

Abstract
Purpose
Therapeutic strategies to treat ischemic stroke are limited due to the heterogeneity of cerebral ischemic injury and the mechanisms that contribute to the cell death. Since oxidative stress is one of the primary mechanisms that cause brain injury post-stroke, we hypothesized that therapeutic targets that modulate mitochondrial function could protect against reperfusion-injury after cerebral ischemia, with the focus here a mitochondrial protein, mitoNEET, that modulates cellular bioenergetics.
Method
In this study, we evaluated the pharmacology of the mitoNEET ligand NL-1 in an in vivo therapeutic role for NL-1 in a C57Bl/6 murine model of ischemic stroke.
Results
NL-1 decreased hydrogen peroxide production with an IC50 of 5.95 μM in neuronal cells (N2A). The in vivo activity of NL-1 was evaluated in a murine 1h transient middle cerebral artery occlusion (t-MCAO) model of ischemic stroke. We found that mice treated with NL-1 (10 mg/kg, i.p.) at time of reperfusion and allowed to recover for 24 hours showed a 43% reduction in infarct volume and 68% reduction in edema compared to sham-injured mice. Additionally, we found that when NL-1 was administered 15 min post-t-MCAO, the ischemia volume was reduced by 41%, and stroke-associated edema by 63%.
Conclusion
As support of our hypothesis, as expected, NL-1 failed to reduce stroke infarct in a permanent photothrombotic occlusion model of stroke. This report demonstrates the potential therapeutic benefits of using mitoNEET ligands like NL-1 as novel mitoceuticals for treating reperfusion-injury with cerebral stroke.
Keywords: OXPHOS, iron-sulfur, CDGSH, bioenergetics, t-MCAO, PAMPA
INTRODUCTION
Stroke is the fifth leading cause of death, and the leading cause of permanent disability in the nation (1–3). Stroke occurs due to an occlusion in the vasculature supplying oxygen and nutrients to the brain, causing an energy crisis in that region which rapidly progresses into tissue death (4). The current treatment strategy for stroke is aimed at removing the occlusion to blood flow and restoring blood supply to the ischemic penumbra in a timely manner. To date, the only therapeutic targeting is to restore blood supply with thrombolytic agents like tissue plasminogen activator (t-PA) and endovascular intervention (thrombectomy) are the most common and only one approaches to this effect, however they do not address local and systemic neuroprotection in brain stroke pathology (3,5). However, when the blood supply is restored and the ischemic tissue is re-perfused, secondary tissue damage is observed, which is generally referred to as ischemia-reperfusion (IR) injury (6–9). A key player in the IR injury are the mitochondria, organelles that play important roles in cellular energetics, apoptosis and reactive oxygen species (ROS) generation (10).
MitoNEET (gene: CISD1) is a recently discovered novel mitochondrial protein located on the outer surface of mitochondria (11). MitoNEET belongs to a group of zinc-finger proteins, although instead of zinc, two redox active iron-sulfur clusters [2Fe-2S] are seen in the crystal structure, acting as redox and pH sensor for mitochondria. Several studies have shown that when mitoNEET is knocked-down or knocked-out, significant effects are seen in the bioenergetic functions of mitochondria including the oxidative phosphorylation (OXPHOS). For instance, mitochondria from mitoNEET knockout mice striatum show increased production of ROS and decreased ATP production. Additionally, when mitoNEET is overexpressed in the adipose of mice, there is a dramatic shift in lipid metabolism with improvements in systemic inflammation and glucose utilization (12–16).
MitoNEET ligands represent a new class of mitochondrially active compounds, which have been shown to have protective effects in models of stroke and traumatic brain injury (17). These include the mixed Peroxisome proliferator-activated receptor gamma (PPAR-γ)/mitoNEET ligand pioglitazone, and recently discovered TT01001 (18). We developed a pioglitazone derivative NL-1, which we have previously shown is a mitoNEET ligand and has neuroprotective activity in Parkinson’s disease model (19) and traumatic brain injury (TBI) (17). Furthermore, we recently published findings that the mitoNEET ligand NL-1 is able to prevent the reperfusion-injury with an in vitro model of oxygen glucose deprivation (20). This in vitro model of ischemia and reperfusion known as the Oxygen-Glucose Deprivation and Reperfusion (OGDR) was employed for in vitro efficacy studies using NL-1. This widely used model mimics ischemic conditions when the cells are cultured in a glucose-free medium such as HBSS and incubated in an environment devoid of oxygen (21–23). Reintroduction of the cells into the normal medium and environment mimics the reperfusion after ischemia. These promising reports led us to explore the effect of NL-1 on mitochondrial function and ischemic brain damage following IR injury of a stroke murine model since attenuation of mitochondrial dysfunction will allow for improved recovery (24,25). Here we hypothesized that NL-1 will be neuroprotective in a reperfusion-injury model of stroke but fail to be protective in a permanent occlusion model of stroke. The work presented here further supports that the mitoNEET class of compounds, including NL-1, can attenuate tissue damage following reperfusion injury after a transient occlusion of the middle cerebral artery (tMCAO) as new class of therapeutics agent for improving mitochondrial function after stroke.
MATERIALS AND METHODS
Experimental animals
All mice were housed in the vivarium facilities, and all experiments were done in accordance with the West Virginia University Institutional Animal Care and Use Committee, and according to the Guide for the Care and Use of Laboratory Animals as adopted and promulgated by the U.S. National Institutes of Health. Male C57BL/6J mice (3-4 months) were used for these assays. Each group of mice was randomly assigned to treatment group, with number of animals ranging 4-5 in each group.
PPAR-γ activating assay
PPAR-γ activation of NL-1 was determined in vitro using a PPAR-γ activity assay (IndigoBiosciences). Neuronal cells containing a PPAR-γ luciferase construct were reconstituted in cell recovery medium and 100 μL of reconstituted cell suspension was seeded on to white 96 well plates. The plated cells were treated with the positive control rosiglitazone at 4 μM as recommended by the manufacturer to ensure reproducible activation, and NL-1, tested at 10 μM, which was the standard concentration evaluated in vitro. Plates were incubated at 37°C for 24 hours. After 24 hours, cell media was aspirated and 100 μL of luciferase detection reagent was added to each well, and the plates allowed to rest at room temperature for 5 minutes. Luciferase activity was measured by quantifying luminescence using a plate reader.
Glucose tolerance test
Glucose tolerance test (GTT) was performed in C57BL/6J mice by intraperitoneal (i.p.) injection of glucose (1.5 g/kg of bodyweight) after overnight fasting. The mice were then administered DMSO (vehicle control) or the test compound, NL-1 (10 mg/kg; i.p.). DMSO was used as the vehicle as it was the drug solvent, to ensure the effects observed were due to drug alone. Blood was collected from the tail vein at the time points of 30, 60, 90 and 120 minutes following glucose injection. Glucose levels were measured using a OneTouch® glucometer (LifeScan, Milipitas, CA).
Kinase Panel
NL-1 was screened against a panel of kinases from Reaction Biology at 10 μM (www.reactionbiology.com). The panel included a set of 349 typical kinases, while 20 atypical kinases were tested for inhibition. NL-1 was tested at 10 μM in DMSO, and the concentration of ATP used for the kinase screen was 10 μM. Staurosporine served as a positive control.
Electron Transport Chain Complex Activities
Electron transport chain (ETC) complexes I and IV activities were measured spectrophotometrically as previously described (27). Briefly, brain was resected from mice. Mitochondria were isolated from organs using BioVision Mammalian Mitochondria Isolation Kit for Tissue & Cultured Cells (Catalog #: K288), resuspended in 1 mL KME buffer each, and stored at −80°C. Complex I (NADH dehydrogenase) activity was determined by measuring NADH oxidation at 340nm. Complex IV (cytochrome c oxidase) activity was evaluated by measuring the oxidation of cytochrome c at 550 nm. Each treatment group was comprised of 8 replicate wells in a 96-well plate. Bradford assay was used to measure protein concentration in isolated mitochondria from brain and liver that was used when performing calculations to obtain measurements expressed as nanomoles substrate converted/min/mg of protein. The mean measurement of each treatment group was compared with the mean of the control group (DMSO-only) with a One-Way ANOVA using GraphPad Prism 8 software program following the exclusion of outliers as determined by ROUT (Q = 1%) outlier method.
Mitochondrial membrane potential
The effect of NL-1 on mitochondrial membrane potential was assessed using previously published method (28). Briefly, N2A cells were seeded into black 96 well plates. The next day, this was followed by the addition of ursodeoxycholic acid (500 nM) as a positive control, and NL-1 at 10 μM for 30 minutes, followed by the addition of TMRM (10 nM final concentration) for an additional 10 minutes. Brilliant Black (1 mg/ml stock) was added the plates to quench background fluorescence. The plate was read using a BioTek Cytation 3 plate reader at 540 nm excitation and 590 nm emission wavelengths.
Cellular respiration
The Seahorse assay using the Seahorse XF96 (Agilent Technologies, Santa Clara, CA) was used to assess the impact of NL-1 treatment on cellular respiration and glycolysis. N2a cells were seeded into collagen-coated Seahorse 96-well XF Cell culture microplate at a density of 20000 cells per well. Cells reached confluence after 24 hours and were then subjected to treatment with vehicle control or 10 μM NL-1 for either 3 or 24 hours. The Seahorse XF96 measured oxygen consumption rate (OCR) under different conditions using sequential addition of oligomycin, carbonyl cyanide p-trifluoromethoxyphenylhydrazone (FCCP), antimycin A and rotenone. The assay provided information about the parameters of mitochondrial respiration such as basal respiration, ATP-linked respiration, proton link, maximal respiratory capacity, spare respiratory capacity and non-mitochondrial respiration. The second measurement performed by Seahorse XF96 was that of the extracellular acidification rate (ECAR), which is a measure of cellular glycolysis. The Seahorse XF96 measured ECAR under different conditions using sequential addition of glucose, oligomycin and 2-deoxyglucose. The assay provided information about the parameters such as glycolysis, glycolytic capacity, glycolytic reserve and non-glycolytic acidification.
Oxygen glucose deprivation and reperfusion (OGDR)
An in vitro cerebral ischemia and reperfusion model was employed by exposing the cells to oxygen-glucose deprivation and reperfusion (OGDR). Cell culture medium was replaced with the glucose deficient Hank’s Balanced Salt Solution (HBSS), containing 10% FBS and transferred to the BioSpherix X3 Xvivo System hypoxia chamber, with 0.1-0.2% oxygen, 5% carbon dioxide. The temperature of hypoxia chamber was maintained at 37°C. The cells were incubated in hypoxia chamber for 3 hours. After the desired period of hypoxia, the glucose-free medium was replaced with normal cell growth medium and cells were incubated in normal growth environment for 24 hours, mimicking the reperfusion following cerebral ischemia (29). The in vitro efficacy of NL-1 in reducing generation of ROS was studied using Amplex Red assay (Thermo Scientific). N2a cells were plated in a black 96-well plate at a density of 10,000 cells per well. After 24 hours, cells were subjected to OGDR. The cells were treated with NL-1 drug during the period of recovery. After the 24-hour recovery, 50 μL of Amplex Red working reagent was added to each well and allowed to react in dark at room temperature for 30 minutes. The amount of hydrogen peroxide in each well was measured by fluorimetry using the BioTek Synergy H1 plate reader (BioTek Instruments, Winooski, VT), at an excitation and emission wavelength of 530 nm and 590 nm, respectively. The data of each plate were normalized for the control wells of that plate, and then compared with other plate replicates.
Brain distribution/pharmacokinetic study
A pharmacokinetic study to determine the distribution profile of NL-1 in support of the stroke studies, were done to determine brain exposure of NL-1 (30). NL-1 was dissolved in DMSO and injected at 10 mg/kg i.p. (1 μL per gram body weight) into C57BL/6J mice, and after a specified period of time euthanized to remove the brain and plasma. The brains were weighed and 1 part (v/w) PBS pH 7.4 was added, and homogenized using a Teflon tipped homogenizer. Blood samples were collected in heparinized tubes, and centrifuged at 5,000 rpm for 10 min. NL-1 was extracted by adding an equal volume of acetonitrile and vortexing the samples, and centrifuging for 10 min. at 10,000 rpm. Samples were analyzed by LC-MS/MS to determine concentration of NL-1 in brain with an ABSciex 5500 LC-MS/MS as described before (20,30). To determine the [brain]/[plasma] ratio of NL-1, 10 mg/kg was delivered I.V and the brain and plasma concentration determined after 30 min of exposure.
Artificial membrane permeability assay
To predict the in vitro BBB permeability of NL-1, an artificial membrane permeably assay was performed as described earlier (31). Briefly, a solution of hexadecane in hexane (5% v/v) was added (15 mL) to a donor plate containing a polycarbonate (3 μm) filter membrane, and allowed to evaporate for 45 min in a fume hood. A stock concentration of NL-1 in phosphate buffered saline (PBS) pH 7.4 was made at 500 μM final concentration, by adding 7 μL of a 10 mM NL-1 stock in pure DMSO to the PBS. A 150 μL of this 500 μM NL-1 solution was added to the donor well. The acceptor well contained PBS with 5% DMSO to match the concentration of DMSO in the top well, and the donor plate placed on top of the acceptor plate. After a 5-hour incubation period, the solutions were analyzed with LC-MS/MS. Propranolol was used as control. Data was presented as logPe.
Transient middle cerebral artery occlusion (t-MCAO)
In vivo cerebral ischemia was induced as described in detail previously (32). Briefly, C57BL/6J mice were subjected to 1h of transient middle cerebral artery occlusion (t-MCAO). Mice were anesthetized with 3 % isoflurane and maintained at 1-1.5% isoflurane during surgery. During the procedure, body temperature was maintained at 37 °C by a thermostatic blanket, and cerebral blood flow was monitored by laser Doppler flowmetry (Moor Instruments, Wilmington, DE). For drug treatment, NL-1 (10 mg/kg, i.p.) was injected at the time of reperfusion. For a post reperfusion study, NL-1 was administered 15 minutes after reperfusion of brain tissue was carried out to mimic aggressive clinical manipulation required in management of brain stroke (post-treatment). Vehicle (DMSO) treated mice served as a control. After 24 hours, the mice were deeply anaesthetized with isofluorane (4%) and euthanized by decapitation. The brains were quickly removed and sectioned into coronal slices of 1-mm thickness (McIlwain Tissue Chopper, Redding CA). Slices were incubated in a 1% solution of 2,3,5-triphenyltetrazolium chloride (TTC) in phosphate buffered saline at 37°C (water bath) for 15 min and then fixed by immersion in 4% buffered formaldehyde solution. Images were acquired by digital camera (Nikon Coolpix, Melville, NY), and areas of both hemispheres and the infarcted regions were quantified for each slice using image analysis software (Image J, NIH). We measured the areas of infarction (volume), and brain edema (tissue swelling) by area increase in the ipsilateral (ischemic) hemisphere, and compared these with the contralateral (non-ischemic) hemisphere. This allowed for a direct comparison of areas of ischemia (volume) with brain edema and allowed us to use each animal as its own control. Quantification of infarct volume and edema formation were done according to previously published protocols. Measurements were made on each brain slice to calculate the volume of the lesion and to correct for overestimation (33) due to the effects of brain edema (brain swelling) where: a = area of infarct (mm2), b = area of the infarcted (ipsilateral) hemisphere slice (mm2), c = area of the non-infarcted (contralateral) hemisphere slice (mm2), d = brain swelling (mm2) = (b−c). The area (Al) of the lesion (mm2), corrected for swelling, was derived from the equation Al = a−d. The edema (swelling) area was designated Ae and quantified by determining the ratio between the areas of the infracted and non-infarcted hemisphere slices, thus: Ae = b−c.
Photothrombic stroke model
Rose Bengal (1 mg/mL in saline) was administered to C57BL/6 mice via IV tail vein as described before (34). The skin was surgically opened exposing the mouse skull. A laser light with 532 nm green laser was used to illuminate the brain for 15 minutes after injection of Rose Bengal. If the mice were injected with a NL-1 or vehicle, treatment occurred 15 min after Rose Bengal administration.
Immunohistochemistry
Immunofluorescence staining was performed on mouse brain coronal sections. The slides with were allowed to thaw at room temperature for about 15 minutes. The sections were then fixed with addition of methanol for 3 minutes, upon which, the slides washed once with 1X PBS. The tissue permeabilization was carried out in a solution of 4% v/v horse serum and 1% w/v TritonX-100 in PBS for one hour, followed by washing thrice with PBS. The slides were then stained for 4-hydroxynonenal (4HNE) (Novus biologicals, mouse, 1:200 dilution), an ROS marker. The primary antibody incubation was carried out in the dark at 4°C, overnight. The following day, the primary antibody was removed and the slides were washed thrice with PBS, prior to secondary antibody incubation. AlexaFluor 568 (Invitrogen, 1:1000 dilution) was used as the fluorescent secondary antibody and the incubation was carried out for 2 hours in the dark at room temperature, following which, the slides were washed thrice with PBS. After the final wash, the slides were cover-slipped using the Flouromount-G (Southern Biotech) mounting medium, and allowed to dry prior to imaging. The slides were imaged by fluorescence microscopy using an Olympus Slide Scanner microscope.
Statistical analysis
Data were analyzed using GraphPad Prism 6.0 (GraphPad Software, La Jolla, CA) were each experiment had technical repeats. Differences between two or more groups were assessed using Student’s t-test and one-way ANOVA, respectively. Data are expressed as mean ± standard deviation. Values of P < 0.05 were considered statistically significant in all experiments. Number of mice used for the study was done based on our previous published data (35).
RESULTS
PPAR-γ activation by NL-1
Figure 1B shows the results of luminescence intensity induced by the activation of PPAR-γ for vehicle, the positive control rosiglitazone and the test compound NL-1. These results indicate that NL-1 (10 μM) did not activate PPAR-γ at the dose tested. The choice of 10 μM for NL-1 was based on our evaluation of NL-1 in other in vitro assay including the mitochondrial function.
Fig. 1:
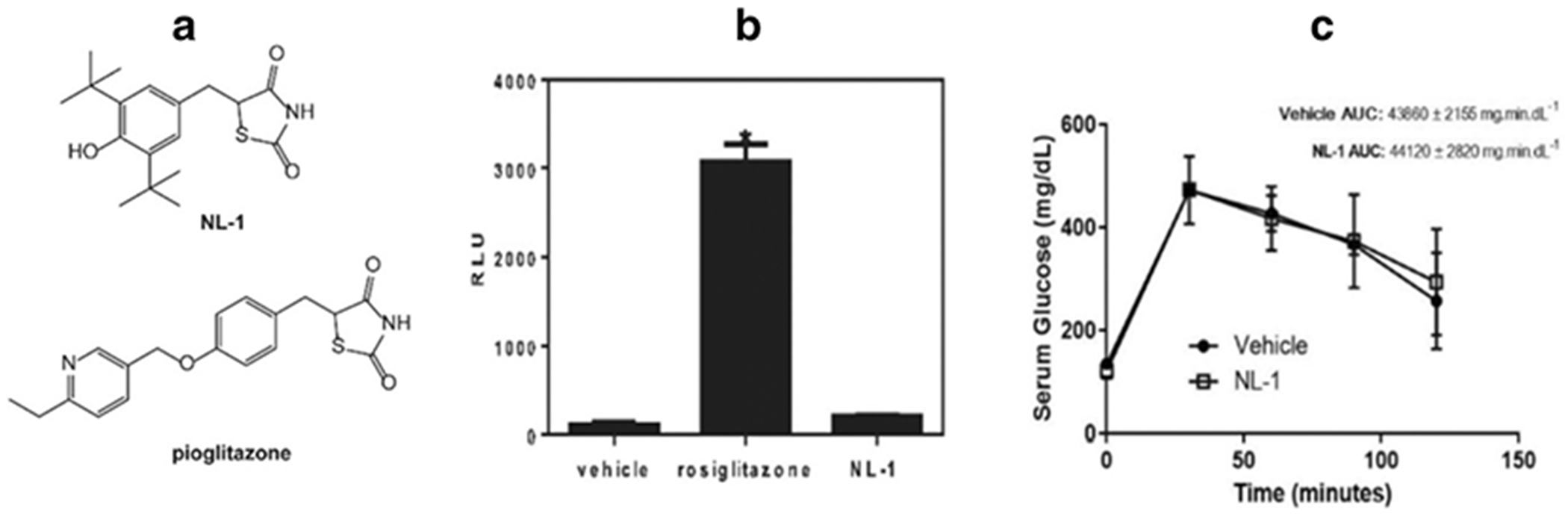
(A) Structures of NL-1 and pioglitazone (B) The mitoNEET ligand NL-1 does not activate PPAR-gamma receptor. The positive control rosiglitazone had significantly higher bioluminescence activity compared to control and NL-1 (P<0.05) N = 4. (C) NL-1 at 10 mg/kg did not lower blood glucose after glucose tolerance test. Each data point represents mean ± S.D. where N = 4.
NL-1 effect on blood glucose levels
The ability of NL-1 to affect blood glucose levels was tested using an intraperitoneal glucose tolerance test. Figure 1C shows a glucose concentration (mg/dL) versus time plot for vehicle and NL-1 treated mice. NL-1 did not have a significant effect on altering blood glucose levels when dosed at 10 mg/kg. The AUC values for vehicle and NL-1 treated groups were found to be 43860 ± 2155 mg.min.dL−1 and 44120 ± 2820 mg.min.dL−1, respectively.
NL-1 effect on kinases
To test the kinase inhibition of NL-1, we used a kinase panel consisting of wild type and atypical kinases. NL-1 did not appreciably inhibit any of the kinases except for the WNK family, WNK1 (76.44% of control), WNK2 (66.89% of control) and WNK3 (54.75% of control) (figure 2A) and PDK/PDHK family, in which inhibition was observed for PDK2/PDHK2 (65.5% of control) and PDK3/PDHK3 (73.8% of control) (Figure 2B).
Fig. 2: Effect of NL-1 treatment on kinase activity.
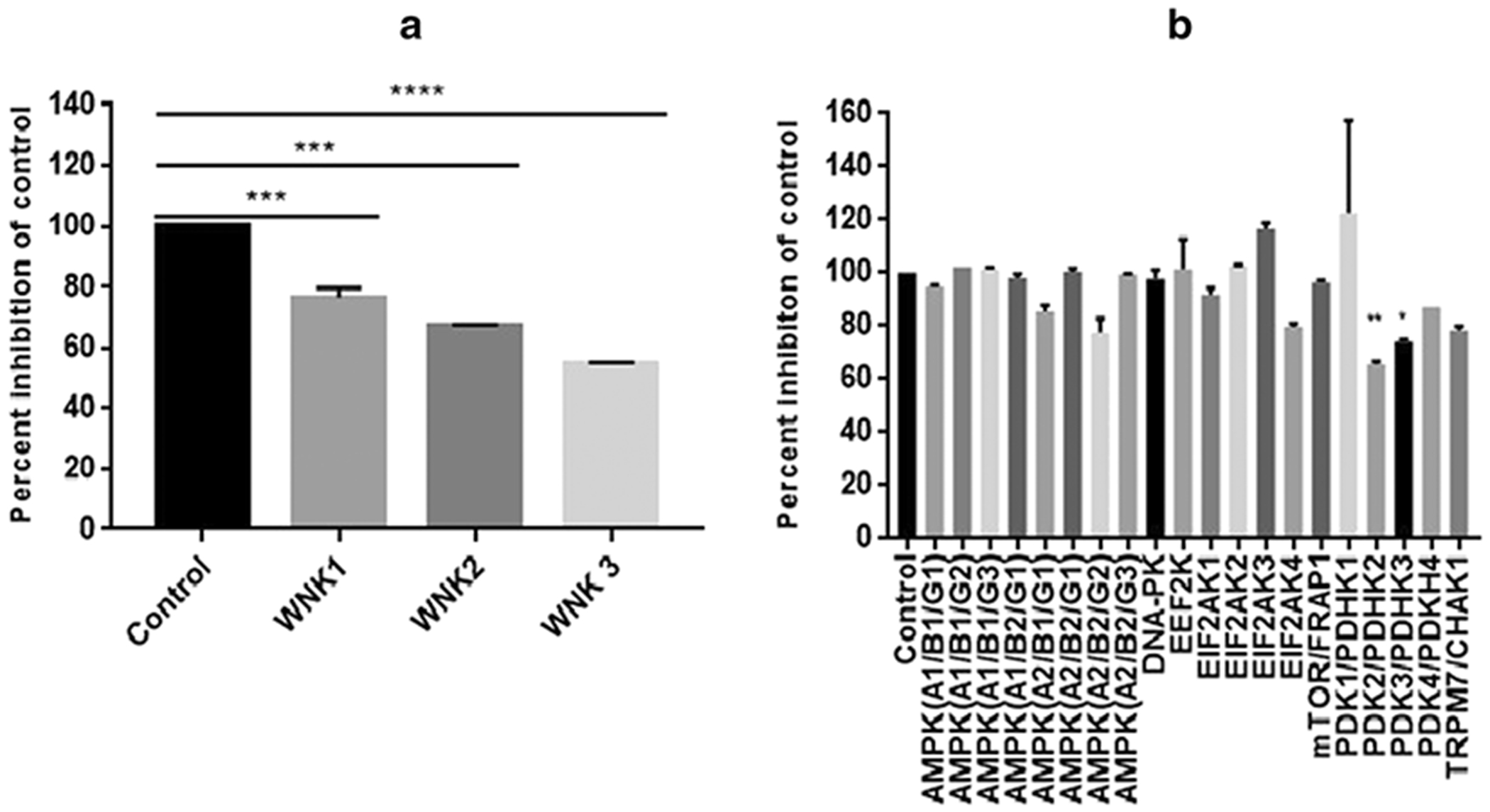
(A) Typical kinase panel shows that the only notable inhibition for the WNK kinase family, out of 349 kinases, and (B) Atypical kinase panel showing significant inhibition of PDK2 and PDK3. Results are expressed as the percent inhibition of control (*p<0.05, **p<0.01, ***p<0.001, ****p<0.0001).
NL-1 effect on ETC
We evaluated the effect of NL-1 treatment on mitochondrial function. NL-1 interactions with the components of electron transport chain (ETC) such as complex I (NADH dehydrogenase) and complex IV (cytochrome c oxidase) were studied my measuring the concentrations of NADH and cytochrome c, which are the substrates of these enzymes. At 10 μM, NL-1 was found to significantly increase Complex I (figure 4A) function by about 33% of DMSO control. At the same concentration, NL-1 had no significant effect on the complex IV activity (figure 4B).
Fig. 4: Effect of NL-1 on mitochondrial complexes of the electron transport chain.
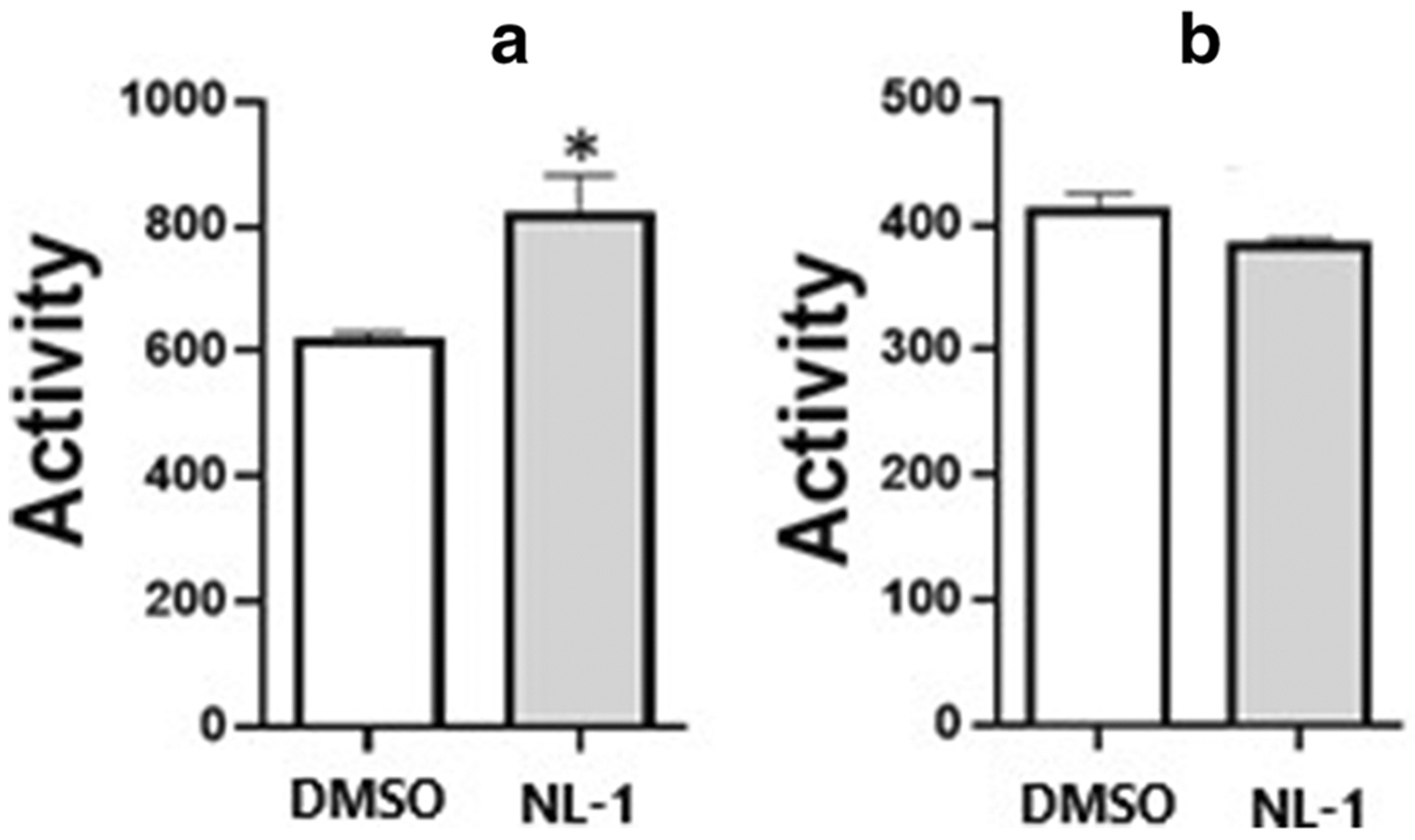
(A) NL-1 (10 μM) stimulates complex I activity in isolated mitochondria; (B) NL-1 does not affect complex IV in isolated mitochondria. Each bar represents average +/− S.D, where N = 3 (*p<0.05).
NL-1 effect on mitochondrial membrane potential
Mitochondrial membrane potential was measured using the tetramethyl rhodamine methyl ester (TMRM) assay (36). Ursodeoxycholic acid at 500 nM was used as a positive control. Figure 5 shows that fluorescence intensities of ursodeoxycholic acid and NL-1 were found to be significantly higher than that of the vehicle treated cells. Thus, showing that NL-1 changed mitochondrial membrane potential at the 10 μM dose tested.
Fig. 5:
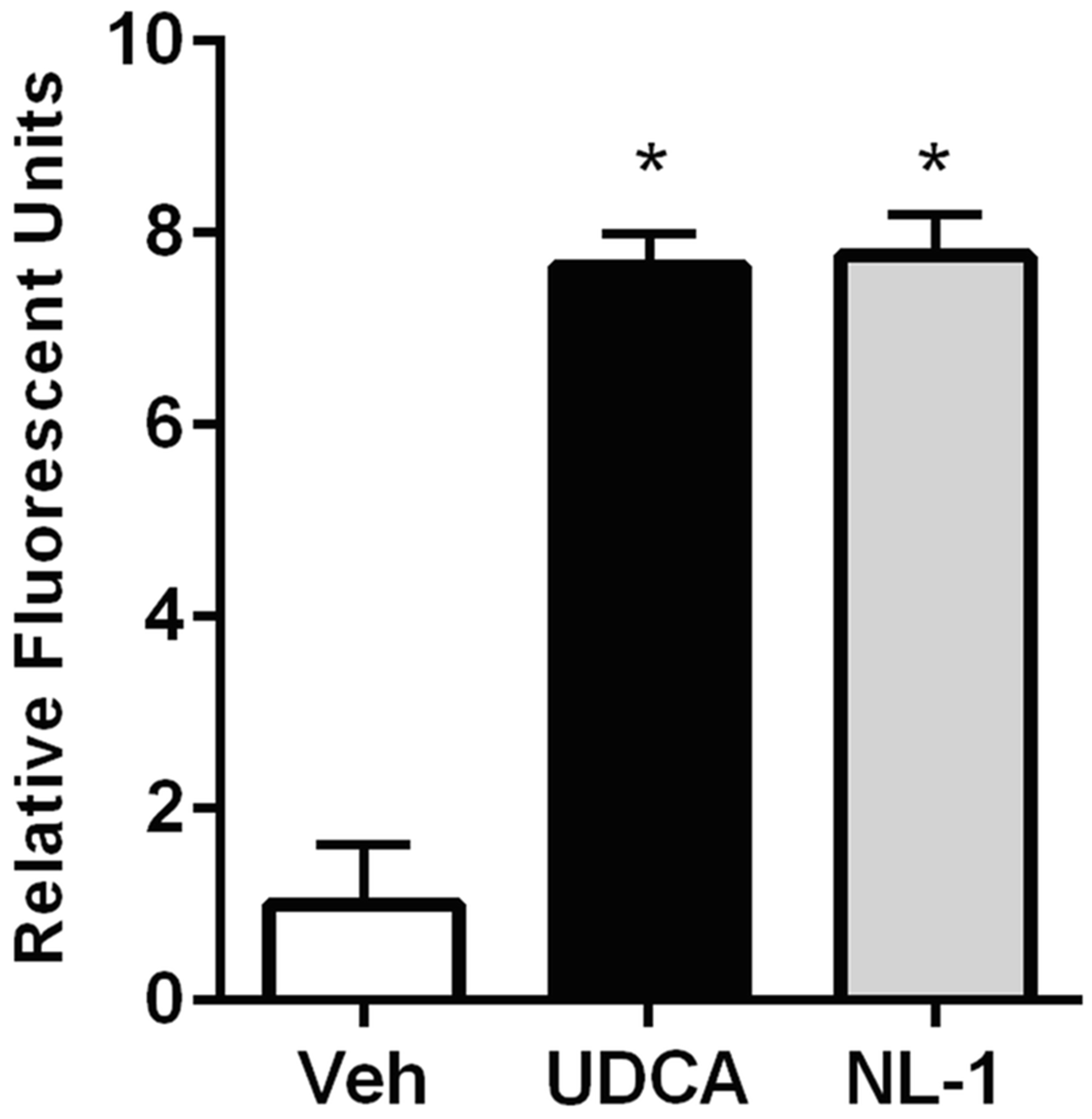
NL-1 (10 μM) stimulates mitochondrial membrane potential in N2A cells with ursodeoxycholic acid used as control (500 nM). Each bar represents average +/− S.D, where N = 3 (*p<0.05).
NL-1 effect on mitochondrial respiration
The data were obtained for mitochondrial respiration which is expressed as the OCR plotted as a function of time and sequential addition of drugs that modulate respiration (figure 7A and 7C). The OCR for different parameters of mitochondrial respiration obtained was plotted as a comparison between DMSO control and 10 μM NL-1 (figure 7B and 7D). For the acute treatment period of 3 hours, NL-1 was found to significantly change the maximal respiration and spare respiratory capacity of N2a cells (figure 7B). For a prolonged drug exposure of 24 hours, NL-1 treatment also significantly improved the cellular basal respiration along with the maximal respiration and spare respiratory capacity (figure 7D). Similarly, the ECAR was plotted as a function of time and sequential addition of drugs that modulate glycolysis (figure 8A and 8C). The ECAR for glycolytic parameters obtained was plotted as a comparison between DMSO control and 10 μM NL-1 (figure 8B and 8D). NL-1 acute treatment (3 hours) had no significant impact on the glycolysis in N2a cells, while prolonged exposure of 24 hours changed the cellular glycolytic capacity.
Fig. 7: Measurement of extracellular acidification rate (ECAR) of N2a cells as a function of glycolysis.
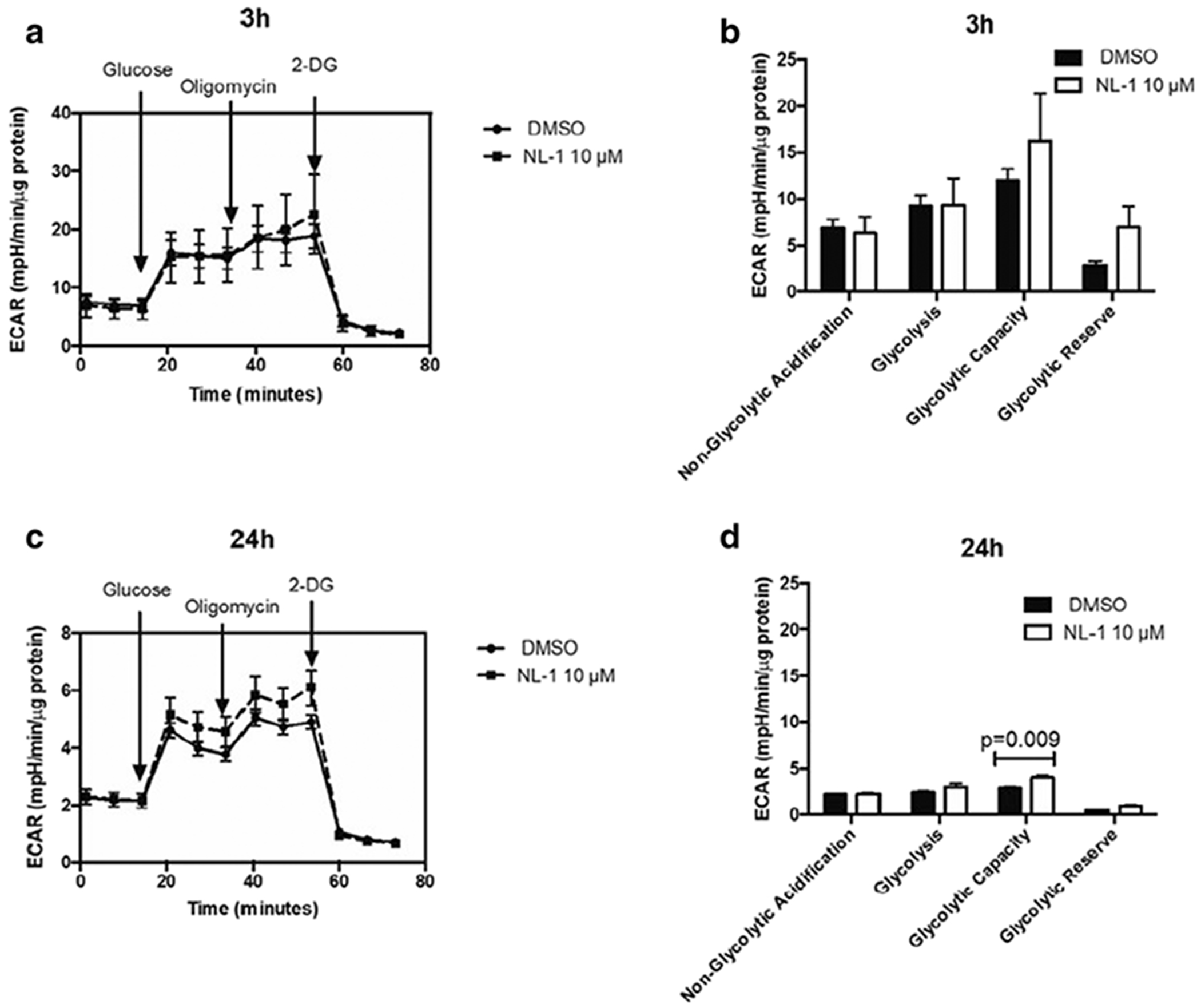
The ECAR is plotted over time with sequential addition of modulators of glycolysis for (A) 3 hour and (C) 24 hour treatment. The ECAR for parameters of glycolysis are compared for control and 10 μM NL-1 for (B) 3 hour and (D) 24 hour treatment.
Fig. 8: NL-1 protects neuronal cells against oxygen glucose deprivation and reperfusion (OGDR).
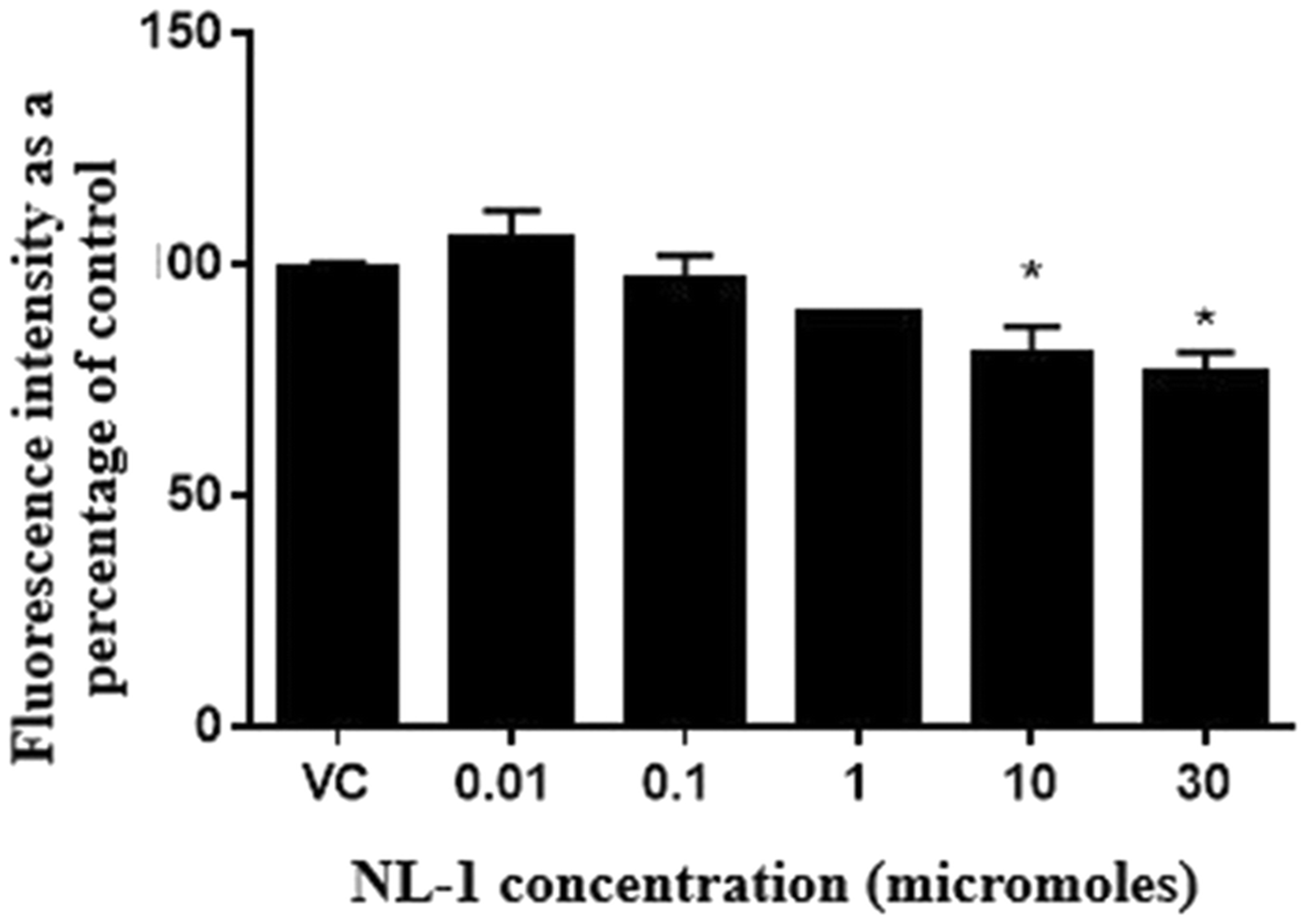
N2a cells were exposed to OGDR conditions for 3 hours and reperfused for 24 hours. The fluorescence intensity from Amplex Red assay is plotted as a percentage on control cells for a series of NL-1 concentrations versus vehicle control (*p<0.05).
NL-1 effect on hydrogen peroxide generation following OGDR
The efficacy of NL-1 in reducing peroxide generation was studied by subjecting N2a cells to OGDR conditions for a period of 3 hours, and a reperfusion period of 24 hours (figure 9). The hydrogen peroxide produced was measured using the Amplex Red assay, and each treatment was compared to control. It was found that NL-1 significantly reduced the peroxide produced at a dose of 10 and 30 μM, for a stroke period of 3 hours.
Fig. 9: Brain and plasma distribution pharmacokinetic profiles of NL-1 into the brain of male C57BL/6J mice (30).
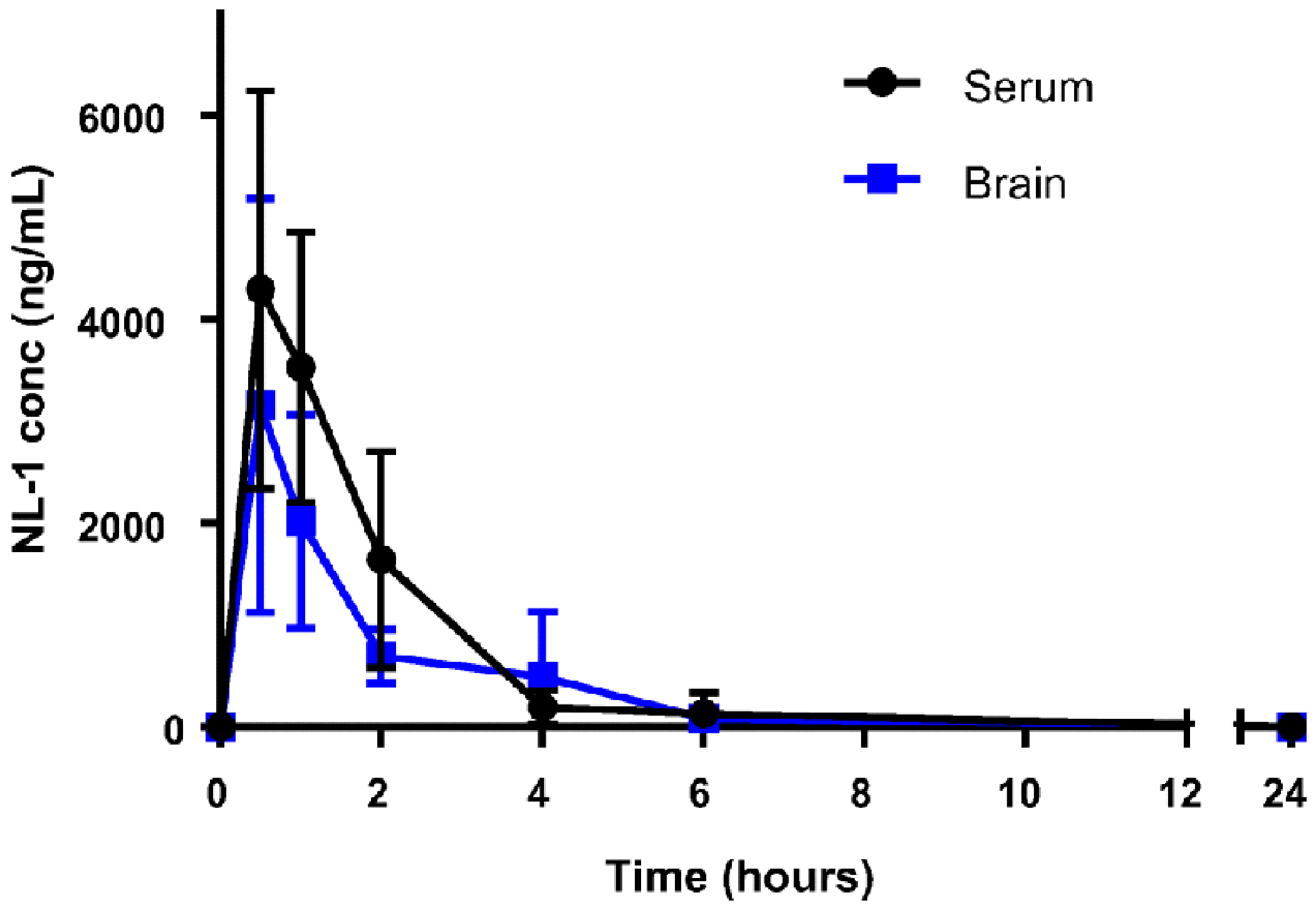
NL-1 (10 mg/kg) was injected i.p. Each data point represents average ± S.D. where N = 6-8 male mice.
Pharmacokinetic study of NL-1
To predict the permeability of NL-1 through the BBB, we evaluated it in an artificial membrane permeability assay found the −logPe of NL-1 as −3.71, while the control compound propranolol was −logPe of −3.94, with a −logPe>−5 considered to be permeable (31). The accumulation of NL-1 in the brain, after an extravascular dose, was evaluated using a preliminary pharmacokinetic study using C57BL/6J mice. The time points of 30 minutes, 1, 2, 6 and 24 hours were used. In figure 10, the concentration of NL-1 extracted from the brain of mice has been plotted for each time point. We found that NL-1 concentrations in the brain reached a maximum at 30 minutes post administration i.p. of the drug with a maximum concentration in the brain of 3154 ± 676 ng/mL NL-1. Subsequently, there was a steady decline in NL-1 levels until 6 hours, beyond which NL-1 brain concentration had reduced to baseline levels. The calculated t1/2 for NL-1 in the plasma was 2.27 hours. A follow-up study to determine the ratio of brain/plasma, we found that the brain/plasma ratio was 1.07, after IV injection, with a logBB of 0.029. Typical classification of molecules to cross the BBB is logBB>0.3 for compounds crossing readily, and logBB< −1 are compounds which poorly distribute to the brain (37,38). Taken together, the PK curve of figure 10, NL-1 has moderate ability to reach the CNS.
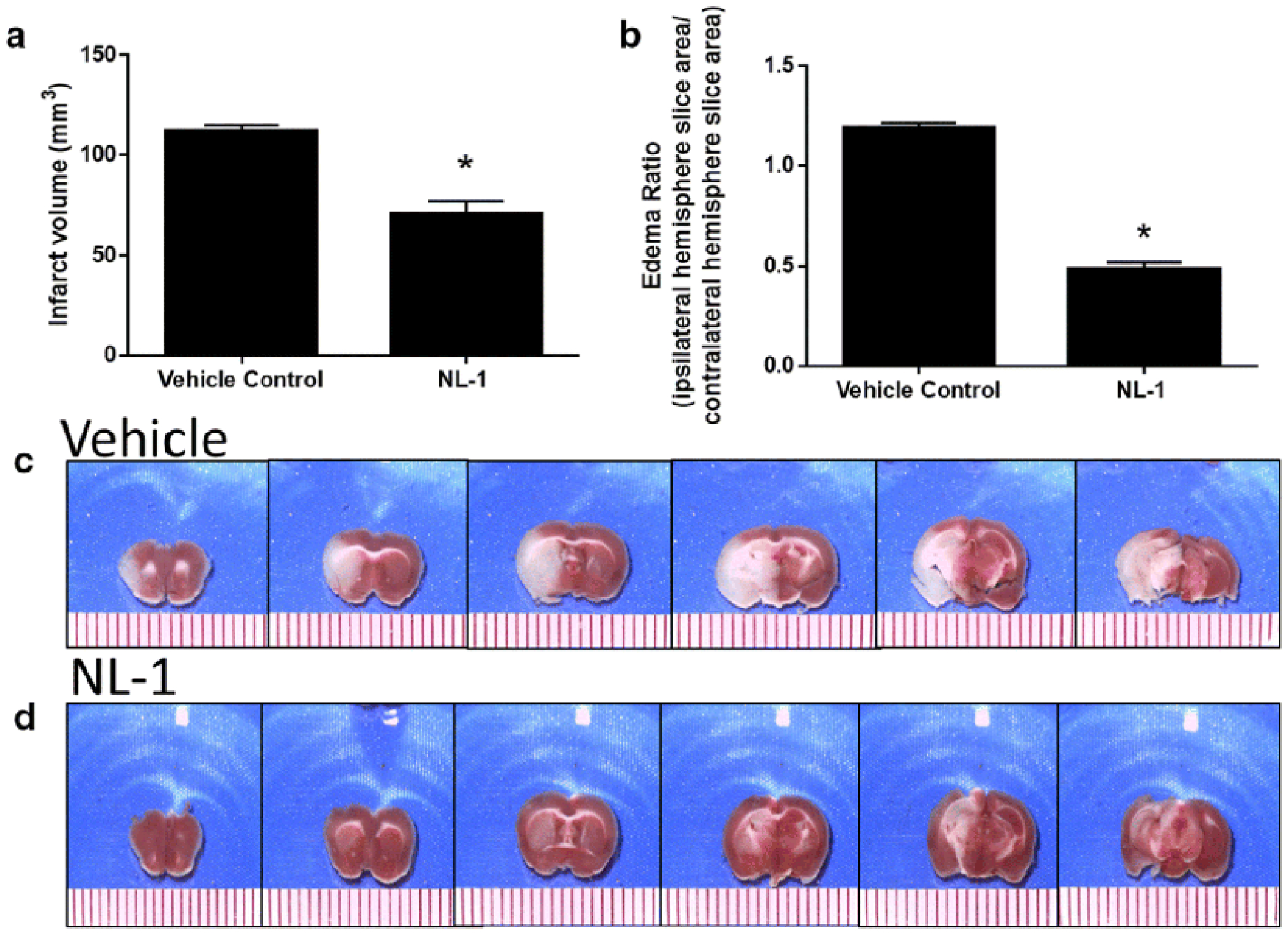
Fig. 10: Administration of NL-1 protects against brain ischemic reperfusion injury.
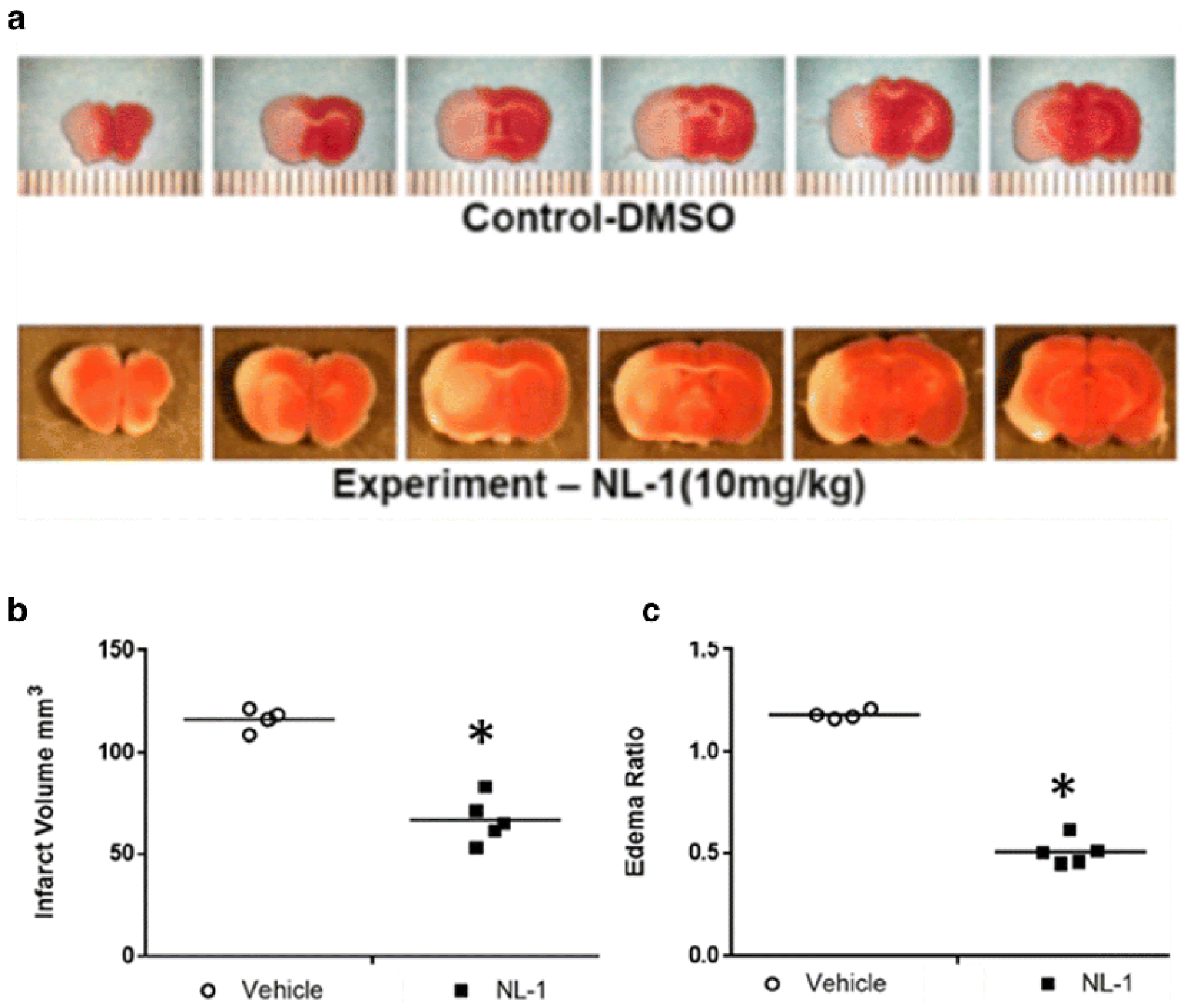
NL-1 was administered to mice at the time of reperfusion, and mice were protected from transient brain ischemic-reperfusion injury. (A) TTC staining, where red areas are live and white areas are ischemic tissue. Effect of NL-1 treatment (10 mg/kg) on the infarct volume (B) and brain edema (C) of mice treated with NL-1 compared to vehicle treated mice. Data shown as mean ± S.D. where N = 3-5 mice. Statistical significance *P<0.05.
NL-1 protects against ischemia-reperfusion injury
Figure 11 shows that NL-1 (10 mg/kg, i.p.) was neuroprotective in a murine t-MCAO ischemic stroke model. NL-1 was administered at the time of reperfusion and the results of the TTC-stained brain sections showed that infarct volumes were larger in control-treated mice compared to NL-1 treated mice (Figure 11). The quantification of infarct volumes demonstrated that NL-1 administration significantly reduced ischemia/reperfusion-injury in vivo via reduction in infarct volume (Vehicle: 116.42 ± 6.73; NL-1: 66.87 ± 11.25) and edema (Vehicle: 1.18 ± 0.026; NL-1: 0.509 ± 0.065). Mice treated with NL-1 (10 mg/kg) 15 min after reperfusion, showed a reduction in infarct volume (Vehicle: 111.88 ± 3.10; NL-1: 70.06 ± 6.80; Figure 12) and edema (Vehicle: 1.19 ± 0.029; NL-1: 0.48 ± 0.037). As control experiment, we used a permanent occlusion model with Rose Bengal clot formation under light activation, which permanently prevents blood flow to the infarct area. We found that the average infract size for the vehicle was 14.89 ± 5.02 mm3, and with NL-1 (10 mg/kg) 12.94 ± 6.39 mm3 (N = 15), which was not significantly reduced in infract size (Figure 13).
Fig. 11. Post-reperfusion administration of NL-1 protects against brain ischemic reperfusion injury.
NL-1 was administered to mice 15 min post-reperfusion protected mice from transient brain ischemic-reperfusion injury as seen in brain slices with TTC staining, where red areas are live and white areas are ischemic tissue. Effect of NL-1 treatment (10 mg/kg) on the infarct volume (A) and brain edema (B) of mice treated with NL-1 compared to vehicle treated mice. (C) Vehicle treated tMCAO; (D) NL-1 (10 mg/kg) with tMCAO. Data shown as mean ± S.D. where N = 4-5 mice. Statistical significance *P<0.05.
Fig. 12: Permanent occlusion ischemic stroke is not protected by NL-1.
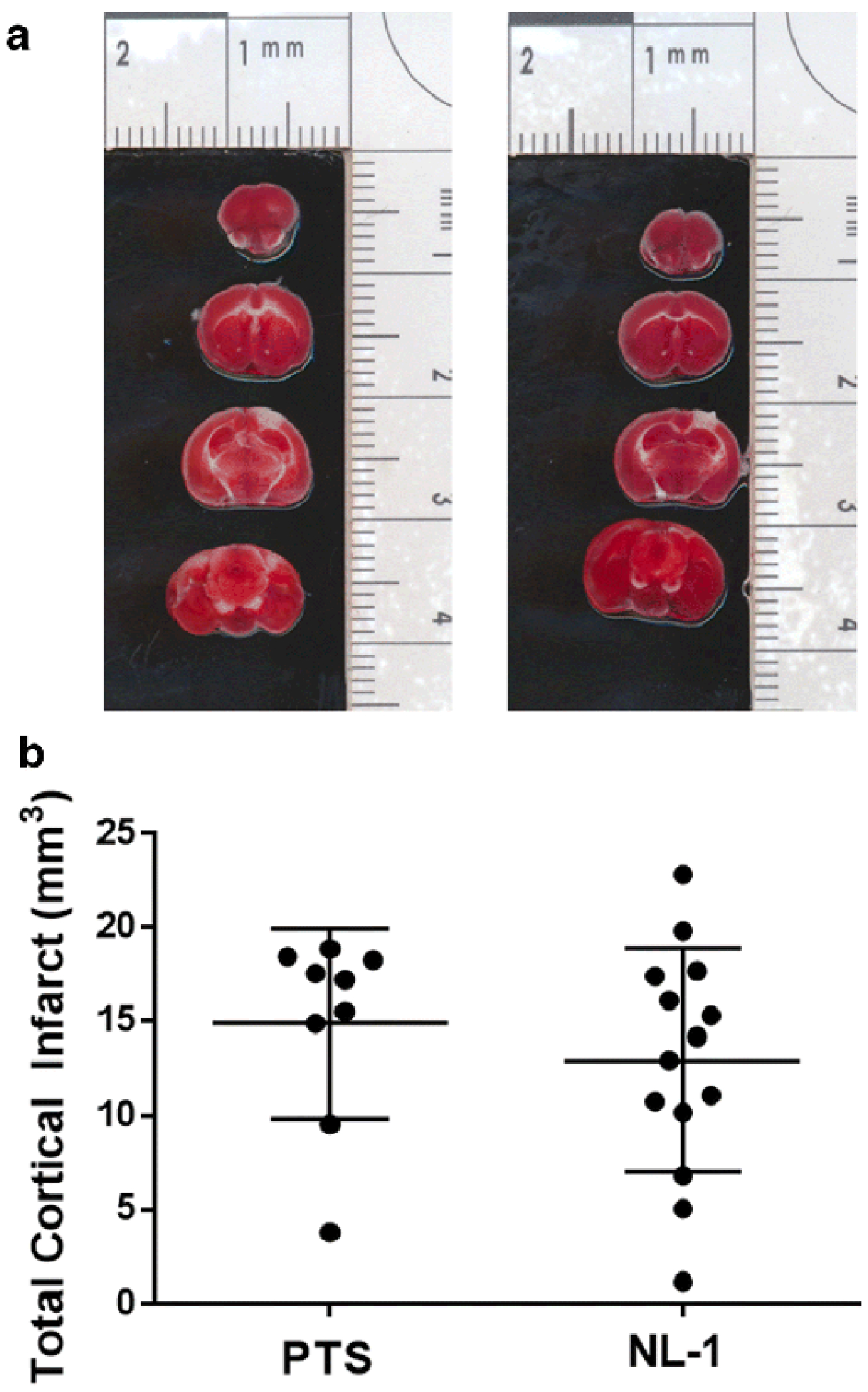
NL-1 was administered to mice 1 hour post a photothrombotic induced stroke (10 mg/kg). (A) TTC brain sections with tissue infarct; (B) no statistical significant reduction in infarct size; N = 10-15 mice.
Fig. 13: Oxidative stress reduction by NL-1 in ischemic reperfusion injury.
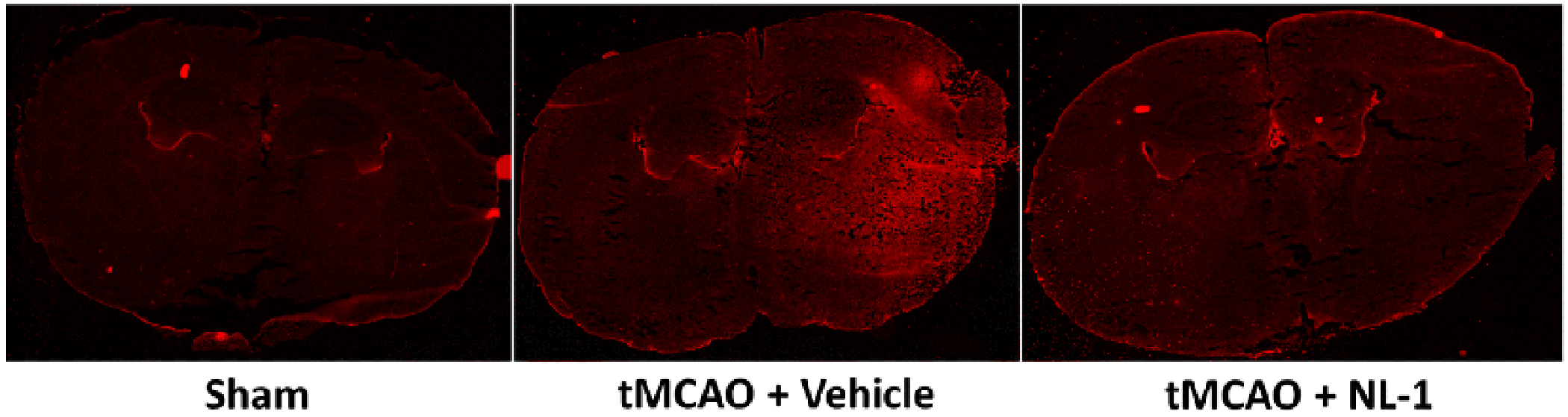
NL-1 reduced the levels of the oxidative stress marker 4-hydroxynonenal in tMCAO mice when treated with 10 mg/kg NL-1.
NL-1 reduces levels of oxidative stress marker 4-HNE
Figure 14 shows the effect of treatment of NL-1 (10 /mg) on the oxidative stress marker 4-HNE. As can be seen, there is decrease in 4-HNE staining, a technique recently published to study mitoNEET redox activity (39). Human recombinant mitoNEET was treated with FMN in the presence of blue light, and showed a decrease in monomer occurrence at 12 kDa. NL-1 treatment increased the presence of the monomer in a dose-dependent manner.
DISCUSSION
The goal of this study was to investigate the mitoNEET ligand, NL-1, as a possible therapeutic strategy for cerebral ischemia and reperfusion injury treatment. Here we evaluated the pharmacological properties of NL-1, a prototype mitoNEET ligand (40), on its ability to interact with the cellular mitochondrial system, and we found that NL-1 can be used as a potential neuroprotective agent for mitigating the pathology of cerebral ischemia and reperfusion injury (41,42).
MitoNEET belongs to the iron-sulfur protein family (gene: cisd), and contains 2Fe-2S clusters, which have been reported to be redox active (43). The natural endogenous ligand of mitoNEET remains to be elucidated, but some studies have indicated that it may play a role in glutathione and other free thiol mediated redox reactions (44–46), as well as interacting with related metabolic systems including adiponectin (47) and GDH (48). MitoNEET was first identified from the binding of pioglitazone, a type II diabetic glitazone (11). The effect pioglitazone exhibits on lowering of lipid levels were independent of activation of the PPARγ receptor. Investigation into its off-target actions led to the discovery of the mitochondrial protein mitoNEET. We have previously shown that pioglitazone has beneficial CNS effects in traumatic brain injury (TBI), and found that first in class mitoNEET ligand NL-1 shared a similar profile of neuroprotection in TBI. Of interest was that the TBI protection was lost in mitoNEET KO mice. Here we further provide additional evidence that NL-1 was not able to activate the PPARγ to any significant extent, therefore, likely not contributing to the activity seen in this stroke study (17).
Calcium homeostasis plays an important role in neurodegeneration following ischemia and reperfusion injury (32,49,50). Dysregulation of intracellular calcium activates the mediators of apoptosis and necrosis and calcium channel blocker are able to mitigate these deleterious effects (35). In mitochondria, excess calcium leads to swelling and a loss of membrane potential which increases the permeability of mitochondrial membranes, promoting cytochrome c release (51,52). Furthermore, mitochondrial dysfunction also leads to disruption of BBB tight junctions, leading to increased permeability (53). Considering the importance of intracellular calcium in regulation of cell survival after stroke, we performed the calcium flux assay to determine if NL-1 itself affected the calcium levels in the cells (54). Potassium chloride was used to depolarize the voltage gated calcium channels (VGCC), allowing for calcium entry into the cells (55,56). We found that NL-1 does not appreciably affect the calcium flux from the VGCC and therefore likely does not contribute to the activities profile seen with the tMCAO mouse model.
Since mitoNEET is a redox and pH sensor for mitochondria, and has been shown to regulate the OXPHOS and bioenergetics, we evaluated the mitochondrial effects of NL-1 at the electron transport chain complex I and IV activities level. Complex I activity has been shown to be decreased following ischemia and is not restored upon reperfusion (57,58). Improvement via pharmacological means in complex I activity contributed to a cerebral-protective effect in a mouse stroke model (58–60), while an impairment in complex I activity exacerbates stroke (61). Since NL-1 treated cells showed improvement in complex I activity, this might in part contribute to the attenuation of tissue loss post stroke. While ischemia is the primary cause of hampered complex I activity, complex IV activity is mainly inhibited by a period of prolonged reperfusion (62). NL-1 caused no significant change in the complex IV activity. Our experiments also indicated that NL-1 primarily helps to improve the mitochondrial respiration, compared to glycolysis with a potential for NL-1 to improve mitochondrial respiration and ATP production in cells that might suffer from a massive ATP deficit following ischemia. Taken together, our findings suggest that NL-1 is likely to be more active during the reperfusion injury phase of stroke.
In the murine stroke models, the mitoNEET ligand NL-1 show improved outcomes following stroke in mice treated with NL-1, to support our primary hypothesis that NL-1 is protective against reperfusion injury (63). Reperfusion-injury is a contributor to secondary tissue damage after a stroke, and mitochondria contribute to this injury. We found that treatment of mice with NL-1 at the time of reperfusion was able to reduce the edema formation and brain tissue damage associated with reperfusion-injury in mice compared to the untreated mice. Additionally, this protection can also be seen when treatment with NL-1 was 15 min post reperfusion, allowing for mitoNEET target engagement between 45-60 min post tissue reperfusion (see pharmacokinetic profile figure 10). Our PK data did indicate that NL-1 does not accumulate in the brain (logBB of 0.029), with lower brain levels than plasma, suggesting that the pharmacological activity profile we observed is during the reperfusion-injury stage. Lastly, to validate our hypothesis that NL-1 is protective in the RI after stroke, we used a permanent clot-based model, the photothrombotic mouse model, where a permanent clot formed prevents the reperfusion of the tissue, and found that NL-1 was unable to significantly reduce the infract size in treated mice as expected. Since NL-1 does not contribute to significantly to changes in calcium flux e.g. excitotoxicity, it would be unlikely that the compound would support protection via the non-reperfusion injury mechanisms such as the calcium excitotoxic mechanisms (64). These data support a reperfusion-injury model of protection by NL-1, which is likely mediated via its ability to interact with mitoNEET on mitochondria. Furthermore, since oxidative stress is a major contributor to the neuronal cell loss post stroke we evaluated the effect NL-1 had on an oxidative stress marker. MitoNEET structure has several lysine residues, and these lysines seem to be susceptible to modification by the oxidative stress by-product of lipid peroxidation, 4-hydoxynonenoal (4-HNE). Proteomics studies indicated that LYS55 is modified by 4-HNE, via an aldehyde covalent bond (65). Preliminary support for this future avenue of investigation is the reduction in 4HNE staining as seen from the IFC staining in stroke. Future studies will be focused on evaluating the possible role NL-1 has on attenuating the modification of mitoNEET lysines, protecting the functionality of these lysines from an 4HNE attack during oxidative stress during reperfusion injury.
To explore the off-target profile of NL-1, an array of kinases present was evaluated. The inhibition of NL-1 towards kinases has been untested and we intended to check for inhibition of kinases by NL-1. The high throughput kinase panel used included members of all major human protein kinase families (66). Staurosporine is a general kinase inhibitor that is widely used as the positive control for a kinase panel (67,68). The only kinase family amongst the typical kinases inhibited by NL-1 (~40% of control) was the WNK kinase family, particularly WNK3. WNK kinases or With-No-Lysine kinases play an important role in body fluid and ion homeostasis and regulates blood pressure (69,70). An inhibitor of WNK kinase family, WNK463, was shown to lower mean arterial pressure, and increase urinary excretion of sodium and potassium (71). It has been recently reported that WNK3 is upregulated upon onset of intracerebral hemorrhage which in turn deteriorates brain injury via the WNK3/SPAK/NKCC1 pathway(72). WNK3 knockout in mouse and rat models exhibit significantly reduced infarct volumes, cerebral edema, axonal demyelination, BBB damage and improved neurocognitive behavior following an ischemic stroke (72–74). Of note is that fact that NL-1 was used at a single concentration of 10 μM, which is a high concentration for kinase inhibition. A more detailed investigation at lower concentrations of NL-1 is necessary to determine its effective inhibitory actions on WNKs and PDKs. Nonetheless, a possible inhibition of WNK3 by NL-1 could contribute to improved outcomes after stroke, and presents a promising mitoNEET-independent axis that warrants further investigation.
A few caveats do exist in our current study. From the kinase panel we found that only kinase family inhibited at 10 μM by NL-1 was the WNK kinase family, particularly WNK3 (69–72). Lastly, we acknowledge that only younger male mice were used in these studies, instead of aged (>16 months of age) mice. We established the pharmacokinetic parameters in this study and initial experimental design in anticipation of repeating these studies in older animals. Cell damage following stroke is multimodal, with a complex interplay occurring between ROS, apoptosis and inflammatory mediators. Further investigation is needed to fully elucidate the pharmacology of NL-1 on biochemical pathways with greater focus on the levels of molecular mediators of neurodegeneration.
CONCLUSION
In summary, we describe studies performed to investigate the viability of using mitoNEET ligands such as NL-1 as a stroke therapeutic in the prevention of tissue loss during cerebral reperfusion injury after a stroke. Our data indicate that use of NL-1 was found to further improve cerebral stroke outcomes following treatment in mice reperfusion injury. This work presents the first report of NL-1 as a possible stroke therapeutic for the novel mitochondrial target mitoNEET in reducing tissue damage in reperfusion injury.
Fig. 3: Effect of NL-1 (10 μM) on endogenous calcium and calcium influx in N2A cells.
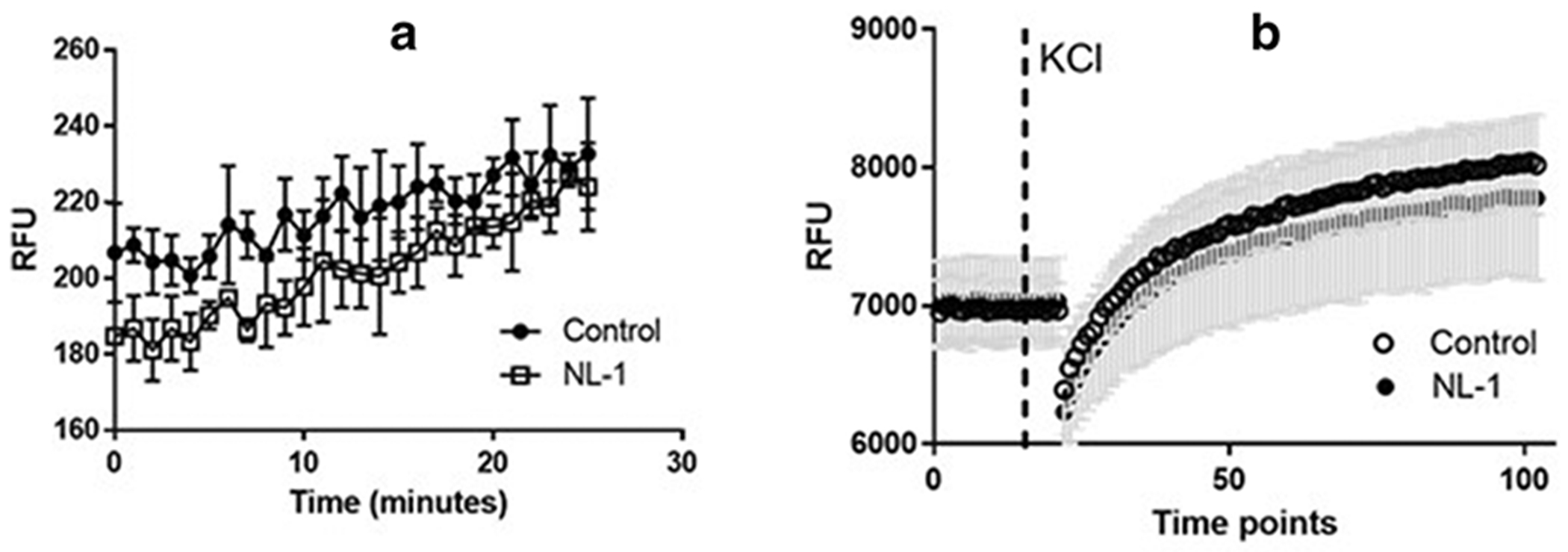
Cells were treated with NL-1 for 15 minutes and the calcium flux measure with the Fluoforte calcium kit. Control cells received DMSO only, with a final concentration of DSMO 1%. (A) Effect of NL-1 on baseline calcium uptake. (B) Effect of NL-1 on cells which have been stimulated by KC1 (50 mM) to stimulate voltage gated ion channels. The cells treated with NL-1 did not show significant effects on calcium flux at the dose tested. Each data point represented by average +/− S.D. were N = 3 -5 for each group. Abbreviation: relative fluorescent units (RFU).
Figure 6: Measurement of oxygen consumption rate (OCR) of N2a cells as a function of mitochondrial respiration.
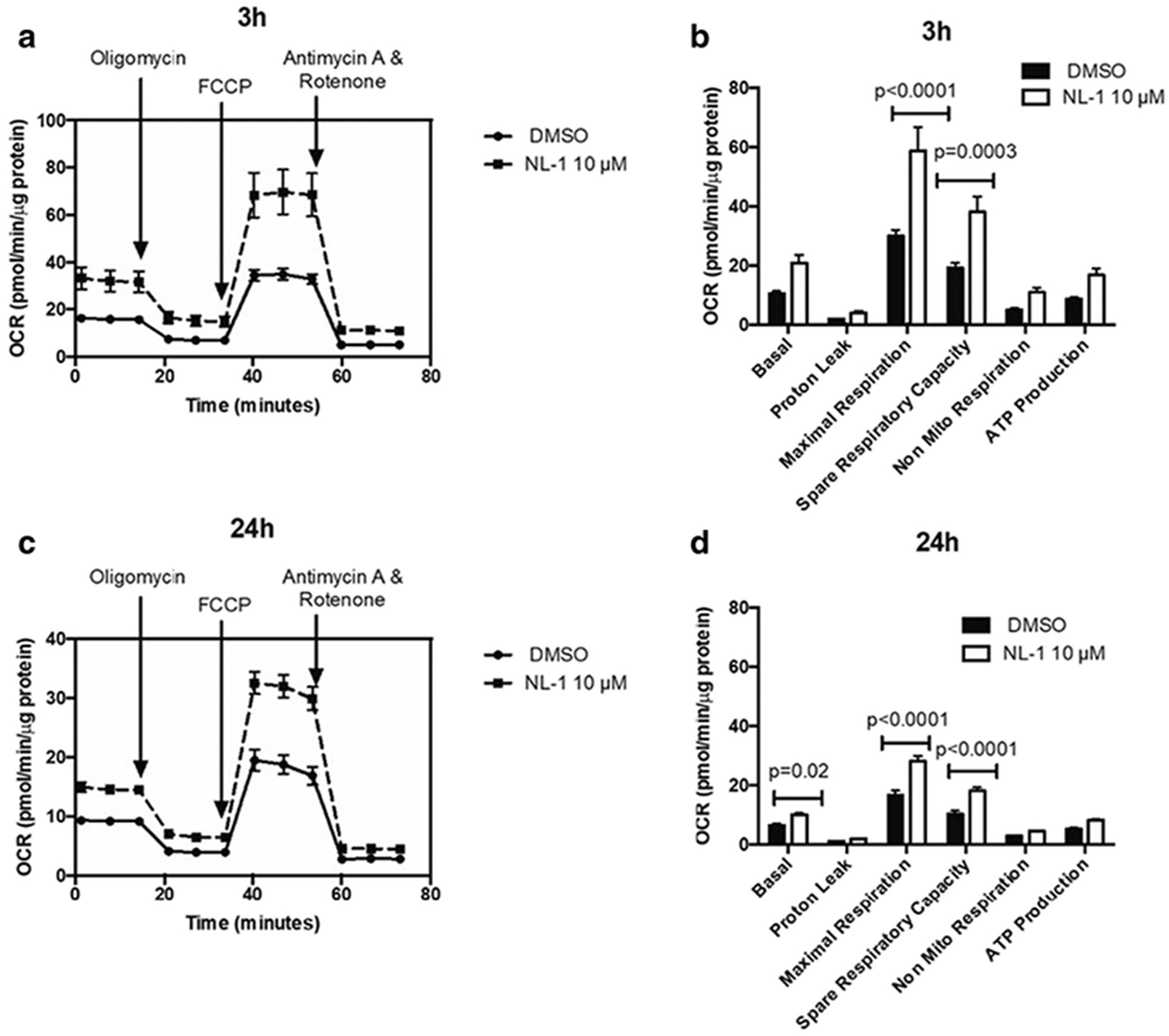
The OCR is plotted over time with sequential addition of modulators of respiration for (A) 3 hour and (C) 24 hour treatment. The OCR for parameters of mitochondrial respiration are compared for control and 10 μM NL-1 for (B) 3 hour and (D) 24 hour treatment.
Acknowledgements.
We are grateful for the technical support from Deborah Corbin in the tissue processing. The project described was supported by the National Institute Of General Medical Sciences, U54GM104942, 1R41NS110070-01 and the WVU Stroke CoBRE Grant 2P20GM109098. The project described was supported by the National Heart, Lung and Blood Institute Grant HL-128485. The project described was supported by the Community Foundation for the Ohio Valley Whipkey Trust. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. This publication was supported by the National Center for Research Resources and the National Center for Advancing Translational Sciences, National Institutes of Health, through Grant UL1TR001998. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Footnotes
Publisher's Disclaimer: This Author Accepted Manuscript is a PDF file of a an unedited peer-reviewed manuscript that has been accepted for publication but has not been copyedited or corrected. The official version of record that is published in the journal is kept up to date and so may therefore differ from this version.
References:
- 1.Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M, et al. Executive summary: Heart disease and stroke statistics-2016 update: A Report from the American Heart Association. Vol. 133, Circulation. Lippincott Williams and Wilkins; 2016. p. 447–54. [DOI] [PubMed] [Google Scholar]
- 2.Price-Haywood EG, Harden-Barrios J, Carr C, Reddy L, Bazzano LA, van Driel ML. Patient-reported outcomes in stroke clinical trials 2002–2016: a systematic review [Internet]. Vol. 28, Quality of Life Research. Springer International Publishing; 2019. [cited 2020 Oct 7]. p. 1119–28. Available from: 10.1007/s11136-018-2053-7 [DOI] [PubMed] [Google Scholar]
- 3.Kim JS. tPA helpers in the treatment of acute ischemic stroke: Are they ready for clinical use? J Stroke [Internet]. 2019. May 1 [cited 2020 Oct 7];21(2):160–74. Available from: /pmc/articles/PMC6549064/?report=abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shi H, Liu KJ. Cerebral tissue oxygenation and oxidative brain injury during ischemia and reperfusion. Front Biosci [Internet]. 2007. [cited 2020 Oct 2]; 12(4):1318–28. Available from: https://pubmed-ncbi-nlm-nih-gov.www.libproxy.wvu.edu/17127384/ [DOI] [PubMed] [Google Scholar]
- 5.Gravanis I, Tsirka SE. Tissue-type plasminogen activator as a therapeutic target in stroke [Internet]. Vol. 12, Expert Opinion on Therapeutic Targets. NIH Public Access; 2008. [cited 2020 Oct 7]. p. 159–70. Available from: /pmc/articles/PMC3824365/?report=abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Choi JH, Pile-Spellman J. Reperfusion Changes After Stroke and Practical Approaches for Neuroprotection [Internet]. Vol. 28, Neuroimaging Clinics of North America. W.B. Saunders; 2018. [cited 2020 Oct 7]. p. 663–82. Available from: https://pubmed-ncbi-nlm-nih-gov.www.libproxy.wvu.edu/30322601/ [DOI] [PubMed] [Google Scholar]
- 7.Wu Meng-Yu, Yiang Giou-Teng, Liao Wan-Ting, Tsai Andy Po-Yi, Cheng Yeung-Leung, Cheng Pei-Wen, Chia-Ying Li C-JL. Current Mechanistic Concepts in Ischemia and Reperfusion Injury. Cell Physiol Biochem. 2018;46(4):1650–67. [DOI] [PubMed] [Google Scholar]
- 8.Raslan A, Bhardwaj A. Medical management of cerebral edema. Vol. 22, Neurosurgical focus. 2007. [DOI] [PubMed] [Google Scholar]
- 9.Jha SK. Cerebral edema and its management. Med J Armed Forces India [Internet]. 2003. [cited 2020 Oct 7];59(4):326—31. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4923559/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Green AR. Pharmacological approaches to acute ischaemic stroke: Reperfusion certainly, neuroprotection possibly. In: British Journal of Pharmacology [Internet]. Wiley-Blackwell; 2008. [cited 2020 Oct 7]. p. S325. Available from: /pmc/articles/PMC2268079/?report=abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Colca JR, McDonald WG, Waldon DJ, Leone JW, Lull JM, Bannow CA, et al. Identification of a novel mitochondrial protein (“mitoNEET”) cross-linked specifically by a thiazolidinedione photoprobe. Am J Physiol - Endocrinol Metab [Internet]. 2003. [cited 2017 Jun 10];286(2). Available from: http://ajpendo.physiology.org/content/286/2/E252.long [DOI] [PubMed] [Google Scholar]
- 12.Paddock ML, Wiley SE, Axelrod HL, Cohen AE, Roy M, Abresch EC, Capraro D, Murphy AN, Nechushtai R, Dixon JE JP. MitoNEET is a uniquely folded 2Fe–2S outer mitochondrial membrane protein stabilized by pioglitazone. Proc Natl Acad Sci U S A. 104(36):14342–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kounosu Asako, Iwasaki T, Baba Seiki,Hayashi-Iwasaki Y, Oshima Tairo, Kumasaka T. Crystallization and preliminary X-ray diffraction studies of the prototypal homologue of mitoNEET (Tth-NEET0026) from the extreme thermophile Thermus thermophilus HB8. Struct Biol Cryst Commun. 2008;64(12):1146–1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nechushtai R, Conlan AR, Harir Y, song L, yogev ohad, Eisenberg-Domovich Y, et al. Characterization of Arabidopsis NEET reveals an ancient role for NEET proteins in iron metabolism. Plant Cell [Internet]. 2012. May [cited 2020 Oct 7];24(5):2139–54. Available from: /pmc/articles/PMC3442592/?report=abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Inupakutika MA, Sengupta S, Nechushtai R, Jennings PA, Onuchic JN, Azad RK, et al. Phylogenetic analysis of eukaryotic NEET proteins uncovers a link between a key gene duplication event and the evolution of vertebrates. Sci Rep [Internet]. 2017. February 16 [cited 2020 Oct 7];7. Available from: https://pubmed.ncbi.nlm.nih.gov/28205535/16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.He Q-Q, Xiong L-L, Liu F, He X, Feng G-Y, Shang F-F, et al. MicroRNA-127 targeting of mitoNEET inhibits neurite outgrowth, induces cell apoptosis and contributes to physiological dysfunction after spinal cord transection. Sci Rep [Internet]. 2016. October 17 [cited 2017 Jan 17];6:35205. Available from: http://www.nature.com/articles/srep35205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yonutas HM, Hubbard WB, Pandya JD, Vekaria HJ, Geldenhuys WJ, Sullivan PG. Bioenergetic restoration and neuroprotection after therapeutic targeting of mitoNEET: New mechanism of pioglitazone following traumatic brain injury. Vol. 327, Experimental Neurology. Academic Press Inc.; 2020. p. 113243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shi Guangyao; Cui Lei; Chen Rui; Liang Shaodong; Wang Chunlei; Wu P. TT01001 attenuates oxidative stress and neuronal apoptosis by preventing mitoNEET-mediated mitochondrial dysfunction after subarachnoid hemorrhage in rats. Neuroreport. 2020;31(11):845–50. [DOI] [PubMed] [Google Scholar]
- 19.Geldenhuys WJ, Benkovic SA, Lin L, Yonutas HM, Crish SD, Sullivan PG, et al. MitoNEET (CISD1) Knockout Mice Show Signs of Striatal Mitochondrial Dysfunction and a Parkinson’s Disease Phenotype. ACS Chem Neurosci. 2017;8(12):2759–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Saralkar P, Arsiwala T, Geldenhuys WJ. Nanoparticle formulation and in vitro efficacy testing of the mitoNEET ligand NL-1 for drug delivery in a brain endothelial model of ischemic reperfusion-injury. Int J Pharm. 2020;578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang L, Niu Y, He G, Wang J. Down-regulation of IncRNA GAS5 attenuates neuronal cell injury through regulating miR-9/FOXO3 axis in cerebral ischemic stroke. RSC Adv [Internet]. 2019. May 23 [cited 2019 Sep 2];9(28):16158–66. Available from: http://xlink.rsc.org/?DOI=C9RA01544B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhao M, Wang J, Xi X, Tan N, Zhang L. SNHG12 Promotes Angiogenesis Following Ischemic Stroke via Regulating miR-150/VEGF Pathway. Neuroscience [Internet]. 2018. October 15 [cited 2019 Sep 2];390:231–40. Available from: https://www.sciencedirect.com/science/article/pii/S0306452218305748 [DOI] [PubMed] [Google Scholar]
- 23.Minaei Beyrami S, Khadem Ansari MH, Rasemi Y, Shakib N, Karimi P. Complete inhibition of phosphatase and tensin homolog promotes the normal and oxygen-glucose deprivation/reperfusion-injured PC 12 cells to cell death. J Cardiovasc Thorac Res [Internet]. 2018. [cited 2019 Sep 2];10(2):83–9. Available from: http://www.ncbi.nlm.nih.gov/pubmed/30116506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Galkin A Brain Ischemia/Reperfusion Injury and Mitochondrial Complex I Damage [Internet]. Vol. 84, Biochemistry (Moscow). Pleiades Publishing; 2019. [cited 2021 Apr 8]. p. 1411–23. Available from: https://link-springer-com.wvu.idm.oclc.org/article/10.1134/S000629791911015431760927 [Google Scholar]
- 25.Ten V, Galkin A. Mechanism of mitochondrial complex I damage in brain ischemia/reperfusion injury. A hypothesis. Vol. 100, Molecular and Cellular Neuroscience. Academic Press Inc.; 2019. p. 103408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Andrabi SS, Ali M, Tabassum H, Parveen S, Parvez S. Pramipexole prevents ischemic cell death via mitochondrial pathways in ischemic stroke. DMM Dis Model Mech [Internet]. 2019. [cited 2021 Apr 7]; 12(8). Available from: /pmc/articles/PMC6737958/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hathaway QA, Nichols CE, Shepherd DL, Stapleton PA, McLaughlin SL, Strieker JC, et al. Maternal-engineered nanomaterial exposure disrupts progeny cardiac function and bioenergetics. Am J Physiol - Hear Circ Physiol [Internet]. 2017. March 1 [cited 2020 Oct 6];312(3):H446–58. Available from: https://europepmc.org/articles/PMC5402018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vongs A, Solly KJ, Kiss L, MacNeil DJ, Rosenblum CI. A miniaturized homogenous assay of mitochondrial membrane potential. Assay Drug Dev Technol [Internet]. 2011. August 1 [cited 2020 Oct 6];9(4):373–81. Available from: https://pubmed-ncbi-nlm-nih-gov.www.libproxy.wvu.edu/21294696/ [DOI] [PubMed] [Google Scholar]
- 29.Saralkar P, Arsiwala T, Geldenhuys WJ. Nanoparticle formulation and in vitro efficacy testing of the mitoNEET ligand NL-1 for drug delivery in a brain endothelial model of ischemic reperfusion-injury. Int J Pharm. 2020. March 30;578:119090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pedada KK, Zhou X, Jogiraju H, Carroll RT, Geldenhuys WJ, Lin L, et al. A quantitative LC?MS/MS method for determination of thiazolidinedione mitoNEET ligand NL-1 in mouse serum suitable for pharmacokinetic studies. J Chromatogr B [Internet]. 2014. January [cited 2017 Jun 14];945–946:141–6. Available from: http://linkinghub.elsevier.com/retrieve/pii/S1570023213006685 [DOI] [PubMed] [Google Scholar]
- 31.Geldenhuys WJ, Kochi A, Lin L, Sutariya V, Dluzen DE, Van Der Schyf CJ, et al. Methyl yellow: A potential drug scaffold for Parkinson’s disease. ChemBioChem [Internet], 2014. July 21 [cited 2021 Apr 12];15(11):1591–8. Available from: https://pubmed-ncbi-nlm-nih-gov.wvu.idm.oclc.org/25045125/ [DOI] [PubMed] [Google Scholar]
- 32.Mdzinarishvili A, Geldenhuys WJ, Abbruscato TJ, Bickel U, Klein J, Van Der Schyf CJ. NGP1-01, a lipophilic polycyclic cage amine, is neuroprotective in focal ischemia. Neurosci Lett. 2005. July 22;383(1–2):49–53. [DOI] [PubMed] [Google Scholar]
- 33.Park CK, Kang SG. Effects of brain oedema in the measurement of ischaemic brain damage in focal cerebral infarction. Acta Neurochir Suppl [Internet], 2000. [cited 2020 Nov 10];76:269–71. Available from: https://link.springer.com/chapter/10.1007/978-3-7091-6346-7_55 [DOI] [PubMed] [Google Scholar]
- 34.Weber RZ, Gronnert L, Mulders G, Maurer MA, Tackenberg C, Schwab ME, et al. Characterization of the Blood Brain Barrier Disruption in the Photothrombotic Stroke Model. Front Physiol [Internet]. 2020. November 12 [cited 2021 Jan 12]; 11. Available from: https://pubmed.ncbi.nlm.nih.gov/33262704/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hao J, Mdzinarishvili A, Abbruscato TJ, Klein J, Geldenhuys WJ, Van der Schyf CJ, et al. Neuroprotection in mice by NGP1-01 after transient focal brain ischemia. Brain Res [Internet], 2008. February 27 [cited 2021 Apr 12]; 1196:113–20. Available from: https://pubmed-ncbi-nlm-nih-gov.wvu.idm.oclc.org/18234166/ [DOI] [PubMed] [Google Scholar]
- 36.Scaduto RC, Grotyohann LW. Measurement of mitochondrial membrane potential using fluorescent rhodamine derivatives. Biophys J. 1999. January 1;76(1 I):469–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vilar S, Chakrabarti M, Costanzi S. Prediction of passive blood-brain partitioning: Straightforward and effective classification models based on in silico derived physicochemical descriptors. J Mol Graph Model [Internet]. 2010. June [cited 2021 Apr 12];28(8):899–903. Available from: https://pubmed.ncbi.nlm.nih.gov/20427217/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Geldenhuys WJ, Bloomquist JR. Development of an a priori computational approach for brain uptake of compounds in an insect model system. Bioorg Med Chem Lett [Internet]. 2021. May [cited 2021 Apr 12];40:127930. Available from: https://pubmed.ncbi.nlm.nih.gov/33711441/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tasnim H, Landry AP, Fontenot CR, Ding H. Exploring the FMN binding site in the mitochondrial outer membrane protein mitoNEET. Free Radic Biol Med. 2020. August 20;156:11–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Geldenhuys WJ, Funk MO, Barnes KF, Carroll RT. Structure-based design of a thiazolidinedione which targets the mitochondrial protein mitoNEET. Bioorganic Med Chem Lett. 2010;20(3):819–23. [DOI] [PubMed] [Google Scholar]
- 41.Geldenhuys WJ, Leeper TC, Carroll RT. mitoNEET as a novel drug target for mitochondrial dysfunction. Drug Discov Today. 2014;19(10):1601–6. [DOI] [PubMed] [Google Scholar]
- 42.Geldenhuys WJ, Yonutas HM, Morris DL, Sullivan PG, Darvesh AS, Leeper TC. Identification of small molecules that bind to the mitochondrial protein mitoNEET. Vol. 26, Bioorganic & Medicinal Chemistry Letters. 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lipper CH, Stofleth JT, Bai F, Sohn YS, Roy S, Mittler R, et al. Redox-dependent gating of VDAC by mitoNEET. Proc Natl Acad Sci U S A. 2019. October 1;116(40):19924–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Landry AP, Ding H. Redox control of human mitochondrial outer membrane protein MitoNEET [2Fe-2S] clusters by biological thiols and hydrogen peroxide. J Biol Chem [Internet]. 2014. February 14 [cited 2017 Jun 11];289(7):4307—15. Available from: http://www.ncbi.nlm.nih.gov/pubmed/24403080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Landry AP, Cheng ZDH. Reduction of mitochondrial protein mitoNEET[2Fe-2S] clusters by human glutathione reductase. Free Radic Biol Med. 81:119–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Habener A, Chowdhury A, Echtermeyer F, Lichtinghagen R, Theilmeier G, Herzog C, et al. MitoNEET Protects HL-1 Cardiomyocytes from Oxidative Stress Mediated Apoptosis in an In Vitro Model of Hypoxia and Reoxygenation. Vanella L, editor. PLoS One [Internet]. 2016. May 31 [cited 2017 Jan 17]; 11(5):e0156054. Available from: http://dx.plos.org/10.1371/journal.pone.015605447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kusminski Christine M, Holland William L, Sun Kai, Park Jiyoung, Spurgin YL Stephen B, Askew G Roger, Simcox Judith A, McClain Don A CL& PES. MitoNEET-driven alterations in adipocyte mitochondrial activity reveal a crucial adaptive process that preserves insulin sensitivity in obesity. Nat Med. 18 (10):1539–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Roberts ME, Crail JP, Laffoon MM, Fernandez WG, Menze MA KM. Identification of disulfide bond formation between MitoNEET and glutamate dehydrogenase 1. Biochemistry [Internet]. 52 (50),:8969–8971. Available from: https://pubs.acs.org/doi/10.1021/bi401038w [DOI] [PubMed] [Google Scholar]
- 49.Carinci M, Vezzani B, Patergnani S, Ludewig P, Lessmann K, Magnus T, et al. Different roles of mitochondria in cell death and inflammation: Focusing on mitochondrial quality control in ischemic stroke and reperfusion [Internet]. Vol. 9, Biomedicines. MDPI AG; 2021. [cited 2021 Apr 12]. p. 1–28. Available from: https://pubmed.ncbi.nlm.nih.gov/33572080/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pittas K, Vrachatis DA, Angelidis C, Tsoucala S, Giannopoulos G, Deftereos S. The Role of Calcium Handling Mechanisms in Reperfusion Injury. Curr Pharm Des [Internet]. 2018. November 29 [cited 2021 Apr 12];24(34):4077–89. Available from: https://pubmed.ncbi.nlm.nih.gov/30465493/ [DOI] [PubMed] [Google Scholar]
- 51.Nagy Z, Nardai S. Cerebral ischemia/repefusion injury: From bench space to bedside. Vol. 134, Brain Research Bulletin. Elsevier Inc.; 2017. p. 30–7. [DOI] [PubMed] [Google Scholar]
- 52.Kobayashi Tohru, Kuroda Satoshi, Tada Mitsuhiro, Houkin Kiyohiro, Yoshinobu Iwasaki HA. Calcium-induced mitochondrial swelling and cytochrome c release in the brain: its biochemical characteristics and implication in ischemic neuronal injury. Brain Res. 2003;960(1–2):62–70. [DOI] [PubMed] [Google Scholar]
- 53.Nagy Z, Goehlert UG, Wolfe LS, Hiittner I. Ca2+ depletion-induced disconnection of tight junctions in isolated rat brain microvessels. Acta Neuropathol [Internet], 1985. March [cited 2020 Oct 1];68(1):48–52. Available from: https://pubmed-ncbi-nlm-nih-gov.www.libproxy.wvu.edu/4050353/ [DOI] [PubMed] [Google Scholar]
- 54.Singh SK, Mishra MK, Eltoum IEA, Bae S, Lillard JW, Singh R. CCR5/CCL5 axis interaction promotes migratory and invasiveness of pancreatic cancer cells. Sci Rep [Internet]. 2018. December 1 [cited 2020 Oct 6];8(1):1–12. Available from: www.nature.com/scientificreports/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Young LM, Geldenhuys WJ, Domingo OC, Malan SF, Van Der Schyf CJ. Synthesis and Biological Evaluation of Pentacycloundecylamines and Triquinylamines as Voltage-Gated Calcium Channel Blockers. Arch Pharm (Weinheim) [Internet]. 2016. April 1 [cited 2020 Oct 6];349(4):252–67. Available from: https://pubmed.ncbi.nlm.nih.gov/26892182/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Geldenhuys WJ, Bezuidenhout LM, Dluzen DE. Effects of a novel dopamine uptake inhibitor upon extracellular dopamine from superfused murine striatal tissue. Eur J Pharmacol. 2009. October 1;619(1–3):38–43. [DOI] [PubMed] [Google Scholar]
- 57.Allen KL, Almeida A, Bates TE, Clark JB. Changes of Respiratory Chain Activity in Mitochondrial and Synaptosomal Fractions Isolated from the Gerbil Brain After Graded Ischaemia. J Neurochem [Internet]. 2002. November 23 [cited 2020 Oct 6];64(5):2222–9. Available from: http://doi.wiley.com/10.1046/j.1471-4159.1995.64052222.x [DOI] [PubMed] [Google Scholar]
- 58.Gaur V, Aggarwal A, Kumar A. Protective effect of naringin against ischemic reperfusion cerebral injury: Possible neurobehavioral, biochemical and cellular alterations in rat brain. Eur J Pharmacol. 2009. August 15;616(1–3):147–54. [DOI] [PubMed] [Google Scholar]
- 59.Miao Y, Zhao S, Gao Y, Wang R, Wu Q, Wu H, et al. Curcumin pretreatment attenuates inflammation and mitochondrial dysfunction in experimental stroke: The possible role of Sirtl signaling. Brain Res Bull. 2016. March 1; 121:9–15. [DOI] [PubMed] [Google Scholar]
- 60.Yang Y, Jiang S, Dong Y, Fan C, Zhao L, Yang X, et al. Melatonin prevents cell death and mitochondrial dysfunction via a SIRT1-dependent mechanism during ischemic-stroke in mice. J Pineal Res [Internet]. 2015. January 1 [cited 2020 Oct 6];58(1):61–70. Available from: https://onlinelibrary.wiley.com/doi/full/10.1111/jpi.12193 [DOI] [PubMed] [Google Scholar]
- 61.Wolff V, Schlagowski A-I, Rouyer O, Charles A-L, Singh F, Auger C, et al. Tetrahydrocannabinol Induces Brain Mitochondrial Respiratory Chain Dysfunction and Increases Oxidative Stress: A Potential Mechanism Involved in Cannabis-Related Stroke. 2015; Available from: 10.1155/2015/323706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Almeida A, Allen KL, Bates TE, Clark JB. Effect of reperfusion following cerebral ischaemia on the activity of the mitochondrial respiratory chain in the gerbil brain. J Neurochem [Internet]. 1995. October 1 [cited 2020 Oct 6];65(4):1698–703. Available from: https://onlinelibrary.wiley.com/doi/full/10.1046/j.1471-4159.1995.65041698.x [DOI] [PubMed] [Google Scholar]
- 63.Nour M, Scalzo F, Liebeskind DS. Ischemia-Reperfusion Injury in Stroke. Interv Neurol [Internet], 2013. September 11 [cited 2021 Apr 12]; 1(3–4):185–99. Available from: https://pubmed.ncbi.nlm.nih.gov/25187778/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Heiss WD, Thiel A, Grond M, Graf R. Which targets are relevant for therapy of acute ischemic stroke? Stroke [Internet]. 1999. [cited 2021 Apr 12];30(7):1486–9. Available from: https://pubmed.ncbi.nlm.nih.gov/10390327/ [DOI] [PubMed] [Google Scholar]
- 65.Arnett D, Quillin A, Geldenhuys WJ, Menze MA, Konkle M. 4-Hydroxynonenal and 4-Oxononenal Differentially Bind to the Redox Sensor MitoNEET. Chem Res Toxicol [Internet]. 2019. June 17 [cited 2020 Oct 6];32(6):977–81. Available from: https://pubmed.ncbi.nlm.nih.gov/31117349/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Anastassiadis T, Deacon SW, Devarajan K, Ma H, Peterson JR. Comprehensive assay of kinase catalytic activity reveals features of kinase inhibitor selectivity. Nat Biotechnol [Internet]. 2011. November 30 [cited 2020 Oct 5];29(11):1039–45. Available from: http://kir.fccc.edu/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Elkamhawy A, Park JE, Cho NC, Sim T, Pae AN, Roh EJ. Discovery of a broad spectrum antiproliferative agent with selectivity for DDR1 kinase: Cell line-based assay, kinase panel, molecular docking, and toxicity studies. J Enzyme Inhib Med Chem [Internet]. 2016. January 2 [cited 2020 Oct 5];31(1):158–66. Available from: http://www.tandfonline.com/doi/full/10.3109/14756366.2015.1004057 [DOI] [PubMed] [Google Scholar]
- 68.El-Deeb IM, Park BS, Jung SJ, Yoo KH, Oh CH, Cho SJ, et al. Design, synthesis, screening, and molecular modeling study of a new series of ROS1 receptor tyrosine kinase inhibitors. Bioorganic Med Chem Lett. 2009. October 1;19(19):5622–6. [DOI] [PubMed] [Google Scholar]
- 69.Shekarabi M, Zhang J, Khanna AR, Ellison DH, Delpire E, Kahle KT. WNK Kinase Signaling in Ion Homeostasis and Human Disease. Vol. 25, Cell Metabolism. Cell Press; 2017. p. 285–99. [DOI] [PubMed] [Google Scholar]
- 70.Veríssimo F, Jordan P. WNK kinases, a novel protein kinase subfamily in multi-cellular organisms. Oncogene [Internet]. 2001. September 6 [cited 2020 Oct 5];20(39):5562–9. Available from: www.hgsc.bcm.tmc.edu/seq_data/. [DOI] [PubMed] [Google Scholar]
- 71.Yamada K, Park HM, Rigel DF, DiPetrillo K, Whalen EJ, Anisowicz A, et al. Small-molecule WNK inhibition regulates cardiovascular and renal function. Nat Chem Biol [Internet]. 2016. November 1 [cited 2020 Oct 5]; 12(11):896–8. [DOI] [PubMed] [Google Scholar]
- 72.Wu D, Lai N, Deng R, Liang T, Pan P, Yuan G, et al. Activated WNK3 induced by intracerebral hemorrhage deteriorates brain injury maybe via WNK3/SPAK/NKCC1 pathway. Exp Neurol. 2020. October 1;332:113386. [DOI] [PubMed] [Google Scholar]
- 73.Begum G, Yuan H, Kahle KT, Li L, Wang S, Shi Y, et al. Inhibition of WNK3 Kinase Signaling Reduces Brain Damage and Accelerates Neurological Recovery after Stroke. Stroke [Internet]. 2015. July 4 [cited 2020 Oct 5];46(7):1956–65. Available from: http://stroke.ahajournals.org/lookup/doi/10.1161/STROKEAHA.115.008939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zhao H, Nepomuceno R, Gao X, Foley LM, Wang S, Begum G, et al. Deletion of the WNK3-SPAK kinase complex in mice improves radiographic and clinical outcomes in malignant cerebral edema after ischemic stroke. J Cereb Blood Flow Metab [Internet]. 2017. February 20 [cited 2020 Oct 5];37(2):550–63. Available from: http://journals.sagepub.com/doi/10.1177/0271678X16631561 [DOI] [PMC free article] [PubMed] [Google Scholar]