

Abstract
Bis(cyclotryptamine) alkaloids have been popular topics of study for many decades. Five possible scaffolds for bis(cyclotryptamine) alkaloids were originally postulated in the 1950s, but only four of these scaffolds have been observed in natural products to date. We describe synthetic access to the elusive fifth scaffold, the piperidinoindoline, through syntheses of compounds now termed “dihydropsychotriadine” and “psychotriadine”. The latter of these compounds was subsequently identified in extracts of the flower Psychotria colorata. Our synthetic route features a stereospecific solid-state photodecarbonylation reaction to introduce the key vicinal quaternary stereocenters.
Since the initial isolation of calycanthine in 1888,1 bis(cyclotryptamine) alkaloids2 have captivated the attention of scientists worldwide. Interest in these natural products has been fueled by a combination of their biological activities and intricate structures. With regard to the latter, the identification and structural elucidation of bis(cyclotryptamine) alkaloids have a rich history.2a,b For example, although calycanthine was isolated in 1888, its structure remained a mystery until 1954, when Robinson and Teuber first proposed a plausible structural identity.3 At that time, they suggested the existence of five possible distinct ring systems, depicted as 1–5, arising from common biosynthon 6 (Figure 1). On the basis of degradation studies, piperidinoindoline 5 was postulated as a constitutional isomer of calycanthine. However, in 1960, studies by Woodward4 and Hamor5 identified bridged bicycle 1 as the correct structure.
Figure 1.
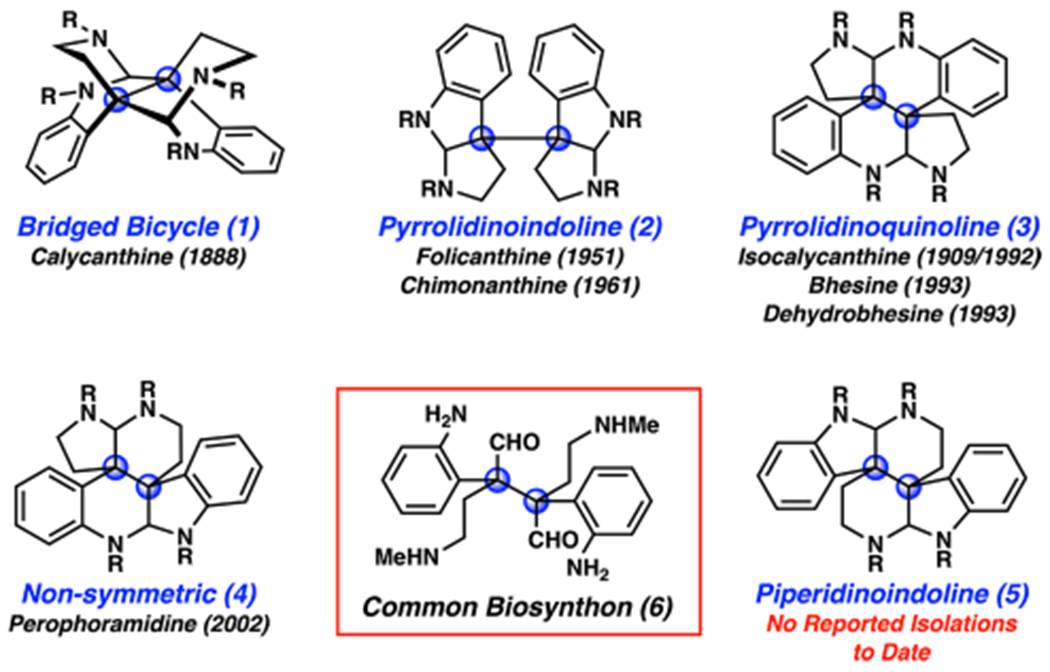
Possible bis(cyclotryptamine) alkaloid constitutional isomers 1–5 arising from common biosynthon 6.
Over the subsequent six decades, many isolation reports,6 biosynthetic studies,7 and synthetic efforts have been disclosed.2 This has led to the discovery of more than 20 bis(cyclotryptamine) alkaloids to date.2 Interestingly, of the five possible isomeric scaffolds originally proposed, only four have been confirmed to exist (i.e., 1–4) in isolated natural products.6 With regard to synthetic studies, completed total syntheses of natural products bearing scaffolds 1, 2, and 4 have been most common over the past few decades. Efforts to access piperidinoindoline scaffold 5 have been rare. In a seminal study, Scott and co-workers are believed to have accessed a compound bearing scaffold 5 in 1967.8 More recently, compounds bearing substituted piperidinoindoline scaffolds have been accessed in the context of communesin studies, as shown independently by our group and Tang’s group9 and by Movassaghi’s group.10 Scaffold 5 has not been observed naturally.
Like many laboratories, we have been drawn to the bis(cyclotryptamine) alkaloids because of their remarkable structures. These compounds typically feature four nitrogen atoms, vicinal quaternary stereocenters (arising biosynthetically from the dimerization of a tryptamine derivative2a–c,7), and six interwoven rings. With the aim of potentially accessing the various isomeric members of the family, we targeted biosynthon 6. Overman,11 Movassaghi,12 and others2,13 have elegantly demonstrated the success of this general approach to access pyrrolidinoindoline isomer 2 from preformed indoline (or related) ring systems.14 In this Communication, we demonstrate an alternative approach to 6 that relies on the stereospecific photodecarbonylation of a crystalline ketone to access the requisite vicinal quaternary centers, ultimately leading to the synthesis of an alkaloid bearing the elusive piperidinoindoline scaffold 5 and its identification in Psychotria colorata flower extracts.
Our retrosynthetic approach targeted biosynthon 6 as a potential means to access various bis(cyclotryptamine) scaffolds (Scheme 1). As 6 itself would not be isolable, we targeted a synthetic equivalent or congener by reduction of bis(lactam) 7 and late-stage C–N bond formation. In turn, brominated compound 7 would arise from ketone 9 via a solid-state photodecarbonylation reaction. This key step would proceed by Norrish type I photodecarbonylation of 9 followed by coupling of radical pair 8. Because of conformational restrictions imposed by the rigid reaction cavity of the crystal lattice, as illustrated by the dotted lines, the conversion of 9 to 7 was expected to occur with retention of stereochemistry. We have previously shown the success of such solid-state photodecarbonylation reactions in simpler systems.15–17 Moreover, this key step would complement the elegant radical-based approach for accessing cyclotryptamine alkaloids pioneered by Movassaghi and co-workers.12 Ketone 9 would be prepared from acid chloride 10 and enolate 11.18
Scheme 1.

Retrosynthetic Analysis of Biosynthon 6 with Key Stereospecific Radical Combination in the Crystalline State
Scheme 2 summarizes our synthesis of ketone substrate 9 and the attempted photodecarbonylation reaction. Arylmalonic ester 12 was converted to pyrrolidinone 13 through alkylation with bromoacetonitrile followed by reductive cyclization. Subsequent methylation furnished pyrrolidinone 14, which served as a point of divergence. In one pathway, 14 was converted to acid chloride 10 through a two-step sequence involving saponification and treatment of the resultant carboxylic acid with oxalyl chloride and catalytic DMF. In the other sequence, 14 was saponified and then thermally decarboxylated to provide amide 15 in 79% yield. To unite the fragments, amide 15 was converted to its lithium enolate by deprotonation with LiHMDS. In situ trapping with acid chloride 10 delivered ketone 9, the desired substrate for photodecarbonylation, as validated by X-ray crystallography.19 Of note, only the d,l-diastereomer of 9 was observed, which we attribute to a highly ordered transition state mediated by Li+ chelation on the basis of prior literature reports.16b With crystalline substrate 9 in hand, we attempted the key solid-state photodecarbonylation. However, only a small quantity of the desired product 7 was formed. Instead, the mass balance was attributed to competitive disproportionation, giving products 15 and 16, as well as substantial nonspecific decomposition.20 Although the yield of 7 was low, thus limiting late-stage efforts, the formation of 7 served as a proof of principle that a solid-state photodecarbonylation could forge the critical vicinal quaternary stereocenters with easily modifiable functional groups in place on the aromatic rings.
Scheme 2.

Synthesis and Photodecarbonylation of Ketone 9
To improve the efficiency of the photodecarbonylation reaction, we explored structural derivatives of ketone substrate 9. Our most promising findings are shown in Figure 2.17 In four linear steps, pyrrolidinone 13 was converted to ketone 17 bearing removable p-methoxybenzyl (PMB) protecting groups (Figure 2A; see the Supporting Information for details). With the hope of being able to introduce other N-substituents and identify a crystalline substrate, we then attempted to enact oxidative cleavage of the PMB moieties using ceric ammonium nitrate (CAN). However, this led to the formation of imide products 18 and 19. Given that both compounds were high-melting crystalline solids, we tested them in the solid-state photodecarbonylation reaction (Figure 2B). Whereas symmetrical ketone 18 proved completely unreactive, even under prolonged irradiation, we were delighted to find that hemiacyl ketone 19 underwent the desired reaction to furnish 21 after N-deprotection. Of note, despite going through the intermediacy of a radical pair with no configurationally inert stereocenters, this decarbonylative C–C bond-forming reaction proceeded with high diastereoselectivity and established the vicinal quaternary stereocenters present in biosynthon 6.16c,21
Figure 2.

Preparation of substrates 18 and 19, solid-state photodecarbonylation studies, and explanation for reaction outcomes (the R groups on imides 18 and 19 have been removed from the X-ray renderings for clarity).
The dramatically different reactivities of ketones 18 and 19 can be rationalized by the analysis shown in Figure 2C and inspection of the single-crystal X-ray structures (see Figure 2B)19 for the two compounds. Solid-state photodecarbonylation requires stabilization of the breaking C–C σ-bonds by neighboring π-systems.15,22 The extent of these hyper-conjugative interactions in substrates 18 and 19 can be correlated to the dihedral angle between the breaking C–C σ bond and the nearest C–C-bond of the aromatic π-system (see bonds highlighted in blue in Figure 2C). A dihedral angle of 90° is ideal, allowing for maximum orbital overlap. Alternatively, when the dihedral angle is 0°, the C–C σ-bond and π-system are orthogonal, resulting in no electronic stabilization. In considering ketone 18, the two relevant dihedral angles are 85° and 20°, the latter of which presumably leads to negligible orbital overlap and failed bond homolysis. On the other hand, the relevant dihedral angles in ketone 19 are 69° and 68°, which we surmise provide sufficient orbital overlap to facilitate decarbonylation.
Having installed the key vicinal quaternary stereocenters, we turned our attention to the elaboration of 21 to a bis(cyclotryptamine) alkaloid (Scheme 3). N-Methylation of 21 proceeded smoothly to furnish 7 in 84% yield. Next, several attempts to effect double C–N bond formation were put forth, but most were deemed unsuccessful, presumably because of the highly sterically hindered nature of the C–Br bonds in 7. Eventually, we found that a modification of Ma’s copper-catalyzed azidation procedure could be implemented to furnish bis(azide) 22.23 Bis(azide) 22 could not be isolated cleanly, despite significant effort, and had to be used directly in the subsequent step.24 With the requisite nitrogen atoms installed, we then attempted a challenging reduction cascade by treating 22 with LiAlH4 at 90 °C. To our surprise, this led to the formation of 25 bearing the elusive piperidinoindoline scaffold.25,26 The structure of 25, a compound we have termed “dihydropsychotriadine”, was ultimately confirmed by single-crystal X-ray diffraction.19 Interestingly, bhesine (26), or variants thereof, were not observed. One plausible pathway from 22 to 25 involves double azide reduction to furnish intermediate 23, double 5-exo-trig cyclization/transamidation to give 24, double cyclization to give the piperidine rings,27 and single amidine reduction.28 Despite the mechanistic possibilities for the formation of other isomers (e.g., scaffolds 1–4) during the reduction of 22, we did not observe any major byproducts by 1H NMR analysis. However, the formation of other isomeric products cannot be ruled out at this time.
Scheme 3.
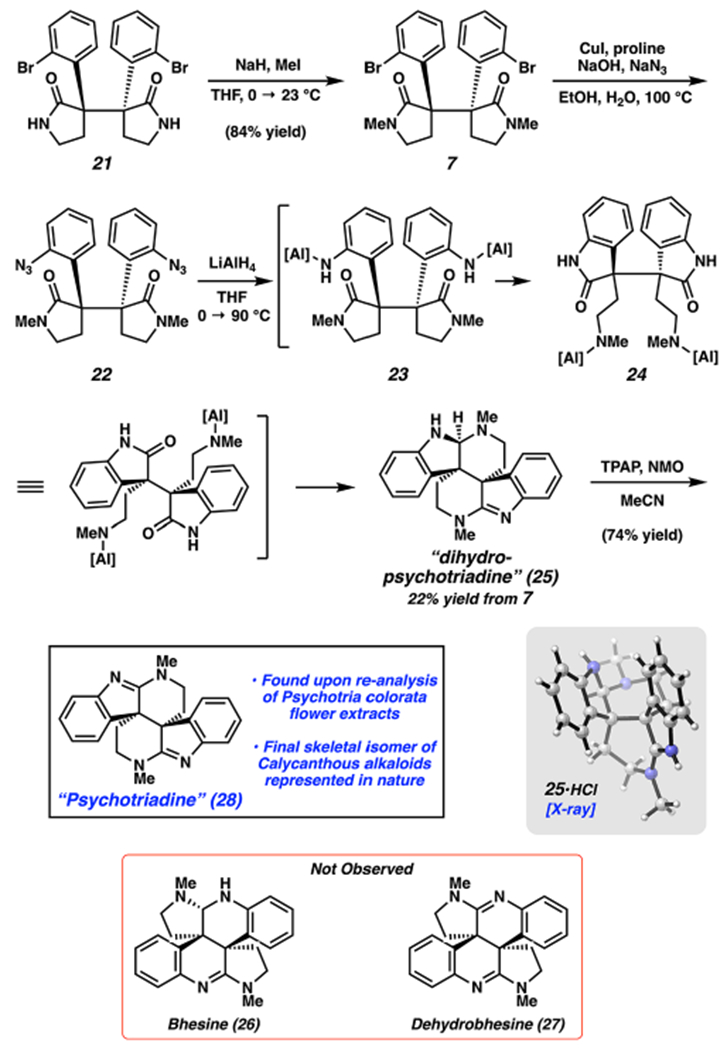
Total Synthesis of “Psychotriadine” (28) Bearing the Piperidinoindoline Scaffold (the Chloride Counterion of the X-ray Structure of 25 Has Been Omitted for Clarity)
Prior to unambiguously establishing the structure of 25 by X-ray diffraction, we had surmised that 25 could be an aminal stereoisomer of 26. Therefore, 25 was treated under Ley–Griffith oxidation conditions to ablate the aminal stereocenter.29 The product, which we obtained in 74% yield, was compared to an authentic sample of dehydrobhesine (27) obtained from extracts from P. colorata.6a Although our synthetic sample did not match 27, the isolation sample also contained a previously unidentified compound representing ~10% of the sample mass. This compound was found to spectroscopically match our synthetic oxidation product. On the basis of the crystallographic characterization of 25 and NMR analysis of the oxidation product, we propose the depicted piperidinoindoline structure for compound 28. Because of its presence in the extracts from P. colorata, 28 is presumed to be a naturally occurring metabolite that we have now termed “psychotriadine”.30
In summary, we have developed a synthetic route to access “psychotriadine”, a previously unknown bis(cyclotryptamine) alkaloid bearing the elusive piperidinoindoline scaffold. Our approach features a stereospecific solid-state photodecarbonylation reaction to convert fully substituted ketone substrate 19 into 21 bearing vicinal quaternary stereocenters. The success or failure of this key step correlates to the solid-state geometry of the ketone substrate. Following late-stage C–N bond formation and a reduction cascade, the piperidinoindoline framework could be accessed. Reanalysis of P. colorata flower extracts revealed the presence of “psychotriadine”, suggesting that it is likely a naturally occurring alkaloid. These studies not only underscore the value of solid-state photodecarbonylation chemistry in total synthesis but also demonstrate that all five of the distinct bis(cyclotryptamine) alkaloid frameworks originally proposed are represented in nature.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank the National Science Foundation (CHE-1855342 for M.G.G.) and the Trueblood Family (for N.K.G.) for financial support. J.J.D. acknowledges the UCLA Graduate Division for a Dissertation Year Fellowship. We thank Professor Mohammad Movassaghi (MIT) for insightful discussions, Ieva Liepuoniute (UCLA) for computing the relative energies of isomers 27 and 28, Dr. Saeed Khan (UCLA) for X-ray analysis, and Professor Luisella Verotta (University of Milan, Italy) for helpful discussions and for providing authentic samples of 27 and 28. These studies were supported by shared instrumentation grants from the NSF (CHE-1048804) and the National Center for Research Resources (S10RR025631).
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.0c04760.
Detailed experimental procedures and compound characterization data (PDF)
Crystallographic data for 18 (CIF)
Crystallographic data for 9 (CIF)
Crystallographic data for 19 (CIF)
Crystallographic data for 25 (CIF)
Complete contact information is available at: https://pubs.acs.org/10.1021/jacs.0c04760
The authors declare no competing financial interest.
Contributor Information
Jordan J. Dotson, Department of Chemistry and Biochemistry, University of California, Los Angeles, California 90095, United States
J. Logan Bachman, Department of Chemistry and Biochemistry, University of California, Los Angeles, California 90095, United States.
Miguel A. Garcia-Garibay, Department of Chemistry and Biochemistry, University of California, Los Angeles, California 90095, United States.
Neil K. Garg, Department of Chemistry and Biochemistry, University of California, Los Angeles, California 90095, United States.
REFERENCES
- (1).Eccles RG Calycanthine. Drug. Circ. Chem. Gaz 1888, 32, 65. [Google Scholar]
- (2).For reviews, see:; (a) May JA; Stoltz B The Structural and Synthetic Implications of the Biosynthesis of the Calycanthaceous Alkaloids, the Communesins, and Nomofungin. Tetrahedron 2006, 62, 5262–5271. [Google Scholar]; (b) Schmidt MA; Movassaghi M New Strategies for the Synthesis of Hexahydropyrroloindole Alkaloids Inspired by Biosynthetic Hypotheses. Synlett 2008, 2008, 313–324. [Google Scholar]; (c) Steven A; Overman LE Total Synthesis of Complex Cyclotryptamine Alkaloids: Stereocontrolled Construction of Quaternary Carbon Stereocenters. Angew. Chem., Int. Ed 2007, 46, 5488–5508. [DOI] [PubMed] [Google Scholar]; (d) Trost BM; Osipov M Recent Advances on the Total Synthesis of Communesin Alkaloids and Perophoramidine. Chem. - Eur. J 2015, 21, 16318–16343. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Xu J-B; Cheng K-J Studies on the Alkaloids of the Calycanthaceae and Their Syntheses. Molecules 2015, 20, 6715–6738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).(a) Robinson R; Teuber HJ Reactions with Nitrosodisulfonate. IV. Calycanthine and Calycanthidine. Chem. Ind. (London) 1954, 783–784. [Google Scholar]; (b) Manske RH The Alkaloids of Calycanthacaea. Alkaloids 1965, 8, 581–589. [Google Scholar]
- (4).Woodward RB; Yang NC; Katz TJ; Harley-Mason J; Ingelby RF; Sheppard N Calycanthine, The Structure of the Alkaloid and Its Degradation Product, Calycanine. Proc. Chem. Soc., London 1960, 76–78. [Google Scholar]
- (5).Hamor TA; Robertson JM; Shrivastava HN; Silverton JV The Structure of Calycanthine. Proc. Chem. Soc., London 1960, 78–80. [Google Scholar]
- (6).For selected examples of bis(cyclotryptamine) alkaloid isolations, see:; (a) Verotta L; Pilati T; Tató M; Elisabetsky E; Amador TA; Nunes DS Pyrrolidinoindoline Alkaloids from Psychotria colorata. J. Nat. Prod 1998, 61, 392–396. [DOI] [PubMed] [Google Scholar]; (b) Balayer A; Sévenet T; Schaller H; Hadi AHA; Chiaroni A; Riche C; Païs M Dihydroquinoline-Type Alkaloids from Bhesea paniculate, celastraceae. Nat. Prod. Lett 1993, 2, 61–67. [Google Scholar]; (c) Adjibade Y; Weniger B; Quirion JC; Kuballa B; Cabalion P; Anton R Dimeric Alkaloids from Psychotria forsteriana. Phytochemistry 1992, 31, 317–319. [Google Scholar]; (d) Verbitski SM; Mayne CL; Davis RA; Concepcion GP; Ireland CM Isolation, Structure Determination, and Biological Activity of a Novel Alkaloid, Perophoramidine, from the Philippine Ascidian Perophoranamei. J. Org. Chem 2002, 67, 7124–7126. [DOI] [PubMed] [Google Scholar]; (e) Gordin HM On the Crystalline Alkaloid of Calycanthus Glaucus. Third Paper. – On Isocalycanthine, Isomeric with Calycanthine. J. Am. Chem. Soc 1909, 31, 1305–1312. [Google Scholar]
- (7).Kirby GW; Shah SW; Herbert EJ Biosynthesis of Chimonanthine from [2-3H]Tryptophan and [2-3H]Tryptamine. J. Chem. Soc. C 1969, 1916–1919. [DOI] [PubMed] [Google Scholar]
- (8).Hall ES; McCapra F; Scott AI Biogenetic-Type Synthesis of the Calycanthaceous Alkaloids. Tetrahedron 1967, 23, 4131–4141. [DOI] [PubMed] [Google Scholar]
- (9).Lin H-C; McMahon TC; Patel A; Corsello M; Simon A; Xu W; Zhao M; Houk KN; Garg NK; Tang Y P450-Mediated Coupling of Indole Fragments to Forge Communesin and Unnatural Isomers. J. Am. Chem. Soc 2016, 138, 4002–4005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Pompeo MM; Cheah JH; Movassaghi M Total Synthesis and Anti-Cancer Activity of All Known Communesin Alkaloids and Related Derivatives. J. Am. Chem. Soc 2019, 141, 14411–14420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).For selected studies from the Overman group pertaining to 1 and 2, see:; (a) Link JT; Overman LE Stereocontrolled Total Synthesis of meso-Chimonanthine and meso-Calycanthine via a Novel Samarium Mediated Reductive Dialkylation. J. Am. Chem. Soc 1996, 118, 8166–8167. [Google Scholar]; (b) Overman LE; Paone DV; Stearns BA Direct Stereo- and Enantiocontrolled Synthesis of Vicinal Stereogenic Quaternary Carbon Centers. Total Synthesis of meso- and (−)-Chimonanthine and (+)-Calycanthine. J. Am. Chem. Soc 1999, 121, 7702–7703. [Google Scholar]; (c) Overman LE; Larrow JF; Stearns BA; Vance JM Enantioselective Construction of Vicinal Stereogenic Quaternary Centers by Dialkylation; Practical Total Synthesis of (+)- and meso-Chimonanthine. Angew. Chem., Int. Ed 2000, 39, 213–215. [DOI] [PubMed] [Google Scholar]; (d) Hoyt SB; Overman LE Investigation of a Dialkylation Approach for Enantioselective Construction of Vicinal Quaternary Stereocenters. Org. Lett 2000, 2, 3241–3244. [DOI] [PubMed] [Google Scholar]; (e) Lebsack AD; Link JT; Overman LE; Stearns BA Enantioselective Total Synthesis of Quadrigemine C and Psycholeine. J. Am. Chem. Soc 2002, 124, 9008–9009. [DOI] [PubMed] [Google Scholar]; (f) Overman LE; Peterson EA Enantioselective Total Synthesis of the Cyclotryptamine Alkaloid Idiospermuline. Angew. Chem. Int. Ed 2003, 42, 2525–2528. [DOI] [PubMed] [Google Scholar]; (g) Kodanko JJ; Overman LE Enantioselective Total Synthesis of the Cyclotryptamine Alkaloids Hodgkinsine and Hodgkinsine B. Angew. Chem., Int. Ed 2003, 42, 2528–2531. [DOI] [PubMed] [Google Scholar]; (h) Ellis JM; Overman LE; Tanner HR; Wang J A Versatile Synthesis of Unsymmetrical 3,3′-Bioxindoles: Stereoselective Mukaiyama Aldol Reactions of 2-Siloxyindoles with Isatins. J. Org. Chem 2008, 73, 9151–9154. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Canham SM; Hafensteiner BD; Lebsack AD; May-Dracka TL; Nam S; Stearns BA; Overman LE Stereocontrolled Enantioselective Total Synthesis of the [2 + 2] Quadrigemine Alkaloids. Tetrahedron 2015, 71, 6424–6436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).For selected studies from the Movassaghi group pertaining to 1 and 2, see:; (a) Movassaghi M; Schmidt MA Concise Total Synthesis of (−)-Calycanthine, (+)-Chimonanthine, and (+)-Folicanthine. Angew. Chem., Int. Ed 2007, 46, 3725–3728. [DOI] [PubMed] [Google Scholar]; (b) Movassaghi M; Schmidt MA; Ashenhurst JA Concise Total Synthesis of (+)-WIN 64821 and (−)-Ditryptophenaline. Angew. Chem., Int. Ed 2008, 47, 1485–1487. [DOI] [PubMed] [Google Scholar]; (c) Kim J; Ashenhurst JA; Movassaghi M Total Synthesis of (+)-11,11’-Dideoxyverticillin A. Science 2009, 324, 238–241. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Movassaghi M; Ahmad OK; Lathrop SP Directed Heterodimerization: Stereocontrolled Assembly via Solvent-Caged Unsymmetrical Diazene Fragmentation. J. Am. Chem. Soc 2011, 133, 13002–13005. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Lathrop SP; Movassaghi M Application of Diazene-Directed Fragment Assembly to the Total Synthesis and Stereochemical Assignment of (+)-Desmethyl-meso-Chimonanthine and Related Heterodimeric Alkaloids. Chem. Sci 2014, 5, 333–340. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Lindovska P; Movassaghi M Concise Synthesis of (−)-Hodgkinsine, (−)-Calycosidine, (−)-Hodgkinsine B, (−)-Quadrigemine C, and (−)-Psycholeine via Convergent and Directed Modular Assembly of Cyclotryptamines. J. Am. Chem. Soc 2017, 139, 17590–17596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).For selected syntheses of calycanthine and chimonanthine, see:; (a) Hendrickson JB; Göschke R; Rees R Total Synthesis of the Calycanthous Alkaloids. Tetrahedron 1964, 20, 565–579. [Google Scholar]; (b) Snell RH; Woodward RL; Willis MC Catalytic Enantioselective Total Synthesis of Hodgkinsine B. Angew. Chem. Int. Ed 2011, 50, 9116–9119. [DOI] [PubMed] [Google Scholar]; (c) Mitsunuma H; Shibasaki M; Kanai M; Matsunaga S Catalytic Asymmetric Total Synthesis of Chimonanthine, Folicanthine, and Calycanthine Through Double Michael Reaction of Bisoxindole. Angew. Chem., Int. Ed 2012, 51, 5217–5221. [DOI] [PubMed] [Google Scholar]; (d) Trost BM; Osipov M Palladium-Catalyzed Asymmetric Construction of Vicinal All-Carbon Quaternary Stereocenters and Its Application to the Synthesis of Cyclotryptamine Alkaloids. Angew. Chem., Int. Ed 2013, 52, 9176–9181. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Ding M; Liang K; Pan R; Zhang H; Xia C Total Synthesis of (+)-Chimonanthine, (+)-Folicanthine, and (−)-Calycanthine. J. Org. Chem 2015, 80, 10309–10316. [DOI] [PubMed] [Google Scholar]; (f) Babu KN; Roy A; Singh M; Bisai A Thiourea-Catalyzed Enantioselective Malonate Addition onto 3-Sulfonyl-3′-Indolyl-2-Oxindoles: Formal Total Synthesis of (−)-Chimonanthine, (−)-Folicanthine, and (+)-Calycanthine. Org. Lett 2018, 20, 6327–6331. [DOI] [PubMed] [Google Scholar]; (g) Gentry EC; Rono LJ; Hale ME; Matsuura R; Knowles RR Enantioselective Synthesis of Pyrroloindolines via Noncovalent Stabilization of Indole Radical Cations and Applications to the Synthesis of Alkaloid Natural Products. J. Am. Chem. Soc 2018, 140, 3394–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Kumar N; Das MK; Ghosh S; Bisai A Development of Catalytic Deacylative Alkylations (DaA) of 3-acyl-2-oxindoles: Total Synthesis of meso-Chimonanthine and Related alkaloids. Chem. Commun 2017, 53, 2170–2173. [DOI] [PubMed] [Google Scholar]
- (14).A common related approach involves preformation of oxindoles instead of indoline ring systems (see ref 2).
- (15).(a) Dotson JJ; Perez-Estrada S; Garcia-Garibay MA Taming Radical Pairs in Nanocrystalline Ketones: Photochemical Synthesis of Compounds with Vicinal Stereogenic All-Carbon Quaternary Centers. J. Am. Chem. Soc 2018, 140, 8359–8371. [DOI] [PubMed] [Google Scholar]; (b) Natarajan A; Ng D; Yang Z; Garcia-Garibay MA Parallel Synthesis of (+)- and (−)-α-Cuparenone by Radical Combination in Crystalline Solids. Angew. Chem., Int. Ed 2007, 46, 6485–6487. [DOI] [PubMed] [Google Scholar]
- (16).For selected examples of solid-state photodecarbonylation reactions with retention of stereochemistry, see:; (a) Hérnandez-Linares MGH; Guerrero-Luna G; Pérez-Estrada S; Ellison M; Ortin M-M; Garcia-Garibay MA Large-Scale Green Chemical Synthesis of Adjacent Quaternary Chiral Centers by Continuous Flow Photodecarbonylation of Aqueous Suspensions of Nanocrystalline Ketones. J. Am. Chem. Soc 2015, 137, 1679–1684. [DOI] [PubMed] [Google Scholar]; (b) Resendiz MJE; Natarajan A; Garcia-Garibay MA Diastereoselective Synthesis and Spin-Dependent Photodecarbonylation of Di(3-Phenyl-2-Pyrrolidinon-3-yl)Ketones: Synthesis of Nonadjacent and Adjacent Stereogenic Quaternary Centers. Chem. Commun 2008, 193–195. [DOI] [PubMed] [Google Scholar]; (c) Resendiz MJE; Family F; Fuller K; Campos LM; Khan SI; Lebedeva NV; Forbes MDE; Garcia-Garibay MA Radical Reactions with Double Memory of Chirality (2MOC) for the Enantiospecific Synthesis of Adjacent Stereogenic Quaternary Centers in Solution: Cleavage and Bonding Faster Than Radical Rotation. J. Am. Chem. Soc 2009, 131, 8425–8433. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Resendiz M Photochemical Decarbonylation of Ketones in the Solid State and in Solution. Progress towards the synthesis of natural products. Ph.D. Thesis, UCLA, 2008. [Google Scholar]
- (17).Although a non-brominated analogue of 9 has been shown to undergo solid-state photodecarbonylation (see ref 16b), we have found that the presence of certain ortho substituents on the aromatic rings is problematic for the photodecarbonylation reaction.
- (18).An alternative strategy was explored involving the use of phosgene and 2 equiv of an enolate species. However, this protocol was found to be unsuccessful. For the parent transformation being carried out on a non-brominated starting material, see refs 16b,c.
- (19).X-ray structures were rendered using CYLview. See:; Legault CY CYLview, ver. 1.0b; Université de Sherbrooke: Quebec, 2009; http://www.cylview.org. [Google Scholar]
- (20).To our knowledge, this is the first documented case of disproportionation in a crystalline solid-state photodecarbonylation reaction. An explanation for this reactivity is currently under investigation and will be reported in due course.
- (21).Although we cannot rule out the formation of the corresponding meso isomer of 21 to a minor extent, we estimate the selectivity to be >9:1.
- (22).(a) Yang Z; Ng D; Garcia-Garibay MA Engineering Reactions in Crystalline Solids: Photochemical Generation of Secondary and Tertiary Enol Radical Pairs from Crystalline Ketodiesters. J. Org. Chem 2001, 66, 4468–4475. [DOI] [PubMed] [Google Scholar]; (b) Campos LM; Dang H; Ng D; Yang Z; Martinez HL; Garcia-Garibay MA Engineering Reactions in Crystalline Solids: Predicting Photochemical Decarbonylation from Calculated Thermochemical Parameters. J. Org. Chem 2002, 67, 3749–3754. [DOI] [PubMed] [Google Scholar]; (c) Ng D; Yang Z; Garcia-Garibay MA Engineering Reactions in Crystals; gem-Dialkoxy Substitution Enables the Photodecarbonylation of Crystalline 2-Indanone. Tetrahedron Lett. 2002, 43, 7063–7066. [Google Scholar]
- (23).Zhu W; Ma D Synthesis of Aryl Azides and Vinyl Azides via Proline-Promoted CuI-Catalyzed Coupling Reactions. Chem. Commun 2004, 888–889. [DOI] [PubMed] [Google Scholar]
- (24).Optimization efforts for this step were regrettably cut short because of COVID-19-related laboratory shutdowns, although preliminary optimization studies showed that the double azidation step could be achieved in 51% yield (according to 1H NMR analysis with an external standard, average of two experiments).
- (25).Although the two-step yield from 7 to 25 proceeds with a 78% loss of mass balance, we surmise that most of this (>65%) occurred during the cross-coupling step (based on 1H NMR analysis of crude 22). Therefore, if isomeric products resulted from the reduction of 22 they were present in < 13% yield.
- (26).“Dihydropsychotriadine” (25) has not yet been found in nature. Whether this compound is naturally occurring remains an open question.
- (27).Presumed intermediate 24 could plausibly undergo cyclization to form either a six-membered ring (observed) or a five-membered ring. It is surprising that the latter was not observed, as related reductive cyclizations have been reported to form the five-membered ring under similar conditions (see refs 11a–c). Of note, the studies disclosed in refs 11a–c, which include alkyl substituents on the oxindole nitrogens, presumably involve reduction of the oxindole to the aldehyde oxidation state followed by a reversible, thermodynamically controlled cyclization to form a five-membered ring. We speculate that the system disclosed herein may undergo a kinetically controlled cyclocondensation of the amine onto the oxindole to form a six-membered ring.
- (28).Other possible mechanisms exist for the conversion of 22 to 25 that do not involve formation of bis(oxindole) 24 (see Scheme S5).
- (29).Oxidation conditions adapted from:; Higuchi K; Sato Y; Tsuchimochi M; Sugiura K.; Hatori M; Kawasaki T First Total Synthesis of Hinckdentine A. Org. Lett 2009, 11, 197–199. [DOI] [PubMed] [Google Scholar]
- (30).Calculations suggest that 28 is 8.7 kcal/mol higher in energy than 27 (ωB97XD/6-31G(d,p)). As such, it is unlikely that 27 spontaneously rearranges to 28 during isolation. It is plausible that the substrate in a lower oxidation state, “tetrahydropsychotriadine”, could readily isomerize to give calycanthine or chimonanthine. Related scaffolds reminiscent of “tetrahydropsychotriadine” have only been isolated previously when constrained by the presence of additional ring systems (see refs 9 and 10). Preliminary efforts aimed at reducing 25 to the corresponding geminal diamine either were met with decomposition or led to recovered starting material (see Figure S4).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.