

Summary
Autoimmune cholestatic liver diseases are rare hepato-biliary disorders characterized by a progressive, inflammatory destruction of bile ducts. Primary biliary cholangitis (PBC) and primary sclerosing cholangitis (PSC) are the main autoimmune cholestatic liver diseases. Both may evolve into secondary biliary cirrhosis and its complications. Therapeutic options are limited and liver transplantation remains the only definitive treatment for PBC and PSC.
Most PBC and PSC patients have a typical presentation, which does not require liver biopsy. However, in routine clinical practice, important variants or specific subgroups that benefit from liver biopsy for proper management may be observed. Herein, we provide a general overview of clinical and pathological characteristic of PBC and PSC, highlighting the most important features for routine diagnostic practice.
Key words: immune-mediated cholangitis, autoimmune cholangitis, PBC, PSC, overlap variants
Introduction
Immune-mediated cholangiopathies are chronic cholestatic disorders whose development is driven by auto- and allo-immunity 1,2. They include primary biliary cholangitis (PBC), primary sclerosing cholangitis (PSC), IgG4-related sclerosing cholangitis, graft-versus-host disease and hepatic allograft rejection, even if some studies also consider biliary atresia as an altered immunity-associated disease 1-5. These diseases affect the biliary tree at different levels and for different extent, and they may also involve the peribiliary glands 6.
Immune-mediated cholangiopathies are characterized by an accumulation of activated auto- or allo-reactive T lymphocytes at the site of bile duct destruction, as the regulatory T cell is the main effector in the initiation of the process 6-8.
In this review we will focus on PBC and PSC, which are considered the main autoimmune cholestatic liver diseases.
Primary biliary cholangitis
PBC, formerly known as primary biliary cirrhosis, is a chronic inflammatory autoimmune cholestatic liver disease characterized by the destruction of small intrahepatic bile ducts, leading to fibrosis and eventually cirrhosis and its complications 9,10. In 1950, Ahrens et al. defined PBC in a series of 25 cases of diagnosed chronic intrahepatic biliary obstruction with xanthomatosis. However, Addison and Gull had described the very first case of PBC almost 100 years earlier 10.
EPIDEMIOLOGY
PBC is mainly diagnosed in women (92% of patients), with a female:male ratio of about 10:1, and a mean age at presentation of 55 years. Population-based epidemiological studies across Europe, North America, Asia, and Australia have revealed an incidence of 0.9 to 5.8 per 100,000 people per year. Some groups reported an increasing incidence over time, but whether this denotes a true rise or is biased by advances in diagnosis, is still unclear 9,10. The prevalence of the disease ranges from 1.9 to 40.2 per 100,000 people, and has also grown over time. The increased prevalence is probably attributable to a combination of better disease recognition and data collection, and increased survival after the introduction of ursodeoxycholic acid (UDCA). PBC is characterized by a significant geographical discrepancy, suggesting a possible role of environmental triggers in the development of disease 10.
OVERVIEW ON CLINICAL AND LABORATORY FEATURES
Fatigue and pruritus are the most common symptoms in PBC patients, and often have a quite negative effect on quality of life 9,10. Patients with pruritus might have excoriations or bleeding as a result of chronic scratching. Melanin deposition may occur, causing hyperpigmentation of the skin in up to 50% of cases. Long-term complications of the disease include osteopenia and osteoporosis, hyperlipidemia and vitamin deficiencies 10. In late stages, typical signs of cirrhosis and portal hypertension (spider nevi, palmar erythema, ascites, splenomegaly and muscle wasting) might be present, as well as an increased risk of hepatocellular carcinoma (HCC) 9,10. In the cirrhotic stage of the disease, radiological HCC surveillance is therefore mandatory 10. Patients with PBC, particularly women, have a higher likelihood of other concomitant autoimmune disorders, and up to 55% have an additional ongoing autoimmune process, such as autoimmune Hashimoto’s thyroiditis, Sjogren’s syndrome and Raynaud’s disease 3,10-12.
PBC patients typically present with a chronic cholestatic biochemical profile: alkaline phosphatase (ALP) levels are increased to 2 or more times the upper limit of normal, and this increase is usually sustained for 6 months or longer; similarly, γ-glutamyl transferase levels are increased to 5 or more times the upper limit of normal. Total bilirubin is usually normal in early stages, while abnormal values should raise concern for advanced disease. Serum aminotransferases might be slightly elevated 9. The serological hallmark of PBC is the presence of anti-mitochondrial antibodies (AMA), highly disease-specific antibodies identified in about 95% of PBC patients 9,10. AMA in a patient with raised ALP is diagnostic of PBC, after the exclusion of other intrahepatic and extrahepatic causes of cholestasis. AMA-negative PBC occurs in 5% of patients with an otherwise typical clinical, biochemical and histological profile 3,10. The presence of isolated AMA positivity in the absence of clinically apparent liver disease or abnormal liver function tests is seen in up to 0.5% of otherwise healthy individuals. This finding may be a hallmark of preclinical disease, and up to 17% will develop PBC over 5 years 3,10. Other autoantibodies are often identified in PBC patients, particularly anti-nuclear antibodies (ANA). Anti-Sp100 and anti-gp210 ANA have a high specificity for PBC, and could be of help in the diagnosis when AMA are negative. Moreover, the presence of anti-Sp100 or anti-gp210 is associated with a more clinically aggressive disease 9,10. IgM levels are often high, and this is useful in the differential diagnosis with autoimmune hepatitis (AIH) in which high IgG levels are typical, while IgM levels are low. Hyperlipidemia is also a common finding 9,10.
PBC may coexist with other liver autoimmune disorders, particularly AIH (see the “Overlap variants” paragraph).
The goals of treatment and management are the prevention of end-stage liver disease, and the improvement of associated symptoms. The natural history of the disease has changed substantially with the introduction of UDCA, as it has significantly ameliorated transplant-free survival in PBC patients, especially when started early in the disease course 9,10,13,14. However, about 40% of patients do not have a biochemical response to UDCA and would probably benefit from new therapies, such as obeticholic acid and off-label therapies (fibric acid derivatives and budesonide) 9,10,14-16. Orthotopic liver transplantation (OLT) is life-saving with excellent outcomes for those with decompensated cirrhosis10. Recurrent PBC after OLT occurs in up to 25% of patients 10.
In the last few years, efforts have been made to improve prognostic tools in PBC patients. The Global PBC Study Group and the UK-PBC consortium developed two new continuous scoring systems, the GLOBE Score and the UK-PBC Risk Score, considering both measures of treatment response and parameters of disease severity 10,17,18. Both showed better performance in the prediction of death or OLT than previous criteria 9,10.
In the presence of a cholestatic liver profile and AMA positivity, liver biopsy is not necessary to establish the diagnosis of PBC. However, biopsy is mandatory when AMA are absent, if the biochemical profile shows a mixed cholestatic and hepatocellular pattern, or when other co-existent liver diseases, such as AIH or non-alcoholic steatohepatitis (NASH), are suspected 9,10,19. Even if liver biopsy remains the gold standard, non-invasive methods to assess hepatic fibrosis are currently being considered, such as transient elastography, that has demonstrated more than 90% of sensitivity and specificity for detecting advanced fibrosis in PBC patients 10.
PATHOGENESIS
PBC seems to be related to complex interactions between genetic predisposition and environmental triggers. Geographical clustering of cases has been reported, and different factors have been associated with the disease, such as infectious agents, hair dyes, nail polish and cigarette smoking, even if their role has not been yet defined. The prevalence of disease is higher in families with an affected member, and several North American and European studies described a strong link between HLA alleles and PBC. Recent molecular studies reported that PBC shares some risk alleles with other autoimmune diseases. These risk alleles seem to occur in genes associated with immune function, potentially affecting different immune pathways, although the mechanisms by which they affect phenotype are not yet known 10,20-22.
The target cell in PBC is the cholangiocyte, which seems to express T-cell ligands essential for the induction of biliary epithelial autolysis. Moreover, cholangiocytes may act as antigen-presenting cells, thus amplifying the immune response 10,21,23-25.
Farnesoid X receptors are nuclear hormone receptors which are essential in bile acid metabolism. They play a role in cholestatic liver diseases and may represent new potential therapeutic targets 10.
HISTOLOGY
None of the histological changes seen in PBC samples are pathognomonic, thus histological features need to be interpreted in the light of clinical and laboratory findings, as in most liver disease settings.
The florid duct lesion, also referred to as non-suppurative destructive cholangitis, is the distinctive histological lesion in PBC. However, even if considered typical, it is seen in only about 10% of biopsy specimens from PBC patients, usually at early disease stages 6,9. The florid duct lesion is characterized by a chronic, intense, inflammatory infiltrate which surrounds and eventually destroys the bile ducts 6,9,26 (Fig. 1). Inflammatory cells classically infiltrate between adjacent cholangiocytes, destroying the bile duct basement membrane (Fig. 2). The inflammatory activity of PBC affects the small interlobular and septal bile ducts, with sparing of the large and extrahepatic ducts. In the affected bile ducts, cholangiocytes show swelling, vacuolated or eosinophilic cytoplasm and pyknotic nuclei. The biliary epithelium also shows proliferative changes with stratification 27. The rupture of the bile duct basement membrane may lead to bile duct ectasia 6. The inflammatory infiltrate consists mainly of CD4+ and CD8+ T lymphocytes, with a variable number of other inflammatory cells 6. Inflammatory cells may sometimes coalesce to form an epithelioid non-caseating granuloma, which is a typical finding in PBC samples, particularly when intimately associated with a damaged bile duct (Fig. 3). Infections and sarcoidosis are the main differential diagnoses in the presence of granulomas in a liver specimen. Special stains for microorganisms as well as a close correlation with clinical data are required to reach a correct diagnosis 6,10. Bile duct damage is usually associated with ductular reaction along the periphery of portal tracts, mainly at early disease stages 20,28 (Fig. 4).
Figure 1.
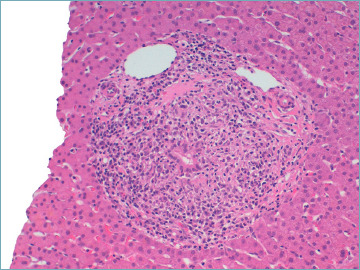
A florid duct lesion in a primary biliary cholangitis patient. A chronic inflammatory infiltrate made of lymphocytes and plasma cells surrounds and destroys the bile duct (hematoxylin-eosin; original magnification 20x).
Figure 2.
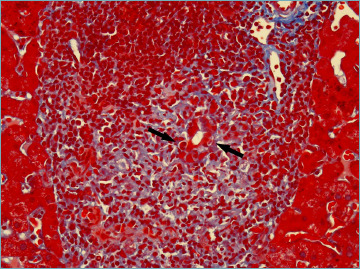
Lymphocytes infiltrate between adjacent cholangiocytes, destroying the bile duct basement membrane (arrows) (Masson’s trichrome stain; original magnification 40x).
Figure 3.
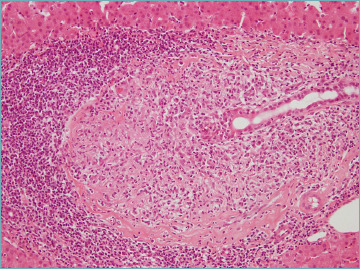
A typical epithelioid non-caseating granuloma in a portal tract, intimately associated with a damaged bile duct (hematoxylin-eosin; original magnification 20x).
Figure 4.
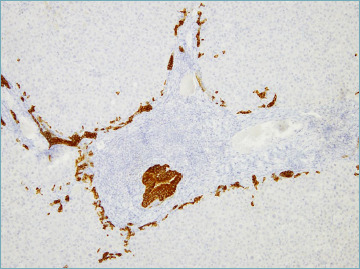
A portal tract showing a damaged bile duct and diffuse ductular reaction (cytokeratin 7 immunostain; original magnification 10x).
Portal tract (PT) inflammation accompanies bile duct damage. Lymphocytes are the predominant cells, but plasma cells are often numerous and this should not be misinterpreted as a sign of AIH. A coronal arrangement of plasma cells around the bile duct has been considered distinctive for PBC 6,29. Eosinophils may also be numerous, particularly at earlier stages (Fig. 5). Lymphoid follicles are often seen in portal tracts of PBC patients, both at early and late stages 10,20.
Figure 5.
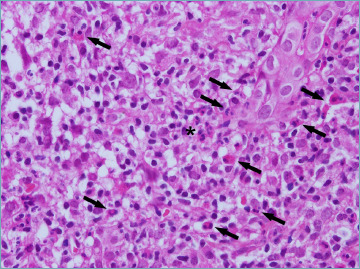
A portal tract with dense chronic inflammatory infiltrate, with numerous eosinophils (arrows), focally arranged in clusters (asterisk) (hematoxylin-eosin; original magnification 40x).
PT inflammation is often associated with interface hepatitis, which is usually mild. If interface hepatitis dominates the histological picture, an alternative diagnosis of AIH or PBC-AIH variant should be considered 6.
Parenchymal necro-inflammatory changes, including hepatocyte necrosis, acidophilic bodies, Kupffer cell hyperplasia and sinusoidal infiltration by lymphocytes and pigment-laden macrophages, may be seen, but they are generally mild in severity 9. Severe necro-inflammatory lobular damage should always raise the suspicion of AIH or PBC-AIH variant.
In advanced disease stages, the toxic effect of hydrophobic bile acids causes the so-called feathery degeneration of hepatocytes (Fig. 6). Bile stasis also leads to deposition of copper or copper-associated protein granules 6.
Figure 6.
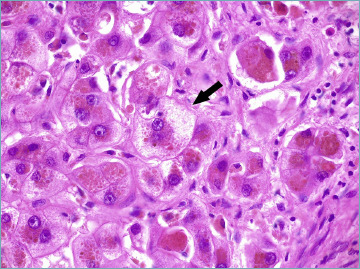
A liver biopsy from a primary biliary cholangitis patient showing diffuse hepatocellular cholestasis, associated with feathery degeneration of hepatocyte cytoplasm (arrow) (hematoxylin-eosin; original magnification 63x).
As the disease progresses, bile duct changes evolve, till only remnants of biliary epithelium can be visible or identifiable by immunohistochemical stain for cytokeratin (CK) 7. In the end, bile duct remnants disappear and only lymphocytic aggregates or granulomas persist. The smallest branches are always the first to disappear. Since the bile ducts typically run parallel to hepatic artery branches, the finding of unpaired arteries is a presumptive evidence of vanishing bile ducts and it is useful in the routine practice to evaluate the extent of bile duct loss (BDL), a parameter included in the current staging system 6 (see below). Persistent ductal and portal inflammation and bile duct damage lead to ductopenia (Fig. 7), periportal or diffuse metaplastic hepatocytes (Fig. 8), and portal and periportal collagen deposition, eventually with bridging fibrosis and cirrhosis development 10,20 (Fig. 9). The biliary-type fibrosis is dense, scar-like, with edema at the periphery, giving rise to the typical halo effect 6.
Figure 7.
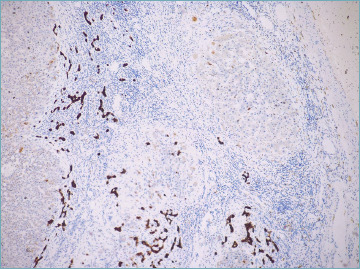
Ductopenia in a patient with advanced primary biliary cholangitis. Cytokeratin 7 stain shows diffuse ductular reaction, with no native bile ducts left (cytokeratin 7 immunostain; original magnification 5x).
Figure 8.
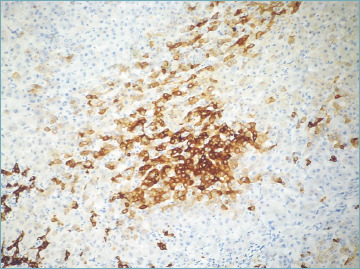
Cytokeratin 7 stain highlights biliary metaplasia of hepatocytes (cytokeratin 7 immunostain; original magnification 10x).
Figure 9.
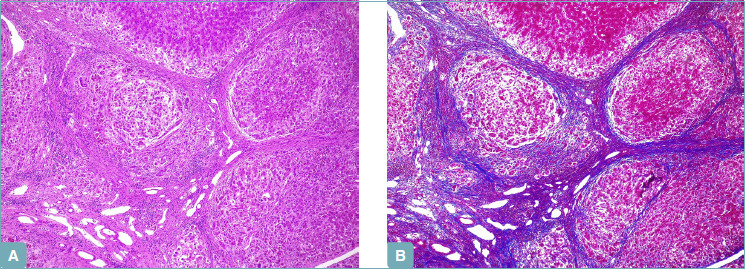
Biliary-type cirrhosis is characterized by parenchymal nodules with edema at the periphery, giving rise to the so-called halo effect (A). Trichrome stain shows complete dense fibrous septa, and better highlights the halo effect at the parenchyma-septum border (B) (A: hematoxylin-eosin; original magnification 5x; B: Masson’s trichrome stain; original magnification 5x).
A premature ductopenic variant has been described in about 5-10% of patients, and it is characterized by a rapid onset of BDL in the absence of significant fibrosis, icteric cholestasis, and a rapid progression towards cirrhosis 3,30.
Foci of concentric periductal fibrosis may be seen in about 20% of PBC patients, and may cause problems in the differential diagnosis with PSC. A cholangiogram is necessary in such cases, since extrahepatic bile ducts are healthy in PBC 6.
It has been demonstrated that the canals of Hering, which connect bile canaliculi to the interlobular bile ducts and are identifiable by CK19 immunostaining in normal liver, are decreased in number in all stages of PBC, suggesting that they are destroyed together with small bile ducts 31. A study by Khan et al. found that this loss may be a very early manifestation of PBC that precedes BDL and can be observed in liver biopsies otherwise normal or nearly normal, defining the so-called ‘minimal change’ PBC 32 (Fig. 10). However, the role of this lesion in the natural history of PBC is still debated 31,33.
Figure 10.
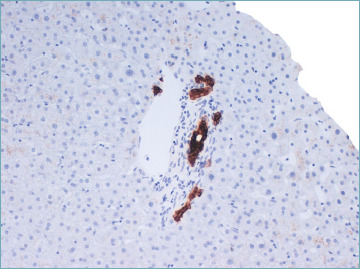
Minimal change primary biliary cholangitis. Cytokeratin 19 immunostaining shows a decreased number of canals of Hering in an otherwise normal portal tract (cytokeratin 19 immunostain; original magnification 20x).
In patients with portal hypertension, a reticulin stain can exclude the presence of nodular regenerative hyperplasia, a known complication of non-cirrhotic PBC 10,34. Moreover, in non-cirrhotic PBC stages, small-cell change of periportal hepatocytes may occur, but it is considered a regenerative phenomenon rather than a pre-malignant lesion 6,35.
STAGING SYSTEMS
Many histological staging systems for PBC have been developed over time, including the Ludwig and Scheuer’s staging systems, which were widely used in the past 36,37 (Tab. I). Recently, a new approach to stage PBC has been proposed by Nakanuma et al., who also introduced the concept of PBC grading. This scoring system demonstrated the strongest prognostic value compared to previous methods, with a more accurate prediction of patient outcome at 10 years, particularly in the development of cirrhosis and its complications 38,39. Therefore, the use of Nakanuma’s staging system should replace the old systems in everyday routine. This system establishes the grade of the disease by evaluating chronic cholangitis activity and hepatitis activity, the latter determined by the presence and extent of interface and lobular hepatitis 38 (Tab. II, Fig. 11). On the other hand, the stage of the disease is determined by the evaluation of two or three criteria: the presence and extent of fibrosis, the presence and extent of BDL, and the presence and extent of orcein-positive granule deposition (Tab. III, Fig. 12). The third criterion is not essential, since an accurate and reproducible staging can be obtained even with the two-criteria method 38.
Table I.
Ludwig and Scheuer’s staging systems for PBC.
| Stage | Ludwig System | Scheuer System |
|---|---|---|
| I | Portal inflammation with bile duct damage, with or without florid duct lesions | Portal inflammation with bile duct damage, with or without florid duct lesions |
| II | Periportal inflammation/ Interface hepatitis |
Ductular reaction |
| III | Bridging fibrosis | Bridging fibrosis |
| IV | Biliary cirrhosis | Biliary cirrhosis |
Table II.
Grading of chronic cholangitis activity and hepatitis activity, according to Nakanuma’s system.
| Chronic cholangitis activity (CA) | |
|
CA 0
(no activity) CA 1 (mild activity) CA 2 (moderate activity) CA 3 (marked activity) |
No cholangitis, mild biliary epithelial damage 1 BD with evident chronic cholangitis ≥ 2 BDs with evident chronic cholangitis ≥ 1 florid duct lesion |
| Hepatitis activity (HA) | |
|
HA 0
(no activity) HA 1 (mild activity) HA 2 (moderate activity) HA 3 (marked activity) |
No IH and no or minimal LH IH in ≥ 10 continuous hepatocytes in 1 PT and mild to moderate LH IH in ≥ 10 continuous hepatocytes in ≥ 2 PTs and mild to moderate LH IH in ≥ 20 continuous hepatocytes in ≥ 1/2 PTs and moderate LH or bridging/zonal necrosis |
BD: bile duct; IH: interface hepatitis; LH: lobular hepatitis; PT: portal tract.
Figure 11.
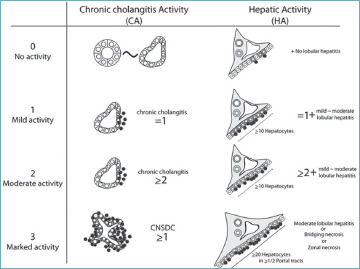
Representation of grading of cholangitis and hepatitis activity, according to Nakanuma’s system 38.
Table III.
Nakanuma’s staging system.
| Fibrosis | ||
|
Score 0 Score 1 Score 2 Score 3 |
No portal fibrosis or fibrosis limited to PTs Portal and periportal fibrosis ± fibrous septa Bridging fibrosis with variable lobular disarray Liver cirrhosis |
|
| Bile duct loss (BDL) | ||
|
Score 0 Score 1 Score 2 Score 3 |
No BDL BDL in <1/3 of PTs BDL in 1/3 to 2/3 of PTs BDL in >2/3 of PTs |
|
| Deposition of orcein-positive granules (DOG) | ||
|
Score 0 Score 1 Score 2 Score 3 |
No DOG DOG in a few periportal hepatocytes in <1/3 of PTs DOG in several periportal hepatocytes in 1/3 to 2/3 of PTs DOG in many hepatocytes in >2/3 of PTs |
|
| Stage | 3 criteria | 2 criteria |
|
Stage 1
(no progression) Stage 2 (mild progression) Stage 3 (moderate progression) Stage 4 (advanced progression) |
0 1-3 4-6 7-9 |
0 1-2 3-4 5-6 |
PT: portal tract.
Figure 12.
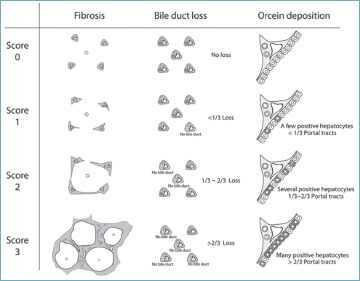
Representation of disease staging, according to Nakanuma’s system 38.
DIFFERENTIAL DIAGNOSES
Bile duct damage may be seen in inflamed portal tracts of patients with viral hepatitis, particularly in chronic hepatitis C, while lymphocytic cholangitis has also been described in viral hepatitis E. However, this does not represent a real problem in routine practice, since a diagnosis of viral hepatitis is easily obtained by serological tests, and a liver biopsy is nowadays rarely required in this setting.
Bile duct inflammatory changes are also seen in AIH, while interface hepatitis (the hallmark of AIH) and some degree of lobular necro-inflammation are common in PBC6. The most important clues for a correct differential diagnosis are: i) BDL and ductopenia are not typical AIH features, since the bile duct is not the target of the disease in AIH 40,41; ii) metaplastic changes of hepatocytes (easily highlighted by CK7 stain) and orcein periportal deposits are signs of chronic cholestasis, and are not observed in AIH specimens, unless extensive fibrosis or cirrhosis is present 42; iii) interface hepatitis is minimal to mild in PBC, and the moderate to severe necro-inflammatory lobular lesions typical of AIH are never present 43. Plasma cells may be numerous in both PBC and AIH, although the presence of plasma cell clusters (defined as collections of ≥ 5 contiguous plasma cells) in the lobule is considered highly sensitive for AIH 29,44.
ROLE OF LIVER BIOPSY IN PBC
As mentioned above, liver biopsy is no longer required for the diagnosis of PBC in most cases, i.e. when a cholestatic liver profile is associated with AMA positivity. As a general rule, liver biopsy should be performed only when the initial diagnostic workout is not able to identify the cause of cholestasis. Liver biopsy is clearly mandatory whenever AMA are not detected, and to rule out the diagnosis of AMA-negative PBC. As reported above, up to 10% of patients may fall into this category 9,10. Suspected PBC-AIH variant is a further indication for liver biopsy. In this setting, a histological diagnosis is mandatory since patients with this variant may benefit from immunosuppressive therapy 3. Again, liver biopsy is necessary whenever other co-existent liver diseases, particularly NASH, are suspected in patients with an established PBC diagnosis 9,10. Of note, NASH has become prevalent in the same population at risk for PBC, probably due to the pandemic diffusion of NASH risk factors. However, a link between NASH and PBC has not been yet demonstrated 45. Finally, liver biopsy remains the gold standard in assessing hepatic fibrosis, even if non-invasive methods are currently being used, with a good performance 10,19.
Primary sclerosing cholangitis
PSC is a chronic cholestatic liver disease in which inflammation and fibrosis lead to multifocal biliary strictures and progression to end-stage liver disease. Intra- and extrahepatic bile ducts are primarily affected. The close association with inflammatory bowel diseases (IBDs) is a hallmark of the condition, affecting about two-thirds of the patients 46,47.
Until the 1960s, most published articles about PSC were case reports, but implementation of endoscopic retrograde cholangiography in the 1970s led to an increasing recognition of the condition 46.
PSC is associated with a considerable risk of gastrointestinal malignancies, mainly cholangiocarcinoma and colorectal cancer 46. Disease progression and end-stage liver disease are inevitable in most patients, and OLT is the only curative treatment available, even if post-transplant recurrence may occur 46.
EPIDEMIOLOGY
PSC is a rare disease, as its incidence, although varies geographically, is as high as 1.3 per 100,000 people per year in Northern Europe. Prevalence is also variable, and it reaches 16.2 per 100,000 people in some studies. Both incidence and prevalence of the disease are increasing, but it is not clear whether the increase is real or due to a better detection of disease 47,48. Although PSC affects both sexes and all age groups, more than 60% of patients are males, and the median age at onset is 30-40 years. Patients are usually non-smokers, and about 2/3 have a coexistent IBD (75% ulcerative colitis) 47.
More than 50% of patients require OLT within 10-15 years of symptom development, as a result of reduced quality of life and increased disease-related complications and comorbidity 47.
OVERVIEW ON CLINICAL AND LABORATORY FEATURES
PSC is insidious, and about half of patients do not have symptoms. Abnormal liver function tests are often the only signs at diagnosis, while hepatomegaly is present in 44% of patients and splenomegaly in 39% of them. When symptoms are present, they include abdominal pain (20% of patients), pruritus (10%), jaundice (6%) and fatigue (6%) 48. Even if the actual risk of PSC in IBD patients is not yet known, probably no more than 10% of patients with colitis will develop PSC. About 10-20% of patients have dominant bile duct strictures and many of them have recurrent bacterial cholangitis. This subgroup of patients seems to have a worse survival and a higher risk of development of malignancies 47.
Typically, ALP is elevated (two to three times the upper limit of normal), along with aspartate and alanine aminotransferases. Different non-specific autoantibodies have been correlated with PSC, such as ANA, anti-cardiolipin autoantibodies, anti-thyroperoxidase autoantibodies, rheumatoid factor and anti-smooth muscle autoantibodies. Even if none is liver specific, their detection may suggest an underlying immune dysregulation 49.
PSC is suspected when increased ALP levels persist for more than 6 months, and diagnosis relies on cholangiographic findings of bile-duct strictures detected by either magnetic resonance cholangio-pancreatography or endoscopic retrograde cholangio-pancreatography. Exclusion of causes of secondary sclerosing cholangitis is mandatory to diagnose PSC, and include biliary stones, cholangiocarcinoma, biliary tract surgery, Caroli’s disease, chronic biliary infection, biliary toxin exposure, chronic portal-vein thrombosis, ischemic stricturing and liver diseases that can cause biliary injury, such as cholestatic drug-induced liver injury 47,50,51.
Magnetic resonance elastography and transient liver elastography are non-invasive diagnostic tools to assess liver fibrosis, even if their role in staging PSC is still under debate 48,50-52.
A variety of coexisting conditions are associated with PSC. Due to the frequent association with IBDs, colonoscopy is warranted in all patients who receive a new PSC diagnosis, since the risk of colon cancer among patients with PSC and concomitant IBD is 4 times higher than the risk among patients with IBD alone and 10 times higher than the risk in the general population 47,48. Gallbladder diseases (stones, polyps, and cancer) are also common in patients with PSC 48.
In developed countries, PSC is the most common risk factor for cholangiocarcinoma, which is 400 times more frequent than in the general population. Among PSC patients, the annual risk of cholangiocarcinoma is 2%, and the 30-year cumulative incidence is 20%. Therefore, annual ultrasonography of the liver and serum testing for CA19-9 are recommended 48.
PSC is a slowly progressive disease, with variable outcomes 48. Several attempts to develop a PSC-specific prognostic model have been made over time 46. The PSC specific revised Mayo risk score and the Amsterdam-Oxford Model are the most widely used models 48,53,54.
Caring for patients with PSC is challenging and requires treatment of the primary liver disease and the coexisting conditions, as well as subsequent therapy for potential complications of end-stage liver disease 48,55. No effective medical therapies exist for PSC. Treatment guidelines for PSC are conflicting: some of them support the use of UDCA, whereas others endorse the use of only moderate doses 46,48. Several new treatments, such as obeticholic acid and simtuzumab, are under investigation and clinical trials are ongoing 48,55. Patients with both PSC and IBD should be treated according to relevant guidelines. Annual colonoscopy with surveillance biopsies should be performed even in patients who underwent OLT, given the constant increased risk of colon cancer. PSC patients without IBD should undergo colonoscopy every 5 years. An annual ultrasonography for the evaluation of the gallbladder is also recommended. Because of the risk of cancer, patients with gallbladder masses of any size should undergo cholecystectomy 48.
Given the progressive nature of PSC, 40% of patients will eventually require OLT. The 1-year and 5-year survival rates are 85% and 72%. However, the disorder may recur in 25% of cases 48.
Small duct PSC (sdPSC) is a rare PSC variant (3-5% of patients) that affects only small intrahepatic bile ducts, sparing the large ones. It is characterized by clinical and histological features of classical large duct PSC, lacking typical cholangiographic changes in extra- and/or intrahepatic bile ducts (normal cholangiographic findings). In a large clinical PSC study, sdPSC was associated with a better prognosis compared to large duct PSC, with a lower risk of OLT, liver-related death and hepato-biliary malignancy. Nevertheless, sdPSC may progress to large duct PSC, developing typical cholangiographic lesions 3,47,48.
PATHOGENESIS
The pathogenesis of PSC is still unknown, but several mechanistic theories have been proposed, and both genetic and environmental factors seem to be involved in disease initiation 46.
Different genes have been studied, and a strong association with HLA alleles has been described 48. Several different possible environmental triggers have been investigated over time, such as exposure to farm animals during childhood, use of contraceptive hormones, urinary tract infections, dietary intake and methods of food preparation, but none was clearly associated with disease development. Of interest, smoking appears to be protective 48.
The strong association between PSC and IBDs led to the “microbiota hypothesis”, supported by observations both in vitro and in animal models. According to this hypothesis, microbial molecules from intestinal dysbiosis reach the liver through the portal circulation and initiate an aberrant cholangiocytic response, including the induction of senescence 23,48,56,57. An additional theory regarding PSC pathogenesis is the “gut lymphocyte homing” hypothesis. It is supposed that intestinal T cells are stimulated within intestine-associated lymphoid tissue and then recruited to the liver thanks to an abnormal expression of adhesion molecules on periportal endothelial cells. Once in the liver, activated T cells from the intestine can initiate immune-mediated damage 23,48.
It is also believed that cholangiocytes themselves may be actively involved in the pathogenesis of PSC, by secreting pro-inflammatory cytokines and mediating recruitment and stimulation of T cells or acquiring a senescent phenotype 23,24,48.
HISTOLOGY
The presence of an inflammatory infiltrate in a large intra- and extra-hepatic bile duct wall, with an obliterative concentric periductal loose fibrosis (so called onion skin fibrosis, Fig. 13), is traditionally considered the pathological hallmark of PSC. Cholangiocytes show features of degeneration and atrophy. The lesion leads to biliary strictures and eventually occlusions (so called bile duct scars, Fig. 14) 6,47,58.
Figure 13.
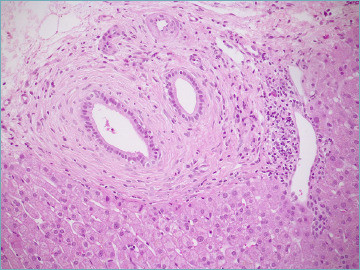
Large intrahepatic bile ducts showing obliterative concentric periductal fibrosis, also called onion skin fibrosis, in a liver biopsy from a primary sclerosing cholangitis patient (hematoxylin-eosin; original magnification 20x).
Figure 14.
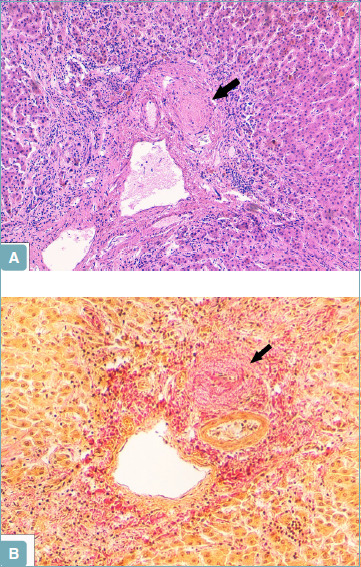
A portal tract showing a bile duct scar (arrow) in a liver biopsy from a primary sclerosing cholangitis patient (A). Van Gieson stain confirms that the bile duct has been replaced by fibrous tissue (arrow) (B) (A: hematoxylin-eosin; original magnification 10x; B: Van Gieson stain; original magnification 20x).
Since needle liver biopsies usually include only the small most peripheral portal tracts, the histologic appearance of PSC varies from normal liver tissue to the presence of only indirect signs of large duct obstruction. Normal tissue may be seen at very early stages, and it does not preclude the diagnosis of PSC if clinical and radiologic findings are otherwise typical. In early disease, the changes are confined to portal tracts, with a mild mixed inflammatory cell infiltrate of lymphocytes, plasma cells and neutrophils, usually more intense around bile ducts. Lymphoid follicles or aggregates may be present 6.
The presence of bile duct fibrotic lesions, accompanied by reduced numbers of interlobular bile ducts, is virtually diagnostic of PSC, but it is present in less than 20% of biopsy specimens 6. The absence of typical onion skin fibrosis does not exclude PSC diagnosis. Of note, although a typical sign of PSC, onion skin fibrosis may be also seen in secondary sclerosing cholangitis.
A significant bile duct basement membrane thickening, displayed with a periodic acid-Schiff stain with diastase, has been recently reported in PSC specimens (Fig. 15). It demonstrated a specificity of 95% and a good agreement among pathologists, making it an important diagnostic tool, particularly in sdPSC 59.
Figure 15.
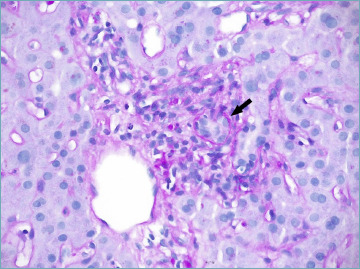
Periodic acid-Schiff stain with diastase shows a moderate bile duct basement membrane thickening (arrow) in primary sclerosing cholangitis (PAS-diastase stain; original magnification 40x).
Recent studies in end-stage PSC livers reported vascular lesions, particularly enlarged arterioles with thickened walls and arterial fibrointimal hyperplasia (Fig. 16), but the functional significance of these changes in the pathogenesis of PSC is unknown 60,61. Persistent portal inflammation, bile duct destruction and periportal fibrosis lead to BDL, disorganized ductular reaction, periportal or diffuse hepatocyte metaplasia, bridging fibrosis and cirrhosis, as in PBC. Histologically, inflammation and fibrogenesis may not be closely associated, and the risk of biliary dysplasia and malignancy seems to be unrelated to disease duration or biliary fibrosis severity 47,58. Histologic changes are unevenly distributed in the liver, and this may explain the high degree of sampling variability in needle and wedge liver biopsies 6. Generally, the pattern of cirrhosis in PSC is more irregular than in PBC, reflecting the larger caliber of the involved ducts, and segmental cirrhosis or hemi-cirrhosis are occasionally observed 6.
Figure 16.
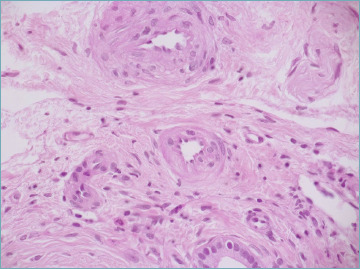
Enlarged arterioles with thickened wall and arterial fibrointimal hyperplasia are common findings in primary sclerosing cholangitis (hematoxylin-eosin; original magnification 40x).
The histologic distinction between PSC and PBC can be difficult, particularly in liver biopsy. However, cholestasis-associated changes are more frequent and tend to appear earlier in PSC than in PBC, periductal fibrosis and bile duct scars favor PSC, while granulomas are most commonly found in PBC 6. As PBC, many histologic staging systems have been developed in the past. However, the new grading and staging system proposed by Nakanuma et al. for PBC has been tested also in PSC patients, and demonstrated the strongest prognostic value compared to previous methods, even in this scenario 38,62 (see above).
ROLE OF LIVER BIOPSY IN PSC
Magnetic resonance cholangio-pancreatography or endoscopic retrograde cholangio-pancreatography are the gold standard for PSC diagnosis, and liver biopsy is not indicated when cholangiographic findings are typical. However, liver biopsy retains an important diagnostic role whenever a high clinical suspicion for PSC is associated to a normal cholangiogram. In such cases, a diagnosis of sdPSC relies on histologic findings. Moreover, a liver biopsy is mandatory whenever a PSC-AIH variant or the presence of other comorbidities are clinically suspected, as in PBC 48,50-52.
“Overlap” variants
PBC-AIH VARIANT
About 10-20% of PBC patients may present AIH, either simultaneously or consecutively, and a small proportion of patients with well-established AIH may develop PBC 3,9. This condition was formerly called “PBC-AIH overlap syndrome”, but the use of the term “overlap” has been discouraged, since not all the patients concurrently present both diseases at the same time, and replaced with “variant”, which seems more proper to describe all the possible clinical scenarios 3,63. However, the term “overlap” is still very popular in the routine practice.
PBC-AIH variant diagnosis is challenging in everyday routine, and no updated consensus criteria for its definition exist. Yet, in clinical practice, the Paris criteria are still in use (Table IV), since previous studies demonstrated a good sensitivity (92%) and specificity (97%) for diagnosing this variant 3,9,64,65. However, these criteria were published 20 years ago, and seem to be very strict and largely different from the separate diagnostic criteria of each disease, with a consequent underestimation of affected patients 3,9. For this reason, changes in the criteria have been proposed, but still not universally approved 3,66,67.
Table IV.
Paris criteria for PBC-AIH variant diagnosis.
| At least 2 out of 3 criteria for each disease | |
|---|---|
| Primary biliary cholangitis | Autoimmune hepatitis |
| ALP > 2x ULN or γGT > 5x ULN | ALT > 5x ULN |
| AMA > 1:40 | IgG serum levels > 2x ULN or ASMA positivity |
| Florid duct lesions on histology | Moderate or severe IH on histology |
ALP: alkaline phosphatase; ULN: upper limit of normal; γGT: γ-glutamyltranspeptidase; AMA: anti-mitochondrial antibodies; ALT: alanine aminotransferase; ASMA: anti-smooth muscle antibodies; IH: interface hepatitis.
Liver biopsy is a fundamental tool in the diagnosis of the PBC-AIH variant, and all national and international guidelines recommend it 3,9,14,68. Of note, specimens should be evaluated by an expert liver pathologist, since there are important therapeutic implications 3. In fact, PBC-AIH variant should not be overdiagnosed to avoid side effects from immunosuppression in PBC patients, though neither underestimated, to correctly identify PBC patients who would benefit from immunosuppressive treatment. The typical histologic picture consists of PBC bile duct lesions in a classical AIH background. However, histologic biliary damage in AIH has been reported in up to 24-60% of well-defined AIH patients, with destructive cholangitis being present in 3-7% 40. Moreover, a recent study reported that bile duct injury and ductular reaction is a frequent phenomenon in AIH and it does not always point toward a PBC-AIH variant 41. Thus, some degree of biliary involvement in AIH does not necessarily lead to a change in the diagnosis. Nevertheless, finding histologic features of chronic cholestasis, such as copper deposition, CK7-positive periportal hepatocytes, and ductopenia in AIH specimens without bridging fibrosis, is not typical and should warrant the hypothesis of a PBC-AIH variant 42. Still, a variable degree of inflammation is commonly present in PBC alone, but interface hepatitis and hepatocellular damage is typically minimal, while acidophil bodies are few or absent 43. Thus, in PBC patients with moderate/severe interface hepatitis, the hypothesis of a PBC-AIH variant should be taken into account, and treatment with immunosuppressive agents should be considered 9. The immunohistochemical assessment of IgG and IgM has been proposed to help in the differential diagnosis between AIH, PBC and PBC-AIH variant, but with conflicting results 69,70.
A reduced 5 year event-free survival in PBC-AIH variant patients, compared to PBC patients (56% vs 81%) 9,71 has been reported.
The therapeutic approach to PBC-AIH variant consists in treating both diseases with a combination of UDCA and immunosuppression, since combination therapy proved more effective than UDCA alone in slowing fibrosis progression 3,72,73.
PSC-AIH VARIANT
As for PBC-AIH variant, the previously used term “overlap” has been abandoned, since PSC-AIH variant reflects a PSC subphenotype instead of a separate clinical entity 3,63,74. The PSC-AIH variant is present in 6-14% of PSC patients, and seem to have less concurrent ulcerative colitis 3,75.
As previously stated, no defined diagnostic criteria exist 3. In clinical practice, PSC-AIH variant diagnosis is based on typical cholangiographic or histologic characteristics of PSC in combination with AIH features 3. In the absence of an abnormal cholangiogram, a typical histological AIH picture with periductal inflammation and fibro-obliterative, onion skin-type, fibrosis is suggestive of a concomitant sdPSC, while the additional loss of interlobar ducts makes the whole scenario diagnostic of a sdPSC-AIH variant, which represents about 16% of the cases 3,76.
A different specific variant, called autoimmune sclerosing cholangitis, has been described in at least 50% of children and adolescents with well-established AIH and sclerosing cholangitis manifestations 3,77,78.
Patients with PSC-AIH variant have a similar risk of liver disease progression compared to those with classic PSC, but they seem to have a lower risk of hepato-biliary malignancy development 3,75.
In PSC-AIH variant, treatment is based on a combination of immunosuppression and UDCA, just like in the PBC-AIH variant 3.
PBC-PSC VARIANT
This variant is exceptionally uncommon, and less than 10 cases have been described in the literature, so far 3,79,80. It consists in a syndrome with clinical and laboratory features of both PBC and PSC, including the sdPSC variant. Because of its rarity, very few data are available, and its existence is still debated 3,80,81.
Conclusions
The role of histology in diagnosing PBC and PSC has changed profoundly in the last 20 years, paralleling a better standardization of clinical, laboratory and imaging diagnostic criteria. Liver biopsy is not currently part of the routine workup in diagnosing PBC and PSC, but it retains a crucial role when patients have an “atypical” presentation or when non-invasive tests fail to reach a definite diagnosis. It is therefore crucial for pathologists to become familiar with histologic lesions of PBC and PSC.
As a general rule, liver biopsy interpretation should always consider the clinical scenario, since different diseases share similar histologic changes. It is of paramount importance for the pathologist to be familiar with clinical and laboratory features of patients with PBC and PSC, in order to reach a correct and clinically useful diagnosis.
Figures and tables
References
- 1.Lazaridis KN, LaRusso NF. The cholangiopathies. Mayo Clin Proc 2015;90:791-800. https://doi.org/10.1016/j.mayocp.2015.03.017 10.1016/j.mayocp.2015.03.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lazaridis KN, Strazzabosco M, Larusso NF. The cholangiopathies: disorders of biliary epithelia. Gastroenterology 2004;127:1565-1577. https://doi.org/10.1053/j.gastro.2004.08.006 10.1053/j.gastro.2004.08.006 [DOI] [PubMed] [Google Scholar]
- 3.Dalekos GN, Gatselis NK. Variant and specific forms of autoimmune cholestatic liver diseases. Arch Immunol Ther Exp (Warsz) 2019;67:197-211. https://doi.org/10.1007/s00005-019-00550-9 10.1007/s00005-019-00550-9 [DOI] [PubMed] [Google Scholar]
- 4.Harada K, Sato Y, Itatsu K, et al. Innate immune response to double-stranded RNA in biliary epithelial cells is associated with the pathogenesis of biliary atresia. Hepatology 2007;46:1146-1154. https://doi.org/10.1002/hep.21797 10.1002/hep.21797 [DOI] [PubMed] [Google Scholar]
- 5.Nakanuma Y, Zen Y. Pathology and immunopathology of immunoglobulin G4-related sclerosing cholangitis: the latest addition to the sclerosing cholangitis family. Hepatol Res 2007;37:S478-486. https://doi.org/10.1111/j.1872-034X.2007.00243.x 10.1111/j.1872-034X.2007.00243.x [DOI] [PubMed] [Google Scholar]
- 6.Zen Y, Hübscher SG, Nakanuma Y. Bile duct diseases. Burt AD, Ferrell LD, Hübscher SG, eds. MacSween’s pathology of the liver. 7th ed. Philadelphia, PA: Elsevier; 2018, p. 515. [Google Scholar]
- 7.Zhang W, Sharma R, Ju ST, et al. Deficiency in regulatory T cells results in development of antimitochondrial antibodies and autoimmune cholangitis. Hepatology 2009;49:545-552. https://doi.org/10.1002/hep.22651 10.1002/hep.22651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sasaki M, Ikeda H, Sawada S, et al. Naturally-occurring regulatory T cells are increased in inflamed portal tracts with cholangiopathy in primary biliary cirrhosis. J Clin Pathol 2007;60:1102-1107. https://doi.org/10.1136/jcp.2006.044776 10.1136/jcp.2006.044776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.European Association for the Study of the Liver. EASL Clinical practice guidelines: the diagnosis and management of patients with primary biliary cholangitis. J Hepatol 2017;67:145-172. https://doi.org/10.1016/j.jhep.2017.03.022 10.1016/j.jhep.2017.03.022 [DOI] [PubMed] [Google Scholar]
- 10.Carey EJ, Ali AH, Lindor KD. Primary biliary cirrhosis. Lancet 2015;386:1565-1575. https://doi.org/10.1016/S0140-6736(15)00154-3 10.1016/S0140-6736(15)00154-3 [DOI] [PubMed] [Google Scholar]
- 11.Chalifoux SL, Konyn PG, Saab S, et al. Extrahepatic manifestations of primary biliary cholangitis. Gut Liver 2017:11;771-780. https://doi.org/10.5009/gnl16365 10.5009/gnl16365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Floreani A, Cazzagon N. PBC and related extrahepatic diseases. Best Pract Res Clin Gastroenterol 2018;34-35:49-54. https://doi.org/10.1016/j.bpg.2018.05.013 10.1016/j.bpg.2018.05.013 [DOI] [PubMed] [Google Scholar]
- 13.Harms MH, van Buuren HR, van der Meer AJ, et al. Ursodeoxycholic acid therapy and liver transplant-free survival in patients with primary biliary cholangitis. J Hepatol 2019;71:357-365. https://doi.org/10.1016/j.jhep.2019.04.001 10.1016/j.jhep.2019.04.001 [DOI] [PubMed] [Google Scholar]
- 14.Hirschfield GM, Dyson JK, Jones DEJ, et al. The British Society of Gastroenterology/UK-PBC primary biliary cholangitis treatment and management guidelines. Gut 2018;67:1568-1594. https://doi.org/10.1136/gutjnl-2017-315259 10.1136/gutjnl-2017-315259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Floreani A, Mangini C. Primary biliary cholangitis: old and novel therapy. Eur J Intern Med 2018;47:1-5. https://doi.org/10.1016/j.ejim.2017.06.020 10.1016/j.ejim.2017.06.020 [DOI] [PubMed] [Google Scholar]
- 16.Gao L, Wang L, Gershwin ME, et al. Clinical management of primary biliary cholangitis - Strategies and evolving trends. Clin Rev Allergy Immunol 2020;59:175-194. https://doi.org/10.1007/s12016-019-08772-7 10.1007/s12016-019-08772-7 [DOI] [PubMed] [Google Scholar]
- 17.Lammers WJ, Hirschfield GM, Corpechot C, et al. on behalf of the Global PBC Study Group . Development and validation of a scoring system to predict outcomes of patients with primary biliary cirrhosis receiving ursodeoxycholic acid therapy. Gastroenterology 2015;149:1804-1812.e4. https://doi.org/10.1053/j.gastro.2015.07.061 10.1053/j.gastro.2015.07.061 [DOI] [PubMed] [Google Scholar]
- 18.Carbone M, Sharp SJ, Flack S, et al. on behalf of the UK-PBC Consortium . The UK-PBC risk scores: derivation and validation of a scoring system for long-term prediction of end-stage liver disease in primary biliary cholangitis. Hepatology 2016;63:930-950. https://doi.org/10.1002/hep.28017 10.1002/hep.28017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Alvaro D, Carpino G, Invernizzi P, et al. Primary biliary cholangitis management: controversies, perspectives and daily practice implications from an expert panel. Liver Int 2020;40:2590-2601. https://doi.org/10.1111/liv.14627 10.1111/liv.14627 [DOI] [PubMed] [Google Scholar]
- 20.Tsuneyama K, Baba H, Ogawa H, et al. Primary biliary cholangitis: its pathological characteristics and immunopathological mechanisms. J Med Invest 2017;64:7-13. https://doi.org/10.2152/jmi.64.7 10.2152/jmi.64.7 [DOI] [PubMed] [Google Scholar]
- 21.Terziroli Beretta-Piccoli B, Mieli-Vergani G, Gershwin ME, et al. The challenges of primary biliary cholangitis: What is new and what needs to be done. J Autoimmun 2019;105:102328. https://doi.org/10.1016/j.jaut.2019.102328 10.1016/j.jaut.2019.102328 [DOI] [PubMed] [Google Scholar]
- 22.Yokoda RT, Rodriguez EA. Review: Pathogenesis of cholestatic liver diseases. World J Hepatol 2020;12:423-435. https://doi.org/10.4254/wjh.v12.i8.423 10.4254/wjh.v12.i8.423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yan C, Koda S, Wu J, et al. Roles of trained immunity in the pathogenesis of cholangiopathies: a therapeutic target. Hepatology 2020;72:1838-1850. https://doi.org/10.1002/hep.31395 10.1002/hep.31395 [DOI] [PubMed] [Google Scholar]
- 24.Banales JM, Huebert RC, Gores GJ, et al. Cholangiocyte pathobiology. Nat Rev Gastroenterol Hepatol 2019;16:269-281. doi:10.1038/s41575-019-0125-y 10.1038/s41575-019-0125-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ronca V, Mancuso C, Invernizzi P, et al. Immune system and cholangiocytes: a puzzling affair in primary biliary cholangitis. J Leukoc Biol 2020;108:659-671. https://doi.org/10.1002/JLB.5MR0320-200R 10.1002/JLB.5MR0320-200R [DOI] [PubMed] [Google Scholar]
- 26.Rubin E, Schaffner F, Popper H. Primary biliary cirrhosis: chronic non-suppurative destructive cholangitis. Am J Pathol 1965;46:387-407. [PMC free article] [PubMed] [Google Scholar]
- 27.Nakanuma Y, Harada K. Florid duct lesion in primary biliary cirrhosis shows highly proliferative activities. J Hepatol 1993;19:216-221. https://doi.org/10.1016/s0168-8278(05)80574-4 10.1016/s0168-8278(05)80574-4 [DOI] [PubMed] [Google Scholar]
- 28.Carpino G, Cardinale V, Gaudio E, et al. Hepatic stem/progenitor cell activation differs between primary sclerosing and primary biliary cholangitis. Am J Pathol 2018;188:627-639. https://doi.org/10.1016/j.ajpath.2017.11.010 10.1016/j.ajpath.2017.11.010 [DOI] [PubMed] [Google Scholar]
- 29.Takahashi T, Miura T, Nakamura J, et al. Plasma cells and the chronic nonsuppurative destructive cholangitis of primary biliary cirrhosis. Hepatology 2012; 55:846-855. https://doi.org/10.1002/hep.24757 10.1002/hep.24757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nakanuma Y, Hoso M, Mizuno Y, et al. Pathologic study of primary biliary cirrhosis of early histologic stages presenting cholestatic jaundice. Liver 1988;8:319-324. https://doi.org/10.1111/j.1600-0676.1988.tb01010.x 10.1111/j.1600-0676.1988.tb01010.x [DOI] [PubMed] [Google Scholar]
- 31.Theise ND, Crawford JM, Nakanuma Y, et al. Canal of Hering loss is an initiating step for primary biliary cholangitis (PBC): a hypothesis. Med Hypotheses 2020;140:109680. https://doi.org/10.1016/j.mehy.2020.109680 10.1016/j.mehy.2020.109680 [DOI] [PubMed] [Google Scholar]
- 32.Khan FM, Komarla AR, Mendoza PG, et al. Keratin 19 demonstration of canal of Hering loss in primary biliary cirrhosis: “minimal change PBC”? Hepatology 2013;57:700-707. https://doi.org/10.1002/hep.26020 10.1002/hep.26020 [DOI] [PubMed] [Google Scholar]
- 33.Kakuda Y, Harada K, Nakanuma Y. Canals of Hering loss relates to the progression of the histological stages of primary biliary cirrhosis. J Clin Pathol 2015;68:141-147. https://doi.org/10.1136/jclinpath-2014-202417 10.1136/jclinpath-2014-202417 [DOI] [PubMed] [Google Scholar]
- 34.Colina F, Pinedo F, Nevado M, et al. Nodular regenerative hyperplasia of the liver in early histological stages of primary biliary cirrhosis. Gastroenterology 1992;102:1319-1324. [PubMed] [Google Scholar]
- 35.Nakanuma Y, Hirata K. Unusual hepatocellular lesions in primary biliary cirrhosis resembling but unrelated to hepatocellular neoplasms. Virchows Arch A Pathol Anat Histopathol 1993;422:17-23. https://doi.org/10.1007/BF01605128 10.1007/BF01605128 [DOI] [PubMed] [Google Scholar]
- 36.Ludwig J, Dickson ER, McDonald GS. Staging of chronic nonsuppurative destructive cholangitis (syndrome of primary biliary cirrhosis). Virchows Arch A Pathol Anat Histol 1978;379:103-112. https://doi.org/10.1007/BF00432479 10.1007/BF00432479 [DOI] [PubMed] [Google Scholar]
- 37.Scheuer P. Primary biliary cirrhosis. Proc R Soc Med 1967;60:1257-1260 [PMC free article] [PubMed] [Google Scholar]
- 38.Harada K, Hsu M, Nakanuma Y, et al. Application and validation of a new histologic staging and grading system for primary biliary cirrhosis. J Clin Gastroenterol 2013;47:174-181. https://doi.org/10.1097/MCG.0b013e31827234e4 10.1097/MCG.0b013e31827234e4 [DOI] [PubMed] [Google Scholar]
- 39.Kakuda Y, Harada K, Sawada-Kitamura S, et al. Evaluation of a new histologic staging and grading system for primary biliary cirrhosis in comparison with classical systems. Hum Pathol 2013;44:1107-1117. https://doi.org/10.1016/j.humpath.2012.09.017 10.1016/j.humpath.2012.09.017 [DOI] [PubMed] [Google Scholar]
- 40.Czaja AJ, Carpenter HA. Autoimmune hepatitis with incidental histologic features of bile duct injury. Hepatology 2001;34:659-665. https://doi.org/10.1053/jhep.2001.27562 10.1053/jhep.2001.27562 [DOI] [PubMed] [Google Scholar]
- 41.Verdonk RC, Lozano MF, Gouw ASH, et al. Bile ductal injury and ductular reaction are frequent phenomena with different significance in autoimmune hepatitis. Liver Int 2016;36:1362-1369. https://doi.org/10.1111/liv.13083 10.1111/liv.13083 [DOI] [PubMed] [Google Scholar]
- 42.Balitzer D, Shafizadeh N, Kakar S, et al. Autoimmune hepatitis: review of histologic features included in the simplified criteria proposed by the international autoimmune hepatitis group and proposal for new histologic criteria. Mod Pathol 2017;30:773-783. https://doi.org/10.1038/modpathol.2016.267 10.1038/modpathol.2016.267 [DOI] [PubMed] [Google Scholar]
- 43.Goet JC, Harms MH, Hansen BE, et al. Risk stratification and prognostic modelling in primary biliary cholangitis. Best Pract Res Clin Gastroenterol 2018;34-35:95-106. https://doi.org/10.1016/j.bpg.2018.06.006 10.1016/j.bpg.2018.06.006 [DOI] [PubMed] [Google Scholar]
- 44.Kobayashi M, Kakuda Y, Harada K, et al. Clinicopathological study of primary biliary cirrhosis with interface hepatitis compared to autoimmune hepatitis. World J Gastroenterol 2014;20:3597-3608. https://doi.org/10.3748/wjg.v20.i13.3597 10.3748/wjg.v20.i13.3597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tan D, Goodman ZD. Liver biopsy in primary biliary cholangitis: indications and interpretation. Clin Liver Dis 2018;22:579-588. https://doi.org/10.1016/j.cld.2018.03.008 10.1016/j.cld.2018.03.008 [DOI] [PubMed] [Google Scholar]
- 46.Karlsen TH, Folseraas T, Vesterhus M, et al. Primary sclerosing cholangitis - a comprehensive review. J Hepatol 2017;67:1298-1323. https://doi.org/10.1016/j.jhep.2017.07.022 10.1016/j.jhep.2017.07.022 [DOI] [PubMed] [Google Scholar]
- 47.Hirschfield GM, Karlsen TH, Adams DH, et al. Primary sclerosing cholangitis. Lancet 2013;382:1587-1599. https://doi.org/10.1016/S0140-6736(13)60096-3 10.1016/S0140-6736(13)60096-3 [DOI] [PubMed] [Google Scholar]
- 48.Lazaridis KN, LaRusso NF. Primary sclerosing cholangitis. N Engl J Med 2016;375:1161-1170. https://doi.org/10.1056/NEJMra1506330 10.1056/NEJMra1506330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Angulo P, Peter JB, Gershwin ME, et al. Serum autoantibodies in patients with primary sclerosing cholangitis. J Hepatol 2000;32:182-187. https://doi.org/10.1016/s0168-8278(00)80061-6 10.1016/s0168-8278(00)80061-6 [DOI] [PubMed] [Google Scholar]
- 50.Lindor KD, Kowdley KV, Harrison ME. ACG Clinical guideline: primary sclerosing cholangitis. Am J Gastroenterol 2015;110:646-659. https://doi.org/10.1038/ajg.2015.112 10.1038/ajg.2015.112 [DOI] [PubMed] [Google Scholar]
- 51.Chapman MH, Thorburn D, Hirschfield GM, et al. British Society of Gastroenterology and UK-PSC guidelines for the diagnosis and management of primary sclerosing cholangitis. Gut 2019;68:1356-1378. https://doi.org/10.1136/gutjnl-2018-317993 10.1136/gutjnl-2018-317993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Aabakken L, Karlsen TH, Hassan C, et al. Role of endoscopy in primary sclerosing cholangitis: European Society of Gastrointestinal Endoscopy (ESGE) and European Association for the Study of the Liver (EASL) clinical guideline. Endoscopy 2017;49:588-608. https://doi.org/10.1055/s-0043-107029 10.1055/s-0043-107029 [DOI] [PubMed] [Google Scholar]
- 53.Kim WR, Therneau TM, Dickson ER, et al. A revised natural history model for primary sclerosing cholangitis. Mayo Clin Proc 2000;75:688-694. https://doi.org/10.4065/75.7.688 10.4065/75.7.688 [DOI] [PubMed] [Google Scholar]
- 54.de Vries EM, Wang J, Ponsioen CY, et al. A novel prognostic model for transplant-free survival in primary sclerosing cholangitis. Gut 2018;67:1864-1869. https://doi.org/10.1136/gutjnl-2016-313681 10.1136/gutjnl-2016-313681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Iravani S, Dooghaie-Moghadam A, Razavi-Khorasani N, et al. An update on treatment options for primary sclerosing cholangitis. Gastroenterol Hepatol Bed Bench 2020;13:115-124. [PMC free article] [PubMed] [Google Scholar]
- 56.Dean G, Hanauer S, Levitsky J. The role of the intestine in the pathogenesis of primary sclerosing cholangitis: evidence and therapeutic implications. Hepatology 2020;72:1127-1138. https://doi.org/10.1002/hep.31311 10.1002/hep.31311 [DOI] [PubMed] [Google Scholar]
- 57.Shah A, Macdonald GA, Morrison M, et al. Targeting the gut microbiome as a treatment for primary sclerosing cholangitis: a conceptional framework. Am J Gastroenterol 2020;115:814-822. https://doi.org/10.14309/ajg.0000000000000604 10.14309/ajg.0000000000000604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Portmann B, Zen Y. Inflammatory disease of the bile ducts-cholangiopathies: liver biopsy challenge and clinicopathological correlation. Histopathology 2012;60:236-248. https://doi.org/10.1111/j.1365-2559.2011.03853.x 10.1111/j.1365-2559.2011.03853.x [DOI] [PubMed] [Google Scholar]
- 59.Colling R, Verrill C, Fleming K, et al. Bile duct basement membrane thickening in primary sclerosing cholangitis. Histopathology 2016;68:819-824. https://doi.org/10.1111/his.12857 10.1111/his.12857 [DOI] [PubMed] [Google Scholar]
- 60.Fiel MI, Sima HR, Schiano TD, et al. A morphometric study of the hepatic arterioles in end-stage primary sclerosing cholangitis. Virchows Arch 2015;466:143-149. https://doi.org/10.1007/s00428-014-1680-9 10.1007/s00428-014-1680-9 [DOI] [PubMed] [Google Scholar]
- 61.Carrasco-Avino G, Thomas D, Schiano TD, Fiel MI, et al. Primary sclerosing cholangitis: detailed histologic assessment and integration using bioinformatics highlights arterial fibrointimal hyperplasia as a novel feature. Am J Clin Pathol 2015;143:505-513. https://doi.org/10.1309/AJCPVKFVIPRBXQR2 10.1309/AJCPVKFVIPRBXQR2 [DOI] [PubMed] [Google Scholar]
- 62.de Vries EM, de Krijger M, Ponsioen CY, et al. Validation of the prognostic value of histologic scoring systems in primary sclerosing cholangitis: an international cohort study. Hepatology 2017;65:907-919. https://doi.org/10.1002/hep.28963 10.1002/hep.28963 [DOI] [PubMed] [Google Scholar]
- 63.Boberg KM, Chapman RW, Hirschfield GM, et al. International Autoimmune Hepatitis Group . Overlap syndromes: the International Autoimmune Hepatitis Group (IAIHG) position statement on a controversial issue. J Hepatol 2011;54:374-385. https://doi.org/10.1016/j.jhep.2010.09.002 10.1016/j.jhep.2010.09.002 [DOI] [PubMed] [Google Scholar]
- 64.Kuiper EMM, Zondervan PE, van Buuren HR. Paris criteria are effective in diagnosis of primary biliary cirrhosis and autoimmune hepatitis overlap syndrome. Clin Gastroenterol Hepatol 2010;8:530-534. https://doi.org/10.1016/j.cgh.2010.03.004 10.1016/j.cgh.2010.03.004 [DOI] [PubMed] [Google Scholar]
- 65.Chazouilleres O, Wendum D, Serfaty L, et al. Primary biliary cirrhosis-autoimmune hepatitis overlap syndrome: clinical features and response to therapy. Hepatology 1998;28:296-301. https://doi.org/10.1002/hep.510280203 10.1002/hep.510280203 [DOI] [PubMed] [Google Scholar]
- 66.Wang Q, Selmi C, Zhou X, et al. Epigenetic considerations and the clinical reevaluation of the overlap syndrome between primary biliary cirrhosis and autoimmune hepatitis. J Autoimmun 2013;41:140-145. https://doi.org/10.1016/j.jaut.2012.10.004 10.1016/j.jaut.2012.10.004 [DOI] [PubMed] [Google Scholar]
- 67.Zhang W, De D, Bacon BR, et al. New scoring classification for primary biliary cholangitis-autoimmune hepatitis overlap syndrome. Hepatol Commun 2018;2:245-253. https://doi.org/10.1002/hep4.1148 10.1002/hep4.1148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Dalekos GN, Koskinas J, Papatheodoridis GV. Hellenic Association for the Study of the Liver (HASL) clinical practice guidelines: autoimmune hepatitis. Ann Gastroenterol 2019;32:1-23. https://doi.org/10.20524/aog.2018.0330 10.20524/aog.2018.0330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lee H, Stapp RT, Shah VV, et al. The usefulness of IgG and IgM immunostaining of periportal inflammatory cells (plasma cells and lymphocytes) for the distinction of autoimmune hepatitis and primary biliary cirrhosis and their staining pattern in autoimmune hepatitis-primary biliary cirrhosis overlap syndrome. Am J Clin Pathol 2010;133:430-437. https://doi.org/10.1309/AJCPE93GZSHUNTAI 10.1309/AJCPE93GZSHUNTAI [DOI] [PubMed] [Google Scholar]
- 70.Abe K, Takahashi A, Nozawa Y, et al. The utility of IgG, IgM, and CD138 immunohistochemistry in the evaluation of autoimmune liver diseases. Med Mol Morphol 2014;47:162-168. https://doi.org/10.1007/s00795-014-0082-z 10.1007/s00795-014-0082-z [DOI] [PubMed] [Google Scholar]
- 71.Yang F, Wang O, Wang Z, et al. The natural history and prognosis of primary biliary cirrhosis with clinical features of autoimmune hepatitis. Clin Rev Allergy Immunol 2016;50:114-123. https://doi.org/10.1007/s12016-015-8516-5 10.1007/s12016-015-8516-5 [DOI] [PubMed] [Google Scholar]
- 72.Ozaslan E, Efe C, Heurgue-Berlot A, et al. Factors associated with response to therapy and outcome of patients with primary biliary cirrhosis with features of autoimmune hepatitis. Clin Gastroenterol Hepatol 2014;12:863-869. https://doi.org/10.1016/j.cgh.2013.09.021 10.1016/j.cgh.2013.09.021 [DOI] [PubMed] [Google Scholar]
- 73.Zhang H, Li S, Yang J, et al. A meta-analysis of ursodeoxycholic acid therapy versus combination therapy with corticosteroids for PBC-AIH-overlap syndrome: evidence from 97 monotherapy and 117 combinations. Prz Gastroenterol 2015;10:148-155. https://doi.org/10.5114/pg.2015.51187 10.5114/pg.2015.51187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Deneau MR, El-Matary W, Valentino PL, et al. The natural history of primary sclerosing cholangitis in 781 children: a multicenter, international collaboration. Hepatology 2017;66:518-527. https://doi.org/10.1002/hep.29204 10.1002/hep.29204 [DOI] [PubMed] [Google Scholar]
- 75.Weismüller TJ, Trivedi PJ, Bergquist A, et al. International PSC Study Group . Patient age, sex, and inflammatory bowel disease phenotype associate with course of primary sclerosing cholangitis. Gastroenterology 2017;152:1975-1984.e8. https://doi.org/10.1053/j.gastro.2017.02.038 10.1053/j.gastro.2017.02.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Olsson R, Glaumann H, Almer S, et al. High prevalence of small duct primary sclerosing cholangitis among patients with overlapping autoimmune hepatitis and primary sclerosing cholangitis. Eur J Intern Med 2009;20:190-196. https://doi.org/10.1016/j.ejim.2008.06.004 10.1016/j.ejim.2008.06.004 [DOI] [PubMed] [Google Scholar]
- 77.Gregorio GV, Portmann B, Karani J, et al. Autoimmune hepatitis/sclerosing cholangitis overlap syndrome in childhood: a 16-year prospective study. Hepatology 2001;33:544-553. https://doi.org/10.1053/jhep.2001.22131 10.1053/jhep.2001.22131 [DOI] [PubMed] [Google Scholar]
- 78.Rojas CP, Bodicharla R, Campuzano-Zuluanga G, et al. Autoimmune hepatitis and primary sclerosing cholangitis in children and adolescents. Fetal Pediatr Pathol 2014;33:202-209. https://doi.org/10.3109/15513815.2014.898721 10.3109/15513815.2014.898721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Oliveira EMG, Oliveira PM, Becker V, et al. Overlapping of primary biliary cirrhosis and small duct primary sclerosing cholangitis: first case report. J Clin Med Res 2012;4:429-433. https://doi.org/10.4021/jocmr1060w 10.4021/jocmr1060w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Floreani A, Motta R, Cazzagon N, et al. The overlap syndrome between primary biliary cirrhosis and primary sclerosing cholangitis. Dig Liver Dis 2015;47:432-435. https://doi.org/10.1016/j.dld.2015.02.002 10.1016/j.dld.2015.02.002 [DOI] [PubMed] [Google Scholar]
- 81.Sundaram S, K S, Mazumdar S, et al. Overlap syndrome between primary biliary cholangitis and primary sclerosing cholangitis. ACG Case Rep J 2018;5:e54. https://doi.org/10.14309/crj.2018.54 10.14309/crj.2018.54 [DOI] [PMC free article] [PubMed] [Google Scholar]