

Abstract

Herein, we present an undirected para-selective two-step C–H alkylation of complex arenes useful for late-stage functionalization. The combination of a site-selective C–H thianthrenation with palladium-catalyzed reductive electrophile cross-coupling grants access to a diverse range of synthetically useful alkylated arenes which cannot be accessed otherwise with comparable selectivity, diversity, and practicality. The robustness of this transformation is further demonstrated by thianthrenium-based reductive coupling of two complex fragments.
The Csp2–Csp3 coupling, particularly direct aryl C–H alkylation, has gained considerable attention as an attractive strategy for alkylation of arenes. While Csp2–Csp2 cross-coupling reactions have been common, Csp2–Csp3 cross-coupling reactions are less frequently used due to unresolved shortfalls in available methodologies.1 A significant challenge is the regioselective functionalization of structurally complex molecules at a late stage (Scheme 1). Though several methods exist to install alkyl groups via C–H functionalization,2 regioselective alkylations in the absence of directing groups remain problematic.3 The alkylation of arenes via aryl halides is efficient4 but lacks applicability to a wide class of aryl substrates due to the challenging site-selective synthesis of complex aryl halide starting materials.5 Simply put, there is currently no reaction chemistry available to introduce, in high positional selectivity, a diverse set of alkyl groups into complex small molecules.6 Herein, we present a solution to this problem by a Csp2–Csp3 reductive cross-coupling between complex aryl thianthrenium salts and readily available alkyl iodides, bromides, and triflates via a two-step undirected regioselective C–H functionalization/reductive alkylation sequence. We show that it is now possible to rapidly access a wide range of alkylated complex arenes, which cannot be accessed by other undirected C–H alkylation methods with the same selectivity, practicality, and diversity of substrates (Scheme 1b). The reactivity of this transformation is robust and can even be applied to two complex fragments. The reaction is thought to proceed via the in situ formation of an alkylzinc species. Compared to many other related Negishi-type aryl alkylations, thianthrene-based reductive couplings do not require the organometallic zinc to be preformed prior to the cross-coupling event.4a,6a,7 The ability to engage structurally complex arenes at a late stage, a broad selection of alkyl iodides, and excellent functional group tolerance distinguish this protocol to quickly access new value-added chemical entities.
Scheme 1. Strategies for Undirected C–H Alkylation of Arenes.

(a) Conceptual representation of various strategies for undirected C–H alkylation of arenes. (b) Experimental results for palladium-catalyzed aryl C–H alkylation via bromination versus thianthrenation. Two-step yield given for compounds 1 and 2. aProduct not detected by LCMS, GCMS, 1H NMR spectroscopy, and 13C NMR spectroscopy (see the Supporting Information for details).
Selective direct aryl C–H alkylation reactions are difficult. Classical Friedel–Crafts C–H alkylations are limited by harsh conditions, low regioselectivity, and overalkylation (Scheme 1a).8 Synthetically useful regioselective C–H alkylations via transition-metal-catalyzed approaches are restricted to arenes that bear coordinating groups.2b The Negishi reaction is one of the most efficient methods to alkylate aryl (pseudo)halides; however, a long-standing challenge is competitive ß-hydride elimination.4a,9 In addition, the required (pseudo)halides are often not available and can generally not be accessed in high selectivity from complex arenes (Scheme 1).5 Furthermore, the Negishi reaction and many other traditional transition-metal-catalyzed reactions remain constrained by the availability, stability, and reactivity of the organometallic nucleophiles that must be prepared separately from the electrophile prior to the cross-coupling event and may limit the substrates that can be employed. In view of these limitations, Weix,10 Molander,11 Gong,12 MacMillan,13 and others14 independently have achieved considerable progress in the field of reductive electrophile cross-coupling reactions and have successfully demonstrated the possibility of directly engaging two readily available halides for Csp2–Csp3 bond formation in the presence of a sacrificial reductant.15 Nonetheless, current reductive electrophile aryl alkylation reactions utilizing alkyl halides often require the use of an aryl (pseudo)halide, and progress toward complex small molecules has not been widely explored. Because general site-selective halogenation is difficult, the substrate scope consists mainly of simple arenes. Thianthrenium salts are promising electrophilic coupling partners for the late-stage site-selective introduction of alkyl motifs on structurally complex arenes to forge products that are currently challenging to access. Owning in part to their positive charge, they can be easier to reduce than aryl halides, which could present a further advantage.6c We rationalized that the use of aryl thianthrenium salts in reductive alkylation reactions with alkyl halides would provide an unrealized opportunity for a two-step undirected para-selective C–H alkylation of complex arenes which, to date, has not been reported.
We investigated the reaction of aryl thianthrenium salt TT-1 with 1-boc-4-iodopiperidine in the presence of a palladium catalyst and a reducing agent (Table 1). Zinc was found to be crucial for the reaction, and other reducing agents such as manganese and tetrakis(dimethylamino)ethylene did not produce any cross-coupled product, which is consistent with the involvement of an intermediate organozinc species. A preference for polar solvents such as DMF was observed as larger amounts of unreacted alkyl iodide remained when the reaction was conducted in less polar solvents, such as toluene, which could be explained by the faster rate of oxidative addition of zinc into alkyl iodides in polar solvents.16 While nickel is the preferred transition metal for reductive aryl–alkyl bond formations,10a,10b,11−13 we identified palladium to be the metal of choice for the reductive alkylation of aryl thianthrenium salts. A series of bulky phosphine ligands were tested including those that have been successful in previous aryl alkylation reactions to suppress competing ß-hydride elimination (see the Supporting Information, Table S1, entries 8 and 17).7d,14d,17 PdCl2(amphos)2 (Table 1) was found to be pivotal for efficient cross-coupling, and all other catalyst systems resulted in significantly lower yields. Simply replacing the tBu groups in amphos with cyclohexyl groups (L2) decreases the yield to 5%. A general challenge in transition-metal-catalyzed alkylation chemistry is control over regioselectivity due to reversible ß-hydride elimination, which often results in constitutional isomers.4a,9 We investigated the selectivity for the branched versus linear product in the cross-coupling of i-PrI with pyriproxyphen thianthrenium salt TT-3 and found superior selectivities when amphos was used as a ligand (>20:1 of i-Pr product over n-Pr product).7a,7d,7e,18 PdCl2(trio-tolyphosphine) and PdCl2(dppf), for example, only yielded the products in 4.2:1 and 0.84:1 selectivity, respectively (see Supporting Information, Table S2). The high selectivities for the branched product with PdCl2(amphos)2 are worth mentioning as competitive ß-hydride elimination is a reoccurring problem in palladium-catalyzed reactions.7e,14k The importance of steric effects of the ligand on the extent of competing ß-hydride elimination can also be observed in Negishi-type aryl alkylations, which require specialized catalysts in order to minimize undesired ß-hydride elimination.7a,7d,7e,18,19 Noteworthy is also that the preformed PdCl2(amphos)2 complex was more efficient than the complex generated in situ from PdCl2 and amphos (L1) even when higher quantities of both PdCl2 and amphos were used. The presence of pyridine could potentially interfere with complex generation in situ. Nonetheless, pyridine was found to improve the overall yield of the reaction, possibly due to ligation to palladium after oxidative addition or to stabilize the organozinc reagent.14d
Table 1. Optimization of the Reaction Conditionsa.

| change in reaction conditions | yieldb |
|---|---|
| none | 75% |
| Mn instead of Zn | n.o. |
| TDAE instead of Zn | n.o. |
| toluene instead of DMF | 20% |
| no catalyst | n.o. |
| NiBr2 + phen instead of PdCl2(amphos)2 | n.o. |
| PdCl2 instead of PdCl2(amphos)2 | <5% |
| PdCl2(dppf) instead of PdCl2(amphos)2 | 20% |
| 10 mol % PdCl2 + 30 mol % L2 instead of PdCl2(amphos)2 | 5% |
| no pyridine | 66% |
Reactions were carried out on a 0.1 mmol scale.
Yields were determined by 1H NMR spectroscopy with mesitylene as the internal standard. TDAE = tetrakis(dimethylamino)ethylene. n.o. = not observed.
The alkylation of aryl thianthrenium salts occurred efficiently with primary and both cyclic and acylic secondary alkyl iodides; tertiary alkyl iodides could not be engaged (Scheme 3). Arenes as electron-rich as anisole to electron-poor as chlorobenzene were tolerated (see the Supporting Information, compound S5). The ability to employ a wide variety of alkyl substrates in a practical way presents an advantage to our previously published selective aryl C–H alkylations that only engage selected alkylzinc reagents and cannot operate on many complex small molecules.6a,7g We targeted both alkyl and aryl substrates, which contain various functional groups, as high functional group tolerance is relevant for the application of this transformation late stage. Unprotected basic amines, acidic NH groups, strained heterocyclic ring systems (e.g., ß-lactam rings), and a range of basic heterocycles, often considered problematic in transition-metal-catalyzed reactions, did not hamper the reactivity. Despite being under reducing conditions, sulfones could be tolerated and were not reduced. Our catalytic system is also tolerant to sulfonamides, which is noteworthy as organozinc reagents are typically reactive toward such acidic functional groups.20 Alcohols, sulfides, and bromides were not compatible. Furthermore, the efficiency of the cross-coupling was not impeded by ortho-substituents (2, 5, 15, 16, and 20). Also the presence of a ketone on the alkyl halide was compatible with the cross-coupling, which requires protection as acetals in other, related protocols.14k Other organometallic groups such as silyl and boron groups are not activated for transmetalation and can be held intact for potential further functionalization (7 and 8). As exemplified by 1, 2, and 15, undirected selective methylations of aryl thianthrenium salts with methyl iodide instead of methylzinc chloride are now possible, which may have potential for isotopic labeling protocols.6a,7g Alkyl substrates containing ß-σ acceptor substituents, which are difficult to engage by other methods such as SN2-type substitutions, could be coupled efficiently (e.g., 5, 6, 9, 10, 11, 12). Because the introduction of saturated heterocyclic motifs is often challenging, they are typically introduced via a more viable sp2–sp2 coupling followed by hydrogenation.21 In this transformation, a variety of saturated heterocyclic motifs could be successfully engaged such as oxetane, azetidine, piperidine, and oxaspiro[3.3]heptane (5, 10, 11, 12, 14, 17). Radical ring-opening reactions such as observed for compound 13 can give rise to otherwise challenging to access structures. We also show that the thianthrene-based reductive cross-coupling can be successfully employed for the linkage of two complex building blocks (18, 19, 20, 21), as exemplified by the coupling of a sulbactam iodide derivative, a privileged motif in drug discovery, which is found in 30% of the approved ß-lactam antibiotics,22 with nefiractam, a nootropic drug (19). In addition to alkyl iodides, primary alkyl bromides and triflates can be used successfully for the reductive cross-coupling. Secondary alkyl bromides are reactive as well, albeit in lower yields; for example, pyriproxyfen thianthrenium salt TT-3 with 3-bromooxetane and 3-iodooxetane gave product 10 at 26% and 72% yields, respectively (see the Supporting Information, pp S41–S44 for details).
Scheme 3. Substrate Scope for the Alkylation of Aryl Thianthrenium Salt.
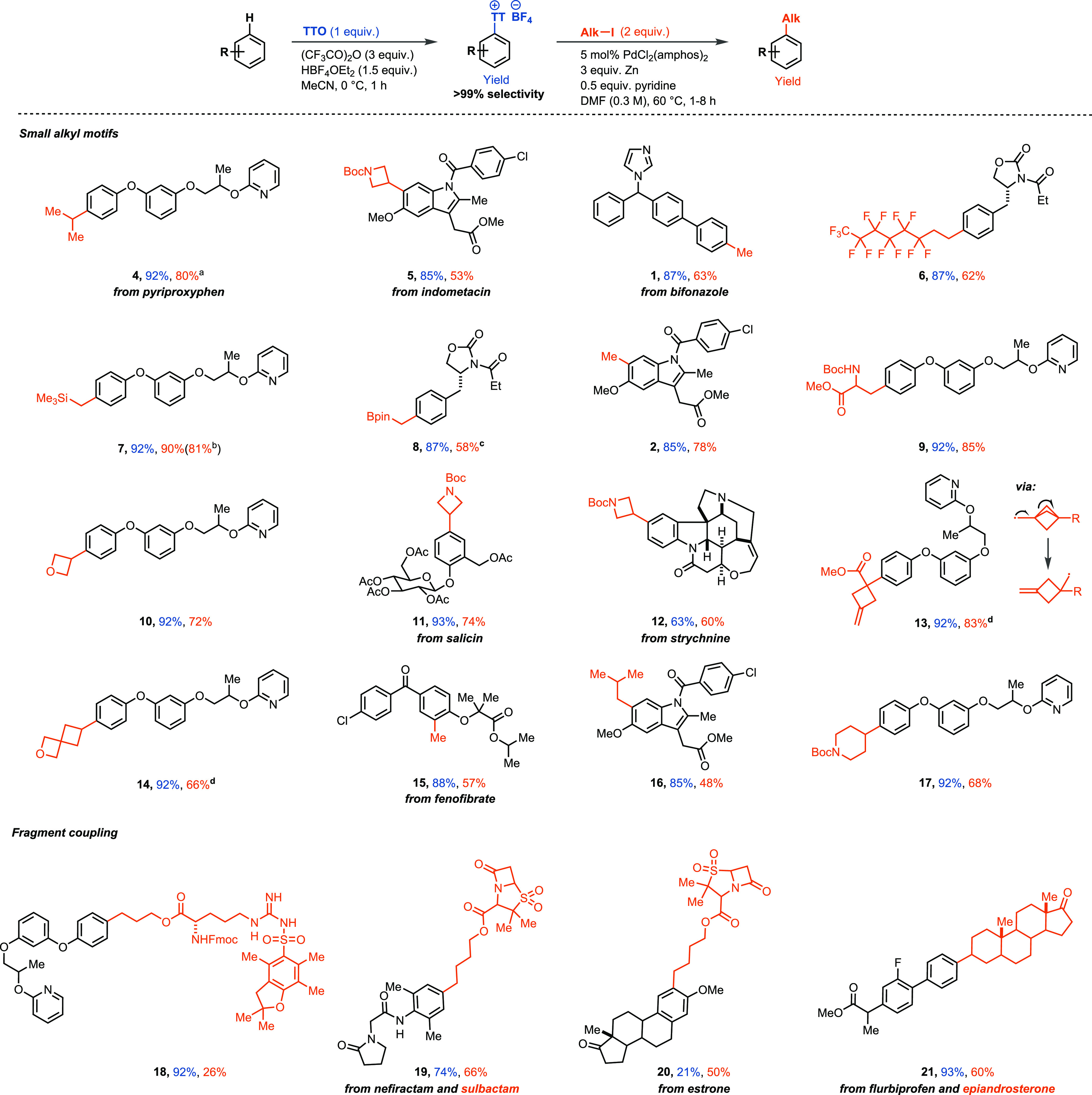
General conditions unless otherwise noted: aryl thianthrenium salt (0.3 mmol), alkyl iodide (0.6 mmol), PdCl2(amphos)2 (15.0 μmol), pyridine (0.15 mmol), DMF (0.3 M). a>20:1 ratio of i-PrAr:n-PrAr product. b3.0 mmol scale. cPyridine was omitted. dReactions carried with aryl thianthrenium salt (0.2 mmol) and MgCl2 (3 equiv) as additive. Yields in blue correspond to yield of C–H thianthrenation. Yields in orange correspond to yield of alkylation of aryl thianthrenium salts. Yields of thianthrenation were obtained from refs (5), (6a), (6b), (6c), (6d), and (6e).
A plausible mechanism hypothesis for this transformation is depicted in Scheme 2d. Though most reductive electrophile cross-coupling reactions with alkyl halides proceed via a radical chain process,4d,10a,15b,23 we believe an oxidative addition-transmetalation-reductive elimination sequence is operative in this transformation. Control experiments showed that, in the presence of zinc and absence of a palladium-catalyst, no cleavage of aryl thianthrenium salts occurred on a time scale compatible with the reductive cross-coupling (Scheme 2a). Instead, the alkyl iodide was hydrodehalogenated with 90% conversion in <5 min (see the Supporting Information). On the basis of redox potentials, aryl thianthrenium salts (E(PhTT+/PhTT•) = −1.5 V vs SCE)6c cannot be reduced by zinc (E(Zn2+/Zn(s)) = −0.76 V vs SCE). Though unactivated alkyl iodides (E(n-BuI/BuI•) = −2.5 V vs SCE)24 are even more difficult to reduce then aryl thianthrenium salts, oxidative addition of zinc is known to proceed via an inner sphere electron transfer involving a bridging ligand.25 Because such a process is more feasible on the alkyl iodide than on the aryl thianthrenium salt,25 selective radical mediated oxidative addition of zinc into the alkyl iodide could take place. Furthermore, since zinc cannot be replaced by an organic reductant, tetrakis(dimethylamino)ethylene (see Table 1), we postulate the intermediacy of an alkylzinc species under our reaction conditions. A radical clock experiment was conducted with TT-2 as a mechanistic probe to distinguish between a concerted oxidative addition and a pathway which involves single electron transfer.6c,26 The observation of noncyclized product 22 in the aryl alkylation of allyl ether thianthrenium salt TT-2 under standard conditions is consistent with a concerted oxidative addition mechanism (Scheme 2b). The aryl 2,2,6,6-tetramethylpiperidin-1-oxyl (TEMPO) adduct was not observed for the aryl alkylation upon addition of TEMPO, which is consistent with the absence of aryl radicals (Scheme 2c). By using amphos, the rate of ß-hydride elimination is slower relative to the rate of reductive elimination, which is consistent with the different product distributions of n-PrAr:i-PrAr when different catalysts are used (see discussion above). The faster reductive elimination from complexes with bulky monodentate ligands instead of bidentate ligands is in agreement with a T-shaped intermediate from which reductive elimination is faster than from a four-coordinate square planar intermediate.27 Though the preliminary mechanistic data presented above is consistent with an oxidative addition–transmetalation–reductive elimination sequence, we cannot exclude a single electron transfer mechanism.
Scheme 2. Mechanistic Investigation.

(a) Zinc insertion experiment. (b) Radical clock cyclization of allyl ether thianthrenium salt TT-2 under standard conditions. (c) Radical trapping experiment with TEMPO under standard conditions. (d) Mechanistic hypothesis. S = solvent or pyridine. L1 = amphos. n.o. = not observed.
In conclusion, we present a method for the site-selective alkylation of aryl thianthrenium salts via a two-step C–H functionalization/reductive alkylation sequence that grants access to alkylated arenes that cannot be obtained with comparable selectivities by other, undirected aryl C–H alkylation methods. By forming the zinc reagent in situ, we bypass the need to preform an organometallic reagent prior to the cross-coupling event, which from a synthetic point of view and in terms of practicality provides an advantage to other, related Negishi-type aryl alkylations, including our previously reported selective aryl alkylations that only work with selected alkylzinc reagents.6a,7g The excellent site-selectivity and robust reactivity enable us to engage complex fragments, which could be of value in medicinal chemistry. We believe this work represents a valuable conceptual extension to existing reductive Csp2–Csp3 cross-coupling reactions with improved efficiency, reactivity, and synthetic utility.
Acknowledgments
We thank the MPI für Kohlenforschung for funding. We are grateful for the help of all analytical departments of the MPI für Kohlenforschung, and we thank the MPI für Kohlenforschung for funding. We thank Daniel Chamier Cieminski for synthesis starting material 20. We thank Dr. Fabio Juliá Hernandez for helpful discussion.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.1c03459.
Experimental procedures and NMR spectra (PDF)
Author Contributions
B.L. conceived the project and optimized the aryl alkylation. B.L. and P.G. expanded the substrate scope. B.L. analyzed the data and wrote the Supporting Information. B.L. and T.R. wrote the manuscript. T.R. directed the project.
The authors declare the following competing financial interest(s): A patent application (number EP18204755.5, Germany) dealing with the use of thianthrene and its derivatives for C-H functionalization has been filed and F.B. and T.R. may benefit from royalty payments.
Supplementary Material
References
- a Walters W. P.; Green J.; Weiss J. R.; Murcko M. A. What do medicinal chemists actually make? A 50-year retrospective. J. Med. Chem. 2011, 54, 6405–6416. 10.1021/jm200504p. [DOI] [PubMed] [Google Scholar]; b Ritchie T. J.; Macdonald S. J.; Peace S.; Pickett S. D.; Luscombe C. N. Increasing small molecule drug developability in sub-optimal chemical space. MedChemComm 2013, 4, 673–680. 10.1039/c3md00003f. [DOI] [Google Scholar]; c Lovering F.; Bikker J.; Humblet C. Escape from flatland: increasing saturation as an approach to improving clinical success. J. Med. Chem. 2009, 52, 6752–6756. 10.1021/jm901241e. [DOI] [PubMed] [Google Scholar]; d Lovering F. Escape from Flatland 2: complexity and promiscuity. MedChemComm 2013, 4, 515–519. 10.1039/c2md20347b. [DOI] [Google Scholar]
- a Dong Z.; Ren Z.; Thompson S. J.; Xu Y.; Dong G. Transition-metal-catalyzed C–H alkylation using alkenes. Chem. Rev. 2017, 117, 9333–9403. 10.1021/acs.chemrev.6b00574. [DOI] [PubMed] [Google Scholar]; b Evano G.; Theunissen C. Beyond Friedel and Crafts: directed alkylation of C–H bonds in arenes. Angew. Chem., Int. Ed. 2019, 58, 7202–7236. 10.1002/anie.201806629. [DOI] [PubMed] [Google Scholar]
- Hartwig J. F. Evolution of C–H bond functionalization from methane to methodology. J. Am. Chem. Soc. 2016, 138, 2–24. 10.1021/jacs.5b08707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Jana R.; Pathak T. P.; Sigman M. S. Advances in transition metal (Pd, Ni, Fe)-catalyzed cross-coupling reactions using alkyl-organometallics as reaction partners. Chem. Rev. 2011, 111, 1417–1492. 10.1021/cr100327p. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Knappke C. E.; Grupe S.; Gärtner D.; Corpet M.; Gosmini C.; Jacobi von Wangelin A. Reductive cross-coupling reactions between two electrophiles. Chem. - Eur. J. 2014, 20, 6828–6842. 10.1002/chem.201402302. [DOI] [PubMed] [Google Scholar]; c Richmond E.; Moran J. Recent advances in nickel catalysis enabled by stoichiometric metallic reducing agents. Synthesis 2018, 50, 499–513. 10.1055/s-0036-1591853. [DOI] [Google Scholar]; d Weix D. J. Methods and mechanisms for cross-electrophile coupling of Csp2 halides with alkyl electrophiles. Acc. Chem. Res. 2015, 48, 1767–1775. 10.1021/acs.accounts.5b00057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang R.-J.; Milcent T.; Crousse B. Regioselective halogenation of arenes and heterocycles in hexafluoroisopropanol. J. Org. Chem. 2018, 83, 930–938. 10.1021/acs.joc.7b02920. [DOI] [PubMed] [Google Scholar]
- a Berger F.; Plutschack M. B.; Riegger J.; Yu W.; Speicher S.; Ho M.; Frank N.; Ritter T. Site-selective and versatile aromatic C–H functionalization by thianthrenation. Nature 2019, 567, 223–228. 10.1038/s41586-019-0982-0. [DOI] [PubMed] [Google Scholar]; b Alvarez E. M.; Plutschack M. B.; Berger F.; Ritter T. Site-Selective C–H Functionalization–Sulfination Sequence to Access Aryl Sulfonamides. Org. Lett. 2020, 22, 4593–4596. 10.1021/acs.orglett.0c00982. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Li J.; Chen J.; Sang R.; Ham W.-S.; Plutschack M. B.; Berger F.; Chabbra S.; Schnegg A.; Genicot C.; Ritter T. Photoredox catalysis with aryl sulfonium salts enables site-selective late-stage fluorination. Nat. Chem. 2020, 12, 56–62. 10.1038/s41557-019-0353-3. [DOI] [PubMed] [Google Scholar]; d Engl P. S.; Häring A. P.; Berger F.; Berger G.; Pérez-Bitrián A.; Ritter T. C–N cross-couplings for site-selective late-stage diversification via aryl sulfonium salts. J. Am. Chem. Soc. 2019, 141, 13346–13351. 10.1021/jacs.9b07323. [DOI] [PubMed] [Google Scholar]; e Sang R.; Korkis S. E.; Su W.; Ye F.; Engl P. S.; Berger F.; Ritter T. Site-Selective C–H Oxygenation via Aryl Sulfonium Salts. Angew. Chem. 2019, 131, 16307–16312. 10.1002/ange.201908718. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Ye F.; Berger F.; Jia H.; Ford J.; Wortman A.; Börgel J.; Genicot C.; Ritter T. Aryl sulfonium salts for site-selective late-stage trifluoromethylation. Angew. Chem. 2019, 131, 14757–14761. 10.1002/ange.201906672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Atwater B.; Chandrasoma N.; Mitchell D.; Rodriguez M. J.; Organ M. G. Pd-PEPPSI-IHeptCl: A General-Purpose, Highly Reactive Catalyst for the Selective Coupling of Secondary Alkyl Organozincs. Chem. - Eur. J. 2016, 22, 14531–14534. 10.1002/chem.201603603. [DOI] [PubMed] [Google Scholar]; b Haas D.; Hammann J. M.; Greiner R.; Knochel P. Recent developments in Negishi cross-coupling reactions. ACS Catal. 2016, 6, 1540–1552. 10.1021/acscatal.5b02718. [DOI] [Google Scholar]; c Sase S.; Jaric M.; Metzger A.; Malakhov V.; Knochel P. One-pot Negishi cross-coupling reactions of in situ generated zinc reagents with aryl chlorides, bromides, and triflates. J. Org. Chem. 2008, 73, 7380–7382. 10.1021/jo801063c. [DOI] [PubMed] [Google Scholar]; d Han C.; Buchwald S. L. Negishi coupling of secondary alkylzinc halides with aryl bromides and chlorides. J. Am. Chem. Soc. 2009, 131, 7532–7533. 10.1021/ja902046m. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Kalvet I.; Sperger T.; Scattolin T.; Magnin G.; Schoenebeck F. Palladium (I) Dimer Enabled Extremely Rapid and Chemoselective Alkylation of Aryl Bromides over Triflates and Chlorides in Air. Angew. Chem., Int. Ed. 2017, 56, 7078–7082. 10.1002/anie.201701691. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Leroux M.; Vorherr T.; Lewis I.; Schaefer M.; Koch G.; Karaghiosoff K.; Knochel P. Late-stage functionalization of peptides and cyclopeptides using organozinc reagents. Angew. Chem., Int. Ed. 2019, 58, 8231–8234. 10.1002/anie.201902454. [DOI] [PubMed] [Google Scholar]; g Serpier F.; Pan F.; Ham W. S.; Jacq J.; Genicot C.; Ritter T. Selective Methylation of Arenes: A Radical C–H Functionalization/Cross-Coupling Sequence. Angew. Chem. 2018, 130, 10857–10861. 10.1002/ange.201804628. [DOI] [PubMed] [Google Scholar]
- Friedel C.; Crafts J. Organic chemistry. J. Chem. Soc. 1877, 32, 725–791. 10.1039/js8773200725. [DOI] [Google Scholar]
- a Rudolph A.; Lautens M. Secondary Alkyl Halides in Transition-Metal-Catalyzed Cross-Coupling Reactions. Angew. Chem., Int. Ed. 2009, 48, 2656–2670. 10.1002/anie.200803611. [DOI] [PubMed] [Google Scholar]; b Frisch A. C.; Beller M. Catalysts for cross-coupling reactions with non-activated alkyl halides. Angew. Chem., Int. Ed. 2005, 44, 674–688. 10.1002/anie.200461432. [DOI] [PubMed] [Google Scholar]
- a Everson D. A.; Jones B. A.; Weix D. J. Replacing conventional carbon nucleophiles with electrophiles: nickel-catalyzed reductive alkylation of aryl bromides and chlorides. J. Am. Chem. Soc. 2012, 134, 6146–6159. 10.1021/ja301769r. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Kim S.; Goldfogel M. J.; Gilbert M. M.; Weix D. J. Nickel-Catalyzed Cross-Electrophile Coupling of Aryl Chlorides with Primary Alkyl Chlorides. J. Am. Chem. Soc. 2020, 142 (22), 9902–9907. 10.1021/jacs.0c02673. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Everson D. A.; Shrestha R.; Weix D. J. Nickel-catalyzed reductive cross-coupling of aryl halides with alkyl halides. J. Am. Chem. Soc. 2010, 132, 920–921. 10.1021/ja9093956. [DOI] [PubMed] [Google Scholar]
- a Molander G. A.; Traister K. M.; O’Neill B. T. Reductive cross-coupling of nonaromatic, heterocyclic bromides with aryl and heteroaryl bromides. J. Org. Chem. 2014, 79, 5771–5780. 10.1021/jo500905m. [DOI] [PubMed] [Google Scholar]; b Molander G. A.; Traister K. M.; O’Neill B. T. Engaging nonaromatic, heterocyclic tosylates in reductive cross-coupling with aryl and heteroaryl bromides. J. Org. Chem. 2015, 80, 2907–2911. 10.1021/acs.joc.5b00135. [DOI] [PubMed] [Google Scholar]
- a Liu J.; Gong H. Stereoselective preparation of α-C-vinyl/aryl glycosides via nickel-catalyzed reductive coupling of glycosyl halides with vinyl and aryl halides. Org. Lett. 2018, 20, 7991–7995. 10.1021/acs.orglett.8b03567. [DOI] [PubMed] [Google Scholar]; b Wang S.; Qian Q.; Gong H. Nickel-catalyzed reductive coupling of aryl halides with secondary alkyl bromides and allylic acetate. Org. Lett. 2012, 14, 3352–3355. 10.1021/ol3013342. [DOI] [PubMed] [Google Scholar]; c Wang X.; Wang S.; Xue W.; Gong H. Nickel-catalyzed reductive coupling of aryl bromides with tertiary alkyl halides. J. Am. Chem. Soc. 2015, 137, 11562–11565. 10.1021/jacs.5b06255. [DOI] [PubMed] [Google Scholar]; d Wang X.; Ma G.; Peng Y.; Pitsch C. E.; Moll B. J.; Ly T. D.; Wang X.; Gong H. Ni-catalyzed reductive coupling of electron-rich aryl iodides with tertiary alkyl halides. J. Am. Chem. Soc. 2018, 140, 14490–14497. 10.1021/jacs.8b09473. [DOI] [PubMed] [Google Scholar]
- a Zhang P.; Le C. C.; MacMillan D. W. Silyl radical activation of alkyl halides in metallaphotoredox catalysis: a unique pathway for cross-electrophile coupling. J. Am. Chem. Soc. 2016, 138, 8084–8087. 10.1021/jacs.6b04818. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Sakai H. A.; Liu W.; Le C. C.; MacMillan D. W. Cross-electrophile coupling of unactivated alkyl chlorides. J. Am. Chem. Soc. 2020, 142, 11691–11697. 10.1021/jacs.0c04812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Czaplik W. M.; Mayer M. Jacobi von Wangelin, A. Domino Iron Catalysis: Direct Aryl–Alkyl Cross-Coupling. Angew. Chem., Int. Ed. 2009, 48, 607–610. 10.1002/anie.200804434. [DOI] [PubMed] [Google Scholar]; b Czaplik W. M.; Mayer M.; von Wangelin A. J. Direct cobalt-catalyzed cross-coupling between aryl and alkyl halides. Synlett 2009, 2009, 2931–2934. 10.1055/s-0029-1218012. [DOI] [Google Scholar]; c Durandetti M.; Nédélec J.-Y.; Périchon J. Nickel-catalyzed direct electrochemical cross-coupling between aryl halides and activated alkyl halides. J. Org. Chem. 1996, 61, 1748–1755. 10.1021/jo9518314. [DOI] [PubMed] [Google Scholar]; d Krasovskiy A.; Duplais C.; Lipshutz B. H. Zn-mediated, Pd-catalyzed cross-couplings in water at room temperature without prior formation of organozinc reagents. J. Am. Chem. Soc. 2009, 131, 15592–15593. 10.1021/ja906803t. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Amatore M.; Gosmini C. Direct Method for Carbon–Carbon Bond Formation: The Functional Group Tolerant Cobalt-Catalyzed Alkylation of Aryl Halides. Chem. - Eur. J. 2010, 16, 5848–5852. 10.1002/chem.201000178. [DOI] [PubMed] [Google Scholar]; f Bhonde V. R.; O’Neill B. T.; Buchwald S. L. An Improved System for the Aqueous Lipshutz–Negishi Cross-Coupling of Alkyl Halides with Aryl Electrophiles. Angew. Chem. 2016, 128, 1881–1885. 10.1002/ange.201509341. [DOI] [PubMed] [Google Scholar]; g Kumar G. S.; Peshkov A.; Brzozowska A.; Nikolaienko P.; Zhu C.; Rueping M. Nickel-Catalyzed Chain-Walking Cross-Electrophile Coupling of Alkyl and Aryl Halides and Olefin Hydroarylation Enabled by Electrochemical Reduction. Angew. Chem., Int. Ed. 2020, 59, 6513–6519. 10.1002/anie.201915418. [DOI] [PubMed] [Google Scholar]; h Truesdell B. L.; Hamby T. B.; Sevov C. S. General C (sp2)–C (sp3) Cross-Electrophile Coupling Reactions Enabled by Overcharge Protection of Homogeneous Electrocatalysts. J. Am. Chem. Soc. 2020, 142, 5884–5893. 10.1021/jacs.0c01475. [DOI] [PubMed] [Google Scholar]; i Perkins R. J.; Pedro D. J.; Hansen E. C. Electrochemical nickel catalysis for sp2-sp3 cross-electrophile coupling reactions of unactivated alkyl halides. Org. Lett. 2017, 19, 3755–3758. 10.1021/acs.orglett.7b01598. [DOI] [PubMed] [Google Scholar]; j Jiao K. J.; Liu D.; Ma H. X.; Qiu H.; Fang P.; Mei T. S. Nickel-Catalyzed Electrochemical Reductive Relay Cross-Coupling of Alkyl Halides to Aryl Halides. Angew. Chem., Int. Ed. 2020, 59, 6520–6524. 10.1002/anie.201912753. [DOI] [PubMed] [Google Scholar]; k Zhang K. F.; Christoffel F.; Baudoin O. Barbier–Negishi Coupling of Secondary Alkyl Bromides with Aryl and Alkenyl Triflates and Nonaflates. Angew. Chem. 2018, 130, 2000–2004. 10.1002/ange.201711990. [DOI] [PubMed] [Google Scholar]
- a Wang X.; Dai Y.; Gong H. Nickel-catalyzed reductive couplings. Top. Curr. Chem. 2017, 61–89. 10.1007/978-3-319-49784-6_3. [DOI] [PubMed] [Google Scholar]; b Gu J.; Wang X.; Xue W.; Gong H. Nickel-catalyzed reductive coupling of alkyl halides with other electrophiles: concept and mechanistic considerations. Org. Chem. Front. 2015, 2, 1411–1421. 10.1039/C5QO00224A. [DOI] [Google Scholar]; c Poremba K. E.; Dibrell S. E.; Reisman S. E. Nickel-Catalyzed Enantioselective Reductive Cross-Coupling Reactions. ACS Catal. 2020, 10, 8237–8246. 10.1021/acscatal.0c01842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Majid T. N.; Knochel P. A new preparation of highly functionaized aromatic and heteroaromatic zinc and copper organometallics. Tetrahedron Lett. 1990, 31, 4413–4416. 10.1016/S0040-4039(00)97635-4. [DOI] [Google Scholar]; b Amano M.; Saiga A.; Ikegami R.; Ogata T.; Takagi K. Synthesis of o-arylenedizinc compounds from 1-iodo-2-trifluoromethylsulfonyloxybenzenes and zinc powder and their synthetic application. Tetrahedron Lett. 1998, 39, 8667–8668. 10.1016/S0040-4039(98)01867-X. [DOI] [Google Scholar]; c Jubert C.; Knochel P. Preparation of new classes of aliphatic, allylic, and benzylic zinc and copper reagents by the insertion of zinc dust into organic halides, phosphates, and sulfonates. J. Org. Chem. 1992, 57, 5425–5431. 10.1021/jo00046a026. [DOI] [Google Scholar]
- Hayashi T.; Konishi M.; Kobori Y.; Kumada M.; Higuchi T.; Hirotsu K. Dichloro[1,1’-bis (diphenylphosphino)ferrocene]palladium (II): an effective catalyst for cross-coupling of secondary and primary alkyl Grignard and alkylzinc reagents with organic halides. J. Am. Chem. Soc. 1984, 106, 158–163. 10.1021/ja00313a032. [DOI] [Google Scholar]
- Yang Y.; Niedermann K.; Han C.; Buchwald S. L. Highly selective palladium-catalyzed cross-coupling of secondary alkylzinc reagents with heteroaryl halides. Org. Lett. 2014, 16, 4638–4641. 10.1021/ol502230p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai C.; Fu G. C. The first general method for palladium-catalyzed Negishi cross-coupling of aryl and vinyl chlorides: use of commercially available Pd (P(t-Bu)3)2 as a catalyst. J. Am. Chem. Soc. 2001, 123, 2719–2724. 10.1021/ja003954y. [DOI] [PubMed] [Google Scholar]
- a Manolikakes G.; Schade M. A.; Hernandez C. M.; Mayr H.; Knochel P. Negishi cross-couplings of unsaturated halides bearing relatively acidic hydrogen atoms with organozinc reagents. Org. Lett. 2008, 10, 2765–2768. 10.1021/ol8009013. [DOI] [PubMed] [Google Scholar]; b Joshi-Pangu A.; Ganesh M.; Biscoe M. R. Nickel-catalyzed Negishi cross-coupling reactions of secondary alkylzinc halides and aryl iodides. Org. Lett. 2011, 13, 1218–1221. 10.1021/ol200098d. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Cheng J.-P.; Zhao Y. Homolytic bond cleavage energies of the acidic N–H bonds in dimethyl sulfoxide solution and properties of the corresponding radicals and radical cations. Tetrahedron 1993, 49, 5267–5276. 10.1016/S0040-4020(01)82376-0. [DOI] [Google Scholar]
- a Marsilje T. H.; Pei W.; Chen B.; Lu W.; Uno T.; Jin Y.; Jiang T.; Kim S.; Li N.; Warmuth M. Synthesis, structure–activity relationships, and in vivo efficacy of the novel potent and selective anaplastic lymphoma kinase (ALK) inhibitor 5-Chloro-N 2-(2-isopropoxy-5-methyl-4-(piperidin-4-yl) phenyl)-N 4-(2-(isopropylsulfonyl) phenyl) pyrimidine-2, 4-diamine (LDK378) currently in phase 1 and phase 2 clinical trials. J. Med. Chem. 2013, 56, 5675–5690. 10.1021/jm400402q. [DOI] [PubMed] [Google Scholar]; b Mesaros E. F.; Thieu T. V.; Wells G. J.; Zificsak C. A.; Wagner J. C.; Breslin H. J.; Tripathy R.; Diebold J. L.; McHugh R. J.; Wohler A. T. Strategies to mitigate the bioactivation of 2-anilino-7-aryl-pyrrolo [2, 1-f][1, 2, 4] triazines: identification of orally bioavailable, efficacious ALK inhibitors. J. Med. Chem. 2012, 55, 115–125. 10.1021/jm2010767. [DOI] [PubMed] [Google Scholar]
- Vitaku E.; Smith D. T.; Njardarson J. T. Analysis of the structural diversity, substitution patterns, and frequency of nitrogen heterocycles among US FDA approved pharmaceuticals: miniperspective. J. Med. Chem. 2014, 57, 10257–10274. 10.1021/jm501100b. [DOI] [PubMed] [Google Scholar]
- a Everson D. A.; Weix D. J. Cross-electrophile coupling: principles of reactivity and selectivity. J. Org. Chem. 2014, 79, 4793–4798. 10.1021/jo500507s. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Biswas S.; Weix D. J. Mechanism and selectivity in nickel-catalyzed cross-electrophile coupling of aryl halides with alkyl halides. J. Am. Chem. Soc. 2013, 135, 16192–16197. 10.1021/ja407589e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y.; Cornella J.; Juliá-Hernández F.; Martin R. Visible-light-promoted atom transfer radical cyclization of unactivated alkyl iodides. ACS Catal. 2017, 7, 409–412. 10.1021/acscatal.6b03205. [DOI] [Google Scholar]
- Guijarro A.; Rosenberg D. M.; Rieke R. D. The reaction of active zinc with organic bromides. J. Am. Chem. Soc. 1999, 121, 4155–4167. 10.1021/ja9844478. [DOI] [Google Scholar]
- Creutz S. E.; Lotito K. J.; Fu G. C. Peters, Photoinduced Ullmann C–N coupling: demonstrating the viability of a radical pathway. Science 2012, 338, 647–651. 10.1126/science.1226458. [DOI] [PubMed] [Google Scholar]
- Tatsumi K.; Hoffmann R.; Yamamoto A.; Stille J. K. Reductive elimination of d8-organotransition metal complexes. Bull. Chem. Soc. Jpn. 1981, 54, 1857–1867. 10.1246/bcsj.54.1857. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.