

Abstract
Cardiac fibrosis remains an unresolved problem in heart diseases. After initial injury, cardiac fibroblasts (CFs) are activated and subsequently differentiate into myofibroblasts (myoFbs) that are major mediator cells in the pathological remodeling. MyoFbs exhibit proliferative and secretive characteristics, and contribute to extracellular matrix (ECM) turnover, collagen deposition. The persistent functions of myoFbs lead to fibrotic scars and cardiac dysfunction. The anti-fibrotic treatment is hindered by the elusive mechanism of fibrosis and lack of specific targets on myoFbs. In this review, we will outline the progress of cardiac fibrosis and its contributions to the heart failure. We will also shed light on the role of myoFbs in the regulation of adverse remodeling. The communication between myoFbs and other cells that are involved in the heart injury and repair respectively will be reviewed in detail. Then, recently developed therapeutic strategies to treat fibrosis will be summarized such as i) chimeric antigen receptor T cell (CAR-T) therapy with an optimal target on myoFbs, ii) direct reprogramming from stem cells to quiescent CFs, iii) “off-target” small molecular drugs. The application of nano/micro technology will be discussed as well, which is involved in the construction of cell-based biomimic platforms and “pleiotropic” drug delivery systems.
Keywords: Cardiac fibrosis, Cardiac fibroblast, Myofibroblasts, Myocardial remodeling, Reprogramming, Drug delivery systems
1. Introduction
Cardiovascular diseases (CVDs) cause approximately 31% of all deaths worldwide [1], and cardiac fibrosis contributes to end-stage extracellular matrix (ECM) remodeling and heart failure [2]. Cardiac fibroblasts (CFs) are not only the source of ECM in healthy heart, they are also critical mediator cells in response to cardiac pathological changes. During injury, functional CFs differentiate into myofibroblasts (myoFbs) that are typical cells secreting contractile proteins [3]. In response to disease stimuli, myoFbs exhibit proliferative and invasive properties, and initiate the reparative would healing response. MyoFbs also remodel the interstitium by secreting ECM-degrading metalloproteinases (MMPs) and increasing collagen turnover. However, myoFbs show a persistent proliferation feature, which augments pro-inflammatory responses and causes collagen net formation [4].
The limited understanding of fibrosis is the cause of slow progress in anti-fibrosis therapies. Specifically, it is complicated to clarify the fibrotic mechanism since fibrosis occurs in various types of cardiac disorders. MyoFbs, the most specialized cells in fibrosis, not only come from resident CFs and endothelial cells, they also differentiate from circulating cells that migrate into the injured heart. The diversity of myoFb origination is a barrier for optimizing specific markers on this type of cell. Recently, stem cells have been propelled to the forefront of cardiac regeneration, presenting unexpected anti-fibrosis capabilities. Prescribed therapeutic agents for CVDs treatment such as Angiotensin II (Ang II) inhibitors have shown “off-target” effects on fibrosis alleviation by inhibiting the activation and proliferation of myoFbs [3]. However, new drug screening is difficult due to lack of cell models to mimic complex cellular interactions in vivo. Nano/micro-technology has been extensively used to deliver drugs, cells and genes via negative or positive targeting. Nano/micro-scale delivery systems such as polymeric nanoparticles and liposomes show multifunctional characteristics including bio-mimic, biocompatible and degradable natures [5–7]. Not confined to the heart, activated fibroblasts participate in fibrosis and adverse remodeling in many tissues like liver, lung and tumor, which allows for the development of fibroblast-targeted therapeutics [8–10]. For instance, nanoscale drug delivery systems have been developed to target tumor-associated fibroblasts for cancer therapy [11,12]. In addition, compelling evidence has shown enhanced targeting and retention efficacies of stem cells in cardiac tissues by nano/micro-technologies [13,14].
In this review, we will give a detailed insight into the progress of fibrosis, myoFb origination and differentiation, and myoFb-mediated fibrotic remodeling in CVDs. The communication between myoFbs and other cell types will also be discussed, elucidating potential approaches to fibrotic inhibition. The investigation of myoFb’s role in responding to disorder stimuli serves to highlight myoFb’s potential as a therapeutic target. Additionally, we will summarize emerging strategies for direct or indirect attenuation of fibrosis. For instance, a specific marker on myoFbs has been discovered, and the engineered chimeric antigen receptor T cell (CAR-T) has be used to target myoFbs and eliminate fibrosis [15]. Unlike previous cell reprogramming of CFs into cardiomyocytes, single cell ribonucleic acid (RNA) sequencing has been used to indicate a new method that induces stem cells into quiescent CFs for fibrosis treatment [16]. Notably, nano/micro-technology-mediated bio-mimic cell delivery systems will also be briefly discussed.
2. Cardiac fibrosis
2.1. Cardiac fibrosis characteristics
Cardiac fibrosis, a scar event in the cardiac muscle, occurs in almost all types of heart diseases including myocardial infarction (MI), hypertrophic cardiomyopathy, dilated cardiomyopathy, diabetic cardiomyopathy and aortic stenosis [17,18]. Turnover of ECM components plays a prominent role in fibrosis that is pathologically characterized by increased deposition of collagens (mainly type I and III) [19,20]. During the process of fibrosis, CF, the key cell type, becomes activated and differentiate into the myoFb [21]. This pathological remodeling results in increased matrix stiffness and abnormal cardiac function, leading to heart failure with reduced ejection fraction [22]. Cardiac fibrosis also causes heart failure with preserved ejection fraction since fibrosis causes myocardial stiffness, impacting filling capacity of the heart and compromising distensibility of ventricles [23].
Myocardial fibrosis presents as either perivascular, focal, replacement, or interstitial depending on the disease. Interstitial fibrosis can be further divided into reactive and infiltrative fibrosis [24]. Reactive interstitial fibrosis can be induced by increased ECM deposition without a significant loss of cardiomyocytes. Therefore, this type of fibrosis normally leads to pressure overload and cardiomyopathies. In comparison, infiltrative interstitial fibrosis occurs in patients with Fabry disease, a rare genetic disease characterized by dysfunctional catabolism of sphingolipids [21,25]. Replacement fibrosis occurs in cardiac injury like MI where cardiomyocytes are damaged and replaced by activated fibroblasts with formation of predominant scar containing type I collagen. In human fibrotic hearts, histological staining of type I collagen has indicated four types of texture: compact, interstitial, patchy and diffuse [26]. Compact fibrosis is deposition of dense and large collagens, where cardiomyocytes are totally devoid. By contrast with compact fibrosis, collagens in interstitial fibrosis deposit in between cells. Patchy fibrosis is characterized by long collagen fiber strands, while diffuse fibrosis exhibits short stretches of fibrosis [26].
Collagen deposition is a critical feature of cardiac fibrosis, specifically the expression of type I and III collagens. For instance, an upregulation of type I collagen was observed in the fibrosis of MI model [27,28], while the expression of type III collagen was increased in patients with ischemic cardiomyopathy [27,29]. Type I and III collagen significantly increase CF proliferation but do not affect myoFb differentiation [30]. The activation of extracellular signal-regulated protein kinases 1 and 2 (ERK1/2), a major mitogenic signal in CFs, has been reported to be involved in the mechanism of type I collagen-induced CF proliferation. However, type III and VI collagen had little or no effect on ERK1/2 phosphorylation [30]. Type VI collagen has been reported to facilitate myoFb differentiation, though the mechanism remains unclear [27]. Specifically, in vitro incubation of type VI collagen with myoFb induced differentiation and had negligible effects on CF proliferation [30]. Bryant et al. found that type VI collagen was significantly elevated in both non-infarcted (1.48 ± 0.13 fold) and infarcted (2.27 ± 0.13 fold) regions when compared to sham operated control 7 days after MI. As for the possible mechanism, they further found that the type VI collagen interacted with α-integrin receptor in CFs, and the blockade of α-integrin receptor-attenuated type VI collagen induced myoFb differentiation [31]. Interestingly, the deletion of type VI collagen in the knockout MI model played a protective role by limiting infarct size and fibrosis [32]. Another feature of cardiac fibrosis is the collagen maturation and fibrotic scar formation with crosslinking density, which leads to an increase in tensile strength of those scars [1,33]. These scar tissues impact on the extent of cardiac relaxation and contractility, which limits cardiac function [1]. Recently, fibrillar type V collagen has been reported to limit scar size after ischemic cardiac injury. Lacking type V collagen enabled scars to initiate integrin-dependent mechanosensitive feedback on CFs, leading to the augment of CF activation, ECM secretion and scar size increase [34]. In detail, the Col5a1 gene in CFs has been shown to regulate type V collagen secretion in CFs. A Col5a1 knockout led to the deficiency of type V collagen, which caused scar size increase and grossly abnormal scar architecture. The altered mechanical features of scar further increased the expression of αvβ3 and αvβ5 integrins, which acted as feedback cues to drive myoFb differentiation. To develop novel fibrosis therapies, further studies are needed to explore the biology of collagen formation and the crosstalk between collagens and CFs.
2.2. Biological pathogenesis in cardiac fibrosis
The pathogenesis of cardiac fibrosis remains unclear. It is well-established that the inflammation response plays a prominent role in cardiac fibrosis where the nucleotide-binding domain and leucine-rich repeat containing the PYD-3 (NLRP3) inflammasome are critical determinants [35]. Activation of the NLRP3 inflammasome can promote TGF-β signaling and drive fibrosis by inducing maturation of interleukin (IL)-1β (IL-1β) and IL-18 in CFs instead of in cardiomyocytes [36,37]. A recent report revealed that yes-associated protein (YAP) was a regulator of macrophage-mediated pro-inflammatory response after MI and cardiac fibrosis [38].
Recently, there is growing evidence to indicate the central role of mitochondrial dysfunction in fibrosis [39]. Mitochondrial dysfunction is mainly characterized by mitochondrial structure and gene damage, as well as changes in cellular oxidative protein activities. Mitochondria serves as the main source of reactive oxygen species (ROS) that is a byproduct of oxygen metabolism. The imbalance between biogenesis and scavenging of ROS results in oxidative damage to mitochondrial proteins, genes and lipids [40,41]. In cardiac fibrosis, ROS may directly regulate interstitial ECM production by modulating the expression and metabolism of matrix proteins [42]. In a recent study, peroxisome proliferator activated receptor gamma coactivator 1-alpha (PGC-1α), a master metabolic regulator, was upregulated in cardiac diseases and altered mitochondrial biogenesis, causing myocardial fibrosis [43]. Ca2+/calmodulin-dependent protein kinase II (CaMKII) functions as a cellular ROS sensor, which may be activated in the presence of ROS [44]. CaMKII is also activated in cardiac diseases with inflammatory environments such as MI and ischemia/reperfusion injury. It regulates pro-inflammatory signaling nuclear factor kappa-B (NF-κB), thus contributing to pathological cardiac remodeling [45]. Recently, mitochondria-targeted strategies have been proposed. For example, antioxidant enzyme CAT alleviated mitochondrial oxidative damage [46], while mitoquinone also inhibited fibrosis in pressure-overloaded hearts [47]. Notably, the dysfunction of metabolic-associated enzymes and the oxidative respiratory chain also serves as a critical factor for cardiac fibrosis. The ablation of nuclear-encoded mitochondrial inorganic pyrophosphatase (PPA2) has been reported to contribute to fibrosis, via the mechanism of activity reduction of the respiratory chain complex I and IV [48].
Further, endoplasmic reticulum (ER) stress and activation of the unfolded protein response also act as pro-fibrotic stimuli. Studies on the fibrotic remodeling via ER stress contain the activation of pro-apoptotic pathways, epithelial-to-mesenchymal transition, and induction of pro-inflammatory responses [49]. Luo et al. demonstrated that the administration of 4-phenylbutyric acid (4-PBA), an ER stress attenuation agent, could effectively alleviate post-MI complications [50]. Additionally, obesity is also a trigger for cardiac fibrosis, which is highly associated with metabolic dysfunction and the inflammatory response [51]. The excessive synthesis of aldosterone is a remarkable characteristic of obesity. Adipocyte-derived hormone leptin has been reported to be a direct regulator in aldosterone secretion, which promotes endothelial dysfunction followed by cardiac fibrosis [52]. Both type I and II diabetes are related with cardiac fibrosis as well. Hyperglycemia directly activates a fibrogenic program, resulting in accumulation of advanced glycation end-products (AGEs) and activation of AGE receptor-mediated pathways. Diabetes-associated fibrosis is mainly characterized by CF activation. Generated AGEs activate resident CFs and induce a matrix-synthetic phenotype [53]. Growing evidence has shown that peroxisome proliferator-activated receptor α (PPARα), a ligand-activated transcription factor, regulates fibrosis by modulating endothelial nitric oxide synthase and anti-inflammatory signaling pathways. Currently, novel PPARα drugs are used to alleviate fibrosis [54].
3. The role of myofibroblasts in fibrosis and cardiac diseases
3.1. The origin of pro-fibrotic myofibroblasts
MyoFbs are rarely observed in healthy cardiac tissues, while these cells replace damaged non-regenerated cardiomyocytes in injured tissues. Except for resident CFs, epithelial-derived cells (EPDCs) and endothelial-derived cells potentially adopt the myoFb phenotype via the process of epithelial-to-mesenchymal transition and endothelial-to-mesenchymal transition, respectively [55,56]. In vivo differentiation of these epithelial and endothelial-derived cells can be achieved by undergoing a defined sequence of events that allows them to present the fibrotic phenotype. These processes are strictly regulated by a sequence of coordinated expression of numerous growth factors such as fibroblast growth factor (FGF) and platelet-derived growth factor (PDGF) [55,57]. It needs to be mentioned that during embryonic development, EPDCs can alternate between spindle-shaped CFs and α-smooth muscle actin (α-SMA)+ phenotypes, which can then be activated to myoFbs in adult life during injury-induced pathological remodeling [58]. Specifically, endothelial-to-mesenchymal transition of resident endothelial cells was reported to generate 70% of the myoFbs in the heart with overload pressure [56,59]. Pericytes, which acts as contractile cells with mesenchymal origin and wraps around the microvasculature, were also proposed to adopt newly generated α-SMA+ myoFbs [60]. Kramann et al. found that resident Gli1+ mesenchymal stem cell (MSC)-like cells in the perivascular niche expressed typical MSC markers and presented trilineage differentiation capacity, contributing to cardiac fibrosis. Genetic lineage tracing analysis revealed that rather than circulating Gli1+ cells, it was the tissues-resident Gli1+ cells that proliferated to generate myoFbs after heart injury [60]. Furthermore, bone marrow-derived stem cells (BMSCs) have a fibroblast-myoFb-like phenotype by the name of fibrocytes, which has been suggested to be recruited into the injured heart. These circulating fibrocytes express characteristics of fibroblasts and play a major role in scar formation during wound healing [61–64]. Using gene labeling, Amerongen et al. confirmed that 21% of BMSCs in the infarct area were myoFbs with peak numbers on day 7 post-MI. These bone marrow-derived myoFbs presented in the infarct area and actively secreted collagen I [62]. The fibroblast lineage and signals that triggers the differentiation into myoFb are not well understood.
More methods have been used to investigate myoFb origins. Using a single-cell engraftment mice model, Norris et al. have demonstrated two extracardiac sources of CFs: the embryonic proepicardial organ and the recruitment of circulating BMSCs of hematopoietic stem cell (HSC) origin. Interestingly, periostin, a profibrogenic matricellular protein, was highly expressed in pathological remodeling and heart failure, and induces the differentiation of nonmyocyte progenitor cells into myoFbs [58]. Kanisicak et al. developed postn (periostin) gene-targeted mice containing a tamoxifen-inducible Cre for cellular lineage-tracing analysis. Their research revealed the postn-expressing myoFbs in the heart mainly derived from tissue-resident fibroblasts of transcription factor 21 (Tcf21) lineage, but not from endothelial, immune, or smooth muscle cells [65]. Taken together, more detailed studies on the myoFb origin and triggers are needed, and the deep exploration on differentiated myoFb activities are essential for developing cardiac repair therapies.
3.2. The differentiation from cardiac fibroblasts to myofibroblasts
CFs are the most prevalent cell types in the heart and play an essential role in regulating normal structure and function. One of important stimuli for the phenotype transition of CFs into myoFbs is a change of mechanical microenvironment. In intact healthy tissues, CFs are protected from stress by the crosslinked ECM framework. However, in an injury microenvironment, there is loss of architectural integrity allowing mechanical stress to reach fibroblasts and induce differentiation into proto-myoFbs [66]. Proto-myoFbs are identified by stress fibers, including cytoplasmic β-actin and γ-actin. When exposed to transforming growth factor-β (TGF-β, which is produced by CFs), those proto-myoFbs are induced to fully differentiate into myoFbs with the aid of the ED-A splice variant of fibronectin [67,68]. The assessment of RNA expression between CFs and myoFbs revealed a large difference between them, specifically the overexpression of actin alpha 2 and Postn with reduced expression of Tcf21 (a significant marker for quiescent fibroblasts) in myoFbs [69]. Different expression of said markers provide a pathway for assessment of the phenotype of active or quiescent fibroblasts. Nowadays, there are few unique markers of myoFbs, one of the reasons being that many cell types exist in the differentiation process (eg. CF-myoFb transition phenotype). Another reason is the fact that myoFb markers are also expressed on other cell types such as endothelial cells, mesenchymal cells and smooth muscle cells (SMCs), which are all sources of myoFbs. It is widely accepted that overexpressed α-SMA and other SMCs differentiation markers are characteristic markers for fully differentiated myoFbs (Fig. 1). Another differentiation marker is the cytoskeletal protein smoothelin, which was reported for contractile SMCs and can be used to distinguish SMCs from myoFbs [70]. However, smoothelin was also observed in lung myoFbs after the treatment of TGF-β1, making it an unreliable target [71]. Similarly, TGF-β1-treated myoFbs expressed of 4Ig paladin which is also expressed in SMCs [72,73]. The lack of specific myoFb markers still hinders the advancement of targeted therapies. Interestingly, fibroblast activation protein (FAP) is specifically expressed on the surface of differentiated myoFbs, and is currently being studied as a potential marker [15]. Moreover, a recent report shows that cardiac ECM in the failing heart could activate CFs via hyperactivated YAP signaling [74]. The discovery and optimization of unique identifiers of myoFbs can facilitate the development of targeted drug delivery systems. MyoFbs can further induce the activation of CFs via autocrine signaling, which provides positive feedback to increase fibroblast activation [75], making delivery of myoFbs even more beneficial.
Fig. 1. Surface markers on differentiated myoFbs.
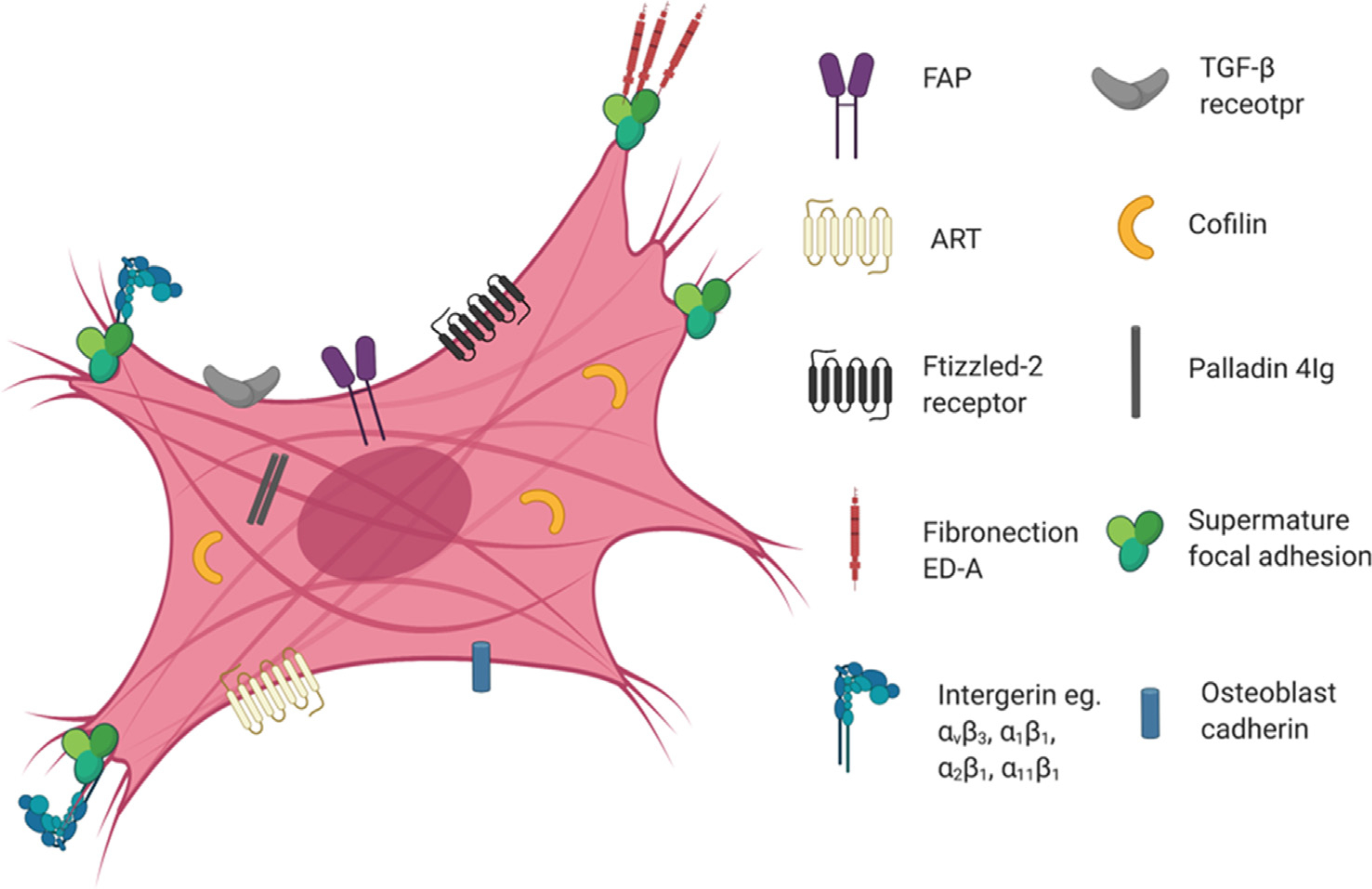
Due to their extracellular nature, FAP, Frizzled-2 receptor, ATR and TGF-β receptor are reasonable target candidates for the design of myoFb-targeted systems. Interestingly, αvβ3 integrins expressed in supermature focal adhesions and their combination are also potential targets. However, because many of these markers are also expressed on other cell types, the specificity needs to be optimized with further studies. Abbreviations: myoFbs, myofibroblasts; FAP, fibroblast activation protein; ATR, angiotensin II receptor; TGF-β, transforming growth factor-β.
3.3. The role of myofibroblasts and their crosstalk with other cell types
It has been established that the phenotypically transformed myoFbs are found at infarct sites where synthesis and deposition of collagens cause scar formation and fibrosis. Chronic or repeated injury enables fibrous tissue deposition to be persistent. MyoFbs can persist at infarct scars and have been observed at six months post-MI in a rat model [76]. Generally, myoFbs express type I collagen, TGF-β, α-SMA, and other factors, which in turn stimulates other cells to become myoFbs and promote fibrotic effects. Ang II, which can be secreted by myoFbs, enhances the synthesis of collagens in infarct sites especially via the activation of type 1 angiotensin receptor (AT1R) [3]. It is also reported that Ang II stimulates TGF-β expression, revealing the downstream role of TGF-β [77]. Smad proteins are part of the classical signaling cascade after the stimulation of TGF-β receptor, which can also be regulated by Ang II [78]. In this case, the Smad signaling pathway may be directly inhibited for deactivating myoFbs and reducing fibrosis. Moreover, myoFbs contribute to ECM synthesis and deposition, secreting collagens, MMP and other proteins [79]. These responses can also be induced by inflammation related factors in injured cardiac tissues. To date, myoFbs have been highly investigated, indicating the role of myoFb as a mediator for adverse remodeling. The most obvious role is the crosstalk between myoFbs and cardiomyocytes. Injury to the heart modifies the microenvironment by increasing expression of ECM proteins, cytokines, and exosomes, which leads to a fibrotic heart. Under this injured condition, cardiomyocytes correspondingly alter paracrine signaling, which augments activation and trans-differentiation of myoFbs [80].
Chronic activation of the myocardial renin angiotensin system increases local Ang II expression, leading to cardiac hypertrophy. In detail, Ang II has been reported to induce myoFbs to secrete exosomes containing miR-21 and miR-423, which resulted in overexpression of AT1R and Ang II receptor type 2 (AT2R) in cardiomyocytes via the activation of mitogen-activated protein kinases (MAPKs) [81]. In cardiac fibrosis, cardiomyocytes can even secrete miR-208a-containing exosomes into CFs, leading to myoFb differentiation followed by fibrosis development [82]. More miRNAs and cytokines have been identified as paracrine signaling factors between cardiomyocytes and CFs in cardiac diseases. For instance, miR-133 expression was decreased during hyperglycemia from diabetic cardiomyopathy, and overexpressed miR-133a prevented ERK1/2 and Smad2 phosphorylation [83]. MiR-378 has been reported to be secreted from cardiomyocytes during mechanical stress, which inhibited the excessive cardiac fibrosis through the suppression of p38 MAPK phosphorylation [84]. Interestingly, CF-specific p38 MAPK in turn induced cardiomyocyte hypertrophy via secreting paracrine factors including IL 6. The IL 6 signaling between fibroblasts and cardiomyocytes further caused cardiac dysfunction such as cardiac hypertrophy [85]. A current report indicated that in transverse aortic constriction (TAC) induced pressure overload, cardiomyocytes activated CFs followed with fibrosis via Wnt5a and Wnt11 signaling. Overexpression of cardiomyocyte-specific LRP6 interacted with the protease cathepsin D and facilitated the degradation of Wnt5a and Wnt11 [86]. Macrophage-secreted exosomes have also been reported to affect myoFbs and fibrosis [87]. For example, macrophages transferred miR-155 abundant exosomes into myoFbs to decrease collagen production [88]. In addition, intra-cardiac injection of stem cells has been shown to induce regional accumulation of macrophages. These accumulated macrophages downregulated CF activity, reduced ECM deposition, and enhanced cardiac function [89]. Together, these data highlight the critical role of cellular paracrine communication in cardiac fibrosis and pathology (Fig. 2)
Fig. 2. Representative crosstalk between myoFbs and other cells in cardiac fibrosis.

During cardiac injury, CFs differentiate into myoFbs, leading to ECM deposition and cardiac interstitium expansion. Many molecules are involved in the fibrosis process, including proteins, exosomes and cytokines. For instance, highly expressed Ang II binds ATR in cardiomyocytes and worsens their hypertrophy. Hypertrophic or injured cardiomyocytes alter paracrine signaling which facilitates myoFbs differentiation, while myoFbs in turn also aggravate cardiomyocyte injury. Pro-inflammatory macrophages aggravate fibrosis, while pro-resolution subtypes block myoFb differentiation via paracrine signaling. Abbreviations: CFs, cardiac fibroblasts; myoFbs, myofibroblasts; ECM, extracellular matrix; Ang II, Angiotensin II; ATR, Ang II receptor.
4. Therapeutic strategies targeting myofibroblasts
4.1. Direct reprogramming
The regeneration of adult mammalian hearts is highly limited due to the low regenerative capacity of cardiomyocytes. Endogenous CFs, which accounts for a significant portion of the mammalian heart, are a potential source of cardiomyocytes for regeneration therapy. A promising method to reprogram fibroblasts into induced cardiac-like myocytes (iCLMs) has been developed. Those iCLMs are expected to act as cardiomyocytes to improve cardiac function. To achieve this goal, different combinations of cardiac-specific factors are being explored, mainly transcription factors, fibroblast activation inhibitors, small molecules, and microRNAs (miRNAs). Viral and non-viral vectors, especially nano/micro delivery systems, have been developed to deliver this cargo into CFs to initiate reprogramming.
4.1.1. Direct reprogramming of murine fibroblasts
In 2010, Srivastava et al. found that three transcription factors: Gata4, Mef2c and Tbx5(GMT) enabled postnatal cardiac and dermal fibroblasts to be reprogrammed into iCLMs in vitro [90]. They further demonstrated successful reprograming of murine CFs into iCLMs via retroviral delivery of GMT, leading to reduced infarct size and fibrosis [91]. GMT were the core components for direct programming [90], and Tbx5 was further reported to promote the differentiation of transfected cells into beating cardiomyocytes [92]. Following studies from Olson et al. demonstrated enhanced reprogramming capacity of CFs both in vitro and in vivo after adding Hand2 in combination with GMT, which increases cardiac troponin T (cTnT) and tropomyosin expression [93]. These findings showed the feasibility of gene therapy for heart regeneration. Recently, various methods have been explored to increase GMT reprogramming efficacy. Qian et al. developed a complex set of polycistronic constructs containing GMT with identical 2A sequences in a single messenger RNA (mRNA). Adjusted ratio expression of Gata4: Mef2c:Tbx5 led to distinct protein expression and altered differentiation efficacy [94]. Inhibitors are alternative adjuvants for the induced expression of cardiac transcription factors. For instance, SB431542, an inhibitor of TGF-β pathway, increased the conversion rate of embryonic fibroblasts into iCLMs. The reprogramming efficacy of SB431542 was up to 5-fold higher than that of GMT, Hand2 and Nkx2.5 combined as measured by Ryr2 gene expression [95]. When TGF-β and Rho-associated kinase pathways are inhibited, a high percentage of embryonic fibroblasts were converted into iCLMs, and about 60% expressed cTnT [96]. In addition, iCLMs could also be inducted and matured using various cytokines such as FGF2, FGF10, and vascular endothelial growth factor (VEGF) [97]. The conversion into iCLMs is a complex process since there are several barriers in epigenetic modulation like Bmi1. Reduced expression of Bmi1 changes chromatin modification at cardiogenic loci by increasing the active histone marker H3K4me3 and decreasing repressive H2AK119ub [98]. This means that cardiogenic gene expression is derepressed in the conversion process of iCLMs. These results reveal that, in addition to transcription factors, many other factors influence fibroblast reprogramming, and inhibitors with sequential addition are promising adjuvants for optimizing the induction efficiency of functional iCLMs.
MiRNAs are alternative families with potential for reprogramming since they can enhance the expression of transcription factors such as GMT and Nkx2.5. MiRNAs participate in every aspect of cardiac development, making them promising therapeutics for functional iCLM reprogramming. The representative miRNA is miR-1, a cardiac muscle-specific molecule, that accounts for about 40% of total miRNAs in the mammalian heart [99]. The primary role of miR-1 is to promote cardiomyocyte proliferation and inhibit apoptosis [100]. MiR-133 also plays an essential role in promoting cardiomyocyte proliferation via miR-133-mediated Snai1 repression [101]. Recently, similar to the application of transcription factors, combination therapy of miRNAs has been explored. For instance, a “miRNA combo” (miR-1, miR-133, miR-208, miR-499) was assembled to convert CFs into functional iCLMs both in vitro and in vivo [102,103]. Furthermore, in a three-dimensional environment, such as hydrogel, the “miRNA combo” enhanced neonatal CF reprogramming [104]. In terms of mechanism, the combination of these miRNAs contributed to the altered expression of H3K27 methyltransferase and demethylase [105]. What is more, associated virus (AAV) vectors act as excellent vehicles for miRNAs, making them more accessible, and the small size of miRNAs potentially enables the convenient loading and delivery of a “miRNA combo” using one AAV vector [98].
Nano and micro delivery systems are alternative non-viral based systems for gene loading and delivery. Although viral vectors have been widely used for miRNA delivery, side effects have been reported, such as genetic aberration and alternative gene expression. Notably, cancer has been confirmed as an induced side effect [106]. In comparison, non-viral vehicles exhibit reduced immunogenic response and high safety properties. For instance, lipofectamine, an excellent lipid nano/micro-reagent, has been commercially approved and presents with low toxicity. The application of nanoparticles was firstly developed by Monica et al. to directly target and reprogram CFs. Dual small molecules were loaded into dextran-functionalized nanoparticles and delivered into CFs, leading to efficient CF reprogramming to iCLMs [107]. Notably, Muniyandi et al. loaded poly(lactide-co-glycolide) (PLGA) microparticles with two miRNAs (miR-1 and miR-133a) for direct CF reprogramming. In detail, polyetherimide, a most commonly used cationic vector for gene transfection, was used to compress these two miRNAs and form a core. The core was then encapsulated by outer biodegradable PLGA nanospheres. Mature late-stage markers, including troponin T and α-actinin, were both screened and had significant enhancement, revealing successful reprogramming into iCLMs [108]. Typical methods to reprogram murine fibroblasts are summarized in Table 1.
Table 1.
Representative factors and results in murine and human cardiac fibroblast reprogramming.
| Combination of factors | Original cell types | Markers and efficiencies | AP | Beating |
|---|---|---|---|---|
| GMT [90] | MCF, TTDF | cTnT+: ~30% of α-MHC cells | + | + |
| GMT [91] | MCF | α-MHC-EYFP+: ~40% at border zone | + | + |
| GHMT [116] | MEF | Sarcomere+:~32% | + | + |
| GHMT, MyoD domain [117] | MHF, MLBF, MTTF | cTnT+: ~4.9% | ND | + |
| GHMT, myoD domain[118] | MHF | cTnT+: ~19% | ND | + |
| GHMT, SB431542 [119] 19 | MCF | Activity of Troponin T-GCaMP5+: 9.27% | ND | − |
| GMT, Mesp1, Myocd, miR-133 [101] | MEF, MCF | α-MHC-GFP & cTnT+: 8.1%; α-actinin+: 19.9% | ND | − |
| GMT mRNA, C_lipo [120]17 | MCF | α-MHC-GFP+: 0.5% of total MCF | ND | − |
| GHMT, miR-1, miR-133, Y-27632, A83–01 [121] | MEF, MAF | cTnT+: ~60% (with A83–01); α-actinin+: ~60% (with A83–01) | + | + |
| OSKM, PEG hydrogel [122] | MEF; MTTF | α-actinin+: 1.72 fold to control | ND | + |
| Hand2, Nkx2.5, Gata4, Mef2c, Tbx5 [123] | MEF, MCF | Activity of Troponin T-GCaMP5+: 1.6% | ND | + |
| miR-1, miR-133, miR-208, miR-499 [124] | MCF | tdTomato+ Troponin T+: ~12% | + | + |
| CHIR99021, RepSox, Forskolin, VPA [125] | MEF, MTTF | α-actinin+: 14.5%; α-MHC+: 9% | + | + |
| OSKM, Ascorbic acid [126] | MEF | Gata4+: ~40%; MHC+: ~24% | + | + |
| GMT, MESP1, Myocd [127] | HCF | cTnT+: 5.9%; α-actinin+: 5.5%; | + | + |
| GMT, Mesp1, Myocd, Esrrg, Zfpm2 [111] | HESC | α-MHC-mCherry+: 15.8% | + | ND |
| GMT, Mesp1, Myocd, miR-133 [101] | HCF | cTnT+: 27.8%; α-actinin+: 8% | ND | + |
| Gata4, Hand2, Tbx5, Myocd, miR-1, miR-133 [128] | HFF | cTnT+: 34.1% | ND | + |
Different combinations of diverse factors and original cells for fibroblast reprogramming. Results are assessed by the expression of cardiac markers, electrophysiological characteristics and beating abilities.
GMT, Gata4, Mef2c, Tbx5; GHMT, Gata4, Hand2, Mef2c, Tbx5; MCF, murine cardiac fibroblast; HCF, human cardiac fibroblast; MTTDF, murine tail-tip dermal fibroblast; MTTF, murine tail tip fibroblast; cTnT, cardiac troponin T; MHF, murine head fibroblast; LBF, murine low body fibroblast; MEF, mouse embryonic fibroblast; MAF, murine adult fibroblast; AP, action potential; HESC, human embryonic stem cell; HFF, human foreskin fibroblast; ND, not detected.
4.1.2. Direct reprogramming of human fibroblasts
Reprogramming therapy can only be translated into clinics once it succeeds on human fibroblasts. Compared with murine fibroblasts, the process of human fibroblast reprogramming is difficult and time-consuming. In 2013, Nam et al. discovered the partial conversion from fibroblasts into iCLMs using the combination of Gata4, Hand2, Tbx5, Myocd (myocardin), miR-1 and miR-133. The milestone discovery was the expression of active cardiac markers [109]. Meanwhile, Wade et al. reported that cell morphology changed from a spindle shape to a rod-like shape when they reprogrammed fibroblasts with the “transcription factor combo” of Gata4, Mef2c, Tbx5, and Myocd (referred to Gmtm) [110]. Despite the reprogramming success on murine fibroblasts, Srivastava and colleagues found that it was insufficient to reprogram human fibroblasts using only GMT. The addition of ESRRG and MESP1 to GMT enabled induced iCLMs to express cardiac-specific genes and form sarcomeres. Interestingly, when adding Myocd and Zep-m2 to GMT, more cardiomyocytes-like features were achieved, such as phenotypic shift to the cardiac state and global cardiac gene expression [111].
Further advancement has appeared with the application of small molecules. Inspired by the reprogramming ability of small molecules [112–114], Ding and colleagues discovered a combination of nine small molecules which programmed human somatic cells into iCLMs in 2016. The molecules contained CHIR99021, A83–01, BIX01294, AS8351, SC1, Y27632, OAC2, SU16F, and JNJ10198409 [115]. After their induction, these iCLMs exhibited naïve human cardiomyocytes properties in terms of their transcriptome, epigenetics and electrophysiology. On this basis, the researchers confirmed the successful conversion of human fibroblasts into iCLMs, showing enhanced cardiac function in the infarcted heart [115]. It is noteworthy that these findings demonstrate great potential for clinical application, considering that low conversion rates can be further overcome in future studies. We summarized representative methods for human fibroblast reprogramming in Table 1.
4.2. Chimeric antigen receptor T cell (CAR-T) based immunotherapies
Fibroblast-targeting therapies remain limited due to the difficulty of identifying fibroblasts. The obvious obstacle is the lack of specific markers for activated myoFbs only when compared to other types of cells. On this end, engineered T cells have caused breakthroughs in cancer immunotherapy [15]. After being modified with a specific T cell receptor or a chimeric antigen receptor, cytotoxic T cells are redirected in vivo to recognize specific antigens on cancer cells and ablate them [129,130]. Through expression analysis of gene signatures, Aghajanian et al. recently discovered FAP, an endogenous protein marker, that was overexpressed on myoFbs when compared with quiescent CFs. They engineered CAR-T against FAP in vivo, which resulted in dramatical reduction of cardiac fibrosis at injured mice (Fig. 3) [15]. These results reveal a proof-of-concept which makes the use of CAR-T to target cardiac fibrosis, though more studies are needed to optimize therapeutic efficacy [131]. On one hand, continued search for alternative, unique antigens that are expressed solely on myoFbs may yield more targets or target combinations for fibrosis ablation. On the other, even when targeting the same antigen, specific CAR-T was able to more significantly affect targeting affinities and anti-fibrotic efficacies [132–134].
Fig. 3. FAP CAR-T targets cardiac fibrosis.

a, Scheme of experiments for CAR-T that targets FAP expressing cells. b, Top, Picro-sirium red staining of heart coronal sections in mice to evaluate fibrosis (red) with treatments of saline (left), Ang II/PE (center) or FAP CAR-T cells with Ang II/PE (right). Bottom, magnification of top results to evaluate left ventricular fibrosis. Scale bar, 100 μm. c, Quantitative results of cardiac fibrosis. ****P < 0.0001; one-way ANOVA between groups, P < 0.0001; post hoc multiple comparisons, Tukey’s test; n = 10, 9, and 7 biologically independent mice, from left to right. Abbreviations: Ang II, angiotensin II; PE, phenylephrine [15]. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
5. Other therapeutic strategies against fibrosis
5.1. Stem cell-based therapies
The application of stem cells with or without biomaterial scaffolds is another direction for the treatment of cardiac fibrosis and heart failure. Cells that have been explored in cardiac regeneration mainly include cardiac-derived stem/stromal cells (CSCs), BMSCs (eg. HSCs, MSCs, endothelial precursor cells (EPCs)), and induced pluripotent stem cells (iPSCs) [135,136], which can be delivered into heart tissue via intramyocardial, intrapericardial or intravenous injection [137–139]. Clinical trials of stem cell therapy continue, especially in this decade [140]. Unfortunately, no significant benefits have been observed, probably due to the variation between trials or exceptional end points, such as death [22]. One reason for modest clinical success is poor survival and engraftment of injected stem cells [136]. Another reason is negligible differentiation capabilities of stem cells towards fully functional cardiomyocytes [22] especially HSCs [141]. Additional studies suggest that stem cells achieve cardiac repair by secreting paracrine factors or interacting with other cells, such as macrophages [89,136,142]. For instance, Ronald J. et al. recently found that transplanted stem cells initiated an acute immune response and wound healing process via facilitating regional accumulation of CCR2+ and CX3CR1+ macrophages. Macrophages with these alternated phenotypes further reduced border zone ECM contents and alleviated fibrosis [89]. Notably, Wu et al. found specific transcriptome profiles using single-cell RNA-sequencing, which allowed for successful induction of human iPSCs to quiescent CFs for anti-fibrosis therapy (Fig. 4) [16]. This developed protocol helps deepen studies on the signaling pathway involved in cardiac fibrosis and also sheds light on the potential role of IPSCs as a platform to screen anti-fibrotic drugs in the future.
Fig. 4. Flow diagram illustrating the process and protocol of inducing human iPSCs into quiescent CFs.

Using mouse single-cell transcriptomic data, tissue-specific marker gene expressing in fibroblast subpopulations are revealed and used to differentiate human iPSCs-CFs. DEG analysis highlights the critical role of tissue-specific transcription factors in regulating the developmental trajectories of fibroblast subpopulations. Finally, human iPSCs-CFs were generated by sequentially differentiation of intermediate cell types including cardiac progenitor and epicardial cells. Abbreviations: iPSC, induced pluripotent stem cell; CF, cardiac fibroblast; DEG, differentially expressed genes [16].
After being intravenously injected, stem cells are likely to aggregate in blood circulation with a high lung homing property [143]. Without shielding, which is the protection of cells by the use of an external vehicle, the shape, size and location of stem cells in heart tissues are difficult to control. These challenges lead to suboptimal cardiac repair. Recently, biomaterial-based scaffolds have been developed to load and deliver stem cells for cardiac regeneration. Using biomaterials, especially biodegradable polymers, it is easy to construct multifunctional nano/micro-scale delivery systems with high cell loading efficacies. As local delivery systems, patches [144] and hydrogels [145] present good niches for CSCs. Microneedles, which are potential in situ or transdermal therapeutic systems [146–148], were also used to form CSC-loaded patches for cardiac repair [149]. Moreover, hydrogels can not only act as vehicles for epicardial placement of stem cells [150], but also could be injectable delivery systems for cardiac healing [151,152]. Recently, platelets have been used for cardiac homing for stem cells. Myocardial ischemia induces endothelial denudation and inflammation, recruiting circulating platelets to injured heart sites such as vessel walls [153] and endothelial cells [154]. Using this cardiac homing property, platelet membranes have been conjugated onto CSC surfaces to boost the injured heart targeting efficacy of CSCs [154]. In addition, antibody systems have been designed to recognize platelets and stem cells simultaneously as a novel cell therapy strategy. For instance, circulating stem cells can be delivered to the injured heart via pre-targeting polymer modified CD34 antibodies which recognize circulating EPCs, and CD41 antibodies which bind circulating platelets [155]. Further, inhaled bispecific antibodies (BsAbs) were designed to redirect stem cells from the lungs to the heart [143]. In detail, the lung has been reported to be a reservoir for HSCs and platelets [156]. Based on this, Liu et al. constructed CD34-CD42b BsAbs to catch HSCs and platelets simultaneously in the lungs, leveraging the lung’s biogenesis to alleviate cardiac fibrosis [143]. Another advancement are scaffold-free cell sheets, which were developed by culturing cells in a dish grafted with thermo-sensitive polymers like poly(N-isopropylacrylamide), and provide the advantage of solely delivering cells [157]. Representative biomaterial-based cell therapies are summarized in Table 2.
Table 2.
Representative cell and derivates loaded nano/micro delivery systems for cardiac fibrosis ablation.
| Vesicle | Biomaterials | Cell type/factors | Delivery route | Efficiencies Potency against fibrosis |
|---|---|---|---|---|
| Vascularized cardiac Patch [144] | Biomimetic micro-vessels (BMVs), which are encapsulated in a fibrin gel spiked with human CSCs | CSCs | In situ delivery | NM |
| Cardiac patch [158] | Biomimetic micro-vessels (BMVs) | CSCs | In situ delivery | Reduced scar size |
| Microneedle patch [149] | PVA Microneedle | CSCs | In situ delivery | Reduced scar fibrosis |
| Hydrogels [150] | Poly(2-alkyl-2-oxazoline) (POx) derivative | MSCs | In situ delivery | Reduced interstitial fibrosis |
| Nanogel [145] | Thermosensitive poly(N-isopropylacrylamine-co-acrylic acid) or P(NIPAM-AA) nanogel | CSCs | In situ gelation | Reduced fibrosis |
| Hydrogels [151] | Chitosan/dextran/beta-glycerophosphate based hydrogels | MSCs | Intravenous injection | Reduced fibrosis area |
| Platelet membrane coated CSCs [154] | Decorating platelet nanovesicles onto the surface of CSCs | CSCs | Intravenous injection | Reduced fibrosis |
| Biorthogonal conjugation system [155] | CD41attached DBCO polymer; azide modified CD34 antibody | Catching bone marrow derived EPCs in blood circulation | Intravenous injection | Attenuated interstitial and perivascular fibrosis compared with control-injected hearts |
| CD34 antibody attached platelet [159] | DSPE-PEG-CD34 antibodies, which were bonded to the surface of platelets | Catching bone marrow derived MSCs in blood circulation | Intravenous injection | NM |
| Bispecific antibodies (CD34-CMHC) [160] | Anti-CD34-F(ab’)2, which were bonded to anti-CMHC- F(ab’)2 | Redirecting BMSCs to injured myocardial cells | Intravenous injection | NM |
| Bispecific antibodies (CD42b-CD34) [143] | Biorthogonal reaction between TCO modified CD34 antibodies and TZ modified CD42b antibodies | Combining HSCs and platelets in the lung & Redirecting HSCs to the heart injury | Inhalation | Reduced scar fibrosis; reduced α-SMA protein expression |
| Cell sheets [161] | NM | Adipocyte cells | In situ transplantation | Inhibit fibrosis |
| Platelet fibrin Patch [162] | Double-lumen syringe including platelet-rich plasma and calcium-containing media solution | platelet-rich plasma | Local syringe | Reduced scar fibrosis |
| Artificial patch [163] | Myocardial extracellular matrix scaffold | CSC secreted factors | Reduced amount of scarring in rat MI model; less myocardial fibrosis in pig MI model. | |
| Synthetic MSCs [164] | PLGA microparticles loading MSC secreted factors, which are coated with MSC membrane | MSC secreted factors | Intramyocardial injection | Reduced scar fibrosis |
| Synthetic CSCs [165] | PLGA microparticles loading CSC secreted factors, which are coated with CSC membrane | CSC-conditioned media with grow factors | Intramyocardial injection | Reduced scar size |
| Platelet membrane coating nanoparticles [166] | CSC secretome loaded nanoparticles, coating with PEG2 conjugated platelet membrane | CSC secretome | Intravenous injection | Reduced scar size |
| Monocyte mimic [167] | Monocyte membrane | MSC derived EVs | Intravenous injection | Alleviated fibrosis remodeling |
Abbreviation: CSC, Cardiac stem/stromal cell; MSC, mesenchymal stem cell; PVA, poly(vinyl alcohol); PLGA, poly(lactic-co-glycolic acid); EPC, endothelial progenitor cell; DBCO, dibenzocyclooctyne; DSPE-PEG, 1, 2-Distearoyl-sn-glycero-3-phosphoethanolamine-Poly(ethylene glycol); BMSC, bone marrow-derived stem cell; CMHC, cardiac myosin heavy chain; TCO, trans-cyclooctene; TZ, tetrazine; HSC, hematopoietic stem cell; α-SMA, α-smooth muscle actin; EV, extracellular vesicle; NM: not mentioned.
5.2. Therapeutic strategies based on stem cell secreted factors
Besides stem cells for therapy, using stem cell-secreted factors is a cost-effective alternative that has been used to treat cardiac injury. These therapies consist mainly of stem cell-derived extracellular vesicles (EVs), secretomes, exosomes, proteins and cytokines [168]. Effective exosomes or secretomes come from stem cells (MSCs, CSCs, ESCs, etc.), or from cells exposed to pathological environments. These exosomes or secretomes contain growth factors and miRNAs that exert anti-apoptosic, anti-fibrotic, and proangiogenic effects [169]. This therapeutic cargo can be loaded into bio-mimicking nano/micro-particles for cardiac repair [170]. One such example is that Tang et al. constructed a regenerative patch by spraying platelet-rich plasma. The cardiac patch could release growth factors, such as VEGF, at the MI site, leading to reduced scar fibrosis and attenuated left ventricular remodeling [162]. Another acellular patch made of a porcine extracellular-matrix scaffold was developed to repair myocardial tissues, which can be notably maintained at the targeted site for up to 28 days [163]. In addition, cell membrane-coated nano/microparticles have emerged as novel targeting platforms, exhibiting homing capabilities to cardiac injury [166]. For instance, cell-mimicking microparticles were fabricated by coating MSC and CSC membranes onto the surface of degradable PLGA microparticles and loading MSC and CSC secretomes [164,165]. Platelet membrane-coated nanoparticles conjugated with PEG2 exhibited desirable injured cardiomyocytes (PEG2 receptors) targeted efficiencies [166]. Additionally, monocyte-mimicking systems have been designed as novel EV delivery vehicles for heart repair [167]. Representative biomaterials-based cell-free therapies are summarized in Table 2.
5.3. Therapeutic strategies developed from “off-target” drugs
Compelling evidence shows the reversion abilities of the myoFb phenotype into quiescent fibroblasts [65,171,172]. Therefore, a potential strategy is to target signaling pathways associated with fibroblast-to-myoFb phenotype differentiation. Interestingly, therapeutic agents against CVDs have “off-target” effects on CFs, which may underlie their benefits on other cells or their unknown effects on myoFbs. These unexpected anti-fibrotic effects are considered as “pleiotropic”. Small molecules presenting “pleiotropic” effects on myoFbs mainly contain anti-hypertensive agents such as angiotensin converting enzyme (ACE) inhibitors, angiotensin receptor blockers (ARBs) (especially Ang II blocker) and lipid-lowering drugs, and pharmacological agents, such as thiazolidinediones (TZDs), with the aim to lower insulin resistance in diabetic patients) [3].
The major task of ACE inhibitors and ARBs is to antagonize Ang II effects. Normally, Ang II binds and activates AT1R and AT2R followed with the activation of downstream pathways. Stimulated by growth factors and cytokines like IL-1 and tumor necrosis factor-α (TNF-α), myoFbs exhibit increased expression of AT1R and AT2R in heart failure when compared to normal CFs in healthy cardiac tissue [173]. MyoFbs also express the required components to the Ang II synthesis [174]. ACE inhibitors specifically inhibit ACE-mediated cleavage of Ang I and thereby block Ang II synthesis, whereas ARBs are used to inhibit AT1R [175]. TGF-β-induced myoFb differentiation also increases ACE expression, which makes TGF-β inhibition an alternated anti-fibrosis mechanism of ACE inhibitors [176]. Besides differentiation, the pro-fibrotic ability of Ang II is also characterized by an increase in ECM protein synthesis, reducing MMP activity expression of tissue inhibitor of metalloproteinase (TIMP). Imidaprilat, an ACE inhibitor, was shown to reduce the expression of IL-1 and MMP-2 in human CFs [177]. Curcumin can attenuate myocardial fibrosis by modulating AT1R and AT2R in rats [178]. Interestingly, another ACE inhibitor lisinopril can reduce TGF-β-induced collagen synthesis in rat CFs [176].
Moreover, statins are inhibitors of 3-hydroxy-3-methylglutaryl coenzyme A (HMG-coA) and are involved in the established treatment of hypercholesterolaemia, where they exhibit anti-fibrotic effects. For instance, simvastatin inhibits TNF-α induced MMP-9 secretion in human myoFbs [179], and pravastatin exhibits MMP-3 and MMP-9 inhibition abilities in Ang II-overexpressed murine CFs [180]. Pravastatin also had anti-proliferation effects on myoFbs by inhibiting TGF-β signaling [181]. In addition, TZDs, agonists of PPARγ, reduced insulin resistance but increased vascular benefits in patients with type-2 diabetes mellitus [182]. Another candidate is the first-in-class thienodiazepine small molecule JQ1, an inhibitor of bromodomain-containing protein 4 (BRD4) [183]. In heart failure, JQ1 not only treated cardiac hypertrophy [69], but also reduced myoFb activation [184] via inhibition of BRD4 signaling. Moreover, as inflammation is a fibrosis pathogenesis factor, colchicine acts as an anti-inflammatory agent and has been reported to exhibit fibrosis alleviation properties [185].
6. Clinical trials for the alleviation of cardiac fibrosis
Recently, new mediators such as miRNAs and cytokines have emerged to alleviate cardiac fibrosis. However, most of them have not been clinically translated. In current clinical trials, anti-fibrosis results trials are disappointing, with promising data resulting mainly from treatments with renin-angiotensin-aldosterone system (RAAS) inhibitors [186]. In general, the first family of antifibrotic drugs against cardiac fibrosis is that of angiotensin II inhibitors. For instance, ACE inhibitors have been reported to exhibited anti-fibrotic benefits not only in animals (as mentioned in 5.3), but also in human patients [186]. In addition, drugs like inflammatory modulators and relaxin also exhibited fibrosis attenuation effects in clinical studies. Some currently representative clinical trials against cardiac fibrosis are shown in Table 3. The effective drugs in clinical studies, such as RAAS, only show modest regression of cardiac fibrosis [186]. Patients with heart failure still encounter persistent cardiac fibrosis even when receiving official standard of care [187]. The failure of anti-fibrotic drugs clinical trials suggests that translating research data from animal models to humans requires further development due to the vast genetic differences between animals and humans.
Table 3.
Representative anti-fibrotic therapies against cardiac fibrosis in clinical trials.
| Study | Agent | Length of treatment | Patient included (n) | Discoveries |
|---|---|---|---|---|
| RAAS inhibitor [188] | Candesartan | 24 months | 153 | PIINP in patients with atrial fibrillation were reduced. |
| RAAS inhibitor [189] | Spironolactone | 6 months | 80 | PICP and PIINP in patients with metabolic syndrome were reduced. |
| RAAS inhibitor [190] | Eplerenone | 6 months | 44 | PINP and PICP in patients with heart failure (ejection fraction was preserved) were reduced. |
| RAAS inhibitor [191] | Losartan | 12 months | 20 | The progression of cardiac fibrosis was attenuated in patients with nonobstructive hypertrophic cardiomyopathy. |
| RAAS inhibitor [192] | Eplerenone | 6 months | 113 | PICP and PIINP was reduced while myocardial deformation was improved in patients with obesity and mild LV diastolic dysfunction. |
| Inflammation modulator [193] | Atorvastatin | 6 months | 56 | PIINP was decreased in patients with heart failure. |
| Endothelin inhibitor [194] | Enrasentan | 6 months | 72 | LVEDVI was improved in asymptomatic patients with LV dysfunction after the treatment of enrasentan compared to that with enalapril treatment. |
| Loop diuretic [195] | Torsemide | 8 months | 36 | PICP and CVF were both reduced in hypertensive patients with symptomatic heart failure after the treatment of Torsemide. |
| Loop diuretic [196] | Torsemide | 8 months | 22 | PCP was decreased in patients with chronic heart failure. |
| Loop diuretic [197] | Torsemide | 8 months | 24 | Collagen crosslinking was enhanced accompanied with normalization of LV chamber stiffness in patients with heart failure. |
| Cyclic GMP-specific phosphodiesterase type-5A inhibitor [198] | Sildenafil | 3 months | 59 | LV contraction parameters were improved while TGF-β and MCP-1 were reduced in patients with diabetic cardiomyopathy. |
| Cyclic GMP-specific phosphodiesterase type-5A inhibitor [199] | Sildenafil | 24 weeks | 216 | No obvious improvement in cardiac activities in patients with heart failure (ejection fraction was preserved). |
| Pre-relaxin-AHF [200] | Relaxin | 48 h | 234 | Cardiovascular deaths were lowered in patients with acute heart failure. |
Abbreviations: PIINP, the amino-terminal peptide of type III procollagen; PICP, the carboxy-terminal peptide of procollagen type I; PINP, the amino-terminal peptide of type I procollagen; LV, left ventricular; LVEDVI, LV end diastolic volume index; CVF, collagen volume fraction; PCP, procollagen type I carboxy-terminal proteinase; TGF-β, transforming growth factor-β.
7. Conclusion
The pathological remodeling of cardiac disorders normally accompanies cardiac fibrosis. Anti-fibrotic therapies are still limited due to the fact that various factors are involved in the pathogenesis of fibrosis, such as inflammation and mitochondrial dysfunction. Different cell types also contributed to the fibrotic progress, with comprehensive crosslinks, especially interactions between myoFbs and cardiomyocytes. In the latest literature, single-cell RNA-sequencing offers a promising platform for identifying novel cellular and molecular protagonists that possibly initiate cardiac fibrosis [201]. For instance, activated CFs in MI mice were found to have high expression of collagen triple helix repeat containing 1 (Cthrc1), which played an crucial role in the process of cardiac fibrosis [202]. These results suggest a potential application of single-cell RNA-sequencing for further investigation on fibrosis and the development of anti-fibrotic strategies.
MyoFbs are dominating cells in cardiac fibrosis which regulate cytokine secretion, collagen synthesis, and non-functional scar formation [203–205]. The secretion of myoFbs can further activate CFs. In myoFb-based therapeutic strategies, the latest decades have seen anti-fibrosis advances with reprogramming and cell engineering. However, further studies are needed since myoFb function and its role in fibrosis is still not entirely understood. Specifically, studies can be conducted on the biology of myoFb activation and signaling pathways, which facilitates the design of myoFb deactivation strategies. Secondly, compared with quiescent fibroblasts, discovery of more myoFb-specific markers would faccilitate the design of myoFb-targeted therapeutics. Lastly, studies can be carried on to investigate the crosstalk between myoFbs and other cell types like macrophages, cardiomyocytes and inflammatory cells. Notably, exosomes exert pleiotropic repair effects using miRNAs, so exploration on specific miRNA for fibrosis inhibition would be beneficial. Specifically, precise anti-fibrotic therapy can be achieved by delivering specific miRNA-loaded nanoparticles to myoFbs. Finally, given the “off-target” anti-fibrotic effects of drugs, an impeccable platform is needed to design drug screening. In addition to therapy, there is also a demand for developing diagnostic tools which enable early and reliable fibrosis detection.
Acknowledgements
This work was supported by grants from the National Institutes of Health (HL123920, HL137093, HL144002, HL146153, HL147357, and HL149940 to K.C.) and the American Heart Association (18TPA34230092 and 19EIA34660286 to K.C.). There is no conflict of interest in this review.
Abbreviations:
- CFs
cardiac fibroblasts
- myoFbs
myofibroblasts
- ECM
extracellular matrix
- CAR-T
chimeric antigen receptor T cell
- CVDs
cardiovascular diseases
- MMPs
metalloproteinases
- Ang II
Angiotensin II (Ang II)
- RNA
single cell ribonucleic acid
- MI
myocardial infarction
- IL
interleukin
- ERK1/2
extracellular signal-regulated protein kinases 1 and 2
- YEP
yes-associated protein
- ROS
reactive oxygen species
- PGC-1α
peroxisome proliferator activated receptor gamma coactivator 1-alpha
- CaMKII
Ca2+/calmodulin-dependent protein kinase II
- NF-κB
nuclear factor kappa-B
- ER
endoplasmic reticulum
- 4-PBA
4-phenylbutyric acid
- AGEs
advanced glycation end-products
- PPARα
peroxisome proliferator-activated receptor α
- EPDCs
epithelial-derived cells
- FGF
fibroblast growth factor (FGF)
- PDGF
platelet-derived growth factor
- α-SMA
α-smooth muscle actin
- MSC
mesenchymal stem cell
- BMSCs
bone-marrow derived stem cells (BMSCs)
- HSC
hematopoietic stem cell
- Tcf21
transcription factor 21
- TGF-β
transforming growth factor-β
- SMCs
smooth muscle cells
- FAP
fibroblast activation protein
- ATR
angiotensin II receptor
- AT1R
type 1 angiotensin receptor
- AT2R
Ang II receptor type 2
- MAPKs
mitogen-activated protein kinases
- iCLMs
induced cardiac-like myocytes
- GMT
Gata4, Mef2c and Tbx5
- cTnT
cardiac troponin T
- mRNA
messenger RNA
- miRNAs
microRNAs
- VEGF
vascular endothelial growth factor
- AAV
ssociated virus
- PLGA
poly(lactide-co-glycolide)
- CSCs
cardiac-derived stem/stromal cells
- EPCs
endothelial precursor cells
- iPSCs
induced pluripotent stem cells
- DEG
differentially expressed genes
- BsAbs
bispecific antibodies
- EVs
extracellular vesicles
- ACE
angiotensin converting enzyme
- ARBs
angiotensin receptor blockers
- TZDs
thiazolidinediones
- TNF-α
tumor necrosis factor-α
- RRAS
renin-angiotensin-aldosterone system
- Cthrc1
collagen triple helix repeat containing 1
Footnotes
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- [1].Murtha LA, Schuliga MJ, Mabotuwana NS, Hardy SA, Waters DW, Burgess JK, Knight DA, Boyle AJ, The processes and mechanisms of cardiac and pulmonary fibrosis, Front. Physiol. 8 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M, Das SR, de Ferranti S, Despres JP, Fullerton HJ, Howard VJ, Huffman MD, Isasi CR, Jimenez MC, Judd SE, Kissela BM, Lichtman JH, Lisabeth LD, Liu SM, Mackey RH, Magid DJ, McGuire DK, Mohler ER, Moy CS, Muntner P, Mussolino ME, Nasir K, Neumar RW, Nichol G, Palaniappan L, Pandey DK, Reeves MJ, Rodriguez CJ, Rosamond W, Sorlie PD, Stein J, Towfighi A, Turan TN, Virani SS, Woo D, Yeh RW, Turner MBC, Amer Heart Assoc Stat, S. Stroke Stat, Heart disease and stroke statistics-2016 update a report from the American Heart Association, Circulation 133 (2016) E38–E360. [DOI] [PubMed] [Google Scholar]
- [3].Porter KE, Turner NA, Cardiac fibroblasts: at the heart of myocardial remodeling, Pharmacol. Ther. 123 (2009) 255–278. [DOI] [PubMed] [Google Scholar]
- [4].Jellis C, Martin J, Narula J, Marwick TH, Assessment of nonischemic myocardial fibrosis, J. Am. Coll. Cardiol. 56 (2010) 89–97. [DOI] [PubMed] [Google Scholar]
- [5].Hajipour MJ, Mehrani M, Abbasi SH, Amin A, Kassaian SE, Garbern JC, Caracciolo G, Zanganeh S, Chitsazan M, Aghaverdi H, Kamali Shahri SM, Ashkarran A, Raoufi M, Bauser-Heaton H, Zhang J, Muehlschlegel JD, Moore A, Lee RT, Wu JC, Serpooshan V, Mahmoudi M, Nanoscale technologies for prevention and treatment of heart failure: challenges and opportunities, Chem. Rev. 119 (2019) 11352–11390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Liu MR, Khan AR, Ji JB, Lin GM, Zhao XG, Zhai GX, Crosslinked self-assembled nanoparticles for chemo-sonodynamic combination therapy favoring antitumor, antimetastasis management and immune responses, J. Control. Release 290 (2018) 150–164. [DOI] [PubMed] [Google Scholar]
- [7].Jiao Y, Pang X, Liu MR, Zhang BM, Li LB, Zhai GX, Recent progresses in bioadhesive microspheres via transmucosal administration, Colloids Surf. B-Biointerfaces 140 (2016) 361–372. [DOI] [PubMed] [Google Scholar]
- [8].Yazdani S, Bansal R, Prakash J, Drug targeting to myofibroblasts: implications for fibrosis and cancer, Adv. Drug Deliv. Rev. 121 (2017) 101–116. [DOI] [PubMed] [Google Scholar]
- [9].Chen ZJ, Jain A, Liu H, Zhao Z, Cheng K, Targeted drug delivery to hepatic stellate cells for the treatment of liver fibrosis, J. Pharmacol. Exp. Ther. 370 (2019) 695–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Hu M, Wang Y, Liu Z, Yu Z, Guan K, Liu M, Wang M, Tan J, Huang L, Hepatic macrophages act as a central hub for relaxin-mediated alleviation of liver fibrosis, Nat. Nanotechnol. (2021). [DOI] [PubMed] [Google Scholar]
- [11].Liu MR, Song WT, Huang L, Drug delivery systems targeting tumor-associated fibroblasts for cancer immunotherapy, Cancer Lett. 448 (2019) 31–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Miao L, Liu Q, Lin CM, Luo C, Wang YH, Liu LN, Yin WY, Hu SH, Kim WY, Huang L, Targeting tumor-associated fibroblasts for therapeutic delivery in desmoplastic tumors, Cancer Res. 77 (2017) 719–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Zhu K, Wu MY, Lai H, Guo CF, Li J, Wang YL, Chen Y, Wang CS, Shi JL, Nanoparticle-enhanced generation of gene-transfected mesenchymal stem cells for in vivo cardiac repair, Biomaterials 74 (2016) 188–199. [DOI] [PubMed] [Google Scholar]
- [14].Kharaziha M, Memic A, Akbari M, Brafman DA, Nikkhah M, Nano-enabled approaches for stem cell-based cardiac tissue engineering, Adv. Healthcare Mater. 5 (2016) 1533–1553. [DOI] [PubMed] [Google Scholar]
- [15].Aghajanian H, Kimura T, Rurik JG, Hancock AS, Leibowitz MS, Li L, Scholler J, Monslow J, Lo A, Han W, Wang T, Bedi K, Morley MP, Linares Saldana RA, Bolar NA, McDaid K, Assenmacher C-A, Smith CL, Wirth D, June CH, Margulies KB, Jain R, Puré E, Albelda SM, Epstein JA, Targeting cardiac fibrosis with engineered T cells, Nature 573 (2019) 430–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Zhang H, Tian L, Shen M, Tu C, Wu H, Gu M, Paik DT, Wu JC, Generation of quiescent cardiac fibroblasts from human induced pluripotent stem cells for in vitro modeling of cardiac fibrosis, Circ. Res. 125 (2019) 552–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Weber KT, Sun Y, Bhattacharya SK, Ahokas RA, Gerling IC, Myofibroblast-mediated mechanisms of pathological remodelling of the heart, Nat. Rev. Cardiol. 10 (2013) 15–26. [DOI] [PubMed] [Google Scholar]
- [18].Segura AM, Frazier OH, Buja LM, Fibrosis and heart failure, Heart Fail. Rev. 19 (2014) 173–185. [DOI] [PubMed] [Google Scholar]
- [19].Schellings MWM, Pinto YM, Heymans S, Matricellular proteins in the heart: possible role during stress and remodeling, Cardiovasc. Res. 64 (2004) 24–31. [DOI] [PubMed] [Google Scholar]
- [20].Rienks M, Papageorgiou A-P, Frangogiannis NG, Heymans S, Myocardial extracellular matrix an ever-changing and diverse entity, Circ. Res. 114 (2014) 872–888. [DOI] [PubMed] [Google Scholar]
- [21].Disertori M, Mase M, Ravelli F, Myocardial fibrosis predicts ventricular tachyarrhythmias, Trends Cardiovasc. Med. 27 (2017) 363–372. [DOI] [PubMed] [Google Scholar]
- [22].Hinderer S, Schenke-Layland K, Cardiac fibrosis - A short review of causes and therapeutic strategies, Adv Drug Deliv Rev 146 (2019) 77–82. [DOI] [PubMed] [Google Scholar]
- [23].Tschoepe C, Lam CSP, Diastolic heart failure: what we still don’t know Looking for new concepts, diagnostic approaches, and the role of comorbidities, Herz 37 (2012) 875–879. [DOI] [PubMed] [Google Scholar]
- [24].Herum KM, Lunde IG, McCulloch AD, Christensen G, The soft-and hard-heartedness of cardiac fibroblasts: mechanotransduction signaling pathways in fibrosis of the heart, J. Clin. Med. 6 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Graham-Brown MPM, Patel AS, Stensel DJ, March DS, Marsh AM, McAdam J, McCann GP, Burton JO, Imaging of myocardial fibrosis in patients with end-stage renal disease: current limitations and future possibilities, Biomed. Res. Int. 2017 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].de Jong S, van Veen TAB, van Rijen HVM, de Bakker JMT, Fibrosis and cardiac arrhythmias, J. Cardiovasc. Pharmacol. 57 (2011) 630–638. [DOI] [PubMed] [Google Scholar]
- [27].Kong P, Christia P, Frangogiannis NG, The pathogenesis of cardiac fibrosis, Cell. Mol. Life Sci. 71 (2014) 549–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Mukherjee D, Sen S, Alteration of cardiac collagen phenotypes in hypertensive hypertrophy - role of blood-pressure, J. Mol. Cell. Cardiol. 25 (1993) 185–196. [DOI] [PubMed] [Google Scholar]
- [29].Mukherjee D, Sen S, Alteration of collagen phenotypes in ischemic cardiomyopathy, J. Clin. Invest. 88 (1991) 1141–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Naugle JE, Olson ER, Zhang X, Mase SE, Pilati CF, Maron MB, Folkesson HG, Horne WI, Doane KJ, Meszaros JG, Type VI collagen induces cardiac myofibroblast differentiation: implications for postinfarction remodeling, American journal of physiology, Heart Circ. Physiol. 290 (2006) H323–H330. [DOI] [PubMed] [Google Scholar]
- [31].Bryant JE, Shamhart PE, Luther DJ, Olson ER, Koshy JC, Costic DJ, Mohile MV, Dockry M, Doane KJ, Meszaros JG, Cardiac myofibroblast differentiation is attenuated by alpha(3) integrin blockade: potential role in post-MI remodeling, J. Mol. Cell Cardiol. 46 (2009) 186–192. [DOI] [PubMed] [Google Scholar]
- [32].Luther DJ, Thodeti CK, Shamhart PE, Adapala RK, Hodnichak C, Weihrauch D, Bonaldo P, Chilian WM, Meszaros JG, Absence of type VI collagen paradoxically improves cardiac function, structure, and remodeling after myocardial infarction, Circ Res 110 (2012) 851–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Brauchle E, Kasper J, Daum R, Schierbaum N, Falch C, Kirschniak A, Schaeffer TE, Schenke-Layland K, Biomechanical and biomolecular characterization of extracellular matrix structures in human colon carcinomas, Matrix Biol. 68–69 (2018) 180–193. [DOI] [PubMed] [Google Scholar]
- [34].Yokota T, McCourt J, Ma F, Ren S, Li S, Kim TH, Kurmangaliyev YZ, Nasiri R, Ahadian S, Nguyen T, Tan XHM, Zhou Y, Wu R, Rodriguez A, Cohn W, Wang Y, Whitelegge J, Ryazantsev S, Khademhosseini A, Teitell MA, Chiou PY, Birk DE, Rowat AC, Crosbie RH, Pellegrini M, Seldin M, Lusis AJ, Deb A, Type V collagen in scar tissue regulates the size of scar after heart injury, Cell 182 (2020) 545–562 e523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Che H, Wang Y, Li H, Li Y, Sahil A, Lv J, Liu Y, Yang Z, Dong R, Xue H, Wang L, Melatonin alleviates cardiac fibrosis via inhibiting lncRNA MALAT1/miR-141-mediated NLRP3 inflammasome and TGF-β1/Smads signaling in diabetic cardiomyopathy, FASEB J.: Off. Publ. Feder. Am. Soc. Exp. Biol. 34 (2020) 5282–5298. [DOI] [PubMed] [Google Scholar]
- [36].Pan XC, Liu Y, Cen YY, Xiong YL, Li JM, Ding YY, Tong YF, Liu T, Chen XH, Zhang HG, Dual role of triptolide in interrupting the NLRP3 inflammasome pathway to attenuate cardiac fibrosis, Int. J. Mol. Sci. 20 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Tao Y, Wang N, Qiu T, Sun X, The role of autophagy and NLRP3 inflammasome in liver fibrosis, Biomed. Res. Int. 2020 (2020) 7269150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Mia MM, Cibi DM, Abdul Ghani SAB, Song W, Tee N, Ghosh S, Mao J, Olson EN, Singh MK, YAP/TAZ deficiency reprograms macrophage phenotype and improves infarct healing and cardiac function after myocardial infarction, PLoS Biol. 18 (2020) e3000941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Li X, Zhang W, Cao Q, Wang Z, Zhao M, Xu L, Zhuang Q, Mitochondrial dysfunction in fibrotic diseases, Cell Death Discovery 6 (2020) 80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Dan Dunn J, Alvarez LA, Zhang X, Soldati T, Reactive oxygen species and mitochondria: a nexus of cellular homeostasis, Redox Biol. 6 (2015) 472–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Zorov DB, Juhaszova M, Sollott SJ, Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release, Physiol. Rev. 94 (2014) 909–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Siwik DA, Colucci WS, Regulation of matrix metalloproteinases by cytokines and reactive oxygen/nitrogen species in the myocardium, Heart Fail Rev. 9 (2004) 43–51. [DOI] [PubMed] [Google Scholar]
- [43].Caravia XM, Fanjul V, Oliver E, Roiz-Valle D, Morán-Álvarez A, Desdín-Micó G, Mittelbrunn M, Cabo R, Vega JA, Rodríguez F, Fueyo A, Gómez M, Lobo-González M, Bueno H, Velasco G, Freije JMP, Andrés V, Ibáñez B, Ugalde AP, López-Otín C, The microRNA-29/PGC1α regulatory axis is critical for metabolic control of cardiac function, PLoS Biol. 16 (2018) e2006247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Luczak ED, Anderson ME, CaMKII oxidative activation and the pathogenesis of cardiac disease, J. Mol. Cell Cardiol. 73 (2014) 112–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Rusciano MR, Sommariva E, Douin-Echinard V, Ciccarelli M, Poggio P, Maione AS, CaMKII activity in the inflammatory response of cardiac diseases, Int. J. Mol. Sci. 20 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Dai DF, Johnson SC, Villarin JJ, Chin MT, Nieves-Cintrón M, Chen T, Marcinek DJ, Dorn GW 2nd, Kang YJ, Prolla TA, Santana LF, Rabinovitch PS , Mitochondrial oxidative stress mediates angiotensin II-induced cardiac hypertrophy and Galphaq overexpression-induced heart failure, Circ. Res. 108 (2011) 837–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Goh KY, He L, Song J, Jinno M, Rogers AJ, Sethu P, Halade GV, Rajasekaran NS, Liu X, Prabhu SD, Darley-Usmar V, Wende AR, Zhou L, Mitoquinone ameliorates pressure overload-induced cardiac fibrosis and left ventricular dysfunction in mice, Redox Biol. 21 (2019) 101100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Kennedy H, Haack TB, Hartill V, Matakovic L´, Baumgartner ER, Potter H, Mackay R, Alston CL, O’Sullivan S, McFarland R, Connolly G, Gannon C, King R, Mead S, Crozier I, Chan W, Florkowski CM, Sage M, Höfken T, Alhaddad B, Kremer LS, Kopajtich R, Feichtinger RG, Sperl W, Rodenburg RJ, Minet JC, Dobbie A, Strom TM, Meitinger T, George PM, Johnson CA, Taylor RW, Prokisch H, Doudney K, Mayr JA, Sudden cardiac death due to deficiency of the mitochondrial inorganic pyrophosphatase PPA2, Am. J. Hum. Genet. 99 (2016) 674–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Tanjore H, Lawson WE, Blackwell TS, Endoplasmic reticulum stress as a pro-fibrotic stimulus, BBA 2013 (1832) 940–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Luo T, Kim JK, Chen B, Abdel-Latif A, Kitakaze M, Yan L, Attenuation of ER stress prevents post-infarction-induced cardiac rupture and remodeling by modulating both cardiac apoptosis and fibrosis, Chem. Biol. Interact. 225 (2015) 90–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Cavalera M, Wang J, Frangogiannis NG, Obesity, metabolic dysfunction, and cardiac fibrosis: pathophysiological pathways, molecular mechanisms, and therapeutic opportunities, Transl. Res.: J. Lab. Clin. Med. 164 (2014) 323–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Huby AC, Antonova G, Groenendyk J, Gomez-Sanchez CE, Bollag WB, Filosa JA, Belin de Chantemèle EJ, Adipocyte-derived hormone leptin is a direct regulator of aldosterone secretion, which promotes endothelial dysfunction and cardiac fibrosis, Circulation 132 (2015) 2134–2145. [DOI] [PubMed] [Google Scholar]
- [53].Russo I, Frangogiannis NG, Diabetes-associated cardiac fibrosis: Cellular effectors, molecular mechanisms and therapeutic opportunities, J. Mol. Cell Cardiol. 90 (2016) 84–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Li S, Yang B, Du Y, Lin Y, Liu J, Huang S, Zhang A, Jia Z, Zhang Y, Targeting PPARα for the treatment and understanding of cardiovascular diseases, Cell. Physiol. Biochem.: Int. J. Exp. Cell. Physiol., Biochem., Pharmacol. 51 (2018) 2760–2775. [DOI] [PubMed] [Google Scholar]
- [55].Kalluri R, Neilson EG, Epithelial-mesenchymal transition and its implications for fibrosis, J. Clin. Invest. 112 (2003) 1776–1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Zeisberg EM, Tarnavski O, Zeisberg M, Dorfman AL, McMullen JR, Gustafsson E, Chandraker A, Yuan X, Pu WT, Roberts AB, Neilson EG, Sayegh MH, Izumo S, Kalluri R, Endothelial-to-mesenchymal transition contributes to cardiac fibrosis, Nat. Med. 13 (2007) 952–961. [DOI] [PubMed] [Google Scholar]
- [57].Wessels A, Pérez-Pomares JM, The epicardium and epicardially derived cells (EPDCs) as cardiac stem cells, Anatom. Rec. Part A, Discov. Mol., Cell., Evolut. Biol. 276 (2004) 43–57. [DOI] [PubMed] [Google Scholar]
- [58].Norris RA, Borg TK, Butcher JT, Baudino TA, Banerjee I, Markwald RR, Neonatal and adult cardiovascular pathophysiological remodeling and repair: developmental role of periostin, Ann. N. Y. Acad. Sci. 1123 (2008) 30–40. [DOI] [PubMed] [Google Scholar]
- [59].Widyantoro B, Emoto N, Nakayama K, Anggrahini DW, Adiarto S, Iwasa N, Yagi K, Miyagawa K, Rikitake Y, Suzuki T, Kisanuki YY, Yanagisawa M, Hirata K, Endothelial cell-derived endothelin-1 promotes cardiac fibrosis in diabetic hearts through stimulation of endothelial-to-mesenchymal transition, Circulation 121 (2010) 2407–2418. [DOI] [PubMed] [Google Scholar]
- [60].Kramann R, Schneider RK, DiRocco DP, Machado F, Fleig S, Bondzie PA, Henderson JM, Ebert BL, Humphreys BD, Perivascular Gli1+ progenitors are key contributors to injury-induced organ fibrosis, Cell Stem Cell 16 (2015) 51–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Bucala R, Spiegel LA, Chesney J, Hogan M, Cerami A, Circulating fibrocytes define a new leukocyte subpopulation that mediates tissue-repair, Mol. Med. 1 (1994) 71–81. [PMC free article] [PubMed] [Google Scholar]
- [62].van Amerongen MJ, Bou-Gharios G, Popa E, van Ark J, Petersen AH, van Dam GM, van Luyn MJ, Harmsen MC, Bone marrow-derived myofibroblasts contribute functionally to scar formation after myocardial infarction, J. Pathol. 214 (2008) 377–386. [DOI] [PubMed] [Google Scholar]
- [63].Crawford JR, Haudek SB, Cieslik KA, Trial J, Entman ML, Origin of developmental precursors dictates the pathophysiologic role of cardiac fibroblasts, J. Cardiovasc. Transl. Res. 5 (2012) 749–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Haudek SB, Xia Y, Huebener P, Lee JM, Carlson S, Crawford JR, Pilling D, Gomer RH, Trial J, Frangogiannis NG, Entman ML, Bone marrow-derived fibroblast precursors mediate ischemic cardiomyopathy in mice, Proc. Natl. Acad. Sci. U S A 103 (2006) 18284–18289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Kanisicak O, Khalil H, Ivey MJ, Karch J, Maliken BD, Correll RN, Brody MJ, Sc JL, Aronow BJ, Tallquist MD, Molkentin JD, Genetic lineage tracing defines myofibroblast origin and function in the injured heart, Nat. Commun. 7 (2016) 12260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Hinz B, Gabbiani G, Mechanisms of force generation and transmission by myofibroblasts, Curr. Opin. Biotechnol. 14 (2003) 538–546. [DOI] [PubMed] [Google Scholar]
- [67].Wang J, Chen H, Seth A, McCulloch CA, Mechanical force regulation of myofibroblast differentiation in cardiac fibroblasts, Am. J. Physiol.-Heart Circ. Physiol. 285 (2003) H1871–H1881. [DOI] [PubMed] [Google Scholar]
- [68].Serini G, Bochaton-Piallat ML, Ropraz P, Geinoz A, Borsi L, Zardi L, Gabbiani G, The fibronectin domain ED-A is crucial for myofibroblastic phenotype induction by transforming growth factor-beta 1, J. Cell Biol. 142 (1998) 873–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Duan Q, McMahon S, Anand P, Shah H, Thomas S, Salunga HT, Huang Y, Zhang R, Sahadevan A, Lemieux ME, Brown JD, Srivastava D, Bradner JE, McKinsey TA, Haldar SM, BET bromodomain inhibition suppresses innate inflammatory and profibrotic transcriptional networks in heart failure, Sci. Transl. Med. 9 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].van Eys GJ, Niessen PM, Rensen SS, Smoothelin in vascular smooth muscle cells, Trends Cardiovasc. Med. 17 (2007) 26–30. [DOI] [PubMed] [Google Scholar]
- [71].Chambers RC, Leoni P, Kaminski N, Laurent GJ, Heller RA, Global expression profiling of fibroblast responses to transforming growth factor-beta(1) reveals the induction of inhibitor of differentiation-1 and provides evidence of smooth muscle cell phenotypic switching, Am. J. Pathol. 162 (2003) 533–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Ronty MJ, Leivonen S-K, Hinz B, Rachlin A, Otey CA, Kahari V-M, Carpen OM, Isoform-specific regulation of the actin-organizing protein palladin during TGF-ss 1-induced myofibroblast differentiation, J, Invest. Dermatol. 126 (2006) 2387–2396. [DOI] [PubMed] [Google Scholar]
- [73].Mykkanen OM, Gronholm M, Ronty M, Lalowski M, Salmikangas P, Suila H, Carpen O, Characterization of human palladin, a microfilament-associated protein, Mol. Biol. Cell 12 (2001) 3060–3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Perestrelo AR, Silva AC, Oliver-De La Cruz J, Martino F, Horváth V, Caluori G, Polansky O´, Vinarsky V´, Azzato G, de Marco G, Žampachová V, Skládal P, Pagliari S, Rainer A, Pinto-do-Ó P, Caravella A, Koci K, Nascimento DS, Forte G, Multiscale analysis of extracellular matrix remodeling in the failing heart, Circ. Res. 128 (2021) 24–38. [DOI] [PubMed] [Google Scholar]
- [75].Bomb R, Heckle MR, Sun Y, Mancarella S, Guntaka RV, Gerling IC, Weber KT, Myofibroblast secretome and its auto-/paracrine signaling, Expert Rev. Cardiovasc. Therapy 14 (2016) 591–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Sun Y, Weber KT, Infarct scar: a dynamic tissue, Cardiovasc. Res. 46 (2000) 250–256. [DOI] [PubMed] [Google Scholar]
- [77].Fisher SA, Absher M, Norepinephrine and ang ii stimulate secretion of tgf-beta by neonatal rat cardiac fibroblasts in-vitro, Am. J. Physiol.-Cell Physiol. 268 (1995) C910–C917. [DOI] [PubMed] [Google Scholar]
- [78].Moustakas A, Souchelnytskyi S, Heldin CH, Smad regulation in TGF-beta signal transduction, J. Cell Sci. 114 (2001) 4359–4369. [DOI] [PubMed] [Google Scholar]
- [79].Vasquez C, Benamer N, Morley GE, The cardiac fibroblast: functional and electrophysiological considerations in healthy and diseased hearts, J. Cardiovasc. Pharmacol. 57 (2011) 380–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Ranjan P, Kumari R, Verma SK, Cardiac fibroblasts and cardiac fibrosis: precise role of exosomes, Front. Cell Dev. Biol. 7 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Lyu L, Wang H, Li B, Qin Q, Qi L, Nagarkatti M, Nagarkatti P, Janicki JS, Wang XL, Cui T, A critical role of cardiac fibroblast-derived exosomes in activating renin angiotensin system in cardiomyocytes, J. Mol. Cell. Cardiol. 89 (2015) 268–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Yang J, Yu XF, Xue FT, Li YY, Liu W, Zhang S, Exosomes derived from cardiomyocytes promote cardiac fibrosis via myocyte-fibroblast cross-talk, Am. J. Transl. Res. 10 (2018) 4350–4366. [PMC free article] [PubMed] [Google Scholar]
- [83].Chen S, Puthanveetil P, Feng B, Matkovich SJ, Dorn GW 2nd, Chakrabarti S, Cardiac miR-133a overexpression prevents early cardiac fibrosis in diabetes, J. Cell Mol. Med. 18 (2014) 415–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Yuan J, Liu H, Gao W, Zhang L, Ye Y, Yuan L, Ding Z, Wu J, Kang L, Zhang X, Wang X, Zhang G, Gong H, Sun A, Yang X, Chen R, Cui Z, Ge J, Zou Y, MicroRNA-378 suppresses myocardial fibrosis through a paracrine mechanism at the early stage of cardiac hypertrophy following mechanical stress, Theranostics 8 (2018) 2565–2582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Bageghni SA, Hemmings KE, Zava N, Denton CP, Porter KE, Ainscough JFX, Drinkhill MJ, Turner NA, Cardiac fibroblast-specific p38α MAP kinase promotes cardiac hypertrophy via a putative paracrine interleukin-6 signaling mechanism, FASEB J.: Off. Publ. Feder. Am. Soc. Exp. Biol. 32 (2018) 4941–4954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Wang X, Zou Y, Chen Z, Li Y, Pan L, Wang Y, Liu M, Yin C, Wu J, Yang C, Zhang L, Li C, Huang Z, Wang D, Qian J, Ge J, Zou Y, Gong H, Low-density lipoprotein receptor-related protein 6 regulates cardiomyocyte-derived paracrine signaling to ameliorate cardiac fibrosis, Theranostics 11 (2021) 1249–1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].de Couto G, Macrophages in cardiac repair: Environmental cues and therapeutic strategies, Exp. Mol. Med. 51 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Wang C, Zhang C, Liu L, Xi A, Chen B, Li Y, Du J, Macrophage-derived mir-155-containing exosomes suppress fibroblast proliferation and promote fibroblast inflammation during cardiac injury, Mol. Ther. 25 (2017) 192–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Vagnozzi RJ, Maillet M, Sargent MA, Khalil H, Johansen AKZ, Schwanekamp JA, York AJ, Huang V, Nahrendorf M, Sadayappan S, Molkentin JD, An acute immune response underlies the benefit of cardiac stem cell therapy, Nature 577 (2019) 405–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Ieda M, Fu J-D, Delgado-Olguin P, Vedantham V, Hayashi Y, Bruneau BG, Srivastava D, Direct reprogramming of fibroblasts into functional cardiomyocytes by defined factors, Cell 142 (2010) 375–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Qian L, Huang Y, Spencer CI, Foley A, Vedantham V, Liu L, Conway SJ, Fu JD, Srivastava D, In vivo reprogramming of murine cardiac fibroblasts into induced cardiomyocytes, Nature 485 (2012) 593–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Vaseghi H, Liu J, Qian L, Molecular barriers to direct cardiac reprogramming, Protein Cell 8 (2017) 724–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Song K, Nam YJ, Luo X, Qi X, Tan W, Huang GN, Acharya A, Smith CL, Tallquist MD, Neilson EG, Hill JA, Bassel-Duby R, Olson EN, Heart repair by reprogramming non-myocytes with cardiac transcription factors, Nature 485 (2012) 599–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Wang L, Liu Z, Yin C, Asfour H, Chen O, Li Y, Bursac N, Liu J, Qian L, Stoichiometry of Gata4, Mef2c, and Tbx5 influences the efficiency and quality of induced cardiac myocyte reprogramming, Circ. Res. 116 (2015) 237–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Ifkovits JL, Addis RC, Epstein JA, Gearhart JD, Inhibition of TGFbeta signaling increases direct conversion of fibroblasts to induced cardiomyocytes, PLoS ONE 9 (2014) e89678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Zhao Y, Londono P, Cao Y, Sharpe EJ, Proenza C, O’Rourke R, Jones KL, Jeong MY, Walker LA, Buttrick PM, McKinsey TA, Song K, High-efficiency reprogramming of fibroblasts into cardiomyocytes requires suppression of pro-fibrotic signalling, Nat. Commun. 6 (2015) 8243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Yamakawa H, Muraoka N, Miyamoto K, Sadahiro T, Isomi M, Haginiwa S, Kojima H, Umei T, Akiyama M, Kuishi Y, Kurokawa J, Furukawa T, Fukuda K, Ieda M, Fibroblast growth factors and vascular endothelial growth factor promote cardiac reprogramming under defined conditions, Stem Cell Rep. 5 (2015) 1128–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Chen Y, Yang Z, Zhao ZA, Shen Z, Direct reprogramming of fibroblasts into cardiomyocytes, Stem Cell Res. Ther. 8 (2017) 118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Rao PK, Toyama Y, Chiang HR, Gupta S, Bauer M, Medvid R, Reinhardt F, Liao R, Krieger M, Jaenisch R, Lodish HF, Blelloch R, Loss of cardiac microRNA-mediated regulation leads to dilated cardiomyopathy and heart failure, Circ. Res. 105 (2009) 585–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Liu L, Yuan Y, He X, Xia X, Mo X, MicroRNA-1 upregulation promotes myocardiocyte proliferation and suppresses apoptosis during heart development, Mol. Med. Rep. 15 (2017) 2837–2842. [DOI] [PubMed] [Google Scholar]
- [101].Muraoka N, Yamakawa H, Miyamoto K, Sadahiro T, Umei T, Isomi M, Nakashima H, Akiyama M, Wada R, Inagawa K, Nishiyama T, Kaneda R, Fukuda T, Takeda S, Tohyama S, Hashimoto H, Kawamura Y, Goshima N, Aeba R, Yamagishi H, Fukuda K, Ieda M, MiR-133 promotes cardiac reprogramming by directly repressing Snai1 and silencing fibroblast signatures, EMBO J. 33 (2014) 1565–1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Jayawardena TM, Egemnazarov B, Finch EA, Zhang L, Payne JA, Pandya K, Zhang Z, Rosenberg P, Mirotsou M, Dzau VJ, MicroRNA-mediated in vitro and in vivo direct reprogramming of cardiac fibroblasts to cardiomyocytes, Circ. Res. 110 (2012) 1465–1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Jayawardena TM, Finch EA, Zhang L, Zhang H, Hodgkinson CP, Pratt RE, Rosenberg PB, Mirotsou M, Dzau VJ, MicroRNA induced cardiac reprogramming in vivo: evidence for mature cardiac myocytes and improved cardiac function, Circ. Res. 116 (2015) 418–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Li Y, Dal-Pra S, Mirotsou M, Jayawardena TM, Hodgkinson CP, Bursac N, Dzau VJ, Tissue-engineered 3-dimensional (3D) microenvironment enhances the direct reprogramming of fibroblasts into cardiomyocytes by microRNAs, Sci. Rep. 6 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Dal-Pra S, Hodgkinson CP, Mirotsou M, Kirste I, Dzau VJ, Demethylation of H3K27 is essential for the induction of direct cardiac reprogramming by miR combo, Circ. Res. 120 (2017) 1403–+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Nayak S, Herzog RW, Progress and prospects: immune responses to viral vectors, Gene Ther. 17 (2010) 295–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Ferreira MPA, Talman V, Torrieri G, Liu D, Marques G, Moslova K, Liu Z, Pinto JF, Hirvonen J, Ruskoaho H, Santos HA, Dual-drug delivery using dextran-functionalized nanoparticles targeting cardiac fibroblasts for cellular reprogramming, Adv. Funct. Mater. 28 (2018). [Google Scholar]
- [108].Muniyandi P, Palaninathan V, Mizuki T, Maekawa T, Hanajiri T, Mohamed MS, Poly(lactic-co-glycolic acid)/polyethylenimine nanocarriers for direct genetic reprogramming of MicroRNA targeting cardiac fibroblasts, ACS Appl. Nano Mater. 3 (2020) 2491–2505. [Google Scholar]
- [109].Nam YJ, Song K, Luo X, Daniel E, Lambeth K, West K, Hill JA, DiMaio JM, Baker LA, Bassel-Duby R, Olson EN, Reprogramming of human fibroblasts toward a cardiac fate, Proc. Natl. Acad. Sci. U S A 110 (2013) 5588–5593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Wada R, Muraoka N, Inagawa K, Yamakawa H, Miyamoto K, Sadahiro T, Umei T, Kaneda R, Suzuki T, Kamiya K, Tohyama S, Yuasa S, Kokaji K, Aeba R, Yozu R, Yamagishi H, Kitamura T, Fukuda K, Ieda M, Induction of human cardiomyocyte-like cells from fibroblasts by defined factors, Proc. Natl. Acad. Sci. U S A 110 (2013) 12667–12672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Fu JD, Stone NR, Liu L, Spencer CI, Qian L, Hayashi Y, Delgado-Olguin P, Ding S, Bruneau BG, Srivastava D, Direct reprogramming of human fibroblasts toward a cardiomyocyte-like state, Stem Cell Rep. 1 (2013) 235–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Li K, Zhu S, Russ HA, Xu S, Xu T, Zhang Y, Ma T, Hebrok M, Ding S, Small molecules facilitate the reprogramming of mouse fibroblasts into pancreatic lineages, Cell Stem Cell 14 (2014) 228–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Zhu S, Ambasudhan R, Sun W, Kim HJ, Talantova M, Wang X, Zhang M, Zhang Y, Laurent T, Parker J, Kim HS, Zaremba JD, Saleem S, Sanz-Blasco S, Masliah E, McKercher SR, Cho YS, Lipton SA, Kim J, Ding S, Small molecules enable OCT4-mediated direct reprogramming into expandable human neural stem cells, Cell Res. 24 (2014) 126–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Wang HX, Cao N, Spencer CI, Nie BM, Ma TH, Xu T, Zhang Y, Wang XJ, Srivastava D, Ding S, Small molecules enable cardiac reprogramming of mouse fibroblasts with a single factor, Oct4, Cell Rep. 6 (2014) 951–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Cao N, Huang Y, Zheng JS, Spencer CI, Zhang Y, Fu JD, Nie BM, Xie M, Zhang ML, Wang HX, Ma TH, Xu T, Shi GL, Srivastava D, Ding S, Conversion of human fibroblasts into functional cardiomyocytes by small molecules, Science 352 (2016) 1216–1220. [DOI] [PubMed] [Google Scholar]
- [116].Nam Y-J, Lubczyk C, Bhakta M, Zang T, Fernandez-Perez A, McAnally J, Bassel-Duby R, Olson EN, Munshi NV, Induction of diverse cardiac cell types by reprogramming fibroblasts with cardiac transcription factors, Development 141 (2014) 4267–4278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Hirai H, Katoku-Kikyo N, Keirstead SA, Kikyo N, Accelerated direct reprogramming of fibroblasts into cardiomyocyte-like cells with the MyoD transactivation domain, Cardiovasc. Res. 100 (2013) 105–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Hirai H, Kikyo N, Inhibitors of suppressive histone modification promote direct reprogramming of fibroblasts to cardiomyocyte-like cells, Cardiovasc. Res. 102 (2014) 188–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Ifkovits JL, Addis RC, Epstein JA, Gearhart JD, Inhibition of TGF beta signaling increases direct conversion of fibroblasts to induced cardiomyocytes, PLoS ONE 9 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Lee K, Yu P, Lingampalli N, Kim HJ, Tang R, Murthy N, Peptide-enhanced mRNA transfection in cultured mouse cardiac fibroblasts and direct reprogramming towards cardiomyocyte-like cells, Int. J. Nanomed. 10 (2015) 1841–1854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Zhao Y, Londono P, Cao Y, Sharpe EJ, Proenza C, O’Rourke R, Jones KL, Jeong MY, Walker LA, Buttrick PM, McKinsey TA, Song K, High-efficiency reprogramming of fibroblasts into cardiomyocytes requires suppression of pro-fibrotic signalling, Nat. Commun. 6 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Smith AW, Hoyne JD, Nguyen PK, McCreedy DA, Aly H, Efimov IR, Rentschler S, Elbert DL, Direct reprogramming of mouse fibroblasts to cardiomyocyte-like cells using Yamanaka factors on engineered poly (ethylene glycol) (PEG) hydrogels, Biomaterials 34 (2013) 6559–6571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Addis RC, Ifkovits JL, Pinto F, Kellam LD, Esteso P, Rentschler S, Christoforou N, Epstein JA, Gearhart JD, Optimization of direct fibroblast reprogramming to cardiomyocytes using calcium activity as a functional measure of success, J. Mol. Cell. Cardiol. 60 (2013) 97–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Jayawardena TM, Finch EA, Zhang L, Zhang H, Hodgkinson CP, Pratt RE, Rosenberg PB, Mirotsou M, Dzau VJ, MicroRNA induced cardiac reprogramming in vivo evidence for mature cardiac myocytes and improved cardiac function, Circ. Res. 116 (2015) 418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Fu Y, Huang C, Xu X, Gu H, Ye Y, Jiang C, Qiu Z, Xie X, Direct reprogramming of mouse fibroblasts into cardiomyocytes with chemical cocktails, Cell Res. 25 (2015) 1013–1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Talkhabi M, Pahlavan S, Aghdami N, Baharvand H, Ascorbic acid promotes the direct conversion of mouse fibroblasts into beating cardiomyocytes, Biochem. Biophys. Res. Commun. 463 (2015) 699–705. [DOI] [PubMed] [Google Scholar]
- [127].Wada R, Muraoka N, Inagawa K, Yamakawa H, Miyamoto K, Sadahiro T, Umei T, Kaneda R, Suzuki T, Kamiya K, Tohyama S, Yuasa S, Kokaji K, Aeba R, Yozu R, Yamagishi H, Kitamura T, Fukuda K, Ieda M, Induction of human cardiomyocyte-like cells from fibroblasts by defined factors, Proc. Natl. Acad. Sci. U S A 110 (2013) 12667–12672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Nam Y-J, Song K, Luo X, Daniel E, Lambeth K, West K, Hill JA, DiMaio JM, Baker LA, Bassel-Duby R, Olson EN, Reprogramming of human fibroblasts toward a cardiac fate, Proc. Natl. Acad. Sci. U S A 110 (2013) 5588–5593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Schmitt TM, Ragnarsson GB, Greenberg PD, T cell receptor gene therapy for cancer, Hum. Gene Ther. 20 (2009) 1240–1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].June CH, O’Connor RS, Kawalekar OU, Ghassemi S, Milone MC, CAR T cell immunotherapy for human cancer, Science 359 (2018) 1361–1365. [DOI] [PubMed] [Google Scholar]
- [131].Fischbach MA, Bluestone JA, Lim WA, Cell-based therapeutics: the next pillar of medicine, Sci. Transl. Med. 5 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Tran E, Chinnasamy D, Yu Z, Morgan RA, Lee C-CR, Restifo NP, Rosenberg SA, Immune targeting of fibroblast activation protein triggers recognition of multipotent bone marrow stromal cells and cachexia, J. Exp. Med. 210 (2013) 1125–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Wang L-CS, Lo A, Scholler J, Sun J, Majumdar RS, Kapoor V, Antzis M, Cotner CE, Johnson LA, Durham AC, Solomides CC, June CH, Pure E, Albelda SM, Targeting fibroblast activation protein in tumor stroma with chimeric antigen receptor T cells can inhibit tumor growth and augment host immunity without severe toxicity, Cancer Immunol. Res. 2 (2014) 154–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Lo A, Wang L-CS, Scholler J, Monslow J, Avery D, Newick K, O’Brien S, Evans RA, Bajor DJ, Clendenin C, Durham AC, Buza EL, Vonderheide RH, June CH, Albelda SM, Pure E, Tumor-promoting desmoplasia is disrupted by depleting FAP-expressing stromal cells, Cancer Res. 75 (2015) 2800–2810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Banerjee MN, Bolli R, Hare JM, Clinical studies of cell therapy in cardiovascular medicine: recent developments and future directions, Circ Res 123 (2018) 266–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Nguyen PK, Rhee JW, Wu JC, Adult stem cell therapy and heart failure, 2000 to 2016: a systematic review, JAMA Cardiol. 1 (2016) (2000) 831–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Forrester JS, Price MJ, Makkar RR, Stem cell repair of infarcted myocardium: an overview for clinicians, Circulation 108 (2003) 1139–1145. [DOI] [PubMed] [Google Scholar]
- [138].Blazquez R, Sanchez-Margallo FM, Crisostomo V, Baez C, Maestre J, Garcia-Lindo M, Uson A, Alvarez V, Casado JG, Intrapericardial administration of mesenchymal stem cells in a large animal model: a bio-distribution analysis, PLoS ONE 10 (2015) e0122377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Rupp H, Rupp TP, Alter P, Jung N, Pankuweit S, Maisch B, Intrapericardial procedures for cardiac regeneration by stem cells: need for minimal invasive access (AttachLifter) to the normal pericardial cavity, Herz 35 (2010) 458–465. [DOI] [PubMed] [Google Scholar]
- [140].Fernandez-Aviles F, Sanz-Ruiz R, Climent AM, Badimon L, Bolli R, Charron D, Fuster V, Janssens S, Kastrup J, Kim HS, Luscher TF, Martin JF, Menasche P, Simari RD, Stone GW, Terzic A, Willerson JT, Wu JC, T.W. Group, C. Authors/Task Force Members, S. Basic Research, S. Translational Research, S. Challenges of Cardiovascular Regenerative Medicine, S. Tissue Engineering, N.T. Delivery, S. Assessment, S. Clinical Trials, Regulatory, s. funding strategies, N.T. Delivery, S. Assessment, Global position paper on cardiovascular regenerative medicine, Eur. Heart J, 38 (2017) 2532–2546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Murry CE, Soonpaa MH, Reinecke H, Nakajima H, Nakajima HO, Rubart M, Pasumarthi KB, Virag JI, Bartelmez SH, Poppa V, Bradford G, Dowell JD, Williams DA, Field LJ, Haematopoietic stem cells do not transdifferentiate into cardiac myocytes in myocardial infarcts, Nature 428 (2004) 664–668. [DOI] [PubMed] [Google Scholar]
- [142].Lim SY, Cho DI, Jeong HY, Kang HJ, Kim MR, Cho M, Kim YS, Ahn Y, Adjuvant role of macrophages in stem cell-induced cardiac repair in rats, Exp. Mol. Med. 50 (2018) 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Liu M, Lutz H, Zhu D, Huang K, Li Z, Dinh PC, Gao J, Zhang Y, Cheng K, Bispecific Antibody Inhalation Therapy for Redirecting Stem Cells from the Lungs to Repair Heart Injury, Adv. Sci. (Weinheim, Baden-Wurttemberg, Germany), 8 (2020) 2002127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Su T, Huang K, Daniele MA, Hensley MT, Young AT, Tang J, Allen TA, Vandergriff AC, Erb PD, Ligler FS, Cheng K, Cardiac stem cell patch integrated with microengineered blood vessels promotes cardiomyocyte proliferation and neovascularization after acute myocardial infarction, ACS Appl. Mater. Interfaces 10 (2018) 33088–33096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Tang J, Cui X, Caranasos TG, Hensley MT, Vandergriff AC, Hartanto Y, Shen D, Zhang H, Zhang J, Cheng K, Heart repair using nanogel-encapsulated human cardiac stem cells in mice and pigs with myocardial infarction, ACS Nano 11 (2017) 9738–9749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Yu J, Zhang Y, Ye Y, DiSanto R, Sun W, Ranson D, Ligler FS, Buse JB, Gu Z, Microneedle-array patches loaded with hypoxia-sensitive vesicles provide fast glucose-responsive insulin delivery, Proc. Natl. Acad. Sci. U S A 112 (2015) 8260–8265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Prausnitz MR, Microneedles for transdermal drug delivery, Adv. Drug Deliv. Rev. 56 (2004) 581–587. [DOI] [PubMed] [Google Scholar]
- [148].Chiappini C, De Rosa E, Martinez JO, Liu X, Steele J, Stevens MM, Tasciotti E, Biodegradable silicon nanoneedles delivering nucleic acids intracellularly induce localized in vivo neovascularization, Nat. Mater. 14 (2015) 532–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Tang J, Wang J, Huang K, Ye Y, Su T, Qiao L, Hensley MT, Caranasos TG, Zhang J, Gu Z, Cheng K, Cardiac cell-integrated microneedle patch for treating myocardial infarction, Sci. Adv. 4 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [150].You Y, Kobayashi K, Colak B, Luo P, Cozens E, Fields L, Suzuki K, Gautrot J, Engineered cell-degradable poly(2-alkyl-2-oxazoline) hydrogel for epicardial placement of mesenchymal stem cells for myocardial repair, Biomaterials (2020), 120356–120356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].Hua C, Liu J, Hua X, Wang X, Synergistic fabrication of dose-response chitosan/dextran/beta-glycerophosphate injectable hydrogel as cell delivery carrier for cardiac healing after acute myocardial infarction, Dose-Response 18 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].Si R, Gao C, Guo R, Lin C, Li J, Guo W, Human mesenchymal stem cells encapsulated-coacervated photoluminescent nanodots layered bioactive chitosan/collagen hydrogel matrices to indorse cardiac healing after acute myocardial infarction, J. Photochem. Photobiol. B-Biol. 206 (2020). [DOI] [PubMed] [Google Scholar]
- [153].Guo HD, Wang HJ, Tan YZ, Wu JH, Transplantation of Marrow-derived cardiac stem cells carried in fibrin improves cardiac function after myocardial infarction, Tissue Eng. Part A 17 (2011) 45–58. [DOI] [PubMed] [Google Scholar]
- [154].Tang J, Su T, Huang K, Dinh PU, Wang Z, Vandergriff A, Hensley MT, Cores J, Allen T, Li T, Sproul E, Mihalko E, Lobo LJ, Ruterbories L, Lynch A, Brown A, Caranasos TG, Shen D, Stouffer GA, Gu Z, Zhang J, Cheng K, Targeted repair of heart injury by stem cells fused with platelet nanovesicles, Nat. Biomed. Eng. 2 (2018) 17–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [155].Li Z, Shen D, Hu S, Su T, Huang K, Liu F, Hou L, Cheng K, Pretargeting and bioorthogonal click chemistry-mediated endogenous stem cell homing for heart repair, ACS Nano 12 (2018) 12193–12200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].Lefrancais E, Ortiz-Munoz G, Caudrillier A, Mallavia B, Liu F, Sayah DM, Thornton EE, Headley MB, David T, Coughlin SR, Krummel MF, Leavitt AD, Passegue E, Looney MR, The lung is a site of platelet biogenesis and a reservoir for haematopoietic progenitors, Nature 544 (2017) 105–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [157].Guo R, Morimatsu M, Feng T, Lan F, Chang D, Wan F, Ling Y, Stem cell-derived cell sheet transplantation for heart tissue repair in myocardial infarction, Stem Cell Res. Ther. 11 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [158].Su T, Huang K, Mathews KG, Scharf VF, Hu S, Li Z, Frame BN, Cores J, Dinh PU, Daniele MA, Ligler FS, Cheng K, Cardiac stromal cell patch integrated with engineered microvessels improves recovery from myocardial infarction in rats and pigs, ACS Biomater. Sci. Eng. 6 (2020) 6309–6320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [159].Shen D, Li Z, Hu S, Huang K, Su T, Liang H, Liu F, Cheng K, Antibody-armed platelets for the regenerative targeting of endogenous stem cells, Nano Lett 19 (2019) 1883–1891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [160].Huang K, Li Z, Su T, Shen D, Hu S, Cheng K, Bispecific antibody therapy for effective cardiac repair through redirection of endogenous stem cells, Adv. Therap. 2 (2019). [Google Scholar]
- [161].Imanishi Y, Miyagawa S, Maeda N, Fukushima S, Kitagawa-Sakakida S, Daimon T, Hirata A, Shimizu T, Okano T, Shimomura I, Sawa Y, Induced adipocyte cell-sheet ameliorates cardiac dysfunction in a mouse myocardial infarction model: a novel drug delivery system for heart failure, Circulation 124 (2011) S10–S17. [DOI] [PubMed] [Google Scholar]
- [162].Tang J, Vandergriff A, Wang Z, Hensley MT, Cores J, Allen TA, Dinh P-U, Zhang J, Caranasos TG, Cheng K, A regenerative cardiac patch formed by spray painting of biomaterials onto the heart, Tissue Eng. Part C: Methods 23 (2017) 146–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [163].<Ke huang Sience translational.pdf>.
- [164].Luo L, Tang J, Nishi K, Yan C, Dinh PU, Cores J, Kudo T, Zhang J, Li TS, Cheng K, Fabrication of synthetic mesenchymal stem cells for the treatment of acute myocardial infarction in mice, Circ. Res. 120 (2017) 1768–1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [165].Tang J, Shen D, Caranasos TG, Wang Z, Vandergriff AC, Allen TA, Hensley MT, Dinh P-U, Cores J, Li T-S, Zhang J, Kan Q, Cheng K, Therapeutic microparticles functionalized with biomimetic cardiac stem cell membranes and secretome, Nat. Commun. 8 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [166].Su T, Huang K, Ma H, Liang H, Dinh P-U, Chen J, Shen D, Allen TA, Qiao L, Li Z, Hu S, Cores J, Frame BN, Young AT, Yin Q, Liu J, Qian L, Caranasos TG, Brudno Y, Ligler FS, Cheng K, Platelet-inspired nanocells for targeted heart repair after ischemia/reperfusion injury, Adv. Funct. Mater. 29 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [167].Zhang N, Song YA, Huang ZY, Chen J, Tan HP, Yang HB, Fan MK, Li QY, Wang QZ, Gao JF, Pang ZQ, Qian JY, Ge JB, Monocyte mimics improve mesenchymal stem cell-derived extracellular vesicle homing in a mouse MI/RI model, Biomaterials 255 (2020). [DOI] [PubMed] [Google Scholar]
- [168].Huang P, Wang L, Li Q, Tian X, Xu J, Xu J, Xiong Y, Chen G, Qian H, Jin C, Yu Y, Cheng K, Qian L, Yang Y, Atorvastatin enhances the therapeutic efficacy of mesenchymal stem cells-derived exosomes in acute myocardial infarction via up-regulating long non-coding RNA H19, Cardiovasc. Res. 116 (2020) 353–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [169].Singla DK, Stem cells and exosomes in cardiac repair, Curr. Opin. Pharmacol. 27 (2016) 19–23. [DOI] [PubMed] [Google Scholar]
- [170].Yan FL, Zhong ZR, Wang Y, Feng Y, Mei ZQ, Li H, Chen X, Cai L, Li CH, Exosome-based biomimetic nanoparticles targeted to inflamed joints for enhanced treatment of rheumatoid arthritis, Journal of Nanobiotechnology 18 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [171].Driesen RB, Nagaraju CK, Abi-Char J, Coenen T, Lijnen PJ, Fagard RH, Sipido KR, Petrov VV, Reversible and irreversible differentiation of cardiac fibroblasts, Cardiovasc. Res. 101 (2014) 411–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [172].Li CX, Talele NP, Boo S, Koehler A, Knee-Walden E, Balestrini JL, Speight P, Kapus A, Hinz B, MicroRNA-21 preserves the fibrotic mechanical memory of mesenchymal stem cells, Nat Mater 16 (2017) 379–389. [DOI] [PubMed] [Google Scholar]
- [173].Anavekar NS, Solomon SD, Angiotensin II receptor blockade and ventricular remodelling, J. Renin-Angiotensin-Aldosterone Syst. 6 (2005) 43–48. [DOI] [PubMed] [Google Scholar]
- [174].Sanghi S, Kumar R, Smith M, Baker KM, Dostal DE, Activation of protein kinase A by atrial natriuretic peptide in neonatal rat cardiac fibroblasts: Role in regulation of the local renin-angiotensin system, Regul. Pept. 132 (2005) 1–8. [DOI] [PubMed] [Google Scholar]
- [175].Rosenkranz S, TGF-beta(1) and angiotensin networking in cardiac remodeling, Cardiovasc. Res. 63 (2004) 423–432. [DOI] [PubMed] [Google Scholar]
- [176].Lijnen PJ, Petrov VV, Fagard RH, Collagen production in cardiac fibroblasts during inhibition of angiotensin converting enzyme and aminopeptidases, J. Hypertens. 22 (2004), S319–S319. [DOI] [PubMed] [Google Scholar]
- [177].Guo X-G, Uzui H, Mizuguchi T, Ueda T, Chena J-Z, Lee J-D, Imidaprilat inhibits matrix metalloproteinase-2 activity in human cardiac fibroblasts induced by interleukin-1 beta via NO-dependent pathway, Int. J. Cardiol. 126 (2008) 414–420. [DOI] [PubMed] [Google Scholar]
- [178].Pang X-F, Zhang L-H, Bai F, Wang N-P, Garner RE, McKallip RJ, Zhao Z-Q, Attenuation of myocardial fibrosis with curcumin is mediated by modulating expression of angiotensin II AT1/AT2 receptors and ACEACE2 in rats, Drug Design Dev. Therapy 9 (2015) 6043–6054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [179].Porter KE, Turner NA, O’Regan DJ, Ball SG, Tumor necrosis factor alpha induces human atrial myofibroblast proliferation, invasion and MMP-9 secretion: inhibition by simvastatin, Cardiovasc. Res. 64 (2004) 507–515. [DOI] [PubMed] [Google Scholar]
- [180].Chen J, Mehta JL, Angiotensin II-mediated oxidative stress and procollagen-1 expression in cardiac fibroblasts: blockade by pravastatin and pioglitazone, Am. J. Physiol.-Heart Circ. Physiol. 291 (2006) H1738–H1745. [DOI] [PubMed] [Google Scholar]
- [181].Moiseeva OM, Semyonova EG, Polevaya EV, Pinayev GP, Effect of pravastatin on phenotypical transformation of fibroblasts and hypertrophy of cardiomyocytes in culture, Bull. Exp. Biol. Med. 143 (2007) 54–57. [DOI] [PubMed] [Google Scholar]
- [182].Touyz RM, Schifffrin EL, Peroxisome proliferator-activated receptors in vascular biology-molecular mechanisms and clinical implications, Vasc. Pharmacol. 45 (2006) 19–28. [DOI] [PubMed] [Google Scholar]
- [183].Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, Morse EM, Keates T, Hickman TT, Felletar I, Philpott M, Munro S, McKeown MR, Wang Y, Christie AL, West N, Cameron MJ, Schwartz B, Heightman TD, La Thangue N, French CA, Wiest O, Kung AL, Knapp S, Bradner JE, Selective inhibition of BET bromodomains, Nature 468 (2010) 1067–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [184].Stratton MS, Bagchi RA, Felisbino MB, Hirsch RA, Smith HE, Riching AS, Enyart BY, Koch KA, Cavasin MA, Alexanian M, Song K, Qi J, Lemieux ME, Srivastava D, Lam MPY, Haldar SM, Lin CY, McKinsey TA, Dynamic chromatin targeting of BRD4 stimulates cardiac fibroblast activation, Circ. Res. 125 (2019) 662–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [185].Chen Y, Shi J, Zhang Y, Miao J, Zhao Z, Jin X, Liu L, Yu L, Shen C, Ding J, An injectable thermosensitive hydrogel loaded with an ancient natural drug colchicine for myocardial repair after infarction, J. Mater. Chem. B 8 (2020) 980–992. [DOI] [PubMed] [Google Scholar]
- [186].Fang L, Murphy AJ, Dart AM, A clinical perspective of anti-fibrotic therapies for cardiovascular disease, Front. Pharmacol. 8 (2017) 186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [187].Querejeta R, López B, González A, Sánchez E, Larman M, Martínez Ubago JL, Díez J, Increased collagen type I synthesis in patients with heart failure of hypertensive origin: relation to myocardial fibrosis, Circulation 110 (2004) 1263–1268. [DOI] [PubMed] [Google Scholar]
- [188].Kawamura M, Ito H, Onuki T, Miyoshi F, Watanabe N, Asano T, Tanno K, Kobayashi Y, Candesartan decreases type III procollagen-N-peptide levels and inflammatory marker levels and maintains sinus rhythm in patients with atrial fibrillation, J. Cardiovasc. Pharmacol. 55 (2010) 511–517. [DOI] [PubMed] [Google Scholar]
- [189].Kosmala W, Przewlocka-Kosmala M, Szczepanik-Osadnik H, Mysiak A, O’Moore-Sullivan T, Marwick TH, A randomized study of the beneficial effects of aldosterone antagonism on LV function, structure, and fibrosis markers in metabolic syndrome, JACC. Cardiovasc. Imag. 4 (2011) 1239–1249. [DOI] [PubMed] [Google Scholar]
- [190].Deswal A, Richardson P, Bozkurt B, Mann DL, Results of the randomized aldosterone antagonism in heart failure with preserved ejection fraction trial (RAAM-PEF), J. Cardiac Fail. 17 (2011) 634–642. [DOI] [PubMed] [Google Scholar]
- [191].Shimada YJ, Passeri JJ, Baggish AL, O’Callaghan C, Lowry PA, Yannekis G, Abbara S, Ghoshhajra BB, Rothman RD, Ho CY, Januzzi JL, Seidman CE, Fifer MA, Effects of losartan on left ventricular hypertrophy and fibrosis in patients with nonobstructive hypertrophic cardiomyopathy, JACC. Heart Failure 1 (2013) 480–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [192].Kosmala W, Przewlocka-Kosmala M, Szczepanik-Osadnik H, Mysiak A, Marwick TH, Fibrosis and cardiac function in obesity: a randomised controlled trial of aldosterone blockade, Heart (British Cardiac Society) 99 (2013) 320–326. [DOI] [PubMed] [Google Scholar]
- [193].Abulhul E, McDonald K, Martos R, Phelan D, Spiers JP, Hennessy M, Baugh J, Watson C, O’Loughlin C, Ledwidge M, Long-term statin therapy in patients with systolic heart failure and normal cholesterol: effects on elevated serum markers of collagen turnover, inflammation, and B-type natriuretic peptide, Clin. Ther. 34 (2012) 91–100. [DOI] [PubMed] [Google Scholar]
- [194].Prasad SK, Dargie HJ, Smith GC, Barlow MM, Grothues F, Groenning BA, Cleland JG, Pennell DJ, Comparison of the dual receptor endothelin antagonist enrasentan with enalapril in asymptomatic left ventricular systolic dysfunction: a cardiovascular magnetic resonance study, Heart (British Cardiac Society) 92 (2006) 798–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [195].López B, Querejeta R, González A, Sánchez E, Larman M, Díez J, Effects of loop diuretics on myocardial fibrosis and collagen type I turnover in chronic heart failure, J. Am. Coll. Cardiol. 43 (2004) 2028–2035. [DOI] [PubMed] [Google Scholar]
- [196].López B, González A, Beaumont J, Querejeta R, Larman M, Díez J, Identification of a potential cardiac antifibrotic mechanism of torasemide in patients with chronic heart failure, J. Am. Coll. Cardiol. 50 (2007) 859–867. [DOI] [PubMed] [Google Scholar]
- [197].López B, Querejeta R, González A, Beaumont J, Larman M, Díez J, Impact of treatment on myocardial lysyl oxidase expression and collagen cross-linking in patients with heart failure, Hypertension (Dallas Tex.: 1979) 53 (2009) 236–242. [DOI] [PubMed] [Google Scholar]
- [198].Giannetta E, Isidori AM, Galea N, Carbone I, Mandosi E, Vizza CD, Naro F, Morano S, Fedele F, Lenzi A, Chronic Inhibition of cGMP phosphodiesterase 5A improves diabetic cardiomyopathy: a randomized, controlled clinical trial using magnetic resonance imaging with myocardial tagging, Circulation 125 (2012) 2323–2333. [DOI] [PubMed] [Google Scholar]
- [199].Redfield MM, Chen HH, Borlaug BA, Semigran MJ, Lee KL, Lewis G, LeWinter MM, Rouleau JL, Bull DA, Mann DL, Deswal A, Stevenson LW, Givertz MM, Ofili EO, O’Connor CM, Felker GM, Goldsmith SR, Bart BA, McNulty SE, Ibarra JC, Lin G, Oh JK, Patel MR, Kim RJ, Tracy RP, Velazquez EJ, Anstrom KJ, Hernandez AF, Mascette AM, Braunwald E, Effect of phosphodiesterase-5 inhibition on exercise capacity and clinical status in heart failure with preserved ejection fraction: a randomized clinical trial, JAMA 309 (2013) 1268–1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [200].Teerlink JR, Metra M, Felker GM, Ponikowski P, Voors AA, Weatherley BD, Marmor A, Katz A, Grzybowski J, Unemori E, Teichman SL, Cotter G, Relaxin for the treatment of patients with acute heart failure (Pre-RELAX-AHF): a multicentre, randomised, placebo-controlled, parallel-group, dose-finding phase IIb study, Lancet (London, England) 373 (2009) 1429–1439. [DOI] [PubMed] [Google Scholar]
- [201].Krstevski C, Cohen CD, Dona MSI, Pinto AR, New perspectives of the cardiac cellular landscape: mapping cellular mediators of cardiac fibrosis using single-cell transcriptomics, Biochem. Soc. Trans. (2020). [DOI] [PubMed] [Google Scholar]
- [202].Ruiz-Villalba A, Romero JP, Hernández SC, Vilas-Zornoza A, Fortelny N, Castro-Labrador L, San Martin-Uriz P, Lorenzo-Vivas E, García-Olloqui P, Palacio M, Gavira JJ, Bastarrika G, Janssens S, Wu M, Iglesias E, Abizanda G, de Morentin XM, Lasaga M, Planell N, Bock C, Alignani D, Medal G, Prudovsky I, Jin YR, Ryzhov S, Yin H, Pelacho B, Gomez-Cabrero D, Lindner V, Lara-Astiaso D, Prósper F, Single-cell RNA sequencing analysis reveals a crucial role for CTHRC1 (collagen triple helix repeat containing 1) cardiac fibroblasts after myocardial infarction, Circulation 142 (2020) 1831–1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [203].Hu S, Li Z, Shen D, Zhu D, Huang K, Su T, Dinh PU, Cores J, Cheng K, Exosome-eluting stents for vascular healing after ischaemic injury, Nat. Biomed. Eng. (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [204].Li Z, Zhu D, Hui Q, Bi J, Yu B, Huang Z, Hu S, Wang Z, Caranasos T, Rossi J, Li X, Cheng K, Wang X, Injection of ROS-responsive hydrogel loaded with basic fibroblast growth factor into the pericardial cavity for heart repair, Adv. Funct. Mater. 31 (2021). [Google Scholar]
- [205].Zhu D, Li Z, Huang K, Caranasos TG, Rossi JS, Cheng K, Minimally invasive delivery of therapeutic agents by hydrogel injection into the pericardial cavity for cardiac repair, Nat. Commun. 12 (2021) 1412. [DOI] [PMC free article] [PubMed] [Google Scholar]