

Abstract
Atrial fibrillation (AF) is the most common type of arrhythmia. Epidemiological studies have documented a substantial genetic component. More than 160 genes have been associated with AF during the last decades. Some of these were discovered by classical linkage studies while the majority relies on functional studies or genome-wide association studies. In this review, we will evaluate the genetic basis of AF and the role of both common and rare genetic variants in AF. Rare variants in multiple ion-channel genes as well as gap junction and transcription factor genes have been associated with AF. More recently, a growing body of evidence has implicated structural genes with AF. An increased burden of atrial fibrosis in AF patients compared with non-AF patients has also been reported. These findings challenge our traditional understanding of AF being an electrical disease. We will focus on several quantitative landmark papers, which are transforming our understanding of AF by implicating atrial cardiomyopathies in the pathogenesis. This new AF research field may enable better diagnostics and treatment in the future.
Subject terms: Atrial fibrillation, Genetics
Introduction
Atrial fibrillation (AF) is the most common cardiac arrhythmia. It affects more than 33 million people worldwide and is associated with a significant increase in morbidity and mortality. The life time risk has been estimated to 25% [1]. AF causes irregular and often abnormally fast heart rate. Patients with AF have an increased risk of heart failure, stroke, dementia, and death [2]. Risk factors for AF include increasing age, heart disease, high blood pressure, and alcohol overuse [1]. In other instances, no risk factors can be identified, which suggests an underlying genetic predisposition to AF. The pathogenesis of AF remains poorly understood, which, to some degree, compromises the development of effective treatments.
The underlying AF mechanisms have been subject to intense research. AF predominantly arise secondary to hypertension, ischemic and/or structural heart disease [2]. These cardiovascular diseases can influence the electrical and structural remodeling of the atria which is likely to play a central role in the pathogenesis of AF. It has furthermore been recognized that atrial myopathy and fibrosis seem to play a driving role in the development of AF [3, 4].
Arnar et al. performed a population-based cohort study of more than 5000 Icelandic AF patients in 2006. The study demonstrated substantial familial aggregation of AF and a strong likelihood of heritability among AF patients. At the time, this led to the likely assumption that there may be undiscovered genetic variants underlying the risk of common AF in Iceland [5]. In 2009, a large study was completed in 1137 Danish twins, investigating the heredity of AF. The heritability of AF was found to be 62% based on biometric models [6]. In 2012, an epidemiological study investigated whether an individual’s risk of developing AF without having risk factors for AF (previously termed lone AF) before 60 years is associated with familial lone AF. This study found an increased risk of lone AF when having family members with lone AF, with the strongest risk associated with multiple affected relatives and relatives with a young age at AF onset [7]. The epidemiological results support that genetic factors seem to contribute to the risk of AF.
Through linkage and family studies, rare AF-associated variants were initially identified to locate to genes encoding ion channels, such as the potassium and sodium channels, contributing to cardiac depolarization or repolarization. Rare variants in ion and non-ion-channel genes were later identified through candidate gene studies, which recognized several rare variants based on prior knowledge of the specific genes.
Genome-wide association studies (GWAS) on AF have shown that more than a hundred single nucleotide polymorphisms (SNPs) also contribute to the risk of AF [8]. The remaining heritability of AF may be explained by promoter variants, epigenetics, structural variants and undiscovered genetic mechanisms [9].
In this review, we will focus on the genetic basis of AF, the role of both common and rare genetic variants and the link to cardiomyopathy and atrial fibrosis.
Materials and methods
We conducted an extensive review of the literature through the online library PubMed. We performed a systematic literature search with the query ((“Atrial fibrillation” [MeSH]) OR (atrial fibrillation)) AND ((“Genetics” [MeSH]) OR (genetic*)) AND ((“Mutation” [MeSH]) OR (mutation*) OR (“Polymorphism, single nucleotide” [MeSH]) OR (polymorphism, single nucleotide) OR (monogenic*) OR (GWAS)). Articles concerning the genetic basis of AF were included and small studies concerning common variants in genes not associated with AF were excluded from this review. Common and rare genetic variants were investigated in various publicly available gene variant databases; gnomAD, TransVar, Ensembl, UCSC Genome Browser, Mutalyzer and NCBI databases [10–15]. Details on rare genetic variants including annotation, original study type, transcript and country of origin were found in the original studies if present or investigated in the beforementioned databases (see Supplementary Table 2).
Results and discussion
The role of common genetic variants
AF has been subject to intense research and investigators have identified many genetic loci associated with AF [16]. A list of common variants associated with AF is shown in Supplementary Table 1.
The most significantly associated AF SNP in all of these studies is located in a noncoding region of chromosome 4q25, ~150,000 base pairs upstream of the gene encoding the paired-like homeodomain transcription factor pitx2 [8]. PITX2 encodes three different isoforms; pitx2a, pitx2b and pitx2c, where pitx2c is the most prominent isoform in the developing heart [17]. Previous work has indicated that PITX2 expression in humans has been significantly reduced in AF patients, offering a connection between PITX2 loss-of-function and AF [17]. In a recent paper by Collins et al. PITX2 loss-of-function was investigated in CRISPR-Cas9 modified zebrafish. These animals were found to have a compromised sarcomere, an increased amount of fibrosis and a more than fourfold upregulation of the expression of the pacemaker gene HCN4. An increased expression of HCN4 may be part of the explanation for increased ectopic activity seen in AF patients [18].
Another highly significant SNP identified through GWAS is rs2106261 (NC_000016.9 (NM_001164766.2:g.73051620C>T)) located on chromosome 16q22 intronic to the transcription factor gene ZFHX3 [19]. The function of ZFHX3 in cardiac tissue is unknown, but it is expressed in mouse hearts and has been associated with myogenic and neuronal differentiation [20].
The SNP rs13376333 (NC_000001.10 (NM_ 002249.6:g.154814353C>T)) is strongly associated with AF, and has been located to an intron of KCNN3 on chromosome 1q21. This gene encodes a calcium activated potassium channel (the SK3 channel) and is thought to be involved in atrial repolarization [21]. Interestingly, there is currently an ongoing clinical phase II study targeted towards inhibition of SK3 channels. These ion channels are present in the heart, where they play a role in regulating the cardiac rhythm. Blocking these channels leads to an antiarrhythmic effect by prolonging the action potential in the atria selectively. This is a promising approach for a new effective treatment of AF, that might prevent harmful pharmaceutical side effects in the ventricles [22]. Furthermore, the gene KCNN2 encodes the SK2 channels which have been shown to co-assemble with the SK3 channels and the gene has been associated with AF in a recent GWAS [16].
Hyperpolarization-activated cyclic nucleotide-gated (HCN) pacemaker channels are expressed in the heart and the gene HCN4 is the predominant gene encoding the cardiac pacemaker channels [23]. This gene has been associated with AF in GWAS [24].
Another locus associated with AF has been found on chromosome 10q22 (rs10824026; NC_000010.10 (NM_001114133.3:g.75421208G>A)) located 5 kb upstream of SYNPO2L and 20 kb upstream of MYOZ1. The structural proteins encoded by these genes are both expressed in skeletal and cardiac muscle, however it is unknown which of these genes are driving the association [24].
In 2018, Thorolfdottir et al. reported an association between AF and two common variants in the gene RPL3L on chromosome 16 and one variant in the MYZAP gene on chromosome 15. The RPL3L gene encodes a ribosomal protein, primarily expressed in skeletal and heart muscle. MYZAP encodes myozap, which is a myocardial zonula adherence protein, mainly expressed in the human heart. The mouse homolog has been located to the intercalated discs [25]. Seeger et al. were the first to identify myozap and because of both the subcellular localization within the cardiomyocytes and the observation of a severe cardiac phenotype in mutated zebrafish, myozap is thought to be implicated in a subtype of atrial cardiomyopathy [26].
In 2017, Christophersen et al. identified 12 novel AF loci through GWAS, implicating genes involved in cardiac and structural remodeling [27]. Later, an AF GWAS was completed in 2018 by Nielsen et al. identifying one novel risk locus and confirming 13 out of 16 already known AF loci [28]. The most statistically significant novel association was observed at the 2q31 locus harboring seven highly correlated missense variations. These missense variants fell within the I-, A- and M-bands of TTN, a strong biological AF candidate gene because of its role in the structural integrity and muscle elasticity of the heart [28]. We will discuss the role of TTN in more detail in a later section.
Same year, a large AF GWAS meta-analysis was conducted which reported a more than threefold increase in the number of loci associated with AF [29]. The analysis was conducted in over half a million individuals, including 65,446 with AF, from more than 50 studies. Two identified loci were located close to genes that are targets for current antiarrhythmic medications. SCN5A, which will be mentioned in the next section, encodes the Nav1.5 channel which is the target for sodium channel blockers such as flecainide. Similarly, KCNH2 encodes the Kv11.1 channel which is the target for potassium-channel-inhibiting medications such as amiodarone. The GWAS furthermore proved transcriptional regulation to be a key feature in AF by associating loci close to genes encoding transcription factors, e.g., TBX3, TBX5 and NKX2-5, with AF. These genes are involved in the development of the cardiac conduction system [29].
Later in 2018, Nielsen et al. conducted an even larger meta-analysis and identified AF risk variants located close to genes which have been associated with serious heart defects in humans, e.g., GATA4, MYH6, NKX2-5, PITX2, or near genes important in striated muscle function e.g., MYH7 [16]. One-hundred-and-eleven genomic regions were identified with at least one variant associated with AF. Furthermore, the authors found an association between younger age of AF onset and a high genetic burden of AF risk variants [16].
Interestingly, the GWAS approach has implicated many genes already suspected to be involved in AF through candidate gene studies (e.g., GJA5, KCNH2, SCN5A, KCNJ2, KCND3, MYH7, and NKX2-5). AF GWAS have furthermore identified AF-associated genes which have been implicated in a variety of inherited arrhythmias, other conduction diseases and cardiomyopathies. This highlights the pleiotropy of these genes as well as the polygenetic nature of AF.
The role of rare genetic variants
Rare variants in genes encoding ion channels, signaling molecules, accessory subunits and gap junctions have been associated with AF. These variants can lead to AF through different pathways, which is illustrated in Fig. 1. Rare AF-associated variants in different genes are listed in Supplementary Table 2 and will be presented in the following.
Fig. 1. AF mechanisms.
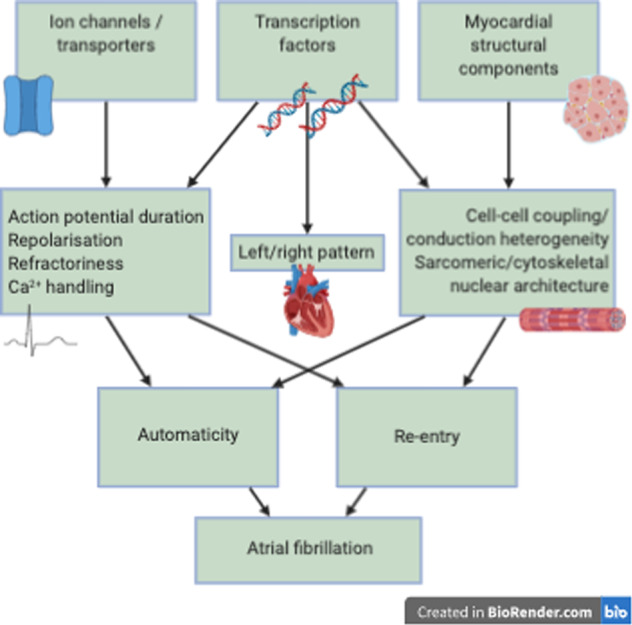
Genetic variants in genes encoding different ion-channels, transcription factors and myocardial structural components predispose to atrial fibrillation (AF) through different pathways that lead to increased automaticity and reentry activity.
Potassium-channel variants
The gene KCNQ1 encodes the pore-forming α-subunit of the cardiac potassium-channel IKs, which is involved in cardiac cell repolarization. The first association between rare variants in KCNQ1 and familial AF was discovered in 2003 and functional analysis of the c.418A>G (p.(Ser140Gly)) variant showed a gain-of-function effect [8]. As opposed to IKs, which has six transmembrane domains, the β-subunits of IKs are single-transmembrane units and are encoded by the KCNE genes, KCNE1-KCNE5. Isolated and familial cases with AF have been shown to carry rare variants in the β-subunits of the IKs channel [8].
The gene KCNH2 encodes the α-subunit of the IKr channel and a rare variant in this gene has been identified in a family with AF and short QT, proposing an overlap in phenotypes [8]. Several other variants have been discovered, which are shown in Supplementary Table 2.
The inward rectifier channel Kir2.1 mediates the IK1 current involved in repolarisation and is encoded by the gene KCNJ2 (Supplementary Material reference 21). A missense variant was found in KCNJ2 in a Chinese AF kindred. Functional analysis demonstrated a gain-of-function variant, and the authors hypothesized that the gene may play a role in initiating and/or maintaining AF [8]. Additional variants are shown in Supplementary Table 2.
A missense variant was found in a cohort of AF patients in the gene KCNJ8, which encodes the cardiac KATP channel Kir6.1 [8]. The authors suggested that the variant was associated with increased susceptibility to AF and the variant has shown to give rise to a gain-of-function effect of the cardiac channel [8].
A particularly interesting case is KCNA5. KCNA5 encodes an atria specific Kv1.5 channel involved in cardiac repolarisation and several loss-of-function variants have been identified and associated with AF [8]. Christophersen et al. identified six novel rare variants in KCNA5 in seven patients with early-onset lone AF. The authors found rare variants in this gene to lead to a gain-of-function effect and hence support the hypothesis that both gain- and loss-of function of the IKur current enhance AF susceptibility [30].
Olson et al. [31] identified a missense variant in ABCC9, which encodes a KATP channel subunit. The missense variant was found in a female with early-onset AF (Supplementary Material reference 1).
Sodium channel variants
SCN5A encodes the cardiac sodium channel Nav1.5 and several SCN5A variants have been reported to associate with AF [32]. In 2012, Olesen et al. identified ten rare variants in SCN5A in a cohort of 192 patients with early-onset AF [33]. Many of these lone AF patients carried an SCN5A variant previously associated with Long QT syndrome (LQTS). Functional investigations of the SCN5A variants revealed both compromised transient peak current and increased sustained current, indicating both gain- and loss-of-function variants in early-onset AF [33]. Several SCN5A variants have subsequently been identified in AF patients and are listed in Supplementary Table 2.
Variants in the four sodium channel ß-subunits, ß1-4, encoded by SCN1-4B, have been identified in AF patients. Loss-of-function variants were identified in SCN1-2B in a cohort of AF patients [34]. Furthermore, another study found three loss-of-function variants in SCN3B in a lone AF cohort, indicating that this gene contribute to the mechanisms behind lone AF [35].
SCN10A encodes the voltage-gated Nav1.8 channel and has been associated with the electrocardiographic (ECG) PR-interval and AF [36]. Jabbari et al. found ten rare missense SCN10A variants in 225 lone AF patients and showed that the common variant c.3218T>C (p.(Val1073Ala)) (rs6795970; NC_000003.11 (NM_001293307.2:g.38766675A>G)) increased the risk of AF [36]. Furthermore, functional characterization of the rare variants revealed reduced activity in Nav1.8, while the common c.3218T>C (p.(Val1073Ala)) variant had a gain-of-function phenotype, which seems to increase the risk of AF. These data on rare variants and the common c.3218T>C (p.(Val1073Ala)) variant suggest that both gain- and loss-of-function of the Nav1.8 current could be involved in the development of AF [36], but further research is needed to confirm the involvement of rare variants in SCN10A. The variants are summarized in Supplementary Table 2.
Non-ion channel variants
Zhang et al. discovered a homozygous variant, c.1172G>A (p.(Arg391His)) in NUP155, that co-segregated in an AF family. Furthermore, heterozygous NUP155(+/−) knockout mice were shown to have AF-like ECG recordings [37].
The NUP155 gene is localized to the chromosome 5p13 and encodes a nucleoporin, a main component of the nuclear pore complexes involved in cytoplasmic transport [37]. Oberti et al. [38] identified a large AF family with an autosomal recessive inheritance pattern and found a co-segregating variant in a locus at chromosome 5p13 (Supplementary Material reference 115). This finding provides evidence that NUP155 might be a disease-causing AF gene.
Hodgson-Zingman et al. studied a family with AF segregating as an autosomal dominant trait and a co-segregating frameshift variant in NPPA [39]. NPPA encodes the atrial natriuretic peptide (ANP). ANP is a circulating hormone secreted from the cardiac atria involved in the regulation of blood pressure [39].
GATA-4 and GATA-6 are cardiac transcription factors involved in myocardial development. In 2010, Posch et al. found variants in the GATA4 gene in a patient with familial lone AF and a second variant in a patient with sporadic AF [40]. Other studies have identified additional GATA4 variants that co-segregate with AF and Yang et al. identified two heterozygous loss-of-function GATA6 variants. These rare variants co-segregated with AF as an autosomal dominant trait [41].
The AF-associated gene, GREM2, encodes the bone morphogenic protein antagonist gremlin-2. Functional studies in zebrafish have revealed that GREM2 is required for cardiac laterality and atrial differentiation during embryonic development [42]. Furthermore, a live heart imaging study in a zebrafish overexpressing the GREM2 variant, c.226C>G (p.(Gln76Glu)), showed an abnormal contraction velocity specifically in atrial cardiomyocytes suggesting that the gene could play a role in AF [42].
Gap-junction variants
Connexin43 and connexin40 are encoded by GJA1 and GJA5, respectively, and are gap-junction proteins in the atrial myocardium. A study done by Thibodeau et al. identified a novel loss-of-function somatic variant in GJA1 associated with lone AF [43]. In contrast to this finding, Gregers et al. investigated the prevalence of somatic variants in 44 AF patients undergoing mitral valve surgery [44]. This much larger study did not identify any somatic variants, indicating that somatic variants do not play a major role in the pathogenesis of AF. Interestingly, a high proportion of the patients in the cohort had rare germline variants indicating that these non-lone AF patients, may also be predisposed to AF by rare germline variants [44]. Several GJA1 and GJA5 variants are shown in Supplementary Table 2.
Variants affecting cardiac structure
Other rare variants in the genes MYH7, MYBPC3, MYL4, and TTN have been associated with AF and cardiomyopathy and will be explained in a later section [45–47].
As described above, rare variants in genes encoding different ion channels, transcription factors as well as structural components of the myocardium have been associated with AF. Figure 1 illustrates how rare variants can lead to atrial pathology e.g., altered sarcomeric architecture, which in turn may lead to arrhythmias through reentry or automaticity.
Genetic overlap with other diseases
AF has been associated with other phenotypes in patients with inherited arrhythmia syndromes, such as Brugada syndrome (BrS) and LQTS, but also with familial cardiomyopathies such as hypertrophic cardiomyopathy (HCM) and dilated cardiomyopathy (DCM).
LQTS is a cardiac repolarization abnormality and has been associated with variants in genes such as KCNQ1, KCNE1-3, KCNH2, KCNJ2, and SCN5A [48]. Patients with genetically proven LQTS have a higher risk of early-onset AF than the rest of the population [49]. Nielsen et al. have previously found that both shortened and prolonged QTc interval durations are risk factors for AF in the general population. The association was strongest in lone AF patients, suggesting a link between an extreme QTc interval and AF [50]. BrS is an inherited syndrome associated with a high incidence of sudden cardiac arrest and has been linked to rare variants in different genes e.g., KCNE3, KCNE5, KCND3, KCNH2, SCN5A, SCN1Bb, and SCN3B [51].
A recent GWAS on coarctation of the aorta among Icelanders (120 cases and 355,166 controls) identified a rare missense variant in MYH6 which also associated with Sick Sinus Syndrome (SSS) and AF [52]. MYH6 encodes an alpha myosin heavy chain subunit (α-MyHC). Myosin is an essential component of the cardiac muscle and α-MyHC is a fast motor protein of the thick filaments of the contractile apparatus in healthy adult atrial muscle [53]. MYH7, on the other hand, encodes a slower MyHC motor protein, β [54]. The β-MyHC is upregulated in heart failure and other cardiac disorders, whereas α-MyHC is downregulated, giving rise to the hypothesis that MyHC isoforms play a role in the determination of cardiac contractility [55]. New research has established a link between an increased MYH7 expression in atrial tissue from patients with chronic AF. Chronic AF myofibrils show biochemical and biophysical differences from healthy myofibrils, potentially as a result of increased MYH7 expression [56].
Another Icelandic study identified a variant in PLEC to associate with both a 55% increased risk of AF and a 64% increased risk of SSS. This gene encodes structural components of the cardiomyocyte [57].
The human genome is contained within the cell nucleus and alterations in the nuclear envelope protein lamin A/C, encoded by the gene LMNA, cause a number of diseases such as cardiomyopathy and muscular dystrophy [58]. A heterozygous missense rare variant was identified in a family with AF, as well as supraventricular tachycardia, ventricular tachycardia and sudden cardiac death, in 2010 by Beckmann et al. [59]. See Supplementary Table 2 for a list of LMNA variants.
In 2018, Bundgaard et al. identified five families with an autosomal dominant cardiac syndrome characterized by uniform ECG changes with persistent non-ischemic ST-segment depressions, AF and ventricular arrhythmias [60]. The ST-segment depression remained stable over time, in contrast to other genetic disorders (BrS and LQTS), that are characterized by dynamic pathognomic ECG changes. Genetic evaluation was performed, however no coding variants were identified to associate with the syndrome [60].
Hyperthyroidism is well known to contribute to cardiovascular morbidity, particularly AF [61]. A mendelian randomization study by Salem et al. suggests a genetically determined variation in thyroid function within a physiologically normal range as a risk factor for AF [61]. In 2019, Ellervik et al. found an association between genetically increased FT3:FT4 ratio and hyperthyroidism and AF [62]. Thyroid hormone replacement for hypothyroidism may increase the AF risk, whereas antithyroid medications to treat hyperthyroidism may reduce the risk of AF. This complicates the treatment of patients with subclinical thyroid disease, and the risk of AF should probably be considered in the clinical decision to treat these patients [61].
Genetic correlation between AF and other traits is illustrated in Fig. 2 (modified from Hadji-Turdeghal et al. [63]). The most significant genetic correlation with AF is heart failure (p = 2 × 10−13). Height was highly correlated with AF (p = 2 × 10−33) and so was hypertension, a risk factor for AF (p = 1 × 10–12). Interestingly, other well-established risk factors for AF such as diabetes type 2 and alcohol dependence, had a non-significant correlation with AF (p = 2 × 10−1).
Fig. 2. Genetic correlation with AF.
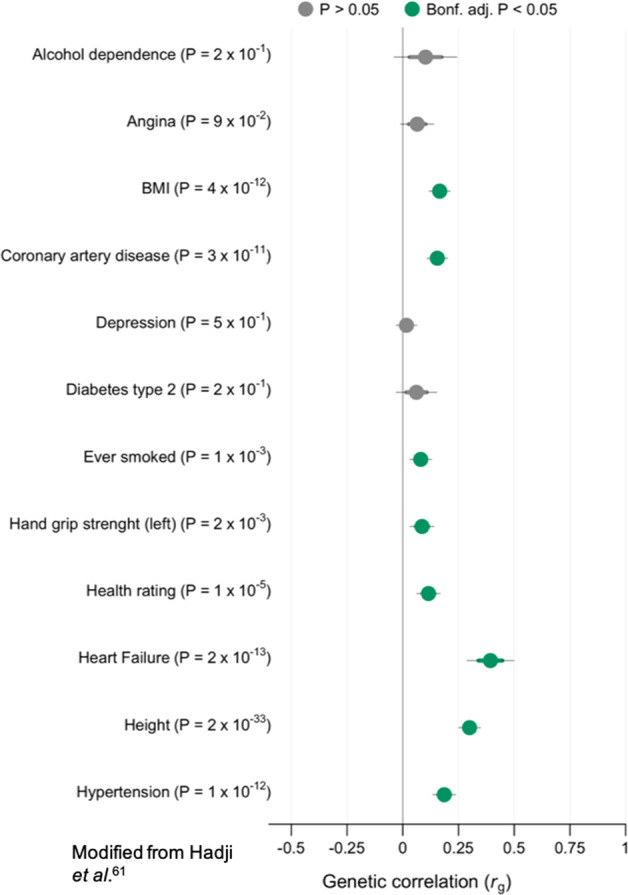
LD score regression revealing genetic correlation between AF (65,446 cases and >500,000 controls) and other phenotypes. Phenotypes with negative log10(P) are displayed on the y-axis. X-axis show genetic correlation (rg). Dots are estimated values with thick lines indicating mean standard error (SE) and thin lines 1.96 SE. Significant association after Bonferroni corrections is denoted with green color. Nominal significance is denoted with violet color and non-significant correlations with grey. SEs and p values were derived from using block jackknife resampling. Modified from Hadji-Turdeghal et al. [63].
Cardiomyopathy
There is an increasing interest in cardiomyopathy and how it is linked to AF. The relationship between genetic variation in HCM and AF has been poorly described, although AF is common in cardiomyopathy patients [64]. Factors such as age and left atrium (LA) size are predictors for AF in HCM patients, where larger LA size may increase the left ventricle (LV) diastolic pressure resulting in diastolic dysfunction [64]. Cardiomyopathies, in particular arrhythmogenic right ventricular cardiomyopathy (ARVC), have been associated with variants in the intercalated disc genes and ARVC patients have an increased risk of AF and ventricular arrhythmias. This is a leading cause of sudden death in young people and athletes [65]. Many rare variants are linked to HCM, but variations in the MYH7 and MYBPC3 genes are the most common [45]. It is still unclear why HCM patients with MYH7 rare variants have a higher risk of AF, but variations in this gene may cause extensive myocardial disease leading to reduced cardiac performance, which may be associated with higher occurrence of AF [64].
Recently, Vad et al. identified six individuals with rare loss-of-function variants in three different DCM genes (DMD, PDLIM3, and FKTN), of which two variants were novel. These data suggest that rare loss-of-function variants in cytoskeletal genes previously associated with DCM may have a role in early-onset AF, perhaps through the development of an atrial cardiomyopathy [66].
Several findings suggest that AF could be caused by atrial cardiomyopathy [67]. Peng et al. showed an association between the MYL4 gene and atrial cardiomyopathy. The MYL4 gene is responsible for the electrical, contractile and structural integrity of the atrium. A loss-of-function variant in the MYL4 gene was found to cause early atrial fibrosis leading to atrial cardiomyopathy and atrial arrhythmia, but also atrial contractile failure and atrial enlargement [46]. Furthermore, two other studies have independently identified an association between early-onset AF and a rare variant in MYL4 [3, 68]. Gudbjartsson et al. [3] identified a frameshift variant in MYL4 associated with a recessive form of AF. The authors found a high risk of stroke in these patients, potentially related to underlying atrial cardiomyopathy. When assessed by the CHAD2DS2-VASc score, the patients were unexpectedly scored to having a low stroke risk, suggesting an alternative stroke mechanism. The results raise the possibility of a genotype-based risk stratification of atrial thrombus formation.
TTN is thought to be an important AF gene, as mentioned previously. The gene encodes a giant sarcomere protein (titin) expressed in all chambers of the human heart and titin-truncating variants (TTNtv) have been shown to predispose directly for AF [47]. TTNtv are known to occur in about 15% of DCM cases and independently predict early arrhythmias in DCM patients [69]. In 2018, Choi et al. found loss-of-function variants in TTN to be associated with early-onset AF. The study supports the role of malfunctioning sarcomeric proteins in the pathogenesis of AF [70]. The occurrence of TTN loss-of-function variants in AF and DCM patients suggests that impaired sarcomere function may be an overlapping pathophysiological mechanism.
Ahlberg et al. found a significant enrichment of rare TTNtv in families diagnosed with AF (n = 399; odds ratio = 36.8; p = 4.13 × 10−6) [47]. Using a zebrafish model carrying a rare variant in ttn, the homologs TTN gene in zebrafish, the authors showed a distinct sarcomere defect in the mutants. Further analysis of the heart revealed an increased amount of fibrosis and a compromised sarcomere structure in the mutant larvae and adult fish, suggesting a predisposition for arrhythmia and conduction disease. The discovery of atrial fibrosis in young zebrafish, indicates that TTNtv predispose to the development of fibrosis in the atria from an early age. The zebrafish findings and the early-onset of AF in the replication cohort both propose a possible link between structural disease and electrical phenotype [47].
The findings of increased atrial fibrosis in zebrafish with TTNtv as well as compromised sarcomere structure associated with both TTN and MYL4 variants, suggest a fundamental role of structural genes in AF and indicate a link between AF and cardiomyopathy. Characterizing genetic subtypes of cardiomyopathy and their associations with AF may help improve our understanding of the AF pathophysiology. Figure 3 illustrates how the AF cardiomyocyte is affected by fibrotic changes.
Fig. 3. AF and atrial cardiomyopathy.

Structural rearrangement in the heart seems to play a key role in atrial cardiomyopathy and in AF, here illustrated with a transmitted electron microscopy (TEM) image of the cardiomyocyte of a patient with AF and atrial cardiomyopathy. A Heart and B cardiomyocyte from AF patient with atrial cardiomyopathy, and C TEM imaging of sarcomere from AF patient with atrial cardiomyopathy affected by fibrotic changes (light areas). The sarcomeres look disrupted with poorly defined M-lines and I-bands, and fuzzy Z-lines (green arrows) and the mitochondria show an increased amount of cristae and ballooning (red arrows). D For comparison, schematic, and TEM imaging of sarcomere from patient without AF. AF; atrial fibrillation.
Our evolving knowledge of the genetic and structural basis of AF has led to new awareness that our current antiarrhythmic drugs might not target the major mechanisms implicated in AF. For many years, AF has been considered an electrical disease, but our understanding of the pathophysiology of AF has improved and it is now considered to be much more than an ion-channel disease. Atrial fibrosis has continuously been reported to be more frequent in patients with AF compared to non-AF patients [71].
In the absence of heart failure, patients with long-standing or persistent AF appear to have increased atrial fibrosis, whereas those with paroxysmal AF do not [72]. Cochet et al. found a higher degree of re-entrant activity in areas with atrial fibrosis in patients with persistent AF, and the authors propose this to be a likely AF mechanism [73].
In line with this, Mahnkopf et al. investigated atrial fibrosis as a marker for structural remodeling and found extensive structural remodeling in lone AF patients compared with non-lone AF patients [74]. In animals with long-standing AF, atrial fibrosis can be prevented by inhibition of the renin–angiotensin system, which appears to significantly reduce the duration of AF [75]. The potential use of angiotensin receptor blockers and angiotensin-converting-enzyme inhibitors could be an interesting option for preventing the promotion of AF by suppressing the development of structural remodeling. Boldt et al. previously reported that angiotensin-converting-enzyme inhibitors reduce fibrosis in patients with lone AF [76].
Conclusion
Although extensive efforts have been made to identify the role of AF genetics in AF pathology, the field continues to grow as we explore new associations. During the last decades, variants in ion-channel genes e.g., in sodium and potassium-channel genes, and in non-ion-channel genes including structural genes have been associated with AF. Recently, both TTN and MYL4 variants have been associated with early-onset AF. The discovery that early atrial fibrosis plays a significant role in atrial cardiomyopathy and in AF has given us a better understanding of the AF pathogenesis. Based on the differences in the pathogenesis of AF, the assumption of a “one-size-fits all” treatment is inadequate. AF as a polygenetic complex disease with a structural component is a new but very promising area of research and might prove very important in the future treatment of AF.
Supplementary information
Acknowledgments
Funding
This work was supported by The John and Birthe Meyer Foundation, Direktør Ib Henriksens fond, The Research Foundation of Rigshospitalet, Villadsen Family Foundation, The Arvid Nilsson Foundation, and The Hallas-Møller Emerging Investigator Novo Nordisk (NNF17OC0031204).
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Julie H. Andersen, Laura Andreasen
Supplementary information
The online version of this article (10.1038/s41431-020-00784-8) contains supplementary material, which is available to authorized users.
References
- 1.Chugh SS, Havmoeller R, Narayanan K, Singh D, Rienstra M, Benjamin EJ, et al. Worldwide epidemiology of atrial fibrillation: a Global Burden of Disease 2010 Study. Circulation. 2014;129:837–47. doi: 10.1161/CIRCULATIONAHA.113.005119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kirchhof P, Benussi S, Kotecha D, Ahlsson A, Atar D, Casadei B, et al. 2016 ESC Guidelines for the management of atrial fibrillation developed in collaboration with EACTS. Eur Heart J. 2016;37:2893–962. doi: 10.1093/eurheartj/ehw210. [DOI] [PubMed] [Google Scholar]
- 3.Gudbjartsson DF, Holm H, Sulem P, Masson G, Oddsson A, Magnusson O, et al. A frameshift deletion in the sarcomere gene MYL4 causes early-onset familial atrial fibrillation. Eur Heart J. 2017;38:27–34. doi: 10.1093/eurheartj/ehw379. [DOI] [PubMed] [Google Scholar]
- 4.Nattel S. Molecular and cellular mechanisms of atrial fibrosis in atrial fibrillation. JACC Clin Electrophysiol. 2017;3:425–35. doi: 10.1016/j.jacep.2017.03.002. [DOI] [PubMed] [Google Scholar]
- 5.Arnar DO, Thorvaldsson S, Manolio TA, Thorgeirsson G, Kristjansson K, Hakonarson H, et al. Familial aggregation of atrial fibrillation in Iceland. Eur Heart J. 2006;27:708–12. doi: 10.1093/eurheartj/ehi727. [DOI] [PubMed] [Google Scholar]
- 6.Christophersen IE, Ravn LS, Budtz-Joergensen E, Skytthe A, Haunsoe S, Svendsen JH, et al. Familial aggregation of atrial fibrillation: a study in Danish twins. Circ Arrhythm Electrophysiol. 2009;2:378–83. doi: 10.1161/CIRCEP.108.786665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Oyen N, Ranthe MF, Carstensen L, Boyd HA, Olesen MS, Olesen SP, et al. Familial aggregation of lone atrial fibrillation in young persons. J Am Coll Cardiol. 2012;60:917–21. doi: 10.1016/j.jacc.2012.03.046. [DOI] [PubMed] [Google Scholar]
- 8.Olesen MS, Nielsen MW, Haunsø S, Svendsen JH. Atrial fibrillation: the role of common and rare genetic variants. Eur J Hum Genet. 2014;22:297–306. doi: 10.1038/ejhg.2013.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Manolio TA, Collins FS, Cox NJ, Goldstein DB, Hindorff LA, Hunter DJ, et al. Finding the missing heritability of complex diseases. Nature. 2009;461:747–53. doi: 10.1038/nature08494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.gnomAD. https://gnomad.broadinstitute.org/. Accessed 18 Feb 2020.
- 11.TransVar. https://bioinformatics.mdanderson.org/transvar/. Accessed 8 Mar 2020.
- 12.Ensembl Genome Browser. http://m.ensembl.org/index.html. Accessed 11 May 2020.
- 13.UCSC Genome Browser Home. Human hg19 chrX:15578261-15621068 UCSC Genome Browser v406. https://genome.ucsc.edu/cgi-bin/hgTracks?db=hg19&lastVirtModeType=default&lastVirtModeExtraState=&virtModeType=default&virtMode=0&nonVirtPosition=&position=chrX%3A15578261%2D15621068&hgsid=962573805_Whh2Ge7we2sIo607ao0M5E8ZxaN5. Accessed 25 May 2020.
- 14.Mutalyzer 2.0.32—Position Converter. https://mutalyzer.nl/position-converter/. Accessed 26 Oct 2020.
- 15.Information NC for B, Pike USNL of M 8600 R, MD B, Usa 20894. National Center for Biotechnology Information. https://www.ncbi.nlm.nih.gov/. Accessed 25 May 2020.
- 16.Nielsen JB, Thorolfsdottir RB, Fritsche LG, Zhou W, Skov MW, Graham SE, et al. Biobank-driven genomic discovery yields new insight into atrial fibrillation biology. Nat Genet. 2018;50:1234–9. doi: 10.1038/s41588-018-0171-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chinchilla A, Daimi H, Lozano-Velasco E, Dominguez JN, Caballero R, Delpón, et al. PITX2 insufficiency leads to atrial electrical and structural remodeling linked to arrhythmogenesis. Circ Cardiovasc Genet. 2011;4:269–79. doi: 10.1161/CIRCGENETICS.110.958116. [DOI] [PubMed] [Google Scholar]
- 18.Collins MM, Ahlberg G, Hansen CV, Guenther S, Marín-Juez R, Sokol AM, et al. Early sarcomere and metabolic defects in a zebrafish pitx2c cardiac arrhythmia model. Proc Natl Acad Sci USA. 2019;116:24115–21. doi: 10.1073/pnas.1913905116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Benjamin EJ, Rice KM, Arking DE, Pfeufer A, Noord CV, Smith AV, et al. Variants in ZFHX3 are associated with atrial fibrillation in individuals of European ancestry. Nat Genet. 2009;41:879–81. doi: 10.1038/ng.416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ido A, Miura Y, Watanabe M, Sakai M, Inoue Y, Miki T, et al. Cloning of the cDNA encoding the mouse ATBF1 transcription factor. Gene. 1996;168:227–31. doi: 10.1016/0378-1119(95)00740-7. [DOI] [PubMed] [Google Scholar]
- 21.Ellinor PT, Lunetta KL, Glazer NL, Pfeufer A, Alonso A, Chung MK, et al. Common variants in KCNN3 are associated with lone atrial fibrillation. Nat Genet. 2010;42:240–4. doi: 10.1038/ng.537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Goldin DinessJonas, Lasse Skibsbye, Rafel Simó-Vicens, Santos JL, Lundegaard P, Citerni C, et al. Termination of vernakalant-resistant atrial fibrillation by inhibition of small-conductance Ca2+-activated K+ channels in pigs. Circ Arrhythm Electrophysiol. 2017;10:e005125. doi: 10.1161/CIRCEP.117.005125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Herrmann S, Layh B, Ludwig A. Novel insights into the distribution of cardiac HCN channels: an expression study in the mouse heart. J Mol Cell Cardiol. 2011;51:997–1006. doi: 10.1016/j.yjmcc.2011.09.005. [DOI] [PubMed] [Google Scholar]
- 24.Ellinor PT, Lunetta KL, Albert CM, Glazer NL, Ritchie MD, Smith AV, et al. Meta-analysis identifies six new susceptibility loci for atrial fibrillation. Nat Genet. 2012;44:670–5. doi: 10.1038/ng.2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Thorolfsdottir RB, Sveinbjornsson G, Sulem P, Nielsen JB, Jonsson S, Halldorsson GH, et al. Coding variants in RPL3L and MYZAP increase risk of atrial fibrillation. Commun Biol. 2018;1:68. doi: 10.1038/s42003-018-0068-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Seeger TS, Frank D, Rohr C, Will R, Just S, Grund C, et al. Myozap, a novel intercalated disc protein, activates serum response factor-dependent signaling and is required to maintain cardiac function in vivo. Circ Res. 2010;106:880–90. doi: 10.1161/CIRCRESAHA.109.213256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Christophersen IE, Rienstra M, Roselli C, Yin X, Geelhoed B, Barnard J, et al. Large-scale analyses of common and rare variants identify 12 new loci associated with atrial fibrillation. Nat Genet. 2017;49:946–52. doi: 10.1038/ng.3843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nielsen JB, Fritsche LG, Zhou W, Teslovich TM, Holmen OL, Gustafsson S, et al. Genome-wide study of atrial fibrillation identifies seven risk loci and highlights biological pathways and regulatory elements involved in cardiac development. Am J Hum Genet. 2018;102:103–15. doi: 10.1016/j.ajhg.2017.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Roselli C, Chaffin MD, Weng L-C, Aeschbacher S, Ahlberg G, Albert CM, et al. Multi-ethnic genome-wide association study for atrial fibrillation. Nat Genet. 2018;50:1225–33. doi: 10.1038/s41588-018-0133-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Christophersen IE, Olesen MS, Liang B, Andersen MN, Larsen AP, Nielsen JB, et al. Genetic variation in KCNA5: impact on the atrial-specific potassium current IKur in patients with lone atrial fibrillation. Eur Heart J. 2013;34:1517–25. doi: 10.1093/eurheartj/ehs442. [DOI] [PubMed] [Google Scholar]
- 31.Olson TM, Alekseev AE, Moreau C, Liu XK, Zingman LV, Miki T, et al. KATP channel mutation confers risk for vein of Marshall adrenergic atrial fibrillation. Nat Clin Pract Cardiovasc Med. 2007;110:110–6. doi: 10.1038/ncpcardio0792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Darbar D, Kannankeril PJ, Donahue BS, Kucera G, Stubblefield T, Haines JL, et al. Cardiac sodium channel (SCN5A) variants associated with atrial fibrillation. Circulation. 2008;117:1927–35. doi: 10.1161/CIRCULATIONAHA.107.757955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Olesen MS, Yuan L, Liang B, Holst AG, Nielsen N, Nielsen JB, et al. High prevalence of long QT syndrome-associated SCN5A variants in patients with early-onset lone atrial fibrillation. Circ Cardiovasc Genet. 2012;5:450–9. doi: 10.1161/CIRCGENETICS.111.962597. [DOI] [PubMed] [Google Scholar]
- 34.Watanabe H, Darbar D, Kaiser DW, Jiramongkolchai K, Chopra S, Donahue BS, et al. Mutations in sodium channel β1- and β2-subunits associated with atrial fibrillation. Circ Arrhythm Electrophysiol. 2009;2:268–75. doi: 10.1161/CIRCEP.108.779181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Olesen MS, Jespersen T, Nielsen JB, Liang B, Møller DV, Hedley P, et al. Mutations in sodium channel β-subunit SCN3B are associated with early-onset lone atrial fibrillation. Cardiovasc Res. 2011;89:786–93. doi: 10.1093/cvr/cvq348. [DOI] [PubMed] [Google Scholar]
- 36.Jabbari J, Olesen MS, Yuan L, Nielsen JB, Liang B, Macri V, et al. Common and rare variants in SCN10A modulate the risk of atrial fibrillation. Circ Cardiovasc Genet. 2015;8:64–73. doi: 10.1161/CIRCGENETICS.113.000442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang X, Chen S, Yoo S, Chakrabarti S, Zhang T, Ke T, et al. Mutation in nuclear pore component NUP155 leads to atrial fibrillation and early sudden cardiac death. Cell. 2008;135:1017–27. doi: 10.1016/j.cell.2008.10.022. [DOI] [PubMed] [Google Scholar]
- 38.Oberti C, Wang L, Li L, Dong J, Rao S, Du W, et al. Genome-wide linkage scan identifies a novel genetic locus on chromosome 5p13 for neonatal atrial fibrillation associated with sudden death and variable cardiomyopathy. Circulation. 2004;110:3753–9. doi: 10.1161/01.CIR.0000150333.87176.C7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hodgson-Zingman DM, Karst ML, Zingman LV, Heublein DM, Darbar D, Herron KJ, et al. Atrial natriuretic peptide frameshift mutation in familial atrial fibrillation. N Engl J Med. 2008;359:158–65. doi: 10.1056/NEJMoa0706300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Posch MG, Boldt L-H, Polotzki M, Richter S, Rolf S, Perrot A, et al. Mutations in the cardiac transcription factor GATA4 in patients with lone atrial fibrillation. Eur J Med Genet. 2010;53:201–3. doi: 10.1016/j.ejmg.2010.03.008. [DOI] [PubMed] [Google Scholar]
- 41.Yang Y-Q, Wang X-H, Tan H-W, Jiang W-F, Fang W-Y, Liu X. Prevalence and spectrum of GATA6 mutations associated with familial atrial fibrillation. Int J Cardiol. 2012;155:494–6. doi: 10.1016/j.ijcard.2011.12.091. [DOI] [PubMed] [Google Scholar]
- 42.Müller II, Melville DB, Tanwar V, Rybski WM, Mukherjee A, Shoemaker MB, et al. Functional modeling in zebrafish demonstrates that the atrial-fibrillation-associated gene GREM2 regulates cardiac laterality, cardiomyocyte differentiation and atrial rhythm. Dis Model Mech. 2013;6:332–41. doi: 10.1242/dmm.010488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Thibodeau IL, Xu J, Li Q, Liu G, Lam K, Veinot JP, et al. Paradigm of genetic mosaicism and lone atrial fibrillation: physiological characterization of a connexin 43-deletion mutant identified from atrial tissue. Circulation. 2010;122:236–44. doi: 10.1161/CIRCULATIONAHA.110.961227. [DOI] [PubMed] [Google Scholar]
- 44.Gregers E, Ahlberg G, Christensen T, Jabbari J, Larsen KO, Herfelt CB, et al. Deep sequencing of atrial fibrillation patients with mitral valve regurgitation shows no evidence of mosaicism but reveals novel rare germline variants. Heart Rhythm. 2017;14:1531–8. doi: 10.1016/j.hrthm.2017.05.027. [DOI] [PubMed] [Google Scholar]
- 45.Maron BJ, Maron MS, Semsarian C. Genetics of hypertrophic cardiomyopathy after 20 years: clinical perspectives. J Am Coll Cardiol. 2012;60:705–15. doi: 10.1016/j.jacc.2012.02.068. [DOI] [PubMed] [Google Scholar]
- 46.Peng W, Li M, Li H, Tang K, Zhuang J, Zhang J, et al. Dysfunction of myosin light-chain 4 (MYL4) leads to heritable atrial cardiomyopathy with electrical, contractile, and structural components: evidence from genetically-engineered rats. J Am Heart Assoc. 2017;6. 10.1161/JAHA.117.007030. [DOI] [PMC free article] [PubMed]
- 47.Ahlberg G, Refsgaard L, Lundegaard PR, Andreasen L, Ranthe MF, Linscheid N, et al. Rare truncating variants in the sarcomeric protein titin associate with familial and early-onset atrial fibrillation. Nat Commun. 2018;9. 10.1038/s41467-018-06618-y. [DOI] [PMC free article] [PubMed]
- 48.Hedley PL, Jørgensen P, Schlamowitz S, Wangari R, Moolman-Smook J, Brink PA, et al. The genetic basis of long QT and short QT syndromes: a mutation update. Hum Mutat. 2009;30:1486–511. doi: 10.1002/humu.21106. [DOI] [PubMed] [Google Scholar]
- 49.Johnson JN, Tester DJ, Perry J, Salisbury BA, Reed CR, Ackerman MJ. Prevalence of early-onset atrial fibrillation in congenital long QT syndrome. Heart Rhythm. 2008;5:704–9. doi: 10.1016/j.hrthm.2008.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nielsen JB, Graff C, Pietersen A, Lind B, Struijk JJ, Olesen MS, et al. J-shaped association between QTc interval duration and the risk of atrial fibrillation: results from the Copenhagen ECG study. J Am Coll Cardiol. 2013;61:2557–64. doi: 10.1016/j.jacc.2013.03.032. [DOI] [PubMed] [Google Scholar]
- 51.Nielsen MW, Holst AG, Olesen S-P, Olesen MS. The genetic component of Brugada syndrome. Front Physiol. 2013;4:179. doi: 10.3389/fphys.2013.00179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bjornsson T, Thorolfsdottir RB, Sveinbjornsson G, Sulem P, Norddahl GL, Helgadottir A, et al. A rare missense mutation in MYH6 associates with non-syndromic coarctation of the aorta. Eur Heart J. 2018;39:3243–9. doi: 10.1093/eurheartj/ehy142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.England J, Loughna S. Heavy and light roles: myosin in the morphogenesis of the heart. Cell Mol Life Sci. 2013;70:1221–39. doi: 10.1007/s00018-012-1131-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Herron TJ, Korte FS, McDonald KS. Loaded shortening and power output in cardiac myocytes are dependent on myosin heavy chain isoform expression. Am J Physiol Heart Circ Physiol. 2001;281:H1217–22. doi: 10.1152/ajpheart.2001.281.3.H1217. [DOI] [PubMed] [Google Scholar]
- 55.Miyata S, Minobe W, Bristow MR, Leinwand LA. Myosin heavy chain isoform expression in the failing and nonfailing human heart. Circ Res. 2000;86:386–90. doi: 10.1161/01.RES.86.4.386. [DOI] [PubMed] [Google Scholar]
- 56.Cañón S, Caballero R, Herraiz-Martínez A, Pérez-Hernández M, López B, Atienza F, et al. miR-208b upregulation interferes with calcium handling in HL-1 atrial myocytes: Implications in human chronic atrial fibrillation. J Mol Cell Cardiol. 2016;99:162–73. doi: 10.1016/j.yjmcc.2016.08.012. [DOI] [PubMed] [Google Scholar]
- 57.Thorolfsdottir RB, Sveinbjornsson G, Sulem P, Helgadottir A, Gretarsdottir S, Benonisdottir S, et al. A Missense Variant in PLEC Increases Risk of Atrial Fibrillation. J Am Coll Cardiol. 2017;70:2157–68. doi: 10.1016/j.jacc.2017.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Holaska JM. Emerin and the nuclear lamina in muscle and cardiac disease. Circ Res. 2008;103:16–23. doi: 10.1161/CIRCRESAHA.108.172197. [DOI] [PubMed] [Google Scholar]
- 59.Beckmann BM, Holinski-Feder E, Walter MC, Haserück N, Reithmann C, Hinterseer M, et al. Laminopathy presenting as familial atrial fibrillation. Int J Cardiol. 2010;145:394–6. doi: 10.1016/j.ijcard.2010.04.024. [DOI] [PubMed] [Google Scholar]
- 60.Bundgaard H, Jøns C, Lodder EM, Izarzugaza JMG, Herrera JAR, Pehrson S, et al. A novel familial cardiac arrhythmia syndrome with widespread ST-segment depression. N Engl J Med. 2018;379:1780–1. doi: 10.1056/NEJMc1807668. [DOI] [PubMed] [Google Scholar]
- 61.Salem J-E, Shoemaker MB, Bastarache L, Shaffer CM, Glazer AM, Kroncke B, et al. Association of thyroid function genetic predictors with atrial fibrillation: a phenome-wide association study and inverse-variance weighted average meta-analysis. JAMA Cardiol. 2019. 10.1001/jamacardio.2018.4615. [DOI] [PMC free article] [PubMed]
- 62.Ellervik C, Roselli C, Christophersen IE, Alonso A, Pietzner M, Sitlani CM, et al. Assessment of the relationship between genetic determinants of thyroid function and atrial fibrillation: a mendelian randomization study. JAMA Cardiol. 2019;4:144–52. doi: 10.1001/jamacardio.2018.4635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hadji-Turdeghal K, Andreasen L, Hagen CM, Ahlberg G, Ghouse J, Bækvad-Hansen M, et al. Genome-wide association study identifies locus at chromosome 2q32.1 associated with syncope and collapse. Cardiovasc Res. 2019. 10.1093/cvr/cvz106. [DOI] [PMC free article] [PubMed]
- 64.Lee S-P, Ashley EA, Homburger J, Caleshu C, Green EM, Jacoby D, et al. Incident atrial fibrillation is associated with MYH7 sarcomeric gene variation in hypertrophic cardiomyopathy. Circ Heart Fail. 2018;11:e005191. doi: 10.1161/CIRCHEARTFAILURE.118.005191. [DOI] [PubMed] [Google Scholar]
- 65.Rampazzo A, Calore M, van Hengel J, van Roy F. Intercalated discs and arrhythmogenic cardiomyopathy. Circ Cardiovasc Genet. 2014;7:930–40. doi: 10.1161/CIRCGENETICS.114.000645. [DOI] [PubMed] [Google Scholar]
- 66.Vad OB, Paludan-Müller C, Ahlberg G, Kalstø SM, Ghouse J, Andreasen L, et al. Loss-of-function variants in cytoskeletal genes are associated with early-onset atrial fibrillation. J Clin Med. 2020;9. 10.3390/jcm9020372. [DOI] [PMC free article] [PubMed]
- 67.Nattel S. Close connections between contraction and rhythm: a new genetic cause of atrial fibrillation/cardiomyopathy and what it can teach us. Eur Heart J. 2017;38:35–7. doi: 10.1093/eurheartj/ehw457. [DOI] [PubMed] [Google Scholar]
- 68.Orr N, Arnaout R, Gula LJ, Spears DA, Leong-Sit P, Li Q, et al. A mutation in the atrial-specific myosin light chain gene (MYL4) causes familial atrial fibrillation. Nat Commun. 2016;7:11303. doi: 10.1038/ncomms11303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tayal U, Newsome S, Buchan R, Whiffin N, Walsh R, Barton PJ, et al. Truncating variants in titin independently predict early arrhythmias in patients with dilated cardiomyopathy. J Am Coll Cardiol. 2017;69:2466–8. doi: 10.1016/j.jacc.2017.03.530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Choi SH, Weng L-C, Roselli C, Lin H, Haggerty CM, Shoemaker MB, et al. Association between titin loss-of-function variants and early-onset atrial fibrillation. JAMA. 2018;320:2354–64. doi: 10.1001/jama.2018.18179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Allessie M, Ausma J, Schotten U. Electrical, contractile and structural remodeling during atrial fibrillation. Cardiovasc Res. 2002;54:230–46. doi: 10.1016/S0008-6363(02)00258-4. [DOI] [PubMed] [Google Scholar]
- 72.Veenhuyzen GD, Simpson CS, Abdollah H. Atrial fibrillation. CMAJ Can Med Assoc J. 2004;171:755–60. doi: 10.1503/cmaj.1031364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Cochet H, Dubois R, Yamashita S, Jefairi NA, Berte B, Sellal J-M, et al. Relationship between fibrosis detected on late gadolinium-enhanced cardiac magnetic resonance and re-entrant activity assessed with electrocardiographic imaging in human persistent atrial fibrillation. JACC Clin Electrophysiol. 2018;4:17–29. doi: 10.1016/j.jacep.2017.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mahnkopf C, Badger TJ, Burgon NS, Daccarett M, Haslam TS, Badger CT, et al. Evaluation of the left atrial substrate in patients with lone atrial fibrillation using delayed-enhanced MRI: implications for disease progression and response to catheter ablation. Heart Rhythm. 2010;7:1475–81. doi: 10.1016/j.hrthm.2010.06.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kumagai K, Nakashima H, Urata H, Gondo N, Arakawa K, Saku K. Effects of angiotensin II type 1 receptor antagonist on electrical and structural remodeling in atrial fibrillation. J Am Coll Cardiol. 2003;41:2197–204. doi: 10.1016/S0735-1097(03)00464-9. [DOI] [PubMed] [Google Scholar]
- 76.Boldt A, Scholl A, Garbade J, Resetar ME, Mohr FW, Gummert JF, et al. ACE-inhibitor treatment attenuates atrial structural remodeling in patients with lone chronic atrial fibrillation. Basic Res Cardiol. 2006;101:261–7. doi: 10.1007/s00395-005-0571-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.