

Abstract
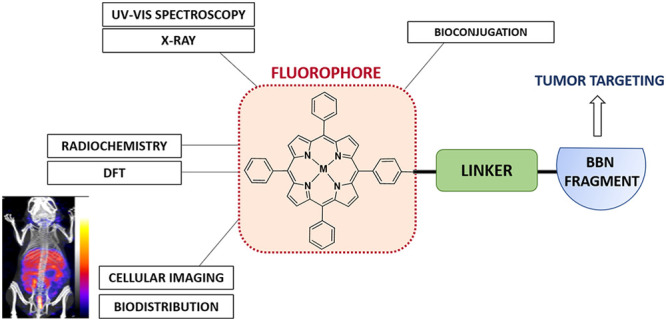
Over the past decade, porphyrin derivatives have emerged as invaluable synthetic building blocks and theranostic kits for the delivery of cellular fluorescence imaging and photodynamic therapy. Tetraphenylporphyrin (TPP), its metal complexes, and related derivatives have been investigated for their use as dyes in histology and as components of multimodal imaging probes. The photophysical properties of porphyrin–metal complexes featuring radiometals have been a focus of our attention for the realization of fluorescence imaging probes coupled with radioimaging capabilities and therapeutic potential having “true” theranostic promise. We report hereby on the synthesis, radiochemistry, structural investigations, and preliminary in vitro and in vivo uptake studies on a range of functionalized porphyrin-based derivatives. In pursuit of developing new porphyrin-based probes for multimodality imaging applications, we report new functionalized neutral, polycationic, and polyanionic porphyrins incorporating nitroimidazole and sulfonamide moieties, which were used as targeting groups to improve the notoriously poor pharmacokinetics of porphyrin tags. The resulting functional metalloporphyrin species were stable under serum challenges and the nitroimidazole and sulfonamide derivatives remained fluorescent, allowing in vitro confocal studies and visualization of the lysosomal uptake in a gallium(III) sulfonamide derivative. The molecular structures of selected porphyrin derivatives were determined by single crystal X-ray diffraction using synchrotron radiation. We also investigated the nature of the emission/excitation behavior of model functional porphyrins using in silico approaches such as TD DFT in simple solvation models. The conjugation of porphyrins with the [7-13] and [7-14] fragments of bombesin was also achieved, to provide targeting of the gastrin releasing peptide receptor (GRPR). Depending on the metal, probe conjugates of relevance for single photon emission computed tomography (SPECT) or positron emission tomography (PET) probes have been designed and tested hereby, using TPP and related functional free base porphyrins as the bifunctional chelator synthetic scaffold and 111In[In] or 68Ga[Ga], respectively, as the central metal ions. Interestingly, for simple porphyrin conjugates good radiochemical incorporation was obtained for both radiometals, but the presence of peptides significantly diminished the radio-incorporation yields. Although the gallium-68 radiochemistry of the bombesin conjugates did not show radiochemical incorporation suitable for in vivo studies, likely because the presence of the peptide changed the behavior of the TPP-NH2 synthon taken alone, the optical imaging assays indicated that the conjugated peptide tags do mediate uptake of the porphyrin units into cells.
1. Introduction
Porphyrins, a family of macrocyclic organic compounds incorporating four pyrrole rings interconnected through methine bridges,1 are naturally occurring molecules and excellent bioligands: a representative example of such conjugated macrocycles is heme, which is involved in several redox reactions and biological functions.2 Due to their stable, planar, and fully conjugated ring system, porphyrins are characterized by aromatic nature following Hückel’s rule (4n + 2). In particular, there are 22 π-electrons in this system, but only 18 are delocalized. Their desirable properties, such as the high stability and unique photophysical features, are attributed to this strong conjugation, especially the intense absorption in both the UV and visible regions of the electromagnetic spectrum. This makes them objects of interest in many biomedical and chemical fields, from cancer treatment and imaging to solar cells, from spin probes for electron paramagnetic resonance (EPR) to gas sensors.3−5 Furthermore, the structural features of the porphyrin core provide a stable coordination environment for metallic radionuclides, such as gallium-68 (t1/2 = 67.7 min), copper-64 (t1/2 = 12.7 h), and more recently manganese-52 (t1/2 = 5.5 days) for positron emission tomography (PET) and indium-111 (t1/2 = 2.8 days) for single photon emission computed tomography (SPECT) imaging.6−8 Research in the field of porphyrins as potential photodynamic therapy (PDT) sensitizers has attracted great interest, as recently reviewed by Abrahamse and Hamblin, and remarkable studies have been focused on the enhancement of porphyrin-based photosensitizers for PDT.9−11 Indeed, their fluorescent properties have made them good candidates for imaging probes, giving information on uptake and localization in the cellular environment.12 Since the porphyrin skeleton is easy to modify, structural changes have been attempted to achieve more efficient porphyrin-based conjugates as photosensitizers. In addition, the tetraphenyl core can be metalated with different metals and axial ligands to develop potential radiolabeled imaging probes, as demonstrated by the incorporation of copper-64 into a porphyrin-peptide-folate (PPF) probe.13 On this basis, imaging combined with PDT could allow more efficient understanding of intracellular mechanisms.14,15 Another example is represented by the synthesis and UV–vis spectroscopy investigation of Ga-acetylide complexes, showing that it is possible to generate new functional structures by using different axial ligands.16 We report here on the synthesis, radiochemistry, structural investigations, and preliminary in vitro and in vivo uptake studies on a range of functionalized porphyrin-based derivatives. TPP 1 and related compounds 2, 3, and 4 (Figure 1) were synthesized as described in the SI and analyzed on a laboratory scale. These simple porphyrin-based structures were selected as starting compounds, because they easily allow chemical modifications and are suitable for auxiliary derivatization to complex building blocks, from the perspective of their potential use for in vitro fluorescence. The purpose was to investigate the effects of different substituents introduced at the outer porphyrin ring in terms of hydrophobicity and biodistribution under physiological conditions. The synthetic methodologies involved hereby are supported by previous studies, which suggested that porphyrin derivatives possess poor intrinsic in vivo pharmacological properties with high nonspecific organ uptake observed with a range of structures.17 For this reason, the inclusion of targeting vectors onto the porphyrin structure was pursued, hereby aiming to improve the selectivity of porphyrin uptake. Prior studies on sulfonamide porphyrins were performed to investigate the binding affinity with several carbonic anhydrase (CA) enzymes.18 A large library of different sulfonamide structures was studied, with data suggesting that the free base porphyrins show medium-strength binding affinity for the cancer related CA IX and CA XII enzymes and favorable selectivity over the ubiquitous CA enzymes, although labeling with indium reduces this specificity. Nitroaromatic porphyrin derivatives have also been previously investigated as hypoxic agents, notably as cytotoxins and radiosensitizers with cobalt complexes of cationic porphyrins, showing selective toxicity toward hypoxic cells.19−22 Following the free base synthesis, selected porphyrin conjugates were subjected to radiolabeling experiments with gallium, zinc, nickel, copper, or indium. The influence of metal chelation on the electronic and structural properties of porphyrins was investigated. Interestingly, zinc metalation led to a simplified purification via column chromatography and limited the interference of the core NH groups in subsequent coupling reactions. Moreover, the corresponding zinc(II) metalated porphyrin was used as the starting material for gallium metalation. The final stage of this study was the coupling attempt between TPP-derived porphyrins and a peptide targeting moiety, following the recent advances in the field of peptide–drug photosensitizers for PDT.23 This was successfully achieved using two truncated analogues of bombesin (BBN), a naturally occurring 14-amino-acid peptide. BBN has been widely investigated for its role in tumor growth and spread.24 Indeed, it targets the well-known gastrin releasing peptide receptor (GRPR), overexpressed in several tumors, such as prostate, breast, small cell lung, and gastrointestinal cancers.25 In addition to the successful attempt to radiolabel the free TPP-NH24, a porphyrin–bombesin free ligand was also submitted to radiochemistry experiments and radiolabeled with gallium-68.
Figure 1.

Structural representation of the basic porphyrin-based building blocks: “cold” metal complexes and their precursors.
2. Results and Discussion
2.1. Choice of Porphyrin Building Blocks and Synthetic Optimizations
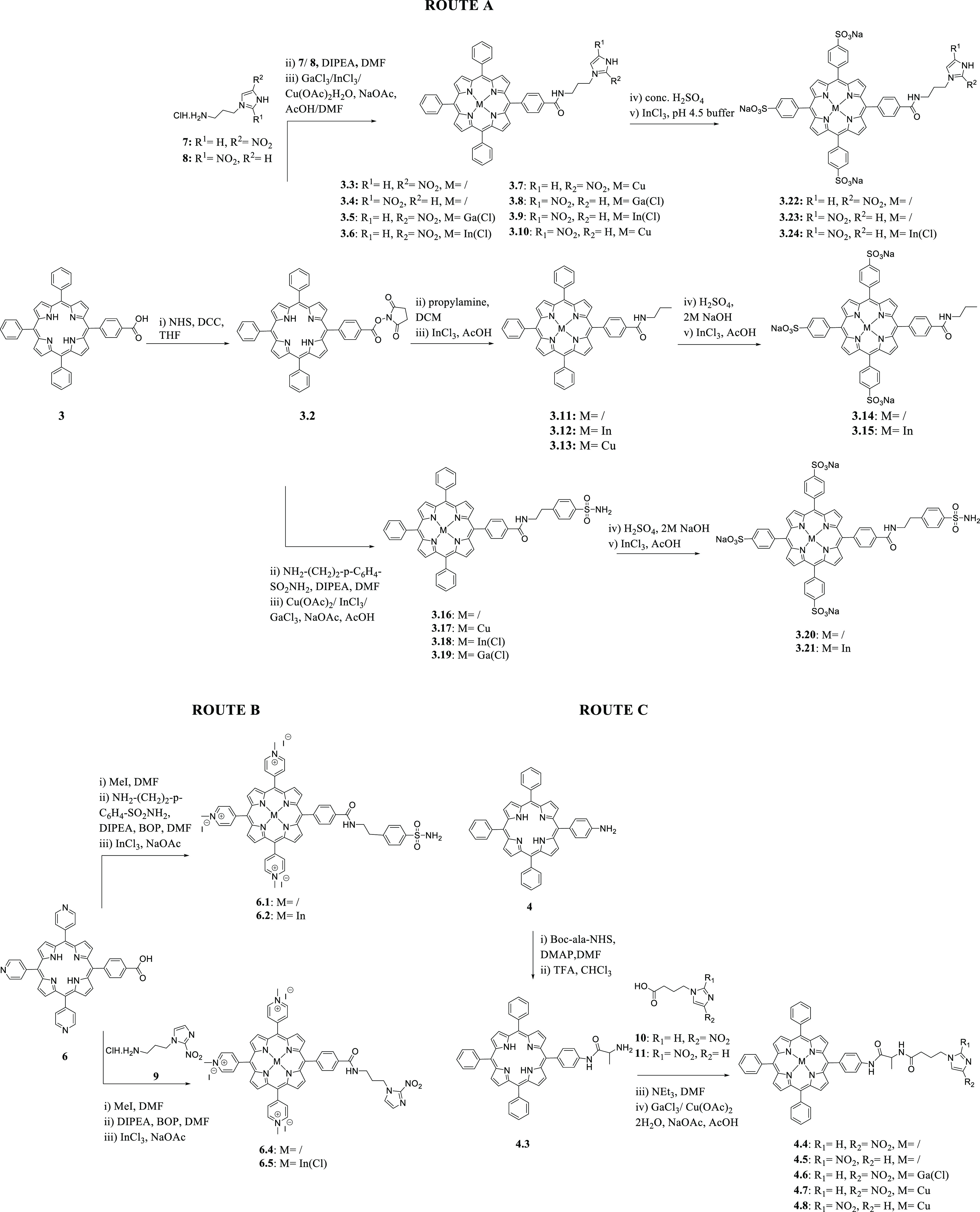
The synthesis of free base TPP 1 was accomplished following one of the most frequently used protocols for synthesizing meso-substituted porphyrins, the Adler-Longo method,26 via condensation of pyrrole and benzaldehyde in propanoic acid under reflux. Esters 2 and 5 were also prepared under Adler-Longo conditions with methyl-4-formylbenzoate or 4-pyridinecarboxaldehyde, respectively. Carboxylic acids 3 and 6 and amine 4 were then obtained as templates to prepare more challenging porphyrin-based structures (polyanionic, neutral, and polycationic species; Figures 1 and 2 and Scheme 1). Compounds 3 and 6 were synthesized through the hydrolysis of the corresponding methyl esters 2 and 5, as described in the literature.27 The synthesis of the monoamino TPP 4 was achieved through a modification of the method of Luguya et al.28 as reported by Dondi et al., from the reduction of the mononitro TPP using Pd/C 5% and sodium borohydride.29 Carboxylic acids 3 and 6 were functionalized with nitroimidazole and sulfonamide moieties. The functionalized porphyrins 3.3–3.24 were obtained from the activated intermediate N-hydroxysuccinimide ester 3.2, while the pyridyl porphyrin 6 was directly coupled with 4-(2-minoethyl)benzene sulfonamide/nitroimidazole moiety using (benzotriazol-1-yloxy)tris(dimethylamino)phosphonium hexafluorophosphate (BOP), as described in the SI. Metal complexation of the obtained porphyrins with GaCl3/InCl3/Cu(OAc)2 was successfully achieved, as confirmed by 1H and 13C NMR spectroscopy, ESI-MS, and HPLC analysis (SI). Furthermore, coupling of 2- and 4-nitroimidazoles 10/11 to monoamino TPP 4 gave porphyrins 4.4 and 4.5. In order to improve the initially poor yields, we decided to introduce an aliphatic linker to obtain a new amine group with increased nucleophilicity. To this purpose, intermediate 4.3 was synthesized through the reaction of the commercially available NHS activated alanine linker in 10-fold excess in DCM with DMAP, and it was used as precursor to the porphyrins 4.4–4.8. The coupling between porphyrin 4.3 and either 2- or 4-nitroimidazoles 10/11 in DMF gave the desired products, 4.4 and 4.5. The obtained ligands were then labeled with GaCl3 and Cu(OAc)2·H2O, as confirmed by 1H NMR spectroscopy, ESI-MS, and HPLC analysis (SI).
Figure 2.

Chemical structures of the most representative synthesized polyanionic, neutral and polycationic porphyrins, and their precursors.
Scheme 1. Synthetic Scheme of Functionalized Porphyrin Species.
Full experimental details and characterization data are given in SI.
2.2. Spectroscopic and Structural Investigations of Building Blocks and Simple Conjugates
UV–vis spectroscopy studies of porphyrin building blocks were recorded in solution and, wherever available, compared with literature examples.30 As expected, all free base porphyrins showed consistent λmax values between 400 and 420 nm and characteristic Q-band regions in the 500–700 nm range. The overlay of the spectra of compounds 2 and 4 indicated that modification of the functional group from −COOH or COMe to −NH2 does not have a significant effect on the UV–vis spectra of the porphyrins (SI). Furthermore, UV–vis spectroscopy provides a useful tool to differentiate between the free base and the metalated porphyrins: a comparison of the spectra of compounds 2 (TPP-OMe), 3 (TPP-COOH), and 3.1 (ZnTPP-COOH) showing that metalation of the porphyrin core had a significant influence on the Q-band region of the spectrum, as expected (SI). Indeed, the normal pattern of peaks IV, III, II, and I of increasing wavelength and decreasing intensity is not seen in the metalated porphyrin 3.1 and only two Q-bands peaks appear in the spectrum. A single peak at similar wavelength to that of Q-band III is observed (586 nm) with another much smaller peak at 30 nm higher wavelength (616 nm). This difference is not unexpected, as the Q-bands are known to be caused by the porphyrin core,30 and despite metalation, the λmax absorption at ca. 410 nm remains unchanged with respect to the free base porphyrins. The newly synthesized class of free base nitroimidazoles, the porphyrin 3.4 and its metal complexes 3.8–3.10 (Figure 3), show a strong Soret absorption band around 418 nm and a series of weaker distinct absorption bands in the Q-bands of the UV–vis spectrum. UV–vis absorption wavelengths (nm), respective molar extinction coefficients for compounds 3.3–3.10, and quantum yields with λex at 418 nm are reported in the SI. The free base porphyrin 3.4 gives a typical four band spectrum in the Q region, which reduces to two bands for the gallium (3.8) and indium (3.9) complexes and to one band for the copper complex (3.10). A red shift from 3.4 of 10 nm is observed in the Soret band for the indium and gallium complexes, while a small blue shift of 3 nm is measured for the copper complexes. This is consistent with the studies of Dorough et al., who report the spectral dependence of metallo-porphyrins on the d electron configuration of the central divalent metal and whether or not a d metal orbital is available for covalent bonding.31 For those metals which have vacant d orbitals, such as copper, silver, and cobalt, a blue shift occurs, while for those metals whose bond is limited to the s and p orbitals, a red shift is registered. Descending down a group, the spectrum shifts to the red while the intensity of the Soret band drops and the intensity of the first excitation band increases, in a similar way to that observed for the gallium and indium spectra 3.8 and 3.9. Changes in the Q-band region have also been reported by a recent UV–vis study, which shows the presence of an additional Q-band in the porphyrin spectra due to the effect of polyynic ligands with silyl end-capping groups (donor groups).16 UV–vis spectroscopy data of axial phenolic and para-aminophenolic incorporated indium(III) analogues also showed significant changes, since the axially ligated metal chelates display improved hyperchromicity of the optical bands in comparison with their free base derivative.32
Figure 3.

Structural representation of compounds 3.4 and 3.8–3.10. (a) Overlay of UV–vis spectra of 3.4 (black), 3.8 (red), 3.9 (blue), and 3.10 (green) (all spectra measured at 0.2 μM in DMF) with expansion of the Q region inset. (b) Overlay of fluorescence spectra of 3.4, 3.8, and 3.9 with λex at 418 nm (all spectra were measured at 0.2 μM in DMF).
Furthermore, UV–vis spectrophotometry was used as tool to assess the kinetic stability of the gallium 3.8 and indium 3.9 complexes prior to in vitro and in vivo radiolabeling studies using established assays.33 The corresponding experimental details are reported in the SI. The stability was modeled on the metalloporphyrins in phosphate buffered solution (PBS) (pH = 7.0) and in serum with respect to the loss of metal ions. By following the changes in absorption of the wavelengths characteristic to both the metal complex and the free base porphyrin over time, it is possible to establish a measure of the stability of the metal species. These studies suggest that the metal porphyrin complexes are stable with respect to loss of the metal ion on aqueous challenge. This is a key feature in the design of radiopharmaceuticals, aiming to ensure that nontarget organ accumulation of the probe is minimized. Following previous studies and interests in the field,6,7,18 we report herein on the new X-ray crystal structure analysis of porphyrins 2, 3.17, and 3.22. Crystals obtained using the vapor diffusion method are shown in the SI with the respective procedures. These new structures were deposited in CCDC CSD (Deposition Number: 2050384 for 1.1, 2050385 for 2, 2050382 for 3.17, and 2050725 for 3.22). Interestingly, we investigated the disposition of different metals inside the porphyrin core, by comparing the structures of copper sulfonamide complex 3.17 and the gallium metalated porphyrin 1.1. The crystal structure of 3.17 reveals the planar binding nature of the copper atom within the porphyrin tetrapyrrole unit, which is in contrast with the gallium metalated porphyrin 1.1 (Figure 4), where the gallium metal ion sits 0.415 Å above the N4 plane. In addition, the M–N bonds are longer for the gallium complex than in the copper species. Crystallographic investigations on simple TPP porphyrin-based systems have been reported over the past years; the CSD search reveals rather limited hits regarding functional metalloporphyrins fully characterized structurally when developed for biological applications, and in many cases synchrotron radiation source was necessary: the small crystal size, large unit cell, and very high degree of disorder within the crystals of such systems generally give rise to very weak scattering. Among them, literature data report the latest insight into indium porphyrin complexes, together with their morphology and aggregation behavior34 showing a pyramidal square geometry structure, in which In3+ ions are bound to the porphyrin core. We discovered this here in solving the synchrotron structure of gallium(III) derivative of TPP (2), where, in particular, this phenomenon called “doming” of the macrocyclic rings occurred, because of the large ionic radius of the metal ion that forces it to lie above the tetrapyrrole plane. Another interesting effect shown through crystallographic studies of certain metal complexes in the literature is represented by a relevant distortion of the plane, due to the four phenyl rings that are twisted by angles of ca. 45° with respect to the N4 core plane and because of steric interactions between the neighboring phenyl protons and β-pyrrole protons,34 which was also found in our investigations here: this may have important implications in the kinetic stability of the metal complexes synthesized. The coordination of a lanthanide ion, such as dysprosium, erbium, lutetium, and holmium (Dy, Er, Lu, Ho) in a tetradentate fashion on porphyrins has been studied, and the distortion from planarity has also been observed.35
Figure 4.
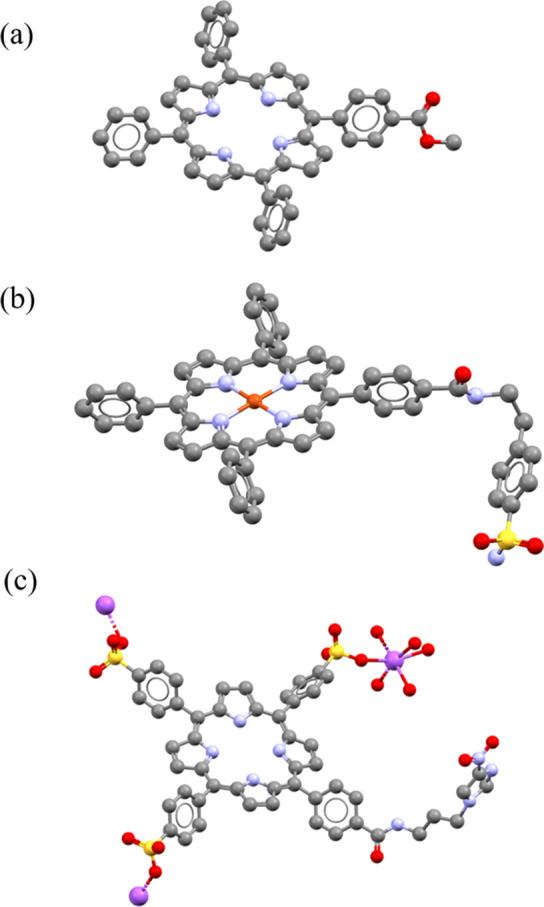
Molecular structures determined from single crystal X-ray diffraction for compounds 2 (a), 3.17 (b), and 3.22 (c). Color code: magenta = Na, blue = N, yellow = S, red = O, gray = C. H atoms and disordered units have been removed. Crystallography details for all structures are given in the SI.
Some relevant short interactions are displayed in the intramolecular network of the crystal structures of complex 3.17, 3.22, and 4.4 (SI). Packing for 3.17 involves H-bonding interactions between the amide oxygen and the sulfonamide nitrogen (O(1)···N(6)* 2.829 Å), and the bond lengths in the sulfonamide unit are in accord with those for similar aromatic sulfonamide species.36 For nitroimidazole compound 3.22, packing is also dominated by H-bonding interactions with H-bonding evident between the imidazole nitrogen and amide NH group (N(5)–H(51)···N(8)″), and nitro group oxygen and amide NH group (N(5)–H(51)···O(3)″) on two adjacent porphyrin conjugates. Weak T-type or edge–face interactions are also present between phenyl rings and pyrrole groups of adjacent molecules, and the partially positively charged hydrogen atom of one of the phenyl rings points perpendicular to the center of the aromatic plane of the pyrrole ring (SI).
2.3. DFT Calculations on the Free Base and Metalated Porphyrins
To gain further insight into the electronic structure of the new porphyrins and to validate the spectroscopic data, we investigated the nature of the porphyrin’s excitation/emission behavior using in silico approaches, using the synchrotron diffraction data of compound 1.1 and the laboratory-source structure of compound 2 as initial starting geometry for geometry optimizations. Density functional theory (DFT) and time dependent density functional theory (TDDFT) calculations on compounds 2, 3, 4, 4.1, and 4.2 and a model (not-synthesized) In(Cl) complex of 4 were performed using the Amsterdam Density Functional (ADF) suite, as described in the SI.37 DFT calculations, extended to the time-dependent DFT approximations with the same basis set, provided a qualitative visualization of the compounds’ molecular orbitals, allowing estimation of the likely influence of substituents on the energy transitions involved, as shown in Figure 5 for 4, 4.1, 4.2, and 4-InCl. This is most relevant to the HOMO and LUMO orbitals, likely participating in the S0 to S1 electronic transition,38−40 and can be used in combination with UV–vis and fluorescence emission data for better understanding of molecule excitation and for establishing the effect of the various functional groups. Geometry optimizations led to geometries well within expectations. Distances and bond angles roughly represented those of the structural data obtained through single crystal X-ray diffraction of related compounds, and the frontier molecular orbitals were modeled. Differences between the orbitals of the protected and free acid porphyrins, compounds 2 and 3, are very limited. By comparing the orbital diagrams of 2 and 3 with that of compound 4, the TPP-NH2 variant, there are some notable differences instead. The carbonyl group of the COOMe/COOH and the phenyl to which it is bonded are involved in neither the HOMO orbital nor any recorded molecular orbitals lower than the HOMO. Conversely, the NH2 group and the phenyl to which it is bonded are involved in neither the LUMO molecular orbital nor any recorded orbitals higher than the LUMO. The energies of the HOMO+1, HOMO, LUMO, and LUMO–1 orbitals in kJ/mol, wavelength, and frequency are shown in the SI. As part of the modeling of molecular orbitals, optimized geometries and predicted bond lengths and angles were obtained for compounds 2, 3, and 4. Directly comparing between the values provided by X-ray crystallography and those modeled for compound 2, all the C–C and C–N lengths are consistent. Therefore, by these models, no significant structural distance between the distinctly functionalized porphyrins should be expected. In addition, the effect of metals coordination was examined, and it was seen that it does not lead to structural distortions, with C–C and C–N bond-lengths predicted to be similar irrespective of the coordinated metal. The free base 4 and the zinc complex 4.1 gave coplanar structures, while the gallium chloride center is predicted to be raised 0.38 Å over the porphyrin plane. The indium chloride center is predicted to be raised 0.58 Å. While the displacement found in crystal structures varies significantly and is dependent on a variety of factors, there are examples of Ga or In TPP crystal structures with values similar to what was found here.41,42 For instance, Enakieva et al. reported an indium chloride porphyrin complex, in which the indium atom was displaced out of the plane by 0.607 Å.43 Transition energies, orbital contributions, and comparison of results obtained using either TD-DFT+TB or Davidson methodology44−46 are described in the SI.
Figure 5.
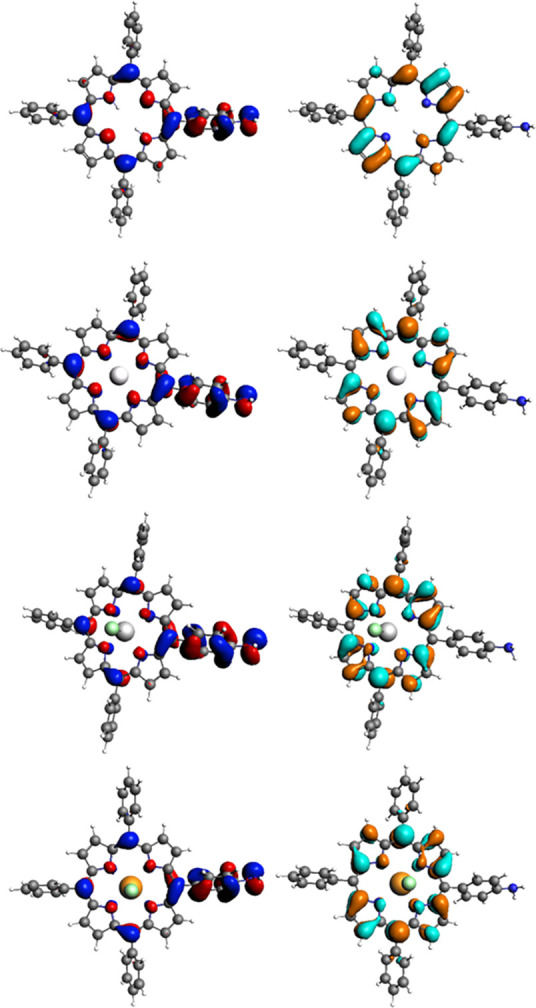
Illustrations of significant frontier orbitals: HOMO (left, blue/red) and LUMO (right, orange/cyan) of 4, 4.1, 4.2, and 4-InCl (top to bottom). Further images are given in SI.
Further details, such as the results of the TDDFT calculations may be found in the SI. As we found that compound 1.1 crystallized as the acetate (1.1-OAc) rather than the chloride salt, we decided to also model this structure by DFT and investigate its transition energies. The structures of 4.2 and 4.2-OAc are depicted below. In addition to the structures depicted above, we decided to calculate the structure of the gallium acetate complex 4.2-OAc. The optimized structures and reconstructed COSMO-surfaces are depicted in Figure 6. The results of the geometry optimization suggest a reduction in the symmetry of the porphyrin. The corresponding TDDFT calculations indicate the occurrence of a number of additional excitation energies, which are likely the result of this reduction in symmetry (see SI for TDDFT spectra).
Figure 6.
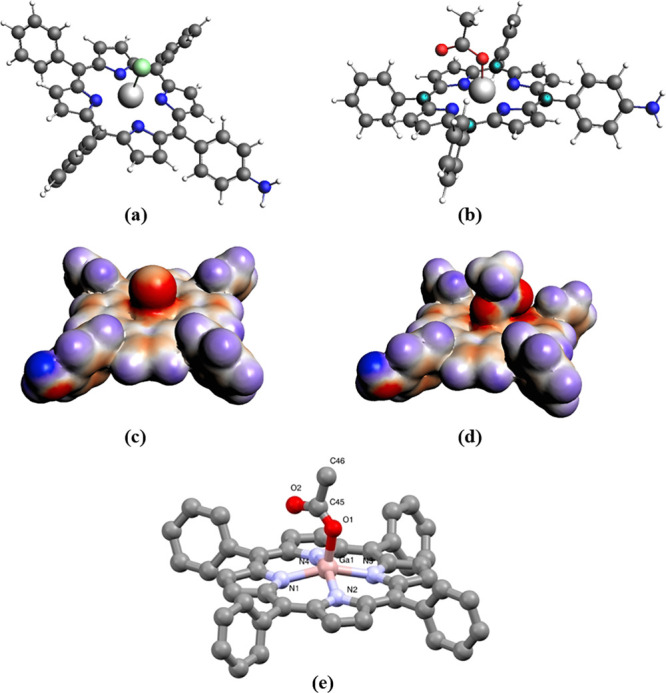
Optimized geometries for model compounds: (a) 4.2-Cl substituted and (b) 4.2-OAc substituted; corresponding COSMO surfaces: (c) 4.2-Cl substituted and (d) 4.2-OAc models; the molecular structure determined from synchrotron single crystal X-ray diffraction analysis for compound 1.1-OAc substituted (e).
2.4. Cellular Imaging Assays
Cellular uptake studies were carried out on PC-3 and HeLa cell lines to investigate the ability of the porphyrins to cross the cell membrane, their distribution, localization and to determine how functionalization can affect their biological behavior. PC-3 cells are a prostate cancer cell line overexpressing the GRPR and are widely used in cancer research. In this study, they were chosen to investigate if the BBN conjugates show enhanced uptake in this cell line, compared to HeLa cells, that do not express the GRPR receptor (Section 2.6). Confocal microscopy and lifetime decay images were used as tools to compare TPP (1), TPP-COOH (3), and TPP-NH2(4). Confocal microscopy images were acquired by excitation with either the 405 nm (blue) or 488 nm (green) lasers, and emissions filtered below 600 nm. Initial cell uptake assays with PC-3 cells over a range of concentrations and incubation times were carried out with porphyrins 2 (TPP-COOMe) and 3 (TPP-COOH) to investigate the potential for these compounds as in vitro fluorescence markers. Poor uptake of both porphyrins after incubation for 1 h was observed (Figure 7). There appears to be some aggregation of porphyrins around the outside of the cell membranes. For porphyrin 2, there are some areas where both porphyrin and nuclear stain are evident, although the cell nuclei no longer appear to be intact. For porphyrin 3, it would appear from the change in morphology that the presence of the porphyrin has triggered cell death, causing the cells to detach from the base of the dish. Both of these porphyrins are poorly soluble in aqueous solvents, and their highly hydrophobic nature may explain their poor uptake.
Figure 7.
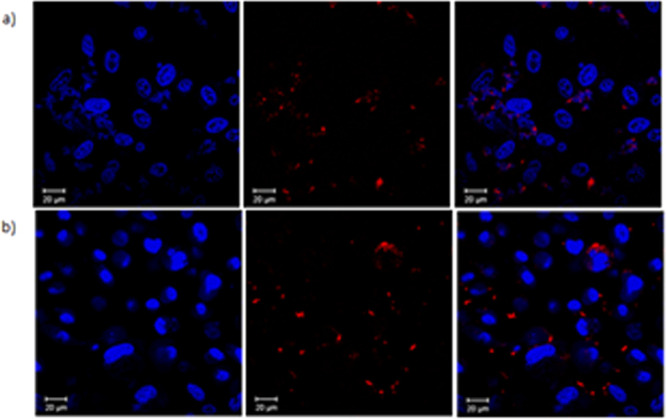
Confocal images of TPP-COOMe (2) and TPP-COOH (3) at 10 μM solution in DMSO (1%) with PC-3 cells after incubation for 1 h at 37 °C. Hoechst nuclear stain was used (1 μg/mL, blue) to visualize the cell nuclei and porphyrins show red fluorescence: (a) Hoechst stained nuclei, porphyrin 2, overlay of blue and red emission channels images; (b) Hoechst stained nuclei, porphyrin 3, overlay of blue and red emission images; Scale bar 20 μm.
For proof of concept, cell uptake studies of TPP-COOH (3) using two-photon FLIM (2P FLIM) were performed on PC-3 cells. The ligand was dissolved in a small amount of DMSO (1%) and added to the cell culture medium to give a concentration of 10 μM. The lifetime distribution profile for the uptake of ligand 3 in PC-3 cells is shown in Figure 8. The high intensity and short lifetime decay is likely due to the tendency of the porphyrins to generate aggregates. Confocal fluorescence imaging on PC-3 cells was performed for TPP-NH2 (4) (Figure 9). The image resolution allows us to observe the porphyrins bound to specific organelles inside the cells. In the red region, porphyrins generate aggregates or punctate regions, leading to high intensity but very short lifetime decay. In the blue region, porphyrins are localized mostly in the cytoplasm. The corresponding FLIM data was unremarkable and very closely matching that of compound 3, yet the intensity images shows a different behavior for TPP-NH2 (4) in comparison with TPP-COOH (3). TPP-NH2 is likely to reside in the cytoplasm, as revealed by bright dots throughout, under pH control, and to be accumulated presumably in the lysosomes. In the case of TPP-COOH, the distribution is probably controlled mainly by the porphyrin core. Colocalization studies are needed to investigate if TPP-COOH preferentially binds to endoplasmic reticulum, as suggested by the subcellular structures observed under fluorescence imaging. The poor solubility and inability to cross cell membranes for free base or metalated porphyrins building blocks 1–4, all necessitating long-term incubation highlighted the necessity to further develop synthetically these porphyrin frameworks.
Figure 8.
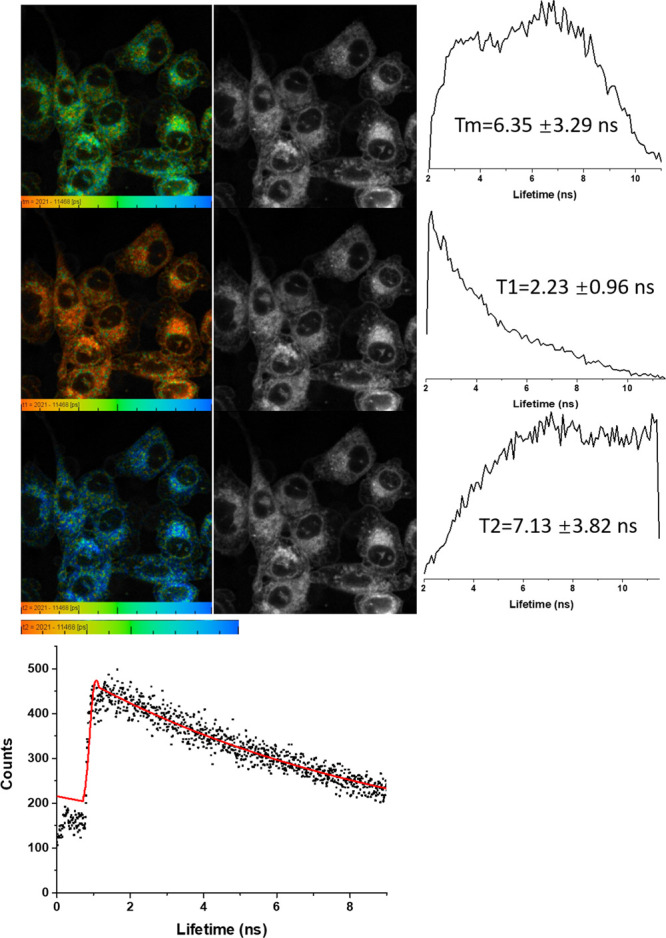
2-Photon FLIM of compound 3 in PC-3 cells after 20 min incubation, 10 μM total conc., DMSO (1%), 3 mW, 810 nm excitation (top three rows). The corresponding solution TCSPC spectra and fitted curve are given: 10 μM in DMSO, χ2 1.65, and τ1 20 ps (38%, indicating aggregation, forming short-lived excimers with lifetime within instrument response) and a porphyrin-characteristic τ2 of 11.37 ns (62%).
Figure 9.
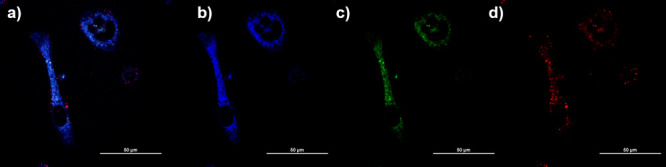
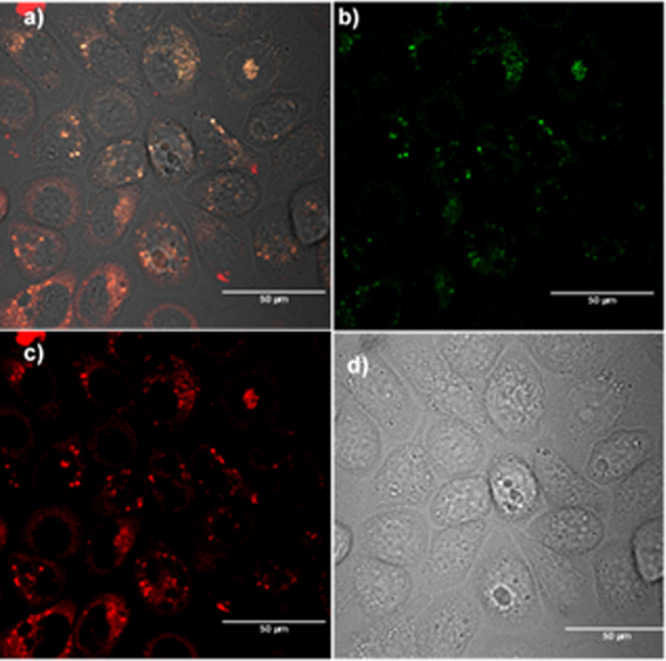
Confocal images for compound 4 in PC-3 cells incubated for 12 h at 10 μm concentration, 37 °C: (a) overlay of blue-green-red channels; (b) blue channel (λem = 417–477 nm); (c) green channel (λem = 500–550 nm); (d) red channel (λem = 570–750 nm) (see SI for additional images and details).
To assess how physicochemical modulations can influence the balance between lipophilicity and hydrophilicity and the consequent cellular internalization, cell uptake studies were carried out on nitroimidazole and sulfonamide derivatives. Incubation of the compound 4.6 with the HeLa cell line over a 4 h period was performed. Compound 4.6 showed strong uptake of the complex with localization in lysosomes apparent from colocalization studies with lysotracker (Figure 10). To assess the binding potential of representative members of the sulfonamide-functionalized porphyrin, and compare the uptake of the free base vs gallium substituted compound in a cell line known to overexpress the CA IX receptors, confocal fluorescence imaging assays were performed with compounds 3.16 and 3.19 using the HCT116 colon carcinoma cell line denoted CAIX positive, which has previously been transfected to overexpress the CA IX enzyme without the need for hypoxic culturing using a known protocol.18 Long-term incubation (over 4 h) with the gallium sulfonamide compound 3.19 and its free base precursor 3.16 in HCT116 (CAIX positive cell line) and subsequent confocal fluorescence imaging showed cytoplasm localization upon λex 405 nm where λem was >600 (Figure 11). There is no distinguishable difference between the uptake in the positive CA IX expressing cell line and the empty vector cell line, in line with previous observations in a related family of compounds. Images shown in Figure 9 are a representative sample of the recorded images (n = 5). The two most notable observations are (a) the free base vs gallium substituted porphyrin species investigated show distinct cellular distributions; (b) in general, cellular imaging suggests that the uptake and localization is strongly driven by the porphyrin unit and not by the nitroimidazole or sulfonamide groups, and the free-base TPP-type derivatives shows strong cytosol uptake with no nuclear staining, as do their indium or gallium derivatives.
Figure 10.
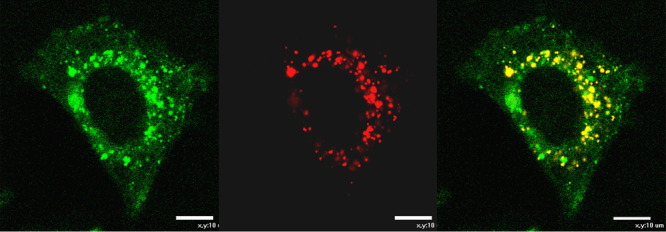
In vitro confocal fluorescence uptake studies of 4.6 (10 μM) in HCT116 (CAIX positive) living cells (4 h incubation, 37 °C, λex = 405 nm, λem = 600 nm); (a) 4.6 uptake; (b) Lysotracker Red uptake; (c) overlay of images recorded after Lysotracker Red dye staining and 4.6 uptake (scale bar 10 μm).
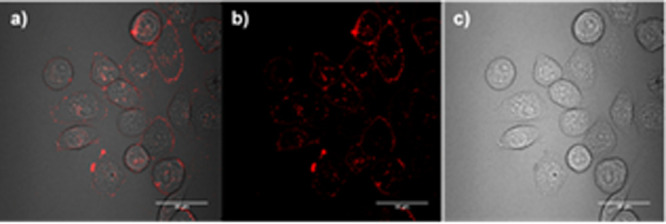
Figure 11.
(a) Confocal fluorescence imaging in living CA IX positive HCT116 cells of the free base precursor 3.16 10 μM 16 h (λex = 410 nm, λem = 625 nm), and (b) corresponding bright field image. (c) Confocal fluorescence imaging in living CA IX positive HCT116 cells of the gallium complex 3.19 16 h, λex = 405 nm, λem = 625 nm and (d) corresponding bright field image; Scale bar = 10 μm.
2.5. Preliminary Radiolabeling Tests
It is well-known that the introduction of a metal, such as gallium or indium, into the porphyrin core causes changes in physicochemical characteristics, including optical properties and lipophilicity.47 Experiments to label the porphyrin core with natural abundance samples of gallium, zinc, nickel, and indium were undertaken as described above, before radiolabeling with gallium-68 and indium-111 was attempted. Experimental details are reported in the SI, with 1H NMR spectroscopy and ESI-MS confirming the production of the “cold” metal complexes.
2.5.1. Radiochemical Experiments with Gallium-68
Experiments to obtain the gallium complex 4.2 first involved natural abundance gallium samples, showing that it is possible to obtain it through a transmetalation reaction from zinc to gallium. The reaction was conducted on a small scale with GaCl3 in methanol and the presence of the desired product was demonstrated by ESI-MS spectrometry (SI). In addition, experiments to directly label various porphyrins with gallium-68 were carried out at different temperatures (95 °C, 100 °C, 120 °C) and using different solvents (H2O, DMSO, EtOH, MeCN, THF). All conditions tested resulted in low radiochemical incorporation (ROI < 20%). The HPLC chromatogram in Figure 12 shows the radiolabeled porphyrin derived from 4, after purification by C18-silica, with a retention time of 10 min, and the free gallium-68 ions peak at 2.17 min. Injection of a product sample purified by reverse-phase chromatography over C18-silica revealed that the product still contained free gallium-68, potentially indicating low kinetic stability of the resulting complex.
Figure 12.

Radiochromatogram of compound [68Ga]4.2 labeled with generator-produced [68Ga]GaCl3 ions in water, after purification by chromatography over a C18-silica cartridge.
In our hands, the radiochemical incorporation dropped significantly (ROI < 1%), when peptide-conjugated porphyrins were used. A reason for this might be that the investigated peptides (bombesin analogues and fragments, vide infra) interfered with the complexation, by interacting with the gallium ions in solution.
2.5.2. Radiochemical Experiments with Indium-111 and Preliminary in Vivo Evaluations
Radiolabeling studies with indium-111, as well as lipophilicity, serum stability, and tumor uptake assays, were undertaken to investigate the pharmacokinetic properties of cationic and negatively charged porphyrins. Radiolabeling studies with indium-111 suggested that both the cationic and negatively charged porphyrin species displayed similar biodistributions and inclusion of a peripheral structure had little effect on the intrinsic biodistribution over 24 h. The nitroimidazole porphyrin free ligand 3.23 was radiolabeled with indium-111 to establish if the nitroimidazole indium complex 3.24 retains any of the hypoxia selectivity of the nitroimidazole vector. Early attempts to radiolabel 3.24 in glass vials gave low radiochemical yields, as a result of the activity becoming associated with the walls of the reaction vial. Subsequent pre-acid washing of the vial improved the yield, while optimum yields were ultimately achieved when the reaction was carried out in a plastic Eppendorf tube. The UV trace of 3.23 was overlaidwith the radio trace of [111In] 3.24 (SI). However, the concentration of the indium-labeled porphyrin was extremely low, and as such, the UV trace of the metal complex was not detectable. In the present study, the free base porphyrin 3.23 has not been separated from the radiolabeled [111In]3.24, and the specific activity of labeled porphyrin remained low. For comparison, 3.15 was radiolabeled following a similar protocol to 3.24 with [111In]InCl3 in 94.5% radiochemical purity (Figure 13), to assess the effect of the nitroimidazole group on the intrinsic uptake properties of the sulfonated phenyl porphyrin. The uptake of fluoromisonidazole [18F] (FMISO) was similarly run in parallel as a positive control for a nitroimidazole compound that displays hypoxia selectivity under the experimental conditions (SI). To investigate the hypoxic uptake, in vitro studies were performed under two different oxygen concentrations.48 Results shown in Figure 13 indicate that the porphyrin [111In]3.15 shows identical uptake under both normoxic and anoxic conditions, while the nitroimidazole functionalized species [111In]3.24 displays a 2-fold higher uptake under anoxic conditions. The differential uptake over the two different gas concentrations of the conjugate is comparable to the FMISO standard run in parallel (SI). These results are strongly encouraging and suggest that specific localization in hypoxic tissue could be possible with this bifunctional porphyrin chelator. However, the general low percentage uptake may suggest that the negative charge of the porphyrin group decreases the cellular inclusion and further work will be required to establish if the neutral or cationic porphyrin analogues show improved hypoxia selectivity. Hypoxia selectivity of porphyrin nitroimidazole was demonstrated in standard cellular assays using HeLa cells cultured under variable O2 concentrations (SI). As expected from their electrochemistry behavior, the nitroimidazole conjugate maintains hypoxia selectivity when labeled with indium-111, whereas the absence of this group markedly diminishes its uptake in anoxic cells. Cyclic voltammetry studies of the nitroimidazole conjugates (SI) also suggest that the reduction potentials of these bioreducible hypoxia selective groups are unperturbed by the porphyrin entity and would, based on electrochemical parameters alone, undergo hypoxic trapping under in vitro conditions.
Figure 13.

(top) Structures of [111In]3.24 (red) and [111In]3.15 (black) with corresponding UV–vis and radio HPLC traces. (bottom) Hypoxia selectivity assay in vitro (HeLa cell line), biodistribution data of [111In]3.15, and SPECT image measured at 24 h of MKN45 tumor bearing mice injected with 3.15 (n = 6). Further details are given in SI.
Serum binding studies of [111In]3.15, [111In]6.6, and [111In]3.20 suggest that the porphyrin conjugates are nearly all completely bound by serum proteins, which may impede the processing of these hypoxic selective groups for bioreductive trapping or for specific receptor binding. In vivo radiolabeling studies of the basic porphyrin core structures based on the tri(sulfonyl-phenyl) and tri(methyl-pyridinium) porphyrin structures have revealed almost identical biodistributions over 24 h incubation with high uptake in liver, kidney, and other nontarget organs such as the spleen (SI). Tumor uptake was reasonable, but in contrast to similar compounds in the literature, poor tumor to blood and tumor to tissue ratios were measured, suggesting that these agents would not be suitable as SPECT imaging agents. Indium-labeled sulfonyl porphyrin sulfonamide species showed similar uptake and biodistributions to the nonsulfonamide porphyrin species, suggesting that the intrinsic background biodistribution is dominated by the porphyrin unit.
2.6. Bioconjugation and Cellular Imaging Protocols for Targeting PC-3 Cells
To extend our toolbox of functional porphyrin probes, preliminary studies were performed to develop several new porphyrin peptide bioconjugates, following the earlier design features whereby the porphyrin would act as a fluorophore, attached to a targeting functionality through a linking moiety.6,7,18 These conjugates are of utmost relevance, since one of the most recently developed applications of porphyrins is their use as multimodal imaging probes and photosensitizers in PDT. In the latter case, conjugation of porphyrins and other tetrapyrrole photosensitizers to antibodies and cell penetrating peptides (CPPs) has been studied to obtain amphiphilic derivatives suitable for PDT in tumor models and other applications in photobiology.29,49−52 CPPs are a family of short (8–30) amino acid sequences able to penetrate across the cell membrane, to facilitate the uptake of a range of molecules, either covalently or noncovalently bonded, and to achieve a targeted drug delivery.53 Early examples of gallium-labeled porphyrin bioconjugates submitted to biological evaluation as therapeutic agents date back to 2015, whereby azide functionalized porphyrins radiolabeled with gallium-68, gallium-69, and gallium-71 were bound to a dodecapeptide and shown to exhibit good affinity for α6β1-integrin.54 A recent example of a porphyrin bioconjugate is a cationic porphyrin bound to a derivative of polymyxin B. It resulted in an efficient photosensitizer in photodynamic antimicrobial chemotherapy (PACT), with improved activity and targeting properties compared to the usual cationic porphyrins.55 For these preliminary studies, we chose two peptides derived from the C-terminal region of bombesin, respectively BBN[7–13] and BBN[7–14], that have been widely studied due to their ability to target GRPR, which is overexpressed in several tumors, especially on human prostate cancer (PC-3) cells.56−58 In order to attach the targeting moieties to the TPP motif, compound 4, with an amino group, was first derivatized to provide compounds suitable for either direct amide bond ligation (4.9), thiol-maleimide ligation (4.11), or coupling via strain-promoted azide–alkyne cycloaddition (SPAAC) (4.13). This was achieved using succinic anhydride, 3-maleimidopropionic acid, and dibenzocyclooctyne acid (DBCO-acid).59 Succinic anhydride and 3-maleimidopropionic acid were chosen for their commercial availability, cost effectiveness, and the simplicity of the coupling reaction to form an amide bond with 4.60 DBCO-acid was selected due to its commercial availability, its usefulness in bioorthogonal ligation, and the increasing reaction rates compared to cyclooctyne reagents without fused pendant phenyl rings.61,62 As shown in Scheme 2, functionalization of 4 with succinic anhydride gave porphyrin 4.9, which was then coupled with BBN[7–13] using PyBOP and DIPEA in DMF at 75 °C, overnight. Thiol-reactive porphyrin 4.11 and azide-reactive porphyrin 4.13 were prepared from 4, following an adapted protocol from Dondi et al.,29 using the relevant carboxylic acid derivatives and 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDC)/1-hydroxybenzotriazole hydrate (HOBt)-mediated coupling. Conjugation of 4.11 with cysteine-functionalized BBN[7–14] and azidoundecanoyl-BBN[7–14] occurred efficiently at room temperature in DMSO. Three TPP-BBN[7, 14] derivatives (4.10, 4.12, 4.14) and a further TPP-BBN[7, 13] conjugate were isolated by semiprep HPLC and characterized by ESI-MS (see SI).
Scheme 2. Coupling Reaction Scheme of Functionalized Porphyrin-Based Conjugates with Peptides.
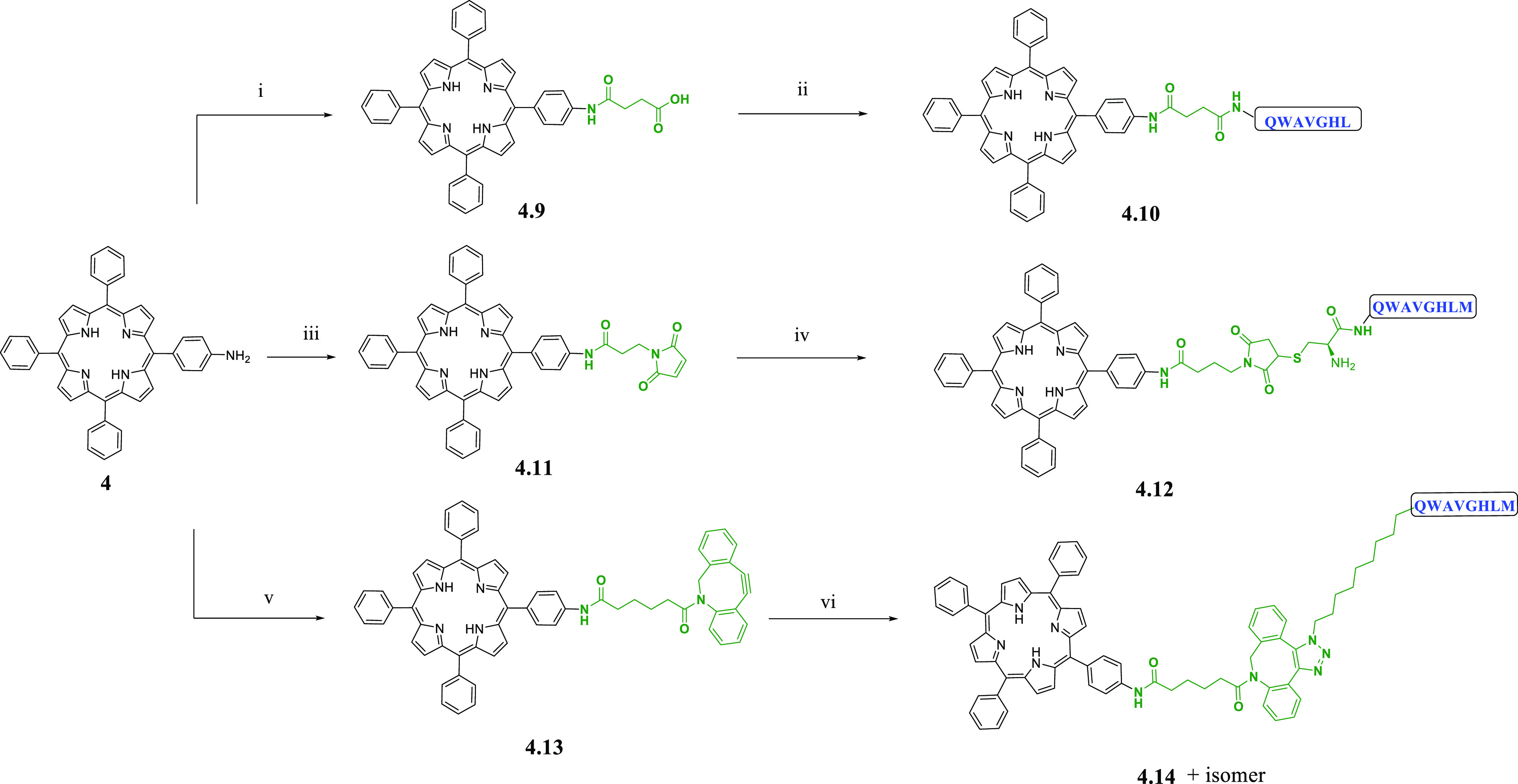
Reagents and conditions: (i) succinic anhydride, TEA, CHCl3; (ii) PyBOP, DIPEA, DMF, BBN[7-13]; (iii) 3-maleimidopropionic acid, HOBt, EDC·HCl, DCM; (iv) Cys-BBN[7-14]-NH2, pyridine, DMSO; (v) dibenzocyclooctyne acid, HOBt, EDC·HCl, DIPEA, DCM; (vi) 11-azido undecanoyl-BBN[7-14]-NH2, DMSO.
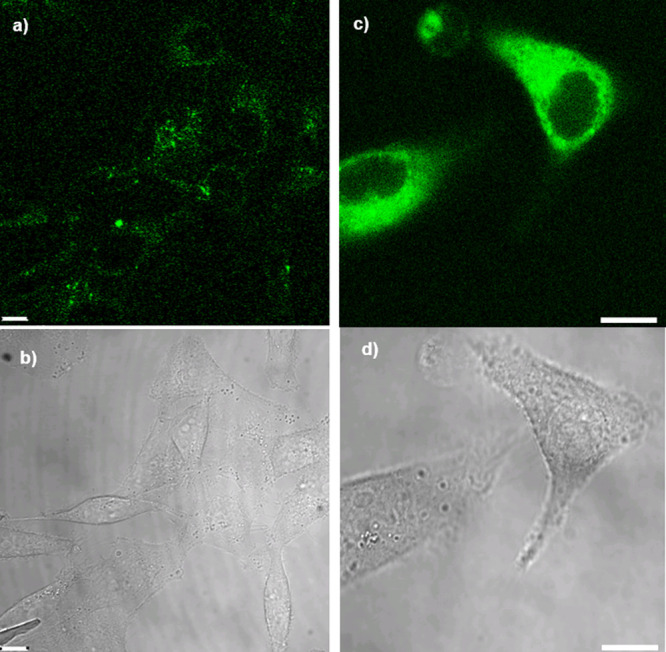
Initial investigations into the cellular uptake of the linker-functionalized porphyrin 4.9 and the porphyrin–bombesin conjugates were performed in PC-3 cells (Figures 14, 15, and 16). Cells were treated with 50 μM final concentration solution of 4.9 (in 1% DMSO serum free medium), washed with PBS, and the medium replaced. The same methodology was applied for 4.10, although the concentration was 200 μM. The images were taken immediately using confocal microscopy and were acquired by excitation at 405 or 488 nm and with emission observed above in the λem = 570–750 nm (red) channels, filtered above 600 nm. Compound 4.9 showed limited uptake into the cells and seems to preferentially localize to the cell membranes. The porphyrin continues to absorb and emit in the red channel, however, demonstrating that there is no impact on the absorbance or emission spectra caused by the addition of the linker functionality. Among the conjugates studied here, compound 4.10 (vide infra) was the only compound that showed absorbance and emission outside the red channel. This can be attributed to the effect of this specific peptide unit, which was shown in control experiments to fluoresce weakly in the green emission channel when excited at 405 nm. In addition, there was no apparent quenching of the porphyrin caused by being linked in close proximity to a relatively large targeting moiety (bombesin-derived peptide). There also appears to be greater uptake and dispersion in the uptake in the cells, with less concentration at the cell membrane; however, the precise intracellular localization could not be assigned with precision. This uptake and nonspecific cytoplasmic distribution could be explained by the presence of the peptide, which altered the uptake characteristics seen for the simple ligand building blocks evaluated (i.e., H2TPP, H2TPPNH2 H2TPPCOOMe, and H2TPPCOOH) and increases the selectivity toward the cells. Interestingly, the H2TPP-linker compound 4.9 (the 4-oxo-4-((4-(10,15,20-triphenylporphyrin-5-yl)phenyl)amino)butanoic acid showed, after only 20 min incubation in PC-3 cells at 50 μM final concentration (on Petri dish) some remarkable membrane-staining effect, which was not previously seen with the simpler building blocks or with the bombesin conjugates. Alternatively, this result can simply be the consequence of altered pharmacokinetic properties due to the relatively hydrophobic nature of the linker and for the porphyrin–peptide conjugates due to the short amino acid chain attached to the porphyrin unit.
Figure 14.
Confocal imaging of the butanoic acid linker-conjugated compound 4.9 (50 μM, 1% DMSO) in PC3 cells after incubation (20 min, 37 °C): λex = 488 nm, λ = 650–700 nm; Scale bar 50 μm.
Figure 15.
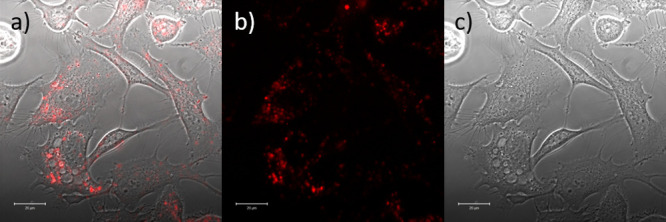
Confocal imaging of the peptide-conjugated compound 4.10 (200 μM, 1% DMSO) in PC3 cells after incubation (20 min, 37 °C). λex = 488 nm, λ = 650–700 nm. Scale bar 50 μm.
Figure 16.
Confocal imaging of the peptide-conjugated compound 4.12 (10 μM, 1% DMSO) in PC3 cells after incubation (20 min, 37 °C). λex = 488 nm, λ = 650–700 nm. Scale bar 20 μm.
Comparative uptake studies of the conjugates 4.10, 4.12, and 4.14 were attempted by confocal microscopy in PC-3 and HeLa cell lines alongside TPP-NH2 (4) and TPP (1). HeLa cells were chosen here to provide insight into the success of this targeting approach, since they do not express the GRP-receptor. The compounds (in three different concentrations regimes, of 10 μM, 50 μM, and 200 μM, in each case with 1% DMSO) were incubated with the cells in RPMI media for timelines ranging between 20 min and 20 h before being imaged by confocal microscopy. Neither the free base H2TPP 1 nor the amino-porphyrin 4 showed any uptake in either cell line at 10 μM concentration regimes, likely due to the highly hydrophobic nature of these compounds and their poor aqueous solubility. Peptide conjugate 4.14 showed no uptake in either cell line and could be seen agglomerating in the cell medium, even after repeated washing with Hank’s Balanced Salt Solution (HBSS) to remove any traces of compound which were not taken into the cells. This is probably due to the poor solubility of the compound in the media due to the long aliphatic chains in the compound. Potentially, the solubility of the conjugate could be improved by sulfonation of the phenyl rings, as shown above for the nitroimidazole class of compounds; however, this avenue was not pursued hereby. Uptake in PC-3 cells was observed for porphyrin conjugate 4.12, yet fluorescence emission observed was weak at 10 μM, indicating the compound was taken up only to a low degree at this low concentration range. A significant cellular uptake in PC-3 was observed for compound 4.10 only upon treatment with an increased concentration (200 μm). Overall, the observations at the uptake of 4.10 and 4.12 in PC-3 cells indicate that the attachment of the otherwise hydrophobic imaging agent (porphyrin) to a tumor-homing peptide is a valuable approach for the future development of in vitro multimodal imaging probes of this type. This shows that conjugation with a tumor-homing peptide such as bombesin could prove to be a very useful approach for improving the localized uptake of other similar hydrophobic imaging agents. Although radiochemistry of these bombesin conjugates did not show radiochemical incorporation (SI) suitable for in vivo studies, likely because the presence of the peptide changed the behavior of the TPP-NH2 or TPP-COOH groups taken alone, the optical imaging studies indicated that porphyrins are good at delivering fluorescence. Uptake in HeLa cells was also attempted; however, a credible selective uptake suggestion for PC-3 vs HeLa cells is not conclusive at this stage, and further studies are necessary. Although the bombesin fragments investigated here were shown to be good at mediating the uptake in cells instead of being dominated by the porphyrin core, future efforts would be focused on improving the yields.
3. Conclusions
The aim of this work was to investigate new bifunctional chelators derived from the tetrapyrrole structure of free base porphyrins for potential applications as PET and SPECT radiopharmaceuticals. This was triggered by the growing research interest that functional porphyrins have attracted over the years as potential therapeutic agents, due to their avid tumor accumulation and photosensitizing ability.63 The aim of research into novel PET/SPECT agents is to achieve a library of diagnostic and therapeutic agents, which can be selected depending on the individual needs of the patient, and as such, there is a need for continual fundamental research into the chemistry and biology of new PET/SPECT radiopharmaceuticals requiring a multidisciplinary approach. A new series of bifunctional chelators based on porphyrin chromophores have been developed and characterized (Schemes 1 and 2). We focused on the elaboration of −COOH or −NH2 tagged porphyrins to provide a method of attaching chromophores to targeting groups. Porphyrins were functionalized with charged groups, such as pyridinium and sulfonate, and with hypoxia-targeting nitroimidazole and sulfonamide vectors, in order to favorably affect their lipophilicity, prevent aggregations, and reduce off-target uptake.64 Charged derivatives based on sulfonic acids and methyl-pyridinium functionalities were successfully synthesized. They allowed aqueous solubility to be conferred on these porphyrin derivatives, with the aim to facilitate radiolabeling protocols with metallic radioisotopes, such as gallium-68, copper-64, and indium-111. Here, we demonstrated that the introduction of tumor targeting vectors onto the porphyrin structure may potentially alter the selectivity of porphyrin uptake in living systems. While displaying promising fluorescence, receptor binding and bioreductive properties, in vitro data suggested that the pharmacodynamics of these bifunctional chelators is dominated by the porphyrin unit. Indeed, general low hypoxia selectivity is conferred to the porphyrin conjugate when nitroimidazoles are incorporated. This highlights the importance of methodologies to address the current challenges in the design of suitable radioisotope chelators for targeted radiotherapy and imaging, though a rational selection of substituents. We also showed that the porphyrin structure provides a stable coordination environment for metallic radionuclides, and it is readily functionalized for structural manipulation, which confers its potential for in vitro fluorescence imaging upon inclusion of biocompatible moieties and bypassing the notorious aggregation and lack of solubility. Previous studies have suggested that porphyrin derivatives possess poor intrinsic in vivo pharmacological properties with high nonspecific organ uptake observed with a range of structures.17,65,66 The use of porphyrins as bifunctional chelators for the specific localization of radioisotopes for nuclear imaging and medicine is not yet well developed for small molecules. No radiolabeled porphyrin-based agent with biodistribution suitable as imaging agent has yet been developed. To date, in all porphyrin-based agents, while tumor uptake is high, large amounts of porphyrins are found to accumulate in the liver and kidneys. While we partly found that this challenge can be overcome by the judicious selection of functional groups, it remains the case that uptake of 111In-radiolabeled functional porphyrins localize in nontarget background organs. In general, metalloporphyrin species have shown to be stable under serum challenges and the nitroimidazole and sulfonamide derivatives remain fluorescent, allowing in vitro confocal studies and visualization of the lysosomal uptake in a gallium(III) sulfonamide derivative. In addition, porphyrin’s functionalization with a small peptide chain (BBN[7–13] and BBN[7–14]) for targeting prostate cancer cells (PC-3) was attempted. Amino-functionalized chromophore building blocks derived from a synthetic porphyrin were prepared in moderate yields. These new porphyrins were elaborated with a maleimide or cyclooctyne moiety to give molecules suitable for bioorthogonal ligation reactions with appropriately modified peptides via thiolmaleimide ligations or SPAAC. The tumor-homing [7–13] or [7–14] amino acid sequences of BBN were prepared by solid-phase Fmoc chemistry on rink-amide resin and post-synthetic modifications on the peptides were achieved with an azido or cysteinyl functionality to complement the chromophores. This resulted in the preparation of 3 different chromophore-bombesin conjugates, 4.10, 4.12, and 4.14. These unprecedented compounds were purified by semipreparative HPLC. Preliminary uptake studies of the new chromophore–bombesin conjugates in PC-3 and GRPR negative HeLa cells showed selective uptake of conjugates in PC-3 cells, but not significantly in HeLa cells compared to the precursor molecules, which generally showed distinct lack of solubility. Surprisingly, conjugate 4.14 showed no uptake in either cell line, and further attempts to improve the solubility of the conjugate via sulfonation of the phenyl rings (which proved successful in the case of simpler nitroimidazole conjugates) might provide a solution for improved internalization. Although promising results were observed in these preliminary investigations, detailed studies should be repeated with variations in incubation times after application of compounds to cells. Furthermore, determination of the highest tolerable concentration of the compound in cells could be determined by MTT colorimetric assays to ensure the compound is nontoxic to cells for use in imaging or only becomes toxic on excitation to cause apoptosis (as in PDT). Small-scale labeling experiments of the conjugates with PET radionuclides gallium-68 have been performed, showing the extremely limited metalation of the porphyrin with the gallium, largely being associated with the peptide tags rather than the porphyrin moiety. The ultimate goal of this work is toward multimodal imaging probes which are highly specific toward biological targets. In addition to BBN, alternative peptides, such as luteinizing hormone releasing hormone (LHRH), or other CPPs could also be investigated to develop novel chromophore–peptide conjugates. The bioorthogonal ligations involving BBN[7–13] and BBN[7–14], studied in this work, show much promise for solubilizing large, hydrophobic fluorescent molecules and for improving uptake into cells. This could be a useful tool in the design of new drug–antibody conjugates, which are highly specific toward biological targets, for nuclear imaging and toward multimodal imaging probes with theranostic potential.
4. Experimental Details
4.1. General Experimental Section
All reagents were purchased from Sigma-Aldrich, Merck Chemicals, Fluorous Technologies, or Alfa-Aesar and were used as supplied without prior purification unless otherwise stated. Thin layer chromatography was performed on Merck Kiesegel 60 F254 0.25 mm precoated aluminum plates. Product spots were visualized under UV light (max = 254 nm) and/or by staining with ceric ammonium molybdate or potassium permanganate. Flash chromatography was performed using silica gel 60 (0.043–0.063 mm, VWR) using head pressure by means of head bellows.
1H NMR spectra were recorded on a Varian Mercury VX300 (300 MHz) spectrometer or a Varian Unity (500 MHz) spectrometer or a Bruker AVC 500 (500 Hz) spectrometer at 298 K and referenced to residual nondeuterated solvent peaks. Chemical shifts are quoted in ppm with resonances reported as either singlet (s), doublet (d), triplet (t), quartet (q), quintet (qt), and multiplet (m) resonances. Coupling constants, J, are measured to the nearest 0.1 Hz. 13C NMR spectra were recorded on a Varian Mercury VX300 (300 MHz) spectrometer or a Varian Unity (500 MHz) spectrometer or a Bruker AVC 500 (500 Hz) spectrometer at 298 K and were referenced to the solvent peak.
Mass spectrometry was performed using a Bruker Micromass Micromass LCT time-of-flight mass spectrometer under conditions of electrospray ionization (ESI-MS). Accurate masses are reported to four decimal places using tetraoctylammonium bromide (466.5352 Da) as an internal reference. Values are reported as a ratio of mass to charge in daltons. Electronic absorption spectroscopy (UV/vis) was performed using a PerkinElmer Lambda 19 spectrometer, running UV Winlab software. Spectra were measured using 1.00 cm quartz cuvettes. HPLC characterization (analytical HPLC) of compounds was performed by one of three methods A, B, C described in the SI.
X-ray crystal structure analyses were performed using the synchrotron radiation source at the SRS Daresbury Laboratory, Station 9.8, Warrington, UK, or measured using an Enraf-Nonius Kappa CCD diffractometer (monochromated Mo Kα radiation, λ = 0.71073 Å). Structures were deposited in CCDC CSD (Deposition Number: 2050384 for 1.1, 2050385 for 2, 2050382 for 3.17, and 2050725 for 3.22) and further details are given in SI.
Density functional theory (DFT) and time-dependent density functional theory (TDDFT) calculations were performed using the Amsterdam Density Functional (ADF) suite.37 All calculations were performed applying the continuous solvation model COSMO (conductor-like screening model). It was used to model DMSO as the solvent (dielectric constant ε = 46.7 F·m–1, radius of the rigid-sphere solvent molecules = 3.04 Å). Following the findings by Rydberg et al., the generalized gradient approximation (GGA) functional BP86 was employed. All calculations were performed utilizing the TZ2P basis set, to accurately describe the metal centers. No frozen cores were applied. In the case of indium chloride, scalar relativistic corrections were applied using the ZORA (zero-order regular approximation) formalism and MAPA (minimum of neutral atomical potential approximation). Geometries were optimized and analytical frequencies calculated,67−69 before allowed singlet–singlet transitions were modeled at the TD-DFT level of theory using the Davidson algorithm (lowest Eigenvalues).
4.2. General Synthetic Protocols
4.2.1. General Amide Coupling Procedures
4.2.1.1. Method A
Amine “A” was added to a solution of Porphyrin “P” in DMF. The mixture was stirred at rt for 12 h where after TLC (CHCl3:MeOH, 95:5) indicated complete consumption of starting material. The solvent was removed under reduced pressure and the residue redissolved in CHCl3. The solution was washed with water and brine and dried over anhydrous magnesium sulfate. The CHCl3 was reduced to minimum volume under reduced pressure and the crude amide was purified by silica gel chromatography (CHCl3:MeOH, x:y).
4.2.1.2. Method B
DIPEA was added to a solution of Porphyrin “P” in DMF and the mixture cooled to 0 °C. BOP was added and the solution stirred at 0 °C for 30 min. Amine “A” was added and the solution allowed to warm to rt and stirred for 12 h. DMF was removed under reduced pressure and the residue redissolved in CHCl3. The solution was washed with 1 M HCl, saturated aqueous Na2CO3, water, and brine, and dried over anhydrous magnesium sulfate. The CHCl3 was reduced to minimum volume under reduced pressure and the crude amide was purified by silica gel chromatography (CHCl3:MeOH, x – y% MeOH in CHCl3).
4.2.2. General Sulfonation Procedure
Porphyrin “P” was dissolved in conc. H2SO4 and heated at 75 °C for 4 h. The reaction mixture was cooled to rt and acetone added to precipitate out a green solid. The solid was filtered and washed with further acetone. The residue was dissolved in a minimum amount of water and the pH adjusted to pH 7 by addition of 2 M NaOH(aq.). A C18 Sep-Pak cartridge supplied by Millipore Ltd. was then preconditioned by elution with MeOH (2 mL) and water (5 mL) and the aqueous porphyrin solution loaded on to the cartridge. Water-soluble impurities were removed from the cartridge by elution with water (5 × 2 mL) and the absorbed porphyrin was eluted using MeOH (3 × 3 mL). The solvent was removed under reduced pressure to leave the sodium salt of the sulfonated porphyrin as a purple solid.
4.2.3. General Pyridine Alkylation Procedure
Porphyrin “P” was dissolved in DMF and MeI added. The solution was stirred with heating at 35 °C for 1 h. The reaction mixture was cooled to rt and Et2O added to precipitate out a brown solid. The solid was dissolved in MeOH and reprecipitated with Et2O. The solid was filtered, washed with Et2O and hexane, and dried under vacuum to leave the tri-iodide salt of the methylpyridinium porphyrin as a brown solid.
4.2.4. General EDC/HOBt Coupling Procedure
The appropriate carboxylic acid was dissolved in anhydrous DMF at 0 °C. The resulting solution was treated with 1.2 mol equiv each of EDC·HCl and (HOBt). The solution was stirred for 45 min before adding either 1 or 2 (1.1 equiv) and N,N-diisopropylethylamine (DIEA, 3 equiv). The reaction was warmed to room temperature and stirred overnight, with complete consumption of starting material generally observed after 18 h. Unless otherwise stated, the resulting solution was diluted with DCM (10 mL) and washed with H2O (10 mL), sat. NaHCO3 (10 mL) and brine (10 mL), dried over Na2SO4, and concentrated to residue. The crude products were purified by flash chromatography.
4.2.5. Preparation of BBN[7–13] and BBN[7–14] Peptides
BBN[7–13] and BBN[7–14] were both prepared by Fmoc solid-phase peptide synthesis on Rink Amide resin using automated solid-support synthesis on an Activotec ACTIVO-P11 peptide synthesizer fitted with a reactor heating jacket.29 Once assembled, cleavage of the peptide was achieved by treatment of the resin with a mixture of TFA/TIS/H2O (95/2.5/2.5) for 4 h in the case of BBN[7–13], or TFA/TIS/H2O plus additional scavengers for BBN[7–14] peptides (detailed in SI).
4.2.6. General Metal Complexation Reactions
4.2.6.1. Method A
Sodium acetate was added to a concentrated solution of a free base porphyrin in acetic acid. The metal salt was added and the solution stirred at reflux for 12 h. The reaction mixture was cooled to rt and neutralized to pH 7 with saturated aqueous Na2CO3 and extracted with CHCl3. The organic layer was washed with saturated aqueous Na2CO3, water, and brine and dried over anhydrous magnesium sulfate. The CHCl3 was removed under reduced pressure and the solid recrystallized from CHCl3:MeOH (9:1)/hexane.
4.2.6.2. Method B
The metal salt was added to a concentrated solution of a free base porphyrin in DMF and the resulting solution was stirred under reflux for 12 h. The reaction mixture was cooled to rt and DMF removed under reduced pressure. The residue was redissolved in CHCl3 and washed with water and brine and dried over anhydrous magnesium sulfate. The CHCl3 was removed under reduced pressure and the solid recrystallized from CHCl3:MeOH (9:1)/hexane.
4.2.6.3. Method C
Water-soluble porphyrin free base was dissolved in pH 4.5 NaOAc buffer and InCl3 added. The solution was then heated under reflux for 2 h. The reaction mixture was cooled to rt and the solution filtered. The filtrate was reduced under vacuum to minimum volume and loaded onto a preconditioned C18 Sep-Pak cartridge (see above). Water-soluble impurities were removed from the cartridge by elution with water (5 × 2 mL) and the absorbed porphyrin was eluted using MeOH (3 × 3 mL). The solvent was removed under reduced pressure to leave a solid residue.
4.2.7. Labeling of Porphyrin 4 with Aqueous [[68Ga]Ga(III)]
A sample of generator-produced aqueous 68Ga harvested in a borosilicate vial recovered in a solution of acetone (98%)/HCl (0.02 M) and was let for 15 min at 92 °C under nitrogen in order to dry in the same container (<70 MBq per experiment). Then, 20 μL of a TPP-NH2 (mg/mL in DMSO) solution along with EtOH (0.5 mL) and NaHCO3 buffer, pH: 4 (0.4 mL) were added to the vial, and the slurry was left at 100 °C for 60 min. Then, the reaction mixture was cooled and a sample prepared for HPLC analysis, using the reaction mixture (0.01 mL) diluted in EtOH (0.1 mL) and injected to HPLC via 0.2 mL loop. Purification of the labeled compound was followed using a C-18 cartridge. The cartridge was first eluted with EtOH (2 mL), and then was dried with air (5 mL) and washed with H2O (10 mL). Then the reaction solution was diluted with H2O (5 mL), and the mixture was run through the cartridge. The [68Ga]Ga-TPP-NH2 was trapped inside and the free gallium-68 was collected in a separate vial. Then, the cartridge was washed with EtOH (0.5 mL) to recover our labeled compound with a radiochemical purity of 60%.
4.2.8. General [111In]In Radiolabeling Procedure
The ligand or complex was prepared as a 1.0 mg/mL solution in DMSO or distilled water. For indium labeling of water-soluble porphyrins 10 μL of the stock porphyrin solution was diluted with 90 μL of pH 4.5 sodium acetate buffered solution and heated at 115 °C for 30 min in an Eppendorf tube in the presence of 20 μL [111In]InCl3. For indium labeling of tetraphenyl porphyrin derivatives 10 μL of the stock solution was diluted with 90 μL of AcOH and heated at 120 °C for 1 h in the presence 10 μL of a 0.1 M NaOAc (aq.) solution and 20 μL 111InCl3 (<10 MBq per experiment). AcOH was then driven off under nitrogen and the residue resuspended in 50 μL of CHCl3. The CHCl3 layer was washed repeatedly with 100 μL aliquots of distilled water until no further activity was associated with the aqueous washings. For copper-64 radiolabeling, 10 μL of the stock porphyrin solution was diluted with 40 μL of DMSO and 50 μL [64Cu]Cu(OAc)2 was added (<10 MBq per experiment), and the reaction stirred for 20 min at room temperature. On reaction completion, an aliquot (20 μL) was removed via syringe, and analyzed by HPLC (radio and UV detection, in series). In all cases, the 20 min HPLC gradient methods A, B, and C were used for the porphyrin labeling experiments.
4.3. In Vitro and In Vivo and Experimental Details
4.3.1. Cell Culturing Methods
The following methods were followed for HeLa cell culturing and preparation of cell plates for fluorescent studies. Cells were cultured at 37 °C in a humidified atmosphere in air and diluted once confluence had been reached. Cells were cultured in DMEM medium with 10% fetal calf serum (FCS) and 100 U/mL penicillin. Samples for fluorescence were prepared in the following way: surplus supernatant containing dead cell matter and excess protein was discarded; the live adherent cells were then washed with two 7 mL aliquots of Phosphate Buffer Saline solution to remove any remaining medium containing FCS. FCS inhibits resuspension of the cells as it contains protease inhibitors which inactivate trypsin. To resuspend the cells in solution, they were incubated in 2 mL of trypsin/EDTA (500 mg/L Trypsin, 200 mg/L EDTA) solution for 3 min at 37 °C. After trypsinizing, 5 mL of DMEM was added to inactivate the trypsin and the solution was centrifuged for 5 min to remove any remaining dead cell matter. The supernatant liquid was poured off and DMEM added to the cell matter left behind to give a sufficient concentration of cells. The cells were plated in a Petri dish containing a glass coverslip and left for 24 h to adhere. One hour before fluorescence imaging measurements were made, the DMEM was replaced with DMEM containing no fluorescent indicator dyes such as phenol red, therefore making it suitable for fluorescent studies.
4.3.2. Microscopy Investigations in Living Cells
Porphyrin complexes were prepared as 10 mM stock solution in DMSO or distilled water, and diluted to 10 μM with DMEM, and incubated with the cells at 37 °C. Prior to imaging, the solution was replaced with 1 mL fresh DMEM. Background autofluorescence was measured by imaging the cells in 1 mL of DMEM medium only. The fluorescent uptake of porphyrin complexes was imaged by laser-scanning confocal microscopy using a Zeiss LSM 510 META microscope irradiating at 405 nm with emission filtered between 565 and 615 nm for the porphyrin complexes.
PC-3 and HeLa cells were seeded as monolayers in T75 tissue culture falcon flasks, and cultured in Roswell Park Memorial Institute (RPMI) 1640 media supplemented with 10% fetal bovine serum (FBS), l-glutamine, penicillin, and streptomycin. Cells were incubated at 37 °C, under an atmosphere of 5% CO2, and passaged with trypsin when 70–80% confluent. For fluorescence microscopy, cell monolayers were plated in glass dishes 3 days in advance to ensure adhesion to the surface, with an estimated 7.5 × 104 cells per dish. Compound stock solutions (1 mM) were prepared in DMSO. After aspiration, cells were washed 5 times with Hank’s Balanced Salt Solution (HBSS) before adding RPMI (990 μL) and the compound solution (10 mM solution, 10 μL, giving a final concentration of 10 μM). Cells were then incubated for 20 h at 37 °C, washed three times with HBSS to remove traces of non-internalized compounds, recovered with 1 mL of serum free RPMI, and imaged using a Nikon or Zeiss LSMS510META confocal microscope with λex = 405 nm, with emission collected above 600 nm. In each case, a minimum of 5 sets of images were recorded.
Fluorescence imaging assays were also performed on the HCT 116 colon carcinoma cell line denoted CAIX positive which has previously been transfected to overexpress the CA IX enzyme without the need for hypoxic culturing. All cell lines were cultured according to known protocols18 and as described below. Cells were seeded on to glass coverslips and left to adhere for 12 h overnight. Cells were incubated with free-base or metal-substituted porphyrin compounds at final concentrations (on cell plate) of 10, 50, and 200 μM for periods ranging from 2 to 16 h at 37 °C. After incubation, the cells were washed three times with PBS and confocal images recorded using a Zeiss LSM 510 META microscope using the excitation laser of 405 nm with emission collected above 625 nm. In each case, a minimum of 5 sets of images were recorded.
4.3.2.1. Fluorescence Lifetime Imaging Microscopy (FLIM)
Cell uptake studies using two-photon FLIM were performed on living PC-3 cells, adhering to glass bottom Petri dishes and incubated with the compound over 15, 20, and 30 min at 37 °C. Images of cells without probe uptake but in the presence of the DMEM and 1% DMSO alone were recorded as background lifetimes. The ligands or complexes were dissolved in a small amount of DMSO (1%) and added to the cell culture medium to give a concentration of 10 μM. Decay lifetimes were measured for the complete field of view over 5 min intervals. Uptake reached a maximum within 30 min and the images and data presented below were obtained after 20 min. FLIM images were recorded by raster scanning the focused NIR (910 nm) 200 fs pulsed laser light at 76 MHz, through a 60× water immersion objective with an NA or 1.2. a BG39 filter was used to filter the fluorescence following the multiphoton excitation and recorded (using a Hamamatsu R3809U) the point decays at every location within the cell (generating a minimum of 128 × 128) pixel image and lifetimes calculated at each pixel using the standard software Becker and Hickl SPCMImage package (ver. 4).
4.3.3. Phosphate Buffer Stability Tests
Solutions of the indium and gallium complexes for testing were prepared as 200 μM solutions in DMSO and diluted with phosphate buffer (pH = 7.0), giving solutions of final concentration 10 μM. The solutions were kept at 37 °C and UV–vis spectra were recorded at time points 1 min, 30 min, and 1, 2, 4, 8, 12, and 24 h. No significant changes in absorption were observed in any of the porphyrins investigated hereby.
4.3.4. Human Serum Binding Tests
Experiments were performed similarly to the phosphate buffer stability tests, where the phosphate buffer was substituted for human serum (from human male AB plasma, Sigma-Aldrich). The samples were incubated at 37 °C and UV–vis spectra were recorded at selected time points. No significant changes in absorption were observed in any of the porphyrins investigated hereby.
4.3.5. Measurements of LogP
In each case, the indium-111 labeled complex (15 μL, <1 MBq) was added to a mixture of octan-1-ol (0.5 mL) and H2O (0.5 mL). The mixture was shaken for 1 min, then centrifuged for 5 min at 2000 rpm. A 50 μL sample of each layer was taken and counted using a gamma counter. The measurements were performed in triplicate. Log P was calculated using the formula: log P = log(counts(octanol)/counts(H2O)).
4.3.6. Serum Binding Experiments
The indium-111 complex (15 μL, <1 MBq) was added to 400 μ L rat serum and incubated at 37 °C. At each time point (5, 30, 60, and 120 min), a 40 μL aliquot was removed from the serum and added to EtOH (400 μL) to precipitate the proteins. The samples were centrifuged and the pellet and supernatant were separated. The pellet was washed with 400 μL EtOH, spun, and supernatant combined with the first. The counts in the pellet and supernatant were recorded. Counts associated with the pellet were considered as copper-64 bound to the protein. Each sample was performed in triplicate.
4.3.7. Oxygen-Dependent Cell Uptake Experiments
HeLa cells (106/mL) were incubated in a humidified atmosphere at 37 °C, under anoxic (0% O2), hypoxic (0.2% O2), or normoxic (21% O2) conditions with 5% CO2, and the remainder N2. Minimum essential medium (MEM) spinner modification was used, with Earle’s salts and sodium bicarbonate, and without calcium chloride and l-glutamine (supplemented with 0.292 g/L l-glutamine, penicillin–streptomycin). After 1 h, the O2 conditions had reached equilibrium (probed using Oxford optronics Oxylab pO2 tissue oxygenation monitor) and the N2 purged radiotracer was added. A 900 μL sample of the cells was removed at 5, 15, 30, 45, and 60 min and divided to three Eppendorf tubes, and the samples were centrifuged to pellet the cells. The supernatant was separated from the pellet, and both fractions were counted using a PerkinElmer Wizard 1470 automatic γ-counter. The percentage of cell associated activity was then calculated. A control was performed with the radiotracer in MEM using normoxic conditions to test the amount of 64Cu associated with the Eppendorf tubes. This was measured and subtracted from each data point.
4.3.8. SPECT Imaging Experiments
Biodistribution and imaging studies in MKN45 gastric cancer tumor bearing mice were conducted at Barts and the London School of Medicine and Dentistry, where all in vivo experiments were conducted in compliance with British Home Office regulations governing animal experimentation. Tumor xenografts were induced in female CD1 nude mice (Charles River) by subcutaneous injection of 5 × 106 MKN45 cells, and the tumors were allowed to grow until they had reached a size greater than 12 mm (10–15 d). Imaging was performed using a NanoSPECT/CT small animal scanner (Bioscan). [111In]In-labeled porphyrins (0.05 mg of Porphyrin/20 MBq of [111In]In(III)) were injected into the tail vein. SPECT/CT imaging was performed at 0, 3, 5, and 24 h post injection. The mice were anaesthetized with isoflurane (4% induction, 2% maintenance). A 36 pinhole collimator with 1.4 mm pinholes was used for the SPECT acquisitions. Count rates ranged between 50,000–100,000 for each of the 16 projections. Radionuclide images were reconstructed using proprietary HiSPECT (Bioscan) iterative reconstruction and were fused with CT images using proprietary InVivoScope (Bioscan) software. The mice were sacrificed by CO2 euthanasia at either 5 or 24 h after injection. Tumors and other tissues (blood, stomach, spleen, liver, pancreas, kidneys, muscle, tail, heart, lungs, and intestines) were removed and weighed. The tissues were counted in a gamma-counter together with standards of known radioactivity, and the percentage injected dose per gram of tissue (%ID/g) was calculated by dividing the activity in the tissue samples (gamma-counter) by the injected activity (dose calibrator). The unpaired t test (Excel; Microsoft) (significance level, 0.05) was used for statistical analysis.
Acknowledgments
The authors would like to thank Ms. Madeleine Rice and Ms. Sophie Pye (Bath University undergraduate research students) for their dedication in pursuing aspects of this project. We thank the greater Dilworth group for helpful discussions and training in radiochemistry experiments at the commencement of this research programme. We thank Dr. Charareh Pourzand, Bath for assistance with cellular experiments and Mr. Colin Sparrow, Chemistry Research Laboratory, University of Oxford for advanced mass spectrometry investigations. We also thank the following collaborators for technical assistance with some of the microscopy experiments at Bath, Oxford and Research Complex at Harwell: Dr. M. Samalova, Dr. I. Moore, Dr. Alan McIntyre, Group, Dr. G. Churchill, Dr. A. Rodgers. We thank Dr. M. Christlieb and Prof. Adrian Harris for the generous loan of the HCT116 (CAIX positive and negative) cells used in imaging experiments and helpful discussions. We thank Dr. Robert King, Dr. Julie Foster, Dr. Ciara Finucane, and Prof. Stephen Mather at the Cancer Imaging Group, Barts Cancer Institute, Queen Mary University of London for access to facilities and technical assistance with in vivo work as well as helpful discussions. We acknowledge the invaluable support, training, and housing of the Bath/Oxford 68Gallium Generator from Dr. Laurence Carroll and Prof. Eric Aboagye (Imperial College London) and Professors Phil Blower and Frank Aigbirhio for helpful discussions, radiochemistry training, and supply of 64Cu.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.bioconjchem.0c00691.
Author Present Address
¶ Valeria Ciaffaglione, Department of Drug Sciences, University of Catania, viale A. Doria 6, 95125 Catania, Italy
Author Contributions
# The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript. V.C., P.A.W., R.M.E., and F.C.-T. contributed equally to the work and manuscript.
This work was financially supported by Science & Technologies Facilities Council, EPSRC Centre for Doctoral Training Centre for Sustainable Chemical Technologies (EP/G03768X/1), ERC Consolidator grant scheme (O2Sense to S.I.P.) and ERC Proof-of-Concept grant Tools-To-Sense to S.I.P., and University of Bath, EPSRC Mass Spectrometry at Swansea. Also LASERLAB-EUROPE (grant agreement no. 654148), European Union’s Horizon 2020 research and innovation program is acknowledged. University of Catania is acknowledged for a travel and study grant to VC.
The authors declare no competing financial interest.
Supplementary Material
References
- Dolphin D.The Porphyrins Volume VII: Biochemistry, part B. [Google Scholar]
- Urata G. (1995) [The chemistry of porphyrins and their precursors on the heme biosynthetic chain]. Nihon rinsho 53 (6), 1319–28. [PubMed] [Google Scholar]
- Josefsen L. B.; Boyle R. W. (2012) Unique diagnostic and therapeutic roles of porphyrins and phthalocyanines in photodynamic therapy, imaging and theranostics. Theranostics 2 (9), 916–66. 10.7150/thno.4571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higashino T.; Imahori H. (2015) Porphyrins as excellent dyes for dye-sensitized solar cells: recent developments and insights. Dalton transactions 44 (2), 448–63. 10.1039/C4DT02756F. [DOI] [PubMed] [Google Scholar]
- Yan G. P.; Bischa D.; Bottle S. E. (2007) Synthesis and properties of novel porphyrin spin probes containing isoindoline nitroxides. Free Radical Biol. Med. 43 (1), 111–6. 10.1016/j.freeradbiomed.2007.03.032. [DOI] [PubMed] [Google Scholar]
- Waghorn P. A. (2014) Radiolabelled porphyrins in nuclear medicine. J. Labelled Compd. Radiopharm. 57 (4), 304–9. 10.1002/jlcr.3166. [DOI] [PubMed] [Google Scholar]
- Pascu S.; Waghorn P. A.; Conry T.; Lin B.; James C.; Zayed J. M. (2009) Design considerations towards simultaneously radiolabeled and fluorescent imaging probes incorporating metallic species. Adv. Inorg. Chem. 61 (48), 131–178. 10.1016/S0898-8838(09)00203-7. [DOI] [Google Scholar]
- Nappa M.; Valentine J. S. (1978) The influence of axial ligands on metalloporphyrin visible absorption spectra. Complexes of tetraphenylporphinatozinc. J. Am. Chem. Soc. 100 (16), 5075–5080. 10.1021/ja00484a027. [DOI] [Google Scholar]
- Abrahamse H.; Hamblin M. R. (2016) New photosensitizers for photodynamic therapy. Biochem. J. 473 (4), 347–64. 10.1042/BJ20150942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y.; Zhou T.; Bai R.; Xie Y. (2020) Chemical approaches for the enhancement of porphyrin skeleton-based photodynamic therapy. J. Enzyme Inhib. Med. Chem. 35 (1), 1080–1099. 10.1080/14756366.2020.1755669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yap S. Y.; Savoie H.; Renard I.; Burke B. P.; Sample H. C.; Michue-Seijas S.; Archibald S. J.; Boyle R. W.; Stasiuk G. J. (2020) Synthesis of a porphyrin with histidine-like chelate: an efficient path towards molecular PDT/SPECT theranostics. Chem. Commun. 56 (75), 11090–11093. 10.1039/D0CC03958F. [DOI] [PubMed] [Google Scholar]
- Sandland J.; Savoie H.; Boyle R. W.; Murray B. S. (2020) Synthesis and In Vitro Biological Evaluation of a Second-Generation Multimodal Water-Soluble Porphyrin-RAPTA Conjugate for the Dual-Therapy of Cancers. Inorg. Chem. 59 (11), 7884–7893. 10.1021/acs.inorgchem.0c01044. [DOI] [PubMed] [Google Scholar]
- Shi J.; Liu T. W.; Chen J.; Green D.; Jaffray D.; Wilson B. C.; Wang F.; Zheng G. (2011) Transforming a Targeted Porphyrin Theranostic Agent into a PET Imaging Probe for Cancer. Theranostics 1, 363–70. 10.7150/thno/v01p0363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubbiani R.; Wu W.; Naik A.; Larocca M.; Schneider L.; Padrutt R.; Babu V.; Konig C.; Hinger D.; Maake C.; et al. (2020) Studying the cellular distribution of highly phototoxic platinated metalloporphyrins using isotope labelling. Chem. Commun. 56 (92), 14373–14376. 10.1039/D0CC05196A. [DOI] [PubMed] [Google Scholar]
- Simoes J. C. S.; Sarpaki S.; Papadimitroulas P.; Therrien B.; Loudos G. (2020) Conjugated Photosensitizers for Imaging and PDT in Cancer Research. J. Med. Chem. 63 (23), 14119–14150. 10.1021/acs.jmedchem.0c00047. [DOI] [PubMed] [Google Scholar]
- Walter V.; Gao Y.; Grzegorzek N.; Krempe M.; Hampel F.; Jux N.; Tykwinski R. R. (2019) Building from Ga-Porphyrins: Synthesis of Ga-Acetylide Complexes Using Acetylenes and Polyynes. Angew. Chem., Int. Ed. 58 (2), 494–498. 10.1002/anie.201812142. [DOI] [PubMed] [Google Scholar]
- Robinson G. D. Jr.; Alavi A.; Vaum R.; Staum M. (1986) Imaging of lymph node uptake after intravenous administration of indium-111 metalloporphyrins. J. Nuclear Med. 27 (2), 239–42. [PubMed] [Google Scholar]
- Dilworth J. R.; Pascu S. I.; Waghorn P. A.; Vullo D.; Bayly S. R.; Christlieb M.; Sun X.; Supuran C. T. (2015) Synthesis of sulfonamide conjugates of Cu(II), Ga(III), In(III), Re(V) and Zn(II) complexes: carbonic anhydrase inhibition studies and cellular imaging investigations. Dalton transactions 44 (11), 4859–73. 10.1039/C4DT03206C. [DOI] [PubMed] [Google Scholar]
- James B. R.; Meng G. G.; Posakony J. J.; Ravensbergen J. A.; Ware C. J.; Skov K. A. (1996) Porphyrins and metalloporphyrins: potential hypoxic agents. Metal-based drugs 3 (2), 85–9. 10.1155/MBD.1996.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards D. I. (1977) The action of metronidazole on DNA. J. Antimicrob. Chemother. 3 (1), 43–8. 10.1093/jac/3.1.43. [DOI] [PubMed] [Google Scholar]
- Denny W. A. (2001) Prodrug strategies in cancer therapy. Eur. J. Med. Chem. 36 (7–8), 577–95. 10.1016/S0223-5234(01)01253-3. [DOI] [PubMed] [Google Scholar]
- Vullo D.; Innocenti A.; Nishimori I.; Pastorek J.; Scozzafava A.; Pastorekova S.; Supuran C. T. (2005) Carbonic anhydrase inhibitors. Inhibition of the transmembrane isozyme XII with sulfonamides-a new target for the design of antitumor and antiglaucoma drugs?. Bioorg. Med. Chem. Lett. 15 (4), 963–9. 10.1016/j.bmcl.2004.12.053. [DOI] [PubMed] [Google Scholar]
- Linden G.; Vazquez O. (2020) Bioorthogonal Turn-On BODIPY-Peptide Photosensitizers for Tailored Photodynamic Therapy. Chem. - Eur. J. 26 (44), 10014–10023. 10.1002/chem.202001718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno P.; Mantey S. A.; Lee S. H.; Ramos-Alvarez I.; Moody T. W.; Jensen R. T. (2018) A possible new target in lung-cancer cells: The orphan receptor, bombesin receptor subtype-3. Peptides 101, 213–226. 10.1016/j.peptides.2018.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pooja D.; Gunukula A.; Gupta N.; Adams D. J.; Kulhari H. (2019) Bombesin receptors as potential targets for anticancer drug delivery and imaging. Int. J. Biochem. Cell Biol. 114, 105567. 10.1016/j.biocel.2019.105567. [DOI] [PubMed] [Google Scholar]
- Adler A. D.; Longo F. R.; Finarelli J. D.; Goldmacher J.; Assour J.; Korsakoff L. (1967) A Simplified Synthesis for meso-Tetraphenylporphin. J. Org. Chem. 32, 476. 10.1021/jo01288a053. [DOI] [Google Scholar]
- Swamy N.; James D. A.; Mohr S. C.; Hanson R. N.; Ray R. (2002) An estradiol-porphyrin conjugate selectively localizes into estrogen receptor-positive breast cancer cells. Bioorg. Med. Chem. 10 (10), 3237–43. 10.1016/S0968-0896(02)00242-0. [DOI] [PubMed] [Google Scholar]
- Luguya R.; Jaquinod L.; Fronczek F. R.; Vicente M. G. H.; Smith K. M. (2004) Synthesis and reactions of meso-(p-nitrophenyl)porphyrins. Tetrahedron 60 (12), 2757–2763. 10.1016/j.tet.2004.01.080. [DOI] [Google Scholar]
- Dondi R.; Yaghini E.; Tewari K. M.; Wang L.; Giuntini F.; Loizidou M.; MacRobert A. J.; Eggleston I. M. (2016) Flexible synthesis of cationic peptide-porphyrin derivatives for light-triggered drug delivery and photodynamic therapy. Org. Biomol. Chem. 14 (48), 11488–11501. 10.1039/C6OB02135B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHENG W; SHAN N; YU L; WANG X (2008) UV-visible, fluorescence and EPR properties of porphyrins and metalloporphyrins. Dyes Pigm. 77, 153–157. 10.1016/j.dyepig.2007.04.007. [DOI] [Google Scholar]
- Dorough G. D. M. J. R.; Huennekens F. M. (1951) Spectra of the Metallo-derivatives of α,β,γ,δ-Tetraphenylporphine. J. Am. Chem. Soc. 73 (9), 4315–4320. 10.1021/ja01153a085. [DOI] [Google Scholar]
- Dechan P.; Bajju G. D. (2019) Synthesis and spectroscopic properties of axial phenoxide and para amino phenoxide incorporated indium (III) porphyrins. J. Mol. Struct. 1195 (1195), 140–152. 10.1016/j.molstruc.2019.05.120. [DOI] [Google Scholar]
- Barnard P. J.; Bayly S. R.; Holland J. P.; Dilworth J. R.; Waghorn P. A. (2008) In vitro assays for assessing the potential for copper complexes to function as radiopharmaceutical agents. Q. J. Nuclear Med. Mol. Imaging 52 (3), 235–44. [PubMed] [Google Scholar]
- Dechan P.; Bajju G. D.; Sood P.; Dar U. A. (2019) Crystallographic elucidations of indium(III) porphyrin conformations, morphology and aggregation behaviour: Comparative optical study of free base porphyrins and their indium(III) derivatives at varying pH. J. Mol. Struct. 1183, 87–99. 10.1016/j.molstruc.2019.01.064. [DOI] [Google Scholar]
- Tobolkina E. A.; Skripnikova T. A.; Starikova A. A.; Shumilova G. I.; Pendin A. A. (2018) Features of proteolytic properties of tetraphenylporphyrin complex with lanthanide group metals. Spectrochim. Acta, Part A 189, 227–230. 10.1016/j.saa.2017.08.012. [DOI] [PubMed] [Google Scholar]
- Siddiqui W. A.; Ahmad S.; Siddiqui H. L.; Hussain T.; Parvez M. (2010) Synthesis and Crystal Structures of o-[(Phenyl/p-methoxyphenyl)carbamoyl]benzene Sulfonamides. J. Chem. Crystallogr. 40 (2), 116–121. 10.1007/s10870-009-9611-3. [DOI] [Google Scholar]
- te Velde G.; Bickelhaupt F. M.; Baerends E. J.; Fonseca Guerra C.; van Gisbergen S. J. A.; Snijders J. G.; Ziegler T. (2001) Chemistry with ADF. J. Comput. Chem. 22 (9), 931–967. 10.1002/jcc.1056. [DOI] [Google Scholar]
- Shao J.; Sun H.; Guo H.; Ji S.; Zhao J.; Wu W.; Yuan X.; Zhang C.; James T. D. (2012) A highly selective red-emitting FRET fluorescent molecular probe derived from BODIPY for the detection of cysteine and homocysteine: an experimental and theoretical study. Chem. Sci. 3 (4), 1049–1061. 10.1039/C2SC00762B. [DOI] [Google Scholar]
- Santhanamoorthi N.; Lo C. M.; Jiang J. C. (2013) Molecular Design of Porphyrins for Dye-Sensitized Solar Cells: A DFT/TDDFT Study. J. Phys. Chem. Lett. 4 (3), 524–30. 10.1021/jz302101j. [DOI] [PubMed] [Google Scholar]
- Balanay M. P.; Kim D. H. (2008) DFT/TD-DFT molecular design of porphyrin analogues for use in dye-sensitized solar cells. Phys. Chem. Chem. Phys. 10 (33), 5121–7. 10.1039/b806097e. [DOI] [PubMed] [Google Scholar]
- Coutsolelos A. G. R.; Bayeul D.; Lecomte C. (1986) Gallium(III) porphyrins: synthesis and physicochemical characteristics of halogeno gallium(III) porphyrins—X-ray crystal structure of chloro-(5,10,15,20-tetraphenylporphyrinato) gallium(III). Polyhedron 5 (6), 1157–1164. 10.1016/S0277-5387(00)81386-8. [DOI] [Google Scholar]
- Lin S.-J.; Hong T.-N.; Tung J.-Y.; Chen J.-H. (1997) Molecular Structures of Ge(tpp)(OAc)2 and In(tpp)(OAc) and Their Implications: Correlations between the 13C NMR Chemical Shift of the Acetato Ligand and Different Types of Carboxylate Coordination in M(por)(OAc)n {por = tpp (5,10,15,20-Tetraphenylporphyrinate), tmpp (5,10,15,20-Tetrakis(4-methoxyphenyl)porphyrinate), tpyp (5,10,15,20-Tetrakis(4-pyridyl)porphyrinate); M = Ga, In, Tl, Ge, Sn; n = 1, 2}. Inorg. Chem. 36 (18), 3886–3891. 10.1021/ic961304i. [DOI] [Google Scholar]
- Enakieva Y. Y.; Volostnykh M. V.; Nefedov S. E.; Kirakosyan G. A.; Gorbunova Y. G.; Tsivadze A. Y.; Bessmertnykh-Lemeune A. G.; Stern C.; Guilard R. (2017) Gallium(III) and Indium(III) Complexes with meso-Monophosphorylated Porphyrins: Synthesis and Structure. A First Example of Dimers Formed by the Self-Assembly of meso-Porphyrinylphosphonic Acid Monoester. Inorg. Chem. 56 (5), 3055–3070. 10.1021/acs.inorgchem.6b03160. [DOI] [PubMed] [Google Scholar]
- Rosa A.; Baerends E. J.; van Gisbergen S. J. A.; van Lenthe E.; Groeneveld J. A.; Snijders J. G. (1999) Electronic Spectra of M(CO)6 (M = Cr, Mo, W) Revisited by a Relativistic TDDFT Approach. J. Am. Chem. Soc. 121 (44), 10356–10365. 10.1021/ja990747t. [DOI] [Google Scholar]
- van Gisbergen S.J.A.; Snijders J.G.; Baerends E.J. (1999) Implementation of time-dependent density functional response equations. Comput. Phys. Commun. 118 (2), 119–138. 10.1016/S0010-4655(99)00187-3. [DOI] [Google Scholar]
- Ruger R.; van Lenthe E.; Heine T.; Visscher L. (2016) Tight-binding approximations to time-dependent density functional theory — A fast approach for the calculation of electronically excited states. J. Chem. Phys. 144 (18), 184103. 10.1063/1.4948647. [DOI] [PubMed] [Google Scholar]
- Silva A. R. d.; Pelegrino A. C.; Tedesco A. C.; Jorge R. A. (2008) Photodynamic activity of chloro(5,10,15,20-tetraphenylporphyrinato)indium(III). J. Braz. Chem. Soc. 19, 491. 10.1590/S0103-50532008000300017. [DOI] [Google Scholar]
- Oswald J.; Treite F.; Haase C.; Kampfrath T.; Mading P.; Schwenzer B.; Bergmann R.; Pietzsch J. (2007) Experimental hypoxia is a potent stimulus for radiotracer uptake in vitro: comparison of different tumor cells and primary endothelial cells. Cancer Lett. 254 (1), 102–10. 10.1016/j.canlet.2007.02.016. [DOI] [PubMed] [Google Scholar]
- Pereira P. M. R.; Korsak B.; Sarmento B.; Schneider R. J.; Fernandes R.; Tome J. P. C. (2015) Antibodies armed with photosensitizers: from chemical synthesis to photobiological applications. Org. Biomol. Chem. 13, 2518–2529. 10.1039/C4OB02334J. [DOI] [PubMed] [Google Scholar]
- Yaghini E.; Dondi R.; Tewari K. M.; Loizidou M.; Eggleston I. M.; MacRobert A. J. (2017) Endolysosomal targeting of a clinical chlorin photosensitiser for light-triggered delivery of nano-sized medicines. Sci. Rep. 7 (1), 6059. 10.1038/s41598-017-06109-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaghini E.; Dondi R.; Edler K. J.; Loizidou M.; MacRobert A. J.; Eggleston I. M. (2018) Codelivery of a cytotoxin and photosensitiser via a liposomal nanocarrier: a novel strategy for light-triggered cytosolic release. Nanoscale 10 (43), 20366–20376. 10.1039/C8NR04048F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoppenz P.; Els-Heindl S.; Beck-Sickinger A. G. (2020) Peptide-Drug Conjugates and Their Targets in Advanced Cancer Therapies. Front. Chem. 8, 571. 10.3389/fchem.2020.00571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derakhshankhah H.; Jafari S. (2018) Cell penetrating peptides: A concise review with emphasis on biomedical applications. Biomed. Pharmacother. 108, 1090–1096. 10.1016/j.biopha.2018.09.097. [DOI] [PubMed] [Google Scholar]
- Bryden F.; Savoie H.; Rosca E. V.; Boyle R. W. (2015) PET/PDT theranostics: synthesis and biological evaluation of a peptide-targeted gallium porphyrin. Dalton transactions 44 (11), 4925–32. 10.1039/C4DT02949F. [DOI] [PubMed] [Google Scholar]
- Le Guern F.; Sol V.; Ouk C.; Arnoux P.; Frochot C.; Ouk T. S. (2017) Enhanced Photobactericidal and Targeting Properties of a Cationic Porphyrin following the Attachment of Polymyxin B. Bioconjugate Chem. 28 (9), 2493–2506. 10.1021/acs.bioconjchem.7b00516. [DOI] [PubMed] [Google Scholar]
- Reile H.; Armatis P. E.; Schally A. V. (1994) Characterization of high-affinity receptors for bombesin/gastrin releasing peptide on the human prostate cancer cell lines PC-3 and DU-145: internalization of receptor bound 125I-(Tyr4) bombesin by tumor cells. Prostate 25 (1), 29–38. 10.1002/pros.2990250105. [DOI] [PubMed] [Google Scholar]
- Rogers B. E.; Rosenfeld M. E.; Khazaeli M. B.; Mikheeva G.; Stackhouse M. A.; Liu T.; Curiel D. T.; Buchsbaum D. J. (1997) Localization of iodine-125-mIP-Des-Met14-bombesin (7–13)NH2 in ovarian carcinoma induced to express the gastrin releasing peptide receptor by adenoviral vector-mediated gene transfer. J. Nuclear Med. 38 (8), 1221–9. [PubMed] [Google Scholar]
- Mansi R.; Minamimoto R.; Macke H.; Iagaru A. H. (2016) Bombesin-Targeted PET of Prostate Cancer. J. Nucl. Med. 57, 67S–72S. 10.2967/jnumed.115.170977. [DOI] [PubMed] [Google Scholar]
- Debets M. F.; van Berkel S. S.; Schoffelen S.; Rutjes F. P.; van Hest J. C.; van Delft F. L. (2010) Aza-dibenzocyclooctynes for fast and efficient enzyme PEGylation via copper-free (3 + 2) cycloaddition. Chem. Commun. 46 (1), 97–9. 10.1039/B917797C. [DOI] [PubMed] [Google Scholar]
- Ji F.-Y.; Zhu L.-L.; Ma X.; Wang Q.-C.; Tian H. (2009) A new thermo- and photo-driven [2]rotaxane. Tetrahedron Lett. 50 (5), 597–600. 10.1016/j.tetlet.2008.11.080. [DOI] [Google Scholar]
- Sletten E. M.; Bertozzi C. R. (2009) Bioorthogonal chemistry: fishing for selectivity in a sea of functionality. Angew. Chem., Int. Ed. 48 (38), 6974–98. 10.1002/anie.200900942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jewett J. C.; Bertozzi C. R. (2010) Cu-free click cycloaddition reactions in chemical biology. Chem. Soc. Rev. 39 (4), 1272–9. 10.1039/b901970g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carneiro J.; Goncalves A.; Zhou Z.; Griffin K. E.; Kaufman N. E. M.; Vicente M. (2018) Synthesis and in vitro PDT evaluation of new porphyrins containing meso-epoxymethylaryl cationic groups. Lasers Surg. Med. 50 (5), 566–575. 10.1002/lsm.22824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hynek J.; Koncosova M.; Zelenka J.; KriZova I.; Ruml T.; Kubat P.; Demel J.; Lang K. (2018) Phosphinatophenylporphyrins tailored for high photodynamic efficacy. Org. Biomol. Chem. 16 (39), 7274–7281. 10.1039/C8OB01984C. [DOI] [PubMed] [Google Scholar]
- Foster N.; Woo D. V.; Kaltovich F.; Emrich J.; Ljungquist C. (1985) Delineation of a transplanted malignant melanoma with indium-111-labeled porphyrin. J. Nucl. Med. 26 (7), 756–60. [PubMed] [Google Scholar]
- Maric N.; Chan S. M.; Hoffer P. B.; Duray P. (1988) Radiolabeled porphyrin vs gallium-67 citrate for the detection of human melanoma in athymic mice. International journal of radiation applications and instrumentation. Part B, Nuclear medicine and biology 15 (5), 543–51. 10.1016/S0969-8051(88)80013-1. [DOI] [PubMed] [Google Scholar]
- Wolff S. K. (2005) Analytical second derivatives in the Amsterdam density functional package. Int. J. Quantum Chem. 104 (5), 645–659. 10.1002/qua.20653. [DOI] [Google Scholar]
- Berces A.; Dickson R. M.; Fan L.; Jacobsen H.; Swerhone D.; Ziegler T. (1997) An implementation of the coupled perturbed Kohn-Sham equations: perturbation due to nuclear displacements. Comput. Phys. Commun. 100 (3), 247–262. 10.1016/S0010-4655(96)00120-8. [DOI] [Google Scholar]
- Jacobsen H.; Berces A.; Swerhone D. P.; Ziegler T. (1997) Analytic second derivatives of molecular energies: a density functional implementation. Comput. Phys. Commun. 100 (3), 263–276. 10.1016/S0010-4655(96)00119-1. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.