

Abstract
Chemotherapy-resistant breast cancer displays aggressive clinical behavior, is poorly differentiated and is associated with the occurrence of epithelial-mesenchymal transition and the presence of cancer stem cells. The anthelmintic drug niclosamide has been shown to have numerous clinical applications in the treatment of malignant tumors, in addition to its traditional use in tapeworm disease. Our previous study demonstrated that niclosamide had an antiproliferative effect and could inhibit the stem-like phenotype of the breast cancer cells, suggesting that it might have the potential to be used in the treatment of triple-negative breast cancer. However, the specific function and underlying mechanism of action of niclosamide in chemoresistant human epidermal growth factor receptor 2 (HER2)-positive breast cancer remain unknown. The present study aimed to determine whether niclosamide can inhibit cell proliferation, invasion and epithelial-to-mesenchymal transition, as well as the stem-like phenotype in cisplatin-resistant HER2-positive breast cancer. Alamar Blue and Annexin V/7-AAD staining, mammosphere formation and Transwell assays were performed to assess the viability, apoptosis, stem-like phenotype and invasion ability of breast cancer cell lines, respectively. Signaling molecule expression was detected via western blotting and a xenograft model was used to verify the inhibitory effect of niclosamide in vivo. The results from the present study demonstrated that niclosamide inhibited the resistance of HER2-positive breast cancer to cisplatin both in vitro and in vivo. Furthermore, niclosamide combined with cisplatin could inhibit breast cancer cell invasion, epithelial-mesenchymal transition and cell stemness. The inhibitory effect of niclosamide was mediated by apoptosis induction and Bcl-2 downregulation. Taken together, the results of the present study suggested that niclosamide combined with cisplatin may be considered as a novel treatment for chemoresistant HER2-positive breast cancer.
Keywords: niclosamide, human epidermal growth factor receptor 2-positive breast cancer, apoptosis, epithelial-mesenchymal transition, stem-like phenotype
Introduction
Breast cancer is one of the most common female malignancies. Although the prognosis of breast cancer has improved with early detection and advances in systemic treatment, 20–30% of patients still experience distant metastasis. Patients with advanced disease have a median survival of only ~2 years (1). As one of the most aggressive breast malignancies, human epidermal growth factor receptor 2 (HER2)-positive breast cancer exhibits frequent recurrence and metastasis during or after treatment (2). In advanced HER2-positive breast cancer, chemotherapy combined with trastuzumab is currently widely used in practice and is recognized as a valid approach for improving patient survival (1). Although chemotherapy is effective in most patients, some patients relapse and develop resistance (3). Cancer stem cells (CSCs) have numerous abilities, including a self-renewal ability, an invasive ability and resistance ability to numerous antitumor agents, including chemotherapy and targeted therapy, all of which are thought to contribute to recurrence or metastasis and to overall aggressiveness of the disease (4).
Chemotherapy can inhibit tumor growth and also leads to hypoxia, which might induce an increase in the proportion of CSCs. CSCs can survive by increasing the expression of hypoxia-inducible factors (5). Previous studies have demonstrated that following treatment of breast cancer cells with chemotherapy, the surviving cells exhibit enrichment in CSCs (6,7). Li et al (8) reported that CD44+/CD24−/low breast cancer cells could separate from primary breast cancer tissues after chemotherapy. In addition, the proportion of stem cell subsets after chemotherapy increased compared with that before chemotherapy, and the formation of mammospheres was enhanced. Previous studies in breast cancer mouse models also suggested that combining chemotherapy with drugs targeting CSCs or metastatic progression could improve survival (7,9).
Niclosamide is an anthelmintic agent that has been approved by the Food and Drug Administration (FDA) and used in humans for nearly 50 years (10). Chen et al (11) identified niclosamide (trade name, Niclocide) as a potentially effective antitumor drug that could alter the hepatocellular carcinoma gene expression pattern. The underlying mechanism of niclosamide has not been well characterized, although it has been reported that it could inhibit mitochondrial oxidative phosphorylation and stimulate ATPase activity (12). Our previous studies revealed that niclosamide exerts anticancer effects in triple-negative and HER2-positive breast cancers by reversing epithelial-mesenchymal transition (EMT) and inhibiting stem cell phenotype (13,14). Furthermore, we previously demonstrated that niclosamide monotherapy alone or combined with cisplatin can significantly inhibit the Akt, ERK and Src signaling pathways (13,14). Thus, the inhibitory effect of niclosamide on tumor cells is well-documented, although the mechanism has not yet been clarified.
The present study aimed to determine the inhibitory effects of niclosamide on the growth, invasion and EMT of chemoresistant HER2-positive breast cancer cells. The molecular mechanisms of niclosamide-mediated inhibition of Bcl-2 and signal transducer and activator of transcription 3 (STAT3) were also investigated.
Materials and methods
Cell culture and cisplatin treatment
The BT474 cell line, estrogen receptor (ER) and HER2-positive breast cancer cell line, was purchased from the American Type Culture Collection. Cells were cultured in RPMI-1640 medium (Gibco; Thermo Fisher Scientific, Inc.) supplemented with 10% FBS (Gibco; Thermo Fisher Scientific, Inc.) and 1% penicillin/streptomycin in at 37°C in a humidified incubator containing 5% CO2. Cisplatin and niclosamide were purchased from Sigma-Aldrich (Merck KGaA). Cisplatin was prepared in DMSO to obtain a stock solution of 200 mM at −20°C. Niclosamide was prepared in DMSO to obtain a stock solution of 10 mM at −20°C. BT474 cells were continuously treated with increasing concentrations of cisplatin (5-20 µmol/l) for >6 months to acquire a stable cisplatin-resistant cell line (13). Before each experiment, cisplatin was removed from the media for at least 72 h.
Cell viability and combination index (CI) analysis
Alamar Blue (resazurin solution), purchased from Sigma-Aldrich; Merck KGaA, was used to assess cell viability as previously described by Liu et al (13). The results are displayed as the ratio of viable treated cells to viable 1% DMSO control treated cells. CompuSyn software (version 1.0; ComboSyn) was utilized to calculate the CI value for niclosamide and cisplatin. The fraction affected value was calculated as the portion of cells inhibited after drug exposure.
Apoptosis analysis
Cells were exposed to DMSO (1%) control, cisplatin (20 µM) or niclosamide (1 µM) for 48 h. Cells in suspension (100 µl) were stained with 100 µl of Muse Annexin V & Dead Cell reagent (Merck KGaA) according to the manufacturer's instructions and incubated in the dark for 20 min at room temperature. The Muse Cell Analyzer (Merck KGaA) was used to analyze fluorescence.
Western blotting
After cisplatin resistant BT474 cells were treated with 1% DMSO (control), 1 µM niclosamide or 1 µM niclosamide combined with 20 µM cisplatin for 48 h, proteins were extracted using RIPA buffer (cat. no. 89900, Thermo Fisher Scientific, Inc.). The bicinchoninic acid assay was used to measure the protein concentration. Total proteins (20 µg/lane) were separated by SDS-PAGE on a 10% gel. The separated proteins were transferred onto PVDF membranes and blocked with 5% fat-free milk in TBST buffer (0.1% Tween-20) for 2 h at room temperature. The membranes were incubated with primary antibodies (diluted at 1:1,000, except for GAPDH, which was diluted at 1:10,000) overnight at 4°C. Antibodies against E-cadherin (cat. no. 3195), N-cadherin (cat. no. 13116), vimentin (cat. no. 3390), Bcl-2 (cat. no. 4223), phosphorylated (p)-STAT3 (Tyr705) (cat. no. 9145), STAT3 (cat. no. 30835) and GAPDH (cat. no. 8884) were purchased from Cell Signaling Technology, Inc. The membranes were washed three times with TBST (TBS with 0.05% Tween-20), and subsequently incubated with anti-rabbit HRP-conjugated IgG (cat. no. 7074) or anti-mouse HRP-conjugated IgG (cat. no. 7076) secondary antibodies (both diluted at 1:5,000; Cell Signaling Technology, Inc.) at room temperature for 1 h and re-washed. The immunoreactive signals were visualized using a peroxide solution with an electrochemiluminescent HRP substrate and luminol reagent (Merck Millipore; cat. no. WBKLS0500). Data were analyzed using ImageJ software (version 1.52v, National Institutes of Health) and normalized to GAPDH.
Mammosphere formation assay
A total of 3,000 cells/ml were plated in ultralow-attachment plates (Corning). The medium contained serum-free DMEM/F12 supplemented with 20 ng/ml human recombinant epidermal growth factor (Invitrogen; Thermo Fisher Scientific, Inc.), 10 ng/ml basic fibroblast growth factor (Invitrogen; Thermo Fisher Scientific, Inc.) and 1X B27 supplement (Gibco; Thermo Fisher Scientific, Inc.). The mammosphere formation assay procedure was performed as previously described (14). Mammosphere-forming efficiency (MFE) was calculated as the mammosphere number (diameter ≥50 µm) divided by the number of seeded cells.
Cell invasion assay
The cell invasion assay was conducted using Matrigel®-coated invasion chambers (BD Biosciences). A total of 5×104 cells in 0.5 ml serum-free medium were seeded into the top chambers of the transwell. Medium containing 10% FBS medium was then added to the bottom chamber. After 12 h incubation, cells in the top chambers were wiped whereas cells on the bottom of the chamber were fixed with 10% neutral formalin at room temperature for 30 min and stained with 1% crystal violet at room temperature for 30 min. The stained cells were observed under a microscope (Zeiss GmbH) with a 10X objective lens in five random regions per sample. Results were presented as the average cell number across fields of view.
Tumor xenograft model
Xenograft experiments were approved by the Tongji University School of Medicine Committee for the Use and Care of Animals and performed in strict accordance with institutional requirements. A total of 5×106 BT474-resistant cells in 100 µl Matrigel (BD Biosciences) mixed with 100 µl RPMI-1640 medium were subcutaneously injected into 12 four-six-week-old female BALB/c nude mice (Slaccas Laboratory Animal; Shanghai, China). Nude mice were housed in a temperature-controlled (24-25°C and 50% humidity) pathogen-free environment with a 12:12 h light:dark cycle. After 14 days, tumor-bearing mice with tumor volumes between 50 and 100 mm3 were evenly divided into four groups according to tumor size. Each group contained 3 mice, which were injected intraperitoneally from day 14 to day 56 with one of the following treatments: i) Vehicle; ii) cisplatin; iii) niclosamide; or iv) niclosamide with cisplatin. The dosage of niclosamide was 20 mg/kg/day five days a week, and the dosage of cisplatin was 2 mg/kg/week. The mice were sacrificed on day 56 by cervical dislocation. Tumor size was measured every seven days and tumor volume was calculated after collection using the following formula: Volume = length × width2 / 2.
Immunohistochemistry
The immunohistochemistry procedure has been described by Liu et al (13). Antibodies against Ki67 (1:100; cat. no. ab15580), HER2 (1:2,000; cat. no. ab237715) and Bcl-2 (1:250; cat. no. ab32124) were purchased from Abcam. The primary antibody incubation step was skipped in one group as a negative control. Images were acquired using a microscope (magnification, ×40; Zeiss GmbH).
Statistical analysis
Statistical analysis was performed using SPSS software (version 19; IBM Corp.) or GraphPad Prism (version 5; GraphPad Software Inc.). Cell viability, apoptosis, western blotting, mammosphere formation and cell invasion assays were performed in triplicate. Data are presented as the mean ± standard deviation (SD). Statistical significance was analyzed by two-way ANOVA followed by Tukey's post hoc test (Fig. 1) or one-way ANOVA followed by Tukey's post hoc test (Figs. 2–5). Data from the in vivo study were analyzed using mixed two-way ANOVA method regarding time as a repeated measure and Tukey's post hoc test. P<0.05 was considered to indicate a statistically significant difference.
Figure 1.

Niclosamide combined with cisplatin reverses cisplatin resistance. (A) Typical images of BT-CS and BT-CR cells. Images were obtained using a microscope (Zeiss) with a 40X objective lens. (B) Alamar Blue assay was used to assess the cell viability. BT-CS and BT-CR cells were treated with the indicated concentrations of cisplatin for 48 h. (C) BT-CR cells were treated with the indicated concentrations of cisplatin and niclosamide or the combination of these two drugs for 48 h. (D) CI plot obtained from the CompuSyn report for BT-CR cells treated with cisplatin and niclosamide combinations. ***P<0.001. BT-CS, cisplatin sensitive BT474 cells; BT-CR, cisplatin resistant BT474 cells; Ctrl, control; CI, combination index.
Figure 2.

Niclosamide and cisplatin induce apoptosis in resistant cells. (A) Cisplatin resistant BT474 cells were cultured with DMSO (control), cisplatin (20 µM), niclosamide (1 µM) or niclosamide (1 µM) combined with cisplatin (20 µM) for 48 h and then subjected to apoptosis analysis. Lower and upper right quadrants of the scatterplots show the percentages of early and late apoptotic cells, respectively. (B) Percentage of early and late apoptotic cells detected by flow cytometry. n=3. ***P<0.001. Ctrl, control; Cis, cisplatin; Ncm, niclosamide.
Figure 5.

Niclosamide and cisplatin suppress EMT and inhibit the Stat3 signaling pathway. (A) Expression of E-cadherin, N-cadherin, vimentin and Bcl-2 evaluated by western blotting in lysates of BT-CR cells treated with DMSO (control), 1 µM niclosamide or 1 µM niclosamide combined with 20 µM cisplatin for 48 h. Histogram represents the density values of each protein. (B) Expression of p-STAT3 (Tyr705) and STAT 3 evaluated by western blotting in lysates of BT-CR cells treated with DMSO (control), 1 µM niclosamide or the combination of 1 µM niclosamide and 20 µM cisplatin for 48 h. Histogram represents the integrated density values of p-STAT3/STAT3 ratio. *P<0.05; **P<0.01; ***P<0.001. BT-CS, cisplatin sensitive BT474 cells; BT-CR, cisplatin resistant BT474 cells; Ctrl, control; Cis, cisplatin; Ncm, niclosamide; EMT, epithelial-mesenchymal transition; p, phosphorylated; STAT3, signal transducer and activator of transcription 3.
Results
Niclosamide combined with cisplatin reverses cisplatin resistance
BT474 cells, which were originally isolated from a case of HER2-positive breast cancer, were cultured with increasing concentrations of cisplatin (5-20 µM) for >6 months. As presented in Fig. 1A, cisplatin resistant BT474 cells (BT-CR) had a morphology similar to that of cisplatin sensitive BT474 cells (BT-CS) but had more membrane protrusions than BT-CS cells. To verify the difference in cisplatin sensitivity between BT-CS and BT-CR cells, Alamar Blue assay was performed on cells following treatment with different cisplatin concentrations (0, 1.25, 2.5, 5, 10 and 20 µM) for 48 h. The results demonstrated that cisplatin cytotoxicity was significantly inhibited in BT-CR cells compared with the sensitive parental cell line BT-CS (Fig. 1B; P<0.001).
BT-CR cells were treated with the indicated concentrations of cisplatin and niclosamide or a combination of these two drugs (combination group) for 48 h (Fig. 1C). The results demonstrated that both niclosamide and the combination treatment decreased the viability of BT-CR cells (P<0.001). To determine whether the antitumor effect of niclosamide combined with cisplatin was synergistic, additive or antagonistic, the CI was calculated using CompuSyn software. The median inhibitory concentration of cisplatin in BT-CR cells was 169.5 µM, whereas the median inhibitory concentration of niclosamide was 2.4 µM. The CI value of combination treatment with niclosamide and cisplatin was <1 µM, indicating that niclosamide and cisplatin exerted a synergistic effect on the resistant cells BT-CR (Fig. 1D).
Niclosamide and cisplatin induce resistant cell apoptosis
The apoptosis of BT-CR cells treated with cisplatin (20 µM) and niclosamide (1 µM) was evaluated. The niclosamide concentration was selected according to previous research (11). Since the serum concentration of niclosamide is 0.25–6.0 µg/ml (corresponding to 0.76–18.35 µM), the concentration of 1 µM chosen was within the serum concentration range (7). Annexin V/7-AAD staining was detected by flow cytometry. Niclosamide (1 µM) treatment for 48 h increased the apoptosis rate compared with that of 20 µM cisplatin treatment in resistant BT474 cells (Fig. 2A). Furthermore, niclosamide combined with cisplatin significantly increased the apoptosis rate, suggesting that niclosamide alone or combined with cisplatin increased cytotoxicity by inducing apoptosis. The histogram represents the percentage of early and late apoptotic cells detected by flow cytometry (Fig. 2B).
Niclosamide and cisplatin inhibit the stem-like phenotype in resistant cells
The mammosphere assay was used to assess the cell stem-like phenotype of BT-CS and BT-CR cells. As presented in Fig. 3, a prominent decrease in the number and diameter of spheres treated with niclosamide was noticed, either alone or combined with cisplatin, in the BT-CS and BT-CR cell lines. Exposure to niclosamide (P<0.01) or combined with cisplatin (P<0.001) significantly decreased the MFE of both BT-CS and BT-CR cells. This result indicated that niclosamide alone or combined with cisplatin could effectively decrease the cell ability to form mammospheres.
Figure 3.

Niclosamide and cisplatin inhibit the stem-like phenotype. (A) Representative images of mammospheres formed by BT-CS and BT-CR cells treated with DMSO (control) 0.1 µM niclosamide alone or combined with 20 µM cisplatin. Images were captured using the microscope (Zeiss) with a 10X objective lens. (B) Mammosphere-forming efficiency levels of BT-CS and BT-CR cell lines treated by indicated drugs (n=3). **P<0.01; ***P<0.001. BT-CS, cisplatin sensitive BT474 cells; BT-CR, cisplatin resistant BT474 cells; Ctrl, control; Cis, cisplatin; Ncm, niclosamide.
Niclosamide and cisplatin inhibit cell invasion ability
Because niclosamide combined with cisplatin was found to induce apoptosis and significantly decrease the MFE, subsequent experiments were carried out to investigate whether cell invasion was also inhibited by niclosamide or combination treatment. Transwell invasion assays were performed to determine the invasive ability of BT-CS and BT-CR cells. The results demonstrated that BT-CR cells had a higher invasive ability compared with BT-CS cells. Furthermore, niclosamide alone and combined with cisplatin significantly attenuated the invasive ability of BT-CS and BT-CR cells (Fig. 4).
Figure 4.

Niclosamide and cisplatin inhibit cell invasion. (A) Representative images of cells on Transwell filters. The stained cells were captured through the microscope (Zeiss) under a 10X objective lens. (B) Results were quantified by calculating the average number across five random fields of view under a 10X objective lens. Data are presented as the mean ± SD (n=30. **P<0.01; ***P<0.001. BT-CS, cisplatin sensitive BT474 cells; BT-CR, cisplatin resistant BT474 cells; Ctrl, control; Cis, cisplatin; Ncm, niclosamide.
Niclosamide and cisplatin suppress EMT and inhibit the STAT3 signaling pathway
Previous studies demonstrated that EMT is a possible mechanism of cell metastasis (15,16). Niclosamide treatment may also suppress EMT in resistant HER2-positive breast cancer cells. To test this hypothesis, epithelial (E-cadherin) and mesenchymal (N-cadherin and vimentin) markers were detected by western blotting. As presented in Fig. 5A, E-cadherin expression significantly increased whereas N-cadherin and vimentin expression significantly decreased in cells treated with niclosamide alone or combined with cisplatin compared with the control group. Moreover, treatment with niclosamide alone or combined with cisplatin decreased the expression level of the antiapoptotic protein Bcl-2 (Fig. 5A). The phosphorylation level (Tyr705) of STAT3 was analyzed by western blotting and the results demonstrated that p-STAT3 levels significantly decreased following niclosamide treatment (Fig. 5B). These findings suggested that niclosamide may inactivate STAT3 to downregulate the expression of Bcl-2 and increase the cisplatin sensitivity of HER2-positive cells.
Antitumor efficacy of niclosamide in vivo
To verify the inhibitory effect of niclosamide in vivo, xenografts generated from BT474-resistant cells were treated as described in the Materials and methods. As presented in Fig. 6A, niclosamide alone and combined with cisplatin significantly inhibited the growth of xenograft tumors derived from resistant cells compared with cisplatin alone (P<0.05). There was no significant difference between the niclosamide group and the combination treatment group. Tumor tissues were stained for Ki67, HER2 and Bcl-2 by immunohistochemistry. As shown in Fig. 6B, both niclosamide and combination therapy inhibited Ki67 and Bcl-2 expression. Taken together, these findings demonstrated that niclosamide may inhibit cisplatin-resistant BT474 tumor growth in vivo.
Figure 6.
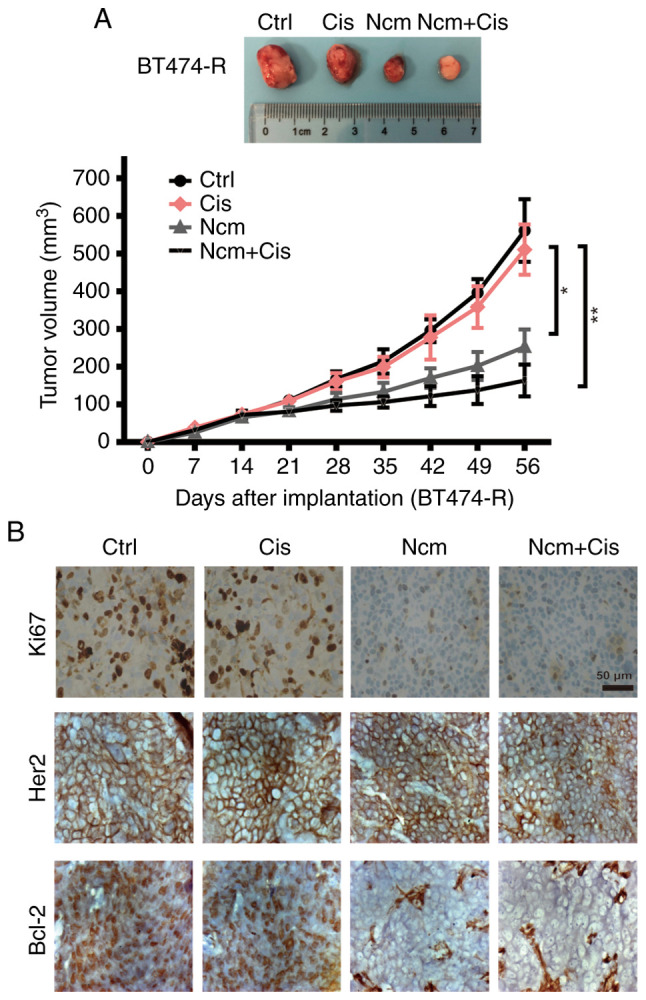
Antitumor efficacy of niclosamide in vivo. (A) Nude mice bearing resistant BT474 (BT474-R) cell-derived xenografts were treated with vehicle control, cisplatin (2 mg/kg/week), niclosamide (20 mg/kg/day) or niclosamide (20 mg/kg/day) combined with cisplatin (2 mg/kg/week) from day 14 to day 56. Tumor volume was calculated as length × width2 / 2. (B) Images of Ki67, HER2 and Bcl-2 immunohistochemistry staining of the xenograft tissues. Scale bar, 50 µm. *P<0.05; **P<0.01. Ctrl, control; Cis, cisplatin; Ncm, niclosamide; HER2, human epidermal growth factor receptor 2.
Discussion
The present study demonstrated that niclosamide, an FDA-approved anthelmintic agent, might also be considered as a potential chemotherapeutic agent for HER2-positive chemoresistant breast cancer. The results demonstrated that niclosamide induced apoptosis and was more effective when combined with cisplatin. Furthermore, niclosamide overcame chemotherapy resistance in HER2-positive cells via Bcl-2 inhibition and STAT3 activation. These results suggested that niclosamide might be considered as a potential antitumor drug and used in combination with chemotherapy in the treatment of HER2-positive breast cancer. A recent study reported that niclosamide, a potent radiosensitizer, acts by inhibiting STAT3 and Bcl-2 and by increasing the production of reactive oxygen species (ROS) in triple-negative breast cancer (17). Another study demonstrated that niclosamide inhibits the growth of melanoma cell lines (A375 and B16-F10) and induces mitochondrial apoptosis, which impairs cell migration and invasion, reduces expression of phosphorylated STAT3 at Tyr705 and inhibits matrix metalloproteinase-2 and −9 expression (18). Conversely, the present study demonstrated that the anthelmintic agent niclosamide may be repurposed to treat chemoresistant HER2-positive breast cancer. To the best our knowledge, the present study was the first to suggest that niclosamide could be used for the treatment of chemoresistant HER2-positive breast cancer.
Niclosamide is administered orally in parasitic patients but is only partially absorbed through the gastrointestinal tract. In anthelmintic therapy, niclosamide is administered orally in adults at a single dose of 2 g. Due to differences in individual absorption rates, the maximum serum concentration of niclosamide in humans is 0.25–6.0 µg/ml (corresponding to 0.76–18.35 µM), which is well within the concentration range of its anticancer activity and within the concentration range we previously observed for the treatment of chemoresistance in HER2-positive breast cancer (10). In addition, in rodent xenograft models, oral administration of niclosamide has been shown to lead to an accumulation of the drug in tumor tissue at concentrations even higher than those in plasma (19). However, additional stage I–III randomized clinical trials are needed to demonstrate the antitumor effect of niclosamide before it could be approved for any cancer treatment.
Niclosamide is increasingly studied in cancer research and has been demonstrated to effectively inhibit a variety of cancer-related signaling pathways. A previous study suggested that niclosamide could inhibit PI3K/Akt, Wnt/β-catenin signaling and β-catenin/T-cell factor complex formation, and promote degradation of the Wnt co-receptor low-density lipoprotein receptor-related protein 6 (20). Jak-STAT and NF-κB signaling have also been identified as potential targets of niclosamide in lung cancer and multiple myeloma (21). Furthermore, niclosamide has been shown to enhance the efficacy of programmed cell death 1/programmed cell death ligand 1 immune checkpoint blockade and cisplatin cytotoxicity in non-small cell lung cancer models (22). Furthermore, Snail family transcriptional repressor 1 is known as a transcriptional repressor of cadherin-1, which encodes E-cadherin. Downregulation of E-cadherin is vital to the migration and invasion of cancer cells. This process might facilitate breast cancer metastasis (23,24). Previous studies have demonstrated that CSCs forming tumor spheres in vitro are more likely to develop tumors in vivo and to become resistant to standard radiation or chemotherapy than differentiated cells (25,26). This, along with our previous studies (13,14), indicates that niclosamide can inhibit EMT in both lapatinib-resistant and chemoresistant breast cancer. In addition, niclosamide alone or in combination with cisplatin successfully decreased MFE in the present study, indicating that niclosamide may be used as an effective anticancer agent by inhibiting EMT and the breast cancer stem-like phenotype.
Cellular metabolic reprogramming is one of the hallmarks of tumor cells, and Dr Otto Warburg reported that tumor cells convert glucose to lactate to support cell growth, even in a normoxic state. This aerobic glycolysis is known as the ‘Warburg effect’ (27). In normal cells, almost all pyruvate produced by glycolysis enters the mitochondria for oxidative phosphorylation, maximizing the energy extraction from glucose, whereas in cancer cells, only ~5% of pyruvate enters the mitochondria (28). Aerobic glycolysis in cancer cells is accompanied by elevated glucose uptake, and only a small fraction of glucose is used for oxidative phosphorylation (27). This explains the energy deficit of cancer cells. Niclosamide is an anthelmintic drug with mitochondrial uncoupling function (29). Previous studies have demonstrated that mitochondrial uncoupling increases the pyruvate influx into mitochondria, upregulates mitochondrial oxidation, reduces lactate production and decreases the biosynthetic pentose phosphate pathway, implying that mitochondrial uncoupling may be considered as an effective way to antagonize aerobic glycolysis and that mitochondrial uncoupling agents could potentially be developed for the treatment of malignancies (29,30). Niclosamide has been shown to be associated with glycolysis and glucose uptake regulation, setting tumor cells at rest. Park et al (31) and Khanim et al (32) confirmed that niclosamide could induce mitochondrial disintegration and induce the production of mitochondrial superoxide, leading to mitochondrial damage. In the present study, niclosamide induced apoptosis in chemoresistant cells by downregulating Bcl-2, which was consistent with previous studies (33,34).
Numerous malignant tumors express activated STAT3 and are resistant to apoptosis induction and chemotherapy (35). A previous study reported that STAT3 may be a key downstream mediator of HER2 signaling (36). Duru et al (37) showed that HER2-STAT3 crosstalk increases aggressiveness and radioresistance in breast CSCs. HER2 also promotes radioresistance in HER2-positive breast cancer through STAT3-survivin regulation (38). Chung et al (39) reported that in HER2-overexpressing breast cancer, STAT3 activation promotes CSC characteristics of which phenotype is associated with tumor drug resistance. In addition, Li et al (40) reported that trastuzumab resistance is regulated by STAT3-dependent feedback activation in HER2-positive breast cancer and gastric cancer. STAT3 activation is therefore a critical pathway for the survival of drug-resistant tumors in HER2-positive breast cancer. A proportion of STAT3 protein is localized in the mitochondria. STAT3 functions as a promoter of the mitochondrial electron transport chain in the mitochondria (41), decreases mitochondrial ROS production and inhibits apoptosis (42). Conversely, a lack of STAT3 decreases ATP levels and increases ROS production in tumor cells (43). Niclosamide has been reported to be a potent STAT3 suppressor that inhibits STAT3 phosphorylation at Tyr705 (44). The inhibitory effect of niclosamide on STAT3 phosphorylation also influences other pathways, including the Wnt/β-catenin pathway, which is relevant to cancer initiation and progression (45). Previously, we reported that niclosamide induces growth inhibition and apoptosis in MDA-MB-231 cisplatin-resistant cells (ER-negative) and suppresses tumor invasion and stem-like phenotype, suggesting that niclosamide might function through the Akt, ERK and Src pathways (13). In the present study, niclosamide induced growth inhibition and apoptosis in ER and HER2-positive cells.
Cisplatin is a chemotherapeutic agent that has not been routinely used in breast cancer; however, an increasing number of studies have reported favorable responses in subgroups of breast cancer (46–48). The National Comprehensive Cancer Network have listed platinum-based agents, such as cisplatin and carboplatin, as the preferred treatment for patients with triple-negative recurrent/stage IV breast cancer who carry the germline BRCA1/2 mutation (49). Cisplatin is a DNA-damaging drug. Based on clinical trials, cisplatin has become the backbone drug for the treatment of primary breast cancer, as well as brain metastases (50). The combination of cisplatin, bevacizumab and etoposide was used in a phase 2 trial in eight patients with breast cancer and brain metastasis in Taiwan, and the treatment was effective in five patients (46). In a previous study on neoadjuvant therapy, the paclitaxel plus cisplatin (PC) regimen combined with trastuzumab was shown to have a high pathological complete response rate in HER2-positive breast cancer (47). In addition, the results from TBCRC009 clinical trial demonstrated that patients with metastatic triple-negative breast cancer receiving cisplatin every 3 weeks had numerically higher objective response rates than those receiving carboplatin (48). Our present study creates the possibility for future clinical trials targeting DNA damage-resistant tumors with niclosamide in the treatment of HER2-positive breast cancer. The combination of these two classes of drug may therefore be considered as a novel option to treat cancer.
In the present study, the sample size of each group in the in vivo experiment was relatively small. When the experiment was conducted, cells were injected into nude mice for 14 days and tumor-bearing mice with tumor volumes of 50–100 mm3 were evenly divided into four groups according to tumor size. Before treatment, the mean tumor volume in each group was consistent and not significantly different. We also controlled factors (mice weight, age, the mean tumor volume and housing conditions) other than the intervention that were perfectly balanced across the groups. The four groups of xenograft tumors showed different growth rates after treatment, which may reflect the antitumor effect of niclosamide in vivo. However, the small sample size remains a limitation of this research. Furthermore, there was no significant difference between the niclosamide group and the combination group. However, there was a trend of higher antitumor effect in the combination group. To determine whether the effect of combination treatment is greater than that of niclosamide alone, clinical trials need to be conducted in cisplatin-resistant patients.
The development and clinical application of oncology drugs is a long and expensive process. New drugs applied in practice account for a very small portion of investigated drugs. Niclosamide is already available in the clinic, and the side effects and other data related to its clinical use are already documented.
In conclusion, the present study offered new ideas for the treatment of chemoresistant HER2-positive breast cancer. The results from this study were in accordance with existing results showing that niclosamide can inhibit EMT and STAT3 phosphorylation, leading to cytotoxicity against chemoresistant cells and breast CSCs. Niclosamide combined with cisplatin may be considered as a novel treatment therapy for chemoresistant HER2-positive breast cancer.
Acknowledgements
Not applicable.
Funding Statement
The present study was supported by the Fundamental Research Funds for the Central Universities (grant no. 22120180601).
Funding
The present study was supported by the Fundamental Research Funds for the Central Universities (grant no. 22120180601).
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author upon reasonable request.
Authors' contributions
JL and JH contributed to the design of the study. HD, HQ and JL performed the experiments. HD and JL analyzed the data. JL drafted the initial manuscript. JL and HD confirmed the authenticity of all the raw data. All authors have read and approved the final manuscript.
Ethics approval and consent to participate
Animal experiments were approved by the Tongji University School of Medicine Committee for the Use and Care of Animals (Shanghai, China; approval no. 11115043) and performed in strict accordance with institutional requirements.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Kennecke H, Yerushalmi R, Woods R, Cheang MC, Voduc D, Speers CH, Nielsen TO, Gelmon K. Metastatic behavior of breast cancer subtypes. J Clin Oncol. 2010;28:3271–3277. doi: 10.1200/JCO.2009.25.9820. [DOI] [PubMed] [Google Scholar]
- 2.Camejo N, Castillo C, Alonso R, Correa F, Rivero E, Mezquita C, Rosich A, Dellacasa F, Silveira L, Delgado L. Effectiveness of Trastuzumab for Human Epidermal Growth Factor Receptor 2-Positive Breast Cancer in a Real-Life Setting: One Decade of Experience Under National Treatment Coverage Regulations. JCO Glob Oncol. 2020;6:217–223. doi: 10.1200/JGO.19.00299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Diaby V, Tawk R, Sanogo V, Xiao H, Montero AJ. A review of systematic reviews of the cost-effectiveness of hormone therapy, chemotherapy, and targeted therapy for breast cancer. Breast Cancer Res Treat. 2015;151:27–40. doi: 10.1007/s10549-015-3383-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Geng SQ, Alexandrou AT, Li JJ. Breast cancer stem cells: Multiple capacities in tumor metastasis. Cancer Lett. 2014;349:1–7. doi: 10.1016/j.canlet.2014.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Semenza GL. Targeting HIF-1 for cancer therapy. Nat Rev Cancer. 2003;3:721–732. doi: 10.1038/nrc1187. [DOI] [PubMed] [Google Scholar]
- 6.Dean M, Fojo T, Bates S. Tumour stem cells and drug resistance. Nat Rev Cancer. 2005;5:275–284. doi: 10.1038/nrc1590. [DOI] [PubMed] [Google Scholar]
- 7.Samanta D, Gilkes DM, Chaturvedi P, Xiang L, Semenza GL. Hypoxia-inducible factors are required for chemotherapy resistance of breast cancer stem cells. Proc Natl Acad Sci USA. 2014;111:E5429–E5438. doi: 10.1073/pnas.1421438111. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 8.Li X, Lewis MT, Huang J, Gutierrez C, Osborne CK, Wu MF, Hilsenbeck SG, Pavlick A, Zhang X, Chamness GC, et al. Intrinsic resistance of tumorigenic breast cancer cells to chemotherapy. J Natl Cancer Inst. 2008;100:672–679. doi: 10.1093/jnci/djn123. [DOI] [PubMed] [Google Scholar]
- 9.Gao J, Liu J, Xie F, Lu Y, Yin C, Shen X. Co-Delivery of Docetaxel and Salinomycin to Target Both Breast Cancer Cells and Stem Cells by PLGA/TPGS Nanoparticles. Int J Nanomedicine. 2019;14:9199–9216. doi: 10.2147/IJN.S230376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Andrews P, Thyssen J, Lorke D. The biology and toxicology of molluscicides, Bayluscide. Pharmacol Ther. 1982;19:245–295. doi: 10.1016/0163-7258(82)90064-X. [DOI] [PubMed] [Google Scholar]
- 11.Chen B, Wei W, Ma L, Yang B, Gill RM, Chua MS, Butte AJ, So S. Computational Discovery of Niclosamide Ethanolamine, a Repurposed Drug Candidate That Reduces Growth of Hepatocellular Carcinoma Cells In Vitro and in Mice by Inhibiting Cell Division Cycle 37 Signaling. Gastroenterology. 2017;152:2022–2036. doi: 10.1053/j.gastro.2017.02.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Al-Hadiya BM. Niclosamide: Comprehensive profile. Profiles Drug Subst Excip Relat Methodol. 2005;32:67–96. doi: 10.1016/S0099-5428(05)32002-8. [DOI] [PubMed] [Google Scholar]
- 13.Liu J, Chen X, Ward T, Pegram M, Shen K. Combined niclosamide with cisplatin inhibits epithelial-mesenchymal transition and tumor growth in cisplatin-resistant triple-negative breast cancer. Tumour Biol. 2016;37:9825–9835. doi: 10.1007/s13277-015-4650-1. [DOI] [PubMed] [Google Scholar]
- 14.Liu J, Chen X, Ward T, Mao Y, Bockhorn J, Liu X, Wang G, Pegram M, Shen K. Niclosamide inhibits epithelial-mesenchymal transition and tumor growth in lapatinib-resistant human epidermal growth factor receptor 2-positive breast cancer. Int J Biochem Cell Biol. 2016;71:12–23. doi: 10.1016/j.biocel.2015.11.014. [DOI] [PubMed] [Google Scholar]
- 15.Taliaferro-Smith L, Oberlick E, Liu T, McGlothen T, Alcaide T, Tobin R, Donnelly S, Commander R, Kline E, Nagaraju GP, et al. FAK activation is required for IGF1R-mediated regulation of EMT, migration, and invasion in mesenchymal triple negative breast cancer cells. Oncotarget. 2015;6:4757–4772. doi: 10.18632/oncotarget.3023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schieber MS, Chandel NS. ROS links glucose metabolism to breast cancer stem cell and EMT phenotype. Cancer Cell. 2013;23:265–267. doi: 10.1016/j.ccr.2013.02.021. [DOI] [PubMed] [Google Scholar]
- 17.Lu L, Dong J, Wang L, Xia Q, Zhang D, Kim H, Yin T, Fan S, Shen Q. Activation of STAT3 and Bcl-2 and reduction of reactive oxygen species (ROS) promote radioresistance in breast cancer and overcome of radioresistance with niclosamide. Oncogene. 2018;37:5292–5304. doi: 10.1038/s41388-018-0340-y. [DOI] [PubMed] [Google Scholar]
- 18.Zhu Y, Zuo W, Chen L, Bian S, Jing J, Gan C, Wu X, Liu H, Su X, Hu W, et al. Repurposing of the anti-helminthic drug niclosamide to treat melanoma and pulmonary metastasis via the STAT3 signaling pathway. Biochem Pharmacol. 2019;169:113610. doi: 10.1016/j.bcp.2019.08.012. [DOI] [PubMed] [Google Scholar]
- 19.Osada T, Chen M, Yang XY, Spasojevic I, Vandeusen JB, Hsu D, Clary BM, Clay TM, Chen W, Morse MA, et al. Antihelminth compound niclosamide downregulates Wnt signaling and elicits antitumor responses in tumors with activating APC mutations. Cancer Res. 2011;71:4172–4182. doi: 10.1158/0008-5472.CAN-10-3978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Londoño-Joshi AI, Arend RC, Aristizabal L, Lu W, Samant RS, Metge BJ, Hidalgo B, Grizzle WE, Conner M, Forero-Torres A, et al. Effect of niclosamide on basal-like breast cancers. Mol Cancer Ther. 2014;13:800–811. doi: 10.1158/1535-7163.MCT-13-0555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.You S, Li R, Park D, Xie M, Sica GL, Cao Y, Xiao ZQ, Deng X. Disruption of STAT3 by niclosamide reverses radioresistance of human lung cancer. Mol Cancer Ther. 2014;13:606–616. doi: 10.1158/1535-7163.MCT-13-0608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Luo F, Luo M, Rong QX, Zhang H, Chen Z, Wang F, Zhao HY, Fu LW. Niclosamide, an antihelmintic drug, enhances efficacy of PD-1/PD-L1 immune checkpoint blockade in non-small cell lung cancer. J Immunother Cancer. 2019;7:245. doi: 10.1186/s40425-019-0733-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schmalhofer O, Brabletz S, Brabletz T. E-cadherin, beta-catenin, and ZEB1 in malignant progression of cancer. Cancer Metastasis Rev. 2009;28:151–166. doi: 10.1007/s10555-008-9179-y. [DOI] [PubMed] [Google Scholar]
- 24.Wu Q, Li J, Zhu S, Wu J, Chen C, Liu Q, Wei W, Zhang Y, Sun S. Breast cancer subtypes predict the preferential site of distant metastases: A SEER based study. Oncotarget. 2017;8:27990–27996. doi: 10.18632/oncotarget.15856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fillmore CM, Kuperwasser C. Human breast cancer cell lines contain stem-like cells that self-renew, give rise to phenotypically diverse progeny and survive chemotherapy. Breast Cancer Res. 2008;10:R25. doi: 10.1186/bcr1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Grimshaw MJ, Cooper L, Papazisis K, Coleman JA, Bohnenkamp HR, Chiapero-Stanke L, Taylor-Papadimitriou J, Burchell JM. Mammosphere culture of metastatic breast cancer cells enriches for tumorigenic breast cancer cells. Breast Cancer Res. 2008;10:R52. doi: 10.1186/bcr2106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Levine AJ, Puzio-Kuter AM. The control of the metabolic switch in cancers by oncogenes and tumor suppressor genes. Science. 2010;330:1340–1344. doi: 10.1126/science.1193494. [DOI] [PubMed] [Google Scholar]
- 28.Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science. 2009;324:1029–1033. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Alasadi A, Chen M, Swapna GVT, Tao H, Guo J, Collantes J, Fadhil N, Montelione GT, Jin S. Effect of mitochondrial uncouplers niclosamide ethanolamine (NEN) and oxyclozanide on hepatic metastasis of colon cancer. Cell Death Dis. 2018;9:215. doi: 10.1038/s41419-017-0092-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lu J, Tan M, Cai Q. The Warburg effect in tumor progression: Mitochondrial oxidative metabolism as an anti-metastasis mechanism. Cancer Lett. 2015;356(2 Pt A):156–164. doi: 10.1016/j.canlet.2014.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Park SJ, Shin JH, Kang H, Hwang JJ, Cho DH. Niclosamide induces mitochondria fragmentation and promotes both apoptotic and autophagic cell death. BMB Rep. 2011;44:517–522. doi: 10.5483/BMBRep.2011.44.8.517. [DOI] [PubMed] [Google Scholar]
- 32.Khanim FL, Merrick BA, Giles HV, Jankute M, Jackson JB, Giles LJ, Birtwistle J, Bunce CM, Drayson MT. Redeployment-based drug screening identifies the anti-helminthic niclosamide as anti-myeloma therapy that also reduces free light chain production. Blood Cancer J. 2011;1:e39. doi: 10.1038/bcj.2011.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yu K, Wang T, Li Y, Wang C, Wang X, Zhang M, Xie Y, Li S, An Z, Ye T. Niclosamide induces apoptosis through mitochondrial intrinsic pathway and inhibits migration and invasion in human thyroid cancer in vitro. Biomed Pharmacother. 2017;92:403–411. doi: 10.1016/j.biopha.2017.05.097. [DOI] [PubMed] [Google Scholar]
- 34.Ye T, Xiong Y, Yan Y, Xia Y, Song X, Liu L, Li D, Wang N, Zhang L, Zhu Y, et al. The anthelmintic drug niclosamide induces apoptosis, impairs metastasis and reduces immunosuppressive cells in breast cancer model. PLoS One. 2014;9:e85887. doi: 10.1371/journal.pone.0085887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yu H, Lee H, Herrmann A, Buettner R, Jove R. Revisiting STAT3 signalling in cancer: New and unexpected biological functions. Nat Rev Cancer. 2014;14:736–746. doi: 10.1038/nrc3818. [DOI] [PubMed] [Google Scholar]
- 36.Duru N, Candas D, Jiang G, Li JJ. Breast cancer adaptive resistance: HER2 and cancer stem cell repopulation in a heterogeneous tumor society. J Cancer Res Clin Oncol. 2014;140:1–14. doi: 10.1007/s00432-013-1494-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Duru N, Fan M, Candas D, Menaa C, Liu HC, Nantajit D, Wen Y, Xiao K, Eldridge A, Chromy BA, et al. HER2-associated radioresistance of breast cancer stem cells isolated from HER2-negative breast cancer cells. Clin Cancer Res. 2012;18:6634–6647. doi: 10.1158/1078-0432.CCR-12-1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kim JS, Kim HA, Seong MK, Seol H, Oh JS, Kim EK, Chang JW, Hwang SG, Noh WC. STAT3-survivin signaling mediates a poor response to radiotherapy in HER2-positive breast cancers. Oncotarget. 2016;7:7055–7065. doi: 10.18632/oncotarget.6855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chung SS, Giehl N, Wu Y, Vadgama JV. STAT3 activation in HER2-overexpressing breast cancer promotes epithelial-mesenchymal transition and cancer stem cell traits. Int J Oncol. 2014;44:403–411. doi: 10.3892/ijo.2013.2195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li G, Zhao L, Li W, Fan K, Qian W, Hou S, Wang H, Dai J, Wei H, Guo Y. Feedback activation of STAT3 mediates trastuzumab resistance via upregulation of MUC1 and MUC4 expression. Oncotarget. 2014;5:8317–8329. doi: 10.18632/oncotarget.2135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wegrzyn J, Potla R, Chwae YJ, Sepuri NB, Zhang Q, Koeck T, Derecka M, Szczepanek K, Szelag M, Gornicka A, et al. Function of mitochondrial Stat3 in cellular respiration. Science. 2009;323:793–797. doi: 10.1126/science.1164551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yang R, Rincon M. Mitochondrial Stat3, the Need for Design Thinking. Int J Biol Sci. 2016;12:532–544. doi: 10.7150/ijbs.15153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gough DJ, Corlett A, Schlessinger K, Wegrzyn J, Larner AC, Levy DE. Mitochondrial STAT3 supports Ras-dependent oncogenic transformation. Science. 2009;324:1713–1716. doi: 10.1126/science.1171721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ren X, Duan L, He Q, Zhang Z, Zhou Y, Wu D, Pan J, Pei D, Ding K. Identification of Niclosamide as a New Small-Molecule Inhibitor of the STAT3 Signaling Pathway. ACS Med Chem Lett. 2010;1:454–459. doi: 10.1021/ml100146z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen H, Yang Z, Ding C, Chu L, Zhang Y, Terry K, Liu H, Shen Q, Zhou J. Discovery of O-Alkylamino Tethered Niclosamide Derivatives as Potent and Orally Bioavailable Anticancer Agents. ACS Med Chem Lett. 2013;4:180–185. doi: 10.1021/ml3003082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wu PF, Lin CH, Kuo CH, Chen WW, Yeh DC, Liao HW, Huang SM, Cheng AL, Lu YS. A pilot study of bevacizumab combined with etoposide and cisplatin in breast cancer patients with leptomeningeal carcinomatosis. BMC Cancer. 2015;15:299. doi: 10.1186/s12885-015-1290-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhou L, Xu S, Yin W, Lin Y, Du Y, Jiang Y, Wang Y, Zhang J, Wu Z, Lu J. Weekly paclitaxel and cisplatin as neoadjuvant chemotherapy with locally advanced breast cancer: A prospective, single arm, phase II study. Oncotarget. 2017;8:79305–79314. doi: 10.18632/oncotarget.17954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Isakoff SJ, Mayer EL, He L, Traina TA, Carey LA, Krag KJ, Rugo HS, Liu MC, Stearns V, Come SE, et al. TBCRC009: A Multicenter Phase II Clinical Trial of Platinum Monotherapy With Biomarker Assessment in Metastatic Triple-Negative Breast Cancer. J Clin Oncol. 2015;33:1902–1909. doi: 10.1200/JCO.2014.57.6660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Silver DP, Richardson AL, Eklund AC, Wang ZC, Szallasi Z, Li Q, Juul N, Leong CO, Calogrias D, Buraimoh A, et al. Efficacy of neoadjuvant Cisplatin in triple-negative breast cancer. J Clin Oncol. 2010;28:1145–1153. doi: 10.1200/JCO.2009.22.4725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shah N, Mohammad AS, Saralkar P, Sprowls SA, Vickers SD, John D, Tallman RM, Lucke-Wold BP, Jarrell KE, Pinti M, et al. Investigational chemotherapy and novel pharmacokinetic mechanisms for the treatment of breast cancer brain metastases. Pharmacol Res. 2018;132:47–68. doi: 10.1016/j.phrs.2018.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets used and/or analyzed during the current study are available from the corresponding author upon reasonable request.