

Abstract
The majority of patients with spinal muscular atrophy (SMA) identified to date harbor a biallelic exonic deletion of SMN1. However, there have been reports of SMA-like disorders that are independent of SMN1, including those due to pathogenic variants in the glycyl-tRNA synthetase gene (GARS1). We report three unrelated patients with de novo variants in GARS1 that are associated with infantile-onset SMA (iSMA). Patients were ascertained during inpatient hospital evaluations for complications of neuropathy. Evaluations were completed as indicated for clinical care and management and informed consent for publication was obtained. One newly identified, disease-associated GARS1 variant, identified in two out of three patients, was analyzed by functional studies in yeast complementation assays. Genomic analyses by exome and/or gene panel and SMN1 copy number analysis of three patients identified two previously undescribed de novo missense variants in GARS1 and excluded SMN1 as the causative gene. Functional studies in yeast revealed that one of the de novo GARS1 variants results in a loss-of-function effect, consistent with other pathogenic GARS1 alleles. In sum, the patients’ clinical presentation, assessments of previously identified GARS1 variants and functional assays in yeast suggest that the GARS1 variants described here cause iSMA. GARS1 variants have been previously associated with Charcot–Marie–Tooth disease (CMT2D) and distal SMA type V (dSMAV). Our findings expand the allelic heterogeneity of GARS-associated disease and support that severe early-onset SMA can be caused by variants in this gene. Distinguishing the SMA phenotype caused by SMN1 variants from that due to pathogenic variants in other genes such as GARS1 significantly alters approaches to treatment.
Keywords: clinical genetics, CMT, GARS, hereditary motor and sensory neuropathy, non-SMN1 SMA
1 |. INTRODUCTION
Spinal muscular atrophy (SMA) and Charcot–Marie-Tooth (CMT) disease are hereditary neuromuscular disorders that lead to progressive weakness and muscle wasting. CMT disease refers to a group of disorders characterized by a progressive chronic motor and sensory polyneuropathy. CMT2, specifically Charcot–Marie–Tooth disease 2D (CMT2D), is the sub-type associated with variants in the glycyl-RNA synthetase gene, GARS1 (also referred to as GARS, which we use throughout the manuscript). CMT2D typically presents with muscle weakness of the hands, characterized by distal motor and sensory neuropathy.
The SMA group of disorders are typically characterized by symmetric proximal muscle weakness and atrophy resulting from progressive degeneration and loss of anterior horn cells in the spinal cord and the brain stem nuclei. The first SMA patients (Russman, 2007) were diagnosed based on clinical and histopathological features. Unified inclusion criteria for SMA were developed by the International SMA Consortium (Munsat, 1991). Patients were stratified by severity based on criteria including clinical features, age of onset and age at death (Drew, Blair, & Nicholson, 2011).
In contrast to the extreme allelic and locus heterogeneity observed in CMT (Timmerman, Strickland, & Züchner, 2014), the vast majority of patients with SMA harbor a biallelic exonic deletion of SMN1 mapping to the long arm of chromosome 5. There has been increasing appreciation of non-SMN1 forms of SMA since the initial reports where it has been estimated that about 5% of SMA patients have a genetic etiology other than SMN1 (Wirth et al., 1999). Over 15 loci have been associated with clinical presentations that mimic SMN1-related SMA. These non-SMN1 SMAs are sometimes referred to as non-5q SMA and typically affect the proximal muscles. On the severe end of the spectrum within the non-5q SMA subgroup is SMA with respiratory distress (SMARD), due to biallelic pathogenic variants in IGHMBP2 (Grohmann et al., 2001; Guenther et al., 2007). Yet another subgroup, SMA type V distal type (dSMAV), presents with distal weakness and no sensory involvement. Both CMT2D and dSMAV are caused by heterozygosity for pathogenic variants in GARS gene, encoding glycyl-tRNA synthetase. In fact, recent literature suggests that dSMAV and CMT2D are on a clinical spectrum of GARS-associated axonal neuropathy, not distinct diseases (Antonellis, Goldfarb, & Sivakumar, 1993). In addition, multiple case reports have described patients with a pathogenic variant in GARS with an iSMA like phenotype (Eskuri, Stanley, Moore, & Mathews, 2012; James et al., 2006; Liao et al., 2015).
In humans, there are 37 genes encoding aminoacyl-tRNA synthetases (aaRS) (Antonellis & Green, 2008). Variants in genes encoding aaRSs have been associated with various human diseases including early onset, multi-system recessive phenotypes and dominant peripheral neuropathy (Fuchs et al., 2019; Meyer-Schuman & Antonellis, 2017). Over the past 16 years, multiple studies have associated defects in aaRS with CMT (Storkebaum, 2016) and dSMAV (Tsai et al., 2017).
The GARS gene was the first aaRS to be implicated in a human inherited disease (Antonellis et al., 2003, 1993). GARS is critical for charging glycine onto cognate tRNA molecules and is required for protein synthesis in the mitochondria and the cytosol (Chihara, Luginbuhl, & Luo, 2007; Qin et al., 2014). The human GARS protein consists of a N-terminal WHEP-TRS domain, a catalytic domain interspersed by three insertion domains and a C-terminal anticodon-binding domain (Antonellis et al., 2003; Qin et al., 2014). The majority of patients reported with GARS-associated dominant neuropathy present with later onset CMT2D or dSMAV (Antonellis et al., 2003). Here we present three patients with severe, early-onset SMA. Each patient carries a de novo pathogenic variant in either the catalytic or anticodon binding domain of GARS. Analysis of these results indicates that pathogenic variants in the anticodon binding and the catalytic domains can result in an early-onset form of SMA.
2 |. PATIENTS AND METHODS
Patients were ascertained in the course of routine clinical care provided at three institutions. Genetic testing, imaging, neurophysiology, and histopathology studies were done on a clinical basis.
Yeast complementation assays were performed as previously described (Oprescu et al., 2017). The p.Ile334Asn GARS variant was modeled in the human wild-type GARS ΔMTSΔWHEP open-reading frame (Chien et al., 2014; Oprescu et al., 2017). Mutagenesis was performed using the Quickchange II XL Site-Directed Mutagenesis Kit, a sequence-validated wild-type GARS ΔMTSΔWHEP pDONR221 construct, and mutation-specific primers (available upon request). Reactions were transformed into Escherichia coli, and plasmid DNA was purified from individual colonies and sequenced to confirm the presence of the mutation and the absence of PCR-induced errors. Sequence-validated constructs for wild-type and p.Ile334Asn GARS ΔMTSΔWHEP were cloned into pYY1 (Chien et al., 2014) using Gate-way cloning technology (Invitrogen). The resulting reactions were transformed into E. coli, and plasmid DNA was purified from individual colonies and digested with BsrGI to confirm proper recombination. To assess the ability of wild-type and p.Ile334Asn GARS ΔMTSΔWHEP to support cellular growth, a previously validated haploid yeast strain with the endogenous GRS1 locus deleted and viability maintained via a pRS316 vector bearing wild-type GRS1 (Turner, Lovato, & Schimmel, 2000) was transformed with wild-type GARS ΔMTSΔWHEP, p.Ile334Asn GARS ΔMTSΔWHEP, or the pYY1 vector with no GARS insert. Transformed yeast cells were grown on solid media lacking leucine (pYY1 harbors the LEU2 gene) and uracil (pRS316 harbors the URA3 gene). Colonies were grown to saturation in 2 mL -leu-ura liquid medium at 30°C and 275 rpm for 48 hr. A 1 mL aliquot from each 2 mL culture was spun down at 10,000 rpm and re-suspended in 50 μL UltraPure RNase/DNase-free water. Undiluted cultures and dilutions of 1:10 and 1:100 were spotted on 0.1% 5-FOA complete solid medium (Teknova, Hollister, CA) to select for cells that have spontaneously lost the URA3-bearing maintenance vector (Boeke, Trueheart, Natsoulis, & Fink, 1987). Yeast viability was assessed by visual inspection after 5 days of incubation at 30°C. Two colonies per transformation were assayed.
3 |. RESULTS
3.1 |. Clinical features of patients
We identified three patients with de novo variants in the GARS gene that led to very early onset neuropathy with respiratory distress, mimicking iSMA. Clinical features of these patients are summarized in Table 1.
TABLE 1.
Clinical features of three patients with de novo GARS variants and infantile SMA presentation with respiratory distress
| Patient 1 | Patient 2 | Patient 3 | |
|---|---|---|---|
| Pathogenic variant | c.1001T>A (p.Ile334Asn) | c.1001T>A (p.Ile334Asn) | c.1954G>C (p.Gly652Arg) |
| Inheritance | de novo | de novo | de novo |
| Age of presentation | 9 weeks | 6 weeks | Day 0 |
| Age at most recent encounter | 6 months | 39 months | 14 months |
| Initial presentation of respiratory distress, poor feeding and muscle weakness | + | + | + |
| Neurologic | |||
| Hypotonia, lower extremities > upper extremities | + | + | + |
| Fasciculations | + | − | + |
| Hyporeflexia | + | + | + |
| Atrophy | − | + | + |
| Pes Cavus | − | + | − |
| Contractures | − | + | + |
| Sensory impairment | a | + | − |
| Respiratory | |||
| Stridor | + | + | + |
| Weak cry | + | + | + |
| Elevated right diaphragm | + | + | b |
| Chronic ventilation | + | + | + |
| Feeding/nutrition | |||
| Poor feeding | + | + | + |
| G-tube dependent | + | + | + |
Absence of sural nerve response.
Poor diaphragm movement noted on ultrasound.
Patient 1 is a former appropriate-for-age (AGA) 40w4d male, born by spontaneous vaginal delivery to a 29-year-old Hispanic female and a 28-year-old Hispanic male. The pregnancy was notable for oligohydramnios with no associated complications. The parents were non-consanguineous and the family history was negative for neuropathy. At birth, the patient had subtle decreased movement in his lower extremities compared to his upper extremities, but without notable hypotonia, respiratory distress, or feeding difficulties.
At approximately 9 weeks of age, he was hospitalized due to respiratory distress, decreased movements, and poor feeding. Initial physical exam findings were significant for an alert infant with a weak cry and associated cyanosis. He progressed to respiratory failure requiring mechanical ventilation. His motor exam was significant for generalized hypotonia with antigravity strength in the upper extremities and inability to overcome gravity in the lower extremities. Proximal muscles were weaker than distal muscles. Muscle stretch reflexes were absent. Tongue fasciculations were present. There was no obvious clinical evidence of sensory impairment, although detailed sensory exam was limited due to his young age and sedation. At 11 months of age, he remains alert, on 24-h chronic ventilation and gastrostomy tube, and requires assistance for all activities of daily living.
His diagnostic evaluation was suggestive of motor neuron involvement in many aspects. MRI of his complete spine showed marked volume loss of ventral cauda equina as well as atrophy of lower lumbar and sacral paraspinal musculature, gluteal and iliopsoas muscles, and pelvic girdle muscles (Figure 1a–c). On nerve conduction study, compound muscle action potentials had severely decreased amplitude with normal latency while some muscles were non-responsive. Sensory nerve action potentials were normal except his right sural nerve that was nonresponsive and bilateral medial plantar nerves that showed mildly prolonged latencies with the decreased amplitude on the right compared to the left. Needle EMG showed decreased insertional activity and recruitment in all tested muscles. Some muscles showed large amplitude motor unit potential, and one muscle with severely decreased recruitment showed polyphasic activity. Biopsy of his quadriceps muscle had marked variation in fiber size with small group and fascicular atrophy (Figure 1d).
FIGURE 1.
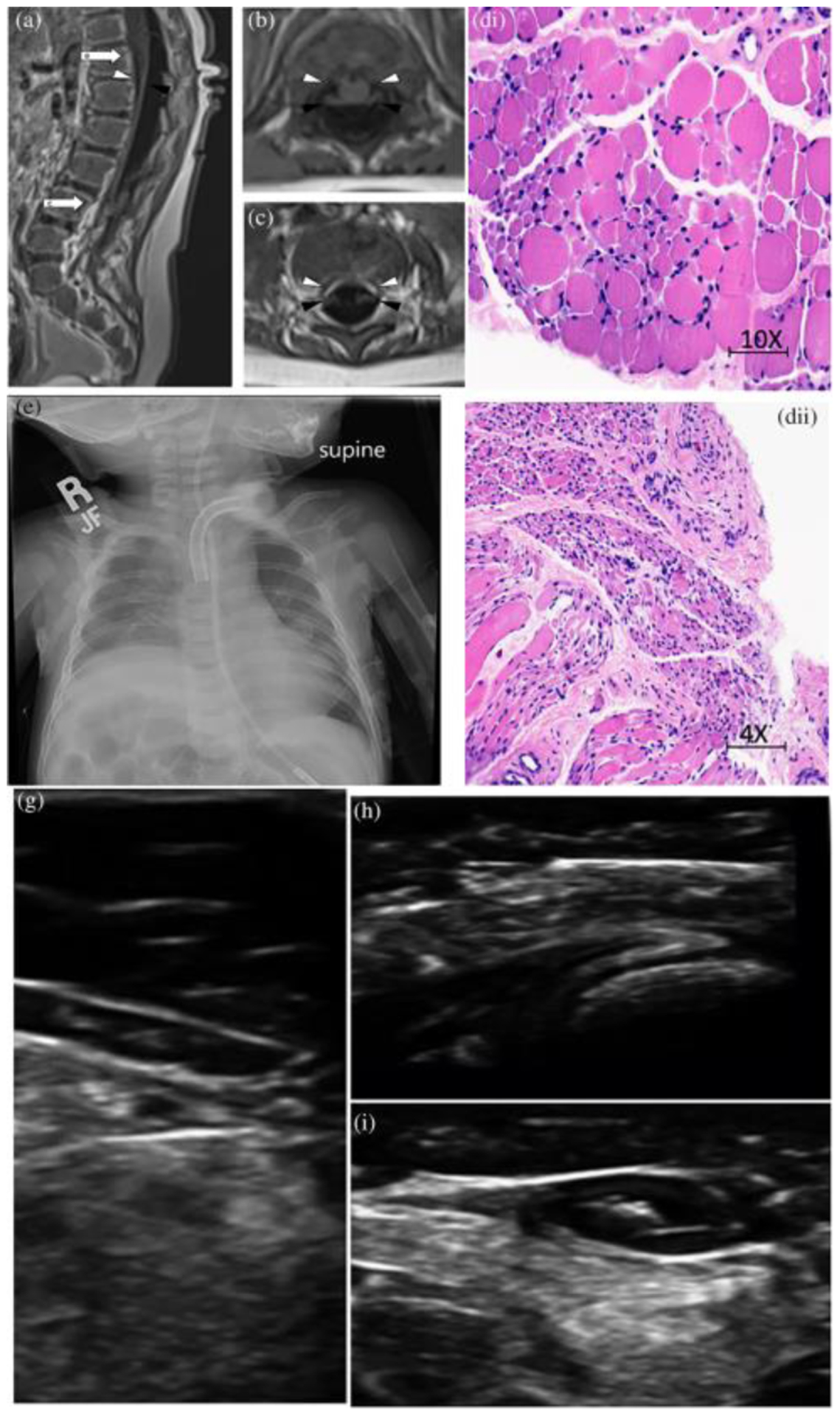
(a) Patient 1 MRI of the spine in sagittal view. Note marked volume loss of ventral cauda equina (white arrowhead) and intact dorsal cauda equina (black arrowhead). White arrows signify the spinal level of images (b) and (c) that show marked volume loss of ventral cauda equina (white arrowhead) and intact dorsal cauda equine (black arrowhead) in axial view. (d) Patient 1, hemotoxylin and eosin stain of quadriceps muscle with marked variation in fiber size with small group and fascicular atrophy typical of SMA1; (e) Patient 2, chest X-ray with elevation of Right hemi-diaaphragm; (f) Patient 2, hemotoxylin and eosin stain of muscle with variation in muscle size, rare myophagocytosis, regenerating fibers (g), (h), and (i) Patient 3, muscle ultrasound with severe atrophy and neurogenic changes of distal > proximal muscles
Patient 2 was born to his Hispanic mother and father at 39 weeks gestation via a repeat C-section. His weight was appropriate for gestational age at 7 pounds. His mother had an uncomplicated pregnancy and he was discharged on day of life 4 without any detected medical issues. There was no consanguinity and no family history of any neuromuscular disorders.
At 6 weeks of age he began having repeated events of respiratory failure requiring multiple intubations and ultimately a tracheostomy ~2 months of age. He has remained on constant mechanical ventilation since that time. His chest X-rays consistently revealed right hemi-diaphragm elevation (Figure 1e). At 4 months of age he had an ultrasound evaluation revealing minimal movement of the right hemi-diaphragm representing likely diaphragmatic paralysis versus significant paresis. A subsequent ultrasound at 9 months of age revealed apparent diminished diaphragmatic motion bilaterally.
He was first evaluated by neurology at 9.5 months of age due to progressive weakness, distal greater than proximal, and respiratory failure. He had significant generalized weakness including lack of antigravity strength proximally and nearly no movement distally. At that time, he had no fixed contractures but hands had claw-like appearance suggestive of significant intrinsic muscle atrophy and bilateral caval deformity of his feet. Reflexes were absent throughout. There were no overt fasciculations. There was no evidence of sensory deficits on clinical exam. He has always been very alert and with apparently normal cognitive function.
He was initially able to roll over around 3 months of age but lost this skill at 18 months of age. He attained the ability to sit at 12 months of age and maintained this at last follow up at 39 months of age but is unable to attain this posture independently. At 39 months of age he remained unable to perform antigravity movements proximally and had very little distal movements. He had significant ankle contractures and has had surgery for his progressive kyphoscoliosis.
His evaluation has included a MRI of the brain and C-spine completed at 9 months of age which revealed slightly increased prominence of the bi-frontal and temporal subarachnoid spaces and mild prominence of the lateral and third ventricles. The cervical spine MRI was normal. At 10 months of age he had a left thigh muscle biopsy with features of denervation atrophy with group atrophy with large groups of atrophic fibers (both type 1 and 2 fibers) as well as features of chronic active myopathy (variation in muscle size, rare myophagocytosis, regenerating fibers, marked endomysial fibrosis, occasional darkly stained hyalinized fibers on trichrome stain, indistinct fiber typing on ATPase pH 9.4, and co-expression of fast and slow myosin) (Figure 1f).
An EMG/NCV was performed at 3 years of age which revealed a severe, active, diffuse, axonal sensory-motor but predominantly motor peripheral neuropathy. Compound muscle action potentials were absent in all tested nerves (right peroneal, tibial, median, ulnar, and musculocutaneous nerves). Sensory nerve action potentials were normal in the median and ulnar nerves but absent in the right medial plantar and sural nerves. Needle EMG revealed numerous fibrillation potentials, complex repetitive discharges, and increased insertional activity. There were a few fasciculation potentials. Activation was examined in a few muscles, which revealed long duration and high amplitude motor units with reduced recruitment.
Patient 3 was the AGA product of a 38-week gestation delivered by spontaneous vaginal delivery to a Caucasian father and French Canadian mother. There was no consanguinity. Pregnancy was uncomplicated and with normal perceived fetal movement. Apgar scores were 8 and 9 at 1 and 5 min, respectively. Inspiratory stridor and hypotonia were noted at birth. He was subsequently diagnosed with vocal cord paralysis. By 1 month of age, he was having difficulty maintaining weight, largely due to poor suck, swallowing difficulties and early fatigue with feeding. He developed more notable respiratory insufficiency at 10 weeks, also with bulbar weakness, including drooling. BiPAP was trialed but following an acute GI bleed he was intubated and subsequently failed multiple extubation attempts. He underwent tracheostomy at 4 months of age and remains ventilator dependent. He had progressive weakness of his extremities, initially with distal weakness that progressed to proximal involvement. At 5 months, he was able to move his legs and at 7 months of age he was still able to move his arms; however, he subsequently lost those abilities. At the time of his last examination, at 14 months of age, he was alert and attentive to his surroundings with marked weakness. He was able to move his shoulders within the plane of gravity, and facial strength was relatively preserved. Tongue movements were limited and he had notable tongue fasciculations.
His evaluation included multiple EMGs, brain MRI and muscle ultrasound. His brain MRI was largely normal. Muscle ultrasound at 13 months of age showed severe atrophy and neurogenic changes in all muscles, more pronounced distally than proximally (Figure 1g–i). In the distal lower extremity, there are dense and severely atrophic muscles with no voluntary or spontaneous activity seen. Triceps were relatively spared compared to the biceps and deltoid muscles, but all showed abnormal echogenicity. There were fasciculations seen in the proximal upper extremities, paraspinal muscles, tongue, and orbicularis oris muscle. The diaphragm muscle appeared very thin and with no spontaneous movement visualized.
Patient 3 had multiple EMGs. His initial EMG was at 2 months of age, which showed a length-dependent, severe, mixed axonal and demyelinating predominantly motor sensorimotor polyneuropathy with active denervation seen on needle EMG exam in proximal and distal muscles of the upper and lower extremities. Repeat EMG at 3 months of age showed absent sensory responses, absent motor responses in the lower extremities, markedly reduced median CMAP motor amplitude (0.6 mV), and needle EMG exam again showed abnormal spontaneous activity suggesting active denervation. Repeat EMG at 13 months of age showed absent sensory and motor responses, no voluntary motor units elicited on needle EMG distally, and evidence of ongoing active denervation in the proximal upper extremity (biceps brachii muscle).
3.2 |. Variants in the catalaytic and anticodon binding domain of GARS leads to early onset SMA
None of the three patients described above harbored the SMN1 gene deletion. Trio exome sequencing of Patient 1 and a hereditary neuropathy gene panel testing of Patient 2 revealed heterozygosity for the same NM_002047.2:c.1001T>A (p.Ile334Asn) missense variant in the catalytic domain of GARS (Figure 2a). This was consistent with the overlapping clinical presentation of the two patients (Table 1). This variant, absent from the gnomAD variant database (Karczewski et al., 2019), affects a highly conserved residue that is invariant from yeast to humans (Figure 2b). The variant identified in both of these patients was a de novo event. Consistent with the involvement of this residue of GARS in hereditary neuropathy, a different missense substitution at this codon (p.Ile334Phe, also reported as p.Ile280Phe) has been identified in patients with features of later onset dSMA (James et al., 2006; Klein et al., 2014). Additionally experimental studies show that this residue is critical for charging tRNA with glycine in vitro (Antonellis et al., 2006; Griffin et al., 2014). Consistent with this we also found that the p.Ile334Asn variant failed to complement the deletion of the yeast ortholog GRS1 in a yeast complementation assay, indicating a loss-of-function effect consistent with other neuropathy-associated GARS variants (Griffin et al., 2014) (Figure 2c).
FIGURE 2.
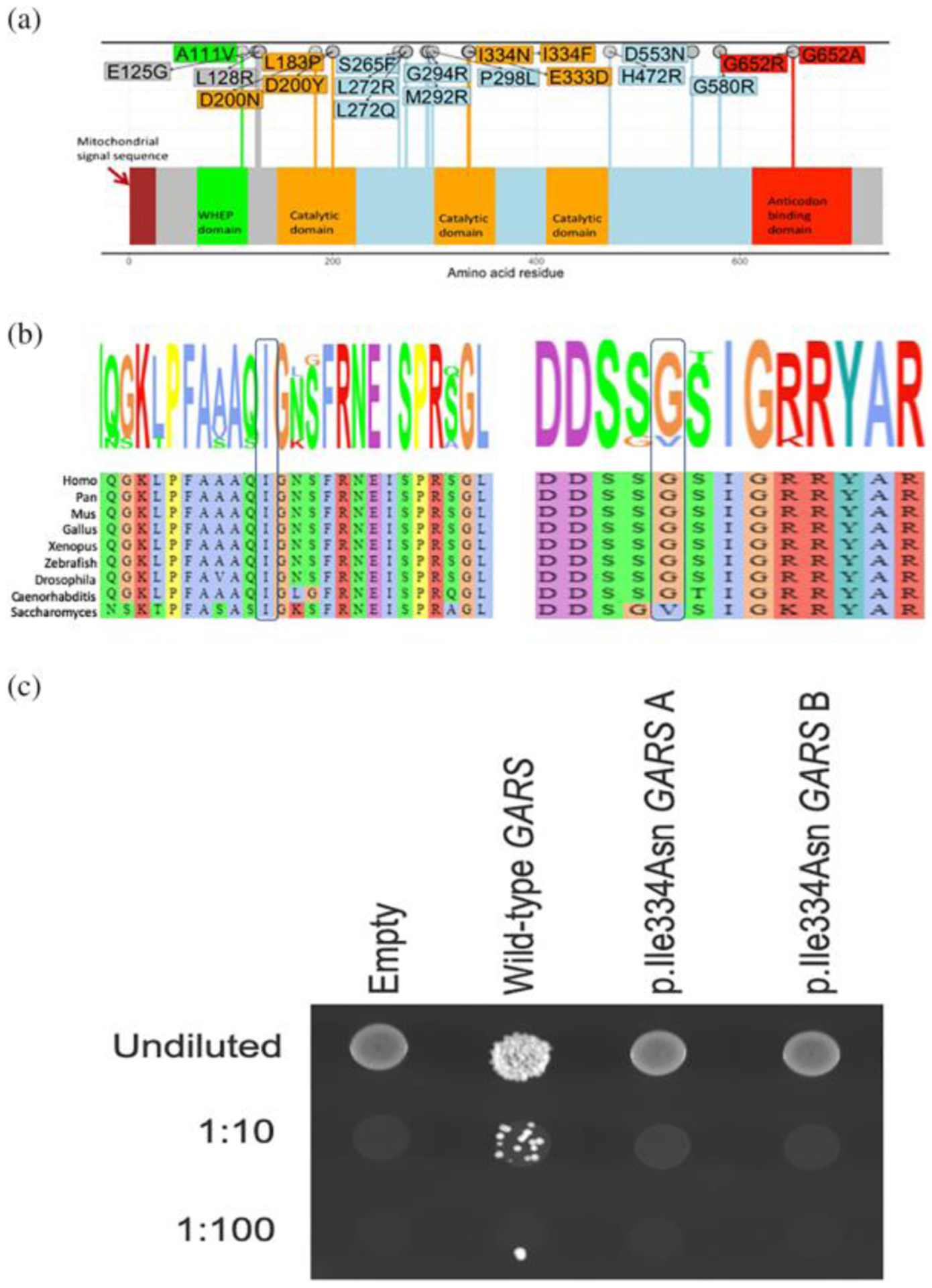
(a) The mutation spectrum of GARS. Missense mutations causing CMT2D/ dSMA are shown on the GARS primary structure. The domains are colored as indicated. (b) Conservation of the p. Ile334 and the p.Gly652 residues. (c) The p.Ile334Asn GARS variant causes a loss-of-function effect in yeast complementation assays. Yeast lacking endogenous GRS1 (the yeast ortholog of GARS) were transformed with vectors containing wild-type GARS ΔMTSΔWHEP or p.Ile334Asn GARS ΔMTSΔWHEP, or a vector with no GARS insert (“Empty”). Resulting cultures were plated undiluted or diluted (1:10 or 1:100) on media containing 5-FOA or media lacking leucine and uracil (−leu-ura) and grown at 30°C. Two independently generated mutant constructs (“a” and “b”) were tested
Trio exome sequencing in Patient 3 revealed heterozygosity for a de novo—and previously unreported- c.1954G>C (p.Gly652Arg) GARS variant. The c.1954G>C (p.Gly652Arg) change is also not present in the gnomAD database; however a p.Gly652Glu variant is present in 1 out of 249,522 alleles. The affected Gly652 residue is conserved between the nematode Caenorhabditis elegans to humans (Figure 2b). Additionally, a different change of the same amino acid (p.Gly652Ala) has been previously reported to cause infantile or early childhood onset axonal CMT and distal spinal muscular atrophy type (Eskuri et al., 2012; James et al., 2006). The p.Gly652Ala change is within the anticodon-binding domain of GARS and mutations within the anticodon-binding domain have been proposed to be more damaging to enzyme function than those within other GARS domains. Taken together we identified two potentially deleterious de novo variants in GARS in three patients presenting with infantile onset SMA.
3.3 |. The I334N GARS variant does not support yeast cell growth in complementation assays
Yeast complementation assays have revealed loss-of-function characteristics for the majority of CMT-associated GARS variants (Antonellis et al., 2006; Griffin et al., 2014) indicating that this model can be employed to predict pathogenicity (Oprescu et al., 2017). GARS with both the mitochondrial targeting sequence (MTS) and WHEP domain deleted (ΔMTSΔWHEP) has been shown to allow robust cell growth (Chien et al., 2014; Oprescu et al., 2017). We therefore compared the ability of wild-type and p.Ile334Asn human GARS ΔMTSΔWHEP to complement the loss of endogenous yeast GRS1 (the yeast ortholog of GARS). Complementation studies were performed in a previously validated haploid yeast strain deleted for endogenous GRS1 and maintained with GRS1 on a URA3-bearing vector (Turner et al., 2000). Wild-type or p.Ile334Asn GARS ΔMTSΔWHEP expression constructs, or constructs with no GARS insert (“Empty” in Figure 2c) were transformed into the yeast strain and grown on 5-FOA, which selects for spontaneous loss of the URA3-bearing maintenance vector (Figure 2c). The empty plasmid did not support yeast growth, consistent with GRS1 being an essential gene (Figure 2c). The wild-type GARS ΔMTSΔWHEP expression construct supported robust yeast growth, which indicates that human GARS can complement the loss of the endogenous GRS1 locus, consistent with previous findings (Chien et al., 2014; Oprescu et al., 2017). The p.Ile334Asn GARS ΔMTSΔWHEP expression construct was unable to support yeast growth (Figure 2c), suggesting that p.Ile334Asn GARS represents a loss-of-function allele.
3.4 |. De novo variants in the GARS anticodon binding domain leads to early-onset SMA
The observation of three patients with de novo variants in different functional domains of GARS and clinical features consistent with early-onset SMA suggested that depending on the nature of the amino-acid change and location of the variant in the GARS protein, the presentation of a patient may mimic infantile onset SMA rather than a later onset CMT2D or dSMAV. Among the patients with dSMA, CMT2D, or infantile SMA and GARS variants published in the literature (n = 45), we found that seven had infantile onset of symptoms (15.5%, age of onset less than 2 years old, Figure 3a,b). The majority of patients are diagnosed with dSMA or CMT2D, particularly when the variants are not in the anticodon-binding or catalytic domain (Chung et al., 2018; Eskuri et al., 2012; James et al., 2006; Liao et al., 2015) (Figure 3b). Our identification of three patients with early-onset SMA adds to the growing appreciation of the broad spectrum of dominant phenotypes associated with GARS pathogenic variants. This includes early onset motor neuropathy that may mimic infantile SMA with respiratory distress, as seen in our patients.
FIGURE 3.
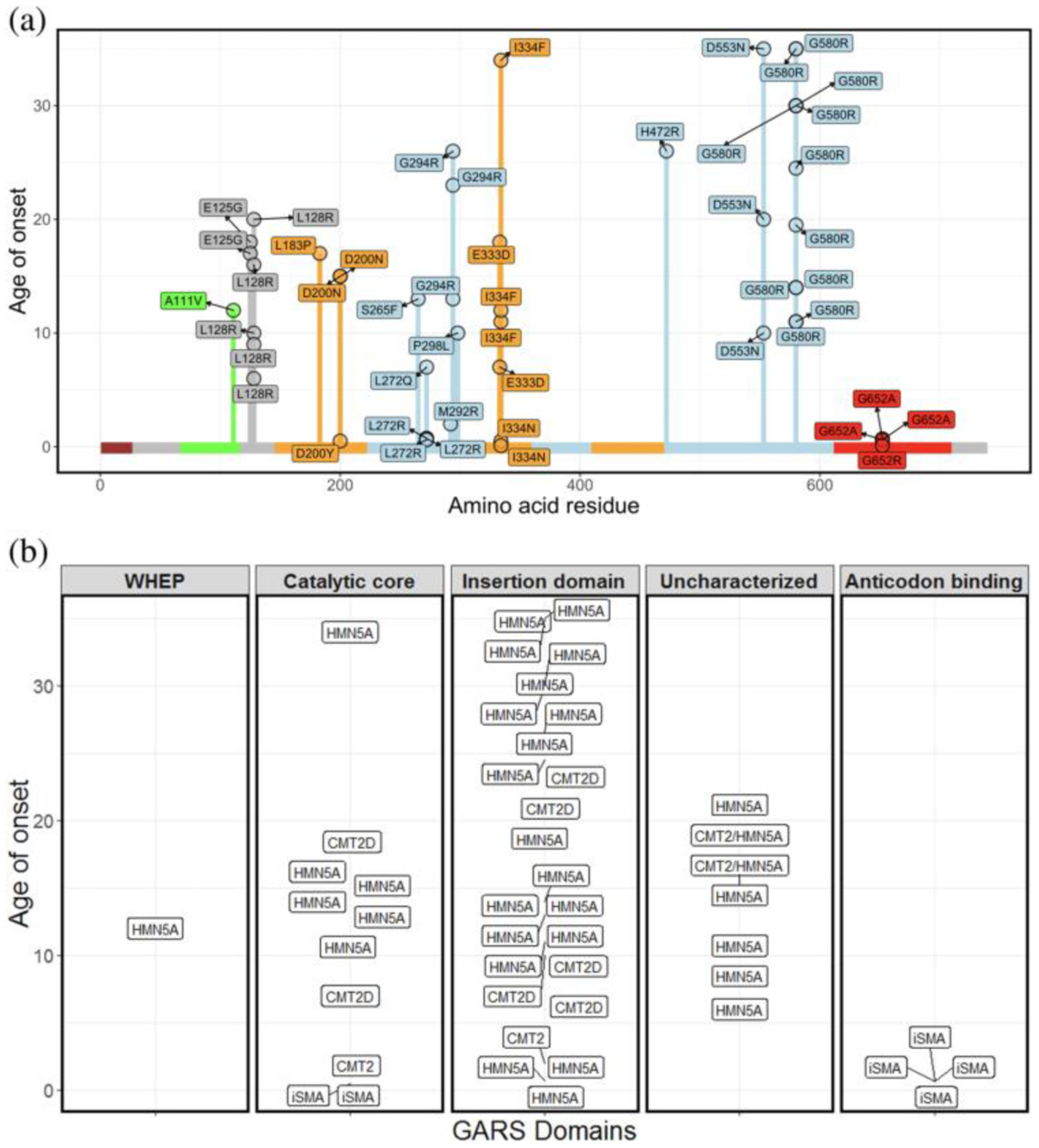
(a) The majority of patients with GARS variants exhibit late-onset clinical features of neuropathy. Plot of age of onset (years) versus amino acid residue for different variants in GARS implicated in different types of neuropathy. The variants reported in this study are indicated by arrows. (b) Age of onset (years) and published diagnosis of patients with putative GARS pathogenic variants. Age of onset is on the y axis and each facet represent the domains of GARS. Each of the rectangles depicts patients and their published diagnosis. Majority of patients are diagnosed as dSMAV or CMT2D. iSMA: infantile SMA
The p.Ile334Asn and p.Gly652Arg GARS variants affect amino acids that are located in the catalytic and the anticodon binding domains of GARS, respectively. Both of these residues with a different amino-acid change were previously reported in patients (Antonellis et al., 2003; Eskuri et al., 2012; James et al., 2006) presenting with neuropathic clinical features. An isoleucine to phenylalanine change at position 334 has been associated with a later onset dSMA in three patients (James et al., 2006; Figure 3a). The isoleucine to asparagine amino-acid change is the first amino acid in this domain that has been associated with early-onset SMA. In contrast, the change from glycine at position 652 to another hydrophobic residue, alanine, has been reported in three patients with early-onset SMA like features. Consistent with the critical nature of this glycine residue, we found that a patient with a hydrophobic to polar amino-acid change (p.Gly652Arg) at this residue presented with early-onset SMA with respiratory distress.
4 |. DISCUSSION
We report three patients with infantile-onset SMA due to pathogenic variants in GARS. This expands the number of patients with GARS-associated, infantile-onset SMA adding support to the idea that deleterious mutations in GARS can lead to infantile SMA. The clinical presentations of patients with pathogenic GARS variants were very similar to that observed in patients with deletion of SMN1. Indeed, in six out of the 10 cases of infantile-onset SMA due to GARS variants, SMN1 testing was ordered as the first line of genetic testing. Given the overlap of clinical features we suggest that in the absence of a homozygous deletion in SMN1, consideration should be given for a compound heterozygous state, as approximately 5% of patients with SMA will have a deletion in one allele and a point mutation in the other. If this were not to identify an etiology, then further genetic analysis should proceed with either a comprehensive neuromuscular panel or whole exome/genome analysis. Based on currently available commercial gene panels, GARS is most often included in those tests targeted at CMT or distal hereditary neuropathies with later rather than early onset. Therefore, the diagnosis of GARS-related disease in patients with infantile onset may be missed. With the addition of SMN1 dosage testing to newborn screening in many states, second-line testing with whole exome sequencing can be expediated in those patients in which SMN1-related disease has already been excluded.
Our patients’ clinical presentations overlap with both SMA and SMA-related disorders. There is overlap with SMA type 1, due to early onset, and dSMAV due to distal weakness greater than proximal. In addition, our patients had varying degrees of sensory impairment, which can be found in CMT2D, a disorder known to overlap with dSMAV. Finally, due to distal greater than proximal weakness and initial presentation of respiratory distress, our patients fit a diagnosis of SMARD1. We highlight the dilemma of diagnosis for our patients that the clinically defined boundaries between these syndromes are not as impermeable as previously thought, consistent with recent literature (Darras, 2011; Peeters, Chamova, & Jordanova, 2014). As we transition from diagnoses hinged purely on clinical features to those based on underlying genomic alterations, these classifications will likely become further refined. For example, to our knowledge IGHMBP2 is the only gene associated with SMARD1; however, based on this report, certain GARS variants may lead to SMARD1-like features. This could potentially alter the current diagnostic criteria for SMARD1, which include pathogenic variants in IGHMBP2.
GARS has been associated with CMT2D and dSMA. Consistent with recent reports (Chung et al., 2018; Eskuri et al., 2012; James et al., 2006; Liao et al., 2015), our results suggest that GARS variants could potentially lead to early onset SMA-like features depending on the nature and location of the amino acid change. Functional studies have shown that GARS consists of multiple domains including the catalytic, anti-codon binding and dimerization domain, and mutations in each of these domains (Antonellis et al., 2003; Griffin et al., 2014) leads to axonal neuropathy. Several disease mechanisms have been proposed based on mice and in vitro studies. Indeed it was recently shown that defective sensory motor connectivity in spinal muscular atrophy results from dysregulation of GARS pathway. This in turn may contribute to molecular and phenotypic overlap between spinal muscular atrophy and CMT2D. (Shorrock et al., 2018). The isoleucine to asparagine variant in our patients is located in the catalytic core of GARS. The critical nature of this residue is evident from functional studies in yeast. An isoleucine to phenylalanine change in this residue resulted in less than 10% aminoacylation activity compared to the wild-type GARS but was able to maintain yeast viability when modeled in the yeast ortholog GRS1 (Griffin et al., 2014). In contrast, the isoleucine to asparagine change that we identified in our patients with a more severe clinical presentation results in a loss-of-function effect in yeast complementation assays. It is therefore likely that the p.Ile334Asn variant in GARS results in reduced enzyme function, which may be particularly detrimental to neurons. The second variant we identified (p.Gly652Arg) affects an amino acid in the anticiodon binding domain. Another variant affecting the same codon, p.Gly652Ala, also found in patients with severe early-onset SMA-like features did not differ from wild-type in a yeast complementation assay (Stum et al., 2011) but was shown to aberrantly interact with proteins involved in axonal transport (Mo et al., 2018). Our finding of a novel variant at the same residue in a patient with severe early-onset SMA supports the idea that depending on the affected domain, different GARS alleles cause axonal neuropathy through different mechanisms that are likely not captured in the yeast assays. These findings continue to expand the spectrum of clinical phenotypes of “GARS-Associated Axonal Neuropathy”. Studies are needed to elucidate the precise mechanisms of how these newly identified variants in GARS lead to axonal neuropathy.
Despite the potential pitfalls of differentiating the different types of SMA-related diseases, due to recent development of treatment for SMN1-related diseases, the differentiation at the genomic level has important management implications. The recent development of antisense oligonucleotide therapy for SMN1-related disease has altered the outcome, especially when given to children early in the disease course. However, since the antisense oligonucleotide and more recently adenovector-based gene therapy are the only therapeutics for SMN1-related disease, it is crucial to delineate SMN1 spinal muscular atrophy from the less common, yet more genetically diverse, non-5q SMA. Specifically, it is important that physicians are aware that non-5q SMA can present as a severe, infantile phenotype, as was the case with our patients. Our results suggests tRNA ligases particularly GARS1 as a key contributor to clinical presentations non 5q SMA. An increased awareness of these non-5q SMA-like patients will help to more rapid diagnosis and provide more optimal disease management. Additionally, this is a diagnosis that should be considered in the case of an infant with rapidly progressive bulbar, axial and appendicular weakness, adding to SMA, SMARD, and pontocerebellar hypoplasias which may have similarly aggressive patterns of weakness. By increasing awareness of these non-5q SMA-like patients, we can more rapidly diagnose and provide optimal treatment to these patients.
ACKNOWLEDGEMENTS
We thank the patients and their families for allowing us to sharing their clinical data. All patients included have provided consent for publications. M.K. is supported by the NIH Medical Scientist Training Program Training Grant (GM007863) and the NIH Cellular and Molecular Biology Training Grant (GM007315) from the National Institute of General Medical Sciences, and an NIH National Research Service Award (F31) from the National Institute of Neurological Disorders and Stroke (NS113515). A.A. is supported by a grant from the National Institute of General Medical Sciences (GM118647).
Funding information
National Institute of General Medical Sciences, Grant/Award Number: GM118647; National Institute of Neurological Disorders and Stroke, Grant/Award Number: NS113515; NIH Cellular and Molecular Biology Training Grant, Grant/Award Number: GM007315; NIH Medical Scientist Training Program Training Grant, Grant/Award Number: GM007863
Footnotes
CONFLICT OF INTEREST
Baylor College of Medicine and Miraca Holdings Inc. have formed a joint venture with shared ownership and governance of Baylor Genetics (BG), formerly the Baylor Miraca Genetics Laboratories, which performs chromosomal microarray analysis and clinical exome sequencing. LP: The Department of Molecular and Human Genetics at Baylor College of Medicine derives revenue from molecular genetic testing offered in the Baylor Genetics Laboratories. The remaining authors did not declare any competing interests.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon request.
REFERENCES
- Antonellis A, Ellsworth RE, Sambuughin N, Puls I, Abel A, Lee-Lin S-Q, … Green ED (2003). Glycyl tRNA synthetase mutations in Charcot-Marie-Tooth disease type 2D and distal spinal muscular atrophy type V. American Journal of Human Genetics, 72, 1293–1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonellis A, Goldfarb LG, & Sivakumar K (1993). GARS-associated axonal neuropathy. In Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJ, Stephens K, & Amemiya A (Eds.), GeneReviews®. Seattle (WA): University of Washington, Seattle. [Google Scholar]
- Antonellis A, & Green ED (2008). The role of aminoacyl-tRNA synthetases in genetic diseases. Annual Review of Genomics and Human Genetics, 9, 87–107. [DOI] [PubMed] [Google Scholar]
- Antonellis A, Lee-Lin S-Q, Wasterlain A, Leo P, Quezado M, Goldfarb LG, … Green ED (2006). Functional analyses of glycyl-tRNA synthetase mutations suggest a key role for tRNA-charging enzymes in peripheral axons. The Journal of Neuroscience, 26, 10397–10406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boeke JD, Trueheart J, Natsoulis G, & Fink GR (1987). 5-Fluoroorotic acid as a selective agent in yeast molecular genetics. Methods in Enzymology, 154, 164–175. [DOI] [PubMed] [Google Scholar]
- Chien C-I, Chen Y-W, Wu Y-H, Chang C-Y, Wang T-L, & Wang C-C (2014). Functional substitution of a eukaryotic glycyl-tRNA synthetase with an evolutionarily unrelated bacterial cognate enzyme. PLoS One, 9, e94659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chihara T, Luginbuhl D, & Luo L (2007). Cytoplasmic and mitochondrial protein translation in axonal and dendritic terminal arborization. Nature Neuroscience, 10, 828–837. [DOI] [PubMed] [Google Scholar]
- Chung P, Northrup H, Azmath M, Mosquera RA, Moody S, & Yadav A (2018). Glycyl tRNA Synthetase (GARS) gene variant causes distal hereditary motor neuropathy V. Case Reports in Pediatrics, 2018, 8516285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darras BT (2011). Non-5q spinal muscular atrophies: The alphanumeric soup thickens. Neurology, 77, 312–314. [DOI] [PubMed] [Google Scholar]
- Drew AP, Blair IP, & Nicholson GA (2011). Molecular genetics and mechanisms of disease in distal hereditary motor neuropathies: Insights directing future genetic studies. Current Molecular Medicine, 11, 650–665. [DOI] [PubMed] [Google Scholar]
- Eskuri JM, Stanley CM, Moore SA, & Mathews KD (2012). Infantile onset CMT2D/dSMA V in monozygotic twins due to a mutation in the anticodon-binding domain of GARS. Journal of the Peripheral Nervous System, 17, 132–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs SA, Schene IF, Kok G, Jansen JM, Nikkels PGJ, van Gassen KLI, … van Hasselt PM (2019). Aminoacyl-tRNA synthetase deficiencies in search of common themes. Genetics in Medicine, 21 (2), 319–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin LB, Sakaguchi R, McGuigan D, Gonzalez MA, Searby C, Züchner S, … Antonellis A (2014). Impaired function is a common feature of neuropathy-associated glycyl-tRNA synthetase mutations. Human Mutation, 35, 1363–1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grohmann K, Schuelke M, Diers A, Hoffmann K, Lucke B, Adams C, … Hübner C (2001). Mutations in the gene encoding immunoglobulin mu-binding protein 2 cause spinal muscular atrophy with respiratory distress type 1. Nature Genetics, 29, 75–77. [DOI] [PubMed] [Google Scholar]
- Guenther U-P, Varon R, Schlicke M, Dutrannoy V, Volk A, Hübner C, … Schuelke M (2007). Clinical and mutational profile in spinal muscular atrophy with respiratory distress (SMARD): Defining novel phenotypes through hierarchical cluster analysis. Human Mutation, 28, 808–815. [DOI] [PubMed] [Google Scholar]
- James PA, Cader MZ, Muntoni F, Childs A-M, Crow YJ, & Talbot K (2006). Severe childhood SMA and axonal CMT due to anticodon binding domain mutations in the GARS gene. Neurology, 67, 1710–1712. [DOI] [PubMed] [Google Scholar]
- Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, … MacArthur DG (2019). Variation across 141,456 human exomes and genomes reveals the spectrum of loss-of-function intolerance across human protein-coding genes. Manuscript submitted for publication. https://www.biorxiv.org/content/10.1101/531210v2. [Google Scholar]
- Klein CJ, Middha S, Duan X, Wu Y, Litchy WJ, Gu W, … Dyck PJ (2014). Application of whole exome sequencing in undiagnosed inherited polyneuropathies. Journal of Neurology, Neurosurgery, and Psychiatry, 85, 1265–1272. [DOI] [PubMed] [Google Scholar]
- Liao Y-C, Liu Y-T, Tsai P-C, Chang C-C, Huang Y-H, Soong B-W, & Lee Y-C (2015). Two novel De novo GARS mutations cause early-onset axonal Charcot-Marie-Tooth disease. PLoS One, 10, e0133423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer-Schuman R, & Antonellis A (2017). Emerging mechanisms of aminoacyl-tRNA synthetase mutations in recessive and dominant human disease. Human Molecular Genetics, 26, R114–R127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mo Z, Zhao X, Liu H, Hu Q, Chen X-Q, Pham J, … Yang X-L (2018). Aberrant GlyRS-HDAC6 interaction linked to axonal transport deficits in Charcot-Marie-Tooth neuropathy. Nature Communications, 9, 1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munsat TL (1991). International SMA collaboration. Neuromuscular Disorders, 1, 81. [DOI] [PubMed] [Google Scholar]
- Oprescu SN, Chepa-Lotrea X, Takase R, Golas G, Markello TC, Adams DR, … Antonellis A (2017). Compound heterozygosity for loss-of-function GARS variants results in a multisystem developmental syndrome that includes severe growth retardation. Human Mutation, 38, 1412–1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peeters K, Chamova T, & Jordanova A (2014). Clinical and genetic diversity of SMN1-negative proximal spinal muscular atrophies. Brain, 137, 2879–2896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin X, Hao Z, Tian Q, Zhang Z, Zhou C, & Xie W (2014). Cocrystal structures of glycyl-tRNA synthetase in complex with tRNA suggest multiple conformational states in glycylation. The Journal of Biological Chemistry, 289, 20359–20369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russman BS (2007). Spinal muscular atrophy: Clinical classification and disease heterogeneity. Journal of Child Neurology, 22, 946–951. [DOI] [PubMed] [Google Scholar]
- Shorrock HK, van der Hoorn D, Boyd PJ, Llavero Hurtado M, Lamont DJ, Wirth B, … Gillingwater TH (2018). UBA1/GARS-dependent pathways drive sensory-motor connectivity defects in spinal muscular atrophy. Brain, 141, 2878–2894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storkebaum E (2016). Peripheral neuropathy via mutant tRNA synthetases: Inhibition of protein translation provides a possible explanation. BioEssays, 38, 818–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stum M, McLaughlin HM, Kleinbrink EL, Miers KE, Ackerman SL, Seburn KL, … Burgess RW (2011). An assessment of mechanisms underlying peripheral axonal degeneration caused by aminoacyl-tRNA synthetase mutations. Molecular and Cellular Neurosciences, 46, 432–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timmerman V, Strickland AV, & Züchner S (2014). Genetics of Charcot-Marie-Tooth (CMT) disease within the frame of the human genome project success. Genes (Basel), 5, 13–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai P-C, Soong B-W, Mademan I, Huang Y-H, Liu C-R, Hsiao C-T, … Lee Y-C (2017). A recurrent WARS mutation is a novel cause of autosomal dominant distal hereditary motor neuropathy. Brain, 140, 1252–1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner RJ, Lovato M, & Schimmel P (2000). One of two genes encoding glycyl-tRNA synthetase in Saccharomyces cerevisiae provides mitochondrial and cytoplasmic functions. The Journal of Biological Chemistry, 275, 27681–27688. [DOI] [PubMed] [Google Scholar]
- Wirth B, Herz M, Wetter A, Moskau S, Hahnen E, Rudnik-Schöneborn S, Zerres K (1999). Quantitative analysis of survival motor neuron copies: Identification of subtle SMN1 mutations in patients with spinal muscular atrophy, genotype-phenotype correlation, and implications for genetic counseling. American Journal of Human Genetics, 64, 1340–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]