

Abstract
Drug-induced pulmonary arterial hypertension (D-PAH) is a form of World Health Organization Group 1 pulmonary hypertension (PH) characterized by severe small vessel loss and obstructive vasculopathy, which leads to progressive right heart failure and death. To date, 16 different compounds have been associated with D-PAH, including anorexinogens, recreational stimulants, and more recently, several Food and Drug Administration–approved medications. While the clinical manifestations, pathology, and hemodynamic profile of D-PAH are indistinguishable from other forms of PAH, its clinical course can be unpredictable and largely dependent on removal of the offending agent. Since only a subset of individuals develop D-PAH, it is likely that genetic susceptibility plays a role in pathogenesis, but characterization of the genetic factors responsible for disease susceptibility remains incomplete. Besides aggressive treatment with PH-specific therapies, the major challenge in the management of D-PAH remains the early identification of compounds capable of injuring the pulmonary circulation in susceptible individuals. Institution of pharmacovigilance, precision medicine strategies, and global warning systems will help facilitate identification of high-risk drugs and institute regulation strategies to prevent further outbreaks of D-PAH.
Keywords: drug-induced pulmonary hypertension, fenfluramine, aminorex, dasatinib, methamphetamine
Pulmonary arterial hypertension (PAH) is a life-threatening disorder associated with elevated pulmonary pressures, which in the absence of therapy leads to right heart failure and death.1 Following its initial description in 1891, “primary” PAH was regarded as an orphan disease of unclear etiology that, given its rarity, many physicians would never encounter in their clinical practice. This all changed in 1965, when numerous cases of PAH were reported in Switzerland, Germany, and Austria.2,3 The link between these cases was exposure to aminorex fumarate, an amphetamine-derived anorexinogen marketed for weight loss and treatment of mood disorders. This “PAH epidemic” caught the attention of the medical community and effectively brought PAH out of the shadows and into the limelight. Despite being banned in 1967, cases of aminorex-induced PAH continued to appear throughout the next decade and led to the first Pulmonary Hypertension World Symposium in 1973, which resulted in the first clinical classification of PAH that would become the blueprint for the development of appropriate approaches to diagnosis and management.
Drug-induced PAH (D-PAH) represents a unique opportunity to understand how genes and environment interact to influence the development of PAH in susceptible individuals. To date, more than 16 different compounds have been linked to risk of D-PAH and it is likely that more agents will continue to be reported (Table 1).1 The clinical presentation and vascular pathology of these patients is indistinguishable from that of idiopathic PAH (Figure 1), which emphasizes the importance of obtaining details concerning the use of medications and illicit drug use as part of the initial medical history. Importantly, recent reports have identified several Food and Drug Administration (FDA)-approved medications as causes of D-PAH and have generated renewed efforts to institute pharmacovigilance to prevent another epidemic of PAH. This review will introduce 3 major examples of D-PAH and summarize our current understanding of the relevant genetic and molecular mechanisms involved in these cases.
Table 1.
Drugs Associated With Risk of Drug-Induced PAH1
| Definite | Likely | Possible |
|---|---|---|
| Aminorex | Amphetamines | Cocaine |
| Fenfluramine | Dasatanib | Phenylpropanolamine |
| Dexfenfluramine | L-tryptophan | St. John’s wort |
| Toxic rapeseed oil | Methamphetamine | Amphetamine-like drugs |
| Benfluorex | Interferon a and b | |
| SSRIs | Selected chemotherapeutics agents (eg, mitomycin) |
Figure 1:
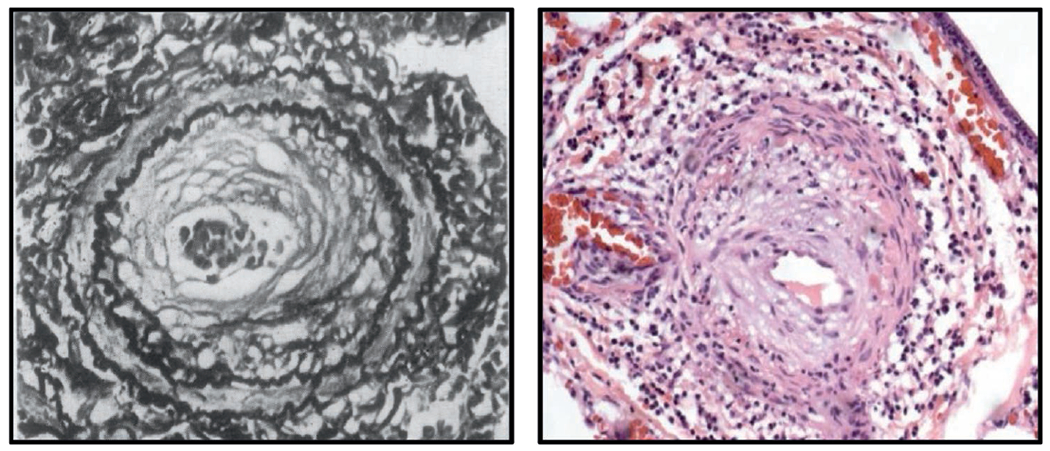
Vascular pathology of aminorex-induced PAH is similar to idiopathic (I) PAH. Left panel shows muscularized artery from a patient with aminorex-induced PAH. Right panel shows similar lesion in an IPAH patient. Aminorex image obtained from Kay JM, Smith P, Heath D. Aminorex and the pulmonary circulation. Thorax. 1971;26(3):262-270.
AMINOREX REDUX: THE RISE AND FALL OF FENFLURAMINES
Obesity is a worldwide epidemic that has been associated with multiple comorbidities such as cardiovascular disease, diabetes, and respiratory disorders, among others.4 Given the magnitude of the problem and the failure of most conventional dietary strategies, there has been an ongoing interest in developing a pharmacological approach to help patients achieve appropriate weight loss. Anorexinogens such as aminorex are amphetamine-derived compounds that facilitate weight loss by suppressing appetite sensation at the level of the central nervous system and increasing catabolism of fat stores in adipose tissue.5 The mechanism of action for most anorexinogens is linked to their capacity for increasing serotonin levels and facilitating catecholamine release from the adrenal glands. However, serotonin can induce changes in the pulmonary vasculature such as smooth muscle growth and increased vasomotor tone that in susceptible individuals could lead to PAH.6–8 Furthermore, serotonin can also induce changes in the heart tissue that compromise heart valve integrity. Tricuspid regurgitation as a result of infiltrative valve disease is a well-described manifestation of excessive serotonin production, a condition associated with carcinoid tumors that produce abnormally elevated amounts of serotonin that are not cleared by the liver.9 Despite this well-known association, it still came as a surprise that similar findings began appearing in patients who were exposed to a newer generation of anorexinogens in the 1990s, which resulted in a second epidemic of anorexinogen-induced PAH.10,11
Fenfluramine-phentermine and dexfenfluramine were introduced as weight loss agents first in Europe and then in the United States with data that supported their use as effective weight loss agents, albeit achieving only a small reduction in body weight (~4%).10 Like aminorex, these agents are serotonin (5-HT) reuptake inhibitors that increase serotonin levels systemically as well as locally in blood vessels. The first warning that fenfluramine derivatives were linked to pulmonary hypertension (PH) came with the publication of the International Primary Pulmonary Hypertension Study in 1995.12 This study documented the characteristics and clinical course of 62 patients with fenfluramine-induced PAH. The majority of patients used fenfluramine derivatives for at least 3 months: about half of the patients used dexfenfluramine alone; 27% used fenfluramine in association with amphetamines; 11% used fenfluramine alone; and 8% used both drugs. The interval between the onset of dyspnea and that of drug intake was approximately 48 months. Compared to idiopathic PAH patients, patients with fenfluramine-induced PAH had similar presentation and prognosis (50% overall survival at 3 years) when treated with available therapies. Based on this evidence, fenfluramine derivatives were banned in Europe in 1997. A study by Humbert et al showed that patients with fenfluramine-induced PAH may be carriers of bone morphogenetic protein receptor type 2 (BMPR2) mutations, and a similar proportion (9%) has been reported in sporadic PAH.13 Interestingly, patients carrying BMPR2 mutations had a significantly lower duration of exposure to fenfluramine than patients without any mutations.
In the United States, it would take 2 more years before these agents were withdrawn from the market following the reports of valvulopathy associated with fenfluramine use. Failure to address this promptly was based on the perception that PAH was still a very rare disease that would not bear consideration in the decision to use these agents in the overweight population.14 To quote the editorial that accompanied the International Society for Heart & Lung Transplantation paper: “Although physicians and patients need to be informed, the possible risks of PH associated with dexfenfluramine is small and appears to be outweighed by the benefits when the drug is used appropriately.”15 This sobering statement suggests the need for pharmacovigilance and advocacy to prevent future outbreaks such as this one.
METHAMPHETAMINE: THE BREAKING BAD CONNECTION
Methamphetamine (METH) is a highly addictive compound whose popularity among young and middle-aged adults has steadily increased worldwide in the past decade. In addition to being a potent neurostimulant, METH can also affect other organs such as the kidneys, heart, and liver, resulting in severe organ dysfunction and premature death.16 While METH use is associated with a higher incidence of cardiovascular disease such as ischemic cardiomyopathy, arrhythmias, and myocardial infarction, only recently has PAH been recognized as a life-threatening complication of METH use. The association between inhaled METH use and PAH was first reported by Schaiberger et al and was further supported by a retrospective study by Chin et al that found significantly higher rates of METH use in patients diagnosed with idiopathic PAH when compared to other PAH groups.17,18 Positron emission tomography (PET) studies have shown that [(11)C] d-METH administered intravenously localizes primarily in the lung tissue, suggesting that the lung is a primary target for METH-related injury.19 On the basis of these studies, METH use was included in the most recent clinical classification of PH as a likely risk factor for D-PAH.
While the true incidence and prevalence of METH-PAH in the United States is unknown, we have seen a disturbing increase in the number of METH-PAH cases diagnosed at the Stanford Adult Pulmonary Hypertension Clinic over the last 10 years. At present, 85% of our drug- and toxin-induced PAH patients carry a diagnosis of METH-PAH, and their median 5-year survival is estimated at 35% (R. Zamanian, personal communication), which is significantly worse compared to that of other PAH patients. Despite the current clinical evidence, it must be emphasized that not all patients with a history of METH use develop PAH. METH-induced cytotoxicity is linked to oxidative injury by its direct activation of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (NOX) and generation of reactive oxygen species (ROS) that cause cellular damage.20
Similar to patients with familial and sporadic PAH, it is possible that variations in certain genes may be required to trigger PAH in a subset of METH users, but no gene candidates have been established. We have recently conducted a whole exome sequencing analysis that led to the identification of carboxylesterase 1 (CES1), a gene involved in drug metabolism, as a candidate gene for METH-PAH. Studies in lung tissue explanted from METH-PAH patients have shown reductions in CES1 expression in pulmonary endothelial cells, and exposure of CES1-deficient pulmonary microvascular endothelial cells (PMVECs) results in increased apoptosis accompanied by concomitant production of ROS and cytoplasmic vacuoles. Our observation of the increased number of vacuoles in CES1-deficient cells raises several interesting mechanistic possibilities that could provide insight into the fundamental mechanism of METH-induced PMVEC apoptosis. One possible explanation is that CES1 could be facilitating the production of autophagosomes and/or altering lysosome function under METH, since CES1 is expressed in both organelles.21,22
Our studies demonstrate that the reduction in CES1 expression and activity increases PMVEC apoptosis alone and in response to METH, but several questions remain concerning the mechanism behind METH-induced ROS generation, initiation of endoplasmic reticulum (ER) stress, and possible impairment of mitochondrial bioenergetics, a well-established feature of other forms of PAH.23 It is worth pointing out that mitochondrial toxicity is also a consequence of METH exposure and could be linked to the autophagy response as these pathways can also interact with the mitochondria to trigger release of cytochrome C and other pro-apoptotic factors.24 Whether pharmacological agents that restore CES1 or the autophagy response could be clinically relevant in the treatment of METH-PAH remains to be determined.
BEYOND STIMULANTS: DASATANIB AND PH
Tyrosine kinase inhibitors (TKIs) are pharmacological agents that are used frequently for the treatment of chronic myelogenous leukemia (CML).25 A target of TKIs is the platelet-derived growth factor (PDGF) pathway, a key player in PAH pathogenesis as evidenced by studies in human samples and animal models of PAH.26,27 Dasatinib is a TKI that is commonly used in the treatment of CML with high affinity for BCR/ABL kinase that targets a wide range of kinases such as the Src family.25 In 2012, a report from the French National Network of PH identified 9 cases of dasatinib-induced PAH.28 Patients presented with severe clinical, functional, and hemodynamic impairment that in some cases required treatment with PAH-specific therapies. Also, 6 out of 9 patients demonstrated bilateral pleural effusions and 3 demonstrated minimal pericardial effusions. Most patients improved after cessation of dasatinib, but the majority of patients failed to fully recover; in fact, 2 patients in this cohort died as a consequence of PAH-related complications. At present, no genetic modifiers have been associated with risk of dasatinib-induced PAH, and the incidence is estimated to be low at approximately 0.45%. The mechanism appears to be related to dasatinib-mediated smooth muscle proliferation and vasoconstriction through inhibition of the Src kinases.29 Until more information is known about the specific risk factors, our recommendation is to avoid using dasatinib for treatment of CML if possible and to choose another TKI for treatment of CML.
PROGNOSIS AND TREATMENT OF D-PAH
Studies looking specifically at the prognosis and treatment of D-PAH are sparse. In general, due to their phenotypic similarities, D-PAH patients are grouped together with patients with idiopathic or hereditary PAH for studies of prognosis and treatment. For example, compared to idiopathic PAH patients, patients with fenfluramine-induced PAH had similar presentation and prognosis (50% overall survival at 3 years) when treated with available therapies.12 Because of this and other observations, the French registry included patients with anorexigen-induced PAH along with idiopathic PAH and hereditary PAH when examining the effects of modern treatment approaches on PAH outcomes.30 Similarly, D-PAH patients may be included with other types of PAH in studies of therapeutic agents (but usually represent a small percentage of the overall study population).
For selected agents, prognostic information is available. For example, most dasatinib-induced PAH patients improve at least in part on cessation of the drug.28 However, PAH-related death has been reported in at least 2 patients. In a recent report of mitomycin-induced pulmonary veno-occlusive disease (PVOD), a fatal course of the disease was observed in 4 of the 7 patients.31 No published data for METH-PAH patients exists; median 5-year survival in these patients is currently estimated at 35% (significantly worse compared to that of other PAH patients). Of note, in patients with METH-, dasatinib-, or mitomycin-induced PAH or PVOD, the prognosis is often negatively affected by concomitant use of other drugs, presence of comorbidities, or presence of an underlying malignancy.
D-PAH patients are currently treated with all available PAH-specific therapies. There is no evidence that one class of agents works better than another, and patients are usually treated based on their risk profile similar to other types of PAH. One notable exception is that placement of permanent intravenous lines is usually avoided in active drug users due to potential for abuse of access for injection of illicit drugs. As in any drug-induced condition, if exposure is ongoing (eg, dasatinib therapy), cessation of the offending drug is of utmost importance. Specific studies of prognosis and therapeutic strategies in this patient population are needed.
FUTURE DIRECTIONS
The cases highlighted here cover approximately 50 years since the first epidemic of drug-induced PAH, and it is highly likely that more drugs will be identified as risk factors for PAH. In the past year, reports of PAH associated with other FDA-approved drugs such as interferon and mitomycin have been published, further illustrating the expanding role of prescription drugs as a cause of PH.31–33 Even so, clearly stating that a certain drug is associated with a higher risk for PAH is a very difficult task because PAH remains a rare disease and associations require demonstration in a large number of patients. As our understanding of PAH pathobiology continues to grow, we must pay attention to pathways and gene modifiers that participate in drug metabolism. Precision medicine tools such as gene sequencing, exosome analysis, and bioinformatics could serve in detecting genetic susceptibility factors that increase the risk of PAH in subsets of patients, but await validation in future studies. In the meantime, physicians must remain vigilant of their patient population and establish close interactions between national drug regulatory agencies, national PH networks, and PAH patient associations to pursue leads and spread the word across the globe. Ultimately, prevention may prove to be the best strategy to deal with this form of PAH, and it will help elevate awareness regarding this terrible disease.
Footnotes
Disclosures: Dr de Jesus Perez has nothing to disclose.
References
- 1.Galiè N, Humbert M, Vachiery JL, et al. 2015 ESC/ERS Guidelines for the Diagnosis and Treatment of Pulmonary Hypertension. Rev Esp Cardiol (Engl Ed). 2016;69(2):177. [DOI] [PubMed] [Google Scholar]
- 2.Follath F, Burkart F, Schweizer W. Drug-induced pulmonary hypertension? Br Med J. 1971;1(5743):265–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kay JM, Smith P, Heath D. Aminorex and the pulmonary circulation. Thorax. 1971;26(3):262–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lavie CJ, De Schutter A, Parto P, et al. Obesity and Prevalence of Cardiovascular Diseases and Prognosis-The Obesity Paradox Updated. Prog Cardiovasc Dis. 2016;58(5):537–547. [DOI] [PubMed] [Google Scholar]
- 5.Rothman RB, Baumann MH. Therapeutic and adverse actions of serotonin transporter substrates. Pharmacol Ther. 2002;95(1):73–88. [DOI] [PubMed] [Google Scholar]
- 6.Adnot S, Houssaini A, Abid S, Marcos E, Amsellem V. Serotonin transporter and serotonin receptors. Handb Exp Pharmacol. 2013;218:365–380. [DOI] [PubMed] [Google Scholar]
- 7.Delaney C, Gien J, Roe G, Isenberg N, Kailey J, Abman SH. Serotonin contributes to high pulmonary vascular tone in a sheep model of persistent pulmonary hypertension of the newborn. Am J Physiol Lung Cell Mol Physiol. 2013;304(12):L894–L901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Johansen AK, Dean A, Morecroft I, et al. The serotonin transporter promotes a pathological estrogen metabolic pathway in pulmonary hypertension via cytochrome P450 1B1. Pulm Circ. 2016;6(1):82–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tanaka M, Matsubara O, Takemura T, et al. Cardiovascular lesion of carcinoid syndrome. An autopsy case of bronchial carcinoid. Acta Pathol Jpn. 1984;34(1):201–213. [DOI] [PubMed] [Google Scholar]
- 10.Fishman AP. Aminorex to fen/phen: an epidemic foretold. Circulation. 1999;99(1):156–161. [DOI] [PubMed] [Google Scholar]
- 11.Gross SB, Lepor NE. Anorexigen-related cardiopulmonary toxicity. Rev Cardiovasc Med. 2000;1(2):80–89, 102. [PubMed] [Google Scholar]
- 12.Abenhaim L, Moride Y, Brenot F, et al. Appetite-suppressant drugs and the risk of primary pulmonary hypertension. International Primary Pulmonary Hypertension Study Group. N Engl J Med. 1996;335(9):609–616. [DOI] [PubMed] [Google Scholar]
- 13.Humbert M, Deng Z, Simonneau G, et al. BMPR2 germline mutations in pulmonary hypertension associated with fenfluramine derivatives. Eur Respir J. 2002;20(3):518–523. [DOI] [PubMed] [Google Scholar]
- 14.Connolly HM, Crary JL, McGoon MD, et al. Valvular heart disease associated with fenfluramine-phentermine. N Engl J Med. 1997;337(9):581–588. [DOI] [PubMed] [Google Scholar]
- 15.Manson JE, Faich GA. Pharmacotherapy for obesity -- do the benefits outweigh the risks? N Engl J Med. 1996;335(9):659–660. [DOI] [PubMed] [Google Scholar]
- 16.Gotway MB, Marder SR, Hanks DK, et al. Thoracic complications of illicit drug use: an organ system approach. Radiographics. 2002;22 Spec No:S119–S135. [DOI] [PubMed] [Google Scholar]
- 17.Chin KM, Channick RN, Rubin LJ. Is methamphetamine use associated with idiopathic pulmonary arterial hypertension? Chest. 2006;130(6):1657–1663. [DOI] [PubMed] [Google Scholar]
- 18.Schaiberger PH, Kennedy TC, Miller FC, Gal J, Petty TL. Pulmonary hypertension associated with long-term inhalation of “crank” methamphetamine. Chest. 1993;104(2):614–616. [DOI] [PubMed] [Google Scholar]
- 19.Volkow ND, Fowler JS, Wang GJ, et al. Distribution and pharmacokinetics of methamphetamine in the human body: clinical implications. PloS One. 2010;5(12):e15269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Park M, Hennig B, Toborek M. Methamphetamine alters occludin expression via NADPH oxidase-induced oxidative insult and intact caveolae. J Cell Mol Med. 2012;16(2):362–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Funakoshi-Hirose I, Aki T, Unuma K, Funakoshi T, Noritake K, Uemura K. Distinct effects of methamphetamine on autophagy-lysosome and ubiquitin-proteasome systems in HL-1 cultured mouse atrial cardiomyocytes. Toxicology. 2013;312:74–82. [DOI] [PubMed] [Google Scholar]
- 22.Tanaka M, Iio T, Tabata T. Purification and characterization of a carboxylesterase from rabbit liver lysosomes. J Biochem. 1987;101(3):619–624. [DOI] [PubMed] [Google Scholar]
- 23.Rehman J, Archer SL. A proposed mitochondrial-metabolic mechanism for initiation and maintenance of pulmonary arterial hypertension in fawn-hooded rats: the Warburg model of pulmonary arterial hypertension. Adv Exp Med Biol. 2010;661:171–185. [DOI] [PubMed] [Google Scholar]
- 24.Senft D, Ronai ZA. UPR, autophagy, and mitochondria crosstalk underlies the ER stress response. Trends Biochem Sci. 2015;40(3):141–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Steinberg M Dasatinib: a tyrosine kinase inhibitor for the treatment of chronic myelogenous leukemia and philadelphia chromosome-positive acute lymphoblastic leukemia. Clin Ther. 2007;29(11):2289–2308. [DOI] [PubMed] [Google Scholar]
- 26.Barst RJ. PDGF signaling in pulmonary arterial hypertension. J Clin Invest. 2005;115(10):2691–2694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schermuly RT, Dony E, Ghofrani HA, et al. Reversal of experimental pulmonary hypertension by PDGF inhibition. J Clin Invest. 2005;115(10):2811–2821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Montani D, Bergot E, Gunther S, et al. Pulmonary arterial hypertension in patients treated by dasatinib. Circulation. 2012;125(17):2128–2137. [DOI] [PubMed] [Google Scholar]
- 29.Guignabert C, Phan C, Seferian A, et al. Dasatinib induces lung vascular toxicity and predisposes to pulmonary hypertension. J Clin Invest. 2016;126(9):3207–3218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Humbert M, Sitbon O, Chaouat A, et al. Survival in patients with idiopathic, familial, and anorexigen-associated pulmonary arterial hypertension in the modern management era. Circulation. 2010;122(2):156–163. [DOI] [PubMed] [Google Scholar]
- 31.Perros F, Gunther S, Ranchoux B, et al. Mitomycin-Induced Pulmonary Veno-Occlusive Disease: Evidence From Human Disease and Animal Models. Circulation. 2015;132(9):834–847. [DOI] [PubMed] [Google Scholar]
- 32.Savale L, Chaumais MC, O’Connell C, Humbert M, Sitbon O. Interferon-induced pulmonary hypertension: an update. Curr Opin Pulm Med. 2016;22(5):415–420. [DOI] [PubMed] [Google Scholar]
- 33.Gagnadoux F, Capron F, Lebeau B. Pulmonary veno-occlusive disease after neoadjuvant mitomycin chemotherapy and surgery for lung carcinoma. Lung Cancer. 2002;36(2):213–215. [DOI] [PubMed] [Google Scholar]