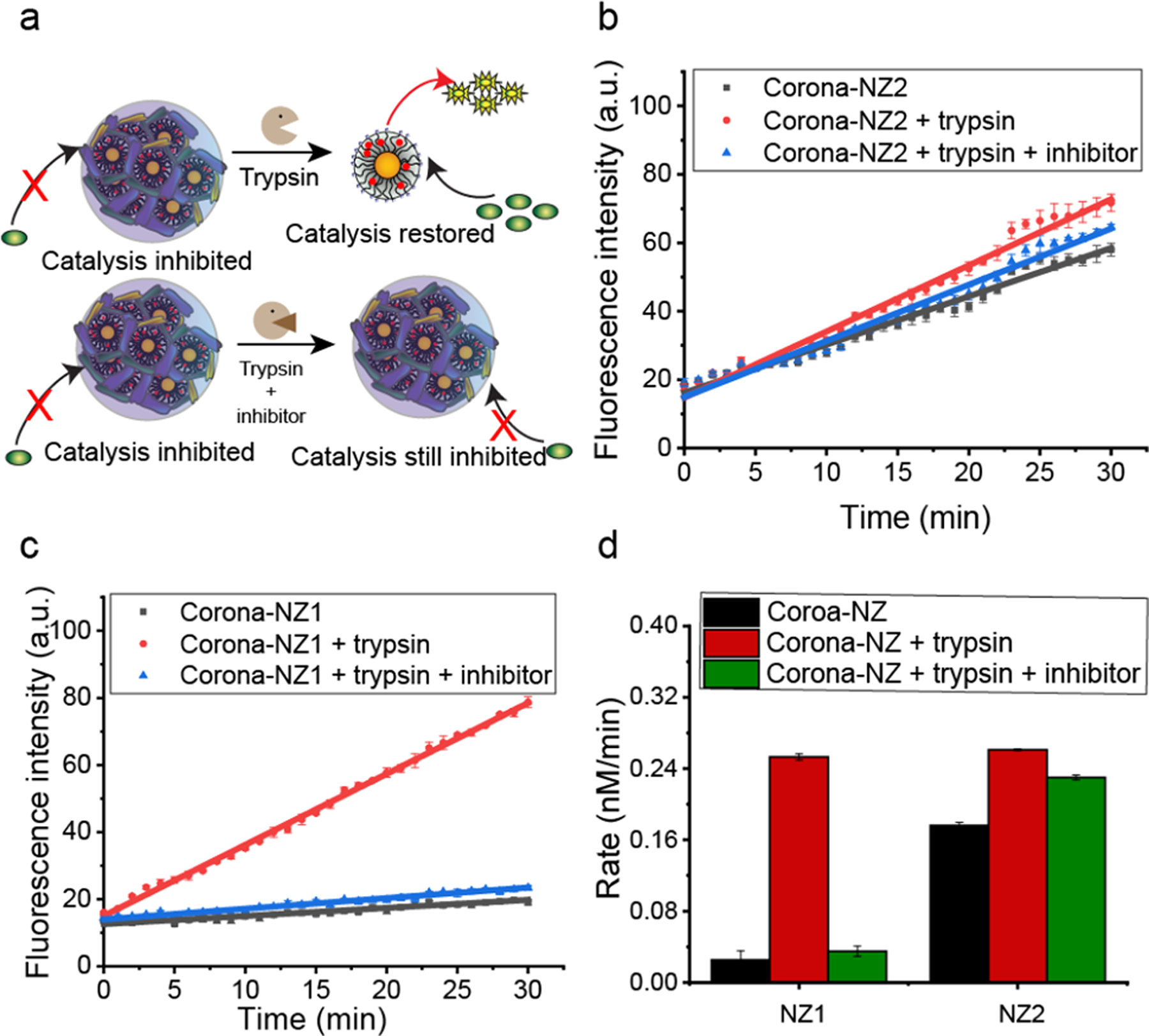
a. Catalytic activity of nanozymes was restored after proteolysis by trypsin, with little restoration observed with inhibitor present. b and c. Kinetic studies of nanozymes (200 nM) in 1% serum with different conditions at 37 °C (Corona-NZs) : 2h incubation in 1% serum; Corona-NZ 1–2 + trypsin: 2h incubation in 1% serum + 30 min Trypsin; and Corona-NZ 1 –2 + trypsin + inhibitor: 2h incubation in 1% serum and inhibitor + 30 min Trypsin.) The results indicated that trypsin rescued nanozymes from deactivation by protein corona. d. Rate of pro-Rho reaction with nanozymes (200nm) under conditions as above. A “turn on” response was observed for NZ1, which confirmed that NZ1 was inhibited by corona formation and reactivated by corona proteolysis.
To further establish that proteolysis is responsible for restoration of catalysis of corona-inhibited nanozymes, we used a protease inhibitor cocktail (P1860,Sigma) to inhibit proteolysis by trypsin. Trypsin was pre-incubated with the protease inhibitor cocktail for 2 h, and corona-nanozymes after (2 h of nanozymes in 1% serum incubation) added and incubated for another 0.5 h. As shown in Figure 5b and c, as expected no significant enhancement of catalytic activity was observed for either; Corona-NZ1 or Corona-NZ2 in the presence of protease inhibitors.
Having studied the catalytic properties of nanozymes in solution, we investigated the intracellular behavior of nanozymes through pro-Rho activation in HeLa cells. Intracellular regeneration of catalysis was predicted due to endogenous endo-some-lysosome mediated proteolysis.48 The cellular internalization of nanozymes (200 nM) was measured by tracking Ru and Au through ICP-MS after 24h incubation. Positively charged nanozymes (NZ1 and NZ2) showed significant cellular uptake while zwitterionic nanozymes (NZ3) had minimal internalization (Figure S13a). HeLa cells incubated with 200nM nanozymes showed no cellular toxicity by Alamar Blue test (Figure S13b).
For pro-Rho activation inside cells, nanozymes (200 nM) were pre-incubated in cell culture media incorporation 10% FBS for 2 h to form a protein corona and then added to HeLa cells along with 100 μM pro-Rho. After 8 h incubation, HeLa cells were imaged under confocal microscopy to observe intracellular catalytic activation. The culture media was also collected, and their fluorescence intensities were measured to quantify catalytic activity in the extracellular space. As expected, both NZ1 and NZ2 had significant green fluorescence (about 30-fold increase) inside cells, and NZ3 had slight green fluorescence) due to low uptake (Figure 6a–c and g). Significantly, no activation of pro-Rho was observed in the supernatant of NZ1, due to blocking the access of substrate to catalyst in presence of the hard corona. NZ2 retained partial catalytic activity in the supernatant, because substrates can gradually diffuse to the catalytic site through the soft corona. Without the for mation of protein corona, NZ3 shown the highest extracellular catalytic activity (Figure 6i). Upon treatment with protease-inhibitor cocktail, both NZ1 and NZ2 lost intracellular catalytic activity (NZ1: ~ 85% and NZ2: ~ 65%), emphasizing the role of intracellular proteases in the restoration of catalysis (Figure 6 d and e). NZ3 retained similar intracellular catalytic activity outside the cells in the presence of inhibitor because of its corona-free property. A limited amount of intracellular fuorescence was observed with NZ3, due to the cell permeability of extracellularly generated Rhodamine.
Figure 6.
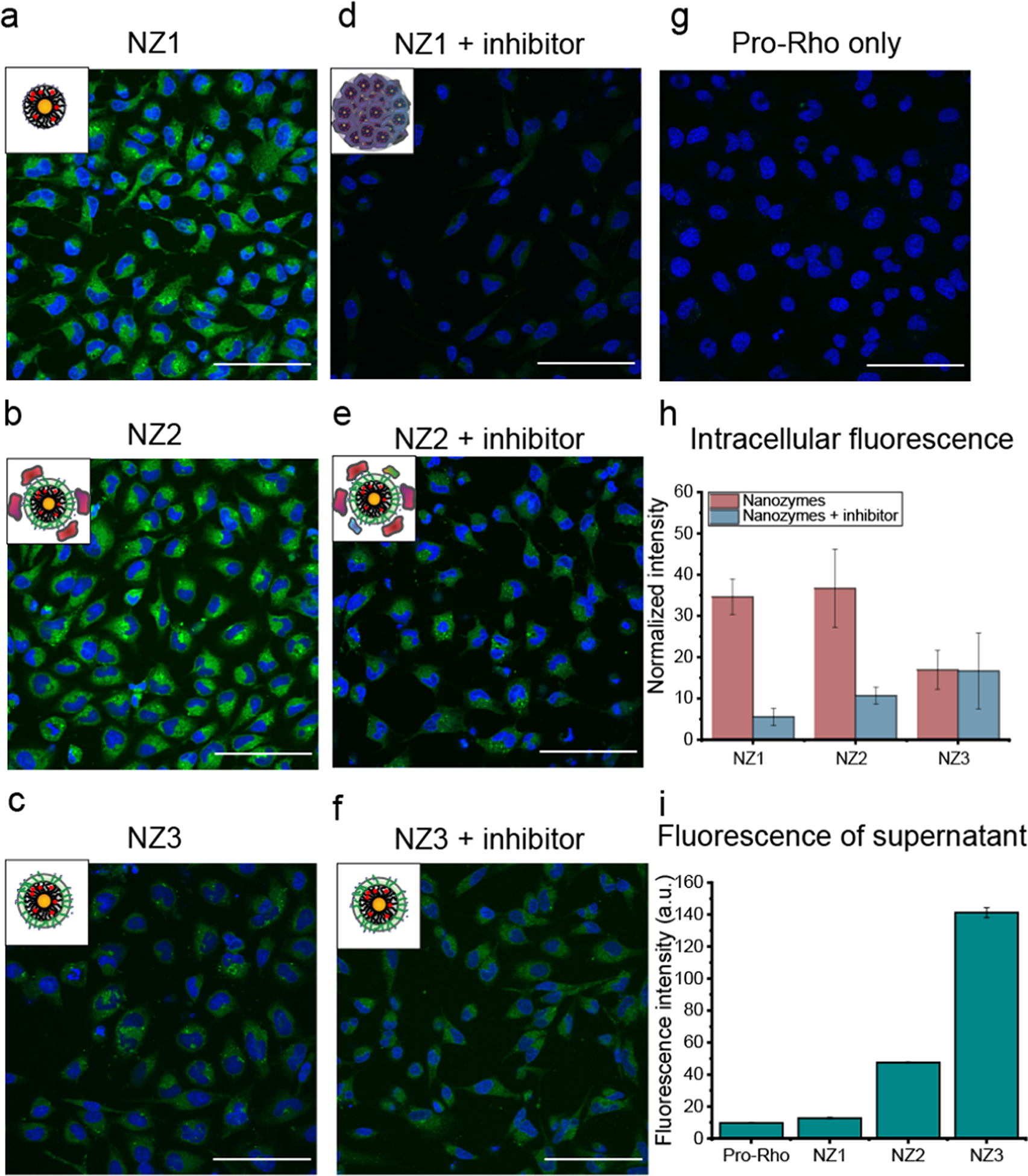
a. to g. Confocal image of HeLa cells after 8h incubation of nanozymes (200 nM) with pro-Rho at 37°C. The result indicated that NZ1 and NZ2 activated pro-Rho inside cells due to the proteolysis by endosomal-lysosomal proteases. NZ2 was still able to perform catalysis in the presence of protease inhibitor while NZ1 showed little to no activation. NZ3 showed limited fluorescence inside cells due to the permeability of extracellularly-generated Rhodamine. Scale bar = 100 μm. h. Quantification of intracellular fluorescence intensity of HeLa cells by ImageJ software after 8h incubation of nanozymes (200 nM) with pro-Rho at 37°C in the presence or without protease-inhibitor cocktail. i. Fluorescence intensity from the supernatant of HeLa cells after 8h incubation of nanozymes (200 nM) with pro-Rho at 37°C. NZ1 remained inactivate outside the cells while NZ2 lose the selectivity and was active both inside and outside the cells, and NZ3 highly active outside the cells due to Rhodamine uptake by the cells.
Conclusion
In this study, we demonstrated a strategy for selective intracellular activation of molecules through endogenous activation of bioorthogonal nanozymes. We observed that the structure of the AuNP ligands dictates the formation of protein coronas and selectively controls catalytic activity of nanozymes. A hard ‘irreversible’ corona (without TEG) deactivated nanozymes through aggregation and steric blocking, while a soft ‘reversible’ corona (with TEG) partially reduced the catalytic activity. The catalytic activity of both soft and hard nanozymes was restored after proteolytic degradation of the protein corona through endogenous proteases present in the endosome and lysosome. Hence, a selective intracellular activation system (without TEG) and an always-on system (with TEG) are obtained by engineering the monolayer of ligands on nanoparticles. This study provides a direct and versatile approach for specific activation of bioorthogonal catalysts through tuning the formation of protein corona on nanozymes. This approach has the potential to reduce off-target effect and extend on-demand generation of imaging agents and localized therapeutics. The generality of this system is suitable for in vivo applications, which are currently under investigations in our group.
Methods
Synthesis of AuNP:
AuNPs with a core diameter of 2 nm the required ligands were synthesized according to previous reports.23 In brief, 2 nm AuNPs were synthesized by Brust-Schiffrin two-phase method to obtain pentanethiol stabilized gold core. Functionalized AuNPs were obtained through ligand exchange reactions in nitrogen atmosphere followed by multiple steps of washing and dialysis. Detailed functionalization and characterization of AuNPs can be found in the supporting information.
Encapsulation of Ru catalyst into the monolayer of AuNP:
1.8 mg (for NZ1), 1.1 mg (for NZ2) or 1.3 (for NZ3) mg Ru catalyst was dissolved in 1 mL acetone solution and added to 1 mL of AuNPs (10 μM) dropwise with continuously stirring. The resulting solution was added into 8 mL of water. Excess catalyst (precipitated) was removed by 0.22 μm PES membrane filter and transferring the filtrate to 10k molecular cutoff ultra-centrifugation tube. The solution was centrifuged at the speed of 7000 min−1 for 5 minutes and washed with Milli-Q water after no color was observed in the filtrate. The concentration of AuNZs was measured by the absorption at 506 nm, and the amount of encapsulated catalysts in AuNPs was measured by ICP-MS by tracking 101Ru and 197Au.
Nanozyme-induced protein (BSA) conformational changes:
1 μM of the respective NZs were incubated with 3.3 μM of bovine serum albumin (BSA) in 5 mM phosphate buffer (pH=7.4) at 37 °C for 2 hours Circular dichroism (CD) experiments were performed on a Jasco J-1500 spectrometer, using a quartz cuvette with a 1 mm path length. Three scans were taken for each sample from 190 to 260 nm at a rate of 20 nm/min. All the experiments were performed at a constant temperature of 20 °C with a 5 min equilibration before the scans.
Kinetic studies in serum:
NZs were pre-incubated with 1% serum at 37 °C for 2 hours. Pro-Rho was used as a substrate to test the catalytic activity of NZs. Substrates were prepared in 96 well black plate, and pre-incubated nanozymes were added obtaining solutions with 5 μM of substrate and 200 nM of NZs, with 5 μM of substrate only was used as negative control. The kinetic results were measured by fluorescence generation (λex = 488 nm, λem = 521 nm, cut off = 515 nm) using a Molecular Devices SpectraMax M2 microplate reader.
For the trypsin studies, pre-incubated NZs were further incubated with trypsin (25 μM) for another 30 min at 37 °C. The kinetic studies were performed under same condition as above (final concentration 5 μM of substrate and 200 nM of NZs) For the trypsin-inhibited study, protease inhibitor cocktail (P1860, Sigma) was diluted to 2% in trypsin solution (25 μM) and incubated for 2 h at 37 °C, and immediately added to the pre-incubated NZs for another 30 min at 37 °C. The kinetic study was performed under same condition as above (5 μM of substrate and 200 nM of NZs) with the presencee of trypsin and inhibitor cocktail.
Confocal Imaging of Catalysis in HeLa cells
HeLa cells were seeded in confocal dishes (100K per dish) 24 h prior to experiments. During the experiment, nanozymes (200nM) were pre-incubated in cell culture media for 2 h, and then transferred to PBS-washed HeLa cells along with 100μM pro-dye and tyhen incubated for 8h. For the protease inhibition study, 100K HeLa cells were seeded and incubated with protease inhibitor cocktail 24 h prior experiments, and then treated as above. All confocal images were obtained after 8h of incubation of NZs with pro-dye under a Nikon A1 spectral detector confocal microscope (A1SP) using a 40X objective. The setting of the confocal microscope: green channel, λex=488 nm and λem = BP 505–530nm; blue channel, λex= 402 nm and λem = BP 450–465 nm (BP=band pass).
Supplementary Material
ACKNOWLEDGMENT
We would thank Dr. Lizz Bartlett for the training on CD. This research was supported by NIH EB022641
Footnotes
Supporting Information
The supporting information is available free of charge on the ACS Publication website.
Preparation and characterization of nanozymes, quantification of Ru and Au using ICP-MS, calibration curve of Rhoda-mine, characterization of protein corona formation and its removal on nanozymes by TEM, and cellular uptake and cell viability of nanozymes (PDF)
REFERENCES
- (1).Sletten EM; Bertozzi CR Bioorthogonal Chemistry: Fishing for Selectivity in a Sea of Functionality. Angew. Chem., Int. Ed 2009, 48, 6974–6998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Bertozzi CR A Decade of Bioorthogonal Chemistry. Acc. Chem. Res 2011, 44, 651–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Prescher JA; Bertozzi CR Chemistry in Living Systems. Nat. Chem. Biol 2005, 1, 13–21. [DOI] [PubMed] [Google Scholar]
- (4).Yusop RM; Unciti-Broceta A; Johansson EMV; Sánchez-Martín RM; Bradley M Palladium-Mediated Intracellular Chemistry. Nat. Chem 2011, 3, 239–243 [DOI] [PubMed] [Google Scholar]
- (5).Streu C; Meggers E Ruthenium-Induced Allylcarbamate Cleavage in Living Cells. Angew. Chem., Int. Ed 2006, 45, 5645–5648 [DOI] [PubMed] [Google Scholar]
- (6).Sasmal PK; Carregal-Romero S; Han AA; Streu CN; Lin Z; Namikawa K; Elliott SL; Köster RW; Parak WJ; Meggers E Catalytic Azide Reduction in Biological Environments. ChemBioChem 2012, 13, 1116–1120 [DOI] [PubMed] [Google Scholar]
- (7).Völker T; Meggers E Transition-Metal-Mediated Uncaging in Living Human Cells-an Emerging Alternative to Photolabile Protecting Groups. Curr. Opin. Chem. Biol 2015, 25, 48–54 [DOI] [PubMed] [Google Scholar]
- (8).Hong V; Steinmetz NF; Manchester M; Finn MG Labeling Live Cells by Copper-Catalyzed Alkyne - Azide Click Chemistry. Bioconjug. Chem 2010, 21, 1912–1916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Li J; Yu J; Zhao J; Wang J; Zheng S; Lin S; Chen L; Yang M; Jia S; Zhang X; Chen PR Palladium-Triggered Deprotection Chemistry for Protein Activation in Living Cells. Nat. Chem 2014, 6, 352–361 [DOI] [PubMed] [Google Scholar]
- (10).Li J; Chen PR Development and Application of Bond Cleavage Reactions in Bioorthogonal Chemistry. Nat. Chem. Biol 2016, 12, 129–137 [DOI] [PubMed] [Google Scholar]
- (11).Bai Y; Chen J; Zimmerman SC Designed Transition Metal Catalysts for Intracellular Organic Synthesis. Chem. Soc. Rev 2018, 47, 1811–1821 [DOI] [PubMed] [Google Scholar]
- (12).Vidal C; Tomás-Gamasa M; Gutiérrez-González A; Mascareñas JL Ruthenium-Catalyzed Redox Isomerizations inside Living Cells. J. Am. Chem. Soc 2019, 141, 5125–5129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Tomás-Gamasa M; Martínez-Calvo M; Couceiro JR; Mascareñas JL Transition Metal Catalysis in the Mitochondria of Living Cells. Nat. Commun 2016, 7, 12538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Vidal C; Destito P; López F; Mascareñas JL Concurrent and Orthogonal Gold(I) and Ruthenium (II) Catalysis inside Living Cells. Nat. Commun 2018, 9, 1913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Davis HJ; Ward TR Artificial Metalloenzymes: Challenges and Opportunities. ACS Cent. Sci 2019, 5, 1120–1136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Jeschek M; Reuter R; Heinisch T; Trindler C; Klehr J; Panke S; Ward TR Directed Evolution of Artificial Metalloenzymes for In Vivo Metathesis. Nature 2016, 537, 661–665 [DOI] [PubMed] [Google Scholar]
- (17).Zhang X; Huang R; Gopalakrishnan S; Cao-milán R; Rotello VM Bioorthogonal Nanozymes: Progress towards Therapeutic Applications. Trends Chem 2019, 1, 90–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Huang Y; Ren J; Qu X Nanozymes: Classification, Catalytic Mechanisms, Activity Regulation, and Applications. Chem. Rev 2019, 119, 4357–4412 [DOI] [PubMed] [Google Scholar]
- (19).Destito P; Sousa-Castillo A; Couceiro JR; López F; Correa-Duarte MA; Mascareñas JL Hollow Nanoreactors for Pd-Catalyzed Suzuki - Miyaura Coupling and O -Propargyl Cleavage Reactions in Bio-Relevant Aqueous Media. Chem. Sci 2019, 10, 2598–2603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Wang F; Zhang Y; Du Z; Ren J; Qu X Designed Heterogeneous Palladium Catalysts for Reversible Light-Controlled Bioorthogonal Catalysis in Living Cells. Nat. Commun 2018, 9, 1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Wang F; Zhang Y; Liu Z; Du Z; Zhang L; Ren J; Qu X A Biocompatible Heterogeneous MOF – Cu Catalyst for In Vivo Drug Synthesis in Targeted Subcellular Organelles. Angew. Chem., Int. Ed 2019, 58, 6987–6992 [DOI] [PubMed] [Google Scholar]
- (22).Bray TL; Salji M; Brombin A; Ana MP; Galbraith LCA; Patton EE; Leung HY; Unciti-Broceta A Bright Insights into Palladium-Triggered Local Chemotherapy. Chem. Sci 2018, 9, 7354–7361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Tonga GY; Jeong Y; Duncan B; Mizuhara T; Mout R; Das R; Kim ST; Yeh YC; Yan B; Hou S; Rotello VM Supramolecular Regulation of Bioorthogonal Catalysis in Cells Using Nanoparticle-Embedded Transition Metal Catalysts. Nat. Chem 2015, 7, 597–603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Cao-milán R; He LD, Shorkey S; Tonga GY; Wang L-S; Zhang X; Uddin I; Das R; Sulakc M; Rotello VM Molecular Systems Design & Engineering Modulating the Catalytic Activity of Enzyme-like Nanoparticles through Their Surface. Mol. Syst. Des. Eng 2017, 2, 624–628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Wu J; Wang X; Wang Q; Lou Z; Li S; Zhu Y; Qin L; Wei H Nanomaterials with Enzyme-like Characteristics (Nanozymes): Next-Generation Artificial Enzymes (II). Chem. Soc. Rev 2019, 48, 1004–1076 [DOI] [PubMed] [Google Scholar]
- (26).Gupta A; Das R; Yesilbag Tonga G; Mizuhara T; Rotello VM Charge-Switchable Nanozymes for Bioorthogonal Imaging of Biofilm-Associated Infections. ACS Nano 2018, 12, 89–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Clavadetscher J; Indrigo E; Chankeshwara SV; Lilienkampf A; Bradley M In-Cell Dual Drug Synthesis by Cancer-Targeting Palladium Catalysts. Angew. Chem., Int. Ed 2017, 129, 6968–6972 [DOI] [PubMed] [Google Scholar]
- (28).Sancho-albero M; Rubio-ruiz B; Pérez-lópez AM; Sebastián V; Martín-duque P; Arruebo M; Santamaría J; Uncitibroceta A Cancer-Derived Exosomes Loaded with Ultrathin Palladium Nanosheets for Targeted Bioorthogonal Catalysis. Nat. Catal 2019, 2, 864–872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Okamoto Y; Kojima R; Schwizer F; Bartolami E; Heinisch T; Matile S; Fussenegger M; Ward TR A Cell-Penetrating Artificial Metalloenzyme Regulates a Gene Switch in a Designer Mammalian Cell. Nat. Commun 2018, 9, 1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Das R; Landis RF; Tonga GY; Cao-mila R; Luther DC; Rotello VM Control of Intra- versus Extracellular Bioorthogonal Catalysis Using Surface- Engineered Nanozymes. ACS Nano 2019, 13, 229–235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Miller MA; Askevold B; Mikula H; Kohler RH; Pirovich D; Weissleder R Nano-Palladium Is a Cellular Catalyst for In Vivo Chemistry. Nat. Commun 2017, 8, 15906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Miller MA; Mikula H; Luthria G; Li R; Kronister S; Prytyskach M; Kohler RH; Mitchison T; Weissleder R Modular Nanoparticulate Prodrug Design Enables Efficient Treatment of Solid Tumors Using Bioorthogonal Activation. ACS Nano 2018, 12, 12814–12826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Rosenblum D; Joshi N; Tao W; Karp JM; Peer D Progress and Challenges towards Targeted Delivery of Cancer Therapeutics. Nat. Commun 2018, 9, 1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Peer D; Karp JM; Hong S; Farokhzad OC; Margalit R; Langer R Nanocarriers as an Emerging Platform for Cancer Therapy. Nat. Nanotechnol 2007, 2, 751–760 [DOI] [PubMed] [Google Scholar]
- (35).Mura S; Nicolas J; Couvreur P Stimuli-Responsive Nanocarriers for Drug Delivery. Nat. Mater 2013, 12, 991–1003 [DOI] [PubMed] [Google Scholar]
- (36).Chen H; Liu D; Guo Z Endogenous Stimuli-Responsive Nanocarriers for Drug Delivery. Chem. Lett 2016, 45, 242–249 [Google Scholar]
- (37).Fleige E; Quadir MA; Haag R Stimuli-Responsive Polymeric Nanocarriers for the Controlled Transport of Active Compounds: Concepts and Applications. Adv. Drug Deliv. Rev 2012, 64, 866–884 [DOI] [PubMed] [Google Scholar]
- (38).De R; Aili D; Stevens MM Enzyme-Responsive Nanoparticles for Drug Release and Diagnostics. Adv. Drug Deliv. Rev 2012, 64, 967–978 [DOI] [PubMed] [Google Scholar]
- (39).Mu J; Lin Jing, Huang Peng, and X. C Development of Endogenous Enzyme-Responsive Nanomaterials for Theranostics. Chem. Soc. Rev 2018, 47, 5554–5573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Hong R; Fischer NO; Verma A; Goodman CM; Emrick T; Rotello VM Control of Protein Structure and Function through Surface Recognition by Tailored Nanoparticle Scaffolds. J. Am. Chem. Soc 2004, 126, 739–743 [DOI] [PubMed] [Google Scholar]
- (41).Worrall JWE; Verma A; Yan H; Rotello VM “Cleaning” of Nanoparticle Inhibitors via Proteolysis of Adsorbed Proteins. Chem. Commun 2006, 22, 2338–2340 [DOI] [PubMed] [Google Scholar]
- (42).Cedervall T; Lynch I; Lindman S; Berggård T; Thulin E; Nilsson H; Dawson KA; Linse S Understanding the Nanoparticle - Protein Corona Using Methods to Quantify Exchange Rates and Affinities of Proteins for Nanoparticles. Proc. Natl. Acad. Sci 2007, 104, 2050–2055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Salvati A; Dawson KA What Does the Cell See? Nat. Nanotechnol 2009, 4, 546–547 [DOI] [PubMed] [Google Scholar]
- (44).Lesniak A; Fenaroli F; Monopoli MP; Christoffer A; Dawson KA; Salvati A Effects of the Presence or Absence of a Protein Corona on Silica Nanoparticle Uptake and Impact on Cells. ACS Nano 2012, 6, 5845–5857 [DOI] [PubMed] [Google Scholar]
- (45).Moyano DF; Saha K; Prakash G; Yan B; Kong H; Yazdani M; Rotello VM Fabrication of Corona-Free Nanoparticles with Tunable Hydrophobicity. ACS Nano 2014, 8, 6748–6755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Saha K; Rahimi M; Yazdani M; Kim ST; Moyano DF; Hou S; Das R; Mout R; Rezaee F; Mahmoudi M; Rotello VM Regulation of Macrophage Recognition through the Interplay of Nanoparticle Surface Functionality and Protein Corona. ACS Nano 2016, 10, 4421–4430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Völker T; Meggers E Chemical Activation in Blood Serum and Human Cell Culture: Improved Ruthenium Complex for Catalytic Uncaging of Alloc-Protected Amines. ChemBioChem 2017, 18, 1083–1086. [DOI] [PubMed] [Google Scholar]
- (48).Chanana M; Rivera-gil P; Correa-Duarte MA; Liz-Marzán LM; Parak WJ Physicochemical Properties of Protein-Coated Gold Nanoparticles in Biological Fluids and Cells before and after Proteolytic Digestion. Angew. Chem., Int. Ed 2013, 52, 4179–4183. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.