

Abstract
The use of prescription opioid analgesics, particularly oxycodone, has dramatically increased, and parallels escalated opioid abuse and drug-related deaths worldwide. Understanding the molecular mechanisms underlying the development of opioid dependence and expanding treatment options to counter prescription opioid abuse has become a critical public health matter. In the present study, we first evaluated the reinforcing effects of oxycodone in a rat model of self-administration and then explored the potential utility of two novel high affinity dopamine D3 receptor (D3R) antagonists/partial agonists, CAB2-015 and BAK4-54, for treatment of prescription opioid abuse and dependence. We found that rats acquired oxycodone self-administration rapidly within a range of unit doses that was similar to that for heroin, confirming that oxycodone has significant abuse potential. Strikingly, pretreatment with either CAB2-015 or BAK4-54 (0.4-10 mg/kg, i.p.) dose-dependently decreased oxycodone self-administration, and shifted the oxycodone dose-response curve downward. Repeated pretreatment with CAB2-015 or BAK4-54 (0.4-4 mg/kg) facilitated extinction and inhibited oxycodone-induced reinstatement of drug-seeking behavior. In addition, pretreatment with CAB2-015 or BAK4-54 (4-10 mg/kg) also dose-dependently decreased oxycodone-enhanced locomotor activity, and oral sucrose self-administration after CAB2-015 only. These data suggest that D3R antagonists may be suitable alternatives or adjunctive to opioid-based medications currently used clinically in treating opioid addiction and that the D3R-selective ligands (CAB2-015 or BAK4-54) provide new lead molecules for development.
Keywords: Dopamine D3 receptor antagonist, CAB2-015, BAK4-54, prescription opioid, oxycodone, self-administration
INTRODUCTION
Prescription opioids such as oxycodone and morphine are effective analgesics in clinical pain management. Serious concerns over preponderant use of these drugs have been raised due to dramatic increases in their nonmedical use and abuse (Cerdá et al., 2015). Negative consequences include increases in emergency room visits, drug-related deaths and admissions to substance-abuse treatment programs, each imposing a significant and negative impact on public health and economic welfare (Meyer et al., 2014). It is estimated that 10.3 million Americans used prescription opioids nonmedically in 2014 and 80% of the population suffering from opioid use disorders are or were once prescription opioid abusers (Compton and Volkow, 2006). Thus, understanding the exceptional reinforcing properties of prescription opioids and developing more effective treatments for opioid addiction and dependence are critical public health challenges.
Methadone and buprenorphine are the approved replacement therapies for the treatment of illicit opioid abuse and dependence. Recently, they have been used for treatment of prescription opioid use disorders in a few open-labeled, small-scale clinical trials, and demonstrated therapeutic efficacy in reducing prescription opioid use and relapse (Fiellin et al., 2014; Nielsen et al., 2016; Potter et al., 2013). However, methadone is a full mu opioid receptor agonist that has significant abuse liability. It is also associated with a number of problems such as withdrawal symptoms after chronic use, reliance on methadone clinics, and drug overdose-induced respiratory depression and death (Novick et al., 2015). Although buprenorphine, a partial μ opioid receptor agonist and κ opioid receptor antagonist, produces limited respiratory depression, extensive clinical studies indicate that methadone may still be superior to buprenorphine in reducing drug use, craving and relapse to drug-seeking behavior (Mattick et al., 2008; Potter et al., 2013). Nevertheless, development of novel safer and more effective non-opioid medications for treatment of prescription opioid abuse poses a significant task.
Compelling preclinical evidence suggests a crucial involvement of brain dopamine D3 receptors (D3R) in drug reward and addiction (Heidbreder and Newman, 2010; Sokoloff and Le Foll, 2017). Different from other dopamine receptor subtypes, D3Rs are localized in brain regions mediating reward, incentive motivation to self-administer addictive drugs, and relapse to drug-seeking behavior (Sokoloff and Le Foll, 2016; Xi and Gardner, 2007). In addition, D3Rs display higher affinity for dopamine (Sokoloff et al., 1990) than the other dopamine receptor subtypes, and are upregulated in reward-related brain regions in cocaine addicts, poly drug abusers (Boileau et al., 2012; Payer et al., 2014) and in laboratory animals following chronic exposure to cocaine (Le Foll et al., 2002; Neisewander et al., 2004). These data suggest that selectively blocking D3Rs may be effective in reducing the rewarding effects of these drugs of abuse with few unwanted side-effects. This is supported by recent preclinical findings that selective D3R antagonists significantly inhibit cocaine or methamphetamine self-administration under progressive ratio reinforcement, conditioned place preference (CPP) to abused drugs, and reinstatement of drug-seeking behavior (Heidbreder and Newman, 2010; Keck et al., 2015; Xi and Gardner, 2007). Although several D3R antagonists such as SB-277011A, NGB2904, and PG01037 have been well characterized in animal models of drug abuse, none of these preclinical tools have translational potential for use in humans due to poor bioavailability, metabolic instability and/or predicted toxicity (Keck et al., 2015; Xi and Gardner, 2007).
We have recently reported that BAK4-54 and CAB2-015 are novel high affinity D3R antagonists/partial agonists with improved metabolic stability over our previous lead compound PG648 (Boateng et al., 2015; Keck et al., 2015). Both compounds have dual functional profiles – as full D3R antagonists at low doses (IC50=8.0 nM or 7.4 nM for BAK4-54 or CAB2-015, respectively) and partial D3R agonists at higher doses (EC50 = 140 nM, 25% stimulation or 20 nM, 31% stimulation for BAK4-54 or CAB2-015, respectively) (Boateng et al., 2015). In radiolabeled binding experiments, both compounds bind to D3R with subnanomolar affinities whereas BAK4-54 is over 100-fold selective for hD3Rs over the highly homologous hD2R, and CAB2-015 is somewhat less D3R-selective (45-fold). These profiles may be important in treating patients with substance use disorders as the partial agonist profile may prevent a full antagonist-induced dysphoria and alleviate drug craving observed during drug withdrawal or abstinence (Pilla et al., 1999).
In the present study, we investigated the potential utility of both D3 receptor antagonists/partial agonists in treatment of prescription opioid abuse and dependence. We first evaluated and compared the reinforcing properties of oxycodone with heroin by allowing groups of rats to self-administer the drug at varying doses. We then explored the capacity of CAB2-015 and BAK4-54 to attenuate oxycodone self-administration and relapse to oxycodone-seeking behavior. The effects of each compound on locomotion and sucrose self-administration were also evaluated following systemic injection alone or in combination with oxycodone. The findings in the present study suggest that D3R antagonism may be a viable mechanism for developing therapeutics as alternatives or adjunctive to opioid-based medications such as methadone or buprenorphine for treating opioid use disorders.
MATERIALS AND METHODS
Male Long-Evans rats (275-325 g) (Charles-River Laboratories, Raleigh, N.C.) were used throughout this study. Upon arrival, they were housed individually in an animal facility under a reversed 12 h light-dark cycle (light on at 7:00 PM) with free access to food and water. They were allowed to acclimatize to the new environment at least for 7 days prior to study initiation. All procedures were approved by the U.S. National Institute on Drug Abuse (NIDA) and were consistent with The Guide for the Care and Use of Laboratory Animals of the U.S. National Research Council.
Surgery
Rats used in oxycodone self-administration experiments were implanted with a microrenathane intravenous (i.v.) catheter (Braintree Scientific Inc., Braintree, MA, USA). Each rat was first anaesthetized with pentobarbital (30 mg/kg i.p.) supplemented with chloral hydrate (140 mg/kg, i.p.) and then a small incision was made to the right of the midline of the neck to expose the external jugular vein. One end of the i.v. catheter was next inserted into the vein with the catheter tip reaching the right atrium. The catheter was then secured to the vein with silk suture and the other end fed subcutaneously around the back of the neck to exit near the back of the skull, connected to a bent 24-gauge stainless steel cannula (Plastics One Inc., Roanoke, VA, USA) with a threaded head used to secure a dummy cannula and, during experimentation, an infusion line. The catheter and the guide cannula were secured to the skull with four stainless steel screws threaded into the skull and dental cement. The incision was then sutured.
After surgery, the catheters were flushed daily with a gentamicin–heparin–saline solution (0.1 mg/ml gentamicin and 30 IU/ml heparin; ICN Biochemicals, Cleveland, OH, USA) as precaution against catheter clogging and infection. The animals were allowed to recover for at least 5 days before behavioral training started.
Self-Administration Apparatus and Training
Oxycodone self-administration training was conducted in an operant conditioning chamber equipped with two response levers and a 15 W house light (Med Associates Inc., Georgia, VT, USA). The two levers were located 6.4 cm above the chamber floor; one lever was active while the other was inactive. A cue light and a speaker were 12 cm above the active lever (Xi and Gardner, 2007).
After full recovery from surgery, the rats were trained to self-administer oxycodone. For this, they were transported on each training day to the test room and connected by polyethylene tubing (protected by a steel coil spring) and a one-channel liquid swivel to a syringe pump (Razel Sci., Stamford, CT, USA) controlled by a microprocessor, and placed in the chamber. Each training session started with the insertion of the response levers into the chamber and the illumination of the house light, which was kept on until the end of each training session. Each rat was trained daily in 3-hr sessions to press the active lever for oxycodone infusions on a fixed ratio-1 (FR-1) schedule of reinforcement. Response on the active lever resulted in the activation of a light-tone compound cue and infusion of an 0.08 ml oxycodone solution over 4.6 sec. This time also served as a timeout period during which the light-tone cue was kept on and the animal’s response on the active lever was recorded but had no scheduled consequence. Each animal’s response on the inactive lever was recorded but not rewarded throughout training and testing.
Locomotor Activity Chamber and Measurement
Locomotor tests were conducted in open-field chambers (L × W × H = 43 × 43 × 30 cm, Accuscan Instruments, Inc., Columbus, OH, USA). Before experiments, rats were first conditioned to the chamber for 2 hr/day for 3 consecutive days. On each conditioning day, the animals were moved to the test room, acclimated for 10 min and then placed in their assigned chambers. The travel distance (cm) of each animal was recorded using VersaMax version 3.0 software (Accuscan Instruments, Inc.).
Oral Sucrose Self-Administration
The procedures for oral sucrose self-administration were identical to those for oxycodone self-administration, except that active lever presses led to delivery of 0.08 ml of 5% sucrose solution into a liquid food tray on the operant chamber wall, and surgery was not performed.
Specific Experiments
Experiment 1: Acquisition of self-administration under various unit doses of oxycodone or heroin.
To test the reinforcing properties of oxycodone under our experimental conditions, we first trained 6 groups of rats to self-administer oxycodone (n=6 /group) or heroin (n=7/group) at different unit doses (25, 50 and 100 μg/kg/infusion, respectively). After recovery from surgery, they were transported daily to the test room, placed in the self-administration chambers and allowed to lever-press for their assigned dose of oxycodone or heroin. Animals’ responses on the levers and oxycodone infusions were recorded; the testing lasted for 10 sessions.
Experiment 2: D3 receptor blockade by CAB2-015 or BAK4-54 on oxycodone self-administration.
Eight groups of rats (n=8/group) were used in these experiments. They were first trained to self-administer oxycodone at a dose of 100 μg/kg/infusion for 2 weeks followed by 50 μg/kg/infusion for the rest of the training sessions and also for the testing session. The daily self-administration of oxycodone at the low dose continued until the average oxycodone infusions/session varied less than 10% over 3 consecutive sessions (normally 6-10 sessions). On the following test day, the groups of rats were first systemically injected with either vehicle (25% 2-hydroxypropyl-β-cyclodextrin, used for dissolving the D3R compounds) or a dose of either CAB2-015 or BAK4-54 (0.4. 4 or 10 mg/kg, i.p.), and 15 min later, the animals were allowed to self-administer oxycodone for another session. Animals’ responses on the levers, and drug infusions were recorded.
Experiment 3: D3 receptor blockade by CAB2-015 or BAK4-54 on self-administration with various oxycodone doses.
Three groups of rats were used to determine whether and how the effects of CAB2-015 and BAK4-54 seen in experiment 2 depended on oxycodone dose. The rats were trained and tested with 6 descending oxycodone doses (100, 50, 25, 12.5, 6.25, and 3.125 μg/kg/infusion). Oxycodone self-administration maintained by each unit dose of oxycodone lasted several days or sessions until stable self-administration was achieved for at least 3 days, and then the effects of CAB2-015 or BAK4-54 pretreatment on each dose oxycodone self-administration were evaluated using a between sessions procedure. One group was repeatedly treated with vehicle while the other two groups were repeatedly treated with either CAB2-015 or BAK4-54 (10 mg/kg) 15 min before each oxycodone test session. The training procedures were the same as described above except that the dose of oxycodone was varied among training phases. The tests were carried out when animals met the training criterion in experiment 2, except at the lowest oxycodone dose which was tested when mean oxycodone self-administration variation reached a criterion of less than 20% between self-administration sessions.
Experiment 4: Repeated treatments with CAB2-015 or BAK4-54 on extinction and on oxycodone-induced reinstatement.
After stable self-administration behavior was established (as described in experiment 2), rats were divided into 8 groups and were tested under extinction conditions for 13 sessions. The extinction sessions were the same as the self-administration sessions except that saline was substituted for oxycodone. Fifteen min prior to each of the 8 initial sessions, rats were given either vehicle (2 groups) or one of 3 doses of either CAB2-015 or BAK4-54 (6 groups, 0.4, 2 or 4 mg/kg, i.p.). Extinction then continued for another 5 sessions without antagonist pretreatments. On the 8th and 13th test days, each animal was challenged with a priming injection of oxycodone (1 mg/kg, i.p.) immediately before starting the session. Animals’ responses on the active and inactive levers throughout the test sessions were recorded.
Experiment 5: D3 receptor blockade by CAB2-015 or BAK4-54 on spontaneous and oxycodone-induced locomotor activity.
Following the completion of tests in experiment 2, the animals were trained to self-administer oxycodone for an additional 4 sessions. They were then rebalanced according to their treatment history and redistributed into 6 groups (n=8/group) and then habituated for 2 hr/day for 3 days in the locomotor test chambers. On the next day, they were pretreated with either vehicle or one dose of their previously treated antagonist (4 or 10 mg/kg, i.p.). Locomotor activity was measured 15 min after injection. The animals were then habituated for an additional session followed by another test session. Fifteen min after vehicle or the antagonist treatment and immediately before the second test session, each rat was challenged with oxycodone (1 mg/kg, i.p.). Locomotor activity was measured for another 2 h.
Experiment 6: D3 receptor blockade by CAB2-015 or BAK4-54 on sucrose self-administration.
Two groups of rats (n=12 each) without surgery were used for in sucrose self-administration training and testing. Sucrose deliveries per session were capped at 120. Following completion of training, each group was tested 3 times. One group was tested following pretreatment (15 min prior to testing) with vehicle, 4 or 10 mg/kg CAB2-015 in a repeated measures design. The other group was tested following pretreatment with vehicle, 4 or 10 mg/kg BAK4-54. The three tests in each group were counterbalanced and were separated by 2 additional training sessions between each 2 tests.
Drugs
CAB2-015 (compound 16 in Boateng et al., 2015; N-(4-(4-(Naphthalen-1-yl)piperazin-1-yl)butyl)quinoline-3-carboxamide oxalate) and BAK4-54 (compound 32 in Boateng et al., 2015; N-(4-(4-(Naphthalen-1-yl)piperazin-1-yl)butyl)-1H-indole-2-carboxamide HCl) were synthesized within the NIDA IRP according to recently published procedures (Boateng et al., 2015). Oxycodone HCl, sucrose and 2-hydroxypropyl-β-cyclodextrin were purchased from Sigma/RBI (St Louis, MO, USA). Oxycodone was dissolved in physiological saline. 2-hydroxypropyl-β-cyclodextrin was dissolved in 25% 2-hydroxypropyl-β-cyclodextrin in distilled water.
Data Analyses
All behavioral data are presented as means ± SEM. One-way or two-way analysis of variance (ANOVA) were used to analyze the data across experiments. Post-hoc multiple comparisons were carried out using the Newman-Keuls test. P<0.05 was considered to indicate statistical significance.
RESULTS
Rats acquired oxycodone self-administration across a wide range of unit doses similarly as for heroin
Figure 1A shows the time course of acquisition to oxycodone self-administration maintained by three different unit doses of oxycodone. Most rats in the 3 groups acquired self-administration behavior after 1-2 days of training as compared to the number of inactive lever pressing, and increased the number of infusions gradually in subsequent training sessions. With the unit dose increase, animals displayed a typical dose-dependent (compensatory) reduction in the numbers of oxycodone infusions per daily session. Two-way ANOVA for repeated measures over time (training sessions) showed a significant time main effect in oxycodone infusions (Fig. 1A, F9,135=4.06, p<0.001), but did not reveal a significant group main effect (Fig. 1A: F2, 15=0.86, p=0.44) or group × time interaction (F18,135= 0.19, p=0.99). Figure 1B shows the oxycodone intake (mg/kg) during the self-administration maintained by the different unit doses of oxycodone. Two-way ANOVA for repeated measures over time showed a significant treatment group main effect in daily oxycodone intake (Fig. 1B: F2,15=20.88, p<0.001), time main effect (F9,133 = 4.06, p<0.001), but not in treatment × time interaction (F18,162 = 0.19, p>0.05). Figure 1-C/D shows the time courses of acquisition for heroin self-administration maintained by the same unit doses and daily heroin intake, demonstrating a similar pattern of heroin-taking behavior as for oxycodone self-administration. Although two-way ANOVA for repeated measures over time did not reveal a significant group main effect in daily heroin infusions (Fig. 1C, F2,18=3.16, p>0.05) or group × time interaction (F18,162 = 0.97, p>0.05), but it revealed a significant time main effect (F9,162 = 9.13, p<0.001). However, two-way ANOVA for repeated measures over time for the heroin intake data revealed a significant group main effect (Fig. 1D, F2,18=7.11, p<0.01), time main effect (F9,162 = 4.64, p<0.001), but not group × time interaction (F18,162 = 0.20, p>0.05).
Figure 1.
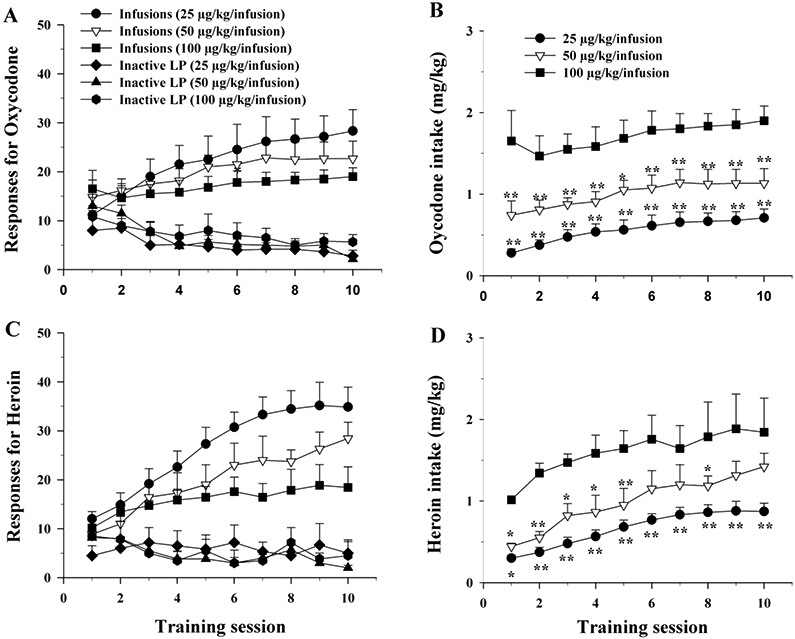
Comparison of the rewarding effects of oxycodone and heroin in rats. A: Time course of the acquisition of oxycodone self-administration maintained by different doses of oxycodone. B: Mean daily oxycodone intake (mg/kg) during the acquisition of oxycodone self-administration. C: Time course of the acquisition of heroin self-administration maintained by three different doses of heroin. D: Mean daily heroin intake (mg/kg) during the acquisition of heroin self-administration. N=6-7/group. Inactive LP – Inactive lever presses. *p<0.05, **p<0.01, compared to 100 μg/kg/infusion group.
CAB2-015 or BAK4-54 decreases oxycodone self-administration
Figure 2 shows the effects of CAB2-015 and BAK4-54 on the maintenance of oxycodone (50 μg/kg/infusion) self-administration. Pretreatment with either CAB2-015 or BAK4-54 (0.4-10 mg/kg, i.p.), produced a dose-dependent reduction in oxycodone self-administration (Fig. 2A-2B, left a/b panels). One-way ANOVA over drug dose revealed a statistically significant drug dose main effect after CAB2-015 (Fig. 2A – Infusions: F3,27= 3.38, p<0.05) or BAK4-54 (Fig. 2B – Infusions, F3,27=4.40, p<0.05). When the drug-induced changes in oxycodone infusions were normalized over the baseline one day before the drug testing (% changes in infusions, Fig. 2A-b, Fig. 2B-b) or calculated into the drug intake (i.e., the number of infusions × unit drug dose, mg/kg, Fig. 2A-c, Fig. 2B-c), one-way ANOVA also revealed a statistically significant reduction % changes in infusion after either CAB2-015 (Fig. 2A-b: F3,27= 11.02, p<0.001) or BAK4-54 (Fig. 2B-b, F3,27= 20.53, p<0.001) or in oxycodone intake after CAB2-015 (Fig. 2B-c, F3,27= 3.38, p<0.05) or after BAK4-54 (Fig. 2B-c, F3,27=4.40, p<0.05). Post-hoc individual group comparisons revealed a significant reduction in oxycodone self-administration after 10 mg CAB2-015 (Fig. 2A), and 4 or 10 mg/kg BAK4-54 pretreatments (Fig. 2B). Pretreatment with either antagonist produced no significant effects on inactive lever-presses.
Figure 2.
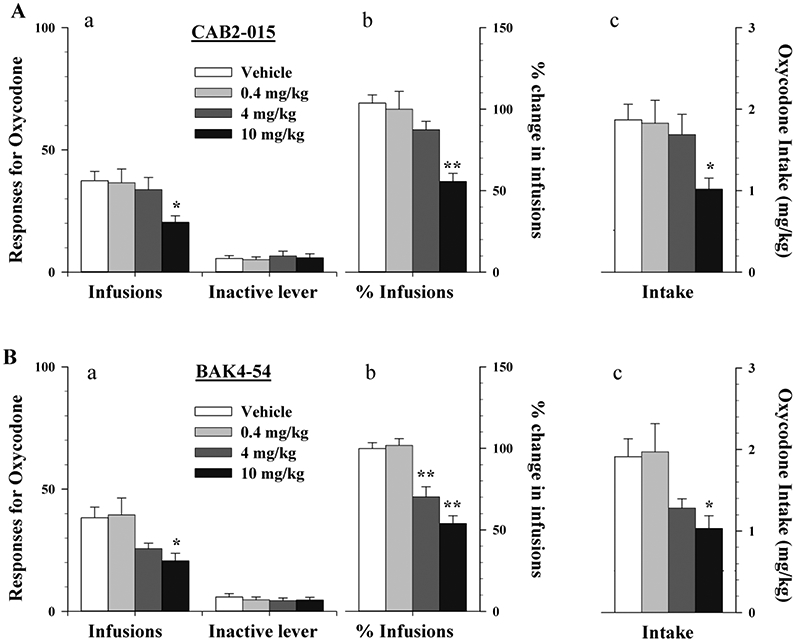
Effects of CAB2-015 or BAK4-54 on the maintenance of oxycodone (50 μg/kg/infusion) self-administration in rats. A: Systemic administration of CAB2-015 (0.4-10 mg/kg, i.p.; 15 min before session) dose-dependently decreased oxycodone self-administration as assessed by the total number oxycodone infusions (a), % change in oxycodone infusions (b), (or oxycodone intake (c).; B: Systemic administration of BAK4-54 (0.4-10 mg/kg, i.p.; 15 min before session) similarly inhibited oxycodone self-administration. *p<0.05, **p<0.01, compared to the vehicle pretreatment group. N=7-8/group.
CAB2-015 or BAK4-54 pretreatment shifts the oxycodone dose-response curves downward
Figure 3 shows that pretreatment with CAB2-015 or BAK4-54 (10 mg/kg) significantly shifted the dose-infusion and dose-intake curves downward. Two-way ANOVA for repeated measures over oxycodone doses and groups (for different D3R antagonist treatment) revealed significant effects of group and oxycodone dose for both oxycodone infusion (Fig. 3A, group main effect, F2, 20=28.18, p<0.001; oxycodone dose main effect, F5, 100=8.02, p<0.001; group × dose interaction, F10, 100=1.87, p>0.05) and intake (Fig. 3B, group main effect, F2, 20=17.32, p<0.001; oxycodone dose main effect, F5, 100=59.11, p<0.001; group × dose interaction ( F10, 100=1.19, p>0.05).
Figure 3.
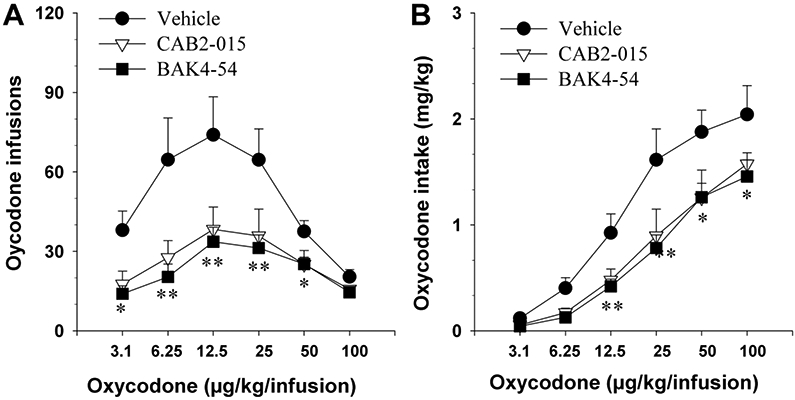
Effects of CAB2-015 or BAK4-54 on oxycodone self-administration dose-response curve in rats. A: Pretreatment with CAB2-015 (10 mg/kg, i.p.) or BAK4-54 (10 mg/kg, i.p.) significantly shifted the dose-response curve downward; B: Calculated oxycodone intake, indicating that CAB2-015 or BAK4-54 significantly shifted the dose-intake curves downward. *p<0.05, **p<0.01, compared to the vehicle group. N=7-8/group.
CAB2-015 or BAK4-54 pretreatment facilitated extinction of oxycodone self-administration and inhibited oxycodone-induced reinstatement of drug-seeking behavior
Three groups of rats were used in this experiment. After stable oxycodone self-administration was achieved, the animals underwent extinction, in which saline was substituted for oxycodone. Repeated pretreatment with either antagonist dose-dependently facilitated extinction of drug-seeking behavior during the extinction phase and significantly attenuated oxycodone-triggered reinstatement of drug-seeking behavior (tested on day 8). After the first oxycodone-induced reinstatement testing, animals continued exhibiting extinction without CAB2-015 or BAK4-54 pretreatment. Strikingly, this inhibitory effect lasted several days. After 4 additional pretreatment-free sessions, all animals received a second oxycodone priming injection. The reinstatement response was still significantly lower in each D3R antagonist pretreatment group than the vehicle control group. Two-way ANOVA for repeated measures over time revealed significant effects of group, time and group × time interaction for both CAB2-015 (Fig. 4A: group, F3, 23=11.89, p<0.001; time, F12, 276=42.30, p<0.001; group × time interaction, F36, 276=2.5, p<0.001) and BAK4-54 (Fig. 4B: group, F3, 22=15.78, p<0.001; time, F12, 264=67.47, p<0.001; group × time interaction, F36, 264=4.70, p<0.001).
Figure 4.
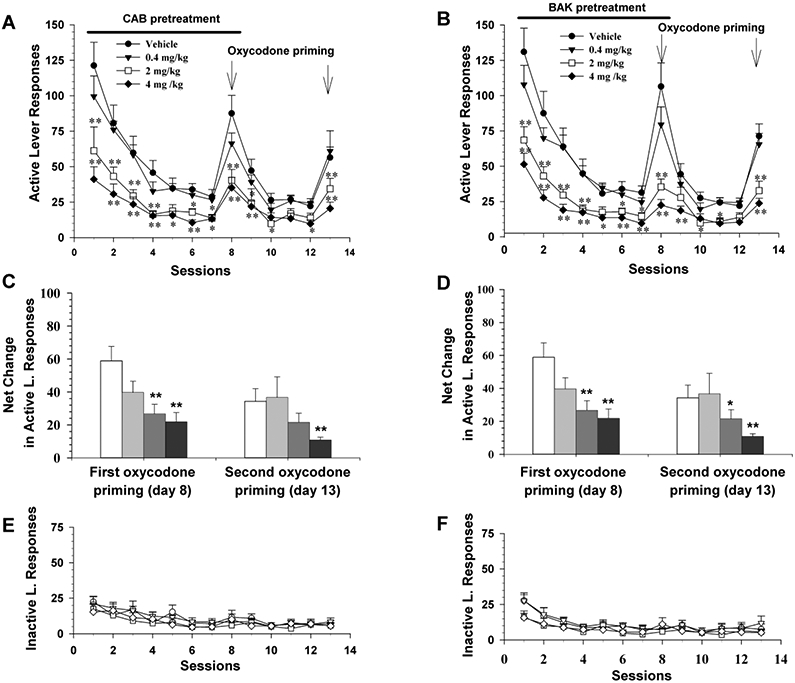
Effects of CAB2-015 and BAK4-54 on drug-seeking behavior observed during extinction and oxycodone-induced reinstatement test. A: Active lever response observed during extinction, illustrating that repeated daily CAB2-015 pretreatment for 8 days significantly decreased oxycodone-seeking (see sessions 1-7) and oxycodone priming-induced reinstatement of drug-seeking behavior (session 8). B: Active lever response observed during extinction, illustrating that repeated daily BAK4-54 pretreatment for 8 days also significantly inhibited oxycodone-seeking (see sessions 1-7) and oxycodone priming-induced reinstatement of drug-seeking behavior (session 8). This inhibitory effect is long-lasting for at least 5 days (sessions 10-14). C/D: Normalized reinstatement response to oxycodone priming by subtraction of basal level of lever response (observed immediately before each oxycodone test session) from oxycodone-induced response on the test day, illustrating that both CAB2-015 and BAK4-54 significantly inhibited oxycodone-induced reinstatement of drug-seeking behavior. E/F: Inactive lever response, illustrating no significant difference between different treatment groups. *p<0.05, **p<0.01, compared to the vehicle group. N=6-7/group.
We note that the basal levels of active lever responses are different before oxycodone priming. To confirm whether repeated daily pretreatment with CAB2-015 or BAK4-54 inhibits oxycodone-induced reinstatement response, we normalized oxycodone-induced changes in drug-seeking to Net Change in drug-seeking behavior by subtraction of the basal level of lever responding (on the session immediately before the oxycodone test session) from the active lever responses on the testing day (Figure 4B). One-way ANOVA for the data on each oxycodone test day revealed a significant treatment main effect after CAB2-015 (Fig. 2C, the first priming test day, F3,23 =5.99, p<0.01; 2nd priming test day, F3,23 = 2.59 p=0.08) or BAK4-54 (Fig. 4D, the first priming test day, F3,22 =11.49, p<0.001; 2nd priming test day, F3,22 = 9.2, p<0.001) pretreatment, suggesting that CAB2-015 or BAK4-54 pretreatment significantly inhibited oxycodone-induced reinstatement of drug-seeking behavior. Pretreatment with either antagonist showed no effects on inactive lever-presses.
CAB2-015 or BAK4-54 pretreatment attenuates oxycodone-induced hyperlocomotion
We also examined whether the pharmacological action produced by CAB2-015 or BAK4-54 in oxycodone self-administration and reinstatement could be generalized to other actions of oxycodone. We found that pretreatment with either CAB2-015 or BAK4-54 (4-10 mg/kg) also dose-dependently decreased the locomotor response to oxycodone (1 mg/kg, i.p., Fig. 5A). Two-way ANOVA for repeated measures over time revealed significant effects of time and time × group interaction for both CAB2-015 (Fig. 5A: time, F5, 105=53.55, p<0.001; interaction, F10, 105=1.97, p<0.05) and BAK4-54 (Fig. 5B: time, F5, 105=77.41, p<0.001; interaction, F10, 105=2.01, p<0.05). CAB2-015 or BAK4-54 alone had no effects on basal locomotor activity (Fig. 5-C/D).
Figure 5.
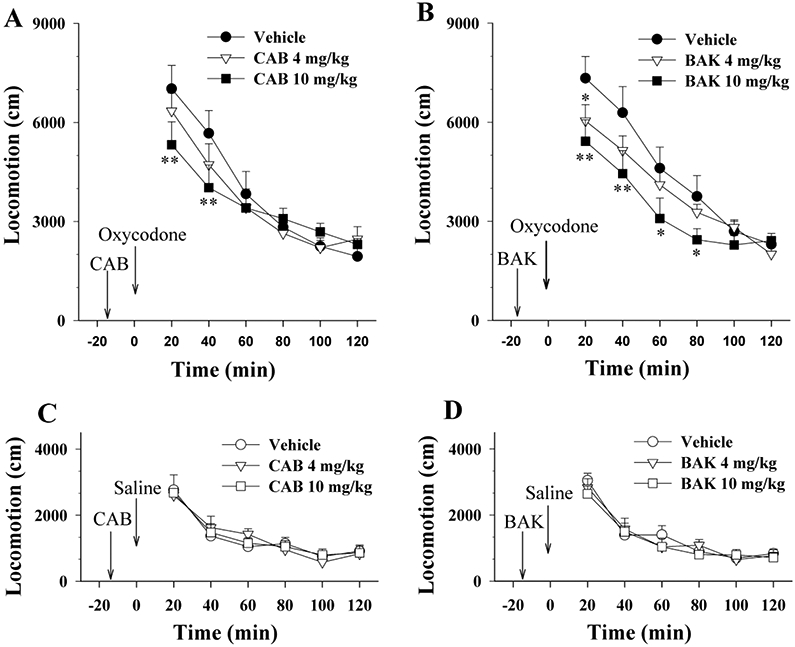
Effects of CAB2-015 or BAK4-54 on basal or oxycodone-enhanced locomotor activity. A: Pretreatment with CAB2-015 (4-10 mg/kg, i.p., 15 min prior to oxycodone injection) dose-dependently inhibited oxycodone-enhanced locomotion. B: Pretreatment with BAK4-54 (4-10 mg/kg, i.p., 15 min prior to oxycodone injection) dose-dependently inhibited oxycodone-enhanced locomotion. C/D: CAB02-15 or BAK4-54 alone, at the same doses, had no effect on basal locomotor activity. *p<0.05, **p<0.01, compared to the vehicle group. N=8 in each group.
CAB2-015, but not BAK4-54, pretreatment inhibits sucrose self-administration
We also observed the effects of both D3R antagonists on oral sucrose self-administration. Figure 6 shows that pretreatment with either CAB2-015 or BAK4-54, at doses that inhibited oxycodone self-administration (4 and 10 mg/kg), failed to significantly decrease sucrose self-administration. All rats reached the maximally allowed 120 sucrose deliveries with different time durations. We therefore normalized data to sucrose deliveries per hour (i.e., the total number of sucrose deliveries divided by the time spent). One-way ANOVA for repeated measures over drug dose did not reveal statistically significant treatment main effects (Fig. 6 – CAB2–015, F2,22 = 3.69, p<0.05; Fig. 6-BAK4-54, F2,22 = 1.12, p>0.05).
Figure 6.
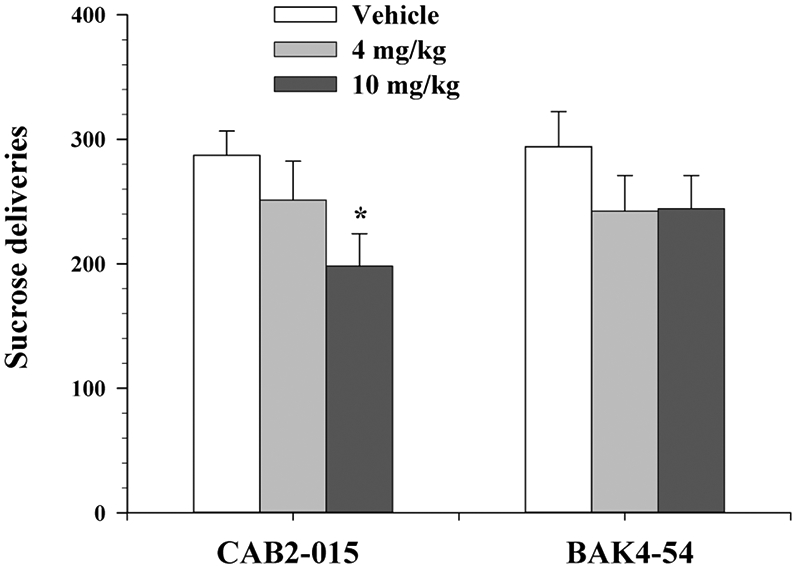
Effects of CAB2-015 or BAK4-54 on oral sucrose self-administration. Pretreatment with CAB2-015, but not BAK4-54, dose-dependently inhibited oral sucrose self-administration behavior. N=12/group. *p<0.05, compared to the vehicle control group..
DISCUSSION
In the present study, we found that oxycodone is highly reinforcing, comparable to heroin, as assessed by rat self-administration procedure. Rats acquired self-administration behavior across a range of oxycodone doses with significantly higher drug intake at the higher doses. The patterns of oxycodone self-administration were also similar to those for heroin self-administration (Lori and Burn, 2005; Martin et al., 2007). Indeed, the dose of oxycodone required to produce maximal self-administration was approximately the same as that required for maximal heroin self-administration, but 20-fold lower than the morphine dose required to produce maximal self-administration (Leri and Burns, 2005) (Martin et al., 2007). These findings suggest that oxycodone has significant abuse potential as other opioids reported previously (Wade et al., 2015). Notably, oxycodone is the most prominent contributor to the current prescription opioid epidemic (Rosenblum et al., 2007), and displays a significantly higher subject rating of “liking” than other opioid analgesics such as morphine and hydrocodone in human nondependent users (Stoops et al., 2010). It has recently been described as producing similar compulsive self-administration and dose escalation to heroin and fentanyl in rats exposed to extended access to these drugs (Wade et al., 2015). The most important finding in the present study is that both the novel DA D3R antagonists/partial agonists CAB2-015 and BAK4-54 appear to be highly effective in reducing oxycodone-taking and oxycodone-seeking behavior as measured in multiple behavioral models, suggesting that both compounds may have therapeutic potential for treatment of opioid use disorders.
Our interest in evaluating D3R antagonists/partial agonists to treat substance use disorders was derived from seminal reports in the1990s, indicating that the D3R played a role in modulating cocaine self-administration (Caine and Koob, 1993). Subsequently, the partial D3R agonist BP 897 was described as being effective in attenuating cocaine seeking behavior in rats (Pilla et al., 1999). Further, the D3R selective antagonist, SB277011A (Reavill et al., 2000; Stemp et al., 2000) was discovered to inhibit cocaine seeking and cocaine-enhanced brain reward (Vorel et al., 2002). Since then, we and others have discovered novel D3R antagonists and compared them to classic D3R antagonists such as BP 897, SB-277011A and NGB-2904. Lead compounds such as PG01037, YQA14, and PG648 (Heidbreder and Newman, 2010; Keck et al., 2015; Micheli and Heidbreder, 2013; Newman et al., 2005) have been developed as potential therapeutics to treat substance use disorders, with a primary focus on psychostimulants. However, it is well known that D3R antagonists are typically unable to block cocaine or methamphetamine self-administration under a low FR schedule of reinforcement (Heidbreder and Newman, 2010; Keck et al., 2015; Xi and Gardner, 2007). This may be related to high extracellular dopamine (DA) levels during active cocaine- or methamphetamine-taking that may compete with antagonist binding to D3Rs, and therefore, attenuate the pharmacological action of a D3R antagonist.
In comparison to psychostimulants, opioids induce relatively moderate increases in extracellular DA within the accumbens during active drug-taking (Badiani et al., 2011; Gratton, 1996; Peng et al., 2010). Thus, we proposed that D3R antagonists may be more effective in reducing opioid use than psychostimulant use. This hypothesis is supported by the finding that D3R antagonists are able to inhibit heroin self-administration in mice (Boateng et al., 2015) and opioid-induced conditioned place preference in rats (Francès et al., 2004). Recently, PF-4363467 was reported to be a D3R antagonist that attenuates opioid drug seeking behavior in rats (Wager et al., 2017). However, the effects of D3R antagonists/partial agonists on prescription opioid use and relapse have not been evaluated.
Both BAK4-54 and CAB2-015 are high affinity D3R ligands with improved metabolic stability over our previous lead compound, PG648 (Boateng et al., 2015). The finding that they are dual D3R antagonists (at lower doses) and partial agonists (at higher doses) (Boateng et al., 2015) may be important in their overall therapeutic profile. As stated above, this profile may be important since full blockade of DA D2-like receptors has been associated with dysphoric effects, while drug craving that occurs during drug withdrawal or abstinence has been associated with reduced DA transmission in the striatum (Volkow et al., 2014). In addition, both compounds have up to 100-fold higher D3R affinity (Ki values of 0.12-0.35 nM) than other D3R antagonists (such as SB-277011A, NGB2904, or YQA14 with Ki values of 1-11 nM) (Song et al., 2012), suggesting that lower drug doses may be sufficient to produce the same therapeutic effects compared to these older D3R antagonists.
CAB2-015 and BAK4-54 each decreased oxycodone self-administration. The dose of BAK4-54 to produce this effect is significantly lower than that of CAB2-015, which is in accordance with the higher affinity of BAK4-54 at D3Rs (Boateng et al., 2015). Pretreatment with CAB2-015 or BAK4-54 shifted the oxycodone dose-response curve downward. These effects are unlikely to have been consequences of the drug’s effects on general motor function since neither antagonist significantly affected inactive lever-presses or basal locomotor activity. We note that BAK4-54, at the same doses that inhibited oxycodone self-administration, had no effect on sucrose self-administration, while CAB2-015 displayed a significant reduction in sucrose self-administration in a dose-dependent manner. This may be related to the fact that CAB2-015 is less selective for D3R over D2R and other off target receptors, compared to BAK4-54 (Boateng et al., 2015). In addition to blockade of D3Rs, CAB 2-015 also has significant binding affinity to hD2Rs (Ki = 15.8 nM) and multiple serotonin receptors (Ki values for 5-HT1A, 5-HT2A and 5-HT2C are 2.5, 0.33, and 0.38 nM, respectively) (Boateng et al., 2015). This is consistent with previous studies demonstrating that selective D3R antagonists typically have no significant effect on food or sucrose self-administration (Heidbreder and Newman, 2010), while the reduction of food or sucrose self-administration produced by high doses of D2-like receptor ligands is most likely mediated by blockade of D2R receptors (Thomsen et al., 2017), as assessed in DA receptor gene-knockout mice (Soto et al., 2016). We also show in the present study that D3R antagonism significantly facilitates extinction of oxycodone-seeking behavior and attenuated oxycodone-induced reinstatement of drug-seeking, even following a treatment-free period, suggesting that D3R antagonism might be a viable strategy in opioid dependence treatment. Conditioned drug craving during extinction is an enduring factor underlying relapse to compulsive drug use (O'Brien et al., 1998). The present findings suggest that D3R antagonism may also interact with the neural processes underlying conditioned drug craving, drug-associated learning and conditioned reward that occur during extinction. Using another newly developed D3R antagonist, we also found that D3R blockade significantly inhibited acquisition of both oxycodone-induced locomotor sensitization and CPP (Kumar et al., 2016). Chronic administration of morphine significantly increased D3R mRNA expression in brain DA system, suggesting increased D3 receptor activity in opioid dependence (Spangler et al., 2003). This may in part explain how D3R antagonism attenuates opioid effects described above.
Finally, the pharmacological action produced by BAK4-54 in the present study appears to be mediated by inhibition of D3Rs since systemic administration of the same doses of BAK4-54 dose-dependently inhibited heroin self-administration only in WT mice, but not in D3-KO mice (Boateng et al., 2015). In contrast to BAK4-54, multiple receptor mechanisms may underlie the actions produced by CAB2-015, since CAB2-015 is not as highly D3R selective. Nevertheless, although in terms of proof-of-concept, we cannot be sure that CAB 2-015’s effects are mediated exclusively by D3R, medications are often poly-mechanistic. A compound such as CAB2-015 with relatively lower selectivity (45-fold) for hD3Rs over hD2Rs may be superior to a highly selective D3R antagonist since D3R blockade plus partial D2R blockade may produce additive therapeutic effects without significant unwanted side-effects produced by full D2R antagonism, as has recently been reported (Peng et al., 2009; Wager et al., 2017). Our present findings also indicate that D3R blockade may be a promising approach for dependence prevention in prescription opioid use for the treatment of chronic pain.
We should point out that although extensive research over the past two decades indicate that D3R antagonists/partial agonists have therapeutic potential for treating substance use disorders, only GSK598,809, a highly D3R selective antagonist, has been tested in humans for treatment of obesity, nicotine or alcohol dependence (Dodds et al., 2012; Mugnaini et al., 2013; Murphy et al., 2017). In one study, a single dose of GSK598,809 produced a significant short-term reduction in craving for cigarette smoking (Mugnaini et al., 2013). This study provided the first clinical evidence supporting the hypothesis that D3R antagonism might be effective in treating substance use disorders. However, plans to evaluate it in cocaine abusers were abandoned when unacceptable increases in blood pressure were observed in the presence of cocaine in dogs (Appel et al., 2015). Of note, GSK598,809 did not cause hypertension on its own, but only in combination with cocaine, which by itself is hypertensive. As the opioids, in general, do not increase blood pressure, we reasoned that this side-effect may not be observed with a D3R antagonist in the presence of an opioid, such as oxycodone. Therefore, we expect D3R selective antagonists/partial agonists may have better translational potential as pharmacotherapeutics to treat opioid use disorders.
In summary, although there are medications available for the treatment of both opioid overdose (e.g., naloxone) and dependence (e.g. methadone or buprenorphine), there are currently no clinically available opioid analgesics without abuse potential or other (deadly) side effects, although promising and novel opioid agonists have been recently reported (Ding et al., 2016; Manglik et al., 2016). Prescription opioids will undoubtedly continue to be prescribed for the treatment of pain. The development of medications that can potentially reduce opioid abuse liability or if dependence does result, can be used for treatment, is critical. D3R antagonists/partial agonists, such as CAB2-015 and BAK4-54 may be the key to significantly reducing prescription opioid abuse liability and addiction-like behaviors. Finally, while others have recently reported that female rats take more oxycodone than male rats under some experimental conditions (Mavrikaki et al., 2017) we only tested male rats in this study. A recent report showed that women are more likely to use opioids to manage stress and fatal opioid overdoses occur more often in women than in men (Chartoff and McHugh, 2016). Thus, further evaluation in female rats will be necessary to determine the effects of these or other novel D3R antagonists/partial agonists on opioid reward and addiction in order to truly evaluate the translational potential of D3R antagonists/partial agonists for treating opioid use disorders in humans.
Highlights:
Rats self-administer oxycodone similar to heroin
The D3R antagonists/partial agonists CAB2-015 and BAK4-54 inhibit oxycodone self-administration
CAB2-015 and BAK4-54 facilitate extinction of oxycodone-seeking behavior
CAB2-015 and BAK4-54 inhibit oxycodone-induced reinstatement of drug-seeking behavior
Funding and Disclosure:
The authors declare that there is no financial conflict of interest. This research was supported by the Intramural Research Program of the National Institute on Drug Abuse, National Institutes of Health, USA, ZIA DA000424.
References
- Appel NM, Li SH, Holmes TH, Acri JB, 2015. Dopamine D3 Receptor Antagonist (GSK598809) Potentiates the Hypertensive Effects of Cocaine in Conscious, Freely-Moving Dogs. J Pharmacol Exp Ther 354, 484–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badiani A, Belin D, Epstein D, Calu D, Shaham Y, 2011. Opiate versus psychostimulant addiction: the differences do matter. Nature Reviews Neuroscience 12, 685–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boateng CA, Bakare OM, Zhan J, Banala AK, Burzynski C, Pommier E, Keck TM, Donthamsetti P, Javitch JA, Rais R, Slusher BS, Xi Z-X, Newman AH, 2015. High Affinity Dopamine D3Receptor (D3R)-Selective Antagonists Attenuate Heroin Self-Administration in Wild-Type but not D3R Knockout Mice. Journal of Medicinal Chemistry 58, 6195–6213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boileau I, Payer D, Houle S, Behzadi A, Rusjan PM, Tong J, Wilkins D, Selby P, George TP, Zack M, Furukawa Y, McCluskey T, Wilson AA, Kish SJ, 2012. Higher binding of the dopamine D3 receptor-preferring ligand [11C]-(+)-propyl-hexahydro-naphtho-oxazin in methamphetamine polydrug users: a positron emission tomography study. J Neurosci 32, 1353–1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caine S, Koob G, 1993. Modulation of cocaine self-administration in the rat through D-3 dopamine receptors. Science 260, 1814–1816. [DOI] [PubMed] [Google Scholar]
- Cerdá M, Santaella J, Marshall BDL, Kim JH, Martins SS, 2015. Nonmedical Prescription Opioid Use in Childhood and Early Adolescence Predicts Transitions to Heroin Use in Young Adulthood: A National Study. The Journal of Pediatrics 167, 605–612.e602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chartoff EH, McHugh RK, 2016. Translational Studies of Sex Differences in Sensitivity to Opioid Addiction. Neuropsychopharmacology 41, 383–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Compton WM, Volkow ND, 2006. Major increases in opioid analgesic abuse in the United States: Concerns and strategies. Drug and Alcohol Dependence 81, 103–107. [DOI] [PubMed] [Google Scholar]
- Ding H, Czoty PW, Kiguchi N, Cami-Kobeci G, Sukhtankar DD, Nader MA, Husbands SM, Ko MC, 2016. A novel orvinol analog, BU08028, as a safe opioid analgesic without abuse liability in primates. Proc Natl Acad Sci U S A 113, E5511–5518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodds CM, O'Neill B, Beaver J, Makwana A, Bani M, Merlo-Pich E, Fletcher PC, Koch A, Bullmore ET, Nathan PJ, 2012. Effect of the dopamine D3 receptor antagonist GSK598809 on brain responses to rewarding food images in overweight and obese binge eaters. Appetite 59, 27–33. [DOI] [PubMed] [Google Scholar]
- Fiellin DA, Schottenfeld RS, Cutter CJ, Moore BA, Barry DT, O'Connor PG, 2014. Primary care-based buprenorphine taper vs maintenance therapy for prescription opioid dependence: a randomized clinical trial. JAMA Intern Med 174, 1947–1954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francès H, Smirnova M, Leriche L, Sokoloff P, 2004. Dopamine D3 receptor ligands modulate the acquisition of morphine-conditioned place preference. Psychopharmacology 175. [DOI] [PubMed] [Google Scholar]
- Gratton A, 1996. In vivo analysis of the role of dopamine in stimulant and opiate self-administration. J Psychiatry Neurosci 21, 264–279. [PMC free article] [PubMed] [Google Scholar]
- Heidbreder CA, Newman AH, 2010. Current perspectives on selective dopamine D(3) receptor antagonists as pharmacotherapeutics for addictions and related disorders. Ann N Y Acad Sci 1187, 4–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keck TM, John WS, Czoty PW, Nader MA, Newman AH, 2015. Identifying Medication Targets for Psychostimulant Addiction: Unraveling the Dopamine D3 Receptor Hypothesis. J Med Chem 58, 5361–5380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar V, Bonifazi A, Ellenberger MP, Keck TM, Pommier E, Rais R, Slusher BS, Gardner E, You Z-B, Xi Z-X, Newman AH, 2016. Highly Selective Dopamine D3Receptor (D3R) Antagonists and Partial Agonists Based on Eticlopride and the D3R Crystal Structure: New Leads for Opioid Dependence Treatment. Journal of Medicinal Chemistry 59, 7634–7650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Foll B, Diaz J, Sokoloff P, 2002. Increased dopamine D3 receptor expression accompanying behavioral sensitization to nicotine in rats. Synapse 47, 176–183. [DOI] [PubMed] [Google Scholar]
- Leri F, Burns LH, 2005. Ultra-low-dose naltrexone reduces the rewarding potency of oxycodone and relapse vulnerability in rats. Pharmacology Biochemistry and Behavior 82, 252–262. [DOI] [PubMed] [Google Scholar]
- Manglik A, Lin H, Aryal DK, McCorvy JD, Dengler D, Corder G, Levit A, Kling RC, Bernat V, Hübner H, Huang X-P, Sassano MF, Giguère PM, Löber S, Da D, Scherrer G, Kobilka BK, Gmeiner P, Roth BL, Shoichet BK, 2016. Structure-based discovery of opioid analgesics with reduced side effects. Nature 537, 185–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin TJ, Kim SA, Buechler NL, Porreca F, Eisenach JC, 2007. Opioid Self-administration in the Nerve-injured Rat. Anesthesiology 106, 312–322. [DOI] [PubMed] [Google Scholar]
- Mattick RP, Kimber J, Breen C, Davoli M, 2008. Buprenorphine maintenance versus placebo or methadone maintenance for opioid dependence. Cochrane Database Syst Rev, CD002207. [DOI] [PubMed] [Google Scholar]
- Meyer R, Patel AM, Rattana SK, Quock TP, Mody SH, 2014. Prescription Opioid Abuse: A Literature Review of the Clinical and Economic Burden in the United States. Population Health Management 17, 372–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Micheli F, Heidbreder C, 2013. Dopamine D3 receptor antagonists: a patent review (2007 - 2012). Expert Opin Ther Pat 23, 363–381. [DOI] [PubMed] [Google Scholar]
- Mugnaini M, Iavarone L, Cavallini P, Griffante C, Oliosi B, Savoia C, Beaver J, Rabiner EA, Micheli F, Heidbreder C, Andorn A, Merlo Pich E, Bani M, 2013. Occupancy of brain dopamine D3 receptors and drug craving: a translational approach. Neuropsychopharmacology 38, 302–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy A, Nestor LJ, McGonigle J, Paterson L, Boyapati V, Ersche KD, Flechais R, Kuchibatla S, Metastasio A, Orban C, Passetti F, Reed L, Smith D, Suckling J, Taylor E, Robbins TW, Lingford-Hughes A, Nutt DJ, Deakin JF, Elliott R, 2017. Acute D3 Antagonist GSK598809 Selectively Enhances Neural Response During Monetary Reward Anticipation in Drug and Alcohol Dependence. Neuropsychopharmacology 42, 1049–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neisewander JL, Fuchs RA, Tran-Nguyen LTL, Weber SM, Coffey GP, Joyce JN, 2004. Increases in Dopamine D3 Receptor Binding in Rats Receiving a Cocaine Challenge at Various Time Points after Cocaine Self-Administration: Implications for Cocaine-Seeking Behavior. Neuropsychopharmacology 29, 1479–1487. [DOI] [PubMed] [Google Scholar]
- Newman AH, Grundt P, Nader MA, 2005. Dopamine D3 Receptor Partial Agonists and Antagonists as Potential Drug Abuse Therapeutic Agents. Journal of Medicinal Chemistry 48, 3663–3679. [DOI] [PubMed] [Google Scholar]
- Nielsen S, Bruno R, Degenhardt L, Demirkol A, Lintzeris N, 2016. Opioid agonist doses for oxycodone and morphine dependence: Findings from a retrospective case series. Drug Alcohol Rev. [DOI] [PubMed] [Google Scholar]
- Novick DM, Salsitz EA, Joseph H, Kreek MJ, 2015. Methadone Medical Maintenance: An Early 21st-Century Perspective. J Addict Dis 34, 226–237. [DOI] [PubMed] [Google Scholar]
- O'Brien CP, Childress AR, Ehrman R, Robbins SJ, 1998. Conditioning factors in drug abuse: can they explain compulsion? Journal of Psychopharmacology 12, 15–22. [DOI] [PubMed] [Google Scholar]
- Payer D, Balasubramaniam G, Boileau I, 2014. What is the role of the D3 receptor in addiction? A mini review of PET studies with [(11)C]-(+)-PHNO. Prog Neuropsychopharmacol Biol Psychiatry 52, 4–8. [DOI] [PubMed] [Google Scholar]
- Peng XQ, Ashby CR Jr., Spiller K, Li X, Li J, Thomasson N, Millan MJ, Mocaer E, Munoz C, Gardner EL, Xi ZX, 2009. The preferential dopamine D3 receptor antagonist S33138 inhibits cocaine reward and cocaine-triggered relapse to drug-seeking behavior in rats. Neuropharmacology 56, 752–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng XQ, Xi ZX, Li X, Spiller K, Li J, Chun L, Wu KM, Froimowitz M, Gardner EL, 2010. Is slow-onset long-acting monoamine transport blockade to cocaine as methadone is to heroin? Implication for anti-addiction medications. Neuropsychopharmacology 35, 2564–2578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilla M, Perachon S, Sautel F, Garrido F, Mann A, Wermuth CG, Schwartz JC, Everitt BJ, Sokoloff P, 1999. Selective inhibition of cocaine-seeking behaviour by a partial dopamine D3 receptor agonist. Nature 400, 371–375. [DOI] [PubMed] [Google Scholar]
- Potter JS, Marino EN, Hillhouse MP, Nielsen S, Wiest K, Canamar CP, Martin JA, Ang A, Baker R, Saxon AJ, Ling W, 2013. Buprenorphine/naloxone and methadone maintenance treatment outcomes for opioid analgesic, heroin, and combined users: findings from starting treatment with agonist replacement therapies (START). J Stud Alcohol Drugs 74, 605–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reavill C, Taylor SG, Wood MD, Ashmeade T, Austin NE, Avenell KY, Boyfield I, Branch CL, Cilia J, Coldwell MC, Hadley MS, Hunter AJ, Jeffrey P, Jewitt F, Johnson CN, Jones DN, Medhurst AD, Middlemiss DN, Nash DJ, Riley GJ, Routledge C, Stemp G, Thewlis KM, Trail B, Vong AK, Hagan JJ, 2000. Pharmacological actions of a novel, high-affinity, and selective human dopamine D(3) receptor antagonist, SB-277011-A. J Pharmacol Exp Ther 294, 1154–1165. [PubMed] [Google Scholar]
- Rosenblum A, Parrino M, Schnoll SH, Fong C, Maxwell C, Cleland CM, Magura S, Haddox JD, 2007. Prescription opioid abuse among enrollees into methadone maintenance treatment. Drug and Alcohol Dependence 90, 64–71. [DOI] [PubMed] [Google Scholar]
- Sokoloff P, Giros B, Martres M-P, Bouthenet M-L, Schwartz J-C, 1990. Molecular cloning and characterization of a novel dopamine receptor (D3) as a target for neuroleptics. Nature 347, 146–151. [DOI] [PubMed] [Google Scholar]
- Sokoloff P, Le Foll B, 2016. The dopamine D3 receptor, a quarter century later. European Journal of Neuroscience 45, 2–19. [DOI] [PubMed] [Google Scholar]
- Sokoloff P, Le Foll B, 2017. The dopamine D3 receptor, a quarter century later. Eur J Neurosci 45, 2–19. [DOI] [PubMed] [Google Scholar]
- Song R, Zhang HY, Li X, Bi GH, Gardner EL, Xi ZX, 2012. Increased vulnerability to cocaine in mice lacking dopamine D3 receptors. Proc Natl Acad Sci U S A 109, 17675–17680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soto PL, Hiranita T, Xu M, Hursh SR, Grandy DK, Katz JL, 2016. Dopamine D(2)-Like Receptors and Behavioral Economics of Food Reinforcement. Neuropsychopharmacology 41, 971–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spangler R, Goddard NL, Avena NM, Hoebel BG, Leibowitz SF, 2003. Elevated D3 dopamine receptor mRNA in dopaminergic and dopaminoceptive regions of the rat brain in response to morphine. Molecular Brain Research 111, 74–83. [DOI] [PubMed] [Google Scholar]
- Stemp G, Ashmeade T, Branch CL, Hadley MS, Hunter AJ, Johnson CN, Nash DJ, Thewlis KM, Vong AKK, Austin NE, Jeffrey P, Avenell KY, Boyfield I, Hagan JJ, Middlemiss DN, Reavill C, Riley GJ, Routledge C, Wood M, 2000. Design and Synthesis oftrans-N-[4-[2-(6-Cyano-1,2,3,4-tetrahydroisoquinolin-2- yl)ethyl]cyclohexyl]-4-quinolinecarboxamide (SB-277011): A Potent and Selective Dopamine D3Receptor Antagonist with High Oral Bioavailability and CNS Penetration in the Rat. Journal of Medicinal Chemistry 43, 1878–1885. [DOI] [PubMed] [Google Scholar]
- Stoops WW, Hatton KW, Lofwall MR, Nuzzo PA, Walsh SL, 2010. Intravenous oxycodone, hydrocodone, and morphine in recreational opioid users: abuse potential and relative potencies. Psychopharmacology 212, 193–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomsen M, Barrett AC, Butler P, Negus SS, Caine SB, 2017. Effects of Acute and Chronic Treatments with Dopamine D2 and D3 Receptor Ligands on Cocaine versus Food Choice in Rats. J Pharmacol Exp Ther 362, 161–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkow ND, Tomasi D, Wang GJ, Logan J, Alexoff DL, Jayne M, Fowler JS, Wong C, Yin P, Du C, 2014. Stimulant-induced dopamine increases are markedly blunted in active cocaine abusers. Mol Psychiatry 19, 1037–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vorel SR, Ashby CR Jr., Paul M, Liu X, Hayes R, Hagan JJ, Middlemiss DN, Stemp G, Gardner EL, 2002. Dopamine D3 receptor antagonism inhibits cocaine-seeking and cocaine-enhanced brain reward in rats. J Neurosci 22, 9595–9603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wade CL, Vendruscolo LF, Schlosburg JE, Hernandez DO, Koob GF, 2015. Compulsive-like responding for opioid analgesics in rats with extended access. Neuropsychopharmacology 40, 421–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wager TT, Chappie T, Horton D, Chandrasekaran RY, Samas B, Dunn-Sims ER, Hsu C, Nawreen N, Vanase-Frawley MA, O'Connor RE, Schmidt CJ, Dlugolenski K, Stratman NC, Majchrzak MJ, Kormos BL, Nguyen DP, Sawant-Basak A, Mead AN, 2017. Dopamine D3/D2 Receptor Antagonist PF-4363467 Attenuates Opioid Drug-Seeking Behavior without Concomitant D2 Side Effects. ACS Chem Neurosci 8, 165–177. [DOI] [PubMed] [Google Scholar]
- Xi Z-X, Gardner EL, 2007. Pharmacological Actions of NGB 2904, a Selective Dopamine D3Receptor Antagonist, in Animal Models of Drug Addiction. CNS Drug Reviews 13, 240–259. [DOI] [PMC free article] [PubMed] [Google Scholar]