

Abstract

Site-selective dihalogenated heteroarene cross-coupling with organometallic reagents usually occurs at the halogen proximal to the heteroatom, enabled by intrinsic relative electrophilicity, particularly in strongly polarized systems. An archetypical example is the Suzuki–Miyaura cross-coupling (SMCC) of 2,4-dibromopyridine with organoboron species, which typically exhibit C2-arylation site-selectivity using mononuclear Pd (pre)catalysts. Given that Pd speciation, particularly aggregation, is known to lead to the formation of catalytically competent multinuclear Pdn species, the influence of these species on cross-coupling site-selectivity remains largely unknown. Herein, we disclose that multinuclear Pd species, in the form of Pd3-type clusters and nanoparticles, switch arylation site-selectivity from C2 to C4, in 2,4-dibromopyridine cross-couplings with both organoboronic acids (SMCC reactions) and Grignard reagents (Kumada-type reactions). The Pd/ligand ratio and the presence of suitable stabilizing salts were found to be critically important in switching the site-selectivity. More generally, this study provides experimental evidence that aggregated Pd catalyst species not only are catalytically competent but also alter reaction outcomes through changes in product selectivity.
Introduction
Dihalogentated organic compounds, particularly heteroarenes, serve as synthetically useful structural templates for increasing molecular complexity. They enable multiple modes of connectivity, providing access to a vast array of compounds with interesting properties, from agrochemicals and pharmaceuticals to advanced materials.1,2 Classical cross-coupling reaction methodologies are powerful tools for enabling site-selective processes to be realized, as outlined in two critical reviews by Fairlamb in 20073 and Spivey et al. in 2017.4 Leading examples are given in Scheme 1, showing the preferred cross-coupling site for a series of dihalogenated heteroarenes. Normally, site-selectivity is seen at halogens activated by the ring heteroatom, either through proximity or favorable bond polarization in the extended π-ring system. Houk et al. explained the origin of normal site-selectivity in the context of the distortion of the C–X bond from a given substrate and interaction energies on approach to the active Pd0Ln catalyst.5 Consideration can further be made for the bond dissociation energies (BDE) at different C–X bonds. Handy et al. demonstrated that cross-coupling site-selectivity could be predicted, with caveats, by comparing the 1H NMR chemical shifts of the parent heteroarene—the most deshielded proton being the typical site for coupling in the corresponding C–X derivative.6 Switching site-selectivity in the cross-coupling reactions of dihalogenated heteroarenes, which effectively possess biased intrinsic reactivity (through relative electrophilicity), is a difficult task. For 2,4-dibromopyridine 1, it is very challenging, as C2 site-selectivity dominates as described in the extensive screening work carried out by Cid7 and Zhou et al.8,9 There are only a few examples where atypical C4 site-selectivity in cross-coupling is known.10 A C4 site-selective Suzuki–Miyaura cross-coupling example on 1 was reported by Hardie and Willans et al.,11 which employs Pd-NHC precatalysts, possessing distinctive ligand architectures. For the best precatalyst, C4:C2 site-selectivity was ∼10:1. However, as is common to an eclectic array of dihalogenated heteroarene substrates, diarylation was found to be a competing process and overall product yields were moderate as a consequence (∼35% for monoarylation product). Dai et al. switched the site-selectivity in Suzuki–Miyaura cross-coupling reactions (SMCCs) involving 2,4-dichloropyridine using a Q-Phos/Pd(OAc)2 precatalyst system, resulting in a marginal bias toward the atypical C4-arylated product, but accompanied by low yields.12 Higher C4-selectivities at 2,4-dichloropyridine were obtained by changes to exogenous ligands at Pd, as reported in 2020 by Yang et al.13
Scheme 1. Site-Selectivity in Suzuki–Miyaura Cross-Couplings of Heteroarenes, Exemplified by Dihalogenated Pyridines and Related Derivatives.

A guiding example, for which many catalyst systems/reaction conditions have been investigated, is given, showing high C2 site-selectivity.
The background literature therefore highlights that switches from typical to atypical site-selectivity are feasible, but that fundamental reasoning is frustratingly lacking—the focus has often been placed on ligand changes, assuming a mononuclear Pd catalyst.14−17 While logical, in our opinion Pd catalyst speciation is a bigger issue, where changes in mechanism might better account for typical to atypical site-selectivity changes.
Our research group has been engaged in understanding the role played by catalytically competent aggregated Pd clusters and nanoparticles in SMCCs, and related cross-couplings, for many years.18−23 We presented the first compelling experimental evidence implicating heterogeneous surface catalysts in SMCCs,24,25 which is supported by recent evidence using time-resolved fluorescence studies26 and surface-enhanced Raman spectroscopic techniques.27
The knowledge outlined above is important in the context of understanding that mononuclear Pd species, generally thought to be the dominant catalytically active species in SMCCs, can aggregate to form higher order Pd nanoparticles that are capable of mediating further substrate turnover. A serious question facing the field of cross-coupling catalysis is the involvement of small Pdn clusters (n < 13), as such species provide a potential bridge from mononuclear Pd1 species to Pd nanoparticles (PdNPs).28 Indeed, in a recent study Li et al.29 presented some evidence that [Pd3(μ-Cl)(μ-PPh2)2(PPh3)3]+30,31 not only was an active Pd catalyst for SMCCs but also appears to invert the order of the oxidative addition and transmetalation steps within the catalytic cycle, proposing the activation of the aryl halide as being less like oxidative addition and more like σ-bond metathesis. Our recent findings showed that similar [Pd3(μ-Cl)(μ-PPh2)2(PPh3)3]X cluster species derive from a Pd3(OAc)6/6PPh3 precatalyst, by reaction of an organohalide (R–X, including 2-bromopyridine) with the intermediate formed PdI dinuclear species.32 The outcome sparked our interest in understanding how higher order Pd species might affect site-selectivity in cross-coupling reactions of 2,4-dibromopyridine 1 with organoboronic acids 2, as well as other nucleophiles, such as Grignard reagents. We were encouraged as [Pd3(μ-Cl)(μ-PPh2)2(PPh3)3]X species were found to be more active in the reported SMCC reactions than Pd0(PPh3)3 (in terms of substrate turnover frequency). [Pd3(μ-X)(μ-PR2)2(PR3)3]X species have been invoked as catalytically relevant species under a range of conditions.29,33,34
Schoenebeck et al. have investigated the use of multinuclear catalysts for chemoselective cross-coupling reactions at substrates containing two or more different (pseudo)halide identities. For example, the reactivity of [Pd(μ-I)(Pt-Bu3)]2 enabled successive selective couplings at Br then OTf then Cl sites on aromatic substrates.35,36 A Pd3 cluster catalyst, derived from highly active [Pd(μ-Br)(Pt-Bu3)]2,37 facilitated selective cross-couplings at aryl iodide over the less activated aryl bromide sites.34 Additionally, a nanoparticulate active catalyst, derived in situ from Pd2(dba)3, was found to enable chemoselective cross-couplings between aryl iodides and arylgermanes.38 Despite the clear synthetic utility of chemoselective reactions at multiply halogenated compounds for rapid molecular diversification, the preferential site of cross-coupling is generally quite clear-cut, defined by the BDE of the C–X bond (e.g., for halides (X), I < Br < Cl ≪ F).3,4 We note that regioselective control of cross-coupling at substrates featuring multiple halogens of the same type (i.e., with similar BDEs) constitutes a greater challenge than a chemoselective approach involving different halogens.
In this paper we examine the behavior of Pd3-type cluster and Pd nanoparticle catalysts that derive from Pd(OAc)2/nPPh3 precatalyst systems under working reaction conditions. Varying the number of PPh3 ligands (relative to Pd) enables us to switch between higher order Pdn catalysis and mononuclear Pd1 catalysis. This has an impact on switching regioselectivity—the reaction outcome—from typical C2 to atypical C4, in 2,4-dibromopyridine 1 cross-couplings with either organoboronic acids 2 (SMCC reactions) or Grignard reagents 5 (Kumada–Corriu type reactions). The activity of PdNPs is modulated by additive stabilizing salts, which proved to be critical in switching catalyst site-selectivity. While PdNPs are established cross-coupling catalysts, this is the first time that site-selectivity in a dihalogenated heteroarene has been reversed through exploitation of conditions that facilitate the in operando (under working reaction conditions) generation of Pd nanoparticles.
Results and Discussion
A benchmark SMCC test reaction [1] is shown in Scheme 2, involving 2,4-dibromopyridine 1 and p-fluorophenyl boronic acid 2a to give three products: 3aC2–Ar, 3aC4–Ar, and 3adiaryl. The calculated bond dissociation energies for the C2–Br and C4–Br bonds in 1 were calculated to be 63.3 and 66.9 kcal mol–1 respectively (determined by Density Functional Theory calculations using the B3LYP/DGTZVP level of theory), which indicate that the C2–Br bond is weaker that the C4–Br bond, mirroring the expected typical site for functionalization.
Scheme 2. Benchmark SMCC of 1 with p-Fluoro-phenylboronic Acid 2a To Give Typical Product 3aC2-Ar, Atypical Product 3aC4-Ar, and Diarylated Product 3adiaryl [1]; the Proposed Equilibrium for 2a and n-Bu4NOH, Which Is Expected to Lie to the Right-Hand Side Is Shown in [2].

The reaction conditions described in Scheme 2 [1] are drawn from our earlier studies,32 informed by the work of Jutand et al.39 The reaction conditions benefit from being homogeneous (THF/H2O/[n-Bu4N]OH base at 40 °C, with a ratio of THF:H2O of 1:1). The high basicity ensures that the dominant boron species present in solution is the aryl boronate species 2a′, stabilized by an nBu4N+ cation [2].40a,40b We anticipated the importance of this in terms of exploiting site-selectivity changes brought about by Pd catalyst speciation, under varying Pd/ligand ratios. Consistent with the findings reported by Cid,7 our reaction conditions employing Pd(PPh3)4 as the catalyst, gave rise to typical C2 site-selectivity at 1 although conversion was low at 40 °C. The latter finding parallels the low reactivity of 2-bromopyridine under identical conditions (i.e., the presence of higher PPh3 equivalents results in lower catalyst efficacy).32
C2-selectivity was also observed when employing Pd2(dba)3·CHCl3 (ca. 93% purity)41 with 2 or 4 equiv of PPh3 under the identical conditions {forming Pd0(dba)3–n/(PPh3)n where n = 1 or 2}. Significant differences in catalyst efficacy were revealed using Pd3(OAc)6/PPh3 precatalyst ratios, hereafter referred to as Pd(OAc)2/nPPh3 (where n = 0.5 to 4) under conditions as summarized in Scheme 2. For each catalytic regime, the conversion of 1 to products 3aC2–Ar, 3aC4–Ar, and 3adiaryl is given in Figure 1 (note that competing homocoupling reactions/protodebromination or protodeborylation were not observable).
Figure 1.
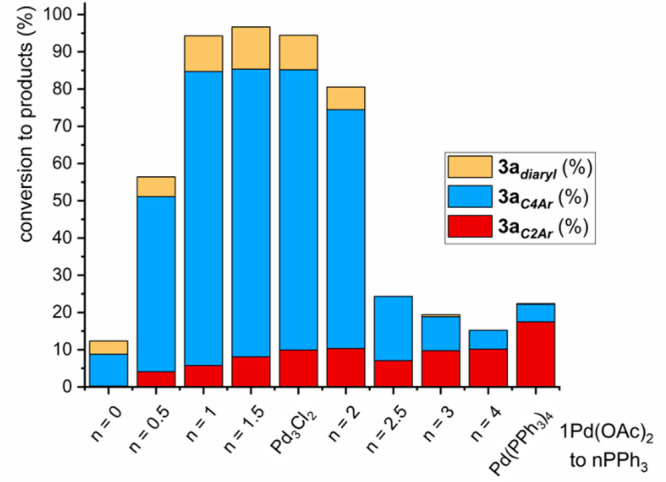
Summarizing Pd catalyst efficacy under different precatalytic Pd:PPh3 regimes, showing reaction conversions of which product selectivities for the SMCC (Scheme 2) of 1 with p-fluoro-phenylboronic acid 2a to give typical product 3aC2–Ar and atypical product 3aC4–Ar and bis-arylated product 3adiaryl.
For the Pd(OAc)2/nPPh3 ratios of 1:3 or 1:4, C2-site selectivity was observed, giving 3aC2–Ar as the major product, an outcome consistent with that observed, including lower product conversions, for the ubiquitous Pd0(PPh3)4 catalyst system. It is well established that Pd0(PPh3)n species (where n = 2 or 3), and/or anionic derivatives, are formed from the Pd(OAc)2/nPPh3 ratios of 1:3 or 1:4, respectively.32,42−45
Altering the Pd(OAc)2/nPPh3 ratio to n = 2.5 results in a switch in site-selectivity to the atypical 3aC4–Ar product. Concomitant with this switch in site-selectivity is an increase in substrate 1 conversion, an outcome particularly evident on lowering nPPh3 in the system to n < 2. The highest catalyst efficacy and C4-site selectivity are seen for Pd(OAc)2/nPPh3, in a 1:1 or 1:1.5 ratio. Of particular note is the activity observed for the [Pd3(μ-Cl)(μ-PPh2)2(PPh3)3]Cl cluster precatalyst (referred to as ‘Pd3Cl2’ in Figure 1). This latter finding correlates with the Pd/P ratio in the Pd3Cl2 cluster which contains three donating PPh3 ligands (1 PPh3 per Pd) and two pseudohalogen-like anionic PPh2 ligands (the average oxidation state per Pd being 4/3). Where n = 0, thus under an exogenous phosphine ligand-free regime, the reactivity drops off significantly, although overall 3aC4–Ar product selectivity is maintained. Hence, under our SMCC conditions, merely changing the Pd(OAc)2/nPPh3 ratio results in a switch in site-selectivity and catalyst efficacy, with markedly increased reaction conversions and higher selectivity for the atypical 3aC4–Ar product.
With the knowledge that Pd(OAc)2 and 1 equiv of PPh3 provided increased C4-site selectivity in SMCC reactions of 1, we investigated whether other aspects of the conditions contributed to the atypical site-selectivities (Scheme 3) of 1 with p-anisylboronic acid (2b), which gave overall 3bC4–Ar selectivity under “benchmark” conditions (entry 1, Table 1).
Scheme 3. Testing Additive and Base Effects for the SMCC between 1 and 2b.

Table 1. Modifying the Base and Additives in the SMCC Reaction between 1 and 2b (Scheme 3).
| Precatalyst | Entry | Base | Additive | Conv (%)a | 3bC2–Ar:3bC4–Ar:3bdiaryla |
|---|---|---|---|---|---|
| Pd(OAc)2/1PPh3 | 1 | n-Bu4NOH | none | 100 | 10:70:20 |
| 2 | KOH | none | 89 | 26:46:28 | |
| 3 | KOH | n-Bu4NBr | 88 | 3:70:26 | |
| Pd(OAc)2/2PPh3 | 4 | n-Bu4NOH | none | 100 | 18:58:24 |
| 5 | KOH | n-Bu4NBr | 100 | 8:79:6 | |
| Pd3Cl2 | 6 | n-Bu4NOH | none | 100 | 15:69:16 |
| 7 | KOH | none | 92 | 38:38:24 | |
| 8 | KOH | n-Bu4NBr | 94 | 9:82:10 | |
| 9 | KOH | n-Oct4NBr | 100 | 7:90:3 |
Determined by 1H NMR analysis of the crude reaction mixture, after 1 h.
Using KOH (aq.) as the base in place of n-Bu4NOH (aq.) (entry 2, Table 1) resulted in a marginal reduction in conversion but, more strikingly, a marked reduction in site-selectivity, as exemplified by the reduced 3bC4–Ar:3bC2–Ar ratio when using a Pd(OAc)2/1PPh3 catalytic system. This observation indicated the cation of the base, n-Bu4NOH(aq.), as a critical factor in the higher site-selectivities observed. Indeed, employing KOH (aq.) base alongside an n-Bu4NBr additive increased the 3bC4–Ar:3bC2–Ar site-selectivity at the expense of relatively higher amounts of 3bdiaryl (entry 3, Table 1). Cations have been shown to be able to influence SMCC reaction rates, principally the transmetalation step,39,46−48 but to our knowledge this is the first example of such a cation affecting the site-selectivity outcome of a cross-coupling reaction involving a dihalogenated heteroarene. Using a Pd(OAc)2/2PPh3 catalytic system alongside a KOH (aq.)/n-Bu4NBr base system similarly boosted C4 selectivity (entries 4 and 5, Table 1). Analogous observations were made employing catalytic Pd3Cl2 (entries 6–9, Table 1). Arguably the best outcome in terms of global 3bC4–Ar product selectivity was obtained using Pd(OAc)2/2PPh3 or Pd3Cl2 along with KOH/n-Bu4NBr (entries 3 and 5 respectively). Switching to the longer-chain quaternary ammonium salt n-octylammonium bromide (n-Oct4NBr) in place of n-Bu4NBr gave the highest product selectivity for 3bC4–Ar (entry 9, Table 1).
An assay was designed to track the product evolution of 3bC4–Ar, 3bC2–Ar, and 3bdiaryl products over time in the SMCC reaction between 1 and p-anisylboronic acid 2b, enabled by the Pd(OAc)2/2PPh3 and Pd3Cl2 catalyst systems and an n-Bu4NOH(aq.) base (Scheme 4, Graphs A and B Figure 2) .
Scheme 4. Conditions, Reagents, and Catalysts Used for Kinetic Product Distribution Analysis in SMCC Reactions of 1.

Figure 2.
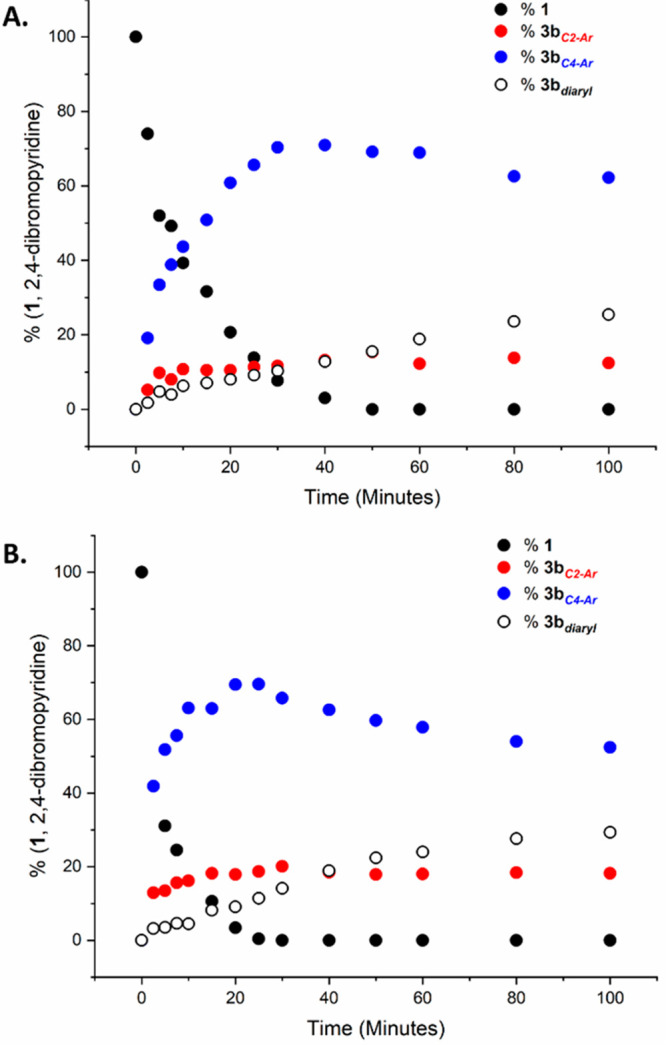
Product distribution of 3bC4–Ar, 3bC2–Ar, and 3bdiaryl as functions of time in the SMCC reaction between 1 and 2b. Using (A) Pd3Cl2 and (B) Pd(OAc)2/2PPh3 as the precatalyst.
Graphs A and B (Figure 2) show that employing Pd3Cl2 or Pd(OAc)2/2PPh3 as a precatalytic system resulted in broadly comparable overall reactivities with time. In both cases 3bC4–Ar was the predominant product, the quantity of which reached a maximum conversion at approximately 35 and 25 min for Pd3Cl2 and Pd(OAc)2/2PPh3, respectively. After this time, 3bC4–Ar was slowly converted into 3bdiaryl while the amount of 3cC2–Ar remained approximately constant. This study indicated that, with the Pd loading fixed at 3 mol %, the Pd(OAc)2/2PPh3 catalyst is marginally more efficacious than Pd3Cl2, accounting for the increased 3bdiaryl conversion observed with Pd(OAc)2/2PPh3, compared with Pd3Cl2 in Table 1.
Given our observations on the importance of the Pd/PPh3 ratio and aliphatic cation n-Bu4N+ as necessary requirements for atypical 3bC4–Ar site-selectivity in SMCCs, it was decided to assess whether such effects emerge in the Kumada cross-coupling of 1 with phenylmagnesium bromide (5) forming 3cC4–Ar, 3cC2–Ar, and 3cdiaryl (Scheme 5).
Scheme 5. Conditions for the Kumada Cross-Coupling of 1 with Phenylmagnesium Bromide 5.

Variables changed are highlighted in bold.
The Pd(OAc)2/nPPh3 ratio and catalyst prestir time (in THF) were altered, with reactions being run in the presence and absence of n-Oct4NBr, enabling conversions of 1 and selectivity changes to monoarylated products: 3cC4–Ar and 3cC2–Ar to be fully assessed (Table 2).
Table 2. Changes in Conversion of 1 and Product Site-Selectivity Outcomes, upon Changing Reaction Variables in Kumada Cross-Couplings (Scheme 5).
| Entry | Pd(OAc)2:nPPh3 | Catalyst prestir time (h) | n-Oct4NBr | Conv (%)a | 3cC2Ar:3cC4Ar:3cdiaryla |
|---|---|---|---|---|---|
| 1 | No cat. | 0.5 | – | 0 | N/A |
| 2 | 1:4 | 0.5 | – | 100 | 83:0:17 |
| 3 | 1:2 | 0.5 | – | 100 | 91:3:6 |
| 4 | 1:1 | 0.5 | – | 99 | 84:6:9 |
| 5 | 1:1 | 24 | – | 85 | 80:11:8 |
| 6 | 1:1 | 0.5 | + | 83 | 21:68:12 |
| 7 | 1:1 | 24 | + | 96 | 15:77:8 |
| 8 | 1:4 | 0.5 | + | 96 | 67:26:7 |
Determined by 1H NMR of the crude reaction mixture after 1 h.
In the absence of n-Oct4NBr, high selectivity for the 3cC2–Ar product was observed (entries 2–4,Table 2). Selectivity for 3cC2–Ar remained, albeit diminishing, when the catalyst prestir time was extended to 24 h (entry 5, Table 2). Employing n-Oct4NBr instigated a switch in site-selectivity favoring 3cC4–Ar as the major product, thus mirroring the requirement for a quaternary ammonium salt observed for C4-site-selectivity in the SMCC regime vide supra.
Lengthening the prestir time to 24 h resulted in a moderate increase in 3cC4–Ar:3cC2–Ar site-selectivity from 3.2:1 to 5.1:1, accompanied by an increase in conversion. The outcome provides an indication that an active and selective Pd catalyst species was generated during this time. The catalyst, generated from Pd(OAc)2 and 4PPh3, prestirred alongside n-Oct4NBr (0.5 h, THF), led to 3cC2–Ar product selectivity, confirming the dual requirement of a high Pd:P ratio as well as an additive salt for overall 3cC4–Ar selectivity under the specified conditions.
To gain insight into the mechanistic dichotomy in site-selectivity seen for 1, the effect of para-aromatic substituents on reaction conversion and site-selectivity was assessed in the SMCC reactions employing appropriate substituted arylboronic acids (Figure 3 and Scheme 6).
Figure 3.

Effect of product selectivities in SMCC reactions as a function of catalyst system employed and para-substituent on the phenylboronic acid substrate. (A) Using Pd2(dba)3·CHCl3/2PPh3. (B) Pd3Cl2. (C) Pd(OAc)2/1PPh3.
Scheme 6. Conditions, Reagents, and Catalysts Used for para-Substituent Analysis of Site-Selective SMCC Reactions at 1.

Determined by 1H NMR analysis of the crude reaction mixture, after 1 h.
The model SMCC reaction was carried out with a series of para Z-substituents to determine whether an electronic contribution influenced the overall site-selectivity, in selecting the C2–Br or C4–Br bonds in 1. Three different precatalytic systems were employed for this part of the study: first, Pd2(dba)3·CHCl3 (∼93% purity) with 2 PPh3, which proved to be an effective 3C2–Ar site-selective catalyst under the conditions. Second, the 3C4–Ar site-selective catalyst systems, namely Pd3Cl2 cluster and Pd(OAc)2/1PPh3, were assessed (Figure 3).
An important observation from this series of experiments is that the greater the electron-withdrawing capacity of the Z-substituent, the higher the selectivity for the atypical 3C4–Ar product. Concomitant with these observations was lower overall product conversions, indicating that the transmetalation step as rate-determining or that, in some form, the Z-substituent influences reaction site-selectivity involving 1. Taking the Pd2(dba)3·CHCl3/2PPh3 catalyst system (Figure 3A), the most active aryl boronic acid is para-anisylboronic acid 2b, affording high selectivity for 3bC2–Ar, although competing 3bdiaryl is apparent. Similar behavior was noted for para-methyl phenylboronic acid 2d and phenylboronic acid 2e.
The response of the Pd3Cl2 cluster catalyst to changes in the para Z-substituents of the phenylboronic acids is predictable, in that high selectivity for the 3C4–Ar products were recorded (Figure 3B). A similar but more subtle response is seen for the Pd(OAc)2/1PPh3 catalyst system (Figure 3C).
A plot of ΔΔG‡ against σP reveals the reaction sensitivity to the phenylboronic acid para-substituent Z (Figure 4). One sees that Pd3Cl2 cluster and Pd(OAc)2/1PPh3 catalyst systems behave quite differently to Pd2(dba)3·CHCl3/2PPh3. The magnitude for the gradient (∼0.24) for the latter catalyst system is in-keeping with the presumption that the aryl boronic acid substituent ought not to affect site-selectivity in 1, as oxidative addition occurs prior to transmetalation for mononuclear Pd catalysts. However, larger gradients are seen for the Pd3Cl2 cluster (∼0.77) and Pd(OAc)2/1PPh3 (∼0.48), providing evidence that these catalyst systems behave in a similar manner.
Figure 4.
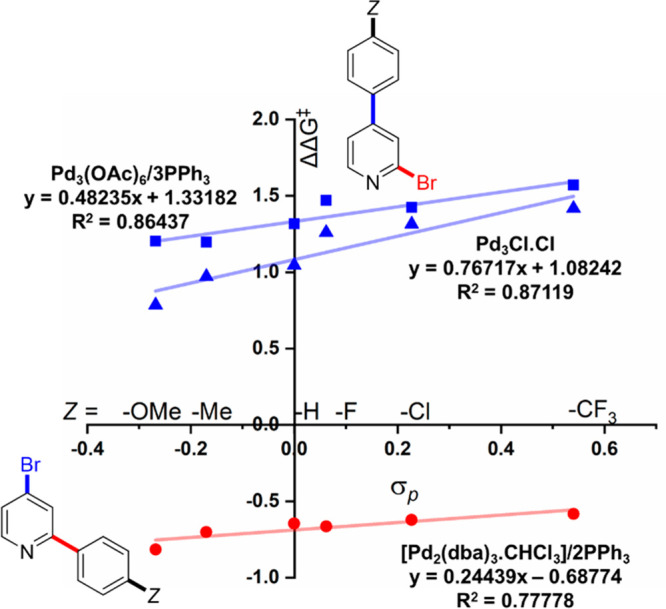
Plot of ΔΔG‡ against σP for para-substituent changes in SMCC reactions of 1 with p-Z-C6H4-B(OH)2 (2a–f).
Given the response of the SMCC reactions of 2,4-dibromopyridine 1 toward the ubiquitous ligand PPh3, we decided to screen other widely used phosphorus-containing ligands (Scheme 7, Figure 5). We tested catalyst mixtures with Pd(OAc)2/ligand ratios of 1:2 in the reaction of 1 with phenylboronic acid 2c to give products 3cC2–Br, 3cC4–Br, and 3cdiaryl. Based on consumption of 1 we see low conversions to the monoarylated products, albeit with a bias toward 3cC4–Ar. However, the dominant product is 3cdiaryl resulting from diarylation.
Scheme 7. Conditions and Reagents Used for Determining the Effects of a Variety of P-Ligands on Site-Selective SMCC Reactions at 1.

Figure 5.
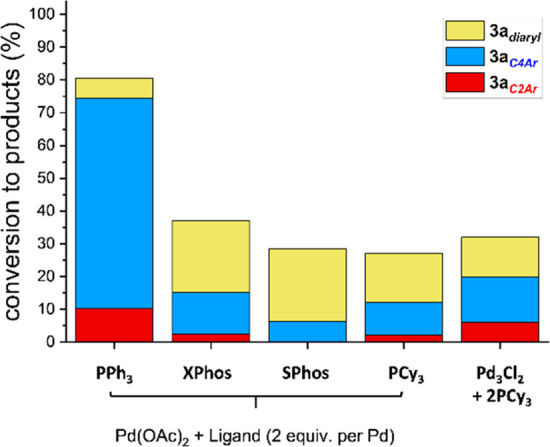
Performance of phosphorus-containing Pd precatalysts systems in site-selective Suzuki–Miyaura cross-coupling of 1.
Post-rationalization and Further Analysis
The important take-home message from the examples presented thus far is that a switch in site-selectivity for the 3C4–Ar product in SMCC and Kumada cross-coupling reactions occurs when a quaternary ammonium salt n-R4NX (R = butyl or octyl, X = Br– or HO–) is employed alongside a low catalytic equivalence of nPPh3 per Pd(OAc)2 (where 0.5 < n ≥ 2.5 in the case of the SMCC reaction). The results point to the existence of different mechanisms being available to Pd, as the Pd(OAc)2/nPPh3 ratios are changed; i.e., the Pd catalyst speciation is different, which is in-keeping with our earlier studies.32 The higher C2 site-selectivity for 3C2–Ar using higher equivalences of PPh3 relative to Pd mirrors that reported by Cid et al.7 using a Pd0(PPh3)4 catalyst which is closely related to the [Pd0(PPh3)n(OAc)]− active species that arises from Pd(OAc)2/≥3 PPh3.32,42−45 Indeed, in our study, in line with observations by Cid et al.,7 we found that the direct reaction of 1 with Pd0(PPh3)4 in toluene at 23 °C (Figure 6) gave the C2-oxidative addition product OAC2–Br as the major regioisomer (OAC2–Br/OAC4–Br ≈ 25:1 by 31P NMR spectral analysis of a crude reaction mixture).
Figure 6.
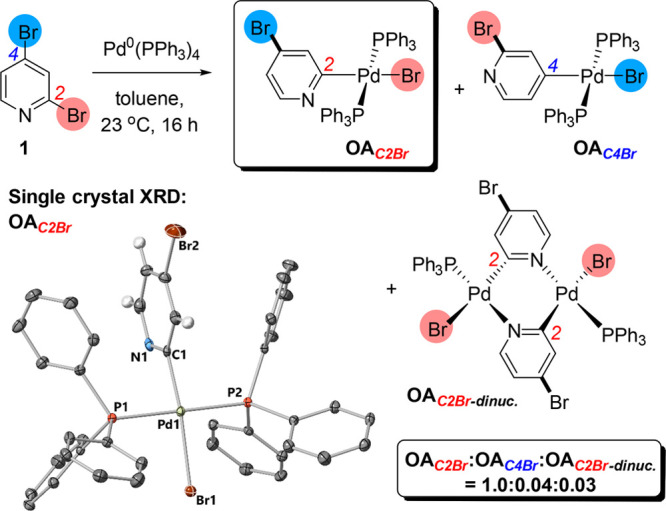
Confirmation of mechanistic reasoning for C2–Br site-selectivity in the reaction of Pd0(PPh3)4 with 1 at 23 °C.
The major regioisomer OAC2–Br was characterized by X-ray diffraction analysis (corroborated by NMR spectroscopic analysis of the single crystal analyzed by X-ray diffraction). Cid et al. characterized the dinuclear Pd complex, OAC2–Br-dinuc (Figure 6), resulting from loss of PPh3 from OAC2–Br and subsequent dimerization of the putative 14-electron PdII species. These results indicate that oxidative addition of Pd0(PPh3)n (n = 2 or 3) is the starting point for the SMCC of 1 when Pd0(PPh3)4 or Pd(OAc)2/≥3PPh3 is used as the precatalyst system, accounting for the overall C2 site-selectivity observed in our study, in addition to the previously reported cross-coupling reactions involving 1.7,8
Further experiments however showed that by stirring a solution of Pd(OAc)2 and 1 PPh3 at 0 °C for 5 min, layering of the solution with hexane and subsequent storage at −18 °C led to the growth of reddish-brown crystals. These have been confirmed by single crystal X-ray diffraction analysis to be the dinuclear PdII complex [PdII(μ2-OAc)(κ-OAc)(PPh3)]2 (4), containing bridging and terminal acetate groups, with one terminal PPh3 ligand at each Pd center (Figure 7A). The structure of 4 was also confirmed by 1H NMR spectroscopic analysis to be the major solution species formed immediately after mixing Pd(OAc)2 and 1 PPh3 (diagnostic peaks for the acetoxy methyl group at δH 1.34 ppm) (Figure 7B). We did not see any evidence of low-ligated phosphine adducts of Pd3(OAc)6.49,50 The broadness of the single methyl resonance (for the OAc ligands) suggests that the two acetate environments are in exchange at 25 °C, supported by the proximal relationship as indicated in the solid-state.
Figure 7.
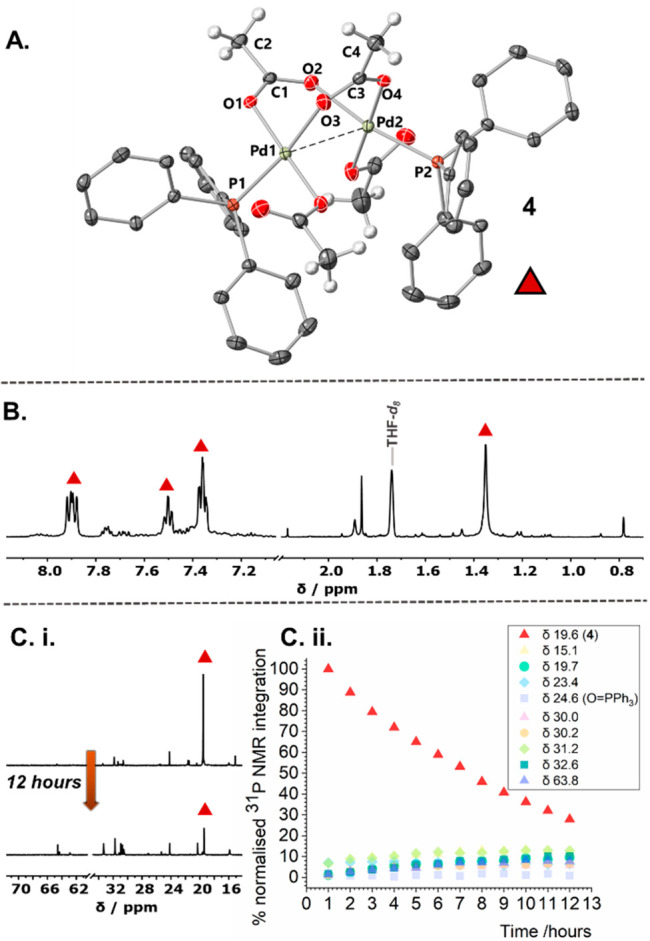
Analysis of the THF-d8. solution arising from the mixture of Pd(OAc)2/1PPh3. (A) XRD structure of a single crystal of 4 is shown (selected atoms). (B) 1H NMR analysis, confirming solution presence of 4 ca. 10 min after mixing at 25 °C. (C. i. and ii.) 31P NMR spectral data and reaction speciation, showing the decay of 4 and growth of multiple P-containing species over 12 h, 25 °C.
Complex 4 was originally reported by Wilkinson et al., who described it as unstable in the solid-state51—we concur with this description but were fortunate in being successful in obtaining a solid-state structure. Interestingly, the reactivity of 4 under (hydrogenative) reducing conditions has been investigated.52,53
We further investigated the solution behavior of 4 in dry d8-THF at room temperature by 31P NMR spectroscopic analysis (Figure 7C). Over time, a darkening of the solution was noted concomitant with the formation of multiple different phosphorus-containing species (Figure 7C. ii.). While Pd(OAc)2/2 or ≥3PPh3 is known to reduce/activate at the expense of concomitant oxidation of PPh3via. trans-[Pd(OAc)2(PPh3)2], in this case, O=PPh3 was only observed as a minor biproduct of the process. 1H NMR spectroscopic analysis of the post reaction solution indicated that Ac2O formed as a major byproduct, alongside AcOH, in a 1:3 ratio, respectively. This observation points toward 4 facilitating a different mechanism for activation of Pd(OAc)2 in the presence of 1 equiv of PPh3, when compared with trans-[Pd(OAc)2(PPh3)2].32,42−45 TEM analysis of the decomposed solution of 4 (after overnight reaction at room temperature) demonstrated the presence of large, spherical Pd particles (micron-sized).32 When Pd(OAc)2 was similarly treated with 2PPh3 at room temperature, a dinuclear PdI species was found to be transiently stable in THF. The observation that optimal catalyst activity and selectivity occur when relatively low precatalytic ratios of PPh3 to Pd(OAc)2 are employed, i.e. enabling formation of aggregated Pd clusters and particles, strongly correlates with the observed reactivity and selectivity involving cross-coupling reactions of 1, in keeping with differences in reactivity seen for the related 2-bromopyridine substrate.32Scheme 8 summarizes our overall findings, linking catalyst speciation under differing Pd(OAc)2/nPPh3 regimes.
Scheme 8. Dichotomy in Site-Selectivity at 1: Different Pd Species Arising from Different Ratios of Pd(OAc)2/nPPh3 Result in Different Cross-Coupling Selectivities under Cross-Coupling Conditions.

In addition to the Pd/P ratio, a key requirement for high 3C4–Ar selectivity is the presence of a quaternary ammonium salt R4NX (R = n-butyl or n-octyl, X = Br–, OH–). The latter requires further comment and experimental corroboration, as there is a wealth of literature that explores the stabilization of highly active anionic PdNP catalyst species by salts. Dupont et al. reported that catalytic Pd particles, generated by in situ reductive activation of a palladacyclic compound, could be stabilized by imidazolium salts for applications in Heck coupling.54 The immediate electropositive outer layer of a metal nanoparticle can be stabilized by anions, the sterics and basicity of which influence PdNP stability.55,56 In a regime analogous to the electrical double layer, the anionic layer can in turn be stabilized by a layer of cations. Astruc et al. explored this electrosteric stabilization in the design of bespoke architectures for the stabilization of PdNPs.57,58 This valuable prior knowledge underpins our hypothesis that electrosteric stabilization of PdNPs is critical to the site-selectivity switch seen in the cross-coupling reactions of 1. Thus, stabilized PdNPs, formed in situ from either precatalysts Pd(OAc)2 and 1PPh3 or Pd3Cl2 by additive or in situ generated salts, are the catalyst species responsible for this atypical selectivity and relatively high activity, compared to that of the dominant mononuclear catalytic species generated from Pd(OAc)2 and ≥3PPh3, Pd(PPh3)4, or Pd2(dba)3·CHCl3/2PPh3.
We have tested our hypothesis further and shown that a tris-imidazolium tribromide salt can effectively stabilize PdNPs enabling a marked rise in site-selectivity at 1, exhibiting a 3bC4–Ar:3bC2–Ar ratio of 17.6:1, with a relatively low formation of 3bdiaryl product (see Supporting Information (SI) for further details).
The notion that changes to Pd catalyst speciation might result in different chemoselectivities has been reported by Schoenebeck et al., elaborating on earlier findings by Fu et al.14,16,59 They rationalized that cross-coupling selectivities at 4-chlorophenyl triflate occurred at the C–Cl site in reactions catalyzed by [Pd0(L)1] and the C–OTf side in reactions catalyzed by the analogous [Pd0(X)(L)]− complex (where X = an anion present in the system, L = PtBu3). In this case, however, both active catalysts were proposed to be mononuclear Pd0 ligated species (based on experimental and computational evidence). Indeed, subsequent work used 4-chlorophenyl triflate as a probe to differentiate between mechanisms arising from a dinuclear PdI precatalyst.60Our work has similarly shown two different mechanisms for the activation of different sites of the dibrominated heterocycle 1. However, in the case of selectivity for the C4 position, under the reaction conditions that we have identified, it is highly unlikely that such mononuclear Pd0 species can be present, an assertion based on what is known about Pd speciation as the Pd(OAc)2/nPPh3 ratio is altered (vide supra).32
Finally, the synthetic utility of the Pd3(OAc)6/3PPh3 catalytic system was demonstrated in the synthesis of a novel 2,4-disubstituted pyridine by successive C4-selective arylation by an SMCC reaction at 1, followed by an Ullman etherification61 at its C2-position (see SI for further details).
Mechanistic Hypotheses
Given that such profound site-selectivity changes are seen for cross-couplings of 2,4-dibromopyridine 1, on changing Pd catalyst speciation in the presence of stabilizing salt additives, a discussion concerning the mechanistic implications is pertinent. If one assumes that only mononuclear Pd species are the relevant catalyst species (dependent on reaction conditions), then selection of the C2–Br over C4–Br bond occurs on activation of 1 by a Pd0(PPh3)n complex (where that n is typically ≥2).7 A neutral pathway is depicted in Scheme 9A. Here, the relative rates of oxidative addition would explain the typical site-selectivity for C2–Br, presuming this step is irreversible and that the associated higher intrinsic electrophilicity of this bond lowers the barrier to its activation. The case for this catalytic cycle has been made strongly elsewhere;7 however, oxidative addition must be reversible (in Scheme 9A) in order to account for our experimental observations. Switching site-selectivity from C2–Br to C4–Br, i.e. 3C4–Ar over 3C2–Ar, arguably requires a quite different ligand environment,14,59,60,62 or a complete change in mechanism. We have not shown anionic mononuclear Pd species here, but clearly in the presence of n-R4NBr, such a pathway could be operative, with n-R4N+ acting as the stabilizing cation.43,45,63
Scheme 9. Mechanistic Hypotheses: (A) Catalytic Cycle Involving Mononuclear Pd Species, via Classical Pd Intermediates or Alternative Route Involving a SNAr-Type Mechanism; (B) Catalyst Cycle Based on That Evidenced by Li et al.29 Involving Pd3 Cluster Species; (C) Proposed Involvement of Higher Order Pd Agglomerates (Note: Only Details of Key Steps Are Shown – trans–cis Isomerizations and Ligand Dissociation/Association Are Involved).

Note: (P) = PPh2; P = PPh3; X = anion, e.g. Br or OH.
Maes and Jutand et al.64 have reported strong evidence for the existence of an SNAr mechanism for the activation of 5-substituted-2-bromo-pyridines, which is therefore shown in A for the C2-arylation pathway, important given the structural similarity to 1.
An alternative mechanism based on the strong experimental support reported by Li et al. “Pd3 cluster” catalysis is shown in Scheme 9B.29 In this case the Pd3Cl2 cluster catalyst, via formation of a Pd3-hydroxo species, was proposed to activate the organoboronic acid first, the adduct of which could then activate the aryl halide. Inversion of the oxidative addition/transmetalation steps could explain the higher than expected Z-substituent sensitivity in the site-selective SMCC reaction involving 1, particularly in the region where Pd3 clusters/Pd nanoparticles are catalytically competent (Figure 3 and Figure 4).
A third scenario (Scheme 9C) highlights the potential role of Pd nanoparticles (agglomerates) in the activation of 1, in essence like the mechanism depicted in Scheme 9B. The Pd nanoparticles are shown ligated by PPh3 and halide ligands, as it is established that such stabilizing surface interactions are important.65,66 In this case, an aryl boronate complex could be activated by the Pd nanoparticle surface, prior to oxidative addition of the C4–Br bond of 1. The interaction of base and anionic aryl boron species at Pd nanoparticle surfaces has been proposed by El-Sayed et al.67 Such a situation aligns with the Z-substituent effect (aryl boronic acid) on reaction efficacy and site-selectivity. The scenario also fits with the observed speciation arising from Pd(OAc)2/1PPh3vide supra—the optimized catalyst system. There can be no doubt that the mechanistic complexity presented in Scheme 9 requires significant independent investigation. (We have embarked on computational studies (DFT) to support the mechanistic hypotheses described in Scheme 9. However, we are yet to obtain reasonable results, as the conformational flexibility in these large Pd3Cl2 cluster species, and related downstream intermediates, is high, leading to local energy minima. We selected to not simplify the Pd3 structural models, as the ligand microenvironment surrounding these is clearly important in stabilization and in controlling how substrates approach the Pd centers and their activation.) We anticipate that specialist experimental methods (real-time fluorescence26 and X-ray absorption spectroscopy24,25) might reveal insight into the underlying catalyst speciation behavior and complexity.
Conclusions
In conclusion, our studies have shown that site-selective cross-couplings of 2,4-dibromopyridine 1 are affected by the type of catalyst system used and catalyst speciation that ultimately results under working reaction conditions. The observations are clear for both SMCC and Kumada cross-coupling reactions. We have confirmed that Pd(OAc)2/≥3PPh3, and related catalyst systems, enable typical C2-selctivity. However, for the Pd(OAc)2/≤2PPh3 catalytic system, atypical C4-selectivity is seen, an outcome that is mirrored using the Pd3Cl2 cluster catalyst. The addition of a quaternary ammonium salt proved to be a critical additive for atypical C4-selectivity, supporting the hypothesis that high site-selectivity is attributable to PdNPs formed in situ, for which the quaternary ammonium salt plays a stabilizing role. The hypothesis was supported using a bespoke tris-imidazolium tribromide salt, capable of stabilizing Pd nanoparticles.54,55,57 Addition of such a salt to the SMCC reaction system led to a significant increase in the C4-selectivity. Our findings mark the first examples of site control of a dihalogenated heteroarene, switching between two halogens of the same type, while using the same Pd source [Pd3(OAc)6] and the same ligand type PPh3. It underlines the importance of controlling precise metal–ligand ratios for optimal catalyst performance. Interestingly, in the context of site-selective SMCCs, Spivey et al.4 stated that “...caution must be applied when trying to rationalise switches in site-selectivities as a function of changes of conditions as the observed products may not arise from the ligated species expected.” We can now confirm that is the case, but that reaction outcomes can be controlled through understanding fundamental changes in Pd catalyst speciation.
More generally our study has demonstrated that the activity of well-established Pd catalyst mixtures can be very easily altered by small changes to the reaction conditions. We can recognize that understanding and controlling catalytic speciation may allow simple Pd catalytic precursors and simple inexpensive ligands (e.g., PPh3) to exhibit unique properties in catalytic cross-coupling chemistries. Such an approach could be potentially exploited to avoid the use of expensive ligand architectures. Furthermore, our approach to understanding the Pd catalyst speciation may serve to complement understanding in other powerful site-selective cross-couplings.12,13,68−71
Acknowledgments
The project was funded by Bayer AG (PhD studentship to NWJS). We thank the University of York for supporting NMR spectrometers & X-ray equipment, and EPSRC for NMR upgrades (EP/K039660/1). C.E.W. acknowledges EPSRC funding (EP/R009406/1). We gratefully acknowledge the efforts of Dr. Peter Karadakov (York) in conducting several exploratory DFT studies to explore our mechanistic hypotheses presented in Scheme 9, and Dr. J. M. Lynam for many discussions regarding this work. We acknowledge the X-ray Diffraction service in York for their valuable work (Dr. Sam Hart and Dr. Rachel Parker).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.1c05294.
Full procedures, compound characterization data, catalysis studies, and X-ray details (PDF)
Accession Codes
CCDC 2060853–2060856 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif, or by emailing data_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223 336033.
The research was principally funded by Bayer AG (CropScience Division), supported by EPSRC grants (EP/K039660/1 and EP/R009406/1).
The authors declare no competing financial interest.
Supplementary Material
References
- Welsch M. E.; Snyder S. A.; Stockwell B. R. Privileged scaffolds for library design and drug discovery. Curr. Opin. Chem. Biol. 2010, 14, 347–361. 10.1016/j.cbpa.2010.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golding W. A.; Pearce-Higgins R.; Phipps R. J. Site-Selective Cross-Coupling of Remote Chlorides Enabled by Electrostatically Directed Palladium Catalysis. J. Am. Chem. Soc. 2018, 140, 13570–13574. 10.1021/jacs.8b08686. [DOI] [PubMed] [Google Scholar]
- Fairlamb I. J. S. Regioselective (site-selective) functionalization of unsaturated halogenated nitrogen, oxygen and sulfur heterocycles by Pd-catalysed cross-couplings and direct arylation processes. Chem. Soc. Rev. 2007, 36, 1036–1045. 10.1039/b611177g. [DOI] [PubMed] [Google Scholar]
- Almond-Thynne J.; Blakemore D. C.; Pryde D. C.; Spivey A. C. Site-selective Suzuki-Miyaura coupling of heteroaryl halides - understanding the trends for pharmaceutically important classes. Chem. Sci. 2017, 8, 40–62. 10.1039/C6SC02118B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legault C. Y.; Garcia Y.; Merlic C. A.; Houk K. N. Origin of regioselectivity in palladium-catalyzed cross-coupling reactions of polyhalogenated heterocycles. J. Am. Chem. Soc. 2007, 129, 12664–12665. 10.1021/ja075785o. [DOI] [PubMed] [Google Scholar]
- Handy S. T.; Zhang Y. A simple guide for predicting regioselectivity in the coupling of polyhaloheteroaromatics. Chem. Commun. 2006, 299–301. 10.1039/B512948F. [DOI] [PubMed] [Google Scholar]
- Sicre C.; Alonso-Gómez J. L.; Cid M. M. Regioselectivity in alkenyl(aryl)-heteroaryl Suzuki cross-coupling reactions of 2,4-dibromopyridine. A synthetic and mechanistic study. Tetrahedron 2006, 62, 11063–11072. 10.1016/j.tet.2006.09.040. [DOI] [Google Scholar]
- Zhou Q.; Zhang B.; Su L.; Jiang T.; Chen R.; Du T.; Ye Y.; Shen J.; Dai G.; Han D.; Jiang H. Palladium-catalyzed highly regioselective 2-arylation of 2,x-dibromopyridines and its application in the efficient synthesis of a 17β-HSD1 inhibitor. Tetrahedron 2013, 69, 10996–11003. 10.1016/j.tet.2013.10.065. [DOI] [Google Scholar]
- Zhang B.; Chen R.; Jiang H.; Zhou Q.; Qiu F.; Han D.; Li R.; Tang W.; Zhong A.; Zhang J.; Yu X. Palladium-catalyzed highly regioselective 2-alkynylation of 2,x-dihalopyridines. Tetrahedron 2016, 72, 2813–2817. 10.1016/j.tet.2016.03.027. [DOI] [Google Scholar]
- Rao M. L. N.; Dhanorkar R. J. Triarylbismuthanes as Threefold Aryl-Transfer Reagents in Regioselective Cross-Coupling Reactions with Bromopyridines and Quinolines. Eur. J. Org. Chem. 2014, 2014, 5214–5228. 10.1002/ejoc.201402455. [DOI] [Google Scholar]
- Fowler J. M.; Britton E.; Pask C. M.; Willans C. E.; Hardie M. J. Cyclotriveratrylene-tethered trinuclear palladium(II)-NHC complexes; reversal of site selectivity in Suzuki-Miyaura reactions. Dalton Trans. 2019, 48, 14687–14695. 10.1039/C9DT03400E. [DOI] [PubMed] [Google Scholar]
- Dai X.; Chen Y.; Garrell S.; Liu H.; Zhang L.-K.; Palani A.; Hughes G.; Nargund R. Ligand-dependent site-selective Suzuki cross-coupling of 3,5-dichloropyridazines. J. Org. Chem. 2013, 78, 7758–7763. 10.1021/jo401096u. [DOI] [PubMed] [Google Scholar]
- Yang M.; Chen J.; He C.; Hu X.; Ding Y.; Kuang Y.; Liu J.; Huang Q. Palladium-Catalyzed C-4 Selective Coupling of 2,4-Dichloropyridines and Synthesis of Pyridine-Based Dyes for Live-Cell Imaging. J. Org. Chem. 2020, 85, 6498–6508. 10.1021/acs.joc.0c00449. [DOI] [PubMed] [Google Scholar]
- Proutiere F.; Schoenebeck F. Solvent Effect on Palladium-Catalyzed Cross-Coupling Reactions and Implications on the Active Catalytic Species. Angew. Chem., Int. Ed. 2011, 50, 8192–8195. 10.1002/anie.201101746. [DOI] [PubMed] [Google Scholar]
- Keylor M. H.; Niemeyer Z. L.; Sigman M. S.; Tan K. L. Inverting Conventional Chemoselectivity in Pd-Catalyzed Amine Arylations with Multiply Halogenated Pyridines. J. Am. Chem. Soc. 2017, 139, 10613–10616. 10.1021/jacs.7b05409. [DOI] [PubMed] [Google Scholar]
- Schoenebeck F.; Houk K. N. Ligand-controlled regioselectivity in palladium-catalyzed cross coupling reactions. J. Am. Chem. Soc. 2010, 132, 2496–2497. 10.1021/ja9077528. [DOI] [PubMed] [Google Scholar]
- Golding W. A.; Schmitt H. L.; Phipps R. J. Systematic Variation of Ligand and Cation Parameters Enables Site-Selective C–C and C–N Cross-Coupling of Multiply Chlorinated Arenes through Substrate–Ligand Electrostatic Interactions. J. Am. Chem. Soc. 2020, 142, 21891–21898. 10.1021/jacs.0c11056. [DOI] [PubMed] [Google Scholar]
- Baumann C. G.; De Ornellas S.; Reeds J. P.; Storr T. E.; Williams T. J.; Fairlamb I. J. S. Formation and propagation of well-defined Pd nanoparticles (PdNPs) during C–H bond functionalization of heteroarenes: are nanoparticles a moribund form of Pd or an active catalytic species?. Tetrahedron 2014, 70, 6174–6187. 10.1016/j.tet.2014.06.002. [DOI] [Google Scholar]
- Hurst E. C.; Wilson K.; Fairlamb I. J. S.; Chechik V. N-Heterocyclic carbene coated metal nanoparticles. New J. Chem. 2009, 33, 1837–1840. 10.1039/b905559b. [DOI] [Google Scholar]
- Williams T. J.; Reay A. J.; Whitwood A. C.; Fairlamb I. J. S. A mild and selective Pd-mediated methodology for the synthesis of highly fluorescent 2-arylated tryptophans and tryptophan-containing peptides: a catalytic role for Pd0 nanoparticles?. Chem. Commun. 2014, 50, 3052–3054. 10.1039/C3CC48481E. [DOI] [PubMed] [Google Scholar]
- Reay A. J.; Neumann L. K.; Fairlamb I. J. S. Catalyst Efficacy of Homogeneous and Heterogeneous Palladium Catalysts in the Direct Arylation of Common Heterocycles. Synlett 2016, 27, 1211–1216. 10.1055/s-0035-1561436. [DOI] [Google Scholar]
- Bray J. T. W.; Ford M. J.; Karadakov P. B.; Whitwood A. C.; Fairlamb I. J. S. The critical role played by water in controlling Pd catalyst speciation in arylcyanation reactions. React. Chem. Eng. 2019, 4, 122–130. 10.1039/C8RE00178B. [DOI] [Google Scholar]
- Reay A. J.; Fairlamb I. J. S. Catalytic C–H bond functionalisation chemistry: the case for quasi-heterogeneous catalysis. Chem. Commun. 2015, 51, 16289–16307. 10.1039/C5CC06980G. [DOI] [PubMed] [Google Scholar]
- Ellis P. J.; Fairlamb I. J. S.; Hackett S. F. J.; Wilson K.; Lee A. F. Evidence for the Surface-Catalyzed Suzuki–Miyaura Reaction over Palladium Nanoparticles: An Operando XAS Study. Angew. Chem., Int. Ed. 2010, 49, 1820–1824. 10.1002/anie.200906675. [DOI] [PubMed] [Google Scholar]
- Lee A. F.; Ellis P. J.; Fairlamb I. J. S.; Wilson K. Surface catalysed Suzuki-Miyaura cross-coupling by Pd nanoparticles: an operando XAS study. Dalton Trans. 2010, 39, 10473–10482. 10.1039/c0dt00412j. [DOI] [PubMed] [Google Scholar]
- Costa P.; Sandrin D.; Scaiano J. C. Real-time fluorescence imaging of a heterogeneously catalysed Suzuki–Miyaura reaction. Nat. Catal. 2020, 3, 427–437. 10.1038/s41929-020-0442-0. [DOI] [Google Scholar]
- Zhao Y.; Du L.; Li H.; Xie W.; Chen J. Is the Suzuki-Miyaura Cross-Coupling Reaction in the Presence of Pd Nanoparticles Heterogeneously or Homogeneously Catalyzed? An Interfacial Surface-Enhanced Raman Spectroscopy Study. J. Phys. Chem. Lett. 2019, 10, 1286–1291. 10.1021/acs.jpclett.9b00351. [DOI] [PubMed] [Google Scholar]
- Eremin D. B.; Ananikov V. P. Understanding active species in catalytic transformations: From molecular catalysis to nanoparticles, leaching, “Cocktails” of catalysts and dynamic systems. Coord. Chem. Rev. 2017, 346, 2–19. 10.1016/j.ccr.2016.12.021. [DOI] [Google Scholar]
- Fu F.; Xiang J.; Cheng H.; Cheng L.; Chong H.; Wang S.; Li P.; Wei S.; Zhu M.; Li Y. A Robust and Efficient Pd3 Cluster Catalyst for the Suzuki Reaction and Its Odd Mechanism. ACS Catal. 2017, 7, 1860–1867. 10.1021/acscatal.6b02527. [DOI] [Google Scholar]
- Coulson D. R. Ready cleavage of triphenylphosphine. Chem. Commun. (London) 1968, 1530–1531. 10.1039/c19680001530. [DOI] [Google Scholar]
- Dixon K. R.; Rattray A. D. Trinuclear palladium clusters: synthesis and phosphorus-31 nuclear magnetic resonance spectra of [Pd3Cl(PPh2)2(PPh3)3][BF4] and related complexes. Inorg. Chem. 1978, 17, 1099–1103. 10.1021/ic50183a001. [DOI] [Google Scholar]
- Scott N. W. J.; Ford M. J.; Schotes C.; Parker R. R.; Whitwood A. C.; Fairlamb I. J. S. The ubiquitous cross-coupling catalyst system ‘Pd(OAc)2’/2PPh3 forms a unique dinuclear PdI complex: an important entry point into catalytically competent cyclic Pd3 clusters. Chem. Sci. 2019, 10, 7898–7906. 10.1039/C9SC01847F. [DOI] [Google Scholar]
- Campos J.; Nova A.; Kolychev E. L.; Aldridge S. A Combined Experimental/Computational Study of the Mechanism of a Palladium-Catalyzed Bora-Negishi Reaction. Chem. - Eur. J. 2017, 23, 12655–12667. 10.1002/chem.201702703. [DOI] [PubMed] [Google Scholar]
- Diehl C. J.; Scattolin T.; Englert U.; Schoenebeck F. C– I-Selective Cross-Coupling Enabled by a Cationic Palladium Trimer. Angew. Chem. 2019, 131, 217–221. 10.1002/ange.201811380. [DOI] [PubMed] [Google Scholar]
- Kalvet I.; Magnin G.; Schoenebeck F. Rapid Room-Temperature, Chemoselective Csp2–Csp2 Coupling of Poly(pseudo)halogenated Arenes Enabled by Palladium(I) Catalysis in Air. Angew. Chem., Int. Ed. 2017, 56, 1581–1585. 10.1002/anie.201609635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keaveney S. T.; Kundu G.; Schoenebeck F. Modular Functionalization of Arenes in a Triply Selective Sequence: Rapid C(sp2) and C(sp3) Coupling of C–Br, C–OTf, and C–Cl Bonds Enabled by a Single Palladium(I) Dimer. Angew. Chem., Int. Ed. 2018, 57, 12573–12577. 10.1002/anie.201808386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stambuli J. P.; Kuwano R.; Hartwig J. F. Unparalleled rates for the activation of aryl chlorides and bromides: coupling with amines and boronic acids in minutes at room temperature. Angew. Chem., Int. Ed. 2002, 41, 4746–4748. 10.1002/anie.200290036. [DOI] [PubMed] [Google Scholar]
- Fricke C.; Sherborne G.; Funes-Ardoiz I.; Senol E.; Guven S.; Schoenebeck F. Orthogonal Nanoparticle Catalysis with Organogermaniums. Angew. Chem., Int. Ed. 2019, 58, 17788–17795. 10.1002/anie.201910060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amatore C.; Jutand A.; Le Duc G. Mechanistic Origin of Antagonist Effects of Usual Anionic Bases (OH–, CO32–) as Modulated by their Countercations (Na+, Cs+, K+) in Palladium-Catalyzed Suzuki-Miyaura Reactions. Chem. - Eur. J. 2012, 18, 6616–6625. 10.1002/chem.201200516. [DOI] [PubMed] [Google Scholar]
- a Pagett A. B.; Lloyd-Jones G. C.. Suzuki–Miyaura Cross-Coupling. In Organic Reactions; Denmark S. E., Ed.; Wiley & Sons: New York, 2019; Vol. 100, pp 547–620. [Google Scholar]; b Payard P.-A.; Bohn A.; Tocqueville D.; Jaouadi K.; Escoude E.; Ajig S.; Dethoor A.; Gontard G.; Perego L. A.; Vitale M.; Ciofini I.; Wagschal S.; Grimaud L. Role of dppf Monoxide in the Transmetalation Step of the Suzuki–Miyaura Coupling Reaction. Organometallics 2021, 40, 1120–1128. 10.1021/acs.organomet.1c00090. [DOI] [Google Scholar]
- Zalesskiy S. S.; Ananikov V. P. Pd2(dba)3 as a Precursor of Soluble Metal Complexes and Nanoparticles: Determination of Palladium Active Species for Catalysis and Synthesis. Organometallics 2012, 31, 2302–2309. 10.1021/om201217r. [DOI] [Google Scholar]
- Amatore C.; Jutand A.; M’Barki M. A. Evidence of the Formation of Zerovalent Palladium from Pd(OAc)2, and Triphenylphosphine. Organometallics 1992, 11, 3009–3013. 10.1021/om00045a012. [DOI] [Google Scholar]
- Amatore C.; Carre E.; Jutand A.; M’Barki M. A.; Meyer G. Evidence for the Ligation of Palladium(0) Complexes by Acetate Ions: Consequences on the Mechanism of Their Oxidative Addition with Phenyl Iodide and PhPd(OAc)(PPh3)2 as Intermediate in the Heck Reaction. Organometallics 1995, 14, 5605–5614. 10.1021/om00012a029. [DOI] [Google Scholar]
- Amatore C.; Carre E.; Jutand A.; M’Barki M. A. Rates and Mechanism of the Formation of Zerovalent Palladium Complexes from Mixtures of Pd(OAc)2 and Tertiary Phosphines and Their Reactivity in Oxidative Additions. Organometallics 1995, 14, 1818–1826. 10.1021/om00004a039. [DOI] [Google Scholar]
- Amatore C.; Jutand A. Anionic Pd(0) and Pd(II) intermediates in palladium-catalyzed Heck and cross-coupling reactions. Acc. Chem. Res. 2000, 33, 314–321. 10.1021/ar980063a. [DOI] [PubMed] [Google Scholar]
- Amatore C.; Jutand A.; Le Duc G. Kinetic data for the transmetalation/reductive elimination in palladium-catalyzed Suzuki-Miyaura reactions: unexpected triple role of hydroxide ions used as base. Chem. - Eur. J. 2011, 17, 2492–2503. 10.1002/chem.201001911. [DOI] [PubMed] [Google Scholar]
- Amatore C.; Jutand A.; Le Duc G. The Triple Role of Fluoride Ions in Palladium-Catalyzed Suzuki–Miyaura Reactions: Unprecedented Transmetalation from [ArPdFL2] Complexes. Angew. Chem., Int. Ed. 2012, 51, 1379–1382. 10.1002/anie.201107202. [DOI] [PubMed] [Google Scholar]
- Amatore C.; Le Duc G.; Jutand A. Mechanism of Palladium-Catalyzed Suzuki-Miyaura Reactions: Multiple and Antagonistic Roles of Anionic “Bases” and Their Countercations. Chem. - Eur. J. 2013, 19, 10082–10093. 10.1002/chem.201300177. [DOI] [PubMed] [Google Scholar]
- Váňa J.; Lang J.; Šoltésová M.; Hanusek J.; Růžička A.; Sedlák M.; Roithová J. The role of trinuclear species in a palladium acetate/trifluoroacetic acid catalytic system. Dalton Trans. 2017, 46, 16269–16275. 10.1039/C7DT03832A. [DOI] [PubMed] [Google Scholar]
- Váňa J.; Hanusek J.; Sedlák M. Bi and trinuclear complexes in palladium carboxylate-assisted C–H activation reactions. Dalton Trans. 2018, 47, 1378–1382. 10.1039/C7DT04269H. [DOI] [PubMed] [Google Scholar]
- Stephenson T. A.; Wilkinson G. Acetato complexes of palladium(II). J. Inorg. Nucl. Chem. 1967, 29, 2122–2123. 10.1016/0022-1902(67)80480-9. [DOI] [Google Scholar]
- Moiseev I. I.; Stromnova T. A.; Busygina I. N.; Tihonova N. Y.; Kozitsyna N. Y.; Ellern A. M.; Antipin M. Y.; Struchkov Y. T. Oxidation of bridging groups by carboxylate coordinated ligands in palladium clusters. J. Cluster Sci. 1992, 3, 411–421. 10.1007/BF00702748. [DOI] [Google Scholar]
- Berenblyum A. S.; Knizhnik A. G.; Mund S. L.; Moiseev I. I. Mechanism of the formation of palladium complexes serving as catalysts in hydrogenation reactions. J. Organomet. Chem. 1982, 234, 219–235. 10.1016/S0022-328X(00)85857-X. [DOI] [Google Scholar]
- Cassol C. C.; Umpierre A. P.; Machado G.; Wolke S. I.; Dupont J. The role of Pd nanoparticles in ionic liquid in the Heck reaction. J. Am. Chem. Soc. 2005, 127, 3298–3299. 10.1021/ja0430043. [DOI] [PubMed] [Google Scholar]
- Özkar S.; Finke R. G. Nanocluster Formation and Stabilization Fundamental Studies: Ranking Commonly Employed Anionic Stabilizers via the Development, Then Application, of Five Comparative Criteria. J. Am. Chem. Soc. 2002, 124, 5796–5810. 10.1021/ja012749v. [DOI] [PubMed] [Google Scholar]
- Ornelas C.; Ruiz J.; Salmon L.; Astruc D. Sulphonated “Click” Dendrimer-Stabilized Palladium Nanoparticles as Highly Efficient Catalysts for Olefin Hydrogenation and Suzuki Coupling Reactions Under Ambient Conditions in Aqueous Media. Adv. Synth. Catal. 2008, 350, 837–845. 10.1002/adsc.200700584. [DOI] [Google Scholar]
- Deraedt C.; Astruc D. Homeopathic” Palladium Nanoparticle Catalysis of Cross Carbon–Carbon Coupling Reactions. Acc. Chem. Res. 2014, 47, 494–503. 10.1021/ar400168s. [DOI] [PubMed] [Google Scholar]
- Chernyshev V. M.; Khazipov O. V.; Eremin D. B.; Denisova E. A.; Ananikov V. P. Formation and stabilization of nanosized Pd particles in catalytic systems: Ionic nitrogen compounds as catalytic promoters and stabilizers of nanoparticles. Coord. Chem. Rev. 2021, 437, 213860. 10.1016/j.ccr.2021.213860. [DOI] [Google Scholar]
- Littke A. F.; Dai C.; Fu G. C. Versatile Catalysts for the Suzuki Cross-Coupling of Arylboronic Acids with Aryl and Vinyl Halides and Triflates under Mild Conditions. J. Am. Chem. Soc. 2000, 122, 4020–4028. 10.1021/ja0002058. [DOI] [Google Scholar]
- Proutiere F.; Aufiero M.; Schoenebeck F. Reactivity and stability of dinuclear Pd(I) complexes: studies on the active catalytic species, insights into precatalyst activation and deactivation, and application in highly selective cross-coupling reactions. J. Am. Chem. Soc. 2012, 134, 606–612. 10.1021/ja209424z. [DOI] [PubMed] [Google Scholar]
- Zhao X.; Chen H.; Xing S.; Yuan W.; Wu L.; Chen X.; Zhan C.-G. Regioselective synthesis of 2- and 4-diarylpyridine ethers and their inhibitory activities against phosphodiesterase 4B. J. Mol. Struct. 2019, 1196, 455–461. 10.1016/j.molstruc.2019.06.086. [DOI] [Google Scholar]
- Kim S.-T.; Kim S.; Baik M.-H. How bulky ligands control the chemoselectivity of Pd-catalyzed N-arylation of ammonia. Chem. Sci. 2020, 11, 1017–1025. 10.1039/C9SC03095F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy A. H.; Hartwig J. F. Oxidative Addition of Aryl Sulfonates to Palladium(0) Complexes of Mono- and Bidentate Phosphines. Mild Addition of Aryl Tosylates and the Effects of Anions on Rate and Mechanism. Organometallics 2004, 23, 194–202. 10.1021/om034187p. [DOI] [Google Scholar]
- Maes B. U. W.; Verbeeck S.; Verhelst T.; Ekomié A.; von Wolff N.; Lefèvre G.; Mitchell E. A.; Jutand A. Oxidative Addition of Haloheteroarenes to Palladium(0): Concerted versus SNAr-Type Mechanism. Chem. - Eur. J. 2015, 21, 7858–7865. 10.1002/chem.201406210. [DOI] [PubMed] [Google Scholar]
- Son S. U.; Jang Y.; Yoon K. Y.; Kang E.; Hyeon T. Facile Synthesis of Various Phosphine-Stabilized Monodisperse Palladium Nanoparticles through the Understanding of Coordination Chemistry of the Nanoparticles. Nano Lett. 2004, 4, 1147–1151. 10.1021/nl049519+. [DOI] [Google Scholar]
- Xue T.; Lin Z.; Chiu C.-Y.; Li Y.; Ruan L.; Wang G.; Zhao Z.; Lee C.; Duan X.; Huang Y. Molecular ligand modulation of palladium nanocatalysts for highly efficient and robust heterogeneous oxidation of cyclohexenone to phenol. Sci. Adv. 2017, 3, e1600615. 10.1126/sciadv.1600615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayanan R.; El-Sayed M. A. FTIR study of the mode of binding of the reactants on the Pd nanoparticle surface during the catalysis of the Suzuki reaction. J. Phys. Chem. B 2005, 109, 4357–4360. 10.1021/jp044659t. [DOI] [PubMed] [Google Scholar]
- Palani V.; Hugelshofer C. L.; Kevlishvili I.; Liu P.; Sarpong R. A Short Synthesis of Delavatine A Unveils New Insights into Site-Selective Cross-Coupling of 3,5-Dibromo-2-pyrone. J. Am. Chem. Soc. 2019, 141, 2652–2660. 10.1021/jacs.8b13012. [DOI] [PubMed] [Google Scholar]
- Strotman N. A.; Chobanian H. R.; He J.; Guo Y.; Dormer P. G.; Jones C. M.; Steves J. E. Catalyst-Controlled Regioselective Suzuki Couplings at Both Positions of Dihaloimidazoles, Dihalooxazoles, and Dihalothiazoles. J. Org. Chem. 2010, 75, 1733–1739. 10.1021/jo100148x. [DOI] [PubMed] [Google Scholar]
- Tang D.-T. D.; Collins K. D.; Glorius F. Completely regioselective direct C–H functionalization of benzo[b]thiophenes using a simple heterogeneous catalyst. J. Am. Chem. Soc. 2013, 135, 7450–7453. 10.1021/ja403130g. [DOI] [PubMed] [Google Scholar]
- Handy S. T.; Zhang Y. A Solvent-Induced Reversal of Regioselectivity in the Suzuki Coupling of Pyrrole Esters. Open Org. Chem. J. 2008, 2, 58–64. 10.2174/1874095200801020058. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.