

Summary
Cancer cells rewire their metabolism and rely on endogenous antioxidants to mitigate lethal oxidative damage to lipids. However, the metabolic processes which modulate the response to lipid peroxidation are poorly defined. Using genetic screens, we compared metabolic genes essential for proliferation upon inhibition of cystine uptake or glutathione peroxidase-4 (GPX4). Interestingly, very few genes were commonly required under both conditions, suggesting that cystine limitation and GPX4 inhibition may impair proliferation via distinct mechanisms. Our screens also identify tetrahydrobiopterin (BH4) biosynthesis as an essential metabolic pathway upon GPX4 inhibition. Mechanistically, BH4 is a potent radical-trapping antioxidant that protects lipid membranes from autoxidation, alone and in synergy with Vitamin-E. Dihydrofolate reductase (DHFR) catalyzes the regeneration of BH4 and its inhibition by methotrexate synergizes with GPX4 inhibition. Altogether, our work identifies the mechanism by which BH4 acts as an endogenous antioxidant and provides a compendium of metabolic modifiers of lipid peroxidation.
Introduction
Reactive oxygen species (ROS) are byproducts of cellular aerobic processes. While ROS may act as signaling molecules regulating processes including proliferation, metabolism, and angiogenesis, some free radicals are cytotoxic and damage nucleic acids, proteins, and membrane lipids1. Cells therefore must maintain a tight balance between ROS generation and elimination. A major toxic effect of excessive ROS production is damage to polyunsaturated fatty acids (PUFAs) in lipid membranes. Upon peroxidation, PUFAs can fragment into toxic byproducts or crosslink, disrupting cellular permeability and membrane function2. To mitigate these toxic effects, mammalian cells rely on lipophilic radical-trapping antioxidants (RTAs), such as tocopherols and coenzyme Q10 (CoQ10)3,4, and on Glutathione Peroxidase-4 (GPX4), a glutathione (GSH)-dependent selenoenzyme that reduces toxic lipid hydroperoxides5,6. If the buffering capacity of these cellular defenses is exceeded, the accumulation and decomposition of polyunsaturated phospholipid peroxides induces ferroptosis, an iron-dependent form of cell death7.
Oxidative damage to the plasma membrane and organellar membranes has been implicated in reperfusion injury, neurodegeneration, and inflammation8. Interestingly, many cancer cells also experience oxidative stress during tumorigenesis and in response to therapy, but display low levels of lipid peroxidation, suggesting the presence of robust antioxidant mechanisms9–11. While this is in part mediated by high GPX4 activity, metabolic factors such as iron and amino acid availability, phospholipid saturation, and antioxidant biosynthesis may impact lipid peroxidation levels9. Despite the critical link between cellular metabolism and ferroptosis, the metabolic players that enable cancer cell proliferation under lipid peroxidation have not been systematically studied.
Using functional genetic screens, we determined distinct metabolic genes that modulate cell proliferation in response to cystine limitation and GPX4 inhibition. In particular, our screens reveal tetrahydrobiopterin (BH4) biosynthesis as an essential metabolic pathway under GPX4 inhibition, but not cystine depletion. BH4 is a potent radical-trapping antioxidant which protects lipids from peroxidation and is regenerated by DHFR. Collectively, our work provides a compendium of metabolic modifiers of ferroptosis and identifies BH4 as an effective endogenous antioxidant for lipid membranes.
Results
CRISPR screens identify metabolic regulators of cystine deprivation and GPX4 inhibition
Both direct inhibition of GPX46 or impairment of its function through cystine starvation and depletion of intracellular GSH pools7 trigger lipid peroxidation and ferroptotic cell death, but whether fundamental differences exist between these two conditions remains poorly understood. While blocking GPX4 function prevents the clearing of lipid hydroperoxides, sustained cystine depletion likely affects diverse processes. Indeed, cystine is necessary for iron sulfur cluster generation, heme biosynthesis, and central carbon metabolism. Additionally, cystine contributes to the production of several intracellular metabolites such as taurine and coenzyme A, raising the possibility that the anti-proliferative effects of cystine depletion may be due to the concomitant impairment of these processes.
To begin to address this, we compared the cytotoxic effects of two small molecules that target distinct nodes of molecular defense against lipid peroxidation: erastin, an inhibitor of the cystine-glutamate antiporter System Xc-, and RSL3, an inhibitor of GPX4 (Fig. 1a). Although both small molecules had anti-proliferative effects on Jurkat T-cell leukemia cells and the pancreatic cancer cell line MIA PaCa-2, addition of the lipophilic antioxidant ferrostatin-1 restored proliferation in RSL3-treated cells but not in those treated with erastin or cultured in cystine-depleted media. (Fig. 1b; Extended Data Fig. 1a). Similarly, the necroptosis inhibitor, necrostatin-1, prevented proliferation arrest only in cells under RSL3 treatment (Extended Data Fig. 1b). Of note, cystine depletion triggered distinct cell death responses in these cell lines. While the apoptosis inhibitor Q-VD-OPh, prevented cell death and caspase-3 cleavage in Jurkat cells upon erastin treatment, ferrostatin-1 blocked cell death in erastin-treated Mia PaCa-2 cells (Extended Data Fig. 1c–e). However, none of these cell death inhibitors restored proliferation in either cell line upon cystine depletion. This suggests that sustained cystine depletion and GPX4 inhibition impact cell proliferation via distinct mechanisms across different cell types.
Figure 1: Metabolism focused CRISPR-Cas9 screens identify metabolic modifiers of lipid peroxidation upon cystine deprivation and GPX4 inhibition.
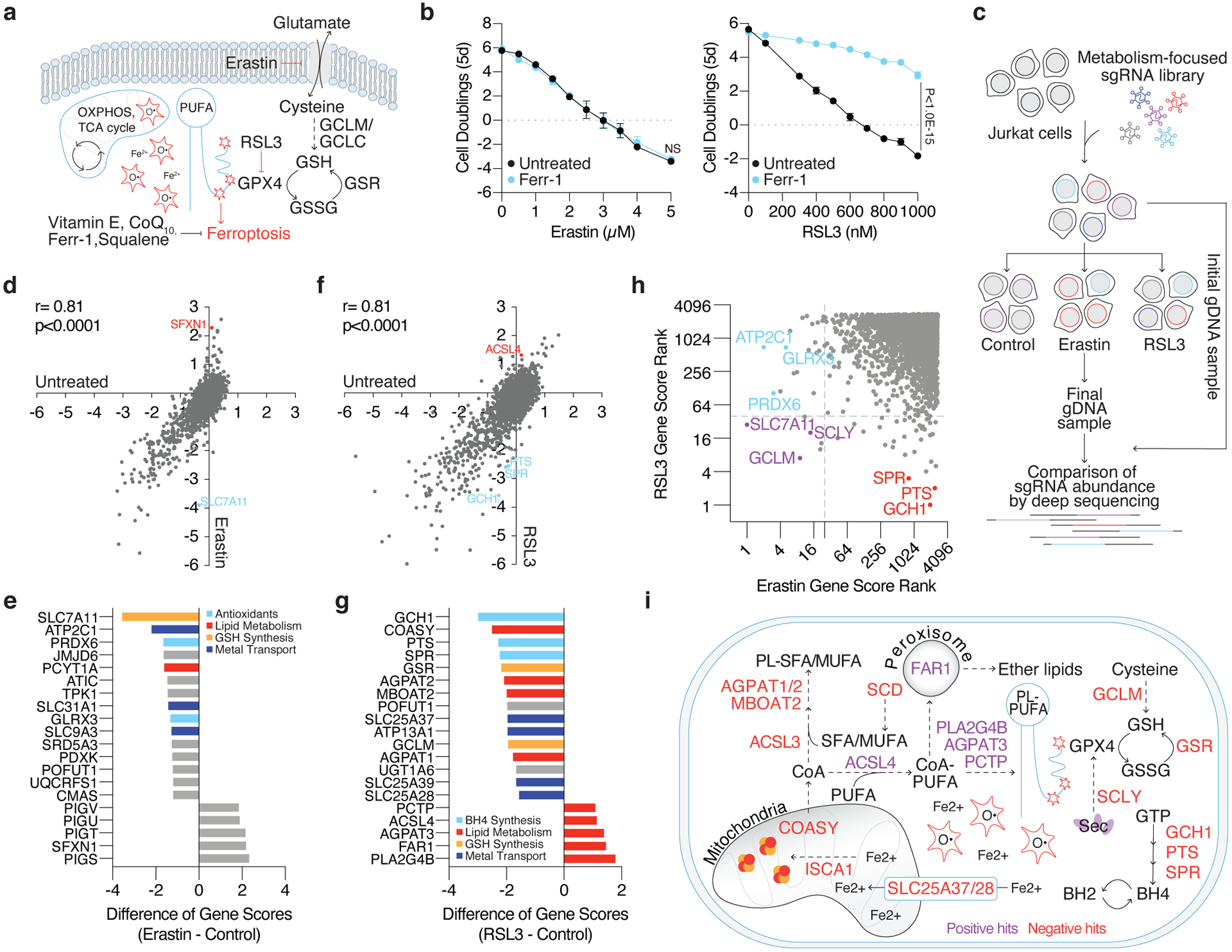
(A) Iron reacts with hydrogen peroxide via the Fenton reaction to produce hydroxyl radicals that damage poly-unsaturated fatty acids (PUFAs). To mitigate lipid peroxidation, cells utilize GPX4, which reduces lipid peroxides at the expense of glutathione (GSH). Alternatively, endogenous antioxidants can quench ROS and lipid peroxides. The accumulation of lipid peroxides either by inhibition of cystine uptake (erastin) or GPX4 (RSL3) induces ferroptosis.
(B) Fold change in cell number (log2) of wild type Jurkat cells treated with erastin (left, black) or RSL3 (right, black) and co-treated with ferrostatin-1 (Ferr-1, blue, 1μM). Data shown as mean ± SD, n=3 biological replicates.
(C) Jurkat cells were transduced with a metabolism-focused library of sgRNAs targeting ~3,000 genes. Transduced cells were cultured without treatment, with erastin (0.5μM), or with RSL3 (0.5μM) for ~14 population doublings. Cells were collected and their genomic DNA (gDNA) isolated. sgRNA abundance was determined by deep sequencing and final sgRNA counts were compared to initial counts to calculate the median differential score for each gene.
(D) Gene scores of untreated (x-axis) and erastin treated (y-axis) Jurkat cells.
(E) Top-scoring genes under erastin treatment. Negative scores represent genes whose loss potentiates erastin toxicity while positive scores represent genes whose loss provides resistance to erastin.
(F) Gene scores of untreated (x-axis) and RSL3 treated (y-axis) Jurkat cells.
(G) Top-scoring genes under RSL3 treatment. Negative scores represent genes whose loss potentiates RSL3 toxicity while positive scores represent genes whose loss provides resistance to RSL3.
(H) Comparison of gene score ranks from erastin and RSL3 screens. Unique hits (p<0.01) in the erastin screen are highlighted (blue) in quadrant II, shared hits are highlighted (purple) in quadrant III, and unique hits in the RSL3 screen are highlighted (red) in quadrant IV.
(I) Summary of positive and negative hits of the RSL3 screen. Red signifies genes essential for survival under GPX4 inhibition (negative hits) and purple signifies genes whose loss provided a proliferative advantage (positive hits).
To further distinguish the metabolic processes regulating cancer cell proliferation under limited cystine from those under GPX4 inhibition, we designed and implemented parallel metabolism-focused CRISPR-Cas9 genetic screens (Fig. 1c). We transduced Jurkat cells with a library of sgRNAs targeting 2,998 metabolic genes and challenged these cells with sublethal doses of erastin or RSL3. After 14 population doublings, we compared the abundance of sgRNAs in erastin- and RSL3- treated cells (Fig. 1d–g). Consistent with the observation that cystine depletion and GPX4 inhibition impair cell proliferation via distinct mechanisms, most of the scoring genes were unique to each condition, except those that encode for glutathione biosynthesis (Fig. 1h, Supplementary Dataset 1).
Notably, the top scoring gene essential for cell proliferation under erastin treatment encodes the cystine transporter, solute carrier family 7 member 11 (SLC7A11) (Fig. 1d–e), likely due to the suboptimal inhibition of cystine transport. Other scoring genes encode glutaredoxin 3, an iron-sulfur cluster binding, glutathione-dependent oxidoreductase, and peroxiredoxin 6 (PRDX6), a non-selenium, glutathione-dependent peroxidase that reduces lipid peroxides (Fig. 1e). PRDX6 has previously been shown to display lysophosphatidylcholine (LPCAT) and lipid peroxidase activity12 and thus may provide alternative means to repair oxidized lipids in the absence of GPX4 activity. These results were recapitulated in an analogous screen performed under low cystine conditions (Extended Data Fig. 2a–b), suggesting that GSH synthesis and cystine transport are the major metabolic limitations for proliferation upon cystine depletion (Extended Data Fig. 2d). Of note, hits unique to the erastin-treated condition could be due to specific effects of the small molecule. Interestingly, the top scoring gene on the positive end of both low and high dose erastin screens and the cystine depletion screen was Sideroflexin 1 (SFXN1), a gene encoding the recently identified mitochondrial serine transporter13 (Fig. 1d–e, Extended Data Fig. 2c).
Polyunsaturated fatty acids (PUFAs), such as arachidonic acid (AA, C20:4) and linoleic acid (LA, C18:2), are the major substrates for lipid peroxidation due to the highly oxidizable methylene group bridging two double bonds2,14 (Fig. 1a). Consistent with this, several phospholipid metabolism genes known to impact membrane saturation scored in the RSL3 screen, but not upon erastin treatment (Fig. 1f–i, Extended Data Fig. 2e). In line with previous studies15–17, loss of long-chain acyl-CoA synthetase 4 (ACSL4), which catalyzes the incorporation of PUFAs into membrane phospholipids, improved cell survival under GPX4 inhibition in our screen (Fig. 1f–g, i). Similarly, loss of the phospholipase PLA2G4B, an enzyme that releases AA from the sn-2 position of phospholipids thereby enabling its lipoxygenase-catalyzed peroxidation, provided resistance to RSL3. Another scoring lipid metabolism gene, PCTP, encodes a phosphatidylcholine (PC) transferase that preferentially binds PUFA-containing PCs. AGPAT3, an enzyme that esterifies PUFAs18 and facilitates their incorporation into phospholipids, also scored positively in contrast to other enzymes of the same family, AGPAT1 and AGPAT2, indicating their substrate preference for saturated fatty acids19 (Fig. 1g, 1i). Loss of FAR1, a critical enzyme in the synthesis of ether glycerolipids, conferred a proliferative advantage under GPX4 inhibition (Fig. 1g, 1i). While a link between plasmalogens and ferroptosis has not been established, ether lipids generally contain unsaturated fatty acids prone to oxidative damage20. Consistent with their role in maintaining membrane fluidity, previous work has shown that plasmalogen synthesis is required for growth under conditions in which saturated fatty acids cause toxicity21,22, indicating potential pro-ferroptotic effects of these lipids. Notably, changes in iron compartmentalization upon loss of the mitochondrial iron transporters SLC25A37, SLC25A39, and SLC25A28 also promoted ferroptosis (Fig. 1g, 1i). Finally, our screen identified GTP cyclohydroxylase-1 (GCH1), 6-pyruvoyltetrahydropterin synthase (PTS), and sepiapterin reductase (SPR), the tetrahydrobiopterin (BH4) biosynthetic pathway, as necessary for proliferation under RSL3 treatment (Fig.1f–i). Indeed, pathway enrichment analysis revealed BH4 biosynthesis and lipid metabolism as the most significantly represented metabolic processes (Extended Data Fig. 2e). Altogether, our screens identify distinct metabolic modifiers of proliferation under cystine depletion and GPX4 inhibition and further suggest that cystine starvation impairs cell proliferation by impacting many other essential processes in addition to GPX4 activity.
SFXN1 loss provides a proliferative advantage under cystine depletion
To understand the mechanisms involved in erastin- and RSL3- induced toxicity, we generated knockout cell lines of scoring genes using CRISPR-Cas9 and assessed their sensitivity to each inhibitor. In line with our screening results, SFXN1 knockout Jurkat cells (Extended Data Fig. 3a) displayed strong resistance to cystine depletion and erastin (Extended Data Fig. 3b). We observed similar responses in HEK 293T cells, indicating a generalized role for SFXN1 across different cell types (Fig. 2a–b).
Figure 2: Loss of SFXN1 enables cell proliferation under cystine depletion.
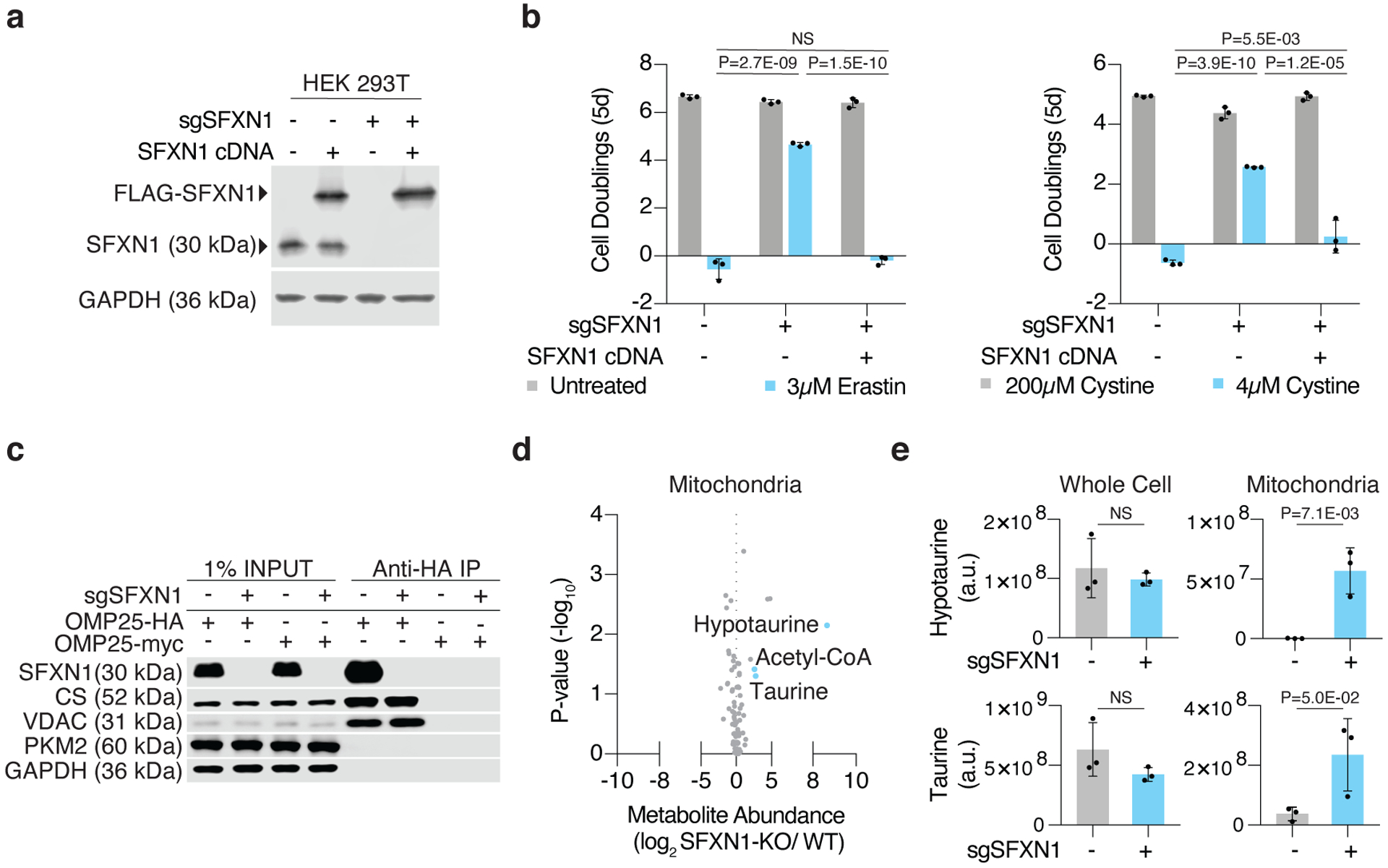
(A) Representative immunoblot analysis of SFXN1 in HEK 293T wild type cells and SFXN1 knockout cells with or without FLAG-SFXN1 cDNA (top). GAPDH was used as a loading control (bottom).
(B) Fold change in cell number (log2) of HEK 293T SFXN1 knockout cells with or without FLAG-SFXN1 cDNA left untreated (gray), treated with erastin (left, blue, 3μM), or cultured in low cystine (right, blue, 4μM). Data shown as mean ± SD, n=3 biological replicates.
(C) Representative immunoblot analysis of cytosolic (PKM2 and GAPDH) and mitochondrial (SFXN1, VDAC, and CS) markers in input, purified mitochondria, or control immunoprecipitates. Lysates were prepared from HEK 293T cells expressing HA-tagged OMP25 or myc-tagged OMP25 (control cells).
(D) Abundance of 83 polar metabolites in mitochondria of HEK 293T cells. Data shown as the ratio of metabolite abundance in SFXN1-null cells to wild type cells (log2, x-axis) versus significance of difference between the median abundances of each metabolite in both groups (−log10p-value, y-axis).
(E) Abundance of hypotaurine (top) and taurine (bottom) in whole cell (left) and mitochondrial (right) lysates from SFXN1-null (blue) or wild type (gray) HEK 293T cells. Data shown as mean ± SD, n=3 biological replicates.
We next focused on determining how SFXN1 mitigates sensitivity to cystine depletion and asked whether loss of SFXN1 affects expression or stabilization of the cystine importer SLC7A11. However, quantification of RNA and immunoblot analysis revealed no changes in SLC7A11 under cystine replete or depleted conditions in wild type or SFXN1-null HEK 293T cells (Extended Data Fig. 3c–d). Glutathione levels and its labeling from cysteine were also unaffected upon SFXN1 loss (Extended Data Fig. 3e). Furthermore, a mass spectrometric analysis of immunoprecipitated proteins from SFXN1 knockout cells expressing 3xFLAG-SFXN1 only revealed an interaction between SFXN1 and its paralog, SFXN3 (Extended Data Fig. 4a), an observation confirmed by immunoblotting experiments (Extended Data Fig. 4b). Consistent with the potential sideroflexin complex and a high degree of homology between SFXN1 and SFXN3 (Extended Data Fig. 4a), expression of SFXN3 restored erastin sensitivity in SFXN1 knockout cells (Extended Data Fig. 4c). These results suggest that the protective effects of SFXN1 deletion are independent of SLC7A11 expression and glutathione synthesis.
SFXN1 is a multi-pass inner mitochondrial membrane protein shown to transport serine13. Thus, we considered that its role in one carbon metabolism might enhance resistance to cystine depletion. However, loss of Serine Hydroxymethyltransferase 2 (SHMT2), the immediate downstream enzyme that catalyzes the conversion of serine to glycine in the mitochondria, did not confer any proliferative advantage under cystine depletion (Extended Data Fig. 4d). This raises the possibility that the phenotype observed upon SFXN1 deletion may not be dependent on its ability to import serine. Rather, our results suggest that loss of SFXN1 may change the compartmentalization of other metabolites relevant to cystine metabolism. To test this, we performed LC-MS based polar metabolomics on whole cells and isolated mitochondria from wild type and SFXN1-null cells (Fig. 2c–e, Extended Data Fig. 4e). While whole cell metabolomics revealed no substantial differences, SFXN1 loss induced profound changes in a subset of mitochondrial metabolites, including an accumulation of acetyl-coenzyme A, taurine, and hypotaurine, two sulfur containing metabolites derived from cysteine (Fig. 2d–e, Extended Data Fig. 4e). Altogether, our results suggest that SFXN1 loss leads to changes in mitochondrial metabolite levels and provides a proliferative advantage under cystine depletion.
BH4 synthesis is necessary and sufficient to protect cells from ferroptosis
Given the high score of the BH4 biosynthetic pathway in the RSL3 screen, we next focused on elucidating the mechanism by which this metabolite mitigates sensitivity to ferroptosis. BH4 is a redox active cofactor for several biosynthetic enzymes including phenylalanine carboxylase and tryptophan hydroxylase23. GCH1 catalyzes the first and rate-limiting step converting GTP to 7,8-dihydroneopterin triphosphate (NH2TP), a pterin phosphate. Two additional enzymes, PTS and SPR subsequently convert NH2TP to BH424. Interestingly, previous studies have shown that BH4 has antioxidant properties in vitro23,24, and a gain of function screen recently identified GCH1 activation as protective during GPX4 inhibition25. However, whether loss of BH4 synthesis sensitizes cells to ferroptosis and the precise mechanism by which BH4 impacts lipid peroxidation remain unknown.
Consistent with the critical role of GCH1 and SPR in BH4 synthesis, CRISPR-mediated deletion of GCH1 or SPR (Fig. 3a), as well as pharmacological inhibition of SPR with the small molecule QM38526, decreased BH4 levels in Jurkat cells (Fig. 3b, Extended Data Fig. 5a). Confirming the screen results, loss of GCH1, SPR, or PTS in Jurkat and Karpas-299 lymphoma cells led to proliferation arrest upon treatment with RSL3 (Fig. 3c, Extended Data Fig. 5b–c). Complementation with an sgRNA-resistant cDNA restored cell proliferation, indicating the on-target effect of the sgRNAs (Fig. 3c). Similarly, chemical inhibition of SPR sensitized parental Jurkat cells to RSL3 (Fig. 3d). Co-treatment with ferrostatin-1 (Fig. 3e) or loss of ACSL4 (Fig. 3f, Extended Data Fig. 5b) reduced sensitivity to GPX4 inhibition in BH4 deficient cells. These results were recapitulated using the alternative GPX4 inhibitor ML162 (Fig. 3g, Extended Data Fig. 5d). However, loss of GCH1 did not sensitize cells to erastin treatment or other ROS-inducing compounds (Fig. 3h), suggesting that BH4 specifically mitigates lipid peroxidation.
Figure 3: BH4 biosynthesis is necessary for cell proliferation upon GPX4 inhibition.
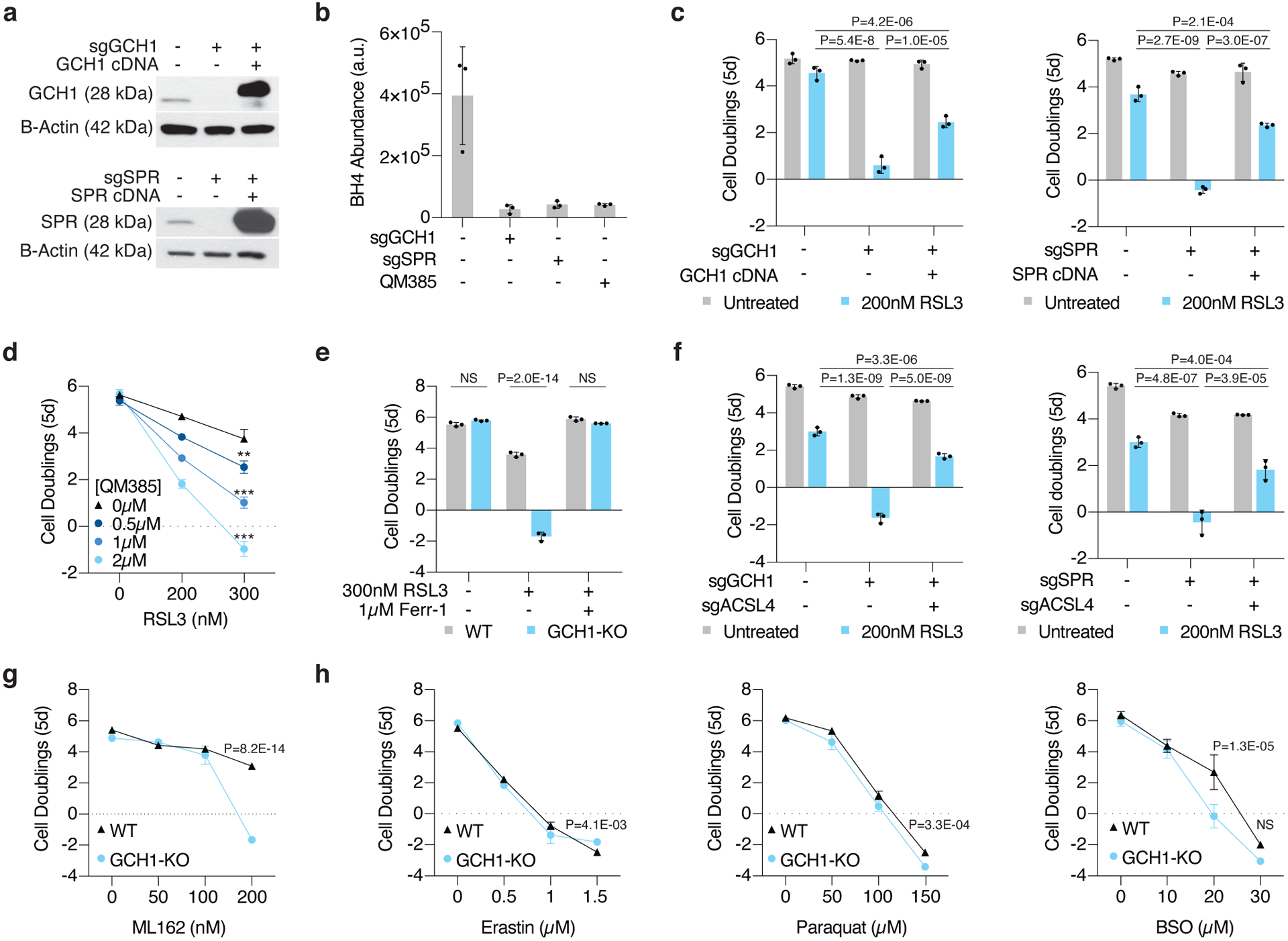
(A) Representative immunoblot analysis of GCH1 in wild type, GCH1 knockout, and GCH1 cDNA expressing Jurkat cells with B-actin used as a loading control (top). Representative immunoblot analysis of SPR in wild type, SPR knockout, and SPR cDNA expressing Jurkat cells with B-actin used as a loading control (bottom).
(B) BH4 abundance in WT, GCH1 knockout, SPR knockout, and QM385-treated Jurkat wild type cells. Data shown as mean ± SD, n=3 biological replicates.
(C) Fold change in cell number (log2) of wild type, GCH1 knockout, and GCH1 cDNA expressing Jurkat cells untreated (gray) or treated with 200nM RSL3 (blue, left). Fold change in cell number (log2) of wild type, SPR knockout, and SPR cDNA expressing Jurkat cells untreated (gray) or treated (blue) with 200nM RSL3 (right). Data shown as mean ± SD, n=3 biological replicates.
(D) Fold change in cell number (log2) of wild type Jurkat cells treated with RSL3 only (black) or cotreated with indicated concentrations of QM385 (blue traces). Data shown as mean ± SD, n=3 biological replicates, **p=2.5E-05, ***p<0.00001 (2.5E-09, 2.1E-11).
(E) Fold change in cell number (log2) of wild type (gray) and GCH1 (blue) knockout Jurkat cells treated with 300nM RSL3 and cotreated with ferrostatin-1 (1μM, Ferr-1). Data shown as mean ± SD, n=3 biological replicates.
(F) Fold change in cell number (log2) of wild type, GCH1 knockout, and GCH1/ACSL4 double knockout Jurkat cells (left) and SPR knockout and SPR/ACSL4 double knockout Jurkat cells (right) left untreated (gray) or treated with 200nM RSL3 (blue). Data shown as mean ± SD, n=3 biological replicates.
(G) Fold change in cell number (log2) of wild type (black trace) and GCH1 knockout (blue trace) Jurkat cells treated with ML162. Data shown as mean ± SD, n=3 biological replicates.
(H) Fold change in cell number (log2) of wild type (black trace) and GCH1 knockout (blue trace) Jurkat cells treated with erastin (left), paraquat (middle) or buthionine sulfoximine (BSO, right). Data shown as mean ± SD, n=3 biological replicates.
To test whether BH4 synthesis protects cells from ferroptosis, we asked if exogenous supplementation of the metabolite would be sufficient to restore proliferation in knockout cells under GPX4 inhibition. As BH4 is easily oxidized in cell culture, we instead used its dehydrogenated product, dihydrobiopterin (BH2), for these experiments. Importantly, supplementing GCH1 knockout cells with BH2 restored endogenous levels of BH4 (Fig. 4a) and abrogated sensitivity to RSL3 and ML210, another inhibitor of GPX4 (Fig. 4b, Extended Data Fig. 6a–b), but not to erastin (Extended Data Fig. 6c). This is consistent with data available from the Cancer Therapeutics Response Portal (CTRP, v2, 2015) which show that GCH1 expression correlates significantly with resistance to GPX4 inhibitors across cancer cell lines (Extended Data Fig. 6d). Consistent with a previous gain of function study25, BH2 supplementation was sufficient to rescue the anti-proliferative effects of genetic deletion of GPX4 in Jurkat (Fig. 4c) and Karpas-299 cells (Extended Data Fig. 6e–f). Ultimately, these experiments show that BH4 availability plays a critical protective role in the cellular response to ferroptosis upon GPX4 inhibition, but not cystine depletion.
Figure 4: GCH1 expression predicts dependence on BH4 upon ferroptosis induction.
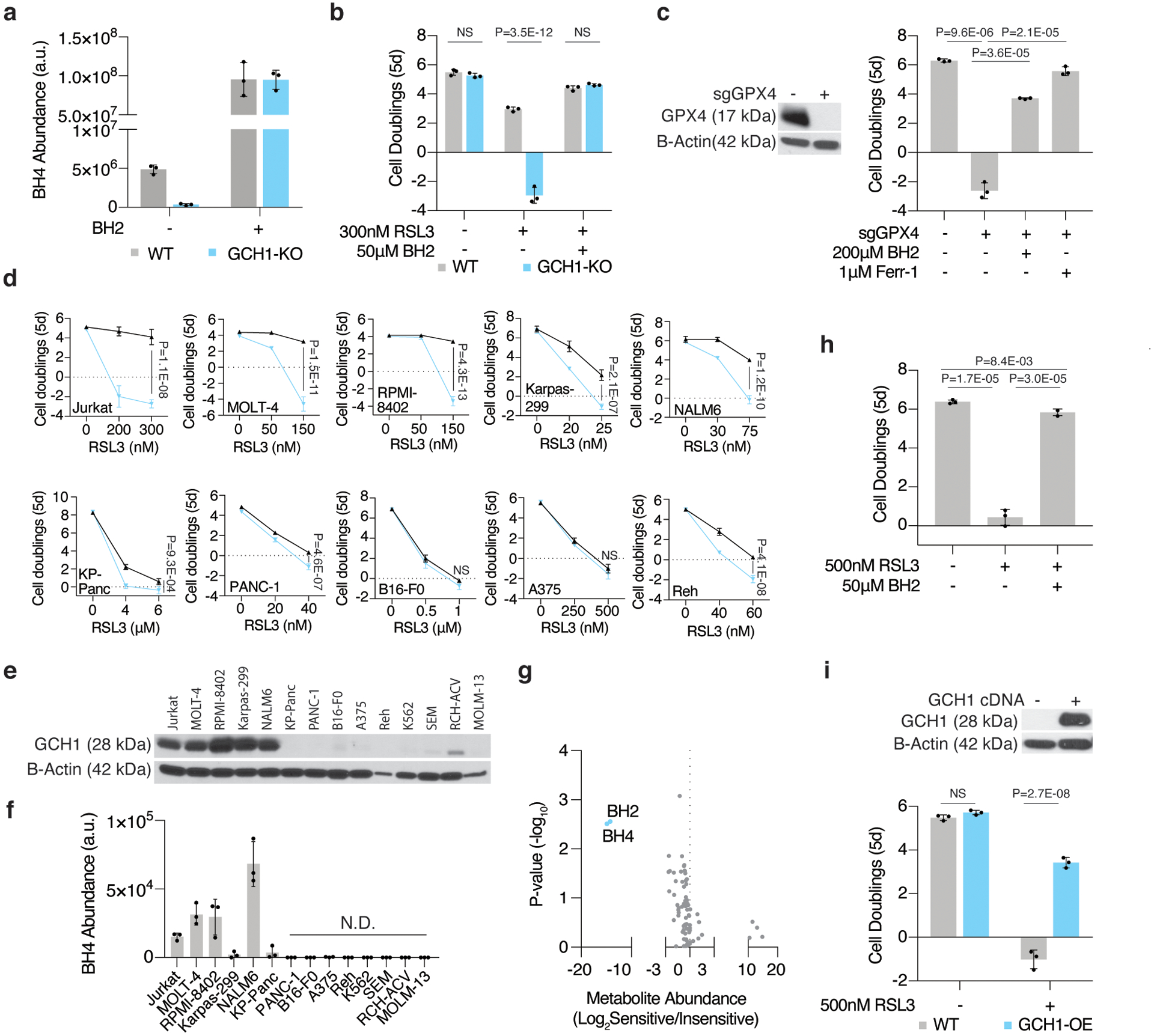
(A) BH4 abundance of wild type (gray) and GCH1 knockout (blue) Jurkat cells at baseline and following BH2 (50μM) supplementation. Data shown as mean ± SD, n=3 biological replicates.
(B) Fold change in cell number (log2) of wild type (gray) and GCH1 knockout (blue) Jurkat cells treated with RSL3 (300nM) and supplemented with BH2 (50μM). Data shown as mean ± SD, n=3 biological replicates.
(C) Representative immunoblot analysis of GPX4 in wild type and GPX4 knockout Jurkat cells (left). Fold change in cell number (log2) of wild type and GPX4 knockout Jurkat cells (right) supplemented with BH2 (200μM) or ferrostatin-1(Ferr-1, 1μM). Data shown as mean ± SD, n=3 biological replicates.
(D) Fold change in cell number (log2) of a panel of cancer cell lines that are sensitive (top) or not (bottom) to GPX4 inhibition upon BH4 depletion (blue traces, cotreated with 3–4μM QM385). Data shown as mean ± SD, n=3 biological replicates.
(E) Representative immunoblot analysis of GCH1 (top) across a panel of cancer cell lines. B-actin was used as a loading control (bottom).
(F) BH4 abundance across a panel of cancer cell lines. Data shown as mean ± SD, n=3 biological replicates.
(G) Metabolomic analysis of 81 polar metabolites. Data shown as the ratio of metabolite abundance in GCH-1 expressing cell lines to GCH1-null cell lines (log2, x-axis) versus significance of difference between the median abundances of each metabolite in both groups (−log10p-value, y-axis).
(H) Fold change in cell number (log2) of wild type A375 cells treated with RSL3 (500nM) and supplemented with BH2 (50μM). Data shown as mean ± SD, n=3 biological replicates.
(I) Representative immunoblot analysis of GCH1 in wild type and GCH1 cDNA expressing A375 cells (GCH1-OE, top). Fold change in cell number (log2, bottom) of wild type and GCH1-OE A375 cells treated with RSL3 (500nM). Data shown as mean ± SD, n=3 biological replicates.
GCH1 expression determines BH4 availability and dependence upon GPX4 inhibition
Because BH4 availability is essential in Jurkat and Karpas-299 cells upon GPX4 inhibition, we next explored whether this metabolic dependence is generalizable across different cancer types. To address this, we tested a diverse panel of cell lines for sensitivity to RSL3 upon SPR inhibition. Surprisingly, only a subset of blood cancer cell lines was acutely sensitive to RSL3 when treated with the SPR inhibitor (Fig. 4d, Extended Data Fig. 6g). In contrast to previous work25, we did not observe any correlation between GCH1 expression and ferroptosis sensitivity. Instead, we found that GCH1 protein levels (Fig. 4e) correlate with increased BH4 availability (Fig. 4f) and dependence on this pathway upon RSL3 treatment (Fig. 4g). Importantly, BH2 supplementation or overexpression of GCH1 in a BH4-deficient cell line was sufficient to phenocopy RSL3 resistance (Fig. 4h–i, Extended Data Fig. 6h). These data show that GCH1 expression is a determinant of BH4 availability and predicts cancer cell line dependence on BH4 upon ferroptosis induction.
BH4 prevents lipid peroxide accumulation independently of its cofactor role
As ferroptosis is driven by the accumulation of phospholipid peroxides, we tested whether BH4 mitigates lipid peroxidation using BODIPY 581/591 C11 staining in wild type and GCH1 knockout cells upon GPX4 inhibition. While we did not observe a significant difference in lipid peroxidation between the two groups at baseline, BH4-depleted cells displayed elevated levels of lipid peroxidation upon GPX4 inhibition compared to wild type cells, a phenotype completely reverted by BH2 supplementation (Fig. 5a, Supplementary Fig. 1). Consistent with this, lipidomic analysis revealed that PUFA-containing lipid species were significantly depleted in SPR knockout cells upon treatment with RSL3, but not at baseline27 (Fig. 5b, Supplementary Table 1).
Figure 5: BH4 is a potent radical-trapping antioxidant in lipid membranes.
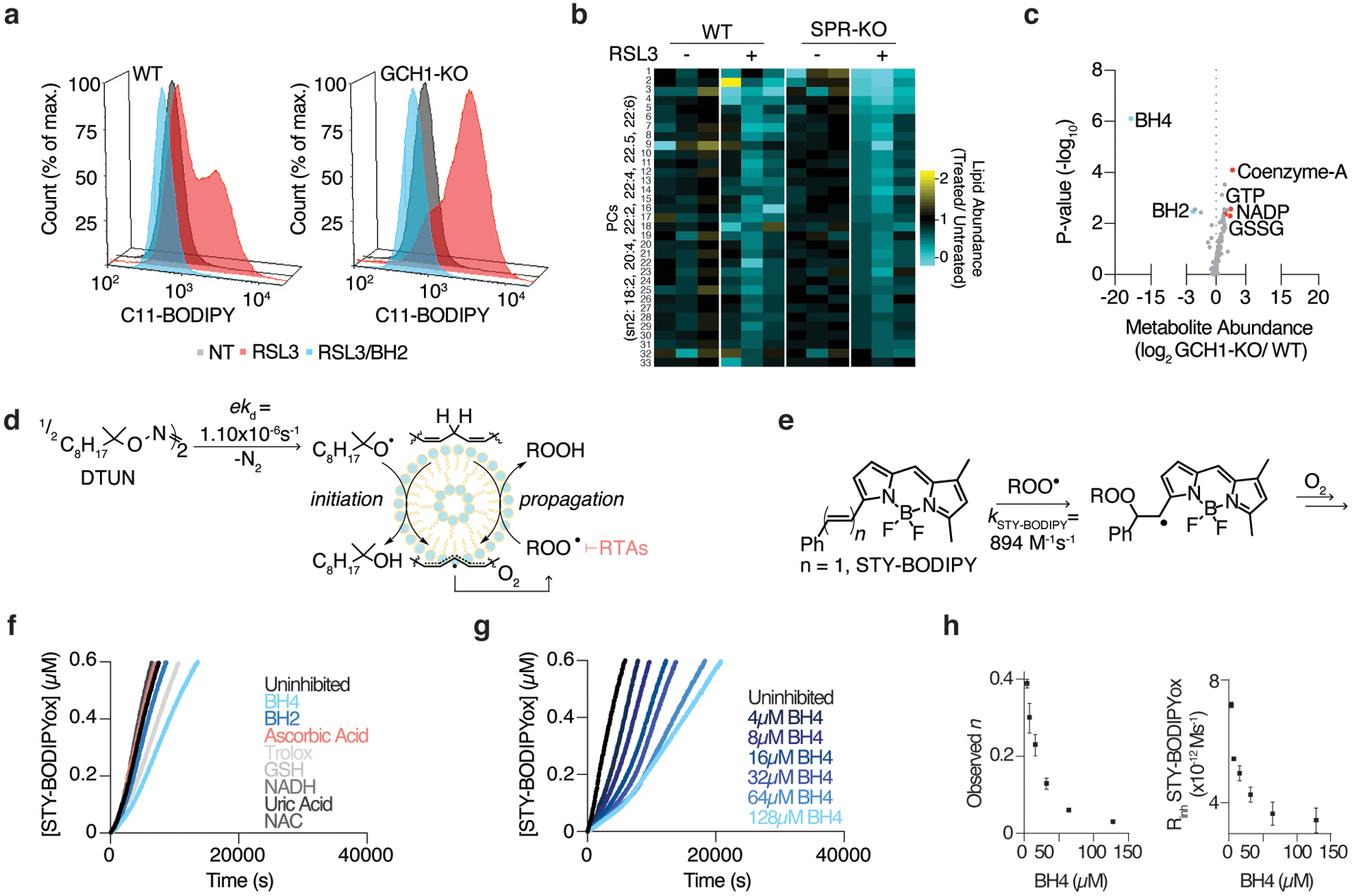
(A) Lipid peroxidation assessed by flow cytometry measuring C11-BODIPY fluorescence in wild type and GCH1-KO Jurkat cells treated with RSL3 (175nM, red) with or without BH2 supplementation (50μM, blue) for 15 hours.
(B) Heatmap showing changes in a subset of PUFA-containing phosphatidylcholines (PCs) in wild type and SPR-KO cells upon RSL3 treatment. Data shown as the relative change in abundance in RSL3-treated cells compared to untreated cells.
(C) Metabolomic analysis of 82 polar metabolites in Jurkat cells. Data shown as the ratio of metabolite abundance in GCH1 knockout to wild type cells (log2, x-axis) versus significance of difference between the median abundances of each metabolite in both cell lines (−log10p-value, y-axis).
(D) The lipophilic hyponitrite DTUN is used to initiate the RTA-inhibited co-autoxidation of the polyunsaturated lipids of egg phosphatidylcholine liposomes and STY-BODIPY.
(E) The initial rate and inhibited period of STY-BODIPY consumption is used to derive the rate constant and stoichiometry of added RTAs.
(F) Representative autoxidations of STY-BODIPY (1μM)-embedded liposomes of egg phosphatidylcholine lipids (1mM, ~100nm particle size) suspended in phosphate-buffered saline pH 7.4 at 37°C initiated by 0.2mM DTUN and inhibited by common water-soluble antioxidants (10μM) and BH4 (4μM).
(G) Representative autoxidation of STY-BODIPY as in (F), but with varying concentrations of BH4.
(H) RTA stoichiometry of BH4 (left) and inhibited rate of STY-BODIPY oxidation with varying concentration of BH4 (right) from the data in (G). Data shown as mean ± SD, n=3 biological replicates.
We next focused on elucidating the mechanism by which BH4 prevents the peroxidation of lipid membranes. In mammalian cells, BH4 serves as a cofactor required for the activity of nitric oxide synthase 3 (NOS3), alkylglycerol monooxygenase (AGMO), and aromatic amino acid hydroxylases. We first considered whether its described role as a cofactor for NOS3 and AGMO might impact redox balance and lipid peroxidation. NOS3 converts arginine and oxygen into citrulline and nitric oxide, oxidizing BH4 to BH2. Low levels of BH4 can cause NOS3 uncoupling, resulting in the formation of superoxide, which can lead to species that initiate lipid peroxidation28,29. AGMO catalyzes the hydroxylation and cleavage of ether lipids, and thus may play a role in the metabolism of polyunsaturated phospholipids30. However, of the known BH4-dependent enzymes, NOS3 is the only one with detectable RNA transcripts in both Jurkat and Karpas-299 cells (Extended Data Fig. 7a–b). Further analysis confirmed that AGMO is not expressed in these cells, while NOS3 is expressed in Jurkat cells (Extended Data Fig. 7c). Notably, chemical inhibition with the pan-NOS inhibitor L-NIO (Extended Data Fig. 7d) and genetic deletion of NOS3 (Extended Data Fig. 7e) failed to protect BH4-depleted cells from GPX4 inhibition, indicating an NOS3-independent mechanism. To determine whether an alternative enzyme relies on BH4 as a cofactor to mitigate lipid peroxidation, we performed a CRISPR-Cas9 genetic screen to identify metabolic genes essential for BH2 to rescue a lethal dose of RSL3 and compared it to a similar screen in which ferrostatin-1 was used to rescue RSL3 treatment. Surprisingly, very few genes differentially scored under each condition, raising the possibility that the cofactor role of BH4 is not relevant in its protective effect against ferroptosis (Extended Data Fig. 7f, Supplementary Dataset 1).
To begin to understand whether BH4 itself is protecting cells from oxidative damage, we performed a polar metabolomic analysis and defined the impact of BH4 availability on whole-cell metabolism and redox-sensitive metabolites. In line with our previous data, BH4 was the most significantly depleted metabolite in GCH1 knockout cells (Fig. 5c). Interestingly, loss of BH4 induced the accumulation of coenzyme A, oxidized glutathione (GSSG), and nicotinamide-adenine dinucleotide phosphate (NADP) (Fig. 5c). Consistent with an observed increase in reduced CoQ10 in GCH1 overexpressing cells25, these results suggest that BH4 availability impacts redox balance in cancer cells. Altogether, these experiments suggest that BH4 regulates cancer cell sensitivity to ferroptosis by preventing the accumulation of PUFA-containing lipid peroxides independent of its role as a cofactor.
BH4 is a potent radical-trapping antioxidant in lipid membranes
Building upon the observation that BH4 triggers changes in redox-sensitive metabolites independently of its cofactor function, we considered whether BH4 could behave as an RTA in mammalian lipid membranes. Indeed, early studies have shown that BH4 reacts with superoxide and hydroxyl radicals24 and can inhibit dopamine autoxidation in vitro31. Similarly, recent work shows that BH4 reduces the stable radical DPPH and AAPH-derived radicals25. However, these experiments do not report on the ability of BH4 to inhibit lipid peroxidation or its potency as a suppressor of ferroptosis32. This is best exemplified by the fact that, in these assays, BH4 was similarly protective to ascorbate, a water-soluble antioxidant that does not inhibit ferroptosis24,25,32. Thus, it remains unclear whether BH4 traps water-soluble radicals or directly inhibits lipid peroxidation similar to ferrostatin-1 and other canonical ferroptosis inhibitors33.
RTAs generally transfer a H-atom to a (phospho)lipid peroxyl radical, preventing it from reacting with another lipid and propagating the lipid peroxidation chain reaction, the process which drives ferroptotic cell death34. To investigate the RTA activity of BH4 in a physiologically relevant system, we carried out co-autoxidations of egg phosphatidylcholine lipids and STY-BODIPY35, a highly fluorescent and oxidizable probe used to monitor the autoxidation reaction and its inhibition by RTAs (Fig. 5d–e, Extended Data Fig. 7g). The rate of the consumption of STY-BODIPY and the duration of the inhibited period was used to derive the rate constant (kinh) and stoichiometry (n) of added RTAs (Extended Data Fig. 7g). Although water-soluble compounds are generally poor inhibitors of phospholipid peroxidation since they are phase-separated from the propagating lipid-peroxyl radicals, BH4 was an effective inhibitor, outperforming all other water-soluble antioxidants tested, enabling the derivation of kinh = 1.7×104 M−1s−1 and n = 0.4 (Fig. 5f). The non-integer stoichiometry reflects the poor oxidative stability of BH4 and suggests that BH4 autoxidizes in parallel with its ability to trap peroxyl radicals, a characteristic of many oxidatively labile RTAs36–38. Indeed, increasing the concentration of BH4 led to further suppression of the rate of oxidation and a corresponding increase in the inhibited period (Fig. 5g) in a non-linear fashion (Fig. 5h). Supplementing the medium with superoxide dismutase and/or catalase did not have a significant impact, indicating that BH4 autoxidizes independently of any superoxide or hydrogen peroxide formed under the experimental conditions (Extended Data Fig. 7h). Importantly, BH4 had similar activity to α-tocopherol (α-TOH), the most potent form of Vitamin E and the most effective endogenous RTA39, which is characterized by kinh = 4.7×103 M−1s−1 and n = 2 (Fig. 6a). These results suggest that although BH4 is water-soluble, it is likely that since it is uncharged at physiological pH40, it can partition into the lipid bilayer where one molecule of BH4 traps one to two lipid-derived peroxyl radicals and subverts lipid peroxidation (Extended Data Fig. 8a–c).
Figure 6: DHFR regenerates BH4, which synergizes with α-tocopherol to suppress ferroptosis.
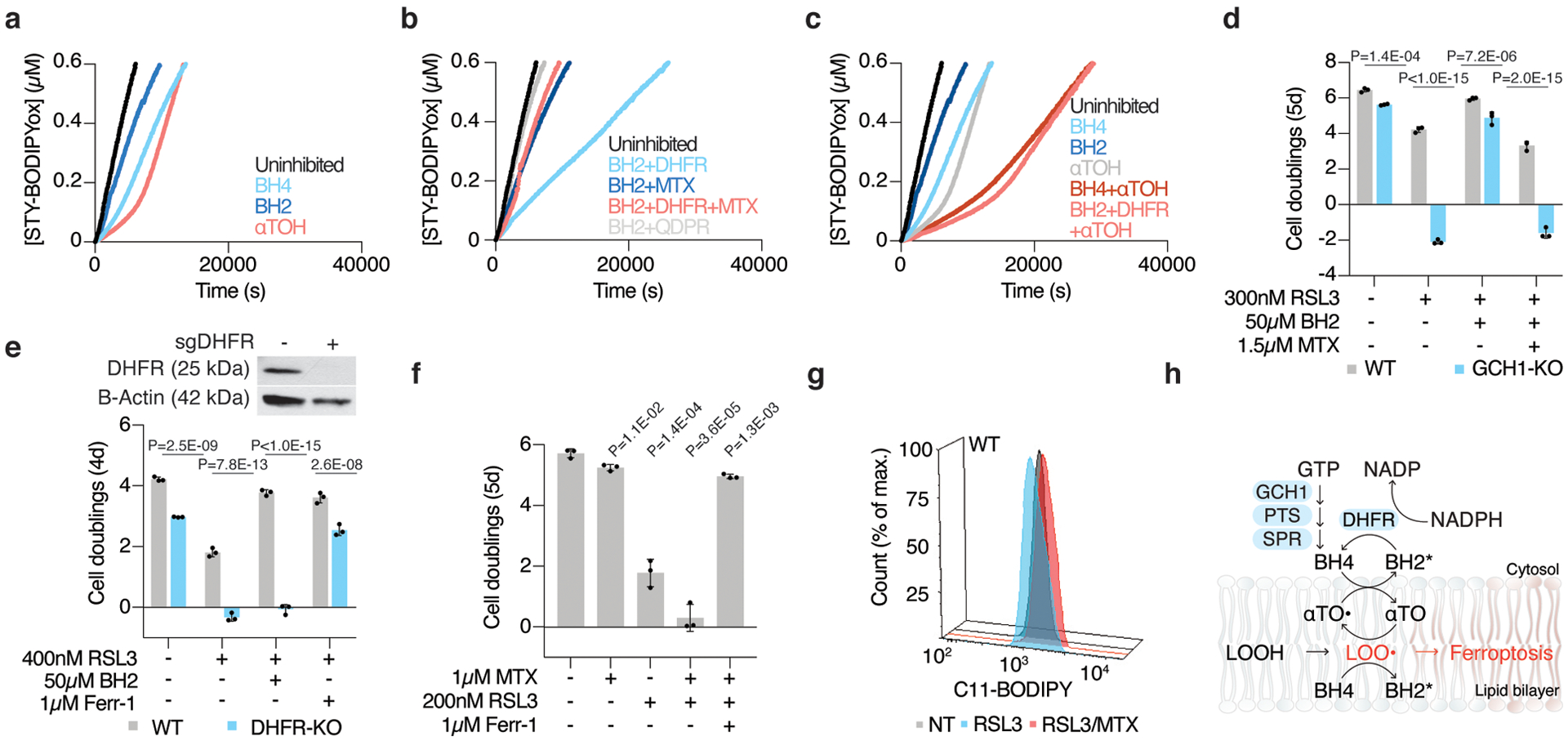
(A) Representative autoxidations of STY-BODIPY (1μM)-embedded liposomes of egg phosphatidylcholine lipids (1mM, ~100nm particle size) suspended in phosphate-buffered saline pH 7.4 at 37°C initiated by 0.2mM DTUN containing 60μM NADPH and either BH4 (4μM), α-tocopherol (5μM), and BH2 (10μM) as indicated.
(B) Representative autoxidation of STY-BODIPY as in (A) but containing 60μM NADPH and BH2 (10μM) with DHFR (50nM), methotrexate (MTX, 100nM), and QDPR (150nM) as indicated.
(C) Representative autoxidation of STY-BODIPY as in (A), but containing 60μM NADPH and BH4 (4μM), BH2 (10μM), α-tocopherol (5μM), and DHFR (50nM) as indicated.
(D) Fold change in cell number (log2) of wild type (gray) and GCH1 knockout Jurkat cells treated with 300nM RSL3, supplemented with 50μM BH2, and cotreated with methotrexate (MTX, 1.5μM). Data shown as mean ± SD, n=3 biological replicates.
(E) Representative immunoblot analysis of DHFR in wild type and DHFR knockout Jurkat cells (top). Fold change in cell number (log2) of wild type (gray) and DHFR knockout (blue) Jurkat cells cultured in nucleoside/ formate supplemented media and treated with 400nM RSL3 and supplemented with 50μM BH2 or cotreated with ferrostatin-1 (Ferr-1, 1μM). Data shown as mean ± SD, n=3 biological replicates.
(F) Fold change in cell number (log2) of wild type Jurkat cells cultured in nucleoside supplemented media treated with methotrexate (1μM) and RSL3 (200nM) and ferrostatin-1 (Ferr-1, 1μM). Data shown as mean ± SD, n=3 biological replicates.
(G) Lipid peroxidation assessed by flow cytometry measuring C11-BODIPY fluorescence in wild type Jurkat cells treated with RSL3 alone (200nM for 5 hours, blue) or with methotrexate (1.5μM for 18 hours, MTX, red)
(H) BH4 synergizes with α-tocopherol to interrupt lipid peroxidation and suppress ferroptosis. BH4 reacts with peroxyl radicals to yield oxidation products (BH2*, which may include BH2, qBH2, BH3, and/or BH4-derived radicals) that may be reduced by DHFR back to BH4.
Dihydrofolate reductase regenerates BH4 and sustains its anti-ferroptotic function
The oxidation of BH4 can yield a complex mixture of products including BH3, BH2, quinoid-BH2 (qBH2), and other BH4-derived radicals41,42. In the liposomal system, at the point where the α-TOH-inhibited oxidation proceeded at the uninhibited rate (corresponding to consumption of α-TOH), the BH4-inhibited oxidation continued at a slightly retarded rate, raising the possibility that a BH4-derived product can inhibit the oxidation, albeit much less efficiently (Fig. 6a). Consistent with the formation of BH2 following BH4 oxidation, BH2 slowed the oxidation reaction at a similar rate (Fig. 6a). Previous in vitro studies suggest that Quinoid dihydropteridine reductase (QDPR) and Dihydrofolate reductase (DHFR) may regenerate BH4 from BH2, but whether the regenerative role of these enzymes is physiologically relevant in the context of lipid peroxidation is unclear43,44. In liposome suspensions, addition of recombinant DHFR to the BH2-inhibited reaction improved its activity dramatically, in line with the formation of BH4 in situ, and this was fully reversed by the addition of methotrexate, an inhibitor of DHFR (Fig. 6b). In contrast, addition of recombinant QDPR did not improve the RTA activity of BH2 in liposomes (Fig. 6b).
Although a previous report suggests that BH4 availability may enhance anti-ferroptotic defenses by impacting the levels of reduced CoQ1025, combining BH4 or BH2+DHFR with CoQ10 did not show any synergy in our liposome assays (Extended Data Fig. 9a). Thus, BH4 mediates ferroptosis likely independently of CoQ10 availability. Indeed, we did not find any scoring genes encoding enzymes involved in CoQ10 metabolism in the BH2 rescue CRISPR screen (Extended Data Fig. 7f, Supplementary Dataset 1). In contrast, the same combinations with α-TOH yielded superior inhibitory activity to that observed for the individual components, indicating a synergistic interaction between BH4 and tocopherol (Fig. 6c). This may result from scavenging of α-TOH derived radicals (α-TO•) by BH4, similar to the synergy observed between α-TOH and ascorbate45 or α-TOH and reduced CoQ10. Indeed, the α-TOH/BH2/DHFR system is reminiscent of the recently characterized α-TOH/CoQ10/FSP13,4 system, in that cytosolic reducing equivalents in the form of NAD(P)H are funneled toward α-TOH, interrupting lipid peroxidation and suppressing ferroptosis in cancer cells (Extended Data Fig. 8a–c).
Consistent with the mechanism derived from the liposome assays, pharmacological inhibition of DHFR with methotrexate precluded rescue by BH2 supplementation in GCH1 knockout Jurkat cells, confirming that BH4, and not BH2, directly mitigates lipid peroxidation (Fig. 6d). Remarkably, genetic deletion or pharmacological inhibition of DHFR in Jurkat and Karpas-299 cells synergized with GPX4 inhibition to induce ferroptosis in a manner independent of DHFR’s role in one carbon metabolism (Fig. 6e–g, Extended Data Fig. 9b). Furthermore, BH2-mediated rescue was independent of QDPR expression (Extended Data Fig. 9c), suggesting that regeneration of BH4 via DHFR is the most efficient pathway in this process (Figure 6h). Altogether, these experiments highlight that the BH4-DHFR axis is essential for mitigating lipid peroxidation and ferroptosis upon GPX4 inhibition in a subset of cancer types.
Discussion
Proliferating cancer cells require substantial metabolic rewiring or endogenous antioxidants to overcome the anti-proliferative effects of lipid peroxidation. While GPX4 mitigates the accumulation of lipid hydroperoxides in most cells, several metabolic factors including iron availability and compartmentalization, lipid saturation status, and antioxidant capacity may also impact the cellular response to lipid peroxidation. Identification of metabolic pathways could provide basic insight into how cells deal with lipid peroxidation and reveal novel therapeutic targets. Using parallel genetic screens, our study provides a compendium of metabolic determinants of cancer cell sensitivity to cystine limitation and GPX4 inhibition. Notably, not all our hits are shared across genetic screens previously performed in different cell lines3,4,46. This suggests that several of the mechanisms identified here or elsewhere may be highly dependent on the cell type or tissue of origin.
Despite the common notion that cystine deprivation inhibits cell growth by increasing lipid peroxidation, our study provides evidence that cystine may be limiting for other metabolic processes in addition to GPX4 activity in cancer cells. Cystine is necessary for protein synthesis, oxidative metabolism and the production of iron sulfur clusters. Our results are also consistent with previous work suggesting that iron sulfur cluster synthesis is impaired in a subset of ovarian cancer cells under cystine deprivation47. As several preclinical strategies using cystine depletion reduce tumor growth48, future work is necessary to understand how these therapies trigger anti-tumor effects in different contexts and cancer types. Notably, our screens identified that loss of SFXN1 allows cells to proliferate under limited cystine conditions. Thus, SFXN1 loss may be one way for cancer cells to develop resistance to these therapies. While we do not completely understand how SFXN1 is involved in cysteine metabolism, in our preliminary work we found an accumulation of two sulfur-containing cysteine-derived metabolites in mitochondria upon SFXN1 loss, namely taurine and hypotaurine, which may act as antioxidants to provide a growth advantage upon cysteine deprivation49. Further investigation is necessary to understand the precise mechanism by which SFXN1 regulates compartmentalized cysteine homeostasis.
A major finding of our work is the identification of the BH4 biosynthesis pathway as an essential regulator of ferroptosis sensitivity in cancer cells. Interestingly, many of the cell lines dependent upon this pathway originate from T and B cell acute lymphoblastic leukemias and lymphomas. As BH4 availability has previously been shown to alter iron metabolism and mitochondrial function in T cells26, the antioxidant capacity of BH4 may be particularly important for T cell function. Despite its relatively polar structure, inhibited autoxidation experiments show that BH4 can act as an endogenous RTA. These findings add BH4 to the list of endogenous antioxidants that cancer cells accumulate to boost antioxidant defenses3,4,46,50. Additionally, our study identifies DHFR as an important regulator of ferroptosis. Indeed, DHFR regenerates oxidized BH4 and blocking DHFR’s function genetically or pharmacologically with methotrexate synergizes with GPX4 inhibition to induce ferroptosis. Since methotrexate is a commonly used chemotherapeutic that targets one carbon metabolism, its combination with potential GPX4 inhibitors or with anti-cancer therapies that induce lipid peroxidation10,11 may have therapeutic value.
Materials and Methods
Compounds
The following compounds were used: polybrene, puromycin, buthionine sulfoxamine, paraquat, L-ascorbic acid (Sigma-Aldrich), blasticidin (Invivogen), RSL3 (Selleckchem), ML162 (Aobious), erastin (Tocris), ML21051 (Selleckchem), dithiothreitol (Biovision), ferrostatin-1 (Tocris), and matrigel (Corning), Egg PC (Sigma), STY-BODIPY35, DTUN32, α-tocopherol (Sigma, purified by flash chromatography before use), CoenzymeQ10 (Alfa Aesar), tetrahydrobiopterin (BH4) and dihydrobiopterin (BH2) (Cayman Chemicals), NADPH (Alfa Aesar), dihydrofolate reductase (R&D Systems), methotrexate (US Biological), superoxide dismutase (Sigma), catalase (Sigma). All purchased compounds had a reported purity value ≥ 98%. QM385 was kindly provided by Dr. Clifford Woolf.
Cell Lines and Cell Culture
All cell lines were purchased from the ATCC, authenticated by STR profiling, and verified to be free of mycoplasma. These and their derivatives were maintained at 37°C and 5% CO2 and cultured in RPMI media (Gibco) supplemented with 2mM glutamine, 10% fetal bovine serum, penicillin, and streptomycin.
Proliferation Assays
Cell lines were cultured in triplicates in 96-well plates at 1,000 (suspension cell lines) or 500 (adherent cell lines) cells per mL in a final volume of 200μL RPMI media and the indicated treatments. An initial time point of untreated cells was used for normalization. After 5 days of growth, 40μl of Cell Titer Glo reagent (Promega) were added to each well and luminescence was read using a SpectraMax M3 plate reader (Molecular Devices). Data is presented as the log2 fold change in luminescence to represent the number of population doublings since the initial timepoint. Where nucleosides were supplemented, thymidine, uridine, adenosine, cytidine, and inosine (Sigma-Aldrich) were added at 250μM in RPMI media unless otherwise indicated. For low cystine experiments, cystine-free RPMI was prepared (U.S. biologicals) and supplemented with 10% dialyzed fetal bovine serum and penicillin/streptomycin with indicated cystine concentrations added to media.
CRISPR-Cas9 Screens
The focused sgRNA library targeting key genes within every mammalian metabolic pathway was designed as previously described52. Oligos (IDT) were cloned into lentiCRISPR-v1 (erastin screen) or lentiCRISPR-v2 (RSL3 screens) using T4 ligase (NEB). Ligation products were cloned into E. coli (NEB) and plasmids were isolated by Miniprep (QUIAGEN). This plasmid pool was used to generate a lentiviral library which was transfected into HEK 293T cells and used to generate viral supernatant as described above. Jurkat and Karpas-299 cells were infected at an MOI of 0.7 and selected with puromycin. An initial sample of 30 million cells was harvested and infected cells were cultured for about 14 population doublings under each condition (sublethal erastin: 0.5μM, sublethal RSL3: 0.5μM, low cysteine: 10μM, lethal erastin: 3μM, lethal RSL3, BH2, ferrsotatin-1: 180nM, 50μM, 1μM). Final samples of 30 million cells were collected. DNA was extracted (DNeasy Blood and Tissue kit, QUIAGEN). sgRNA inserts were PCR amplified and barcoded by PCR, using primers unique to each condition. PCR amplicons were purified and sequenced (NextSeq 500, Illumina). Screens were analyzed using Python (v.2.7.13), R (v.3.3.2), and Unix (v.4.10.0–37-generic x86_64). Gene score for each gene was defined as the median log2 fold change in abundance of each sgRNA targeting that gene. All gene scores for each screen are provided in Supplementary Dataset 1.
Generation of Knockout and cDNA Overexpression Cell Lines
For SFXN1, GCH1, SPR, PTS, QDPR, and DHFR knockout cell lines, sgRNAs targeting each gene of interest were cloned into lentiCRISPR-v2-puro linearized with BsmBI by T4 ligase (NEB). The oligos (IDT) used to generate sgRNAs are available in Supplementary Table 2.
For virus production, the sgRNA-expressing vector and lentiviral packaging vectors (VSV-G and Delta-VPR) were transfected into HEK 293T cells using XtremeGene 9 transfection reagent (Roche). For SFXN1 cDNA expressing cell lines, cDNA was cloned into PMXS-IRES-GFP linearized with BamHI and NotI by Gibson Assembly (NEB). For GCH1 and SPR cDNA expressing cell lines, cDNA cloned into PMXS-IRES-blast linearized with BamHI and NotI by Gibson Assembly (NEB) and transfected along with retroviral packaging plasmids (VSV-G and Gag-pol) into HEK 293T cells. 18 hours after transfection, transfection media was replaced with fresh media. 48 hours after transfection, media containing viral particles was collected and filtered using a 0.45mm filter to exclude HEK 293T cells. 18 hours prior to infection, cells of interest were plated in 6-well tissue culture plates (100,000 cells/well for suspension lines and 50,000 cells/well for adherent lines). Then, infection media and 8μg/mL of polybrene were added. Cells were spin infected by centrifugation at 2,200rpm and 37°C for 1.5 hours. 24 hours after infection, infection media was replaced with fresh media. 48 hours after infection, selection of transduced cells was started by adding puromycin (for sgRNA lentiviral vector) or blasticidin (for overexpression retroviral vectors). For pMXS-SFXN1-IRES-GFP expressing cells, GFP-positive cells were sorted 48–72 hours post transduction. DHFR-KO cells were infected and cultured in media supplemented with 1μM ferrostatin-1, 1mM formate, and 100μM nucleosides.
For Karpas-299 GPX4 knockout cells and for double knockouts (ACSL4, AGMO and NOS3), sgRNAs were cloned into lentiCRISPR-GFP. Cells were infected as above, with the exception that GPX4-KO cells were cultured in the presence of 1μM ferrostatin-1. The top 25% GFP-fluorescent cells were FACS-sorted and collected. For Jurkat single cell GPX4 knockout clones, the top 25% GFP-fluorescent cells were FACS sorted and single cells were plated into a 96-well plate, each well containing 200μL of media with 1μM ferrostatin. All mixed-population knockouts and single cell clones were validated by Western blot with the exception of PTS knockouts due to the lack of a specific antibody. SHMT2 knockout Jurkat cells were a generous gift from Dr. Richard Possemato (NYU Langone Medical Center).
Guide resistant FLAG-SFXN1(Q9H9B4-1) and HA-SFXN3 (Q9H9B4-1) cDNA was designed and codon-optimized. Guide resistant GCH1 (P30793-1) and SPR (P35270-1) cDNA was designed and codon-optimized. The truncated isoform, GCH1–3 (P30793-3), was amplified using Phusion High Fidelity PCR (NEB). Sequences of the primers used for this reaction are available in Supplementary Table 2. PCR-overlap extension and Gibson assembly was used to clone both isoforms cDNA into PMXS-ires-blast.
Immunoblotting
Cell pellets were washed twice with PBS and lysed in standard lysis RIPA buffer (10 mM Tris-Cl pH 7.5, 150 NaCl, 1 mM EDTA, 1% Triton X-100, 1% SDS) supplemented with protease inhibitors (Roche). After a 10-minute incubation on ice, cell lysates were centrifuged for 10 minutes at 10,000×g. Supernatants were collected and protein concentrations were determined using Pierce BCA Protein Assay Kit (Thermo Scientific). Bovine serum albumin was used as a protein standard. Lysates were diluted to a total of 20μg of protein in 20μL of lysis buffer, resolved on a 10–20% SDS-PAGE gradient gel, and analyzed as previously described52. The following antibodies were used: anti-GCH1 (Abnova, H00002643-M01, 1:1000 in 5% milk), anti-beta-Actin (GeneTex, GTX109639, 1:1000 in 5% milk), anti-SPR (Proteintech, C828X82, 1:1000 in 5% milk), anti-ACSL4, (GeneTex, GTX100260, 1:1000 in 5% milk), anti-GPX4 (Abcam, ab41787, 1:1000 in 5% milk), anti-GAPDH (GeneTex, GTX627408, 1:2000 in 5% BSA), anti-SFXN1 (Proteintech, 12296-1-AP, 1:1000 in 5% BSA), anti-SFXN3 (Proteintech, 15156-1-AP, 1:1000 in 5% BSA), anti-SLC7A11 (Cell Signaling Technologies, 12691S, 1:1000 in 5% BSA), anti-SHMT2 (Sigma, HPA020549, 1:1000 in 5% BSA), anti-DHFR (ABclonal, A1607, 1:1000 in 5% milk), anti-AGMO (Novus Biologicals, NBP1-70404, 1:1000 in 5% milk), anti-NOS3 (Santa Cruz, sc-376751, 1:500 in 5% BSA), anti-QDPR (ABclonal, A5733, 1:1000 in 5% milk), anti-HA (Cell Signaling Technologies, #2367, 1:1000 in 5% BSA), anti-VDAC (Cell Signaling Technologies, #4661, 1:1000 in 5% BSA), anti-CS (Cell Signaling Technologies, #14309, 1:1000 in 5% BSA), anti-PKM2 (Cell Signaling Technologies, #4053, 1:1000 in 5% BSA), anti-CTH (Proteintech, 12217-1-AP, 1:1000 in 5% BSA), anti-FLAG (Sigma, F1804, 1:1000 in 5% BSA), HRP-conjugated anti-rabbit and anti-mouse (Cell Signaling Technologies, #7074 and #7076, 1:1000 in 5% milk or BSA). All immunoblots were replicated at least two times with similar results. Full scans of immunoblots are provided.
Real Time qPCR Analysis of SLC7A11
HEK 293T cells were collected, washed once with PBS, and RNA was isolated using the RNeasy Kit (QUIGEN). RNA was spectrophometrically quantified and equal amounts were used to synthesize cDNA using Supercript III RT Kit (Invitrogen). qRT-PCR analysis of gene expression was performed using SYBR green Mastermix (Applied Biosystems) and GAPDH was used as a control. The sequences of the primers used for this experiment are available in Supplementary Table 2.
Network Analysis
Pathway enrichment analysis of the top 50 most differentially essential genes from each screen was determined using the online tool MetaboAnalyst53. Gene sets were derived from the REACTOME pathway database. Redundant gene sets and gene sets made up 35 or more genes were excluded.
Immunoprecipitation and Proteomics
For FLAG-SFXN1 immunoprecipitation followed by mass spectrometry/proteomics, immunoprecipitates were prepared from SFXN1-KO 293T cells expressing 3xFLAG-SFXN1 or 3xFLAG-SLC25A12 (control). For SFXN1/SFXN3 interaction experiments, immunoprecipitation on was performed on SFXN1_KO 293T cells expressing FLAG-SFXN1 cDNA or HA-SFXN3 cDNA. Prior to cell lysis (~30e6 cells) proteins were crosslinked with cell permeable crosslinking agent dithiobis (succinimidyl propionate) (2.5mM) for 15 minutes and quenched with 1M Tris pH 8.5 for 2 minutes. Cells were washed in cold isotonic (150mM NaCl) TBS and lysed in TBS 150 mM NaCl, 1 mM EDTA, 1% DDM. For downstream proteomics experiments, lysates were incubated with 50uL of a 50% slurry of pre-washed Ezview Red ANTI-FLAG M2 Affinity Gel (Sigma Aldrich, F2426) at 4C with rocking for 2–4 hours. For SFXN1 / SFXN 3 interaction experiments, lysates were incubated with 50uL of a 50% slurry of pre-washed anti-FLAG magnetic beads (Sigma-Aldrich M8823) or anti-HA magnetic beads (Thermo Fisher Scientific 88837) at 4C with rocking for 2–4 hours. Beads were washed twice in TBS, 150 mM NaCl, 0. 1% DDM and once with hypertonic (300m) TBS. For downstream proteomics, proteins were eluted off beads using 3x FLAG peptide (50ng/ul) in hypertonic TBS. For interaction experiments, proteins were elutated off beads for 10 minutes by acid elution in 100uL 0.1M glycine, pH 2.0, 1% DDM and followed by neutralization by adding 15ul of 1M Tris pH 8.5. Interaction of FLAG-SFXN1 and HA-SFXN3 was ascertained by immunoblotting as above. Following immunoprecipitation FLAG-SFXN and FLAG-SLC25A12 elutates, were prepared for mass spectrometry as described previously21. Briefly, following cold acetone precipitation, reduction (DTT 10mM), alkylation (40mM iodoacetamide), denaturation (8M urea) and LysC (Wako) and trypsin (Promega) digest, samples were desalted and run on a reversed phase nano-LC-MS/MS using a Q-Exactive Plus (Thermo Scientific).
Data were quantified and searched against a Uniprot human database (March 2016) using ProteomeDiscoverer v.1.4.0.288 (Thermo Scientific) combined with Mascot v. 2.5.1 (Matrix Science). Oxidation of methionine and protein N-terminal acetylation were allowed as variable modifications and all cysteines where treated as carbamidomethylated. Peptide matches were filtered using a Percolator with a calculated false discovery rate of 1%. Contaminants are further removed by comparison to proteins in the CRAPome v1.154 with an average SC of 0.2 and above and a FLAG-GFP control immunoprecipitation.
Rapid Mitochondrial Purification for Metabolite Profiling
Mitochondria were purified from HEK 293T cell expressing HA-OPM25 (mitochondrial isolation) or myc-OPM25 (background control) according to previously described protocol55. Briefly, 30 million cells were collected and washed twice with cold PBS, scraped into 1mL of cold KPBS, and pelleted via centrifugation at 1000×g for 1.5 minutes at 4°C. Cells were resuspended in 1mL of KPBS, 10mL of cells were transferred into 50mL of 1% triton lysis buffer for a whole cell protein sample and 10mL of cells were transferred into 50mL of 80% methanol, containing heavy labeled amino acid standards, for direct extraction of whole cell metabolites. With one set of 20 strokes and another set of 10 strokes, the remaining sample was homogenized using a 2mL homogenizer. After centrifugation, the homogenate was incubated with 200mL of KPBS pre-washed anti-HA magnetic beads on a rotator shaker for 5 minutes at 4°C. Beads were washed 3 times in cold KPBS. 10% of bead volume was lysed with 1% triton buffer for protein extracts and remaining 90% was extracted in 80% methanol containing heavy labeled amino acid standards on a rotator shaker for 10 min at 4°C. Samples were spun down at 20,000×g to remove potential cellular debris or bead contamination prior to LC-MS polar metabolite profiling.
To assess glutathione synthesis from cysteine, cells were cultured in cysteine free media supplemented with 200μM 13C3-Cysteine 24 hours prior to metabolite extraction as detailed above.
Polar Metabolite Profiling
For profiling of polar metabolites, cells were plated 24 hours prior to collection in triplicate at half-max confluency in 6-well plates. Cells were washed three times with 1mL of ice-cold 0.9% NaCl and polar metabolites were extracted in 600μL of ice-cold 80% LC/MS grade methanol containing internal standards (MSK-A2-1.2, Cambridge Isotope Laboratories, Inc.) and 250μM ascorbic acid and 0.5% dithiothreitol to maintain BH4 in its reduced state. Samples were subjected to 10 minutes of vortexing at 4°C and then centrifuged for 10 minutes at 10,000×g and 4°C. The supernatant was collected and samples were nitrogen-dried and stored at −80°C until analyzed.
Polar metabolites were separated on a ZIC-pHILIC 150 × 2.1mm (5μm particle size) column (EMD Millipore) connected to a Thermo Vanquish UPLC system UPLC system and a Q Exactive benchtop orbitrap mass spectrometer equipped with a heated electrospray ionization (HESI) probe. Dried polar samples were resuspended in 6μl of 50% acetonitrile, vortexed for 10 seconds, centrifuged for 15 minutes at (20,000g, 4°C) and 5μl of the supernatant was injected onto the LC-MS in a randomized sequence. Mobile phase A consisted of 20mM ammonium carbonate with 0.1% (v/v) ammonium hydroxide (adjusted to pH 9.3) and mobile phase B was acetonitrile. Chromatographic separation was achieved using the following gradient (flow rate set at 0.15 mL/min): gradient from 90% to 40% B (0–22min), held at 40% B (22–24min), returned to 90% B (24–24.1min), equilibrating at 90% B (24.1–30min). The mass spectrometer was operated in polarity switching mode for both full MS and DDA scans; spray voltage, 3.0kV; capillary temperature, 275°C; source temperature, 250°C; sheath gas flow, 40a.u. auxiliary gas flow, 15a.u. The full MS scan was acquired as such; 70,000 resolution, 1×106 AGC target, 80 ms max injection time, scan range 55–825m/z. The data-dependent MS/MS scans were acquired at a resolution of 17,500, 1×105 AGC target, 50 ms max injection time, 1.6Da isolation width, stepwise normalized collision energy (NCE) of 20, 30, 40 units, 8sec dynamic exclusion, and loop count of 2.
Relative quantitation of polar metabolites was performed using Skyline Daily56 (v.20.1.1.158) with the maximum mass and retention time tolerance set to 2ppm and 12sec, respectively, referencing an in-house library of chemical standards. Metabolite levels were normalized to the total protein amount for each condition.
Lipid Profiling
For lipid metabolite profiling experiments, 3 million wild type and SPR-KO Jurkat cells were cultured in triplicates in 6-well plates and left untreated or treated for 24 hours with 250nM RSL3. After, cells were collected and washed three times with 1mL of ice-cold 0.9% NaCl and polar metabolites were extracted in 600μL of ice-cold 80% LC/MS grade methanol and consecutive addition of 250μL LC/MS grade water followed by 400μL of LC/MS grade chloroform. Samples were vortexed for 10 minutes at 4°C and centrifuged for 10 minutes at 10,000×g and 4°C. After, 350mL of the lower lipid-containing layer was carefully collected and dried under nitrogen. Dried lipid extracts were stored at 80°C until LC/MS analysis.
Lipids were separated on an Ascentis Express C18 2.1mm × 150mm × 2.7μm particle size column (Supelco) connected to a Vanquish UPLC system and a Q Exactive benchtop orbitrap mass spectrometer (Thermo Fisher Scientific), equipped with a heated electrospray ionization (HESI) probe. Dried lipid extracts were reconstituted in 50μl of 65:30:5 acetonitrile:isopropanol:water (v/v/v), vortexed for 10sec, centrifuged for 10min (20,000 g ,4°C) and 5μl of the supernatant was injected into the LC-MS in a randomized order, with separate injections for positive and negative ionization modes. Mobile phase A consisted of 10mM ammonium formate in 60:40 water: acetonitrile (v/v) with 0.1% formic acid, and mobile phase B consisted of 10mM ammonium formate in 90:10 isopropanol:acetonitrile (v/v) with 0.1% formic acid. Chromatographic separation was achieved using the previously described gradient57. The column oven and autosampler were held at 55°C and 4°C, respectively. The mass spectrometer was operated with the following parameters; positive or negative ion polarity; spray voltage, 3500V; heated capillary temperature, 285°C; source temperature, 250°C; sheath gas, 60a.u.; auxiliary gas, 20a.u. External mass calibration was performed every five days using the standard calibration mixture. Mass spectra were acquired, in both positive and negative ionization modes, using a Top15 data-dependent MS/MS method. The full MS scan was acquired as such; 70,000 resolution, 1×106 AGC target, 250ms max injection time, scan range 200–2,000m/z. The data-dependent MS/MS scans were acquired at a resolution of 17,500 AGC target of 1×105, 75ms max injection time, 1.0Da isolation width, stepwise normalized collision energy (NCE) of 20, 30, 40 units and 8sec dynamic exclusion. High-throughput identification and relative quantification of lipids was performed separately for positive and negative ionization mode data, using LipidSearch58,59 software (v.4.2.21, Thermo Fisher Scientific/Mitsui Knowledge Industries) and the default parameters for Q Exactive product search and alignment.
Raw peak areas for all identified lipids were exported to Microsoft Excel and filtered according to the following predetermined quality control criteria: Rej (“Reject” parameter calculated by LipidSearch) equal to 0; PQ (“Peak Quality” parameter calculated by LipidSearch software) greater than 0.85; CV (standard deviation / mean peak area across triplicate injections of a represented (pooled) biological sample) below 0.4; R (linear correlation across a three-point dilution series of the representative (pooled) biological sample) greater than 0.9. Typically, approximately 70% of identified lipids passed all four quality control criteria. Raw peak areas of the filtered lipids were added together to create a total lipid signal for each sample, and individual lipid peak areas were normalized to this total signal as a control for lipid extraction efficiency, as well as sample loading. Lipid groups with CV more than 0.15 were then excluded.
C11-BODIPY FACS Analysis
18 hours prior to collection, Jurkat cells were plated at 250,000 cells/well in 6-well plates. Cells were treated with 175nM RSL3 with or without 50μM BH2 for 15 hours. Cells were collected and washed twice with HBSS (Corning) before incubating in 250μL of 1μM BODIPY 581/591 C11 in HBSS for 15 minutes at 37°C in the dark. Then, cells were collected, washed twice with ice cold HBSS, resuspended in HBSS with 50ng/mL DAPI, and filtered into FACS tubes. Flow cytometry data were collected on a BD LSR II Flow Cytometer (BD Biosciences) by using an excitation wavelength of 488nm and the FL1 collection channel. FCS Express v7 software was used to include only live singlet cells in the analysis. The gating strategy used in this experiment is available in Supplementary Figure 1.
FENIX Assay of EggPC liposomes
These experiments were carried out in a manner described previously32. In brief, EggPC liposomes (extruded to 100nm, 1.031mM), STY-BODIPY (1.031μM) and either an aliquot of α-TOH (5.15μM), Coenzyme Q10 (515 μM) or vehicle (EtOH) were vortexed in PBS (10mM, 150mM NaCl, pH 7.4). 291μL aliquots of liposomes were incubated in 96-well plates (polypropylene, Nunc) in a BioTek Synergy H1 plate reader at 37°C for 20 mins. Thereafter, 3μL aliquots of 100× enzyme (PBS), cofactor mixture (PBS), DTUN (EtOH), or the appropriate vehicle were promptly added. The plate was vigorously mixed for 5 min and kinetic data of [STY-BODIPYox] was acquired at λex = 488 nm and λem = 518 nm as described32. Rate constants and stoichiometry were determined from equations shown in Figure 5e. The data shown are representative results of three independent experiments.
Statistical Analyses and Reproducibility
Sample size, mean, and P values are indicated in the text and figure legends. Error bars represent standard deviation (SD) from triplicate biological samples. Except were specified, all P values were determined by unpaired t-test corrected for multiple comparisons using the Holm-Sidak method. Analyses were performed using GraphPad Prism 7. All experiments reported were independently replicated at least three times with similar results.
Data Availability
All data generated for and reported in this study are available from the corresponding authors upon request. Source data for Figures 1–6 and Extended Data Figures 1–7 are provided with the paper. Gene scores obtained from each screen are available in Supplementary Dataset 1. The Uniprot human database used for proteomic analysis is publicly available at https://www.uniprot.org/proteomes/UP000005640. The REACTOME pathway database is publicly available at https://reactome.org/. The gene expression data in Extended Data Figure 7a–b is publicly available at https://depmap.org/portal/. Data plotted in Extended Data Figure 5d is publicly available at https://portals.broadinstitute.org/ctrp.v2.1/.
Extended Data
Extended Data Figure 1: Cystine depletion and GPX4 inhibition may impact cell proliferation via distinct mechanisms.
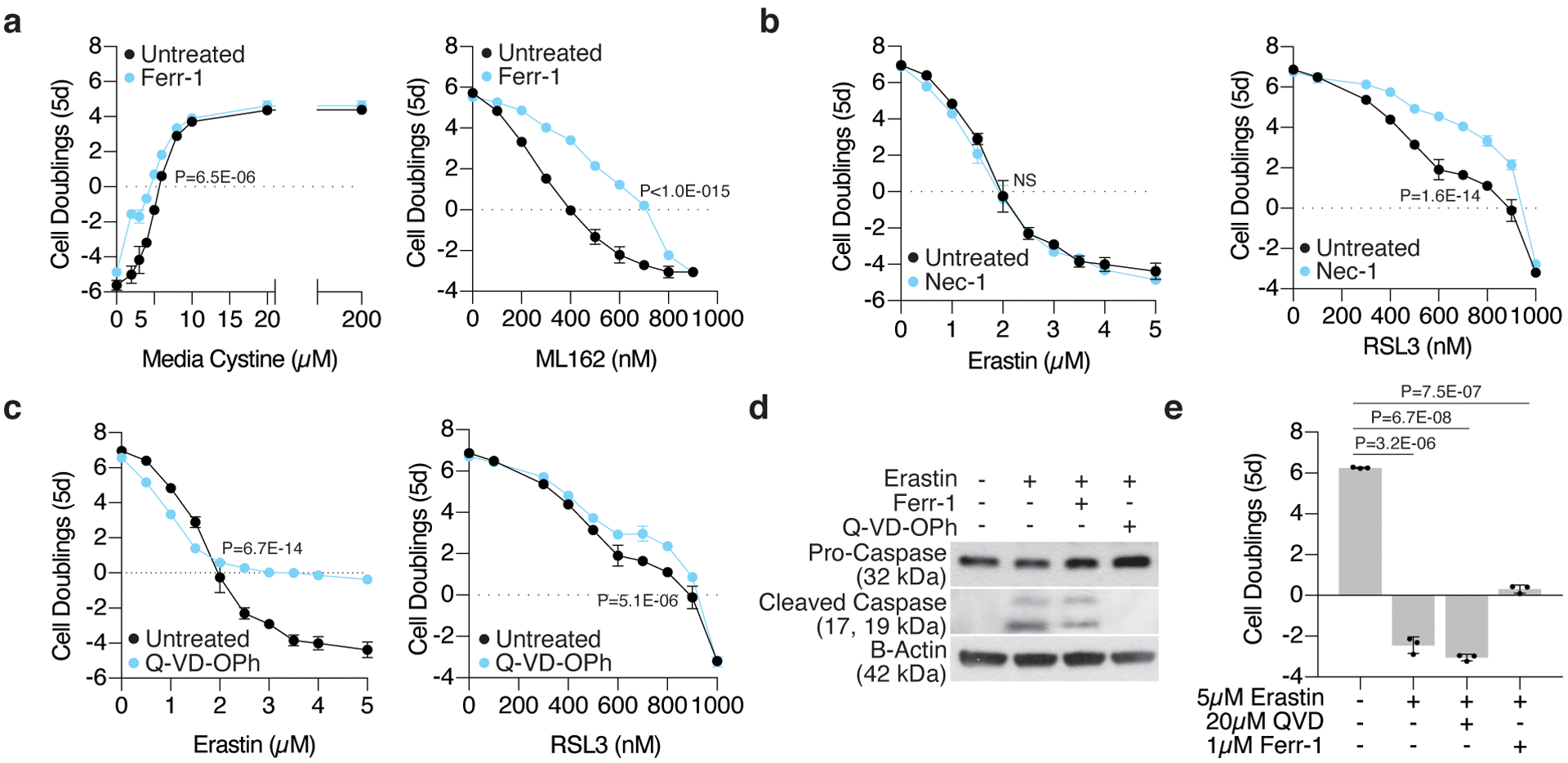
(A) Fold change in cell number (log2) of wild type Jurkat cells cultured with increasing concentrations of cystine (left, black) or ML162 (right, black) and cotreated with ferrostatin-1 (blue, Ferr-1, 1μM). Data shown as mean ± SD, n=3 biological replicates.
(B) Fold change in cell number (log2) of wild type Jurkat cells treated with erastin (right, black) or RSL3 (left, black) and co-treated with necrostatin-1 (blue, Nec-1, 10μM). Data shown as mean ± SD, n=3 biological replicates.
(C) Fold change in cell number (log2) of wild type Jurkat cells treated with erastin (right, black) or RSL3 (left, black) and co-treated with Q-VD-OPh (blue, 20μM). Data shown as mean ± SD, n=3 biological replicates.
(D) Representative immunoblot analysis of procaspase-3 (top) and cleaved caspase-3 (middle) in wild type Jurkat cells left untreated or treated for 12 hours with erastin (50μM) alone or cotreated with ferrostatin-1 (Ferr-1, 1μM) or Q-VD-OPh (20μM). B-actin was used as a loading control (bottom).
(E) Fold change in cell number (log2) of wild type MIA PaCa-2 cells treated with erastin (5μM) and co-treated with Q-VD-OPh (20μM) or ferrostatin-1 (Ferr-1, 1μM). Data shown as mean ± SD, n=3 biological replicates.
Extended Data Figure 2: CRISPR-Cas9 genetic screens identify metabolic regulators of the cellular response to cystine depletion and GPX4 inhibition.
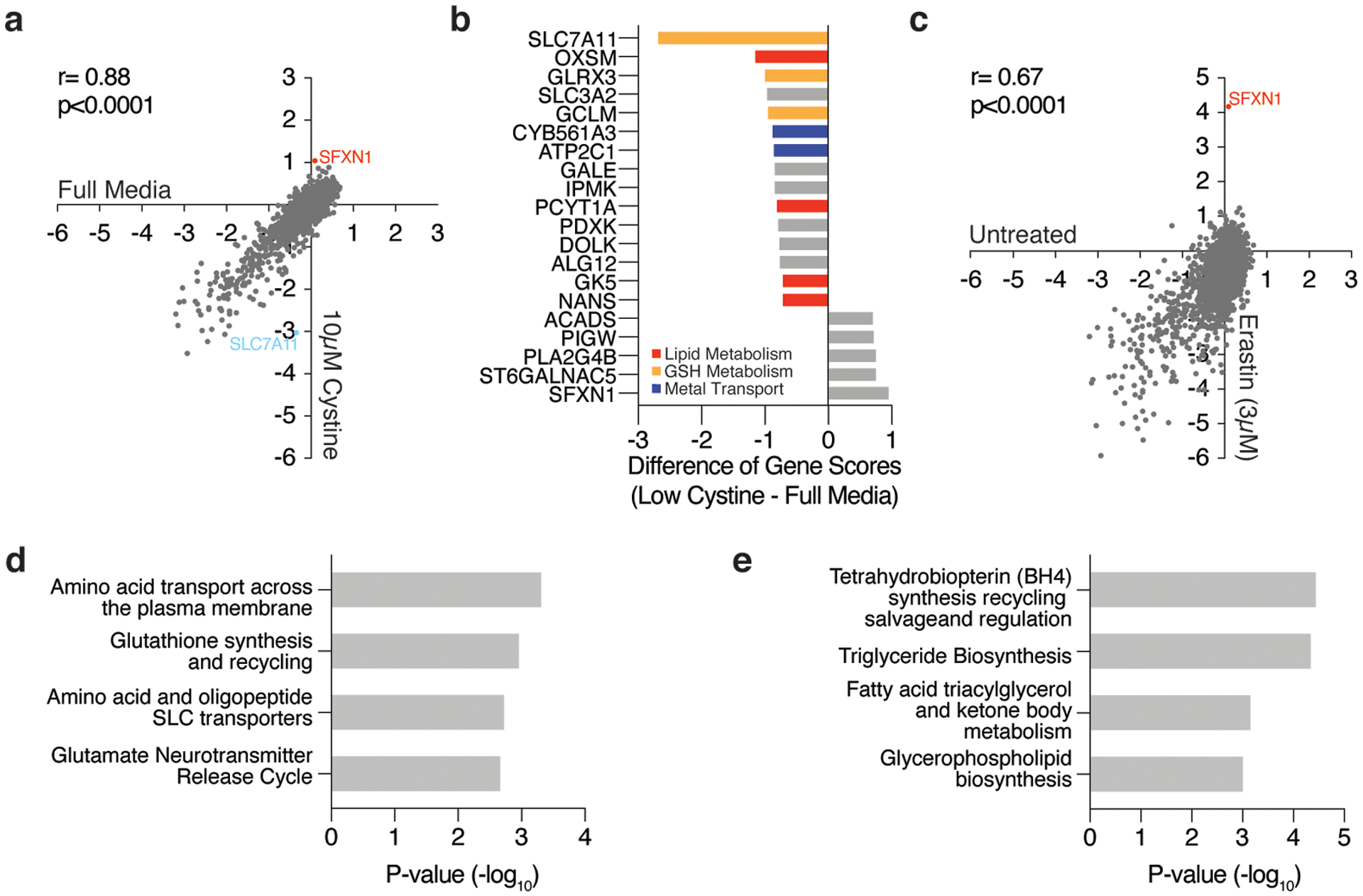
(A) Gene scores of Jurkat cells left untreated (x-axis) or cultured in low cystine (10μM, y-axis).
(B) Top-scoring genes under low cystine conditions. Negative scores represent genes whose loss potentiates low cystine toxicity while positive scores represent genes whose loss provides resistance to low cystine.
(C) Gene scores of untreated (x-axis) and erastin treated (3μM, y-axis) Jurkat cells.
(D) Significantly enriched pathways (REACTOME) represented within the top 50 most essential genes in the low dose erastin screen.
(E) Significantly enriched pathways (REACTOME) represented within the top 50 most essential genes in the RSL3 screen.
Extended Data Figure 3: SFXN1 loss enables cell proliferation under cystine depletion.
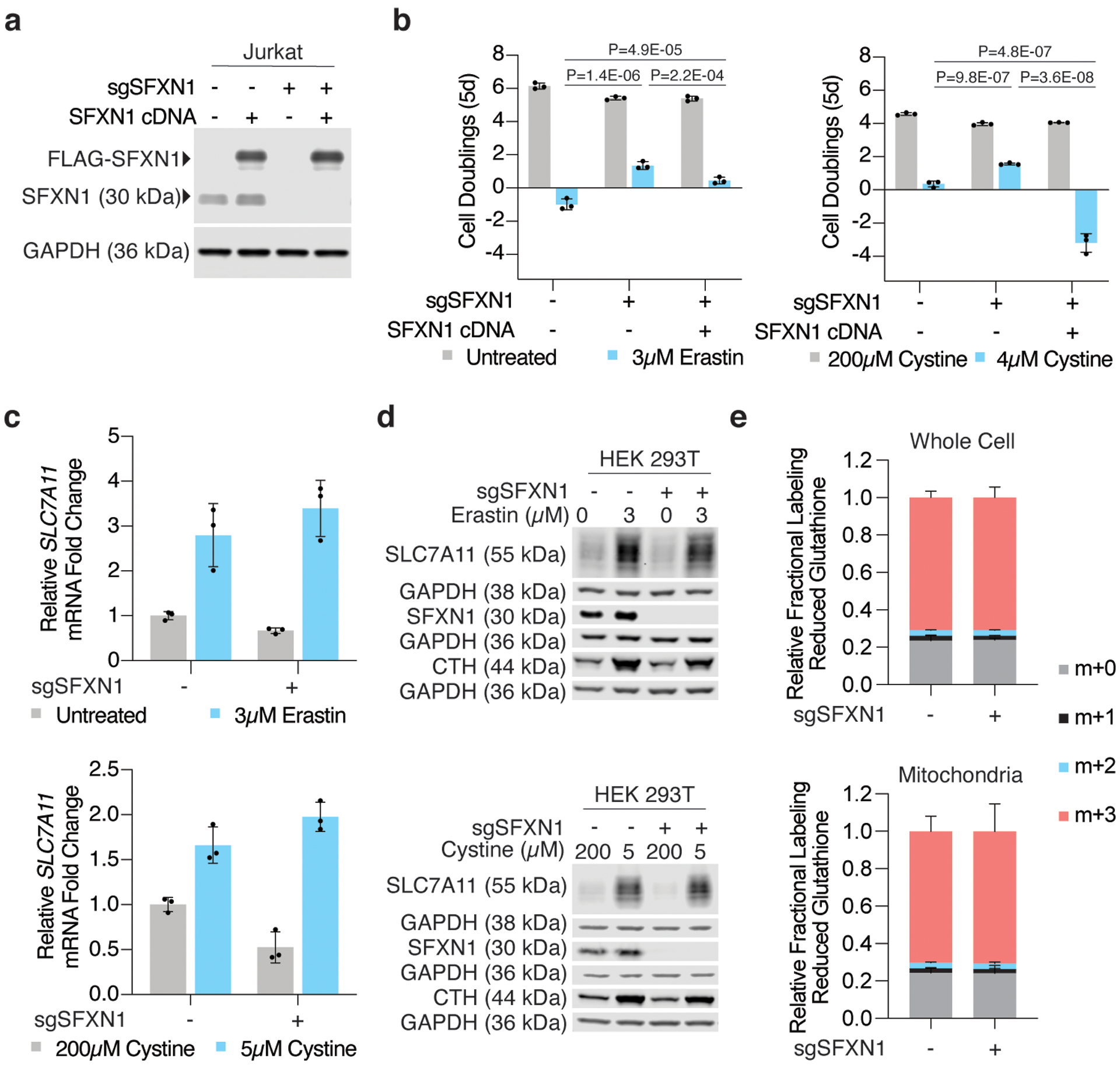
(A) Representative immunoblot analysis of SFXN1 in wild type, SFXN1 knockout, and FLAG-SFXN1 cDNA expressing Jurkat cells (top). GAPDH was used as a loading control (bottom).
(B) Fold change in cell number (log2) of wild type, SFXN1 knockout, and FLAG-SFXN1 cDNA expressing Jurkat cells left untreated (gray) or treated with erastin (left, blue, 3μM) or cultured in low cystine (right, blue, 4μM). Data shown as mean ± SD, n=3 biological replicates.
(C) Relative fold change in SLC7A11 mRNA transcripts in HEK 293T cells left untreated (gray) or treated with erastin (top, blue, 3μM for 24 hours) or cultured in cystine-depleted media (bottom, blue, 5μM for 24 hours). Data shown as mean ± SD, n=3,
(D) Representative immunoblot analysis of SLC7A11 in wild type or SFXN1-null HEK 293T cells left untreated or treated with erastin (top, 3μM for 24 hours) or cultured in cystine-depleted media (bottom, 5μM for 24 hours). CTH was used as a marker of cysteine depletion and GAPDH was used as a loading control.
(E) Fractional labeling of glutathione from 13C3-labeled cysteine in whole cell and mitochondrial lysates obtained from HEK 293T cells. Data shown as mean ± SD, n=3 biological replicates.
Extended Data Figure 4: Characterization of SFXN1 knockout cells.
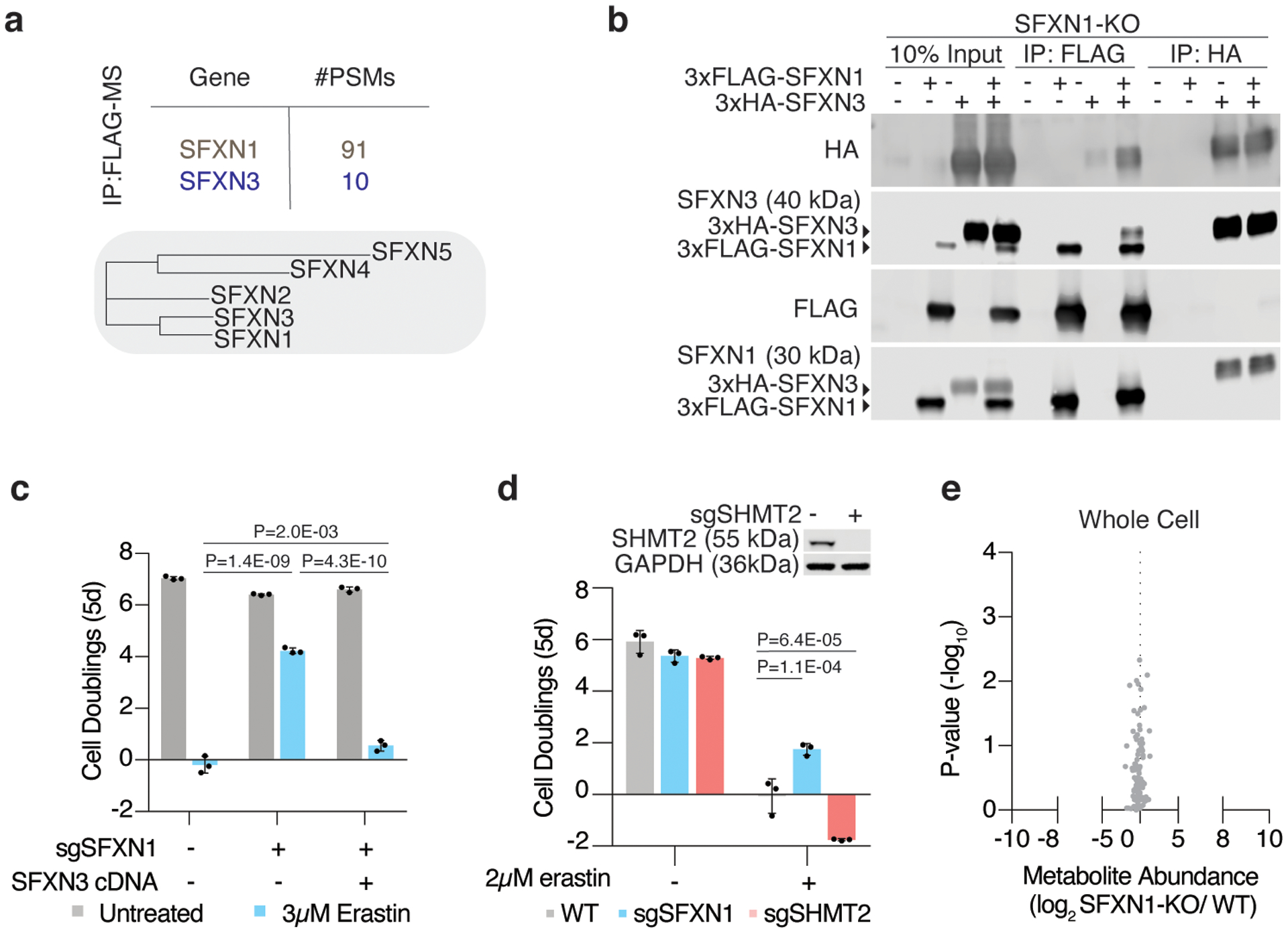
(A) Phylogenic tree of the five human sideroflexin paralogs generated from BIONJ clustering using uncorrected pairwise differences from uncorrected pairwise differences of Clustal Omega alignments. From these analyses, SFXN3 is the closest paralog to SFXN1. Mass spectrometric analyses on 3xFLAG-SFXN1 immunoprecipitants, revealed protein-protein interactions with SFXN1 and SFXN3.
(B) Representative immunoblot analyses of various SFXN1 and SFXN3 constructs from input, FLAG, and HA immunoprecipitants of SFXN1 knockout HEK 293T cells.
(C) Fold change in cell number (log2) of wild type, SFXN1 knockout, and HA-SFXN3 cDNA expressing Jurkat cells untreated (gray) or treated with erastin (blue, 3μM). Data shown as mean ± SD, n=3 biological replicates.
(D) Representative immunoblot analysis of SHMT2 in wild type and SHMT2 knockout Jurkat cells (top). Fold change in cell number (log2) of wild type (gray), SFXN1 knockout (blue), and SHMT2 knockout (red) Jurkat cells untreated or treated with erastin (2μM). Data shown as mean ± SD, n=3 biological replicates.
(E) Abundance of 85 polar metabolites in whole cell lysates of HEK 293T cells. Data shown as the ratio of metabolite abundance in SFXN1-null cells to wild type cells (log2, x-axis) versus significance of difference between the median abundances of each metabolite in both groups (−log10p-value, y-axis).
Extended Data Figure 5: BH4 availability determines cancer cell sensitivity to ferroptosis.
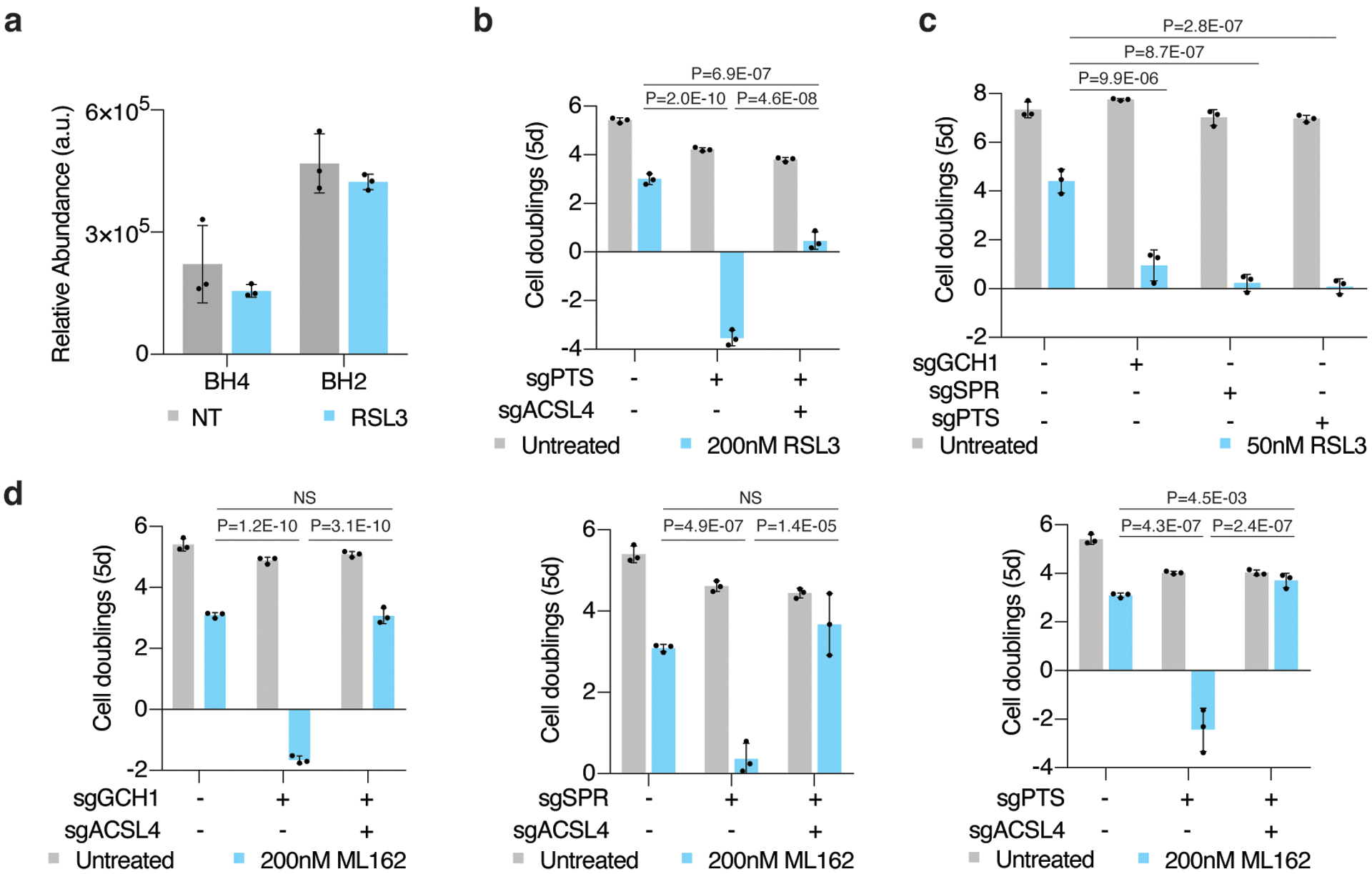
(A)Relative abundance of BH4 and its oxidation product, BH2, in wild type Jurkat cells left untreated (gray) or treated with RSL3 (blue, 175nM for 15 hours). Data shown as mean ± SD, n=3 biological replicates.
(B) Fold change in cell number (log2) of wild type, PTS knockout, and PTS/ACSL4 double knockout Jurkat cells untreated (gray) or treated with 200nM RSL3 (blue). Data shown as mean ± SD, n=3 biological replicates.
(C) Fold change in cell number (log2) of wild type, GCH1, SPR, and PTS knockout Karpas-299 cells untreated (gray) or treated with 50nM RSL3 (blue). Data shown as mean ± SD, n=3 biological replicates.
(D) Fold change in cell number (log2) of wild type, GCH1 (left), SPR (middle), and PTS (right) knockout, and respective ACSL4 double knockout Jurkat cells left untreated (gray) or treated with 200nM ML162 (blue). Data shown as mean ± SD, n=3 biological replicates.
Extended Data Figure 6: GCH1 expression predicts dependence on BH4 upon ferroptosis induction.
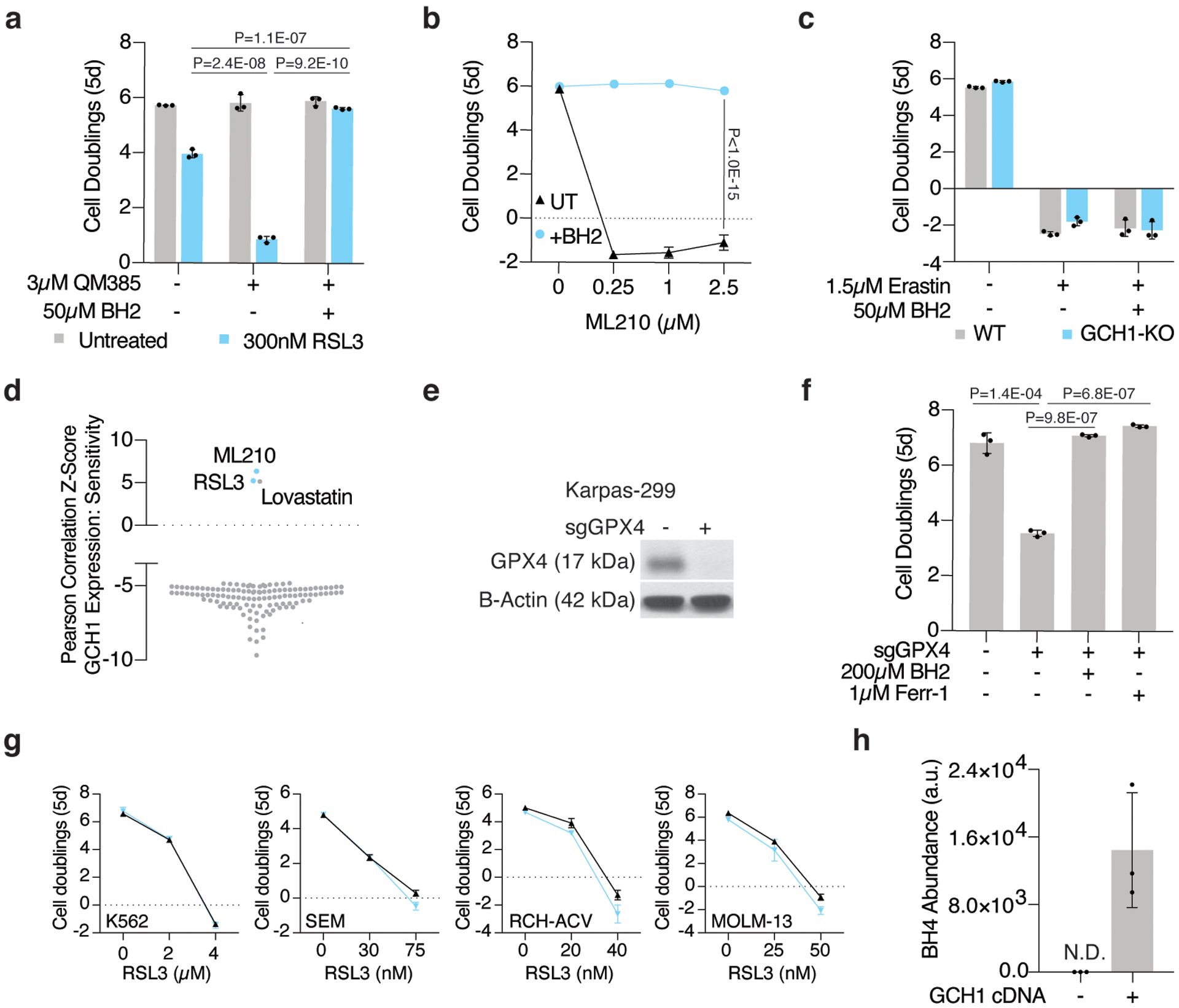
(A) Fold change in cell number (log2) of wild type Jurkat cells left untreated (gray) or treated with RSL3 (300nM, blue), cotreated with or without QM385 (3μM) and supplemented with BH2 (50μM). Data shown as mean ± SD, n=3 biological replicates.
(B) Fold change in cell number (log2) of wild type Karpas-299 cells treated with ML210 only (black trace) or supplemented with BH2 (blue trace, 50μM). Data shown as mean ± SD, n=3 biological replicates.
(C) Fold change in cell number (log2) of wild type (gray) and GCH1-KO (blue) Jurkat cells treated with erastin (1.5μM) and supplemented with BH2 (50μM). Data shown as mean ± SD, n=3 biological replicates.
(D) Z-scores of correlations between GCH1 mRNA levels and resistance to small molecule probes and drugs across cancer cell lines (CTRP v2, 2015).
(E) Representative immunoblot analysis of GPX4 in wild type and GPX4 knockout Karpas-299 cells. B-actin was used as a loading control.
(F) Fold change in cell number (log2) of wild type and GPX4 knockout Karpas-299 cells supplemented with BH2 (200μM) or ferrostatin-1 (Ferr-1, 1μM). Data shown as mean ± SD, n=3 biological replicates.
(G) Fold change in cell number (log2) of a panel of cancer cell lines that are not sensitive to GPX4 inhibition upon BH4 depletion (blue traces, cotreated with 3μM QM385). Data shown as mean ± SD, n=3 biological replicates.
(H) BH4 abundance in wild type and GCH1 over-expressing A375 cells. Data shown as mean ± SD, n=3 biological replicates.
Extended Data Figure 7: BH4 protects cells from ferroptosis in an enzyme-independent manner.
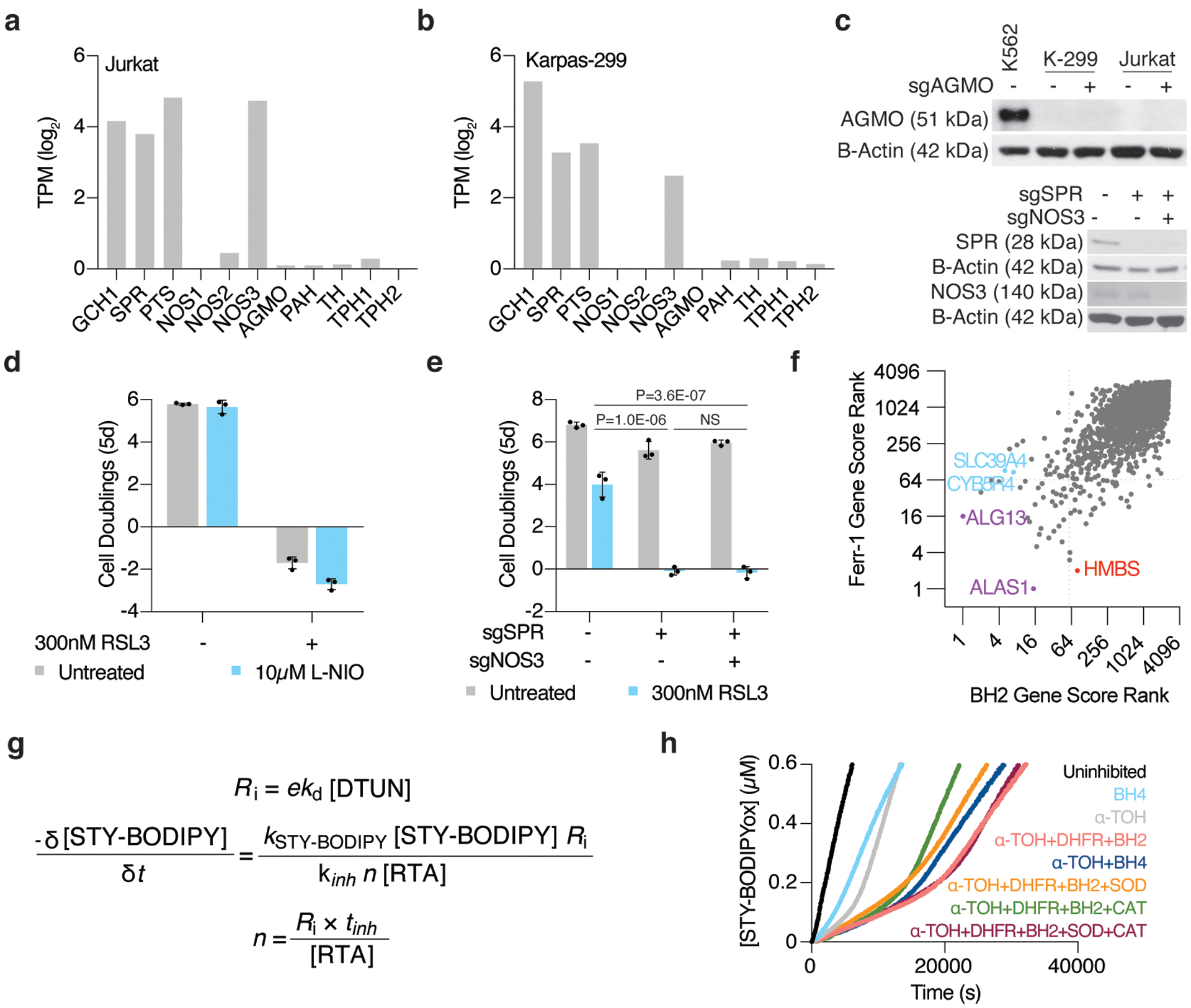
(A) RNAseq gene expression data for BH4-associated enzymes in Jurkat cells. Data shown as log2transcript per million (TPM, DepMap).
(B) RNAseq gene expression data for BH4-associated enzymes in Karpas-299 cells. Data shown as log2transcript per million (TPM, DepMap).
(C) Representative immunoblot analysis of AGMO in Jurkat and Karpas-299 (K-299) cells (top) and of NOS3 in Jurkat cells (bottom). B-actin was used as a loading control and K562 cells were used as a positive control for AGMO expression.
(D) Fold change in cell number (log2) and GCH1 knockout Jurkat cells left untreated (gray) or treated with L-NIO (blue, 10μM) cotreated with or without RSL3 (300nM). Data shown as mean ± SD, n=3 biological replicates.
(E) Fold change in cell number (log2) of wild type, SPR knockout, and SPR/NOS3 double knockout Jurkat cells left untreated (gray) or treated RSL3 (blue, 300nM). Data shown as mean ± SD, n=3 biological replicates.
(F) Comparison of gene score ranks from BH2 and ferrostatin-1 (Ferr-1) rescue screens in Karpas-299 cells. Unique hits (p<0.01) in the BH2 screen are highlighted (blue) in quadrant II, shared hits are highlighted (purple) in quadrant III, and unique hits in the ferrostatin-1 screen are highlighted (red) in quadrant IV.
(G) The initial rate and inhibited period of STY-BODIPY consumption is used to derive the rate constant and stoichiometry of added RTAs.
(H) Representative autoxidations of STY-BODIPY (1μM)-embedded liposomes of egg phosphatidylcholine lipids (1mM, ~100nm particle size) suspended in phosphate-buffered saline pH 7.4 at 37°C initiated by 0.2mM DTUN in the presence of NADPH (60μM) with BH4 (4μM), BH2, α-tocopherol (5μM), DHFR (50nM), and 50 U/mL each of superoxide dismutase (SOD) and catalase (CAT).
Extended Data Figure 8: Possible mechanisms for lipid peroxyl radical trapping by BH4.
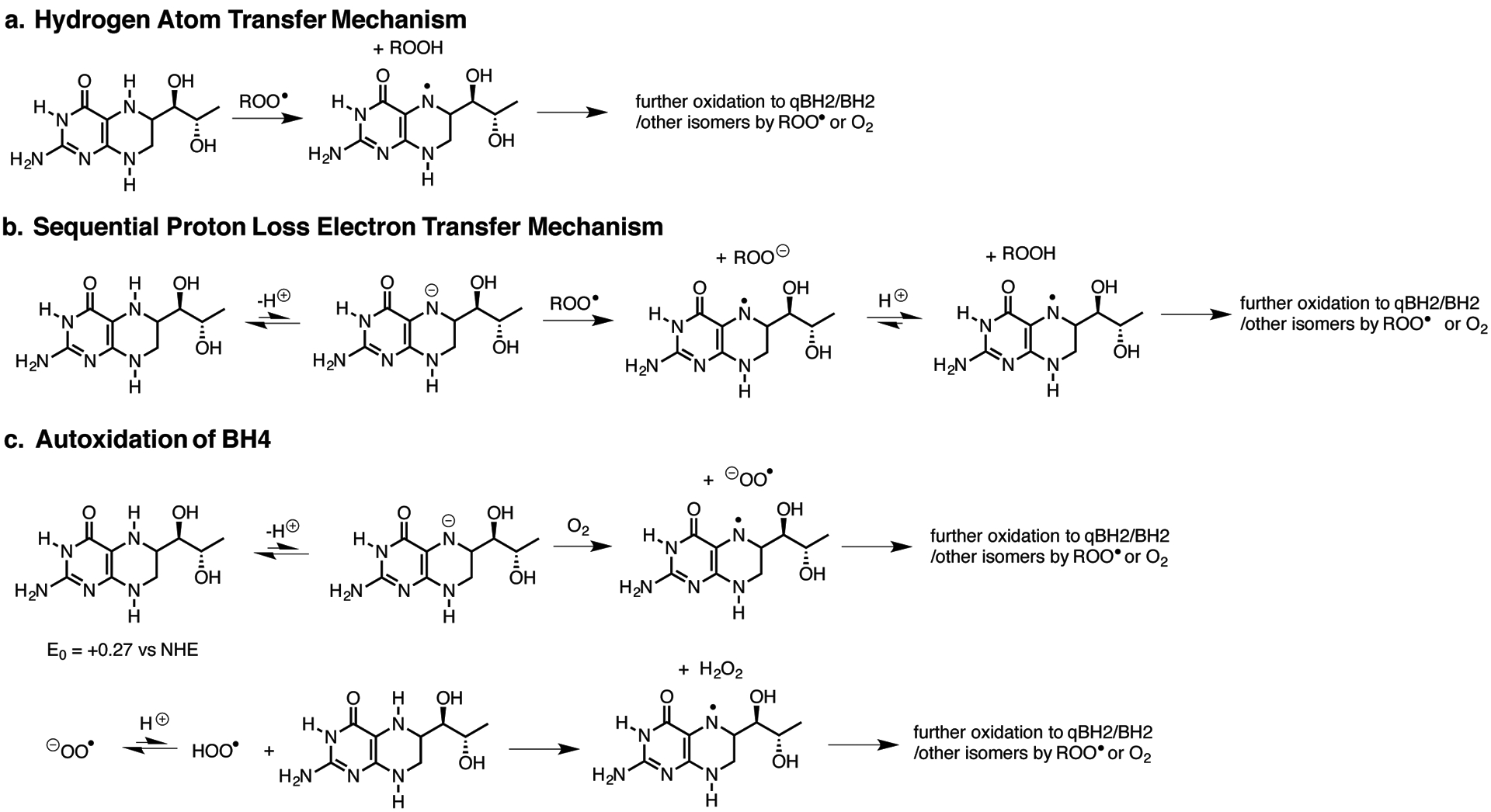
(A) Hydrogen atom transfer from BH4 to yield a BH3 (aminyl) radical that has various fates.
(B) Sequential proton loss electron transfer to yield a BH3 (aminyl) radical that has various fates.
(C) Competing initiation and propagation of BH4 autoxidation.
Extended Data Figure 9: DHFR regenerates BH4 efficiently.
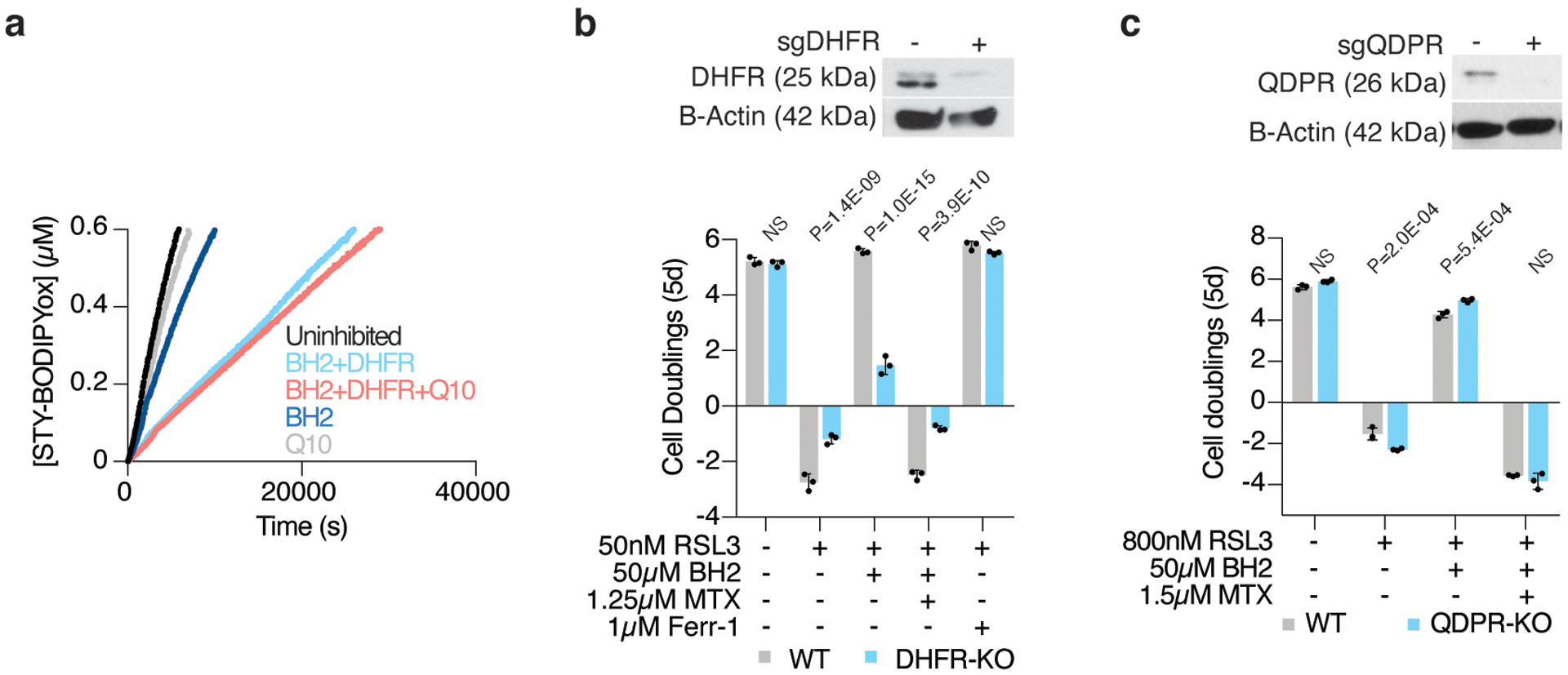
(A) Representative autoxidations of STY-BODIPY (1μM)-embedded liposomes of egg phosphatidylcholine lipids (1mM, ~100nm particle size) suspended in phosphate-buffered saline pH 7.4 at 37°C initiated by 0.2mM DTUN containing 60μM NADPH, BH2 (10μM), CoQ10 (5μM), and DHFR (50nM) as indicated.
(B) Representative immunoblot analysis (top) of DHFR in wild type and DHFR knockout Karpas-299 cells. B-actin was used as a loading control. Fold change in cell number (bottom, log2) of wild type (gray) and DHFR knockout (blue) Karpas-299 cells left untreated or treated with RSL3 (50nM), cotreated with or without methotrexate (MTX, 1.25μM), and supplemented with BH2 (50μM) or ferrostatin-1 (Ferr-1, 1μM). Data shown as mean ± SD, n=3 biological replicates.
(C) Representative immunoblot analysis (top) of QDPR in wild type and QDPR knockout Jurkat cells. B-actin was used as a loading control. Fold change in cell number (bottom, log2) of wild type (gray) and QDPR knockout (blue) Jurkat cells left untreated or treated with RSL3 (800nM), cotreated with or without methotrexate (MTX, 1.5μM), and supplemented with BH2 (50μM). Data shown as mean ± SD, n=3 biological replicates.
Supplementary Material
Acknowledgements
We thank all members of the Birsoy Lab for helpful suggestions. We would like to thank members of the Rockefeller University Genomics Resource Center, Proteomics Resource Center, and the Flow Cytometry Resource Center for their assistance. We also thank Dr. Kai Johnsson (Max Planck Institute of Medical Research) and Dr. Clifford Woolf (Boston Children’s Hospital) for their generous gift of QM385. Research reported in this publication was supported by the National Institute of General Medical Sciences of the National Institutes of Health (T32GM066699; M.S.) and by the NIH Director’s New Innovator Award (DP2 CA228042-01; K.B.), Pershing Square Sohn Foundation (K.B.), AACR NextGen Grant (K.B.), the Natural Sciences and Engineering Research Council of Canada (D.A.P) and Canada Foundation for Innovation (D.A.P). J.G.B. is a Special Fellow of the Leukemia & Lymphoma Society. K.B. is a Searle Scholar, Pew-Stewart Scholar and Sidney Kimmel Scholar.
References
- 1.Sies H, Berndt C & Jones DP Oxidative Stress. Annu. Rev. Biochem 86, 715–748 (2017). [DOI] [PubMed] [Google Scholar]
- 2.Yin H, Xu L & Porter NA Free radical lipid peroxidation: mechanisms and analysis. Chem. Rev 111, 5944–5972 (2011). [DOI] [PubMed] [Google Scholar]
- 3.Bersuker K et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 575, 688–692 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Doll S et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature 575, 693–698 (2019). [DOI] [PubMed] [Google Scholar]
- 5.Ingold KU & Pratt DA Advances in Radical-Trapping Antioxidant Chemistry in the 21st Century: A Kinetics and Mechanisms Perspective. Chem. Rev 114, 9022–9046 (2014). [DOI] [PubMed] [Google Scholar]
- 6.Yang WS et al. Regulation of Ferroptotic Cancer Cell Death by GPX4. Cell 156, 317–331 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dixon SJ et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 149, 1060–1072 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Skouta R et al. Ferrostatins inhibit oxidative lipid damage and cell death in diverse disease models. J. Am. Chem. Soc 136, 4551–4556 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cerutti P Prooxidant states and tumor promotion. Science 227, 375–381 (1985). [DOI] [PubMed] [Google Scholar]
- 10.Lei G et al. The role of ferroptosis in ionizing radiation-induced cell death and tumor suppression. Cell Res 30, 146–162 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang W et al. CD8 + T cells regulate tumour ferroptosis during cancer immunotherapy. Nature 569, 270–274 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lu B et al. Identification of PRDX6 as a regulator of ferroptosis. Acta Pharmacol. Sin 40, 1334–1342 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kory N et al. SFXN1 is a mitochondrial serine transporter required for one-carbon metabolism. Science 362, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Conrad M & Pratt DA The chemical basis of ferroptosis. Nat. Chem. Biol 15, 1137–1147 (2019). [DOI] [PubMed] [Google Scholar]
- 15.Doll S et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat. Chem. Biol 13, 91–98 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zou Y et al. A GPX4-dependent cancer cell state underlies the clear-cell morphology and confers sensitivity to ferroptosis. Nat. Commun 10, 1617 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dixon SJ et al. Human Haploid Cell Genetics Reveals Roles for Lipid Metabolism Genes in Nonapoptotic Cell Death. ACS Chem. Biol 10, 1604–1609 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yuki K, Shindou H, Hishikawa D & Shimizu T Characterization of mouse lysophosphatidic acid acyltransferase 3: an enzyme with dual functions in the testis. J. Lipid Res 50, 860–869 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Aguado B & Campbell RD Characterization of a Human Lysophosphatidic Acid Acyltransferase That Is Encoded by a Gene Located in the Class III Region of the Human Major Histocompatibility Complex. J. Biol. Chem 273, 4096–4105 (1998). [DOI] [PubMed] [Google Scholar]
- 20.Dorninger F et al. Homeostasis of phospholipids — The level of phosphatidylethanolamine tightly adapts to changes in ethanolamine plasmalogens. Biochim. Biophys. Acta 1851, 117–128 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhu XG et al. CHP1 Regulates Compartmentalized Glycerolipid Synthesis by Activating GPAT4. Mol. Cell 74, 45–58.e7 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jain IH et al. Genetic Screen for Cell Fitness in High or Low Oxygen Highlights Mitochondrial and Lipid Metabolism. Cell 181, 716–727.e11 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shen R & Zhang Y Reduced Pterins as Scavengers for Reactive Oxygen Species. in Chemistry and Biology of Pteridines and Folates (eds. Ayling JE, Nair MG & Baugh CM) 351–354 (Springer US, 1993). doi: 10.1007/978-1-4615-2960-6_73. [DOI] [PubMed] [Google Scholar]
- 24.Kojima S et al. Antioxidative Activity of 5,6,7,8-Tetrahydrobiopterin and its Inhibitory Effect on Paraquat-Induced Cell Toxicity in Cultured Rat Hepatocytes. Free Radic. Res 23, 419–430 (1995). [DOI] [PubMed] [Google Scholar]
- 25.Kraft VAN et al. GTP Cyclohydrolase 1/Tetrahydrobiopterin Counteract Ferroptosis through Lipid Remodeling. ACS Cent. Sci 6, 41–53 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cronin SJF et al. The metabolite BH4 controls T cell proliferation in autoimmunity and cancer. Nature 563, 564–568 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kagan VE et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat. Chem. Biol 13, 81–90 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rosen GM et al. The role of tetrahydrobiopterin in the regulation of neuronal nitric-oxide synthase-generated superoxide. J. Biol. Chem 277, 40275–40280 (2002). [DOI] [PubMed] [Google Scholar]
- 29.Kapralov AA et al. Redox lipid reprogramming commands susceptibility of macrophages and microglia to ferroptotic death. Nat. Chem. Biol 16, 278–290 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Watschinger K et al. Tetrahydrobiopterin and alkylglycerol monooxygenase substantially alter the murine macrophage lipidome. Proc. Natl. Acad. Sci. U. S. A 112, 2431–2436 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shen RS Inhibition of dopamine autoxidation by tetrahydrobiopterin and NADH in the presence of dihydropteridine reductase. Neurotoxicology 12, 201–208 (1991). [PubMed] [Google Scholar]
- 32.Shah R, Farmer LA, Zilka O, Van Kessel ATM & Pratt DA Beyond DPPH: Use of Fluorescence-Enabled Inhibited Autoxidation to Predict Oxidative Cell Death Rescue. Cell Chem. Biol 26, 1594–1607.e7 (2019). [DOI] [PubMed] [Google Scholar]
- 33.Zilka O et al. On the Mechanism of Cytoprotection by Ferrostatin-1 and Liproxstatin-1 and the Role of Lipid Peroxidation in Ferroptotic Cell Death. ACS Cent. Sci 3, 232–243 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shah R, Shchepinov MS & Pratt DA Resolving the Role of Lipoxygenases in the Initiation and Execution of Ferroptosis. ACS Cent. Sci 4, 387–396 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Haidasz EA, Van Kessel ATM & Pratt DA A Continuous Visible Light Spectrophotometric Approach To Accurately Determine the Reactivity of Radical-Trapping Antioxidants. J. Org. Chem 81, 737–744 (2016). [DOI] [PubMed] [Google Scholar]
- 36.Chauvin J-PR, Griesser M & Pratt DA Hydropersulfides: H-Atom Transfer Agents Par Excellence. J. Am. Chem. Soc 139, 6484–6493 (2017). [DOI] [PubMed] [Google Scholar]
- 37.Valgimigli L et al. The Unusual Reaction of Semiquinone Radicals with Molecular Oxygen. J. Org. Chem 73, 1830–1841 (2008). [DOI] [PubMed] [Google Scholar]
- 38.Zielinski Z, Presseau N, Amorati R, Valgimigli L & Pratt DA Redox Chemistry of Selenenic Acids and the Insight It Brings on Transition State Geometry in the Reactions of Peroxyl Radicals. J. Am. Chem. Soc 136, 1570–1578 (2014). [DOI] [PubMed] [Google Scholar]
- 39.Burton GW & Ingold KU Vitamin E: application of the principles of physical organic chemistry to the exploration of its structure and function. Acc. Chem. Res 19, 194–201 (1986). [Google Scholar]
- 40.Kallen RG & Jencks WP The Dissociation Constants of Tetrahydrofolic Acid. J. Biol. Chem 241, 5845–5850 (1966). [PubMed] [Google Scholar]
- 41.Davis MD & Kaufman S Products of the tyrosine-dependent oxidation of tetrahydrobiopterin by rat liver phenylalanine hydroxylase. Arch. Biochem. Biophys 304, 9–16 (1993). [DOI] [PubMed] [Google Scholar]
- 42.Patel KB, Stratford MRL, Wardman P & Everett SA Oxidation of tetrahydrobiopterin by biological radicals and scavenging of the trihydrobiopterin radical by ascorbate. Free Radic. Biol. Med 32, 203–211 (2002). [DOI] [PubMed] [Google Scholar]
- 43.Armarego WL, Randles D & Waring P Dihydropteridine reductase (DHPR), its cofactors, and its mode of action. Med. Res. Rev 4, 267–321 (1984). [DOI] [PubMed] [Google Scholar]
- 44.Curtius HC et al. Tetrahydrobiopterin biosynthesis. Studies with specifically labeled (2H)NAD(P)H and 2H2O and of the enzymes involved. Eur. J. Biochem 148, 413–419 (1985). [DOI] [PubMed] [Google Scholar]
- 45.Niki E Interaction of ascorbate and alpha-tocopherol. Ann. N. Y. Acad. Sci 498, 186–199 (1987). [DOI] [PubMed] [Google Scholar]
- 46.Garcia-Bermudez J et al. Squalene accumulation in cholesterol auxotrophic lymphomas prevents oxidative cell death. Nature 567, 118–122 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Novera W et al. Cysteine deprivation targets ovarian clear cell carcinoma via oxidative stress and iron-sulfur cluster biogenesis deficit. Antioxid. Redox Signal (2020) doi: 10.1089/ars.2019.7850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cramer SL et al. Systemic depletion of L-cyst(e)ine with cyst(e)inase increases reactive oxygen species and suppresses tumor growth. Nat. Med 23, 120–127 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Aruoma OI, Halliwell B, Hoey BM & Butler J The antioxidant action of taurine, hypotaurine and their metabolic precursors. Biochem. J 256, 251–255 (1988). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sayin VI et al. Activation of the NRF2 antioxidant program generates an imbalance in central carbon metabolism in cancer. eLife 6, e28083 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Eaton JK et al. Selective covalent targeting of GPX4 using masked nitrile-oxide electrophiles. Nat. Chem. Biol 16, 497–506 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Birsoy K et al. An essential role of the mitochondrial electron transport chain in cell proliferation is to enable aspartate synthesis. Cell 162, 540–551 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chong J et al. MetaboAnalyst 4.0: towards more transparent and integrative metabolomics analysis. Nucleic Acids Res 46, W486–W494 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mellacheruvu D et al. The CRAPome: a contaminant repository for affinity purification-mass spectrometry data. Nat. Methods 10, 730–736 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chen WW, Freinkman E, Wang T, Birsoy K & Sabatini DM Absolute quantification of matrix metabolites reveals the dynamics of mitochondrial metabolism. Cell 166, 1324–1337.e11 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pino LK et al. The Skyline ecosystem: Informatics for quantitative mass spectrometry proteomics. Mass Spectrom. Rev (2017) doi: 10.1002/mas.21540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hu C et al. RPLC-Ion-Trap-FTMS Method for Lipid Profiling of Plasma: Method Validation and Application to p53 Mutant Mouse Model. J. Proteome Res 7, 4982–4991 (2008). [DOI] [PubMed] [Google Scholar]
- 58.Taguchi R & Ishikawa M Precise and global identification of phospholipid molecular species by an Orbitrap mass spectrometer and automated search engine Lipid Search. J. Chromatogr. A 1217, 4229–4239 (2010). [DOI] [PubMed] [Google Scholar]
- 59.Yamada T et al. Development of a lipid profiling system using reverse-phase liquid chromatography coupled to high-resolution mass spectrometry with rapid polarity switching and an automated lipid identification software. J. Chromatogr. A 1292, 211–218 (2013). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated for and reported in this study are available from the corresponding authors upon request. Source data for Figures 1–6 and Extended Data Figures 1–7 are provided with the paper. Gene scores obtained from each screen are available in Supplementary Dataset 1. The Uniprot human database used for proteomic analysis is publicly available at https://www.uniprot.org/proteomes/UP000005640. The REACTOME pathway database is publicly available at https://reactome.org/. The gene expression data in Extended Data Figure 7a–b is publicly available at https://depmap.org/portal/. Data plotted in Extended Data Figure 5d is publicly available at https://portals.broadinstitute.org/ctrp.v2.1/.