

Abstract
Cancer has myriad effects on metabolism that include both rewiring of intracellular metabolism to enable cancer cells to proliferate inappropriately and adapt to the tumor microenvironment, and changes in normal tissue metabolism. With the recognition that fluorodeoxyglucose (FDG) PET imaging is an important tool for the management of many cancers, other metabolites in biological samples have been in the spotlight for cancer diagnosis, monitoring, and therapy. Metabolomics is the global analysis of small molecule metabolites that like other “-omics” technologies can provide critical information about the cancer state that are otherwise not apparent. Here we review how cancer and cancer therapies interact with metabolism at the cellular and systemic level. We provide an overview of metabolomics with a focus on currently available technologies and how they have been applied in the clinical and translational research setting. We also discuss how metabolomics could be further leveraged in the future to improve management of cancer patients. As we move towards personalized targeted therapy for cancer patients, it is important that we understand how metabolism is affected by cancer to incorporate this information into decision making to improve treatment outcomes.
Introduction
In cancer cells, metabolism is dysregulated to support the demands of uncontrolled proliferation [1–3]. This rewiring of cellular metabolism leads to characteristic metabolic phenotypes that can be used for earlier cancer diagnosis, patient selection strategies for clinical trials, and/or as biomarkers of treatment response. Altered metabolism also results in unique metabolic dependencies that in some cases can be targeted with precision medicine and nutrition, including drugs that selectively target metabolic enzymes [4, 5]. Cancer and cancer therapies can also alter metabolism at the whole-body level and interact with the metabolic effects of diet and exercise in complex ways that may affect cancer outcomes and impact a patient’s quality of life.
In the past, much of the assessment of metabolic changes has been limited to measuring individual hormones and metabolites using imaging modalities and standard clinical laboratory tests. In contrast, metabolomics involves the systematic measurement of many metabolites, including nutrients, drugs, signaling mediators and the metabolic products of these small molecules in blood, urine, tissue extracts, or other body fluids [6]. Metabolomics is a powerful tool that can identify cancer biomarkers and drivers of tumorigenesis [7]. This review aims to provide an overview of current and future opportunities of metabolomics to improve the diagnosis, monitoring, and treatment of cancer. We begin with a review of how metabolism and cancer interact at the level of cells, tissues, and the whole body (Figure 1). We then introduce general technical aspects of metabolomics, instrumentation, and source material, including the pros and cons of different approaches. In the final section we provide examples of how metabolomics has been used in the clinical and translational research setting, as a way to guide potential future applications. As the role of metabolism in cancer has been extensively covered, the reader is referred to other excellent reviews cited throughout this manuscript for a more in-depth discussion of specific topics.
Figure 1.
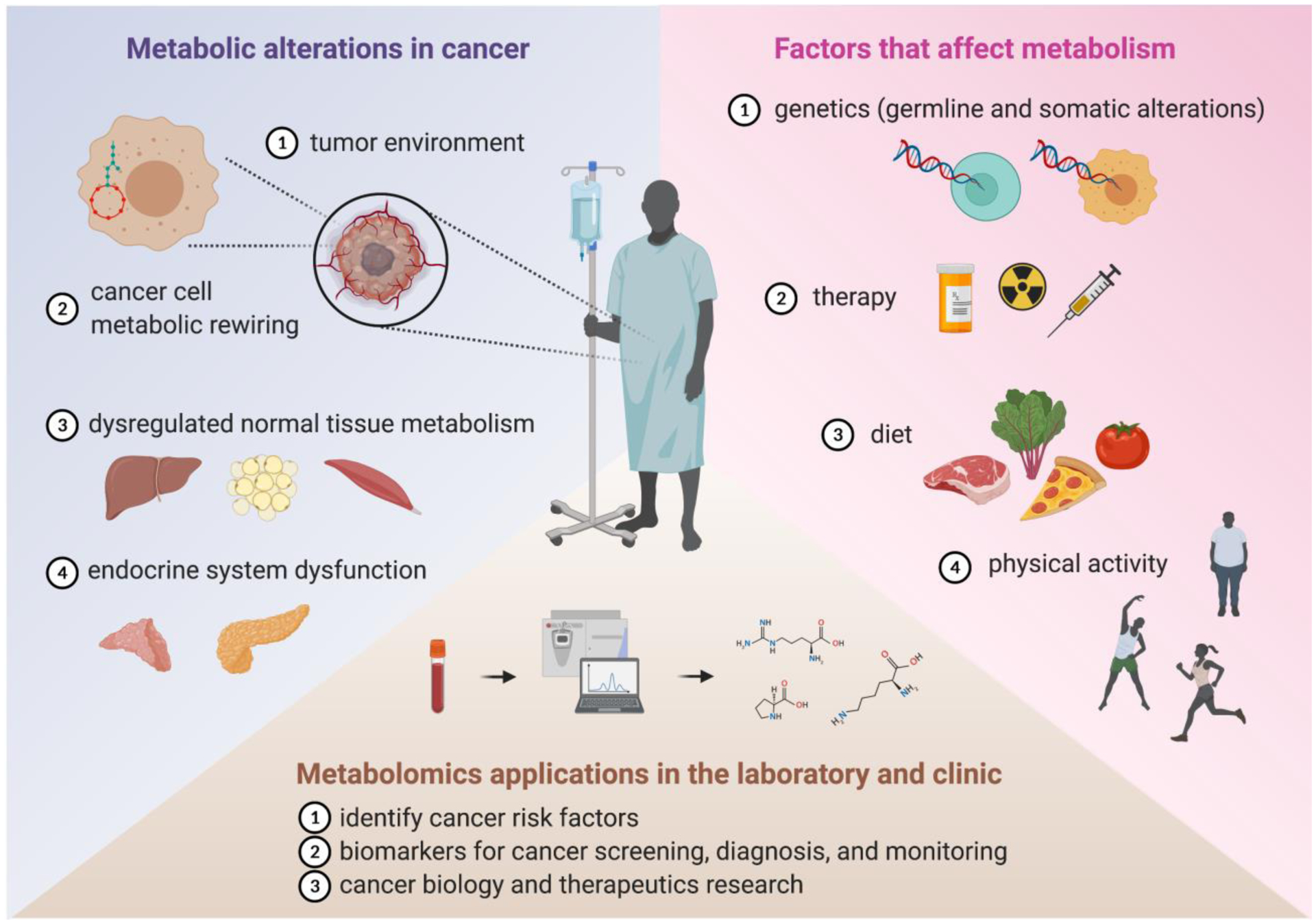
Cancer and metabolism interact at many levels. Cancer causes metabolic alterations in cancer cells and normal tissues which in turn interact with intrinsic and extrinsic factors to affect systemic metabolism. Metabolomics is a systems based approach used to define these complex metabolic interactions for diagnostic and therapeutic gain. See text for details.
Metabolism in Tumors and Cancer Patients
In this section we explore the intersection between metabolism, cancer, and therapy. We begin with a discussion of metabolic alterations in cancer cells and review drugs that target metabolic pathways. We then discuss the effect of cancer and cancer therapy on systemic metabolism. Finally, the role of diet and lifestyle factors in carcinogenesis and response to therapy is reviewed.
Cancer Cell Metabolism
One of the earliest and most recognized metabolic alterations in cancer cells is increased glucose consumption by tumors. Elevated glucose uptake by tumors is detected by FDG PET imaging for initial cancer staging, assessing response to therapy, and surveillance. Beginning with the initial observation by Otto Warburg and others nearly a century ago that tumor cells increase glucose uptake and produce high quantities of lactate even in the presence of oxygen, it has been well established that cancer cells engage in altered metabolism [8–11]. Through much of the genomic era from the inception of gene cloning technologies and subsequent cancer gene discovery, cancer biology was focused on how signaling pathways and transcription factors control cancer growth and the cell cycle. However, in recent years, there has been a renewed interest in understanding how altered metabolism contributes to cancer pathogenesis. Many factors, such as tumor hypoxia, stromal composition, immune cell infiltration, and genetic alterations play critical roles in defining cancer cell metabolism. Genetic and/or epigenetic alterations can provide a crucial survival advantage to cancer cells in a nutrient-starved environment. The renewed interest in cancer cell metabolism came initially from recognizing that dysregulated signaling pathways and transcriptional reprograming result in metabolic alterations in cancer cells [12–15]. Subsequently, it has been shown that the induction of oncogenes and/or loss of tumor suppressors is/are sufficient to drive metabolic changes in cancer cells [16–19]. Indeed, tumor-associated metabolic alterations are recognized as an emerging cancer hallmark [1, 20]. In this section we briefly review select examples of how metabolism is altered in cancer cells to highlight the diversity of mechanisms underlying rewired metabolism in cancer cells. For further information the reader is referred to several excellent reviews of cancer metabolism [12, 13, 15, 21–24].
Glucose is the single most abundant nutrient for most cells and can be a source of biomass and fuel for energy production. Numerous signaling pathways altered in cancer affect glucose metabolism through a variety of mechanisms. In one classic paradigm, receptor tyrosine kinases (RTKs) induced by insulin or other growth factors activate the PI3K-AKT signaling pathway to stimulate glycolysis [25]. AKT (also known as Protein kinase B) is a serine-threonine kinase that can increase glycolytic activity directly by phosphorylating hexokinase (the enzyme that catalyzes the first step in glycolysis) and indirectly via phosphorylation of substrates that regulate trafficking of glucose transporters (GLUT1 and GLUT4) to the plasma membrane [26–28]. PI3K-AKT signaling also induces mammalian target of rapamycin complex 1 (mTORC1), which results in increased expression of hypoxia inducible factor (HIF) 1α [29–32]. HIFs are heterodimeric transcription factors consisting of an α-subunit (HIF1α and HIF2α) that is degraded in the presence of oxygen and a stable β-subunit (HIF1β, also known as ARNT) [33]. In the presence of hypoxia, HIFs are stabilized and activate a transcriptional response that allows adaptation to hypoxic stress including increased expression of glucose transporter 1 and 3 (GLUT1 and GLUT3), hexokinase 2 (HK2), and some isoforms of phosphofructokinase 2 (PFK2) [34]. HIF1α also promotes pyruvate dehydrogenase kinase expression, which inhibits pyruvate oxidation and shunts glucose metabolism toward lactate as an adaptation to hypoxic conditions [35]. HIF2α regulates other aspects of anabolic metabolism and is a therapeutic target in clear cell renal cell carcinoma [36, 37]. Growth factor receptor signaling also activates RAS proteins to promote glucose uptake, glycolysis, and the pentose phosphate pathway (PPP). For example, in pancreatic ductal adenocarcinoma oncogenic KRAS drives glycolytic intermediates into the nonoxidative PPP to support nucleotide production [38].
Energy is maximally released when glucose is completely oxidized to CO2, a process that produces ATP via oxidative phosphorylation (OXPHOS). When oxygen is limiting in cells, glycolysis becomes uncoupled from OXPHOS, and the end product of glycolysis, pyruvate, is instead reduced to lactate. Interestingly, in many cancer cells, glucose is preferentially catabolized via fermentation to lactate even when oxygen is not limiting (the “Warburg effect”). The conversion of pyruvate to lactate appears to be an essential mechanism by which cancer cells maintain an appropriate balance of redox cofactors to support biosynthetic functions [2, 39]. It has been shown that loss of TP53 activity can push cells towards the glycolytic pathway instead of OXPHOS [40]. As first demonstrated nearly a century ago, tumors net dispose of lactate via systemic circulation from where it is taken up by the liver and re-converted to glucose through gluconeogenesis (the “Cori cycle”) [41, 42]. Recently, it has been reported that lactate may also be taken up by some cells in tumors to fuel the TCA cycle [43–45], but how this relates to increased glucose uptake remains an open question [46].
In addition to glucose, increased uptake of other nutrients by cancer cells has been shown to support cancer cell survival, growth, and invasion. Oncogenic MYC has been linked to increased glutaminolysis that results in glutamine addiction in some cancer cells [47]. Glutamine, like glucose, can provide carbons for lipogenesis and MYC has been shown to regulate fatty acid metabolism [48, 49]. Inactivation of the tumor suppressor retinoblastoma protein (RB) has also been shown to increase glutamine utilization due to the upregulation of glutamine transporter ASCT2 in human cancer cells [50]. Although glucose and glutamine are the most abundant nutrients in plasma and tissue culture media, the list of nutrients consumed by cultured cancer cells and tumors is vast. It is likely that in most cases the requirement for specific nutrients depends on tumor type and nutrient environment. For example, dependence on glutamine can be driven by high levels of extracellular cystine [51], and some tumors are less dependent on glutamine metabolism in vivo [52, 53]. As another example, many prostate cancers are not FDG-avid, yet they demonstrate increased uptake of [18F]-fluciclovine (a synthetic analog of leucine) and [11C]-choline used in PET imaging to detect metastases that are otherwise occult by conventional imaging [54, 55]. How tumors balance glucose and glutamine metabolism with other nutrients available in their environment is an area of active investigation.
As demonstrated by the following examples, the cancer genomic era also brought the recognition that some metabolic enzyme are frequently mutated or amplified across a variety of cancer cell types. A recently published study analyzed a TCGA database of over 10,000 tumors across 32 cancer types and found at least one metabolic gene alteration per tumor with a varied number of metabolic gene alterations among cancer types [56]. Germline mutations in two TCA cycle enzymes, fumarate hydratase (FH) and succinate dehydrogenase (SDH), predispose to cancer syndromes hereditary leiomyomatosis and renal cell cancer renal (HLRCC) and hereditary paraganglioma-pheochromocytoma (PGL/PCC) respectively [57, 58]. Affected individuals show loss-of-heterozygosity (or mutation) of the wild-type FH and SDH allele in tumor tissue, which leads to the accumulation of fumarate and succinate, respectively. These metabolites, when present at high concentrations, inhibit DNA and histone demethylases and prolyl hydroxylases, resulting in DNA hypermethylation, chromatin modification, stabilization of hypoxia-inducible factors (HIF1α and HIF2α) and other oncogenic factors [59–61]. Thus, mutations in a single metabolic gene can result in altered metabolite levels, which lead to transcriptional changes in cancer cells that promote uncontrolled growth.
Another prominent example of a metabolic enzyme that is frequently mutated in cancer is isocitrate dehydrogenase (IDH). Mutations in IDH-1 and IDH-2 genes occur in a significant percentage of patients with malignant glioma (60–90%), chondrosarcoma (50–70%), cholangiocarcinoma (10–20%) and acute myeloid leukemia (10–20%), and less frequently in other cancers [62]. In glioma, IDH mutations are prognostic and predictive of response to the DNA alkylating agent, temozolomide [63, 64]. Cancer-associated IDH mutations occur exclusively in the enzyme’s active site, which creates “neomorphic” activity resulting in excessive production of the “oncometabolite” D-2-hydroxyglutarate (D-2-HG) [65, 66]. When D-2-HG levels build up in tumors, it blocks the activity of 2-oxoglutarate dependent DNA and histone demethylases. This is turn leads to hypermethylation and silencing of genes, including the enzyme O6-methylguanine-DNA methyltransferase (MGMT) that reverses DNA damage caused by temozolomide [67–70]. Thus, a metabolite produced in high levels exclusively in IDH-mutant gliomas increases these cancers’ sensitivity to temozolomide.
As pointed out in the previous examples, mutations or deletions in metabolic enzymes can cause abnormal accumulation of intracellular metabolites that lead to altered protein function via allosteric regulation. In addition, genes encoding metabolic enzymes can also undergo amplification via copy number alteration. For example, amplification of phosphoglycerate dehydrogenase (PHGDH), the first rate-limiting enzyme in the serine synthesis pathway, is observed in various cancer types, including a subset of melanomas and triple-negative breast cancers [71, 72]. Serine is a proteinogenic amino acid that is also a significant source of one-carbon units for the folate cycle, which is required for de novo synthesis of purines and thymidine, and in some settings has also been shown to contribute to NADPH production [73]. Some cancer cells require PHGDH amplification to obtain sufficient serine to support cancer cell proliferation [71, 72].
Cell lineage is also recognized as an important determinant of cancer cell phenotypes, including metabolic activity, proliferative index, metastatic potential, and response to therapy. As organs and tissues are formed during development, genetic programs are executed that determine cell fate. These developmental programs ultimately drive progenitor cells to become specialized cell types with unique metabolic activities designed to support the function of a given tissue type. During carcinogenesis, metabolic activity is rewired to support cancer growth, yet how cancer and associated stromal cells adapt their metabolism depends on the tissue and cell of origin of the cancer [74–76]. Cancer cells can maintain specific metabolic signatures, analogous to how cancer cells maintain lineage markers that reflect the tissue/cell type of origin [77]. For example, it has been described that primary prostate cancer and prostate cancer metastases maintain increased citrate production, a metabolite that secreted in high quantities by normal prostate glands [78].
In addition to metabolic alterations that are intrinsic to cancer cells, it is also increasingly recognized that metabolic alterations occur in other cells within the tumor microenvironment; these changes can also contribute to cancer progression (reviewed in [79, 80]). Furthermore, cancer cell heterogeneity and plasticity can also manifest as metabolic heterogeneity. Several studies have attempted to characterize metabolic heterogeneity in cancers [81–83]. However, this remains a challenging area of research given the inherent limitations of assessing metabolism at the single cell level.
Immuno-oncology is an active area of research, which has increasing intersection with the field of metabolism. Significant energy goes into the production of cytokines, chemokines, and immune mediators, immune cell activation, and expansion, suggesting that changes in energy availability can affect the immune response [84, 85]. Clinical studies have shown a clear connection between autoimmune diseases, such as rheumatoid arthritis and systemic lupus erythematosus, and immune cell metabolism [86, 87]. Immune cells rely on specific metabolic pathways for activation, and manipulating these metabolic dependencies could present a unique opportunity to target diseases. For instance, activation of the glycolytic pathway in CD4 T cells promotes an inflammatory phenotype, while increased fatty acid oxidation skews them towards a regulatory phenotype [88]. B cell and T cell receptors directly activate transcription factors, such as c-MYC, HIF1α, PI3K, mTOR, and FOXO1, which play a critical role in immune cell metabolism and subsequent immune response [89–91]. Similarly, M1 macrophages rely on glycolysis and glutaminolysis pathways, while M2 macrophages prefer the oxidative phosphorylation pathway to meet the high demand for energy production during the activation phase [92]. Of clinical interest, it has been shown that blocking glutamine metabolism can enhance anti-tumor immune responses [93].
In summary, there are multiple and often complex interactions between metabolic pathways and signaling pathways that together result in metabolic reprogramming, a fundamental hallmark of cancer. Furthermore, both intrinsic (genomic/epigenomic alterations) and extrinsic factors (nutrients, drugs, hormones, and interactions with stromal cells, extracellular matrix, and the immune system) contribute to the metabolic reprogramming of cancer cells. For further information the reader is referred to recent reviews of these topics [2, 79].
Cancer Therapies that Target Metabolism
While multiple mechanisms can contribute to metabolic reprograming in cancer, regardless of the underlying mechanism(s), the altered activity of metabolic enzymes provides an opportunity for therapeutic intervention if such activity is required for tumor maintenance (often referred to as a metabolic dependency) and if inhibition of the activity can be tolerated by host metabolism. In this section we provide select examples of the major classes of cancer drugs that target metabolic enzymes (Table 1). For a more comprehensive discussion, the reader is referred to other excellent reviews of this topic [4, 94–96].
Table 1.
Cancer therapies/drugs that target metabolic pathways. Key targets of nucleoside analogs are shown; however, most nucleoside analogs inhibit multiple nucleic acid and DNA synthesis/repair enzymes including DNA polymerase.
| Metabolic Pathway | Target | Drug | Stage of Development | References |
|---|---|---|---|---|
| Nucleic acid synthesis | Thymidylate synthase (TS) | 5-fluorouracil Capecitabine Pemetrexed Raltitrexed |
Approved | [162–165] |
| Dihydrofolate reductase (DHFR) | Methotrexate Pemetrexed |
Approved | [164, 166, 167] | |
| Glycinamide ribonucleotide formyltransferase (GARFT) | Pemetrexed |
Approved | [164] | |
| dihydroorotate dehydrogenase (DHODH) | Brequinar Leflunomide |
Phase I/II | [168] | |
| Ribonucleotide reductase (RNR) | Gemcitabine Clofarabine Fludarabine Cladribine Cytarabine |
Approved | [169–173] | |
| 5-phosphoribosyl-1-pyro-phosphatase (PRPP) amidotransferase | Mercaptopurine Thioguanine |
Approved | [174–176] | |
| Glycolysis | GLUT1 | WZB117 BAY-876 |
Pre-clinical | [116, 177] |
| Hexokinase | 2-deoxyglucose |
Phase I/II | [106] | |
| Pyruvate Kinase M2 (PKM2) | TEPP-46 | Pre-clinical | [111] | |
| Lactate dehydrogenase A (LDHA) | Quinoline 3-sulfonamides FX11 PSTMB |
Pre-clinical | [117, 178, 179] | |
| Monocarboxylate transporter 1 (MCT1) | AZD3965 | Phase I | [119] | |
| Glutamine metabolism | Glutaminase 1 (GLS1) | CB-839 IPN60090 |
Phase I/II | [123, 124, 126] |
| ASCT2 (SLC1A5) | GPNA | Pre-clinical | [128, 180] | |
| multiple targets | JHU-083 | Pre-clinical | [93, 181] | |
| Amino acid transport and biosynthesis | Phosphoglycerate dehydrogenase (PHGDH) | CBR-5884 NCT-503 |
Pre-clinical | [131–133] |
| Indoleamine-2,3-dioxygenase-1 (IDO1) | Epacadostat Indoximod |
Phase III | [137] | |
| Circulating asparagine | L-Asparaginase | Approved | [105] | |
| Large neutral amino acid transporter (LAT1) | JPH203 | Pre-clinical | [129, 182] | |
| Mitochondrial metabolism | Pyruvate dehydrogenase (PDH), α-ketoglutarate dehydrogenase | CPI-613 | Phase II | [183] |
| Electron Transport Chain Complex 1 | Metformin IACS-010759 |
Phase I-III | [184, 185] | |
| Lipid metabolism | ATP-citrate lyase (ACLY) | SB-204990 | Pre-clinical | [142, 143] |
| acetyl-CoA carboxylase (ACC) | Soraphen-A | Pre-clinical | [144, 145] | |
| fatty acid synthase (FASN) | TVB-2640 | Phase II | [141] | |
| Enzymes mutated in cancer | Mutant isocitrate dehydrogenase 1 (IDH1) | AG-120 BAY1436032 LY3410738 FT-2102 |
Phase I-III | [156, 157] |
| Mutant isocitrate dehydrogenase 2 (IDH2) | AG-221 | Phase III | [158] |
From the earliest days of cancer research, the increased proliferative index of cancer cells was recognized as a metabolic vulnerability. The observation that rapidly proliferating cells can be killed by agents that interfere with DNA replication led to the development of the first chemotherapeutic agents, including small molecules with direct genotoxic effects such as the nitrogen mustards and drugs targeting nucleotide synthesis [97, 98]. Due to their structural similarity to native metabolites, this latter class, termed antimetabolites, saw early successes that led to the rapid expansion of drugs targeting enzymes involved in nucleotide metabolism. One of the earliest drugs to treat cancer was methotrexate, an anti-folate drug that inhibits thymidine synthesis [99]. Similarly, the related folate analog, aminopterin, inhibits one-carbon metabolism necessary for de novo nucleotide synthesis and was found to be effective in children with acute lymphoblastic leukemia [100]. Early clinical successes with these agents paved the way for rapid development of additional small molecule inhibitors of nucleotide synthesis enzymes, many of which continue to form the backbone of multiagent chemotherapy regimens, such as inhibitors of dihydrofolate reductase and other folate-utilizing enzymes, thymidylate synthase, phosphoribosyl pyrophosphate amidotransferase, ribonucleotide reductase, and other enzymes involved in purine and pyrimidine synthesis and salvage (Table 1 and reviewed in [101–103]).
Apart from targeting nucleotide and DNA synthesis, early cancer treatments have also targeted other metabolic pathways. For instance, it was discovered that acute lymphoblastic leukemia cells rely on exogenous asparagine for growth [104], which led to the use of the bacterial enzyme L-asparaginase to limit the availability of asparagine for leukemia cell growth [105]. More recently, efforts have focused on developing agents to deplete other amino acids and to target central metabolic pathways aberrantly regulated in cancer cells, including glycolysis, the TCA cycle, and lipogenesis. Many of these agents are still in preclinical stages; however, some are now undergoing evaluation in clinical trials (Table 1).
Targeting central metabolic pathways always raises questions about therapeutic window, as alteration of systemic metabolism can have harmful effects. For example, targeting glycolysis directly has proven to be challenging due to the low therapeutic index of glucose uptake inhibitors such as 2-deoxyglucose (2-DG), which affects glucose uptake in both cancer and normal cells [106, 107]. However, the recognition that cancer cells have altered regulation of central metabolic pathways led to renewed interest in this field [108]. An example of this is pyruvate kinase (PK), which catalyzes the last step in the glycolytic pathway. Different tissues express different isoforms of PK [109]. Most cancer cells express a PK isoform (PKM2) that is different from the one expressed in erythrocytes (PKR), liver (PKL), myocytes (PKM1), and brain (PKM1). Multiple mechanisms have been proposed for why PKM2 is advantageous to cancer cells [110]. Preclinical studies have shown that drugs targeting PKM2 can be effective in some cancer types, and early trials of those drugs for other indications have proven safe in patients [111–114]. Other drugs targeting glucose metabolism, such as GLUT1 inhibitors (WZB117 and BAY876), GLUT4 inhibitors (Silibinin and Ritonavir), GLUT2 inhibitor (Quercetin) are in various stages of clinical development [94, 115, 116]. In cancer cells, a significant proportion of glucose is metabolized to lactate and secreted by monocarboxylate transporters (MTCs) present on the plasma membrane. Inhibitors of lactate dehydrogenase A (LDHA) such as Quinoline, 3-sulfonamides, FX11, and PSTMB, are being investigated in preclinical settings [117, 118]. An MCT1 inhibitor, AZD3965, is in early phase clinical trials for patients with solid tumors, diffuse large B cell lymphoma, and Burkitt lymphoma [119]. GAPDH is another target with a therapeutic window determined by the extent of the Warburg effect on glycolysis [120, 121].
Amino acids are also important nutrients that support anabolic metabolism in cancer cells. Glutamine is the most abundant amino acid in the blood, and glutamine dependence has been observed in many cancer cell lines [122]. Although glutamine is a non-essential amino acid, it is an important nitrogen donor for the biosynthesis of diverse compounds, such as glutathione, hexosamine, nucleotides, fatty acids, and non-essential amino acids. Furthermore, glutamine carbon can feed directly into the TCA cycle. In fact, a glutamine antagonist, JHU083, has shown a potent antitumor response in combination with immune checkpoint blockade therapy in an animal model [93]. One route of glutamine catabolism to supply carbon to cells involves the enzyme glutaminase. Drugs targeting glutaminase, such as CB-839 and IPN60090, have been effective in some preclinical models and are now in trials for various malignancies [123–126]. Interestingly, targeting glutaminase appears to cooperate in preclinical settings with immune therapies, including CAR-T cell therapies [127]. In addition, for several glutamine transporters, such as SLC1A5, LAT1, and SLC6A14, inhibition showed promising results in preclinical settings and are being actively pursued for future clinical applications [128, 129]. Serine is another proteinogenic amino acid essential for nucleotide biosynthesis via its role as a donor of one-carbon units for the folate cycle [130]. As previously discussed, PHGDH, the enzyme that catalyzes the first step in the serine biosynthesis pathway, is amplified in some cancers, and in this context is essential for proliferation [71, 72]. Small molecule drugs targeting PHGDH are effective in some preclinical models, including inhibition of brain metastases [131–134]. Indoleamine-2,3-dioxygenase-1 (IDO1) is the critical enzyme in tryptophan catabolism, and elevated levels of IDO1 has been associated with poor outcomes in patients with cervical cancer and glioblastoma multiforme [135, 136]. The inhibitors of IDO1 (epacadostat and indoximod) are in clinical trials in combination with other anti-cancer agents [137].
Compared to most normal tissues, cancer cells have an increased demand for fatty acids to generate lipid membranes and precursors for signaling molecules. Fatty acids can be acquired exogenously through diet or synthesized endogenously from glucose, glutamine, or acetate [138]. Lipogenic enzymes are upregulated in many cancer cells, and de novo fatty acid synthesis has been viewed as a potential therapeutic target in cancer cells [139, 140]. Three lipogenic enzymes have been a particular focus of drug development efforts, including ATP-citrate lyase (ACLY), acetyl-CoA carboxylase (ACC), and fatty acid synthase (FASN) [141–145]. The success of these agents in the clinic will likely depend on identifying tumors that are unable to take up sufficient lipids from their environment and therefore require increased de novo fatty acid synthesis for survival. Although aberrant fatty acid metabolism helps cancer cells satisfy higher energy demand, it is also associated with increased lipotoxicity. Cancer cells need to maintain a proper ratio of saturated to unsaturated fatty acids to avoid mitochondrial dysfunction, excess reactive oxygen species, and endoplasmic reticulum stress [146, 147]. To overcome lipotoxicity, cancer cells overexpress different isoforms of stearoyl-CoA desaturases (SCDs), which converts saturated to monounsaturated fatty acids. SCD1 is overexpressed in various cancer types, and SCD1 inhibitors are under investigation in preclinical studies [148–152].
Although most drugs that target metabolism do not discriminate between cancer and normal cells, in the case of IDH-mutant cancers, there is a unique opportunity to target cancer cells selectively. Indeed, several drugs have been developed that selectively target the mutated enzyme [153–155]. This strategy has been effective in treating IDH-mutated AML [156–158]. IDH inhibitors are also in clinical trials for IDH-mutated solid tumors (glioma, chondrosarcoma, and intrahepatic cholangiocarcinoma); however, preclinical studies have suggested that IDH inhibitors may be less effective in solid tumors and it has been suggested that this may be due to mutant IDH activity being a driver of cancer initiation by altering epigenetic signatures, but may be less important in tumor maintenance at later stages of tumor progression [4, 159–161]. It is worth pointing out that across many solid tumor types, drugs that target metabolic enzymes are effective in slowing tumor growth in preclinical models, yet tumor regression is rarely observed, indicating that some of these agents may be most useful as part of multiagent regimens or as maintenance therapy.
Systemic Metabolic Effects of Cancer
Weight loss is a common presenting symptom in cancer patients, and more than half of patients with cancer experience anorexia at baseline, the etiology of which remains poorly understood [186–188]. Cancer cachexia is a severe form of wasting characterized by loss of lean body mass or sarcopenia with or without loss of fat mass [189]. This contrasts with starvation, where liver and fat mass are lost while lean body mass is initially preserved. Numerous circulating factors, including cytokines, neuropeptides, eicosanoids, and tumor-derived proteins, have been implicated in cachexia’s pathogenesis, and the underlying etiology is likely multifactorial [190–192]. Increased resting energy expenditure has been observed in some cancer patients, and prevalence may depend on cancer type, indicating that hypermetabolism may be a feature of some cancers that contributes to the wasting phenotype [193, 194].
Although anorexia likely contributes to cancer-associated weight loss and cachexia, hyperalimentation with parenteral nutrition does not improve treatment outcomes and only partially reverses the wasting phenotype indicating that the pathophysiology of cachexia is more complex than simply reduced calorie intake secondary to anorexia [195, 196]. In a randomized clinical trial of patients with small-cell lung cancer, parenteral nutrition temporarily improved body fat yet had no effect on lean body mass or survival [197]. Cachexia is an early presenting sign in many patients with pancreatic ductal adenocarcinoma (PDAC), and decreased exocrine function has been implicated as a contributory factor [198–200]. Yet, surprisingly, despite reversing some tissue wasting, replacement of pancreatic enzymes does not improve survival in a murine PDAC model [199]. Furthermore, in PDAC patients, cachexia is not necessarily associated with worse survival [199]. Nevertheless, pre-treatment weight loss is a poor prognostic factor in several cancers [188] and the systemic metabolic effects of cancer are heterogeneous with much left to understand.
Cancer cachexia and weight loss are also associated with systemic metabolic changes, including derangements in glucose, lipid, and protein metabolism [201–204]. These systemic metabolic alterations have been attributed to changes in host metabolism induced by the tumor rather than the metabolic activity of the tumor itself [205]. This contrasts with animal models where the tumor to host mass ratio is often large, and tumor metabolism can cause direct systemic metabolic changes [206]. For the most part, early human studies failed to show significant metabolic changes in patients with early-stage localized cancer leading to the assumption that the metabolic changes were not due to cancer per se but rather a manifestation of cancer-associated wasting phenotype [205]. However, as will be discussed in further detail below, technological advances resulting in improved metabolite detection and resolution have led to the discovery of distinct systemic metabolic signatures in cancer patients even at early stages and, in some cases, even before the disease becomes clinically apparent [207].
Metabolic Effects of Cancer Therapy
In addition to the aforementioned cancer-induced systemic metabolic changes, treatment of cancer with surgery, radiation, systemic or hormonal therapy causes acute and long-term side effects that can also affect metabolism. Not surprisingly, side effects involving the digestive system account for the majority of acute treatment-related toxicities affecting metabolism [208]. Malnutrition and weight loss may result from nausea, vomiting, diarrhea, mucositis, dysgeusia, which are common in patients receiving treatment for head and neck and gastrointestinal malignancies. Resting energy expenditure initially decreases in patients undergoing chemoradiotherapy for head and neck cancer and increases toward the end of treatment [209]. It has been suggested that the increased energy expenditure at the end of therapy is related to stress caused by the cumulative effects of chemoradiotherapy [209]. A systematic review of the literature on energy metabolism in patients treated with chemotherapy for all cancer types showed no universal impact of chemotherapy on energy expenditure but suggested that patients undergoing chemotherapy become hypometabolic during treatment [210]. Most studies have used indirect calorimetry, anthropometry, and routine laboratory tests to assess metabolic status. There is comparatively little metabolomic data on changes that occur during cancer therapy. A small study using stable isotope tracers showed no significant difference in glucose, lipid, or protein metabolism in patients undergoing radiotherapy for head and neck and lung cancer [211]. While combined modality therapy (surgery, radiation, and chemotherapy) for soft tissue sarcoma had significant acute nutritional effects, patients who were disease-free following treatment showed minimal nutritional morbidity [212].
Although acute metabolic side effects of aggressive cancer therapy can be pronounced, fortunately, long-term side effects tend to be more subtle. Incidence and severity are dependent on treatment modality and interval since treatment [213]. Other than long term sequelae of decreased endocrine/exocrine function following surgery or radiation involving the pituitary, thyroid, adrenal, pancreas, and gonads, there is relatively little data on the long-term metabolic effects of these modalities [214]. Nevertheless, even low doses of total body radiation in the pediatric population significantly increase the risk of long-term endocrine disturbance and metabolic abnormalities [215]. In contrast to local therapies, systemic therapy carries a higher risk of long-term metabolic derangements. The long-term metabolic effects of hormonal therapy in breast and prostate cancer patients are well documented, including chronic effects on mineral and lipid metabolism [216–218]. Additionally, prior chemotherapy is associated with chronic weight gain in breast cancer survivors, with the highest risk in premenopausal women [219]. Long-term metabolic effects of therapy in other cancers and potential pathophysiologic mechanisms have been reviewed [213]. Unfortunately, much of the prior data is insufficient to establish causation. Broader use of metabolomic analysis in clinical trials and surveillance protocols could provide a better understanding of pathologic mechanisms and identify opportunities for intervention.
The Role of Diet and Lifestyle Factors in Carcinogenesis and Response to Treatment
Metabolism has also been the focus of research aimed at identifying risk factors for cancer. Epidemiologic studies demonstrate a positive correlation between cancer incidence and deleterious metabolic states, including obesity and diabetes [220–222]. Additionally, dietary factors and physical inactivity have been implicated [223–225]. Furthermore, in the post-diagnosis setting, the relevance of these factors to disease progression, recurrence risk, and mortality has been demonstrated for some cancers [226–233]. Collectively, these data have led to the adoption of guidelines on healthy food choices and physical activity to reduce cancer risk [234].
Conceptually, dietary composition can affect circulating nutrients and metabolic hormones, which may directly affect metabolism within tumor cells [235, 236]. While the effect of diet, exercise, and systemic metabolic status on cancer initiation can be challenging to model experimentally, the impact of diet on tumor progression has been extensively evaluated in mouse models of various cancers, including breast, prostate, lung, pancreas, liver, and others [237]. In line with human data, laboratory studies in rodents generally support the notion that in some cases caloric restriction and ketogenic diet can slow tumor growth, while a high-fat diet can promote tumor progression [238–242]. However, as some studies show, this is a gross oversimplification, and the effect of diet on tumor progression likely also depends on tumor genetics and the tumor microenvironment [243, 244]. Furthermore, mechanistically it has been difficult to ascertain to what extent individual nutrients directly affect tumor growth instead of indirect effects on growth factor signaling. For example, insulin-like growth factor (IGF) signaling has been implicated in many caloric restriction studies, yet direct activation of IGF only partially reverses the effect of caloric restriction on tumor growth [245–247]. Since manipulating one dietary factor or nutrient can affect many others, future studies will need to address the effects of diet and exercise on the metabolomic landscape both systemically and ideally within the tumor microenvironment.
In addition to impacting carcinogenesis, diet and lifestyle factors may also affect cancer therapy [248–250]. A low calorie, low protein diet improved objective tumor response in patients receiving neoadjuvant chemotherapy for breast cancer [251]. In animal models, caloric restriction and other dietary perturbations have been shown to alter the efficacy of chemotherapy and radiotherapy via metabolite-specific effects on IGF signaling, oxidative stress, and nucleotide metabolism [252–254]. While data from human observational studies and mechanistic studies in laboratory animals are compelling, results from human interventional trials have, for the most part, been disappointing and highlight the limitations of a reductionist approach. It has been suggested that a more unified approach combining genomics, metabolomics, and biomarkers may be more effective at identifying modifiable risk factors in the context of heterogeneous dietary patterns and tumor characteristics [255]. A conceptual framework for investigating dietary effects on tumor metabolism has been proposed [256]. The key elements build on clinical and laboratory observations that dietary manipulation can alter nutrients in the tumor environment, and this, in turn can impact tumor metabolism, growth, progression, and response to therapy. Indeed, depletion of specific amino acids can slow growth of some tumors [252, 257–261], and if we extend this framework to account for complex interactions between diet, exercise, stress, inflammation, and the microbiome on micronutrients and growth activating/inhibitory signals in the tumor environment, it is clear that a multi-omics approach is needed to identify actionable diet and lifestyle factors that can impact response to cancer therapy.
Defining Metabolomics
Historically, human disease has been studied using a reductionist approach to separate and identify individual factors that cause or contribute to a pathologic state. In contrast, systems biology seeks to understand complex biological systems by considering the sum of its molecular constituents and how they interact to define a phenotype [262]. At the heart of systems biology is an ability to measure many aspects of cell state, which has been facilitated by advances in genomics, transcriptomics, proteomics, and metabolomics.
In concept, metabolomics stands apart from the other “-omics” in that it provides a functional readout of metabolic processes and therefore is a direct assessment of phenotype (Figure 2). That is to say, metabolomics, if interpreted appropriately, can provide a readout of the sum of alterations occurring at the DNA, RNA, and protein level, and in some cases may be the most sensitive way to identify pathologic variants since even small changes in protein expression or structure can lead to significant changes in protein activity and metabolite levels [263]. Conversely, metabolites can alter protein activity and thereby affect nearly every biological process, including DNA replication, RNA transcription, and translation (Figure 2). The term “epimetabolites” has been used to describe a subset of metabolites that function as active biomarkers and are involved in diverse biological functions including epigenetic regulation, tumorigenesis, cancer cell invasion, cancer stem cell pluripotency, insulin sensitivity, and other cellular processes [264]. Furthermore, metabolomics also considers alterations in the tumor environment, including therapeutic interventions that can exert selective pressures on tumor subclones and thus shape the genome, transcriptome, and proteome [265]. Understanding the metabolic milieu therefore has important implications for interpreting genomic, transcriptomic, and proteomic data.
Figure 2.
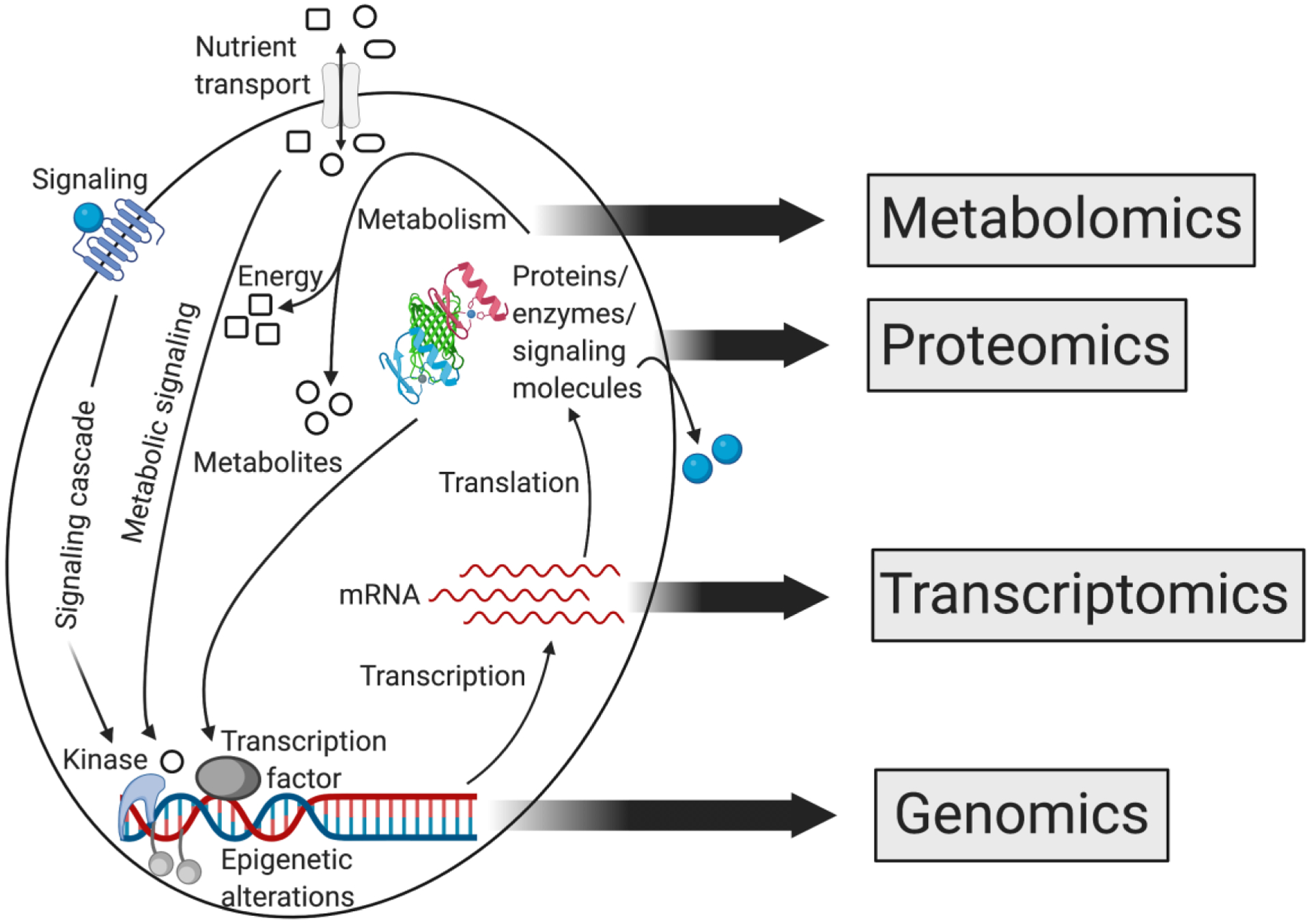
The relationship between ”-omics” approaches of systems biology. Cancer is caused by changes at the genomic level that result in altered RNA transcription, protein expression, and protein function. The metabolome provides a functional readout of these upstream changes. In turn, individual metabolites affect protein activity and thereby alter RNA transcription and DNA replication.
In practice, metabolomics is defined as the analysis of small molecule metabolites (1500 Daltons or less and non-peptide) in a biological specimen [266]. It involves the simultaneous identification of hundreds to thousands of chemicals based in part on the chemical properties and/or weight of atoms within a molecule (described further in the subsequent section). This contrasts with standard clinical measurement of metabolites, such as glucose and urea, which rely on identifying chemicals based on enzymatic reactions, and requires a separate test for each metabolite. The field of metabolomics has benefitted greatly from relatively recent technological advances in instrumentation, resulting in more affordable instruments with a smaller footprint. In addition, vendors have focused on delivering more user-friendly acquisition software and computational analysis tools. Validated datasets have made it easier to quickly and accurately process data. In addition there are many open-source software packages available for analyzing metabolomics data [267]. Nevertheless, appropriate experience and training of staff can still be a bottleneck for smaller academic/clinical laboratories.
As discussed in the next section, multiple methods exist for metabolomics analysis, each with its own advantages and disadvantages; however, the first decision point in selecting the appropriate method is whether an untargeted or targeted approach is desired. Generally, untargeted metabolomics is used for hypothesis generation and is used extensively in biomarker discovery, whereas a targeted approach (that is, defining the metabolites that will be measured prior to performing the analysis) is used when testing a specific hypothesis or in validation and implementation stages [268]. In genomics, transcriptomics, and proteomics, the objective is to identify the macromolecular structure from a sequence of chemical constituents (nucleotides, amino acids) that are well defined and relatively limited in diversity. Since the chemical composition is constrained, a complete analysis is obtained by applying a single protocol. In contrast, metabolomics deals with a chemically diverse and complex set of molecules. Because lipids, sugars, organic acids, and other polar molecules have a wide range of physical characteristics, multiple methods for sample preparation and data acquisition are needed for their analysis. Furthermore, metabolites that are annotated by software in untargeted experiments require subsequent validation with a chemical standard using a targeted method. Consequently, analyzing differences between groups detected by untargeted metabolomics requires considerable expertise and effort, particularly if it is desirable to assign identities to each species measured. Therefore, the usefulness of untargeted metabolomics in aiding biological understanding and interpretation is limited by the ability to identify unknown metabolites [268].
Methodology and Instrumentation
In this section we introduce the major technologies that are used for metabolomics (Figure 3). We focus primarily on methods involving mass spectrometry (MS) and nuclear magnetic resonance (NMR), which can both be used for compound detection in any biofluid or cell/tissue extract in the liquid phase. NMR can also be used with solid-phase samples such as cell membranes or even intact cells and tissues. Other techniques involving analysis of metabolites in situ, such as matrix-assisted laser desorption/ionization mass spectrometric imaging (MALDI-MSI), or NMR-based in vivo imaging, such as magnetic resonance spectroscopic imaging (MRSI), are more limited in their application and have been reviewed elsewhere [269, 270].
Figure 3.
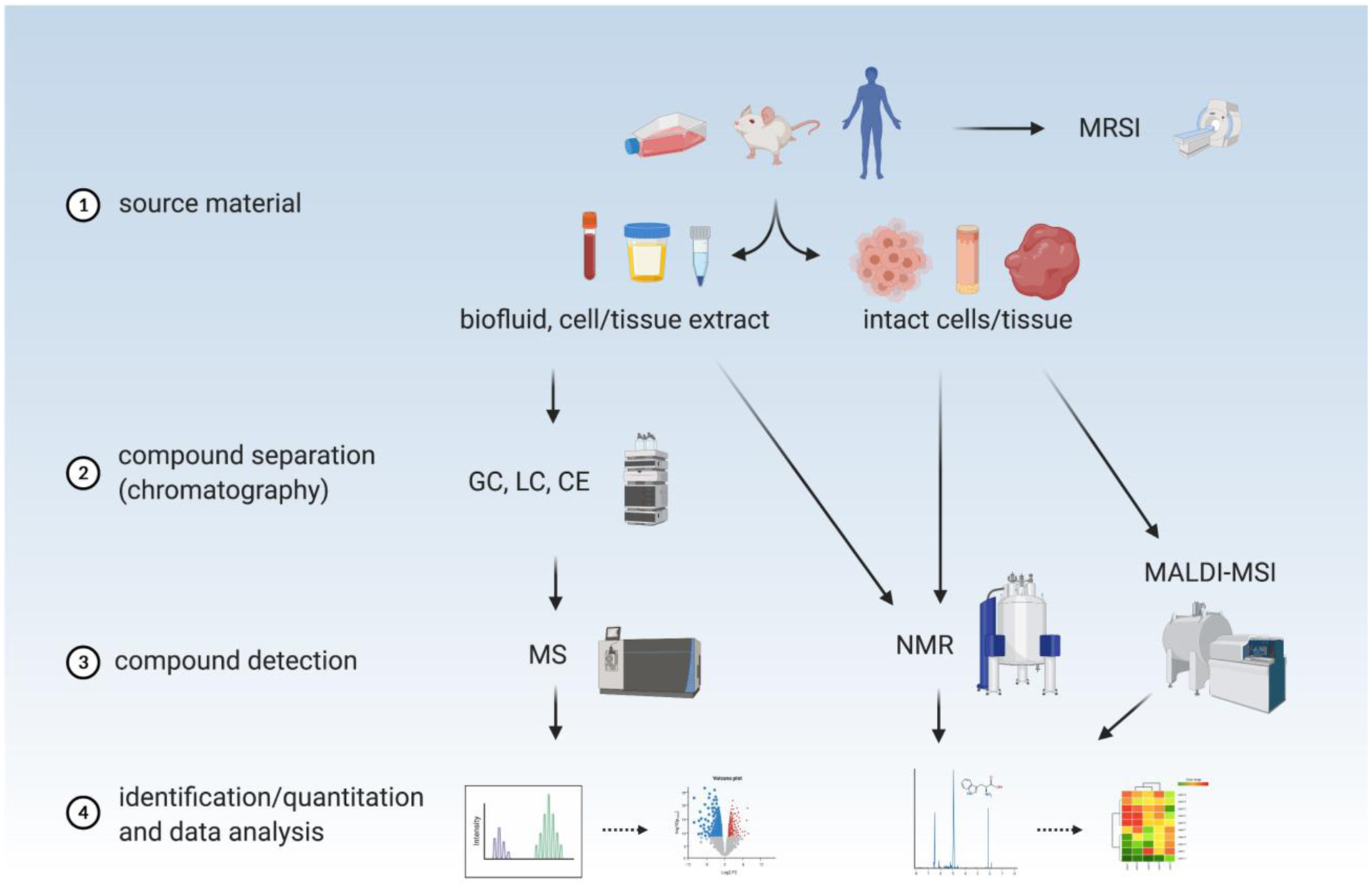
The optimal metabolomics workflow depends on source material and application. Various technologies and methods can be used to acquire raw data, which then provides the starting point for computational analysis. See text for details. MRSI magnetic resonance spectral imaging, GC gas chromatography, LC liquid chromatography, CE capillary electrophoresis, MS mass spectrometry, NMR nuclear magnetic resonance, MALDI-MSI matrix-assisted laser desorption/ionization mass spectrometric imaging.
NMR relies on measuring chemical properties of specific atoms in a molecule. NMR is a versatile method as it can be used with biospecimens in liquid, solid, or gas phase without prior processing. A sample is subjected to a strong magnetic field and then pulsed with radiofrequency waves. The energy from radiofrequency radiation is used to transiently excite certain nuclei in a molecule (such as 1H, 13C, 15N or 31P), causing them to flip their spin state when aligned in a strong magnetic field. As these nuclei relax, they produce a characteristic spectroscopic pattern (chemical shift) that reflects the type, location, and electromagnetic environment of excited atoms in a given molecule. Material is not consumed during NMR spectroscopy and can therefore be used for subsequent analysis. The major drawback is the large footprint of NMR instruments and relatively low sensitivity (micromolar) compared with mass spectrometry (nanomolar) [271]. Since most metabolites are relatively low abundance in biological material, the diversity of chemical species that can be practically measured by NMR is also lower than for mass spectrometry (MS)-based techniques. NMR is capable of resolving several hundred unique metabolites, while MS can be used to identify thousands of features with unique masses.
MS is the most widely used analytical technique in metabolomics. It relies on determining the ratio of mass to charge (m/z) of a molecule and/or its characteristic fragments. A sample in the liquid or gas phase is injected into the mass spectrometer where metabolites become ionized and then separated by their m/z. Sample material is entirely consumed during this process. MS is typically coupled with an initial chromatographic stage, which increases the resolution of isobaric (same mass) compounds and improves the detection of less abundant species by reducing signal suppression by more abundant species. When coupled with gas chromatography (GC-MS), the technique is highly sensitive, but generally requires initial chemical modification (derivatization) of metabolites to increase their volatility and make them suitable for separation in the gas phase. GC-MS can be used to analyze most polar, nonpolar, organic, and inorganic compounds; however, phosphorus-containing compounds (many sugars, lipids, and nucleotides) are challenging to detect via GC-MS due to difficulty generating volatile derivatives. In contrast to GC, liquid chromatography (LC) involves passing a liquid solution of metabolites over a solid-phase column during which they are separated based on their chemical affinity for the solid phase. The chemical composition of the liquid phase is changed until all metabolites are eluted from the column. LC-MS does not typically require chemical modification of analytes and is well suited for the analysis of biofluids and phosphorus-containing compounds; however, it is necessary to use different solid-phase columns and different mobile-phase compositions for the optimal separation of compounds from different chemical classes. For example, lipidomics, which focuses on the lipid subset of the metabolome, involves chromatographic techniques that are fundamentally different from techniques used to analyze polar metabolites such as sugars or organic acids [272]. Nevertheless, given the combination of ease of use with biofluids, high sensitivity, and broad range of metabolites that can be measured, LC-MS is the most comprehensive and widely-used analytical method associated with metabolomics. Capillary electrophoresis-mass spectrometry (CE-MS) is a related technique in which charged compounds are separated based on electrophoretic mobility (charge-to-size ratio) and can therefore be useful to detect intrinsically charged metabolites (e.g., choline) present in low abundance and very small sample volumes [273].
Although compound separation prior to injection into a mass spectrometer significantly increases sensitivity to detect low abundance metabolites, the time needed for sufficient chromatographic separation of a complex biological sample can limit the number of samples that can be analyzed in a finite period of time. The addition of system suitability and quality control samples also increases the run time for untargeted metabolomics experiments [274]. For example, a batch of 50 samples could take several days of instrument time. To increase throughput, biofluids can also be directly injected into a mass spectrometer without prior chromatographic separation. This technique, referred to as flow injection mass spectrometry (FI-MS), substantially increases throughput, allowing hundreds of samples to be run in a single day. The downside of FI-MS is that less abundant species can be missed; therefore, this technique is best suited for high-throughput, low-sensitivity analyses such as metabolic fingerprinting, where the goal is to capture a snapshot of total metabolite content rather than identify individual metabolites. It can also be used as a screening tool for both untargeted metabolomics (when used together with optimized m/z scan ranges [275, 276]), and -lipidomics (termed “shot-gun lipidomics” [277]) . However, ultimately these methods must be combined with traditional LC-MS approaches if identification of the detected features is required. This is especially important in lipidomics, since shot-gun lipidomics cannot separate isobaric/isomeric lipid species, of which there are many.
Source Material
Metabolomics can be performed on a wide range of biological materials, including cells and tissues cultured in the laboratory, specimens collected from laboratory animals, and either freshly obtained or appropriately archived clinical specimens, including tumors and biofluids. The quantity of starting material depends on the technique used. For NMR, samples volumes are typically in the 0.1 to 0.5 mL range for liquid samples. MS-based methods are more sensitive, with 10 to 1000-fold lower limits of detection compared to NMR. Thus, as little as a few microliters or milligrams of starting material can be sufficient to detect many metabolites.
In the laboratory, cells and tissues can be studied under controlled conditions where levels of nutrients in the environment can be manipulated, and isotope labeled nutrients can be used to assess nutrient fate and determine flux through metabolic pathways [278–280]. Samples can be collected at predefined time points and rapidly processed. The acquisition, handling and storage of clinical specimens are more complex and require careful planning and coordination between clinical and laboratory staff; however, with careful protocols even stable isotope-labeled metabolite tracing can be accomplished in cancer patients [83, 281]. The most common types of clinical material that have been used for metabolomics analysis include blood, plasma, serum, urine, and extracts of biopsy or surgical specimens, examples of which are discussed in a following section. However, many other types of biofluids have also been used, including sputum, bronchial washings, saliva, sweat, tears, cerebrospinal fluid, pleural or ascitic effusions, fecal water, bile, breast milk, amniotic fluid, seminal plasma, expressed prostatic secretions, and others [266, 282–286]. Regardless of the material used, the method of collection, storage, and processing must be appropriately selected for the desired analysis. Many chemical species are labile or undergo rapid chemical modification by molecular oxygen or metabolic enzymes. Where applicable, appropriate quality controls are needed to ensure that chemical decomposition is minimized. Guidelines for reporting biospecimen source, collection, and processing have been set forth by the Metabolomics Society [287].
Clinical Implementation of Metabolomics
Though both untargeted and targeted metabolomics have huge potential in biomarker discovery and hypothesis testing in the translational setting, several challenges still need to be overcome before metabolomics can become widespread in clinical research and practice. As described above, multiple complimentary methods are required to cover the entire metabolome. Often, this requires multiple instrumentation platforms, which may not be available in many academic and clinical labs. Furthermore, there are multiple software packages for data processing and analysis, particularly in untargeted metabolomics. Different peak picking algorithms, for example, can yield slightly different results. Analyzing large metabolomics datasets also requires robust experimental design, which facilitates appropriate statistical analysis. At a minimum, a successful metabolomics research study therefore requires the combined efforts and expertise of analytical chemists, statisticians, and biologists. Furthermore, given the multiple methods and instrumentation platforms involved, as well as the various data processing algorithms, it is a logistical challenge to apply “industry standards” to the field in a way that is necessary in a clinical laboratory setting. Another major challenge for the clinical implementation of metabolomics is standardization of data analysis and reporting between institutions. Currently most metabolomics studies yield relative quantification. In order for standardization to happen across platforms absolute quantification would be required. Additional challenges and suggestions for new strategies have been reviewed extensively in [288]. In 2007 the Metabolomics Society launched a standards initiative to define the minimum reporting standards required for metabolomics data [242]. However, many published datasets still fall short of these minimum standards due to lack of consensus across laboratories.
Clinical Applications
Metabolomics seeks to capitalize on the metabolic signature of cancer to assess disease risk or for earlier cancer detection, diagnosis of specific disease subsets, or for treatment monitoring. Metabolomics in principle may also help inform the rational selection of targeted therapies to match the metabolic dependencies of cancer. In this final section we discuss these clinical applications of metabolomics and provide select examples to illustrate how the metabolomics field has opened new opportunities in cancer research and is beginning to impact diagnosis and treatment.
Identifying Cancer Risk Factors
It is widely accepted that tumorigenesis involves sequential accumulation of genetic mutations that ultimately give rise to malignancy [289]. While some oncogenic mutations and predisposing polymorphisms are germline-encoded, environmental factors promote the process of tumorigenesis by both inducing somatic DNA alterations and selecting for transformed cells. Small molecule carcinogens and ultraviolet or ionizing radiation cause DNA alterations by directly interacting with DNA. In contrast, inflammatory and metabolic factors contribute indirectly to tumorigenesis by inducing reactive oxygen species and creating an environment in which oncogenic mutations and epigenetic alterations are selected. In addition, altered metabolism affects availability of substrates used to modify chromatin thereby influencing chromatin dynamics and epigenetic changes that drive tumorigenesis [290]. Untargeted metabolomics on pre-diagnostic serum can uncover metabolic risk factors, but requires access to material from large population-based cohort studies. Several examples are discussed in this section.
Early epidemiology studies focused on the contribution of one or a few metabolic factors on tumorigenesis. In the 1970s, multiple studies linked colorectal cancer to high-fat diet intake, low serum cholesterol, and high fecal bile acids [291, 292]. Analysis of pre-diagnostic serum in prostate cancer cohort studies suggested a positive correlation between omega-6-polyunsaturated fatty acids and cancer risk while omega-3-polyunsaturated fatty acids were inversely correlated with cancer risk [293]. Likewise, some polyunsaturated fatty acids but not others were linked to increased breast cancer risk [294, 295].
More recently, metabolomics has been used to examine the association between a broader range of metabolites in pre-diagnostic serum and cancer risk. Prospective analysis of circulating metabolites in breast cancer patients revealed that acylcarnitine and phosphotidylcholines were strongly associated with risk of breast cancer, regardless of breast cancer subtype, age, fasting status, menopausal status, and adiposity [296]. Similarly, higher circulating lysophosphatidylcholines were found to correlate with lower risks of breast, prostate, and colorectal cancer [297]. Pre-diagnostic serum from the Prostate, Lung, Colorectal, and Ovarian (PLCO) Cancer Screening Trial was the first to identify serum metabolites associated with coffee intake and found that some caffeine-related metabolites were inversely associated with colorectal cancer [298]. Another study examined metabolites in pre-diagnostic serum from postmenopausal invasive breast cancer cases and matched controls from the PLCO trial and found that metabolites related to alcohol, vitamin E, and animal fats were associated with the risk of hormone receptor-positive breast cancer [299]. Metabolomic analysis of fatal prostate cancer cases and controls from the Alpha-Tocopherol, Beta-Carotene Cancer Prevention (ATBC) Study found higher levels of amino acids involved in redox metabolism (thioproline, cystine, and cysteine) were associated with reduced risk of lethal prostate cancer [300]. In contrast, leucylglycine and gamma-glutamyl amino acids were associated with increased risk of terminal prostate cancer, while 3-hydroxybutyrate, acyl carnitines, and dicarboxylic fatty acids were higher in patients presenting with de novo metastatic disease [300]. Other metabolomic studies using serum collected before cancer diagnosis in screening or prevention trials found associations between branched-chain amino acids and pancreatic cancer [207] and pseudouridine and ovarian cancer [301].
Despite these associations, a contentious issue has been whether implicated metabolites play a direct causal role in tumorigenesis or are merely an early manifestation of preclinical disease. For this reason, it has been suggested that incident cases occurring within two years of non-localized cancer diagnosis be excluded from analyses searching for a direct causal role of metabolic factors in tumorigenesis [302].
Identifying Biomarkers for Cancer Screening, Diagnosis, and Monitoring
Cancer detection techniques are essential not only for initial diagnosis, but they also provide effective ways to screen appropriate populations, guide initial treatment strategy, assess treatment efficacy, and track cancer progression over time. In recent years there have been several comprehensive reviews of metabolic biomarker studies in common cancers [303–307]. Here we review some of the metabolite-based tests with established clinical applications and provide examples of metabolomic studies that are expanding the repertoire of metabolic biomarkers for cancer detection and surveillance.
Imaging techniques, including CT, MRI, PET, and radionuclide scans, are used extensively in the clinic for cancer detection and follow-up. One of the earliest metabolic markers used in cancer detection was FDG-PET [308]. It takes advantage of elevated glucose uptake by cancer cells, but since glucose uptake also increases during inflammation this limits the use of FDG-PET in some settings [309]. In addition to FDG, a wide range of radio-labeled carbohydrates, amino acids, and fatty acids have been used in preclinical and clinical settings to take the advantage of the high metabolic rate of tumors as a diagnostic tool. Other 18F-labelled sugars, such as D-mannose, D-lactose, D-fructose, and D-galactose, have been developed and studied as PET tracers in preclinical settings and may allow the detection of some cancer cells that utilize a sugar molecule other than glucose for energy [310–313]. Some cancer cells overexpress specific amino acid transporters such that radiolabeled amino acids can be used to detect these cancers. For instance, amino acid utilization has been evaluated by [11C]-tyrosine PET in soft tissue sarcomas and pituitary adenomas, while [11C]-methionine PET has been evaluated in brain tumors [314–316]. To study the role of glutaminolysis in tumors, a series of glutamine-based PET tracers, including L-[5-11C]-glutamine, [18F]-(2S,4R)-4-fluoroglutamine, and [18F]-(2S,4S)-4-(3-fluoropropyl)glutamine, have been synthesized [317–319]. A recent clinical study has shown high uptake of [18F]-(2S,4R)-4-fluoroglutamine in glioma patients suggesting increased dependence on glutaminolysis [320]. Anti-1-amino-3-18F-fluorocyclobutane-1-carboxylic acid (18F-FACBC), also known as [18F]-fluciclovine is a synthetic analog of the amino acid leucine, whose uptake is facilitated by amino acid transporters and is useful for detection of recurrent prostate cancer [54, 321]. Apart from amino acids and sugars, cancer cells also engage in elevated lipid metabolism to sustain rapid cell proliferation and survival. 11C-fatty acids have been developed and used as PET radiotracers for studying beta-oxidation in animals and humans [322]. Radiolabeled forms of the phospholipid head group choline, [18F]-fluoromethylcholine and [18F]-fluoroethylcholine, are used in the clinic for prostate cancer detection [55, 323].
In addition to PET imaging, MR spectral imaging (MRSI) is also emerging as a tool for non-invasive assessment of tumor metabolism [324]. For example, detection of D-2-HG by MR-spectroscopy in IDH-mutant glioma has been demonstrated in clinical studies and may have a role in monitoring patients receiving IDH-targeted therapy [325, 326]. The use of MRSI in the clinic will likely expand as protocols become more standardized and efficient and as new metabolic biomarkers are identified.
Despite its established role in the clinic, there are several limitations to PET imaging, including limited availability and short half-life of some radiotracers, poor image resolution, inability to detect smaller tumors, and inability to distinguish tumors from non-malignant hypermetabolic processes. For example, a meta-analysis of 45 different studies, which assessed lymph node involvement in non-small cell lung cancer patients, concluded that the sensitivity and specificity of PET-CT is roughly 75% and 90%, respectively [327]. Progress in understanding metabolic differences between tumor, normal tissue, and non-malignant disease states will hopefully facilitate the implementation of tracers with greater diagnostic accuracy.
Compared to metabolic imaging, direct analysis of metabolites in clinical specimens and biofluids has the advantage of increased sensitivity and diversity of chemical species that can be monitored. When surgical specimens are available, metabolomic analysis of tumor and adjacent normal tissue can be used to identify metabolic pathways that are altered in cancer. For evaluating labile metabolites, rapidly collected and processed biopsy tissue is ideal; however, even formalin-fixed paraffin embedded archival tissue has been used for metabolic profiling [328]. In lung cancer, one of the most commonly elevated metabolites in tumor tissue and serum is lactate [329–333]. In fact, one study found that more aggressive lung cancers had higher lactate production, suggesting that lactate levels can be used to assess disease aggressiveness [334]. Similarly, glutamate is elevated in tumor and serum samples of lung cancer patients [329, 331, 333, 335, 336]. Studies have shown that the elevated glutamate likely comes from a dependency of cancer cells for increased glutamine, which provides necessary nitrogen for the synthesis of nucleotides and amino acids [337]. Glutamate enrichment is also seen in breast cancer tissue compared to normal breast tissue [338]. In contrast, glutamine levels are lower in colon and stomach tumor tissue due to the high rate of glutaminolysis driving proliferation [339]. A wide range of other metabolites, including amino acids, purines, pyrimidines, and intermediates of metabolic pathways have been found to be altered in cancer relative to normal tissue. For example, mass spectrometry-based metabolomic analysis of human and mouse colorectal tumors found ten metabolites that were increased in tumor tissues, compared with nontumor tissues (proline, threonine, glutamic acid, arginine, N1-acetylspermidine, xanthine, uracil, betaine, symmetric dimethylarginine, and asymmetric-dimethylarginine); furthermore, these metabolites also showed detectable increases in urine of tumor-bearing mice [340].
Metabolomic analysis of tumor tissue may also allow differentiation between tumor subtypes and aggressiveness of tumors [331, 341–343]. By analyzing tumor tissue, plasma, and urine, sarcosine was identified to play a role in prostate cancer progression [344, 345]. The role of sarcosine as a biomarker of prostate cancer in urine, however, has not been replicated in other studies [346]. In IDH-mutant AML, serum and urine D-2-HG levels have been evaluated as a tool to assess disease activity and therapeutic response [347].
While tissue biopsy is critical for establishing the initial diagnosis, it is neither practical nor desirable for cancer screening, monitoring, or surveillance. In contrast, biofluids, such as blood, serum, urine, saliva, and sweat, are a convenient source of material for biomarker detection. Serum antigen and hormone biomarkers exist for prostate adenocarcinoma, pancreatic ductal adenocarcinoma, ovarian cancer, non-seminomatous germ-cell tumors, thyroid cancers, hepatocellular carcinoma, and others. However, most of these biomarkers are not specific for cancer and therefore must be interpreted together with other clinical or laboratory findings. There is intense interest in developing improved prognostic and diagnostic biomarkers based on molecular analysis of circulating tumor cells and cell-free DNA, RNA, or protein. In recent years, a remarkable number of studies have also explored the use of biofluids as a source of metabolic biomarkers for cancer detection, treatment monitoring, and surveillance. For illustrative purposes we discuss only a few examples and refer the reader to several reviews for more comprehensive coverage of this topic in different cancers [306, 348–350].
In breast cancer, four metabolites (L-octanoylcarnitine, 5-oxoproline, hypoxanthine, and docosahexaenoic acid) were identified as potential serum biomarkers [351]. In pancreatic cancer, a panel of five serum metabolites including glutamate, choline, 1,5-anhydro-D-glucitol, betaine, and methylguanidine was able to distinguish cancer patient from controls [352]. Another study showed that nine serum metabolites (histidine, proline, sphingomyelin d18:2, sphingomyelin d17:1, phosphatidylcholine, isocitrate, sphingosine-1-phosphate, pyruvate, and ceramide) combined with the carbohydrate antigen CA19–9 were able to distinguish between pancreatic cancer and chronic pancreatitis [353]. In a study that focused on free amino acids in plasma, consistent changes were found across patients with lung, gastric, colorectal, breast, and prostate cancer compared to gender- and age-matched controls [354]. Interestingly, elevated plasma levels of branched-chain amino acids are an early event in pancreas cancer development (when disease is still occult) and at the time of diagnosis they are predictive of future tissue wasting [207, 355]. In osteosarcoma patients, serum and urinary metabolomics revealed a distinct phenotype compared to healthy patients, suggesting that downregulation of central carbon metabolism and increased glutathione and polyamine metabolism are characteristic features of osteosarcoma patients [356].
Urine metabolites have also been evaluated as potential biomarkers for cancer detection. For instance, urine samples of bladder cancer patients have a significantly lower level of citrate, 2,5-furandicarboxylic acid, ribitol, and ribonic acid compared to healthy individuals [357–359]. In contrast, two independent studies have shown elevated taurine levels in the urine of bladder cancer patients [359, 360]. Amino acids, such as citrulline, leucine, serine, tryptophan, and tyrosine, were found to be decreased in patients with prostate cancer [361, 362]. Urine metabolite analysis has also been used to detect malignancies that are not in close contact with urine. For example, patients with hepatocellular carcinoma have higher urine creatine and carnitine and lower citrate and glycine compared to a healthy cohort [363, 364]. LC/MS analysis of urine samples collected from lung cancer patients has shown an increased level of amino acids tyrosine, tryptophan, and phenylalanine [365]. Modified nucleosides have also been detected in the urine of patients with a variety of cancer types [366–368].
In addition to tissue and biofluids, exhaled breath has also been explored as a potential source of cancer biomarkers. It has been shown that volatile organic compounds in exhaled breath can be used to differentiate patients with non-small cell lung cancer from non-cancer controls [369–371]. Sensitivity and specificity were 72–90% and 83–94% respectively, which is comparable to the performance of low dose CT in lung cancer screening trials [372].
It is important to acknowledge that a key limitation of assessing global metabolic changes in biofluids is the inability to differentiate cancer from other diseases that present with systemic metabolic alterations. Metabolomic analysis of tumor tissue is less subject to such confounding effects and, where appropriate, can be a valuable source of metabolic biomarkers that can be leveraged in early treatment phases or during disease progression when tumor tissue is available.
Discovery of Targeted Therapies that Interfere with Cancer Metabolism
A growing area of research is the use of metabolomics to uncover metabolic dependencies of cancer that can point to novel drug targets. In this section we discuss a few examples of how metabolomics directly contributed to the discovery of new targets for precision medicine. The reader is also referred to a recent review on this topic [373].
While cancer drivers have been detected through genomic, transcriptomic, or proteomic approaches, understanding which cancer-associated mutations, gene expression changes, and post-translational modifications are functionally relevant is not always straightforward. As previously discussed, changes in metabolite levels reflect the activity of metabolic enzymes in cancer cells and thus could be used to help identify which alterations at the DNA, RNA, and protein levels result in functional changes in cellular activity. A classic example of this is the oncometabolite D-2-hydroxyglutarate (D-2-HG), which was found to be markedly elevated in cells expressing cancer-associated IDH mutations and was subsequently shown to be significantly elevated in cells, tissues, and plasma from cancers with somatic mutations in isocitrate dehydrogenase (IDH) [347, 374, 375]. These and other studies demonstrating that D-2-HG alters the activity of chromatin modifying enzymes and contributes to disease progression led to the development of drugs targeting mutant IDH, which are now in clinical trials (Table 1 and [153, 155]).
In androgen-driven prostate cancer, where hundreds of androgen receptor regulated genes can be differentially regulated in cancer a combination of transcriptomics and metabolomics was used to identify calcium/calmodulin-dependent protein kinase kinase 2 (CAMKK2) as a hormone-dependent modulator of anabolic metabolism [376]. In clear cell renal cell carcinoma (ccRCC), multi-omics datasets were compared to gain molecular insights beyond the sum of individual omics [377]. The analysis revealed crosstalk within and between phosphoproteomics, transcriptomics, and metabolomics, including known ccRCC drug targets [377]. Finally, a comprehensive analysis of expression patterns of metabolic genes across 22 diverse types of human tumors identified hundreds of tumor-specific expression changes, while corresponding changes in metabolite levels were used to highlight enzymes that could potentially serve as drug targets [77].
Stable isotope tracing is another powerful tool to allow functional assessment of tumor metabolism in vivo [281], and when combined with genomics, transcriptomics, and proteomics can inform how altered metabolism relates to molecular drivers of cancer. Stable isotope resolved metabolomic analysis (SIRM) following infusion of 13C-glucose into lung cancer patients showed that tumors had increased glycolysis and Kreb’s cycle activity relative to non-cancer tissue and also processed glycolytic metabolites differently [329]. 13C-glucose infusion also revealed that lung cancers display metabolic heterogeneity in nutrient utilization, which could have important implications for selecting therapies that target metabolism [83]. It is noteworthy that a common theme throughout many of the studies presented here is that the best use of metabolomics is in combination with other “omics” datasets to uncover clinically relevant and actionable drug targets.
Conclusions
While less utilized compared to the other “omics” approaches, metabolomics has the potential to significantly impact core areas of oncology including screening, diagnosis, and therapy. However, such applications require a better understanding of how these measurements are connected to human physiology and cancer biology. In biofluids that are readily accessible clinically, most notably plasma, our understanding of what metabolites can be measured to reflect cancer status is in its very early stages. Although some inroads have been made [207, 378, 379], it is still unclear to what extent a metabolite profile in plasma reveals the metabolic activity of the cancer. Additional studies that conduct metabolomics experiments in fluids that harbor the cancer and connect these measurements to both metabolism and the biology of the tumor is a promising new direction. Much remains to be learned about how to interpret cancer metabolism from these measurements.
One of the challenges with metabolomics is the vast number and chemical complexity of metabolites that exist. For example, plasma metabolite composition is a manifestation of liver, muscle and other organ-level metabolism, dietary intake, activity of the microbiome, and other factors. It is also important to recognize that metabolomics differs from other omics technology in that no one metabolomics approach can be completely comprehensive. We propose that currently the best use of metabolomics in research is in combination with other omics approaches and hypothesis-driven investigation to discover functionally and diagnostically relevant alterations in cancer cells. We anticipate that as standardized protocols, affordable instruments, and user-friendly analysis platforms become more widely available, metabolomics will play an increasingly important role alongside other diagnostic and prognostic tests in the clinic and at the bedside.
Acknowledgements
We wish to thank Chris Nabel for helpful discussion pertaining to nucleotide synthesis inhibitors. Regrettably, due to space limitations we are unable to cite many primary studies that have contributed to our understanding of how metabolism is altered in cancer. We hope the interested reader can discover these studies by exploring other review articles we have cited. D.G.K. is supported by R35CA197616 from the National Cancer Institute at the National Institutes of Health (NCI). D.R.S. is supported by NCATS/NIH KL2 TR002542 administered through the Harvard Catalyst Medical Research Investigator Training Program, and a grant from the Joint Center for Radiation Therapy Foundation. M.G.V.H. acknowledges support from Stand Up to Cancer, the Emerald Foundation, the Lustgarten Foundation, the MIT Center for Precision Cancer Medicine, the Ludwig Center at MIT, a Faculty Scholars grant from HHMI, and the NCI (R35CA242379, R01CA201276, and P30CA14051). J.W.L acknowledges support from the American Cancer Society, the Lustgarten Foundation, and R01CA193256 from NCI. Figures were created with BioRender.com .
Bibliography
- 1.Pavlova NN and Thompson CB, The Emerging Hallmarks of Cancer Metabolism. Cell Metab, 2016. 23(1): p. 27–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vander Heiden MG and DeBerardinis RJ, Understanding the Intersections between Metabolism and Cancer Biology. Cell, 2017. 168(4): p. 657–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Teicher BA, Linehan WM, and Helman LJ, Targeting cancer metabolism. Clin Cancer Res, 2012. 18(20): p. 5537–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Luengo A, Gui DY, and Vander Heiden MG, Targeting Metabolism for Cancer Therapy. Cell Chem Biol, 2017. 24(9): p. 1161–1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vernieri C, et al. , Targeting Cancer Metabolism: Dietary and Pharmacologic Interventions. Cancer Discov, 2016. 6(12): p. 1315–1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu X and Locasale JW, Metabolomics: A Primer. Trends Biochem Sci, 2017. 42(4): p. 274–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rinschen MM, et al. , Identification of bioactive metabolites using activity metabolomics. Nat Rev Mol Cell Biol, 2019. 20(6): p. 353–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Warburg O, Posener K, and Negelein E, Über den Stoffwechsel der Carcinomzelle. Biochem Zeitschrift, 1924. 152: p. 309–44. [Google Scholar]
- 9.Warburg O, On the origin of cancer cells. Science, 1956. 123(3191): p. 309–14. [DOI] [PubMed] [Google Scholar]
- 10.Vander Heiden MG, Cantley LC, and Thompson CB, Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science, 2009. 324(5930): p. 1029–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Koppenol WH, Bounds PL, and Dang CV, Otto Warburg’s contributions to current concepts of cancer metabolism. Nat Rev Cancer, 2011. 11(5): p. 325–37. [DOI] [PubMed] [Google Scholar]
- 12.Hsu PP and Sabatini DM, Cancer cell metabolism: Warburg and beyond. Cell, 2008. 134(5): p. 703–7. [DOI] [PubMed] [Google Scholar]
- 13.Dang CV and Semenza GL, Oncogenic alterations of metabolism. Trends Biochem Sci, 1999. 24(2): p. 68–72. [DOI] [PubMed] [Google Scholar]
- 14.Shaw RJ, LKB1 and AMP-activated protein kinase control of mTOR signalling and growth. Acta Physiol (Oxf), 2009. 196(1): p. 65–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.DeBerardinis RJ and Chandel NS, Fundamentals of cancer metabolism. Sci Adv, 2016. 2(5): p. e1600200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dang CV, MYC, metabolism, cell growth, and tumorigenesis. Cold Spring Harb Perspect Med, 2013. 3(8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kimmelman AC, Metabolic Dependencies in RAS-Driven Cancers. Clin Cancer Res, 2015. 21(8): p. 1828–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vousden KH and Ryan KM, p53 and metabolism. Nat Rev Cancer, 2009. 9(10): p. 691–700. [DOI] [PubMed] [Google Scholar]
- 19.Worby CA and Dixon JE, Pten. Annu Rev Biochem, 2014. 83: p. 641–69. [DOI] [PubMed] [Google Scholar]
- 20.Hanahan D and Weinberg RA, Hallmarks of cancer: the next generation. Cell, 2011. 144(5): p. 646–74. [DOI] [PubMed] [Google Scholar]
- 21.Vander Heiden MG, et al. , Metabolic pathway alterations that support cell proliferation. Cold Spring Harb Symp Quant Biol, 2011. 76: p. 325–34. [DOI] [PubMed] [Google Scholar]
- 22.Schulze A and Harris AL, How cancer metabolism is tuned for proliferation and vulnerable to disruption. Nature, 2012. 491(7424): p. 364–73. [DOI] [PubMed] [Google Scholar]
- 23.Cairns RA, Harris IS, and Mak TW, Regulation of cancer cell metabolism. Nat Rev Cancer, 2011. 11(2): p. 85–95. [DOI] [PubMed] [Google Scholar]
- 24.Hoxhaj G and Manning BD, The PI3K-AKT network at the interface of oncogenic signalling and cancer metabolism. Nat Rev Cancer, 2020. 20(2): p. 74–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Elstrom RL, et al. , Akt stimulates aerobic glycolysis in cancer cells. Cancer Res, 2004. 64(11): p. 3892–9. [DOI] [PubMed] [Google Scholar]
- 26.Roberts DJ, et al. , Akt phosphorylates HK-II at Thr-473 and increases mitochondrial HK-II association to protect cardiomyocytes. J Biol Chem, 2013. 288(33): p. 23798–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sano H, et al. , Insulin-stimulated phosphorylation of a Rab GTPase-activating protein regulates GLUT4 translocation. J Biol Chem, 2003. 278(17): p. 14599–602. [DOI] [PubMed] [Google Scholar]
- 28.Waldhart AN, et al. , Phosphorylation of TXNIP by AKT Mediates Acute Influx of Glucose in Response to Insulin. Cell Rep, 2017. 19(10): p. 2005–2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Duvel K, et al. , Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol Cell, 2010. 39(2): p. 171–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hudson CC, et al. , Regulation of hypoxia-inducible factor 1alpha expression and function by the mammalian target of rapamycin. Mol Cell Biol, 2002. 22(20): p. 7004–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhong H, et al. , Modulation of hypoxia-inducible factor 1alpha expression by the epidermal growth factor/phosphatidylinositol 3-kinase/PTEN/AKT/FRAP pathway in human prostate cancer cells: implications for tumor angiogenesis and therapeutics. Cancer Res, 2000. 60(6): p. 1541–5. [PubMed] [Google Scholar]
- 32.Majumder PK, et al. , mTOR inhibition reverses Akt-dependent prostate intraepithelial neoplasia through regulation of apoptotic and HIF-1-dependent pathways. Nat Med, 2004. 10(6): p. 594–601. [DOI] [PubMed] [Google Scholar]
- 33.Majmundar AJ, Wong WJ, and Simon MC, Hypoxia-inducible factors and the response to hypoxic stress. Mol Cell, 2010. 40(2): p. 294–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Denko NC, Hypoxia, HIF1 and glucose metabolism in the solid tumour. Nat Rev Cancer, 2008. 8(9): p. 705–13. [DOI] [PubMed] [Google Scholar]
- 35.Kim JW, et al. , HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab, 2006. 3(3): p. 177–85. [DOI] [PubMed] [Google Scholar]
- 36.Hoefflin R, et al. , HIF-1alpha and HIF-2alpha differently regulate tumour development and inflammation of clear cell renal cell carcinoma in mice. Nat Commun, 2020. 11(1): p. 4111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ricketts CJ, Crooks DR, and Linehan WM, Targeting HIF2alpha in Clear-Cell Renal Cell Carcinoma. Cancer Cell, 2016. 30(4): p. 515–517. [DOI] [PubMed] [Google Scholar]
- 38.Ying H, et al. , Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell, 2012. 149(3): p. 656–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Luengo A, et al. , Increased demand for NAD(+) relative to ATP drives aerobic glycolysis. Mol Cell, 2021. 81(4): p. 691–707 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Matoba S, et al. , p53 regulates mitochondrial respiration. Science, 2006. 312(5780): p. 1650–3. [DOI] [PubMed] [Google Scholar]
- 41.Cori CF and Cori GT, The Carbohydrate Metabolism of Tumors: II. Changes in the Sugar, Lactic Acid, and CO2-Combining Power of Blodd Passing Through a Tumor. Journal of Biological Chemistry, 1925. 65(2): p. 397–405. [Google Scholar]
- 42.Cori CF and Cori GT, The Carbohydrate Metabolism of Tumors: I. The Free Sugar, Lactic Acid, and Glycogen Content of Malignant Tumors. Journal of Biological Chemistry, 1925. 64(1): p. 11–22. [Google Scholar]
- 43.Hui S, et al. , Glucose feeds the TCA cycle via circulating lactate. Nature, 2017. 551(7678): p. 115–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Faubert B, et al. , Lactate Metabolism in Human Lung Tumors. Cell, 2017. 171(2): p. 358–371 e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sonveaux P, et al. , Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. J Clin Invest, 2008. 118(12): p. 3930–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu S, et al. , Quantitative Analysis of the Physiological Contributions of Glucose to the TCA Cycle. Cell Metab, 2020. 32(4): p. 619–628 e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wise DR, et al. , Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc Natl Acad Sci U S A, 2008. 105(48): p. 18782–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gouw AM, et al. , The MYC Oncogene Cooperates with Sterol-Regulated Element-Binding Protein to Regulate Lipogenesis Essential for Neoplastic Growth. Cell Metab, 2019. 30(3): p. 556–572 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Casciano JC, et al. , MYC regulates fatty acid metabolism through a multigenic program in claudin-low triple negative breast cancer. Br J Cancer, 2020. 122(6): p. 868–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Reynolds MR, et al. , Control of glutamine metabolism by the tumor suppressor Rb. Oncogene, 2014. 33(5): p. 556–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Muir A, et al. , Environmental cystine drives glutamine anaplerosis and sensitizes cancer cells to glutaminase inhibition. Elife, 2017. 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Davidson SM, et al. , Environment Impacts the Metabolic Dependencies of Ras-Driven Non-Small Cell Lung Cancer. Cell Metab, 2016. 23(3): p. 517–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Biancur DE, et al. , Compensatory metabolic networks in pancreatic cancers upon perturbation of glutamine metabolism. Nat Commun, 2017. 8: p. 15965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Savir-Baruch B, Zanoni L, and Schuster DM, Imaging of Prostate Cancer Using Fluciclovine. PET Clin, 2017. 12(2): p. 145–157. [DOI] [PubMed] [Google Scholar]
- 55.Umbehr MH, et al. , The role of 11C-choline and 18F-fluorocholine positron emission tomography (PET) and PET/CT in prostate cancer: a systematic review and meta-analysis. Eur Urol, 2013. 64(1): p. 106–17. [DOI] [PubMed] [Google Scholar]
- 56.Sinkala M, Mulder N, and Patrick Martin D, Metabolic gene alterations impact the clinical aggressiveness and drug responses of 32 human cancers. Commun Biol, 2019. 2: p. 414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Baysal BE, et al. , Mutations in SDHD, a mitochondrial complex II gene, in hereditary paraganglioma. Science, 2000. 287(5454): p. 848–51. [DOI] [PubMed] [Google Scholar]
- 58.Tomlinson IP, et al. , Germline mutations in FH predispose to dominantly inherited uterine fibroids, skin leiomyomata and papillary renal cell cancer. Nat Genet, 2002. 30(4): p. 406–10. [DOI] [PubMed] [Google Scholar]
- 59.Martinez-Reyes I and Chandel NS, Mitochondrial TCA cycle metabolites control physiology and disease. Nat Commun, 2020. 11(1): p. 102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhang J, et al. , VHL substrate transcription factor ZHX2 as an oncogenic driver in clear cell renal cell carcinoma. Science, 2018. 361(6399): p. 290–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hu L, et al. , TBK1 Is a Synthetic Lethal Target in Cancer with VHL Loss. Cancer Discov, 2020. 10(3): p. 460–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cairns RA and Mak TW, Oncogenic isocitrate dehydrogenase mutations: mechanisms, models, and clinical opportunities. Cancer Discov, 2013. 3(7): p. 730–41. [DOI] [PubMed] [Google Scholar]
- 63.Houillier C, et al. , IDH1 or IDH2 mutations predict longer survival and response to temozolomide in low-grade gliomas. Neurology, 2010. 75(17): p. 1560–6. [DOI] [PubMed] [Google Scholar]
- 64.Yan H, et al. , IDH1 and IDH2 mutations in gliomas. N Engl J Med, 2009. 360(8): p. 765–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Parker SJ and Metallo CM, Metabolic consequences of oncogenic IDH mutations. Pharmacol Ther, 2015. 152: p. 54–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Losman JA and Kaelin WG Jr., What a difference a hydroxyl makes: mutant IDH, (R)-2-hydroxyglutarate, and cancer. Genes Dev, 2013. 27(8): p. 836–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hegi ME, et al. , MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med, 2005. 352(10): p. 997–1003. [DOI] [PubMed] [Google Scholar]
- 68.Xu W, et al. , Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell, 2011. 19(1): p. 17–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Figueroa ME, et al. , Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell, 2010. 18(6): p. 553–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chowdhury R, et al. , The oncometabolite 2-hydroxyglutarate inhibits histone lysine demethylases. EMBO Rep, 2011. 12(5): p. 463–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Locasale JW, et al. , Phosphoglycerate dehydrogenase diverts glycolytic flux and contributes to oncogenesis. Nat Genet, 2011. 43(9): p. 869–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Possemato R, et al. , Functional genomics reveal that the serine synthesis pathway is essential in breast cancer. Nature, 2011. 476(7360): p. 346–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Fan J, et al. , Quantitative flux analysis reveals folate-dependent NADPH production. Nature, 2014. 510(7504): p. 298–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gaude E and Frezza C, Tissue-specific and convergent metabolic transformation of cancer correlates with metastatic potential and patient survival. Nat Commun, 2016. 7: p. 13041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Mayers JR, et al. , Tissue of origin dictates branched-chain amino acid metabolism in mutant Kras-driven cancers. Science, 2016. 353(6304): p. 1161–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yuneva MO, et al. , The metabolic profile of tumors depends on both the responsible genetic lesion and tissue type. Cell Metab, 2012. 15(2): p. 157–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hu J, et al. , Heterogeneity of tumor-induced gene expression changes in the human metabolic network. Nat Biotechnol, 2013. 31(6): p. 522–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Marberger H, et al. , Citric acid in human prostatic secretion and metastasizing cancer of prostate gland. Br Med J, 1962. 1(5281): p. 835–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Elia I and Haigis MC, Metabolites and the tumour microenvironment: from cellular mechanisms to systemic metabolism. Nat Metab, 2021. 3(1): p. 21–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lyssiotis CA and Kimmelman AC, Metabolic Interactions in the Tumor Microenvironment. Trends Cell Biol, 2017. 27(11): p. 863–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lau AN, et al. , Dissecting cell-type-specific metabolism in pancreatic ductal adenocarcinoma. Elife, 2020. 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.DeVilbiss AW, et al. , Metabolomic profiling of rare cell populations isolated by flow cytometry from tissues. Elife, 2021. 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hensley CT, et al. , Metabolic Heterogeneity in Human Lung Tumors. Cell, 2016. 164(4): p. 681–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.O’Neill LA, Kishton RJ, and Rathmell J, A guide to immunometabolism for immunologists. Nat Rev Immunol, 2016. 16(9): p. 553–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Geltink RIK, Kyle RL, and Pearce EL, Unraveling the Complex Interplay Between T Cell Metabolism and Function. Annu Rev Immunol, 2018. 36: p. 461–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Yang Z, et al. , Phosphofructokinase deficiency impairs ATP generation, autophagy, and redox balance in rheumatoid arthritis T cells. J Exp Med, 2013. 210(10): p. 2119–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Choi SC, et al. , Inhibition of glucose metabolism selectively targets autoreactive follicular helper T cells. Nat Commun, 2018. 9(1): p. 4369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Michalek RD, et al. , Cutting edge: distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J Immunol, 2011. 186(6): p. 3299–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wang R, et al. , The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity, 2011. 35(6): p. 871–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Phan AT and Goldrath AW, Hypoxia-inducible factors regulate T cell metabolism and function. Mol Immunol, 2015. 68(2 Pt C): p. 527–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Su W, et al. , Protein Prenylation Drives Discrete Signaling Programs for the Differentiation and Maintenance of Effector Treg Cells. Cell Metab, 2020. 32(6): p. 996–1011 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Viola A, et al. , The Metabolic Signature of Macrophage Responses. Front Immunol, 2019. 10: p. 1462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Leone RD, et al. , Glutamine blockade induces divergent metabolic programs to overcome tumor immune evasion. Science, 2019. 366(6468): p. 1013–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Galluzzi L, et al. , Metabolic targets for cancer therapy. Nat Rev Drug Discov, 2013. 12(11): p. 829–46. [DOI] [PubMed] [Google Scholar]
- 95.Bobrovnikova-Marjon E and Hurov JB, Targeting metabolic changes in cancer: novel therapeutic approaches. Annu Rev Med, 2014. 65: p. 157–70. [DOI] [PubMed] [Google Scholar]
- 96.Weinberg SE and Chandel NS, Targeting mitochondria metabolism for cancer therapy. Nat Chem Biol, 2015. 11(1): p. 9–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Chabner BA, et al. , The clinical pharmacology of antineoplastic agents (first of two parts). N Engl J Med, 1975. 292(21): p. 1107–13. [DOI] [PubMed] [Google Scholar]
- 98.Chabner BA, et al. , The clinical pharmacology of antineoplastic agents (second of two parts). N Engl J Med, 1975. 292(22): p. 1159–68. [DOI] [PubMed] [Google Scholar]
- 99.Sun J, et al. , A systematic analysis of FDA-approved anticancer drugs. BMC Syst Biol, 2017. 11(Suppl 5): p. 87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Farber S and Diamond LK, Temporary remissions in acute leukemia in children produced by folic acid antagonist, 4-aminopteroyl-glutamic acid. N Engl J Med, 1948. 238(23): p. 787–93. [DOI] [PubMed] [Google Scholar]
- 101.Scott RB, Cancer chemotherapy--the first twenty-five years. Br Med J, 1970. 4(5730): p. 259–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Chabner BA and Roberts TG Jr., Timeline: Chemotherapy and the war on cancer. Nat Rev Cancer, 2005. 5(1): p. 65–72. [DOI] [PubMed] [Google Scholar]
- 103.Parker WB, Enzymology of purine and pyrimidine antimetabolites used in the treatment of cancer. Chem Rev, 2009. 109(7): p. 2880–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Neuman RE and McCoy TA, Dual requirement of Walker carcinosarcoma 256 in vitro for asparagine and glutamine. Science, 1956. 124(3212): p. 124–5. [DOI] [PubMed] [Google Scholar]
- 105.Clavell LA, et al. , Four-agent induction and intensive asparaginase therapy for treatment of childhood acute lymphoblastic leukemia. N Engl J Med, 1986. 315(11): p. 657–63. [DOI] [PubMed] [Google Scholar]
- 106.Dwarakanath BS, et al. , Clinical studies for improving radiotherapy with 2-deoxy-D-glucose: present status and future prospects. J Cancer Res Ther, 2009. 5 Suppl 1: p. S21–6. [DOI] [PubMed] [Google Scholar]
- 107.Raez LE, et al. , A phase I dose-escalation trial of 2-deoxy-D-glucose alone or combined with docetaxel in patients with advanced solid tumors. Cancer Chemother Pharmacol, 2013. 71(2): p. 523–30. [DOI] [PubMed] [Google Scholar]
- 108.Vander Heiden MG, Targeting cancer metabolism: a therapeutic window opens. Nat Rev Drug Discov, 2011. 10(9): p. 671–84. [DOI] [PubMed] [Google Scholar]
- 109.Christofk HR, et al. , The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature, 2008. 452(7184): p. 230–3. [DOI] [PubMed] [Google Scholar]
- 110.Israelsen WJ and Vander Heiden MG, Pyruvate kinase: Function, regulation and role in cancer. Semin Cell Dev Biol, 2015. 43: p. 43–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Anastasiou D, et al. , Pyruvate kinase M2 activators promote tetramer formation and suppress tumorigenesis. Nat Chem Biol, 2012. 8(10): p. 839–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Parnell KM, et al. , Pharmacologic activation of PKM2 slows lung tumor xenograft growth. Mol Cancer Ther, 2013. 12(8): p. 1453–60. [DOI] [PubMed] [Google Scholar]
- 113.Kung C, et al. , AG-348 enhances pyruvate kinase activity in red blood cells from patients with pyruvate kinase deficiency. Blood, 2017. 130(11): p. 1347–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Grace RF, et al. , Safety and Efficacy of Mitapivat in Pyruvate Kinase Deficiency. N Engl J Med, 2019. 381(10): p. 933–944. [DOI] [PubMed] [Google Scholar]
- 115.Akins NS, Nielson TC, and Le HV, Inhibition of Glycolysis and Glutaminolysis: An Emerging Drug Discovery Approach to Combat Cancer. Curr Top Med Chem, 2018. 18(6): p. 494–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Ma Y, et al. , Ovarian Cancer Relies on Glucose Transporter 1 to Fuel Glycolysis and Growth: Anti-Tumor Activity of BAY-876. Cancers (Basel), 2018. 11(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Kim EY, et al. , A Novel Lactate Dehydrogenase Inhibitor, 1-(Phenylseleno)-4-(Trifluoromethyl) Benzene, Suppresses Tumor Growth through Apoptotic Cell Death. Sci Rep, 2019. 9(1): p. 3969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Qian Y, Wang X, and Chen X, Inhibitors of glucose transport and glycolysis as novel anticancer therapeutics. World J. Transl. Med, 2014. 3: p. 37–57. [Google Scholar]
- 119.Marchiq I and Pouyssegur J, Hypoxia, cancer metabolism and the therapeutic benefit of targeting lactate/H(+) symporters. J Mol Med (Berl), 2016. 94(2): p. 155–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Shestov AA, et al. , Quantitative determinants of aerobic glycolysis identify flux through the enzyme GAPDH as a limiting step. Elife, 2014. 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Liberti MV, et al. , A Predictive Model for Selective Targeting of the Warburg Effect through GAPDH Inhibition with a Natural Product. Cell Metab, 2017. 26(4): p. 648–659 e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Altman BJ, Stine ZE, and Dang CV, From Krebs to clinic: glutamine metabolism to cancer therapy. Nat Rev Cancer, 2016. 16(10): p. 619–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Xiang Y, et al. , Targeted inhibition of tumor-specific glutaminase diminishes cell-autonomous tumorigenesis. J Clin Invest, 2015. 125(6): p. 2293–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Gross MI, et al. , Antitumor activity of the glutaminase inhibitor CB-839 in triple-negative breast cancer. Mol Cancer Ther, 2014. 13(4): p. 890–901. [DOI] [PubMed] [Google Scholar]
- 125.Song M, et al. , Recent Development of Small Molecule Glutaminase Inhibitors. Curr Top Med Chem, 2018. 18(6): p. 432–443. [DOI] [PubMed] [Google Scholar]
- 126.Soth MJ, et al. , Discovery of IPN60090, a Clinical Stage Selective Glutaminase-1 (GLS-1) Inhibitor with Excellent Pharmacokinetic and Physicochemical Properties. J Med Chem, 2020. 63(21): p. 12957–12977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Johnson MO, et al. , Distinct Regulation of Th17 and Th1 Cell Differentiation by Glutaminase-Dependent Metabolism. Cell, 2018. 175(7): p. 1780–1795 e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Yoo HC, et al. , A Variant of SLC1A5 Is a Mitochondrial Glutamine Transporter for Metabolic Reprogramming in Cancer Cells. Cell Metab, 2020. 31(2): p. 267–283 e12. [DOI] [PubMed] [Google Scholar]
- 129.Enomoto K, et al. , A novel therapeutic approach for anaplastic thyroid cancer through inhibition of LAT1. Sci Rep, 2019. 9(1): p. 14616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Locasale JW, Serine, glycine and one-carbon units: cancer metabolism in full circle. Nat Rev Cancer, 2013. 13(8): p. 572–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Wang Q, et al. , Rational Design of Selective Allosteric Inhibitors of PHGDH and Serine Synthesis with Anti-tumor Activity. Cell Chem Biol, 2017. 24(1): p. 55–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Pacold ME, et al. , A PHGDH inhibitor reveals coordination of serine synthesis and one-carbon unit fate. Nat Chem Biol, 2016. 12(6): p. 452–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Mullarky E, et al. , Identification of a small molecule inhibitor of 3-phosphoglycerate dehydrogenase to target serine biosynthesis in cancers. Proc Natl Acad Sci U S A, 2016. 113(7): p. 1778–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Ngo B, et al. , Limited Environmental Serine and Glycine Confer Brain Metastasis Sensitivity to PHGDH Inhibition. Cancer Discov, 2020. 10(9): p. 1352–1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Hascitha J, et al. , Analysis of Kynurenine/Tryptophan ratio and expression of IDO1 and 2 mRNA in tumour tissue of cervical cancer patients. Clin Biochem, 2016. 49(12): p. 919–24. [DOI] [PubMed] [Google Scholar]
- 136.Zhai L, et al. , Infiltrating T Cells Increase IDO1 Expression in Glioblastoma and Contribute to Decreased Patient Survival. Clin Cancer Res, 2017. 23(21): p. 6650–6660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Prendergast GC, et al. , Discovery of IDO1 Inhibitors: From Bench to Bedside. Cancer Res, 2017. 77(24): p. 6795–6811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Vazquez A, et al. , Cancer metabolism at a glance. J Cell Sci, 2016. 129(18): p. 3367–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Menendez JA and Lupu R, Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat Rev Cancer, 2007. 7(10): p. 763–77. [DOI] [PubMed] [Google Scholar]
- 140.Flavin R, P. S Nguyen PL, Loda M, Fatty acid synthase as a potential therapeutic target in cancer. Future Oncol, 2010. 6(4): p. 551–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Mullen GE and Yet L, Progress in the development of fatty acid synthase inhibitors as anticancer targets. Bioorg Med Chem Lett, 2015. 25(20): p. 4363–9. [DOI] [PubMed] [Google Scholar]
- 142.Hatzivassiliou G, et al. , ATP citrate lyase inhibition can suppress tumor cell growth. Cancer Cell, 2005. 8(4): p. 311–21. [DOI] [PubMed] [Google Scholar]
- 143.Shah S, et al. , Targeting ACLY sensitizes castration-resistant prostate cancer cells to AR antagonism by impinging on an ACLY-AMPK-AR feedback mechanism. Oncotarget, 2016. 7(28): p. 43713–43730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Svensson RU, et al. , Inhibition of acetyl-CoA carboxylase suppresses fatty acid synthesis and tumor growth of non-small-cell lung cancer in preclinical models. Nat Med, 2016. 22(10): p. 1108–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Corominas-Faja B, et al. , Chemical inhibition of acetyl-CoA carboxylase suppresses self-renewal growth of cancer stem cells. Oncotarget, 2014. 5(18): p. 8306–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Ackerman D and Simon MC, Hypoxia, lipids, and cancer: surviving the harsh tumor microenvironment. Trends Cell Biol, 2014. 24(8): p. 472–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Ariyama H, et al. , Decrease in membrane phospholipid unsaturation induces unfolded protein response. J Biol Chem, 2010. 285(29): p. 22027–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Fritz V, et al. , Abrogation of de novo lipogenesis by stearoyl-CoA desaturase 1 inhibition interferes with oncogenic signaling and blocks prostate cancer progression in mice. Mol Cancer Ther, 2010. 9(6): p. 1740–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Holder AM, et al. , High stearoyl-CoA desaturase 1 expression is associated with shorter survival in breast cancer patients. Breast Cancer Res Treat, 2013. 137(1): p. 319–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Huang GM, et al. , SCD1 negatively regulates autophagy-induced cell death in human hepatocellular carcinoma through inactivation of the AMPK signaling pathway. Cancer Lett, 2015. 358(2): p. 180–190. [DOI] [PubMed] [Google Scholar]
- 151.von Roemeling CA, et al. , Stearoyl-CoA desaturase 1 is a novel molecular therapeutic target for clear cell renal cell carcinoma. Clin Cancer Res, 2013. 19(9): p. 2368–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Tracz-Gaszewska Z and Dobrzyn P, Stearoyl-CoA Desaturase 1 as a Therapeutic Target for the Treatment of Cancer. Cancers (Basel), 2019. 11(7). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Losman JA, et al. , (R)-2-hydroxyglutarate is sufficient to promote leukemogenesis and its effects are reversible. Science, 2013. 339(6127): p. 1621–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Rohle D, et al. , An inhibitor of mutant IDH1 delays growth and promotes differentiation of glioma cells. Science, 2013. 340(6132): p. 626–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Wang F, et al. , Targeted inhibition of mutant IDH2 in leukemia cells induces cellular differentiation. Science, 2013. 340(6132): p. 622–6. [DOI] [PubMed] [Google Scholar]
- 156.DiNardo CD, et al. , Durable Remissions with Ivosidenib in IDH1-Mutated Relapsed or Refractory AML. N Engl J Med, 2018. 378(25): p. 2386–2398. [DOI] [PubMed] [Google Scholar]
- 157.Heuser M, et al. , Safety and efficacy of BAY1436032 in IDH1-mutant AML: phase I study results. Leukemia, 2020. 34(11): p. 2903–2913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Stein EM, et al. , Enasidenib in mutant IDH2 relapsed or refractory acute myeloid leukemia. Blood, 2017. 130(6): p. 722–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Turcan S, et al. , Efficient induction of differentiation and growth inhibition in IDH1 mutant glioma cells by the DNMT Inhibitor Decitabine. Oncotarget, 2013. 4(10): p. 1729–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Tateishi K, et al. , Extreme Vulnerability of IDH1 Mutant Cancers to NAD+ Depletion. Cancer Cell, 2015. 28(6): p. 773–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Suijker J, et al. , Inhibition of mutant IDH1 decreases D-2-HG levels without affecting tumorigenic properties of chondrosarcoma cell lines. Oncotarget, 2015. 6(14): p. 12505–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Heidelberger C, et al. , Fluorinated pyrimidines, a new class of tumour-inhibitory compounds. Nature, 1957. 179(4561): p. 663–6. [DOI] [PubMed] [Google Scholar]
- 163.Jackman AL, et al. , ICI D1694, a quinazoline antifolate thymidylate synthase inhibitor that is a potent inhibitor of L1210 tumor cell growth in vitro and in vivo: a new agent for clinical study. Cancer Res, 1991. 51(20): p. 5579–86. [PubMed] [Google Scholar]
- 164.Shih C, et al. , LY231514, a pyrrolo[2,3-d]pyrimidine-based antifolate that inhibits multiple folate-requiring enzymes. Cancer Res, 1997. 57(6): p. 1116–23. [PubMed] [Google Scholar]
- 165.Miwa M, et al. , Design of a novel oral fluoropyrimidine carbamate, capecitabine, which generates 5-fluorouracil selectively in tumours by enzymes concentrated in human liver and cancer tissue. Eur J Cancer, 1998. 34(8): p. 1274–81. [DOI] [PubMed] [Google Scholar]
- 166.Meyer LM, et al. , Treatment of acute leukemia with amethopterin (4-amino, 10-methyl pteroyl glutamic acid). Acta Haematol, 1950. 4(3): p. 157–67. [DOI] [PubMed] [Google Scholar]
- 167.Wright JC, et al. , An evaluation of folic acid antagonists in adults with neoplastic diseases: a study of 93 patients with incurable neoplasms. J Natl Med Assoc, 1951. 43(4): p. 211–40. [PMC free article] [PubMed] [Google Scholar]
- 168.Sykes DB, The emergence of dihydroorotate dehydrogenase (DHODH) as a therapeutic target in acute myeloid leukemia. Expert Opin Ther Targets, 2018. 22(11): p. 893–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Xie KC and Plunkett W, Deoxynucleotide pool depletion and sustained inhibition of ribonucleotide reductase and DNA synthesis after treatment of human lymphoblastoid cells with 2-chloro-9-(2-deoxy-2-fluoro-beta-D-arabinofuranosyl) adenine. Cancer Res, 1996. 56(13): p. 3030–7. [PubMed] [Google Scholar]
- 170.Heinemann V, et al. , Inhibition of ribonucleotide reduction in CCRF-CEM cells by 2’,2’-difluorodeoxycytidine. Mol Pharmacol, 1990. 38(4): p. 567–72. [PubMed] [Google Scholar]
- 171.Greene BL, et al. , Ribonucleotide Reductases: Structure, Chemistry, and Metabolism Suggest New Therapeutic Targets. Annu Rev Biochem, 2020. 89: p. 45–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Evans JS, et al. , Antitumor activity of 1-beta-D-arainofuranosylcytosine hydrochloride. Proc Soc Exp Biol Med, 1961. 106: p. 350–3. [PubMed] [Google Scholar]
- 173.Hertel LW, et al. , Evaluation of the antitumor activity of gemcitabine (2’,2’-difluoro-2’-deoxycytidine). Cancer Res, 1990. 50(14): p. 4417–22. [PubMed] [Google Scholar]
- 174.Skipper HE, et al. , Observations on the anticancer activity of 6-mercaptopurine. Cancer Res, 1954. 14(4): p. 294–8. [PubMed] [Google Scholar]
- 175.Atkinson MR and Murray AW, Inhibition of Pruine Phosphoribosyltransferases of Ehrlich Ascites-Tumour Cells by 6-Mercaptopurine. Biochem J, 1965. 94: p. 64–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Hill DL and Bennett LL Jr., Purification and properties of 5-phosphoribosyl pyrophosphate amidotransferase from adenocarcinoma 755 cells. Biochemistry, 1969. 8(1): p. 122–30. [DOI] [PubMed] [Google Scholar]
- 177.Liu Y, et al. , A small-molecule inhibitor of glucose transporter 1 downregulates glycolysis, induces cell-cycle arrest, and inhibits cancer cell growth in vitro and in vivo. Mol Cancer Ther, 2012. 11(8): p. 1672–82. [DOI] [PubMed] [Google Scholar]
- 178.Billiard J, et al. , Quinoline 3-sulfonamides inhibit lactate dehydrogenase A and reverse aerobic glycolysis in cancer cells. Cancer Metab, 2013. 1(1): p. 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Le A, et al. , Inhibition of lactate dehydrogenase A induces oxidative stress and inhibits tumor progression. Proc Natl Acad Sci U S A, 2010. 107(5): p. 2037–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Esslinger CS, Cybulski KA, and Rhoderick JF, Ngamma-aryl glutamine analogues as probes of the ASCT2 neutral amino acid transporter binding site. Bioorg Med Chem, 2005. 13(4): p. 1111–8. [DOI] [PubMed] [Google Scholar]
- 181.Hanaford AR, et al. , Orally bioavailable glutamine antagonist prodrug JHU-083 penetrates mouse brain and suppresses the growth of MYC-driven medulloblastoma. Transl Oncol, 2019. 12(10): p. 1314–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Oda K, et al. , L-type amino acid transporter 1 inhibitors inhibit tumor cell growth. Cancer Sci, 2010. 101(1): p. 173–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Zachar Z, et al. , Non-redox-active lipoate derivates disrupt cancer cell mitochondrial metabolism and are potent anticancer agents in vivo. J Mol Med (Berl), 2011. 89(11): p. 1137–48. [DOI] [PubMed] [Google Scholar]
- 184.Molina JR, et al. , An inhibitor of oxidative phosphorylation exploits cancer vulnerability. Nat Med, 2018. 24(7): p. 1036–1046. [DOI] [PubMed] [Google Scholar]
- 185.Yam C, et al. , Efficacy and safety of the combination of metformin, everolimus and exemestane in overweight and obese postmenopausal patients with metastatic, hormone receptor-positive, HER2-negative breast cancer: a phase II study. Invest New Drugs, 2019. 37(2): p. 345–351. [DOI] [PubMed] [Google Scholar]
- 186.Grosvenor M, Bulcavage L, and Chlebowski RT, Symptoms potentially influencing weight loss in a cancer population. Correlations with primary site, nutritional status, and chemotherapy administration. Cancer, 1989. 63(2): p. 330–4. [DOI] [PubMed] [Google Scholar]
- 187.Ezeoke CC and Morley JE, Pathophysiology of anorexia in the cancer cachexia syndrome. J Cachexia Sarcopenia Muscle, 2015. 6(4): p. 287–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Dewys WD, et al. , Prognostic effect of weight loss prior to chemotherapy in cancer patients. Eastern Cooperative Oncology Group. Am J Med, 1980. 69(4): p. 491–7. [DOI] [PubMed] [Google Scholar]
- 189.Fearon K, et al. , Definition and classification of cancer cachexia: an international consensus. Lancet Oncol, 2011. 12(5): p. 489–95. [DOI] [PubMed] [Google Scholar]
- 190.Tisdale MJ, Cachexia in cancer patients. Nat Rev Cancer, 2002. 2(11): p. 862–71. [DOI] [PubMed] [Google Scholar]
- 191.Argiles JM, et al. , Mediators involved in the cancer anorexia-cachexia syndrome: past, present, and future. Nutrition, 2005. 21(9): p. 977–85. [DOI] [PubMed] [Google Scholar]
- 192.Fearon K, Arends J, and Baracos V, Understanding the mechanisms and treatment options in cancer cachexia. Nat Rev Clin Oncol, 2013. 10(2): p. 90–9. [DOI] [PubMed] [Google Scholar]
- 193.Bosaeus I, Daneryd P, and Lundholm K, Dietary intake, resting energy expenditure, weight loss and survival in cancer patients. J Nutr, 2002. 132(11 Suppl): p. 3465S–3466S. [DOI] [PubMed] [Google Scholar]
- 194.Vazeille C, et al. , Relation between hypermetabolism, cachexia, and survival in cancer patients: a prospective study in 390 cancer patients before initiation of anticancer therapy. Am J Clin Nutr, 2017. 105(5): p. 1139–1147. [DOI] [PubMed] [Google Scholar]
- 195.Klein S, Simes J, and Blackburn GL, Total parenteral nutrition and cancer clinical trials. Cancer, 1986. 58(6): p. 1378–86. [DOI] [PubMed] [Google Scholar]
- 196.McGeer AJ, Detsky AS, and O’Rourke K, Parenteral nutrition in cancer patients undergoing chemotherapy: a meta-analysis. Nutrition, 1990. 6(3): p. 233–40. [PubMed] [Google Scholar]
- 197.Evans WK, et al. , Limited impact of total parenteral nutrition on nutritional status during treatment for small cell lung cancer. Cancer Res, 1985. 45(7): p. 3347–53. [PubMed] [Google Scholar]
- 198.Vujasinovic M, et al. , Pancreatic Exocrine Insufficiency in Pancreatic Cancer. Nutrients, 2017. 9(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Danai LV, et al. , Altered exocrine function can drive adipose wasting in early pancreatic cancer. Nature, 2018. 558(7711): p. 600–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Wigmore SJ, et al. , Changes in nutritional status associated with unresectable pancreatic cancer. Br J Cancer, 1997. 75(1): p. 106–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Schein PS, et al. , Cachexia of malignancy: potential role of insulin in nutritional management. Cancer, 1979. 43(5 Suppl): p. 2070–6. [DOI] [PubMed] [Google Scholar]
- 202.Marks PA and Bishop JS, The glucose metabolism of patients with malignant disease and of normal subjects as studied by means of an intravenous glucose tolerance test. J Clin Invest, 1957. 36(2): p. 254–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Jasani B, et al. , Mechanism of impaired glucose tolerance in patients with neoplasia. Br J Cancer, 1978. 38(2): p. 287–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Fearon KC, et al. , Influence of whole body protein turnover rate on resting energy expenditure in patients with cancer. Cancer Res, 1988. 48(9): p. 2590–5. [PubMed] [Google Scholar]
- 205.Douglas RG and Shaw JH, Metabolic effects of cancer. Br J Surg, 1990. 77(3): p. 246–54. [DOI] [PubMed] [Google Scholar]
- 206.Carrascosa JM, Martinez P, and Nunez de Castro I, Nitrogen movement between host and tumor in mice inoculated with Ehrlich ascitic tumor cells. Cancer Res, 1984. 44(9): p. 3831–5. [PubMed] [Google Scholar]
- 207.Mayers JR, et al. , Elevation of circulating branched-chain amino acids is an early event in human pancreatic adenocarcinoma development. Nat Med, 2014. 20(10): p. 1193–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Donaldson SS and Lenon RA, Alterations of nutritional status: impact of chemotherapy and radiation therapy. Cancer, 1979. 43(5 Suppl): p. 2036–52. [DOI] [PubMed] [Google Scholar]
- 209.Garcia-Peris P, et al. , Prospective study of resting energy expenditure changes in head and neck cancer patients treated with chemoradiotherapy measured by indirect calorimetry. Nutrition, 2005. 21(11–12): p. 1107–12. [DOI] [PubMed] [Google Scholar]
- 210.Van Soom T, et al. , The effects of chemotherapy on energy metabolic aspects in cancer patients: A systematic review. Clin Nutr, 2020. 39(6): p. 1863–1877. [DOI] [PubMed] [Google Scholar]
- 211.Klein S, et al. , Metabolic response to radiation therapy in patients with cancer. Metabolism, 1996. 45(6): p. 767–73. [DOI] [PubMed] [Google Scholar]
- 212.Sloan GM, Maher M, and Brennan MF, Nutritional effects of surgery, radiation therapy, and adjuvant chemotherapy for soft tissue sarcomas. Am J Clin Nutr, 1981. 34(6): p. 1094–102. [DOI] [PubMed] [Google Scholar]
- 213.de Haas EC, et al. , The metabolic syndrome in cancer survivors. Lancet Oncol, 2010. 11(2): p. 193–203. [DOI] [PubMed] [Google Scholar]
- 214.Chemaitilly W, et al. , Endocrine Late Effects in Childhood Cancer Survivors. J Clin Oncol, 2018. 36(21): p. 2153–2159. [DOI] [PubMed] [Google Scholar]
- 215.Meacham LR, et al. , Diabetes mellitus in long-term survivors of childhood cancer. Increased risk associated with radiation therapy: a report for the childhood cancer survivor study. Arch Intern Med, 2009. 169(15): p. 1381–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Faris JE and Smith MR, Metabolic sequelae associated with androgen deprivation therapy for prostate cancer. Curr Opin Endocrinol Diabetes Obes, 2010. 17(3): p. 240–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Goldvaser H, et al. , Toxicity of Extended Adjuvant Therapy With Aromatase Inhibitors in Early Breast Cancer: A Systematic Review and Meta-analysis. J Natl Cancer Inst, 2018. 110(1). [DOI] [PubMed] [Google Scholar]
- 218.Braga-Basaria M, et al. , Metabolic syndrome in men with prostate cancer undergoing long-term androgen-deprivation therapy. J Clin Oncol, 2006. 24(24): p. 3979–83. [DOI] [PubMed] [Google Scholar]
- 219.Tonorezos ES and Jones LW, Energy balance and metabolism after cancer treatment. Semin Oncol, 2013. 40(6): p. 745–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Islami F, et al. , Proportion and number of cancer cases and deaths attributable to potentially modifiable risk factors in the United States. CA Cancer J Clin, 2018. 68(1): p. 31–54. [DOI] [PubMed] [Google Scholar]
- 221.Faulds MH and Dahlman-Wright K, Metabolic diseases and cancer risk. Curr Opin Oncol, 2012. 24(1): p. 58–61. [DOI] [PubMed] [Google Scholar]
- 222.Lauby-Secretan B, et al. , Body Fatness and Cancer--Viewpoint of the IARC Working Group. N Engl J Med, 2016. 375(8): p. 794–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Whiteman DC and Wilson LF, The fractions of cancer attributable to modifiable factors: A global review. Cancer Epidemiol, 2016. 44: p. 203–221. [DOI] [PubMed] [Google Scholar]
- 224.Wilson LF, et al. , How many cancer cases and deaths are potentially preventable? Estimates for Australia in 2013. Int J Cancer, 2018. 142(4): p. 691–701. [DOI] [PubMed] [Google Scholar]
- 225.Michels KB and Ekbom A, Caloric restriction and incidence of breast cancer. JAMA, 2004. 291(10): p. 1226–30. [DOI] [PubMed] [Google Scholar]
- 226.Bassett WW, et al. , Impact of obesity on prostate cancer recurrence after radical prostatectomy: data from CaPSURE. Urology, 2005. 66(5): p. 1060–5. [DOI] [PubMed] [Google Scholar]
- 227.Chlebowski RT, Nutrition and physical activity influence on breast cancer incidence and outcome. Breast, 2013. 22 Suppl 2: p. S30–7. [DOI] [PubMed] [Google Scholar]
- 228.Van Blarigan EL and Meyerhardt JA, Role of physical activity and diet after colorectal cancer diagnosis. J Clin Oncol, 2015. 33(16): p. 1825–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Ornish D, et al. , Intensive lifestyle changes may affect the progression of prostate cancer. J Urol, 2005. 174(3): p. 1065–9; discussion 1069–70. [DOI] [PubMed] [Google Scholar]
- 230.Parsons JK, et al. , Effect of a Behavioral Intervention to Increase Vegetable Consumption on Cancer Progression Among Men With Early-Stage Prostate Cancer: The MEAL Randomized Clinical Trial. JAMA, 2020. 323(2): p. 140–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Kenfield SA, et al. , Physical activity and survival after prostate cancer diagnosis in the health professionals follow-up study. J Clin Oncol, 2011. 29(6): p. 726–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Chan DSM, et al. , Body mass index and survival in women with breast cancer-systematic literature review and meta-analysis of 82 follow-up studies. Ann Oncol, 2014. 25(10): p. 1901–1914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Demark-Wahnefried W, et al. , Weight management and physical activity throughout the cancer care continuum. CA Cancer J Clin, 2018. 68(1): p. 64–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Rock CL, et al. , American Cancer Society guideline for diet and physical activity for cancer prevention. CA Cancer J Clin, 2020. 70(4): p. 245–271. [DOI] [PubMed] [Google Scholar]
- 235.Bose S, Allen AE, and Locasale JW, The Molecular Link from Diet to Cancer Cell Metabolism. Mol Cell, 2020. 78(6): p. 1034–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Sullivan MR, et al. , Quantification of microenvironmental metabolites in murine cancers reveals determinants of tumor nutrient availability. Elife, 2019. 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Lv M, et al. , Roles of caloric restriction, ketogenic diet and intermittent fasting during initiation, progression and metastasis of cancer in animal models: a systematic review and meta-analysis. PLoS One, 2014. 9(12): p. e115147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Hursting SD, et al. , Calorie restriction, aging, and cancer prevention: mechanisms of action and applicability to humans. Annu Rev Med, 2003. 54: p. 131–52. [DOI] [PubMed] [Google Scholar]
- 239.Poff AM, et al. , The ketogenic diet and hyperbaric oxygen therapy prolong survival in mice with systemic metastatic cancer. PLoS One, 2013. 8(6): p. e65522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Weber DD, et al. , Ketogenic diet in the treatment of cancer - Where do we stand? Mol Metab, 2020. 33: p. 102–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Chen M, et al. , An aberrant SREBP-dependent lipogenic program promotes metastatic prostate cancer. Nat Genet, 2018. 50(2): p. 206–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Llaverias G, et al. , A Western-type diet accelerates tumor progression in an autochthonous mouse model of prostate cancer. Am J Pathol, 2010. 177(6): p. 3180–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Kalaany NY and Sabatini DM, Tumours with PI3K activation are resistant to dietary restriction. Nature, 2009. 458(7239): p. 725–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Sullivan MR, et al. , Increased Serine Synthesis Provides an Advantage for Tumors Arising in Tissues Where Serine Levels Are Limiting. Cell Metab, 2019. 29(6): p. 1410–1421 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Pollak M, The insulin and insulin-like growth factor receptor family in neoplasia: an update. Nat Rev Cancer, 2012. 12(3): p. 159–69. [DOI] [PubMed] [Google Scholar]
- 246.Boyd DB, Insulin and cancer. Integr Cancer Ther, 2003. 2(4): p. 315–29. [DOI] [PubMed] [Google Scholar]
- 247.Nogueira LM, et al. , Dose-dependent effects of calorie restriction on gene expression, metabolism, and tumor progression are partially mediated by insulin-like growth factor-1. Cancer Med, 2012. 1(2): p. 275–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Kanarek N, Petrova B, and Sabatini DM, Dietary modifications for enhanced cancer therapy. Nature, 2020. 579(7800): p. 507–517. [DOI] [PubMed] [Google Scholar]
- 249.Gallagher EJ and LeRoith D, Hyperinsulinaemia in cancer. Nat Rev Cancer, 2020. [DOI] [PubMed] [Google Scholar]
- 250.Nencioni A, et al. , Fasting and cancer: molecular mechanisms and clinical application. Nat Rev Cancer, 2018. 18(11): p. 707–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.de Groot S, et al. , Fasting mimicking diet as an adjunct to neoadjuvant chemotherapy for breast cancer in the multicentre randomized phase 2 DIRECT trial. Nat Commun, 2020. 11(1): p. 3083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Gao X, et al. , Dietary methionine influences therapy in mouse cancer models and alters human metabolism. Nature, 2019. 572(7769): p. 397–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Lee C, et al. , Fasting cycles retard growth of tumors and sensitize a range of cancer cell types to chemotherapy. Sci Transl Med, 2012. 4(124): p. 124ra27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Kanarek N, et al. , Histidine catabolism is a major determinant of methotrexate sensitivity. Nature, 2018. 559(7715): p. 632–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Mayne ST, Playdon MC, and Rock CL, Diet, nutrition, and cancer: past, present and future. Nat Rev Clin Oncol, 2016. 13(8): p. 504–15. [DOI] [PubMed] [Google Scholar]
- 256.Lien EC and Vander Heiden MG, A framework for examining how diet impacts tumour metabolism. Nat Rev Cancer, 2019. 19(11): p. 651–661. [DOI] [PubMed] [Google Scholar]
- 257.Maddocks ODK, et al. , Modulating the therapeutic response of tumours to dietary serine and glycine starvation. Nature, 2017. 544(7650): p. 372–376. [DOI] [PubMed] [Google Scholar]
- 258.Maddocks OD, et al. , Serine starvation induces stress and p53-dependent metabolic remodelling in cancer cells. Nature, 2013. 493(7433): p. 542–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Gravel SP, et al. , Serine deprivation enhances antineoplastic activity of biguanides. Cancer Res, 2014. 74(24): p. 7521–33. [DOI] [PubMed] [Google Scholar]
- 260.Guo H, et al. , Therapeutic tumor-specific cell cycle block induced by methionine starvation in vivo. Cancer Res, 1993. 53(23): p. 5676–9. [PubMed] [Google Scholar]
- 261.Hoshiya Y, et al. , Human tumors are methionine dependent in vivo. Anticancer Res, 1995. 15(3): p. 717–8. [PubMed] [Google Scholar]
- 262.Kirschner MW, The meaning of systems biology. Cell, 2005. 121(4): p. 503–504. [DOI] [PubMed] [Google Scholar]
- 263.Griffin JL and Shockcor JP, Metabolic profiles of cancer cells. Nat Rev Cancer, 2004. 4(7): p. 551–61. [DOI] [PubMed] [Google Scholar]
- 264.Showalter MR, Cajka T, and Fiehn O, Epimetabolites: discovering metabolism beyond building and burning. Curr Opin Chem Biol, 2017. 36: p. 70–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Nunes SC, Tumor Microenvironment – Selective Pressures Boosting Cancer Progression, in Tumor Microenvironment : The Main Driver of Metabolic Adaptation, Serpa J, Editor. 2020, Springer International Publishing: Cham. p. 35–49. [Google Scholar]
- 266.Wishart DS, et al. , HMDB 3.0--The Human Metabolome Database in 2013. Nucleic Acids Res, 2013. 41(Database issue): p. D801–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Spicer R, et al. , Navigating freely-available software tools for metabolomics analysis. Metabolomics, 2017. 13(9): p. 106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Schrimpe-Rutledge AC, et al. , Untargeted Metabolomics Strategies-Challenges and Emerging Directions. J Am Soc Mass Spectrom, 2016. 27(12): p. 1897–1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Posse S, et al. , MR spectroscopic imaging: principles and recent advances. J Magn Reson Imaging, 2013. 37(6): p. 1301–25. [DOI] [PubMed] [Google Scholar]
- 270.Crecelius AC, Schubert US, and von Eggeling F, MALDI mass spectrometric imaging meets “omics”: recent advances in the fruitful marriage. Analyst, 2015. 140(17): p. 5806–20. [DOI] [PubMed] [Google Scholar]
- 271.Emwas AH, et al. , NMR Spectroscopy for Metabolomics Research. Metabolites, 2019. 9(7). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Breitkopf SB, et al. , A relative quantitative positive/negative ion switching method for untargeted lipidomics via high resolution LC-MS/MS from any biological source. Metabolomics, 2017. 13(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Ramautar R, Capillary Electrophoresis-Mass Spectrometry for Clinical Metabolomics. Adv Clin Chem, 2016. 74: p. 1–34. [DOI] [PubMed] [Google Scholar]
- 274.Broadhurst D, et al. , Guidelines and considerations for the use of system suitability and quality control samples in mass spectrometry assays applied in untargeted clinical metabolomic studies. Metabolomics, 2018. 14(6): p. 72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Beckmann M, et al. , High-throughput, nontargeted metabolite fingerprinting using nominal mass flow injection electrospray mass spectrometry. Nat Protoc, 2008. 3(3): p. 486–504. [DOI] [PubMed] [Google Scholar]
- 276.Sarvin B, et al. , Fast and sensitive flow-injection mass spectrometry metabolomics by analyzing sample-specific ion distributions. Nat Commun, 2020. 11(1): p. 3186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Han X and Gross RW, Shotgun lipidomics: multidimensional MS analysis of cellular lipidomes. Expert Rev Proteomics, 2005. 2(2): p. 253–64. [DOI] [PubMed] [Google Scholar]
- 278.Metallo CM, Walther JL, and Stephanopoulos G, Evaluation of 13C isotopic tracers for metabolic flux analysis in mammalian cells. J Biotechnol, 2009. 144(3): p. 167–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Alves TC, et al. , Integrated, Step-Wise, Mass-Isotopomeric Flux Analysis of the TCA Cycle. Cell Metab, 2015. 22(5): p. 936–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Buescher JM, et al. , A roadmap for interpreting (13)C metabolite labeling patterns from cells. Curr Opin Biotechnol, 2015. 34: p. 189–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Bruntz RC, et al. , Exploring cancer metabolism using stable isotope-resolved metabolomics (SIRM). J Biol Chem, 2017. 292(28): p. 11601–11609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Chen L, et al. , Characterization of the human tear metabolome by LC-MS/MS. J Proteome Res, 2011. 10(10): p. 4876–82. [DOI] [PubMed] [Google Scholar]
- 283.Wang C, et al. , Metabolomic analysis based on 1H-nuclear magnetic resonance spectroscopy metabolic profiles in tuberculous, malignant and transudative pleural effusion. Mol Med Rep, 2017. 16(2): p. 1147–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Delgado-Povedano MM, et al. , Metabolomics analysis of human sweat collected after moderate exercise. Talanta, 2018. 177: p. 47–65. [DOI] [PubMed] [Google Scholar]
- 285.Gardner A, Carpenter G, and So PW, Salivary Metabolomics: From Diagnostic Biomarker Discovery to Investigating Biological Function. Metabolites, 2020. 10(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Serkova NJ, et al. , The metabolites citrate, myo-inositol, and spermine are potential age-independent markers of prostate cancer in human expressed prostatic secretions. Prostate, 2008. 68(6): p. 620–8. [DOI] [PubMed] [Google Scholar]
- 287.Sumner LW, et al. , Proposed minimum reporting standards for chemical analysis Chemical Analysis Working Group (CAWG) Metabolomics Standards Initiative (MSI). Metabolomics, 2007. 3(3): p. 211–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Pinu FR, Goldansaz SA, and Jaine J, Translational Metabolomics: Current Challenges and Future Opportunities. Metabolites, 2019. 9(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Vogelstein B and Kinzler KW, Cancer genes and the pathways they control. Nat Med, 2004. 10(8): p. 789–99. [DOI] [PubMed] [Google Scholar]
- 290.Reid MA, Dai Z, and Locasale JW, The impact of cellular metabolism on chromatin dynamics and epigenetics. Nat Cell Biol, 2017. 19(11): p. 1298–1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Rose G, et al. , Colon cancer and blood-cholesterol. Lancet, 1974. 1(7850): p. 181–3. [DOI] [PubMed] [Google Scholar]
- 292.Reddy BS, Mastromarino A, and Wynder EL, Further leads on metabolic epidemiology of large bowel cancer. Cancer Res, 1975. 35(11 Pt. 2): p. 3403–6. [PubMed] [Google Scholar]
- 293.Harvei S, et al. , Prediagnostic level of fatty acids in serum phospholipids: omega-3 and omega-6 fatty acids and the risk of prostate cancer. Int J Cancer, 1997. 71(4): p. 545–51. [DOI] [PubMed] [Google Scholar]
- 294.Saadatian-Elahi M, et al. , Serum fatty acids and risk of breast cancer in a nested case-control study of the New York University Women’s Health Study. Cancer Epidemiol Biomarkers Prev, 2002. 11(11): p. 1353–60. [PubMed] [Google Scholar]
- 295.Vatten LJ, et al. , Polyunsaturated fatty acids in serum phospholipids and risk of breast cancer: a case-control study from the Janus serum bank in Norway. Eur J Cancer, 1993. 29A(4): p. 532–8. [DOI] [PubMed] [Google Scholar]
- 296.His M, et al. , Prospective analysis of circulating metabolites and breast cancer in EPIC. BMC Med, 2019. 17(1): p. 178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Kuhn T, et al. , Higher plasma levels of lysophosphatidylcholine 18:0 are related to a lower risk of common cancers in a prospective metabolomics study. BMC Med, 2016. 14: p. 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Guertin KA, et al. , Serum biomarkers of habitual coffee consumption may provide insight into the mechanism underlying the association between coffee consumption and colorectal cancer. Am J Clin Nutr, 2015. 101(5): p. 1000–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Playdon MC, et al. , Nutritional metabolomics and breast cancer risk in a prospective study. Am J Clin Nutr, 2017. 106(2): p. 637–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Huang J, et al. , Prospective serum metabolomic profiling of lethal prostate cancer. Int J Cancer, 2019. 145(12): p. 3231–3243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.Zeleznik OA, et al. , A Prospective Analysis of Circulating Plasma Metabolites Associated with Ovarian Cancer Risk. Cancer Res, 2020. 80(6): p. 1357–1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Kritchevsky SB, et al. , Changes in plasma lipid and lipoprotein cholesterol and weight prior to the diagnosis of cancer. Cancer Res, 1991. 51(12): p. 3198–203. [PubMed] [Google Scholar]
- 303.Bamji-Stocke S, et al. , A review of metabolism-associated biomarkers in lung cancer diagnosis and treatment. Metabolomics, 2018. 14(6): p. 81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304.Gunther UL, Metabolomics Biomarkers for Breast Cancer. Pathobiology, 2015. 82(3–4): p. 153–65. [DOI] [PubMed] [Google Scholar]
- 305.Mahajan UM, et al. , Metabolic Biomarkers of Pancreatic Cancer, in Translational Pancreatic Cancer Research: From Understanding of Mechanisms to Novel Clinical Trials, Michalski CW, et al., Editors. 2020, Springer International Publishing: Cham. p. 83–96. [Google Scholar]
- 306.Kdadra M, et al. , Metabolomics Biomarkers of Prostate Cancer: A Systematic Review. Diagnostics (Basel), 2019. 9(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307.Erben V, et al. , Metabolomics Biomarkers for Detection of Colorectal Neoplasms: A Systematic Review. Cancers (Basel), 2018. 10(8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308.Som P, et al. , A fluorinated glucose analog, 2-fluoro-2-deoxy-D-glucose (F-18): nontoxic tracer for rapid tumor detection. J Nucl Med, 1980. 21(7): p. 670–5. [PubMed] [Google Scholar]
- 309.Fletcher JW, et al. , Recommendations on the use of 18F-FDG PET in oncology. J Nucl Med, 2008. 49(3): p. 480–508. [DOI] [PubMed] [Google Scholar]
- 310.Furumoto S, et al. , In vitro and in vivo characterization of 2-deoxy-2–18F-fluoro-D-mannose as a tumor-imaging agent for PET. J Nucl Med, 2013. 54(8): p. 1354–61. [DOI] [PubMed] [Google Scholar]
- 311.Arumugam T, et al. , Preliminary evaluation of 1’-[(18)F]fluoroethyl-beta-D-lactose ([(18)F]FEL) for detection of pancreatic cancer in nude mouse orthotopic xenografts. Nucl Med Biol, 2014. 41(10): p. 833–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312.Wuest M, et al. , Radiopharmacological evaluation of 6-deoxy-6-[18F]fluoro-D-fructose as a radiotracer for PET imaging of GLUT5 in breast cancer. Nucl Med Biol, 2011. 38(4): p. 461–75. [DOI] [PubMed] [Google Scholar]
- 313.Sorensen M, et al. , The potential use of 2-[(1)(8)F]fluoro-2-deoxy-D-galactose as a PET/CT tracer for detection of hepatocellular carcinoma. Eur J Nucl Med Mol Imaging, 2011. 38(9): p. 1723–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314.Plaat B, et al. , Protein synthesis rate measured with L-[1–11C]tyrosine positron emission tomography correlates with mitotic activity and MIB-1 antibody-detected proliferation in human soft tissue sarcomas. Eur J Nucl Med, 1999. 26(4): p. 328–32. [DOI] [PubMed] [Google Scholar]
- 315.van den Bergh AC, et al. , Tyrosine positron emission tomography and protein synthesis rate in pituitary adenoma: different effects of surgery and radiation therapy. Radiother Oncol, 2011. 98(2): p. 213–6. [DOI] [PubMed] [Google Scholar]
- 316.Bustany P, et al. , Brain tumor protein synthesis and histological grades: a study by positron emission tomography (PET) with C11-L-Methionine. J Neurooncol, 1986. 3(4): p. 397–404. [DOI] [PubMed] [Google Scholar]
- 317.Wu F, et al. , Uptake of 14C- and 11C-labeled glutamate, glutamine and aspartate in vitro and in vivo. Anticancer Res, 2000. 20(1A): p. 251–6. [PubMed] [Google Scholar]
- 318.Lieberman BP, et al. , PET imaging of glutaminolysis in tumors by 18F-(2S,4R)4-fluoroglutamine. J Nucl Med, 2011. 52(12): p. 1947–55. [DOI] [PubMed] [Google Scholar]
- 319.Wu Z, et al. , [(18)F](2S,4S)-4-(3-Fluoropropyl)glutamine as a tumor imaging agent. Mol Pharm, 2014. 11(11): p. 3852–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320.Venneti S, et al. , Glutamine-based PET imaging facilitates enhanced metabolic evaluation of gliomas in vivo. Sci Transl Med, 2015. 7(274): p. 274ra17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321.Odewole OA, et al. , Recurrent prostate cancer detection with anti-3-[(18)F]FACBC PET/CT: comparison with CT. Eur J Nucl Med Mol Imaging, 2016. 43(10): p. 1773–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322.Iozzo P, et al. , Fatty acid metabolism in the liver, measured by positron emission tomography, is increased in obese individuals. Gastroenterology, 2010. 139(3): p. 846–56, 856 e1–6. [DOI] [PubMed] [Google Scholar]
- 323.DeGrado TR, et al. , Synthesis and evaluation of 18F-labeled choline as an oncologic tracer for positron emission tomography: initial findings in prostate cancer. Cancer Res, 2001. 61(1): p. 110–7. [PubMed] [Google Scholar]
- 324.Glunde K and Bhujwalla ZM, Metabolic tumor imaging using magnetic resonance spectroscopy. Semin Oncol, 2011. 38(1): p. 26–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 325.Andronesi OC, et al. , Detection of oncogenic IDH1 mutations using magnetic resonance spectroscopy of 2-hydroxyglutarate. J Clin Invest, 2013. 123(9): p. 3659–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 326.Choi C, et al. , 2-hydroxyglutarate detection by magnetic resonance spectroscopy in IDH-mutated patients with gliomas. Nat Med, 2012. 18(4): p. 624–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327.Schmidt-Hansen M, et al. , PET-CT for assessing mediastinal lymph node involvement in patients with suspected resectable non-small cell lung cancer. Cochrane Database Syst Rev, 2014(11): p. CD009519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 328.Cacciatore S, et al. , Metabolic Profiling in Formalin-Fixed and Paraffin-Embedded Prostate Cancer Tissues. Mol Cancer Res, 2017. 15(4): p. 439–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 329.Fan TW, et al. , Altered regulation of metabolic pathways in human lung cancer discerned by (13)C stable isotope-resolved metabolomics (SIRM). Mol Cancer, 2009. 8: p. 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 330.Kami K, et al. , Metabolomic profiling of lung and prostate tumor tissues by capillary electrophoresis time-of-flight mass spectrometry. Metabolomics, 2013. 9(2): p. 444–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 331.Rocha CM, et al. , NMR metabolomics of human lung tumours reveals distinct metabolic signatures for adenocarcinoma and squamous cell carcinoma. Carcinogenesis, 2015. 36(1): p. 68–75. [DOI] [PubMed] [Google Scholar]
- 332.Deja S, et al. , Metabolomics provide new insights on lung cancer staging and discrimination from chronic obstructive pulmonary disease. J Pharm Biomed Anal, 2014. 100: p. 369–380. [DOI] [PubMed] [Google Scholar]
- 333.Zhang X, et al. , Non-targeted and targeted metabolomics approaches to diagnosing lung cancer and predicting patient prognosis. Oncotarget, 2016. 7(39): p. 63437–63448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 334.Alderton GK, Tumour metabolism: Feeding the TCA cycle in vivo. Nat Rev Cancer, 2016. 16(4): p. 198. [DOI] [PubMed] [Google Scholar]
- 335.Wikoff WR, et al. , Metabolomic markers of altered nucleotide metabolism in early stage adenocarcinoma. Cancer Prev Res (Phila), 2015. 8(5): p. 410–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 336.Puchades-Carrasco L, et al. , Serum metabolomic profiling facilitates the non-invasive identification of metabolic biomarkers associated with the onset and progression of non-small cell lung cancer. Oncotarget, 2016. 7(11): p. 12904–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 337.Zhou W, et al. , Proteomic analysis reveals Warburg effect and anomalous metabolism of glutamine in pancreatic cancer cells. J Proteome Res, 2012. 11(2): p. 554–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 338.Budczies J, et al. , Glutamate enrichment as new diagnostic opportunity in breast cancer. Int J Cancer, 2015. 136(7): p. 1619–28. [DOI] [PubMed] [Google Scholar]
- 339.Hirayama A, et al. , Quantitative metabolome profiling of colon and stomach cancer microenvironment by capillary electrophoresis time-of-flight mass spectrometry. Cancer Res, 2009. 69(11): p. 4918–25. [DOI] [PubMed] [Google Scholar]
- 340.Manna SK, et al. , Biomarkers of coordinate metabolic reprogramming in colorectal tumors in mice and humans. Gastroenterology, 2014. 146(5): p. 1313–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 341.Tayyari F, et al. , Metabolic profiles of triple-negative and luminal A breast cancer subtypes in African-American identify key metabolic differences. Oncotarget, 2018. 9(14): p. 11677–11690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 342.Cheng LL, et al. , Metabolic characterization of human prostate cancer with tissue magnetic resonance spectroscopy. Cancer Res, 2005. 65(8): p. 3030–4. [DOI] [PubMed] [Google Scholar]
- 343.Roig B, et al. , Metabolomics reveals novel blood plasma biomarkers associated to the BRCA1-mutated phenotype of human breast cancer. Sci Rep, 2017. 7(1): p. 17831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 344.Sreekumar A, et al. , Metabolomic profiles delineate potential role for sarcosine in prostate cancer progression. Nature, 2009. 457(7231): p. 910–4. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 345.Khan AP, et al. , The role of sarcosine metabolism in prostate cancer progression. Neoplasia, 2013. 15(5): p. 491–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 346.Ankerst DP, et al. , A case control study of sarcosine as an early prostate cancer detection biomarker. BMC Urol, 2015. 15: p. 99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 347.Fathi AT, et al. , Prospective serial evaluation of 2-hydroxyglutarate, during treatment of newly diagnosed acute myeloid leukemia, to assess disease activity and therapeutic response. Blood, 2012. 120(23): p. 4649–52. [DOI] [PubMed] [Google Scholar]
- 348.Zhang F, et al. , Metabolomics for biomarker discovery in the diagnosis, prognosis, survival and recurrence of colorectal cancer: a systematic review. Oncotarget, 2017. 8(21): p. 35460–35472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 349.Long NP, et al. , A systematic review on metabolomics-based diagnostic biomarker discovery and validation in pancreatic cancer. Metabolomics, 2018. 14(8): p. 109. [DOI] [PubMed] [Google Scholar]
- 350.Armitage EG and Barbas C, Metabolomics in cancer biomarker discovery: current trends and future perspectives. J Pharm Biomed Anal, 2014. 87: p. 1–11. [DOI] [PubMed] [Google Scholar]
- 351.Park J, et al. , Plasma metabolites as possible biomarkers for diagnosis of breast cancer. PLoS One, 2019. 14(12): p. e0225129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 352.Xie G, et al. , Plasma metabolite biomarkers for the detection of pancreatic cancer. J Proteome Res, 2015. 14(2): p. 1195–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 353.Mayerle J, et al. , Metabolic biomarker signature to differentiate pancreatic ductal adenocarcinoma from chronic pancreatitis. Gut, 2018. 67(1): p. 128–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 354.Miyagi Y, et al. , Plasma free amino acid profiling of five types of cancer patients and its application for early detection. PLoS One, 2011. 6(9): p. e24143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 355.Babic A, et al. , Postdiagnosis Loss of Skeletal Muscle, but Not Adipose Tissue, Is Associated with Shorter Survival of Patients with Advanced Pancreatic Cancer. Cancer Epidemiol Biomarkers Prev, 2019. 28(12): p. 2062–2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 356.Zhang Z, et al. , Serum and urinary metabonomic study of human osteosarcoma. J Proteome Res, 2010. 9(9): p. 4861–8. [DOI] [PubMed] [Google Scholar]
- 357.Pasikanti KK, et al. , Noninvasive urinary metabonomic diagnosis of human bladder cancer. J Proteome Res, 2010. 9(6): p. 2988–95. [DOI] [PubMed] [Google Scholar]
- 358.Pasikanti KK, et al. , Urinary metabotyping of bladder cancer using two-dimensional gas chromatography time-of-flight mass spectrometry. J Proteome Res, 2013. 12(9): p. 3865–73. [DOI] [PubMed] [Google Scholar]
- 359.Wittmann BM, et al. , Bladder cancer biomarker discovery using global metabolomic profiling of urine. PLoS One, 2014. 9(12): p. e115870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 360.Srivastava S, et al. , Taurine - a possible fingerprint biomarker in non-muscle invasive bladder cancer: A pilot study by 1H NMR spectroscopy. Cancer Biomark, 2010. 6(1): p. 11–20. [DOI] [PubMed] [Google Scholar]
- 361.Struck-Lewicka W, et al. , Urine metabolic fingerprinting using LC-MS and GC-MS reveals metabolite changes in prostate cancer: A pilot study. J Pharm Biomed Anal, 2015. 111: p. 351–61. [DOI] [PubMed] [Google Scholar]
- 362.Derezinski P, et al. , Amino Acid Profiles of Serum and Urine in Search for Prostate Cancer Biomarkers: a Pilot Study. Int J Med Sci, 2017. 14(1): p. 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 363.Shariff MI, et al. , Urinary metabolic biomarkers of hepatocellular carcinoma in an Egyptian population: a validation study. J Proteome Res, 2011. 10(4): p. 1828–36. [DOI] [PubMed] [Google Scholar]
- 364.Shariff MI, et al. , Characterization of urinary biomarkers of hepatocellular carcinoma using magnetic resonance spectroscopy in a Nigerian population. J Proteome Res, 2010. 9(2): p. 1096–103. [DOI] [PubMed] [Google Scholar]
- 365.An Z, et al. , Integrated ionization approach for RRLC-MS/MS-based metabonomics: finding potential biomarkers for lung cancer. J Proteome Res, 2010. 9(8): p. 4071–81. [DOI] [PubMed] [Google Scholar]
- 366.Sasco AJ, et al. , Breast cancer prognostic significance of some modified urinary nucleosides. Cancer Lett, 1996. 108(2): p. 157–62. [DOI] [PubMed] [Google Scholar]
- 367.Seidel A, et al. , Modified nucleosides: an accurate tumour marker for clinical diagnosis of cancer, early detection and therapy control. Br J Cancer, 2006. 94(11): p. 1726–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 368.Zheng YF, et al. , Urinary nucleosides as biological markers for patients with colorectal cancer. World J Gastroenterol, 2005. 11(25): p. 3871–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 369.Phillips M, et al. , Volatile organic compounds in breath as markers of lung cancer: a cross-sectional study. Lancet, 1999. 353(9168): p. 1930–3. [DOI] [PubMed] [Google Scholar]
- 370.Poli D, et al. , Exhaled volatile organic compounds in patients with non-small cell lung cancer: cross sectional and nested short-term follow-up study. Respir Res, 2005. 6: p. 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 371.Phillips M, et al. , Detection of lung cancer with volatile markers in the breath. Chest, 2003. 123(6): p. 2115–23. [DOI] [PubMed] [Google Scholar]
- 372.National Lung Screening Trial Research, T., et al. , Results of initial low-dose computed tomographic screening for lung cancer. N Engl J Med, 2013. 368(21): p. 1980–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 373.Wishart DS, Emerging applications of metabolomics in drug discovery and precision medicine. Nat Rev Drug Discov, 2016. 15(7): p. 473–84. [DOI] [PubMed] [Google Scholar]
- 374.Dang L, et al. , Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature, 2009. 462(7274): p. 739–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 375.Ward PS, et al. , The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell, 2010. 17(3): p. 225–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 376.Massie CE, et al. , The androgen receptor fuels prostate cancer by regulating central metabolism and biosynthesis. EMBO J, 2011. 30(13): p. 2719–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 377.Dugourd A, et al. , Causal integration of multi-omics data with prior knowledge to generate mechanistic hypotheses. Mol Syst Biol, 2021. 17(1): p. e9730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 378.Locasale JW, et al. , Metabolomics of human cerebrospinal fluid identifies signatures of malignant glioma. Mol Cell Proteomics, 2012. 11(6): p. M111 014688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 379.Liu X, et al. , Metformin Targets Central Carbon Metabolism and Reveals Mitochondrial Requirements in Human Cancers. Cell Metab, 2016. 24(5): p. 728–739. [DOI] [PMC free article] [PubMed] [Google Scholar]